Submitted:
24 September 2024
Posted:
24 September 2024
You are already at the latest version
Abstract
Ferroptosis is a recently identified iron-dependent programmed cell death with lipid peroxides accumulation and condensation and compaction of the mitochondria. Recent study indicated that ferroptosis played a pivotal role in ischemic cardiac injury with the mechanisms remained largely unknown. This study demonstrated that iron overload occurred in the ischemia/reperfusion (I/R) cardiac tissues, which initiated myocardial ferroptosis. The expression levels of mitochondrial inner membrane protein MPV17 were reduced when ferroptosis occurred in cardiomyocytes treated with high levels of iron or in the ischemic cardiac tissues. Overexpression of MPV17 delivered via adenovirus significantly reduced ferroptosis in both cardiomyocytes subject to high levels of iron and cardiac I/R tissues. Mitochondrial glutathione (mtGSH), crucial for reactive oxygen species (ROS) scavenging and mitochondrial homeostasis maintenance, was depleted during myocardial ferroptosis caused by iron overload. The mechanistic study showed that MPV17 could increase mtGSH levels through maintaining the protein homeostasis of SLC25A10, which was a mitochondrial inner-membrane glutathione transporter. The absence of MPV17 during iron overload resulted in the ubiquitination-dependent degradation of SLC25A10, leading to impaired mitochondrial glutathione import. Moreover, we found that MPV17 was the targeted gene of Nrf2, which played a pivotal role in preventing lipid peroxides accumulation and ferroptosis. The decreased expression levels of Nrf2 led to the inactivation of MPV17 during iron overload-induced myocardial ferroptosis. In summary, this study demonstrated the critical role of MPV17 in protecting cardiomyocytes from ferroptosis and elucidated the Nrf2-MPV17-SLC25A10/mtGSH signaling pathway in the regulation of myocardial ferroptosis.
Keywords:
ischemia/reperfusion injury
; iron overload
; myocardial ferroptosis
; MPV17
; mtGSH
1. Introduction
Cardiac diseases, including the ischemic cardiac injury, were principally caused by myocardial cell death [1,2,3,4]. Ferroptosis was a special form of programmed cell death, initially induced by several small compounds such as erastin or RSL3. This type of cell death is also marked by the lipid peroxides accumulation and the mitochondrial shrinkage [5]. Increasing evidences indicated that ferroptosis promoted cardiac injury during the cardiac diseases, and therefore, targeting ferroptosis represented a promising approach for diseases treatment [6,7]. Nonetheless, the mechanisms regulating myocardial ferroptosis remained poorly understood.
Iron overload is considered a critical factor during cardiac pathological processes, including ischemic cardiac injury [8,9,10,11,12,13]. In both the clinical practices and the experimental mice model, ischemic cardiac injury usually accompanied with the iron overload, which was an active participator in ferroptosis initiation [14,15]. Generally, iron overload was associated with lipid peroxides build-up through the Fenton reaction at the onset of ferroptosis [16,17]. Moreover, alterations in antioxidant status had also been implied in the initiation of ferroptosis during the iron overload related pathological processes [18,19]. In cardiopathy induced by doxorubicin (DOX), increased expression of Hmox1 degrades heme, resulting in the release of iron, consequent iron overload, and subsequent myocardial ferroptosis [20]. Here, we would further explore the signaling pathways regulating iron overload-induced ferroptosis during ischemic cardiac injury.
MPV17, a nuclear-encoded transmembrane protein located within the inner mitochondrial membrane, was known to be involved in mitochondrial DNA replication. Mutation in MPV17 usually led to mitochondrial DNA deletion-associated diseases, such as neurohepatopathy [21,22,23]. The function of MPV17 was also closely related with mitochondrial oxidative phosphorylation, mitochondrial permeability transition pore (MPTP) regulation, and mitochondrial ROS metabolism [24]. Additionally, nuclear magnetic resonance (NMR) structural analysis suggested that MPV17 might act as a scaffold protein, whose function had not been fully understood [25]. The deletion of MPV17 did not significantly impact cardiac function under normal conditions; however, MPV17 could improve cardiac functional recovery following I/R injury [24]. This work aimed to delve into the function of MPV17 in myocardial ferroptosis and its underlying molecular mechanisms.
Mitochondria served not only as energy providers but also as signaling centers in various biological processes. Evidences suggested that dysfunction of mitochondria promoted ferroptosis initiation [26]. The mitochondrial ROS generated by the activity of the α-ketoglutarate dehydrogenase complex was a causative factor for ferroptosis initiation in situations where cysteine was deprived. Depletion of mitochondria through Parkin mediated mitophagy in human fibrosarcoma cells showed high resistance to cysteine deprivation-induced ferroptosis [26]. mtGSH functioned as an antioxidant to eliminate oxidized lipids and maintained redox homeostasis in the mitochondria [27]. Evidences indicated that reduction of mtGSH made cardiomyocytes more sensitive to ferroptotic stimuli [28]. However, the mitochondria could not synthesize GSH but imported GSH from the cytosol where all the cellular GSH were synthesized [29]. Solute carrier family 25 member 10 (SLC25A10) is one of the key mtGSH transporters embedded in the inner mitochondrial membrane, responsible for importing GSH from the cytosol into the mitochondrial matrix [28,30]. The protective role of SLC25A10 had been revealed in RSL3-induced myocardial ferroptosis through importing GSH from cytosol into mitochondria [28]. We supposed that SLC25A10 also functioned in iron overload-induced myocardial ferroptosis.
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a transcriptional factor, which plays an important role in the clearance of lipid peroxides through controlling the expression of antioxidant genes [31]. Numerus studies indicated that Nrf2 played a potent role in the inhibition of ferroptosis and targeting Nrf2 was a promising therapeutic approach for treating ferroptosis-driven diseases [32]. However, its targeted genes had not been fully uncovered. This study explored the regulatory effect of Nrf2 on MPV17 expression during iron overload-induced ferroptosis, aiming to identify potential targets of Nrf2 to further explore the mechanism in the inhibition of ferroptosis.
2. Results
2.1. MPV17 Prevented Iron Overload-Induced Myocardial Ferroptosis
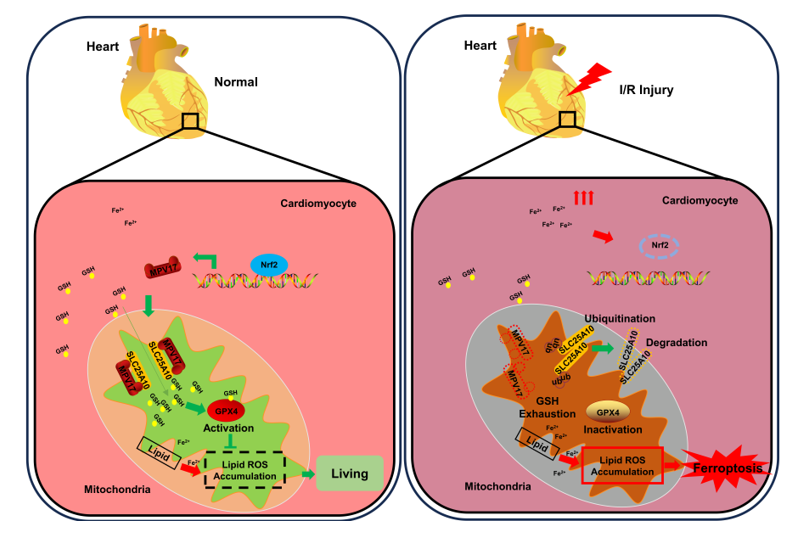
To investigate the molecular mechanism of myocardial ferroptosis induced by iron overload, ferric ammonium citrate (FAC) was introduced into the culture medium to mimic the high-iron condition, thus inducing myocardial ferroptosis [33]. Under the treatment of FAC, the expression levels of MPV17 were decreased in cardiomyocytes (Figure 1A). To investigate the function of MPV17 in regulating ferroptosis, we overexpressed MPV17 using an adenovirus carrying the MPV17 gene in cardiomyocytes (Figure 1B). We found that overexpression of MPV17 significantly prevented iron overload-induced ferroptosis (Figure 1D). Then, siRNA was applied to knockdown the endogenous MPV17 in cardiomyocytes (Figure 1C). The knockdown of MPV17 enhanced ferroptosis induced by FAC in cardiomyocytes (Figure 1E). To further explored the role of MPV17 in myocardial ferroptosis regulation, we detected the impact of MPV17 on lipid peroxide accumulation [34]. Our results demonstrated that treatment with FAC led to lipid peroxide accumulation, which was mitigated by MPV17 overexpression (Figure 1F). Knockdown of MPV17 exacerbated the accumulation of lipid peroxidation (Figure 1G). Next, we detected the impact of MPV17 on the expression of ferroptosis associated proteins including ACSL4, FTH-1 and GPX4 in cardiomyocytes treated with FAC [35]. The increased protein levels of ACSL4 and the decreased protein levels of FTH-1 and GPX4 indicated the initiation of ferroptosis, which were partially restored by MPV17 overexpression (Figure 1H). Moreover, knockdown of MPV17 further promoted the expression of ACSL4 and decreased the expression of GPX4 and FTH-1 in cardiomyocytes treated with FAC (Figure 1I). All these results demonstrated that MPV17 prevented iron overload-induced ferroptosis.
2.2. MPV17 Prevented Myocardial Ferroptosis during Cardiac I/R Injury
The iron levels detected by Prussian Blue staining increased in the I/R cardiac tissues, indicating that iron overload occurred when mice hearts underwent cardiac I/R injury (Figure 2A). Myocardial cell death dramatically increased in the mice hearts underwent I/R injury while the ferroptosis inhibitor ferrostatin-1 (Fer-1) or iron chelator deferoxamine (DFO) treatment decreased this cell death (Figure 2B,C). Then, lipid peroxides accumulation was examined by detecting the malondialdehydes (MDA) levels. The MDA levels were increased during the I/R injury, which was reversed by Fer-1 or DFO treatment. Moreover, the expression of ACSL4 were increased while the expression of FTH-1 and GPX4 were decreased, which ware also reversed by Fer-1 or DFO treatment. All these evidences illustrated that iron overload-induced ferroptosis occurred during cardiac I/R injury. Next, we examined the role of MPV17 in myocardial ferroptosis in mice hearts underwent I/R injury. The PI staining results of the cardiac tissues showed that MPV17 overexpression mice group significantly reduced the myocardial ferroptosis during cardiac I/R injury compared with the β-gal overexpression group (Figure 2E). Moreover, the increased MDA levels in ischemic cardiac tissues were attenuated by MPV17 overexpression (Figure 2G). The increased expression levels of ACSL4 and the decreased expression levels of FTH-1 and GPX4 were also reversed in MPV17 overexpression group (Figure 2H). Myocardial fibrosis is closely related to cardiac remodeling and the recovery of long-term function after I/R injury. We found that collagen areas were decreased by the MPV17 overexpression after cardiac I/R injury, indicating that MPV17 improved cardiac remodeling after I/R injury (Figure 2F). All these evidences demonstrated that MPV17 could prevent myocardial ferroptosis during cardiac I/R injury.
2.3. MPV17 Prevented Iron Overload-Induced Ferroptosis through Maintaining Mitochondrial mtGSH Levels
It was hypothesized that mitochondrial dysfunction was critical in the initiation of ferroptosis. The immunofluorescence staining analysis indicated that MPV17 was localized within the mitochondria, indicating its regulatory role in mitochondrial homeostasis (Figure 3A). Firstly, we detected the mitochondrial membrane potential though the JC-1 staining and flow cytometry. The decreased mitochondrial membrane potential was observed under FAC treatment, while concurrent overexpression of MPV17 partially restored mitochondrial membrane potential (Figure 3B,C). Then, we detected the distribution of lipid peroxides through the C11-BO staining and confocal imaging. It was observed that lipid peroxides mainly accumulated in the mitochondria of cardiomyocytes treated with FAC, which indicating the critical role of mitochondria in ferroptosis initiation (Figure 3D). Overexpression of MPV17 prevented lipid peroxides accumulation in the mitochondria of cardiomyocytes treated with FAC, while knockdown of MPV17 promoted lipid peroxides accumulation in the mitochondria of cardiomyocytes treated with FAC (Figure 3D–F). These results illustrated that MPV17 prevented mitochondrial lipid peroxides accumulation in cardiomyocytes administrated with FAC. Next, we detected the impact of iron overload on mitochondrial respiratory chain. FAC treatment significantly disturbed the activity of mitochondrial respiratory chain (Complex I-V) while overexpression of MPV17 partially recovered the activity of mitochondrial respiratory chain (Figure 3G–K). Although mitochondrial Fe2+ levels were significantly increased detected by the mito-FerroGreen staining after the treatment of FAC, overexpression of MPV17 did not influence the mitochondrial Fe2+ levels (Supplementary Figure S1). Then, we detected its impact on the maintenance of mitochondrial redox homeostasis. mtGSH has been confirmed to play an essential role in preventing ferroptosis initiation28. We found that FAC treatment decreased the levels of mtGSH while overexpression of MPV17 partially recovered mtGSH levels (Figure 2N). Knockdown of MPV17 promoted the exhaustion of mtGSH in cardiomyocytes treated with FAC (Figure 3O). mtGSH was not only an anti-oxidant reagent but also essential for mitochondrial protein glutathionylation which protected proteins from oxidative damage under oxidative stress. We found that mitochondrial protein glutathionylation increased slightly in cardiomyocytes treated with FAC while overexpression of MPV17 further promoted mitochondrial protein glutathionylation. Interestingly, the core enzymes of mitochondrial respiratory chain, NADPH oxidase 4 (nox4) and cytochrome c oxidase 1 (cox1) were also glutathionylated, which were further promoted by MPV17 overexpression. The glutathionylation of nox4 and cox1 protected them from oxidative damages and explained the recovery of mitochondrial respiratory chain activity after MPV17 overexpression. To further confirmed the role of mtGSH in iron-overload induced ferroptosis, we pre-treated the cardiomyocytes with GSH for 6 hours. We found that GSH treatment prevented iron overload-induced ferroptosis and simultaneous overexpression of a mitochondrial GPX4 (mtGPX4) further reduced myocardial ferroptosis (Figure 3M). However, simultaneous overexpression of MPV17 was not as pronounced as simultaneous overexpression of mtGPX4 in the reduction of ferroptosis in cardiomyocytes treated with GSH (Figure 3M). These results indicated that MPV17 functioned upstream of the GSH/GPX4 signaling. In conclusion, iron overload decreased mtGSH which led to mitochondrial lipid peroxides accumulation and mitochondrial respiratory chain dysfunction. MPV17 attenuated iron overload-induced myocardial ferroptosis through maintaining the mtGSH levels.
2.4. SLC25A10 Prevented Myocardial Ferroptosis by Mediating mtGSH Import
SLC25A10 is a mitochondrial inner membrane GSH transporter that enables the import of GSH from the cytosol into the mitochondrial matrix. Immunofluorescence staining results demonstrated that SLC25A10 was localized in the mitochondria (Figure 4A). Additionally, the decreased expression levels of SLC25A10 were observed under the treatment of FAC suggesting that SLC25A10 were involved in myocardial ferroptosis induced by iron overload (Figure 4B). Overexpression of SLC25A10 was performed in cardiomyocytes to examine its role in iron overload-induced myocardial ferroptosis (Figure 4C). The results showed that enhanced expression of SLC25A10 prevented myocardial ferroptosis induced by FAC (Figure 4E). Conversely, silencing SLC25A10 through siRNA facilitated myocardial ferroptosis (Figure 4D and 4F). Subsequently, lipid peroxide accumulation was detected in cardiomyocytes. Overexpression of SLC25A10 decreased lipid peroxides accumulation in cardiomyocytes treated with FAC (Figure 4G). Conversely, the silencing of SLC25A10 led to increased lipid peroxides accumulation in cardiomyocytes treated with FAC (Figure 4H). These evidences demonstrated that SLC25A10 prevented iron overload-induced myocardial ferroptosis.
4.5. MPV17 Prevented Myocardial Ferroptosis through Maintaining the Protein Stability of SLC25A10
We hypothesized that MPV17 and SLC25A10 had a functional connection in regulating myocardial ferroptosis. MPV17 was found to colocalize with SLC25A10 in cardiomyocytes (Figure 5A). Co-immunoprecipitation results showed that MPV17 could interact with SLC25A10 (Figure 5B). FAC treatment decreased the expression levels of SLC25A10 while simultaneous overexpression of MPV17 partially recovered the expression levels of SLC25A10 (Figure 5C). Further studies revealed that FAC treatment led to protein instability and ubiquitination of SLC25A10. Simultaneous overexpression of MPV17 reduced the ubiquitination of SLC25A10 (Figure 5D). These results indicated that MPV17 could stabilize SLC25A10 through interacting with it. Moreover, knockdown of MPV17 aggravated myocardial ferroptosis induced by FAC while simultaneous overexpression of SLC25A10 attenuated these myocardial ferroptosis (Figure 5E). Overexpression of MPV17 attenuated myocardial ferroptosis induced by FAC while simultaneous knockdown of SLC25A10 aggravated these myocardial ferroptosis (Figure 5F). Next, we detected the lipid peroxides levels in FAC treated cardiomyocytes. Knockdown of MPV17 increased lipid peroxides accumulation induced by FAC while simultaneous overexpression of SLC25A10 attenuated these lipid peroxides accumulation (Figure 5G). Overexpression of MPV17 decreased lipid peroxides accumulation while simultaneous knockdown of SLC25A10 increased these lipid peroxides accumulation (Figure 5H). These results indicated that SLC25A10 functioned downstream of MPV17 to regulate iron overload-induced myocardial ferroptosis. Taken together, MPV17 prevented iron overload-induced ferroptosis through maintaining the protein stability of SLC25A10.
2.6. Nrf2 Prevented Iron Overload-Induced Myocardial Ferroptosis through Transcriptionally Activating MPV17
Nrf2 was a core transcriptional factor in preventing ferroptosis through encoding antioxidant genes. Under the treatment of FAC, the expression levels of Nrf2 were decreased in cardiomyocytes (Figure 6A). An Nrf2 binding site was predicted in the promoter region of MPV17 by the JASPAR database, which was confirmed by the Chip assays and the luciferase assays (Figure 6B,C). Moreover, knockdown of Nrf2 decreased the expression levels of MPV17 (Figure 6D), while overexpression of Nrf2 partially recovered the expression levels of MPV17 in cardiomyocytes treated with FAC (Figure 6F). Next, we explored the functional relationship between Nrf2 and MPV17 in myocardial ferroptosis regulation. Knockdown of Nrf2 promoted myocardial ferroptosis while simultaneous overexpression of MPV17 attenuated myocardial ferroptosis induced by FAC (Figure 6E). Overexpression of Nrf2 prevented myocardial ferroptosis while simultaneous knockdown of MPV17 promoted myocardial ferroptosis (Figure 6F). Then, we detected the lipid peroxides levels in FAC treated cardiomyocytes. Knockdown of Nrf2 increased lipid peroxides accumulation induced by FAC while simultaneous overexpression of MPV17 attenuated lipid peroxides accumulation (Figure 7G). Overexpression of Nrf2 decreased lipid peroxides accumulation while simultaneous knockdown of MPV17 increased lipid peroxides accumulation (Figure 7H). These results indicated that Nrf2 prevented iron overload-induced myocardial ferroptosis through transcriptionally activating MPV17.
3. Discussion
Ferroptosis is a newly identified programmed cell death process and plays an active role in several pathological processes [20,36,37]. Although myocardial ferroptosis has been implicated in ischemic cardiac diseases, the underlying mechanisms remained largely unknown. Clinical observations indicated that iron overload occurred in the cardiac ischemic zone,which was a dangerous factor in ferroptosis initiation [15,38]. This study further confirmed the role of iron overload in myocardial ferroptosis initiation during cardiac I/R injury. Moreover, we found an emerging role of MPV17 in protecting cardiomyocytes from iron overload-induced ferroptosis and attenuating cardiac I/R injury through maintaining the protein homeostasis of SLC25A10, which imported GSH from the cytosol into the mitochondria. Nrf2 could prevent myocardial ferroptosis through transcriptionally activating MPV17. This study revealed the Nrf2-MPV17-SLC25A10/mtGSH signaling pathway in myocardial ferroptosis regulation and provided new targets for cardiac diseases treatment.
MPV17 is widely expressed in various tissues and organs with its role little known. The close relationship between MPV17 mutation and human diseases attract broad research interests. Recent studies found that MPV17 actively participate in mitochondrial respiration and ROS metabolism as a scaffold protein [24,39,40]. In this study, MPV17 was indicated to participate in the regulation of mtGSH import. MPV17 had been demonstrated to be a transporter protein and formed nonselective channel in the mitochondrial inner membrane [25,41,42]. However, the regulation of mtGSH import by MPV17 did not depend on its channel function. MPV17 functioned more like a scaffolder protein to interact with the mtGSH transporter SLC25A10. This interaction protected SLC25A10 from ubiquitination and degradation. Absence of MPV17 induced by iron overload led to SLC25A10 instability and mtGSH exhaustion, leading to the final myocardial ferroptosis. These findings not only revealed the molecular mechanism of ferroptosis initiation but also enriched the functional role of MPV17.
Iron overload occurred during both the cardiac ischemic injury and the heart transplant process [38,43]. Mitochondria were an essential organelle in intracellular iron metabolism and played an indispensable role in ferroptosis initiation in several cases [44]. In human HT-1080 cells, elimination of mitochondria protected cells from ferroptosis induction[26]. In DOX induced myocardial ferroptosis, iron chelator targeting mitochondrial Fe2+ prevented myocardial ferroptosis initiation [45]. Free Fe2+ promoted lipid peroxides accumulation and ferroptosis initiation through Fenton reaction [46,47]. The knockdown or overexpression of MPV17 had no effect on the levels of mitochondrial iron although overexpression of MPV17 diminished lipid peroxides accumulation in mitochondria (Supplementary Figure S1). Lipid peroxides accumulation depended on not only the increased lipid oxidation but also the dysfunction of lipid oxidation scavenging system. Our study showed that iron overload could also delete the mtGSH and changed the redox state of mitochondria, leading to lipid peroxides accumulation. However, the cause of iron overload during cardiac injury has not been further explored in this study.
In cysteine deprivation induced ferroptosis, mtGSH was essential for the maintenance of mitochondrial bioenergetic respiration and mitochondrial ROS metabolism [30]. GSH was also an important partner of GPX4, which converted peroxidized phosphatidylethanolamines (PEs) to hydroperoxyl-Pes [48]. GSH deletion would lead to the dysfunction of GPX4 and lipid peroxides accumulation. In this work, decreased mtGSH levels were observed and the dysfunction of GSH/GPX4 lipid oxidation scavenging system was causative for myocardial ferroptosis initiation. Pre-treatment with GSH attenuated myocardial ferroptosis while simultaneous overexpression of mito-GPX4 would further prevent myocardial ferroptosis. Moreover, we also found that glutathionylation insufficiency of the mitochondrial protein occurred because of mtGSH deletion. The glutathionylation of mitochondrial protein usually protected them from oxidative damage under stress condition [49]. Overexpression of MPV17 partially recovered the glutathionylation of mitochondrial protein, including the key enzymes (nox4 and cox1) involved in the mitochondrial respiratory chain (Figure 3L). These findings might explain the facts that the activities of mitochondrial respiration chain were decreased during iron overload-induced myocardial ferroptosis while overexpression of MPV17 could partially recovered these activities. However, the further relationship between mitochondrial protein glutathionylation and ferroptosis initiation had not been further explored in this study.
Nrf2 has long been considered as an important transcriptional regulator in eliminating lipid peroxides, free iron and cystine import and GSH production though its targeted genes [50,51]. However, the transcriptional activity of Nrf2 also depends on the cell types and physical conditions. This study identified MPV17 as a new target of Nrf2 in the regulation of iron overload-induced myocardial ferroptosis. These findings further revealed the mechanisms of Nrf2 in myocardial ferroptosis regulation and enriched the knowledge of molecular biology in cardiac diseases.
4. Materials and Methods
4.1. Cell Treatment
H9c2 cells (American Type Culture Collection) were cultured in the cell incubator with DMEM/F-12 (Gibco, 21331046) contained 10% fetal bovine serum with 5% CO2 at 37 °C. Cells were co-cultured with 500 µM ammonium ferric citrate (FAC) to induce robust cell death. Cells were co-cultured with 100 µM FAC to induce mild cell death.
4.2. Mice Ischemia/Reperfusion Model
Mice underwent ligation with LAD for 30 min and re-perfused for 3 H. Mice (N = 5/group) received Propidium Iodide (PI) injection (10 mg/kg) (Solarbio, P8080) at the end of the surgery to label the ferroptotic cells. The cardiac tissues were frozen with liquid nitrogen and 5 μm sections were cut with an ice slicer (Leica, CM3050S) and counterstained with DAPI to quantitatively detect cell death. All the approaches of involving animals were approved by Ethics Committee of Qingdao Agricultural University.
4.3. Detection of Myocardial Ferroptosis by PI Staining and Flowcytometry
The H9c2 cells were cultured with the treatment of FAC at the indicated times. The cells were digested with pancreatic enzyme (0.25%) for 2 min and resuscitated with PBS in a 1.5 ml tube. Discard the supernatant and PI staining buffer (10 ug/ml) was added into the H9c2 cells. The cells were kept out of light and incubated in an ice bath for 30 min. After three washes with PBS, we used a BD FACS Aria III flow cytometer and the PE channel to detect the PI positive cells.
4.4. Immunofluorescence Staining
The H9c2 cells cultured on the slide were fixed at 4℃ overnight and goat serum was used for blocking. Discard the blocking buffer and added the diluted antibody at the indicated dilution. Incubated the cells at 4℃ overnight. The secondary antibody labeled with FITC or Cy3 were added into the cells after three washes of PBST. The nuclear was labeled with DAPI. Images were captured with a Leica SP5 confocal microscope. The anti-MPV17 antibody was from Proteintech (10310-1-AP); anti-SLC25A10 antibody was from Proteintech (12086-1-AP).
4.5. Prussian Blue Stain and Iron Levels Detection
Prussian blue staining was performed following the instructions (Solarbio, G1420). The free iron was detected following the instructions (Biolab, SNM159).
4.6. Masson Staining and Detection of Myocardial Fibrosis
Myocardial fibrosis was detected by Masson Staining kit following the instructions (Solarbio, G1340).
4.7. Detection of Mitochondrial Respiratory Chain Activity
Isolated mitochondria were used to measure the activity of complex I-V following the instruction of the detection kit (Solarbio BC0515, Solarbio BC3230, Solarbio BC3240, Solarbio BC0945, Solarbio,BC1440) by using the spectrophotometer/microplate reader.
4.8. Detection of Lipids Peroxides in the Cells
The C11 BODIPY 581/591 (C11-BO) staining dye was purchased from the Invitrogen (Invitrogen, Thermo.D3861). The cultured H9c2 cells were plated on 6-well plate or on the slide. After treatment with the FAC, the C11-BO staining dye (2 µM) was added into the cultured cells, which were kept in 37℃,5% CO2 for 30 min. Then the cells were digested with pancreatic enzyme (0.25%) and collected for detection. A BD FACS Aria III flow cytometer was used to detect the fluoresce signaling. FITC channel and PE channel were used for the detection of green fluoresce and red fluoresce respectively. The cultured cells plated on the slide were detected by a Leica SP5 confocal microscope directly after the staining.
4.9. Western Blotting (WB) Assay
The procedures for WB assays were described in the previous work52. Anti-α-actinin antibody was from Sigma (A7732);Anti-β-Tubulin antibody was from Abclonal (A17073); Anti-ACSL4 Antibody was from BOSTER (A04372-2); Anti-GPX4 Antibody was from BOSTER (A02059-1); Anti-Fth1 Antibody was from BOSTER (BM4487); Anti-NOX4 Antibody was from BOSTER (BM4135); Anti-COX-1 Antibody was from BOSTER (PB9002); Anti-Nrf2 Antibody was from Proteintech (16396-1-AP); Anti-GSH antibody was from VIROGEN (101A). Anti-Ubiquitin Antibody was from Santacruz (sc-8017). β-tubulin antibody was used for loading control.
4.10. Immunoprecipitation Assay
The immunoprecipitation assay was performed as previously described [52]. The RIPA buffer with a cocktail (Merk, 539134) was used for protein lysis. 2 µg antibody and 50 µl protein A/G Agarose beads (Biolinkedin, Ik-1004) were used for one sample. The mixtures were slightly rotated at 4℃ overnight. The immunoprecipitants were resuspended in sample buffer with sodium lauryl sulfate (SDS) and boiled at 98 ℃ for 10 min.
4.11. Cell Transfection
The siRNA and plasmid vectors were transfected with lippo3000 following the manual instructions. The siRNA targeting MPV17 were purchased from Origene (SR508989); The siRNA targeting slc25a10 were purchased from Origene (SR509035); The siRNA sequences targeting Nrf2 was 5′-CCGGCATTTCACTGAACACAA-3′, which was validated previously; The open reading frame (ORF) of SLC25A10 (rat) or Nrf2 (rat) were constructed into the eukaryotic vectors pcDNA3.1.
4.12. Detection of Mitochondrial outer Membrane Potential
JC-1 was added into the H9c2 cells and incubated for 30 min in 37 ℃. After three washed, a BD FACS Aria III flow cytometer was used to detect the red/green fluoresce ratio through FITC channel and PE channel respectively.
4.13. Detection of the mtGSH Levels
The mitochondria were isolated by the mitochondria isolation kit following the instructions (Solarbio, SM0020). Then the mtGSH levels were detected by GSH detection kit following the instructions (Nanjing Jiancheng, A006-2-1).
4.14. Detection of the Luciferase Activity
The instructions for luciferase assay were described previously [53].
4.15. Chip Assay
The well-cultured cardiomyocytes were immersed in 1% formaldehyde solution and treated under vacuum for 10 minutes to promote cross-linking. 125mm glycine was applied to interrupt the cross-linking reaction. After 3 times of washes with cooled PBS, the lysis buffer contained protease inhibitor was used for cell lysis. The collected samples were processed by ultrasonic crusher to lyse the cells and release chromatin, which was sheared into 100-1000 BP fragments using an ultrasonic crusher. The un-lysed cell fragments were removed and antibody incubation was performed at 4℃ overnight. The magnetic beads were applied for the binding of antibody at room temperature for 1 hours. Low-salt, high-salt, lithium-chloride and TE washing buffer were used to wash the beads sequentially to remove non-specific binding. 200 mM NaCl was used for reverse cross-linking at 65 ° C for 4 hours. After digestated with proteinase K at 45 ° C for 1 hour, the mixtures were purified by using phenol chloroform. A (BIO-RAD) T100 Thermal Cycler was used for the replication of the aimed DNA fragments.
Chip-PCR primers: F: 5′-GCTAGCCGGGTCATTTTACA-3′, R:5′-CGGAGCTCAGACTTGTTCCA-3′.
4.16. Statistical Analysis
Three independent experiments were performed and one-way ANOVA were applied to estimate variance. ∗, p < 0.05 was statistically significant.
5. Conclusions
In conclusion, we demonstrated the important role of MPV17 in preventing iron overload-induced ferroptosis and delineated the Nrf2-MPV17-SLC25A10/mtGSH signaling pathway in the ferroptosis regulation. These findings provided new strategy for cardiac diseases treatment.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Tao Xu: Methodology; Investigation; Original Draft; Writing; Funding Acquisition. Guilan Chen: Methodology; Formal Analysis. Writing; Review & Editing; Funding Acquisition.
Funding
This work was supported by National Natural Science Foundation of China (82000290) and the Talents of High-Level Scientific Research Foundation of Qingdao Agricultural University (663/1121039 and 663/1120108). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. This work has not been published previously or under consideration for publication elsewhere. Its publication is approved by all authors.
Institutional Review Board Statement
The animal study protocol was approved by the Ethics Committee of Qingdao Agricultural University.
Data Availability Statement
We encourage all authors of articles published in MDPI journals to share their research data. In this section, please provide details regarding where data supporting reported results can be found, including links to publicly archived datasets analyzed or generated during the study. Where no new data were created, or where data is unavailable due to privacy or ethical restrictions, a statement is still required. Suggested Data Availability Statements are available in section “MDPI Research Data Policies” at https://www.mdpi.com/ethics.
Conflicts of Interest
All the authors declared no competing interests.
References
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Ding, W.; Ji, X.Y.; Ao, X.; Liu, Y.; Yu, W.P.; Wang, J.X. Oxidative Stress in Cell Death and Cardiovascular Diseases. Oxidative Med. Cell. Longev. 2019, 2019, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.; Whelan, R.S.; Kitsis, R.N. Mechanisms of Cell Death in Heart Disease. Arter. Throm Vas. 2012, 32, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- Piché, M.E.; Tchernof, A.; Despres, J.P. Obesity Phenotypes, Diabetes, and Cardiovascular Diseases. Circ. Res. 2020, 126, 1477–1500. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg Kathryn, M.; Lamprecht Michael, R.; Skouta, R.; Zaitsev Eleina, M.; Gleason Caroline, E.; Patel Darpan, N.; Bauer Andras, J.; Cantley Alexandra, M.; Yang Wan, S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Fang, X.; Ardehali, H.; Min, J.; Wang, F. The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat. Rev. Cardiol. 2022, 20, 7–23. [Google Scholar] [CrossRef]
- Li, N.; Jiang, W.; Wang, W.; Xiong, R.; Wu, X.; Geng, Q. Ferroptosis and its emerging roles in cardiovascular diseases. Pharmacol. Res. 2021, 166, 105466. [Google Scholar] [CrossRef]
- Díez-López, C.; Comín-Colet, J.; González-Costello, J. Iron overload cardiomyopathy. Curr. Opin. Cardiol. 2018, 33, 334–340. [Google Scholar] [CrossRef]
- Fang, X.; Cai, Z.; Wang, H.; Han, D.; Cheng, Q.; Zhang, P.; Gao, F.; Yu, Y.; Song, Z.; Wu, Q.; et al. Loss of Cardiac Ferritin H Facilitates Cardiomyopathy via Slc7a11-Mediated Ferroptosis. Circ. Res. 2020, 127, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Koerner, M.M.; Tenderich, G.; Minami, K.; Zu Knyphausen, E.; Mannebach, H.; Kleesiek, K.; Meyer, H.; Koerfer, R. Heart transplantation for end-stage heart failure caused by iron overload. Br. J. Haematol. 2003, 97, 293–296. [Google Scholar] [CrossRef]
- Mavrogeni, S.; Kolovou, G.; Bigalke, B.; Rigopoulos, A.; Noutsias, M.; Adamopoulos, S. Transplantation in patients with iron overload: is there a place for magnetic resonance imaging? Heart Fail. Rev. 2018, 23, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Sukumaran, A.; Chang, J.; Han, M.; Mintri, S.; Khaw, B.A.; Kim, J. Iron overload exacerbates age-associated cardiac hypertrophy in a mouse model of hemochromatosis. Sci. Rep. 2017, 7, 5756. [Google Scholar] [CrossRef] [PubMed]
- Wongjaikam, S.; Kumfu, S.; Khamseekaew, J.; Chattipakorn, S.C.; Chattipakorn, N. Restoring the impaired cardiac calcium homeostasis and cardiac function in iron overload rats by the combined deferiprone and N-acetyl cysteine. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.B.; Ren, X.Y.; Wang, Y.; Chen, D.X.; Jiang, L.; Li, X.; Li, T.; Huo, M.F.; Li, Q. Targeting Ferroptosis by Polydopamine Nanoparticles Protects Heart against Ischemia/Reperfusion Injury. Acs Appl. Mater. Inter. 2021, 13, 53671–53682. [Google Scholar] [CrossRef]
- Murphy, C.J.; Oudit, G.Y. Iron-Overload Cardiomyopathy: Pathophysiology, Diagnosis, and Treatment. J. Card. Fail. 2010, 16, 888–900. [Google Scholar] [CrossRef]
- Latunde-Dada, G.O. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Bba-Gen. Subj. 2017, 1861, 1893–1900. [Google Scholar] [CrossRef]
- Feng, Z.; Min, L.X.; Chen, H.; Deng, W.W.; Tan, M.L.; Liu, H.L.; Hou, J.M. Iron overload in the motor cortex induces neuronal ferroptosis following spinal cord injury. Redox Biol. 2021, 43, 101984. [Google Scholar] [CrossRef]
- Wang, X.T.; Wang, Z.X.; Cao, J.; Dong, Y.L.; Chen, Y.X. Melatonin Alleviates Acute Sleep Deprivation-Induced Memory Loss in Mice by Suppressing Hippocampal Ferroptosis. Front. Pharmacol. 2021, 12, 708645. [Google Scholar] [CrossRef] [PubMed]
- Kajarabille, N.; Latunde-Dada, G.O. Programmed Cell-Death by Ferroptosis: Antioxidants as Mitigators. Int. J. Mol. Sci. 2019, 20, 4968. [Google Scholar] [CrossRef]
- Fang, X.X.; Wang, H.; Han, D.; Xie, E.J.; Yang, X.; Wei, J.Y.; Gu, S.S.; Gao, F.; Zhu, N.L.; Yin, X.J.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Wang, J.; Dai, H.; Almannai, M.; Staufner, C.; Alfadhel, M.; Gambello, M.J.; Prasun, P.; Raza, S.; Lyons, H.J.; et al. MPV17-related mitochondrial DNA maintenance defect: New cases and review of clinical, biochemical, and molecular aspects. Hum. Mutat. 2018, 39, 461–470. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Wang, J.L.; Dai, H.Z.; Almannai, M.; Staufner, C.; Alfadhel, M.; Gambello, M.J.; Prasun, P.; Raza, S.; Lyons, H.J.; et al. MPV17-related mitochondrial DNA maintenance defect: New cases and review of clinical, biochemical, and molecular aspects. Hum. Mutat. 2018, 39, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Blakely, E.L.; Butterworth, A.; Hadden RD, M.; Bodi, I.; He, L.; McFarland, R.; Taylor, R.W. MPV17 mutation causes neuropathy and leukoencephalopathy with multiple mtDNA deletions in muscle. Neuromuscul. Disord. 2012, 22, S21–S21. [Google Scholar] [CrossRef]
- Madungwe, N.B.; Feng, Y.S.; Aliagan, A.I.; Tombo, N.; Kaya, F.; Bopassa, J.C. Inner mitochondrial membrane protein MPV17 mutant mice display increased myocardial injury after ischemia/reperfusion. Am. J. Transl. Res. 2020, 12, 3412–3428. [Google Scholar]
- Sperl, L.E.; Hagn, F. NMR Structural and Biophysical Analysis of the Disease-Linked Inner Mitochondrial Membrane Protein MPV17. J. Mol. Biol. 2021, 433, 167098. [Google Scholar] [CrossRef]
- Gao, M.H.; Yi, J.M.; Zhu, J.J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X.J. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–+. [Google Scholar] [CrossRef]
- Haddad, M.; Herve, V.; Ben Khedher, M.R.; Rabanel, J.M.; Ramassamy, C. Glutathione: An Old and Small Molecule with Great Functions and New Applications in the Brain and in Alzheimer's Disease. Antioxid. Redox Sign 2021, 35, 270–292. [Google Scholar] [CrossRef]
- Jang, S.; Chapa-Dubocq, X.R.; Tyurina, Y.Y.; St Croix, C.M.; Kapralov, A.A.; Tyurin, V.A.; Bayir, H.; Kagan, V.E.; Javadov, S. Elucidating the contribution of mitochondrial glutathione to ferroptosis in cardiomyocytes. Redox Biol. 2021, 45, 102021. [Google Scholar] [CrossRef]
- Oestreicher, J.; Morgan, B. Glutathione: subcellular distribution and membrane transport. Biochem. Cell Biol. 2019, 97, 270–289. [Google Scholar] [CrossRef]
- Sreekumar, P.G.; Ferrington, D.A.; Kannan, R. Glutathione Metabolism and the Novel Role of Mitochondrial GSH in Retinal Degeneration. Antioxidants 2021, 10, 661. [Google Scholar] [CrossRef]
- Baiskhanova, D.; Schäfer, H. The Role of Nrf2 in the Regulation of Mitochondrial Function and Ferroptosis in Pancreatic Cancer. Antioxidants 2024, 13, 696. [Google Scholar] [CrossRef] [PubMed]
- Kuosmanen, S.M.; Kansanen, E.; Kaikkonen, M.U.; Sihvola, V.; Pulkkinen, K.; Jyrkkänen, H.-K.; Tuoresmäki, P.; Hartikainen, J.; Hippeläinen, M.; Kokki, H.; et al. NRF2 regulates endothelial glycolysis and proliferation with miR-93 and mediates the effects of oxidized phospholipids on endothelial activation. Nucleic Acids Res. 2018, 46, 1124–1138. [Google Scholar] [CrossRef]
- Xu, T.; Ding, W.; Ao, X.; Chu, X.M.; Wan, Q.G.; Wang, Y.; Xiao, D.D.; Yu, W.P.; Li, M.Y.; Yu, F.; et al. ARC regulates programmed necrosis and myocardial ischemia/reperfusion injury through the inhibition of mPTP opening. Redox Biol. 2019, 20, 414–426. [Google Scholar] [CrossRef]
- Wan, Q.G.; Xu, T.; Ding, W.; Zhang, X.J.; Ji, X.Y.; Yu, T.; Yu, W.P.; Lin, Z.J.; Wang, J.X. miR-499-5p Attenuates Mitochondrial Fission and Cell Apoptosis via p21 in Doxorubicin Cardiotoxicity. Front. Genet. 2019, 9, 734. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.A.; Xiong, W.Y.; Zhang, A.L.; Zhang, H.; Lin, H.; Gao, L.; Ke, J.H.; Huang, S.Y.; Zhang, J.F.; Gu, J.; et al. The Imbalance of p53-Park7 Signaling Axis Induces Iron Homeostasis Dysfunction in Doxorubicin-Challenged Cardiomyocytes. Adv. Sci. 2023, 10. [Google Scholar] [CrossRef]
- Drummen, G.P.; van Liebergen, L.C.; den Kamp JA, O.; Post, J.A. C11-BODIPY581/591, An oxidation-sensitive fluorescent lipid peroxidation probe: (micro)spectroscopic characterization and validation of methodology. Free. Radic. Biol. Med. 2002, 33, 473–490. [Google Scholar] [CrossRef]
- Chen, X.; Comish, P.B.; Tang, D.; Kang, R. Characteristics and Biomarkers of Ferroptosis. Front. Cell Dev. Biol. 2021, 9, 637162. [Google Scholar] [CrossRef]
- Yu, Y.Y.; Jiang, L.; Wang, H.; Shen, Z.; Cheng, Q.; Zhang, P.; Wang, J.M.; Wu, Q.; Fang, X.X.; Duan, L.Y.; et al. Hepatic transferrin plays a role in systemic iron homeostasis and liver ferroptosis. Blood 2020, 136, 726–739. [Google Scholar] [CrossRef]
- Reichert, C.O.; de Freitas, F.A.; Sampaio-Silva, J.; Rokita-Rosa, L.; Barros, P.D.; Levy, D.; Bydlowski, S.P. Ferroptosis Mechanisms Involved in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8765. [Google Scholar] [CrossRef]
- Miyamoto, H.D.; Ikeda, M.; Ide, T.; Tadokoro, T.; Furusawa, S.; Abe, K.; Ishimaru, K.; Enzan, N.; Sada, M.; Yamamoto, T.; et al. Iron Overload via Heme Degradation in the Endoplasmic Reticulum Triggers Ferroptosis in Myocardial Ischemia-Reperfusion Injury. JACC-Basic Transl. Sci. 2022, 7, 801–820. [Google Scholar] [CrossRef]
- Jacinto, S.; Guerreiro, P.; de Oliveira, R.M.; Cunha-Oliveira, T.; Santos, M.J.; Grazina, M.; Rego, A.C.; Outeiro, T.F. MPV17 Mutations Are Associated With a Quiescent Energetic Metabolic Profile. Front. Cell Neurosci. 2021, 15, 641264. [Google Scholar] [CrossRef] [PubMed]
- Krick, S.; Shi, S.; Ju, W.; Faul, C.; Tsai S-y Mundel, P.; Böttinger, E.P. Mpv17l protects against mitochondrial oxidative stress and apoptosis by activation of Omi/HtrA2 protease. Proc. Natl. Acad. Sci. USA 2008, 105, 14106–14111. [Google Scholar] [CrossRef] [PubMed]
- Antonenkov, V.D.; Isomursu, A.; Mennerich, D.; Vapola, M.H.; Weiher, H.; Kietzmann, T.; Hiltunen, J.K. The Human Mitochondrial DNA Depletion Syndrome Gene MPV17 Encodes a Non-selective Channel That Modulates Membrane Potential. J. Biol. Chem. 2015, 290, 13840–13861. [Google Scholar] [CrossRef]
- Mukherjee, S.; Das, S.; Bedi, M.; Vadupu, L.; Ball, W.B.; Ghosh, A. Methylglyoxal-mediated Gpd1 activation restores the mitochondrial defects in a yeast model of mitochondrial DNA depletion syndrome. Bba-Gen. Subj. 2023, 1867, 130328. [Google Scholar] [CrossRef]
- Diez-Lopez, C.; Comin-Colet, J.; Gonzalez-Costello, J. Iron overload cardiomyopathy: from diagnosis to management. Curr. Opin. Cardiol. 2018, 33, 334–340. [Google Scholar] [CrossRef]
- Lill, R.; Hoffmann, B.; Molik, S.; Pierik, A.J.; Rietzschel, N.; Stehling, O.; Uzarska, M.A.; Webert, H.; Wilbrecht, C.; Muhlenhoff, U. The role of mitochondria in cellular iron-sulfur protein biogenesis and iron metabolism. Bba-Mol. Cell Res. 2012, 1823, 1491–1508. [Google Scholar] [CrossRef] [PubMed]
- Tadokoro, T.; Ikeda, M.; Ide, T.; Deguchi, H.; Ikeda, S.; Okabe, K.; Ishikita, A.; Matsushima, S.; Koumura, T.; Yamada, K.; et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight 2020, 5, e132747. [Google Scholar] [CrossRef]
- Zhang, S.; Xin, W.; Anderson, G.J.; Li, R.; Gao, L.; Chen, S.; Zhao, J.; Liu, S. Double-edge sword roles of iron in driving energy production versus instigating ferroptosis. Cell Death Dis. 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Jiao, Q.; Du, X.; Jia, F.; Chen, X.; Yan, C.; Jiang, H. Ferroptosis in Parkinson's disease—The iron-related degenerative disease. Ageing Res. Rev. 2024, 101, 102477. [Google Scholar] [CrossRef]
- Forcina, G.C.; Dixon, S.J. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 2019, 19. [Google Scholar] [CrossRef]
- Vrettou, S.; Wirth, B. S-Glutathionylation and S-Nitrosylation in Mitochondria: Focus on Homeostasis and Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 15849. [Google Scholar] [CrossRef]
- Anandhan, A.; Dodson, M.; Schmidlin, C.J.; Liu, P.; Zhang, D.D. Breakdown of an Ironclad Defense System: The Critical Role of NRF2 in Mediating Ferroptosis. Cell Chem. Biol. 2020, 27, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Lin, B.; Jin, W.; Tang, L.; Hu, S.; Cai, R. NRF2, a Superstar of Ferroptosis. Antioxidants 2023, 12, 1739. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
(a) A Protein levels of MPV17 in cardiomyocytes treated with FAC evaluated by WB; (b) Overexpression of MPV17 in cardiomyocytes was detected by WB; (c) Knockdown of MPV17 in cardiomyocytes by siRNA was detected by WB; (d) cell death labeled with PI staining in each group; (e) Cell death labeled with PI staining in each group; (f) C11-BO was applied to label the lipid peroxides and flow cytometry was used for the detection of green fluoresce in each group; (g) C11-BO was applied to label the lipid peroxides and flow cytometry was used for the detection of green fluoresce in each group; (h) ACSL4,FTH-1 and GPX-4 were evaluated by WB; I ACSL4,FTH-1 and GPX-4 were evaluated by WB.
Figure 1.
(a) A Protein levels of MPV17 in cardiomyocytes treated with FAC evaluated by WB; (b) Overexpression of MPV17 in cardiomyocytes was detected by WB; (c) Knockdown of MPV17 in cardiomyocytes by siRNA was detected by WB; (d) cell death labeled with PI staining in each group; (e) Cell death labeled with PI staining in each group; (f) C11-BO was applied to label the lipid peroxides and flow cytometry was used for the detection of green fluoresce in each group; (g) C11-BO was applied to label the lipid peroxides and flow cytometry was used for the detection of green fluoresce in each group; (h) ACSL4,FTH-1 and GPX-4 were evaluated by WB; I ACSL4,FTH-1 and GPX-4 were evaluated by WB.
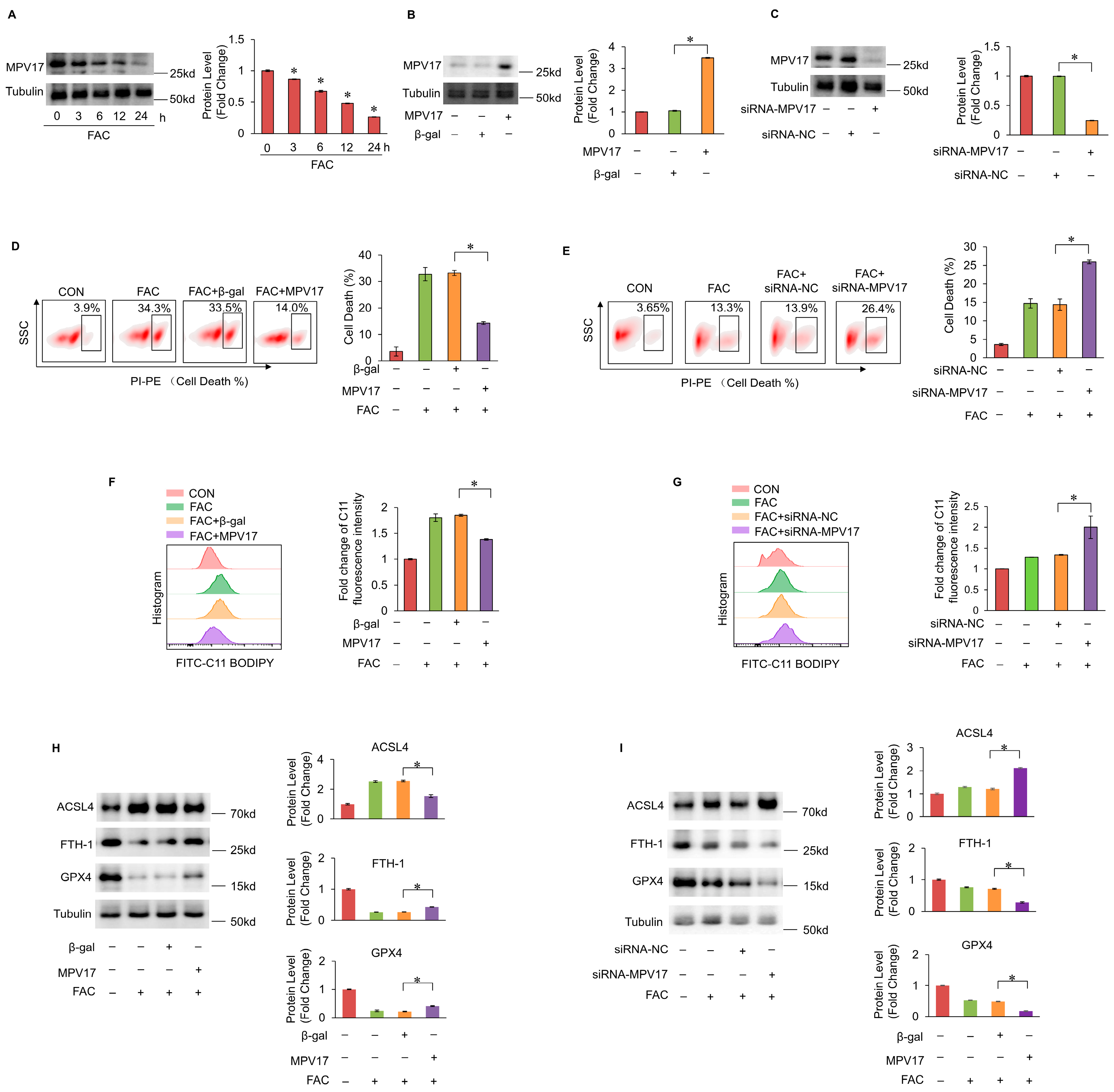
Figure 2.
(a) Prussion Blue staining for the examination of iron levels in cardiac tissues; scale bar, 60 µm; (b) Cell death labeled with PI staining in cardiac tissues after I/R surgery; Red, PI; Blue, DAPI; Green, Actinin; scale bar, 60 µm; (c) Statistic data of the relative MDA levels; (d) The expression of ACSL4,FTH-1 and GPX-4 were checked by WB; (e) Cell death labeled with PI staining in cardiac tissues after I/R surgery; Red, PI; Blue, DAPI; Green, Actinin; scale bar, 60 µm; (f) Masson trichrome staining for the examination of collagen in the heart tissue; (g) The expression of MPV17, ACSL4, FTH-1 and GPX-4 were checked by WB. H Statistic data of relative MDA levels in each group.
Figure 2.
(a) Prussion Blue staining for the examination of iron levels in cardiac tissues; scale bar, 60 µm; (b) Cell death labeled with PI staining in cardiac tissues after I/R surgery; Red, PI; Blue, DAPI; Green, Actinin; scale bar, 60 µm; (c) Statistic data of the relative MDA levels; (d) The expression of ACSL4,FTH-1 and GPX-4 were checked by WB; (e) Cell death labeled with PI staining in cardiac tissues after I/R surgery; Red, PI; Blue, DAPI; Green, Actinin; scale bar, 60 µm; (f) Masson trichrome staining for the examination of collagen in the heart tissue; (g) The expression of MPV17, ACSL4, FTH-1 and GPX-4 were checked by WB. H Statistic data of relative MDA levels in each group.
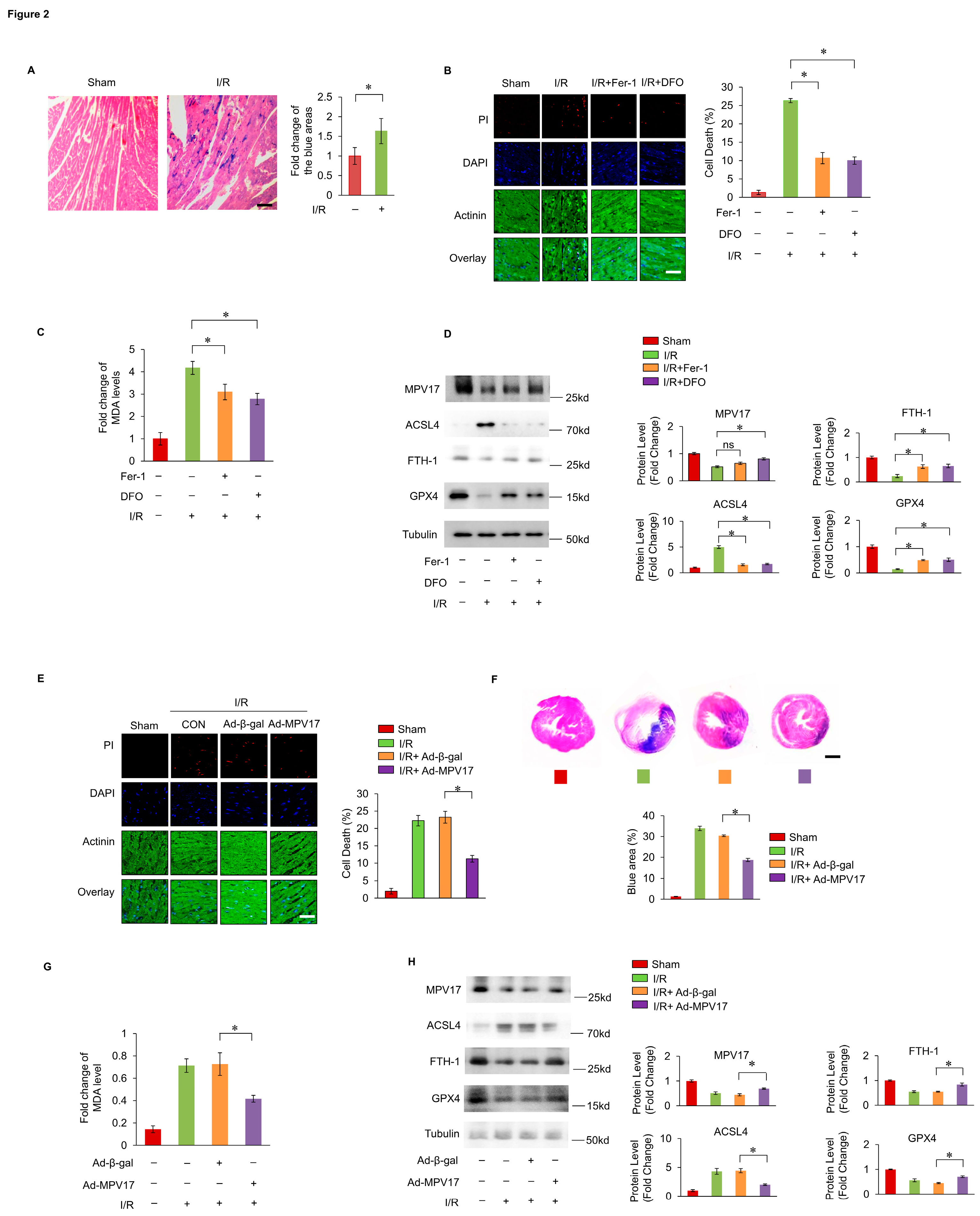
Figure 3.
(a) The localization of MPV17 detected by immunofluorescence and confocal imaging; green, MPV17; red, mitotracker; blue, DAPI; scale bar, 10 µm; (b) JC-1 and flow cytometry were used for the measurement of mitochondrial membrane potential; (c) The ratio of cells with green fluorescence in each group labeled with JC-1; (d) Confocal imaging of the lipid peroxides labeled with C11-BO; green, C11-BO; red,mitotracker; blue, DAPI; scale bar, 20 µm; (e) Fold changes of the green fluorescence intensity of the C11-BO staining in mitochondria in each group; (f)Fold changes of the green fluorescence intensity of the C11-BO in mitochondria in each group. (g-k) Relative activity of the Complex I-V; (l) The glutathionylation of nox1, cox1 and the whole mitochondria lysates (wcl) were evaluated by WB. (m)The percentage of cell death labeled with PI staining in each group. (n) Fold change of the mtGSH levels in the cardiomyocytes in each group. O Fold change of the mtGSH levels in the cardiomyocytes in each group.
Figure 3.
(a) The localization of MPV17 detected by immunofluorescence and confocal imaging; green, MPV17; red, mitotracker; blue, DAPI; scale bar, 10 µm; (b) JC-1 and flow cytometry were used for the measurement of mitochondrial membrane potential; (c) The ratio of cells with green fluorescence in each group labeled with JC-1; (d) Confocal imaging of the lipid peroxides labeled with C11-BO; green, C11-BO; red,mitotracker; blue, DAPI; scale bar, 20 µm; (e) Fold changes of the green fluorescence intensity of the C11-BO staining in mitochondria in each group; (f)Fold changes of the green fluorescence intensity of the C11-BO in mitochondria in each group. (g-k) Relative activity of the Complex I-V; (l) The glutathionylation of nox1, cox1 and the whole mitochondria lysates (wcl) were evaluated by WB. (m)The percentage of cell death labeled with PI staining in each group. (n) Fold change of the mtGSH levels in the cardiomyocytes in each group. O Fold change of the mtGSH levels in the cardiomyocytes in each group.
Figure 4.
(a) The localization of SLC25A10 detected by immunofluorescence and confocal imaging; green, SLC25A10; red, mitotracker; blue, DAPI; scale bar, 20 µm; (b) The protein levels of SLC25A10 in cardiomyocytes treated with FAC for indicated time was evaluated by WB; (c) Overexpression of SLC25A10 by pcDNA3.1 eukaryotic vector in cardiomyocytes; (d) Knockdown of SLC25A10 by siRNA in cardiomyocytes; (e) Cell death labeled with PI staining was detected in each group; (f) Cell death labeled with PI staining was detected in each group; (g) Fold changes of the fluorescence intensity of the C11-BO staining in mitochondria in each group; (h) Fold changes of the fluorescence intensity of the C11-BO staining in mitochondria in each group.
Figure 4.
(a) The localization of SLC25A10 detected by immunofluorescence and confocal imaging; green, SLC25A10; red, mitotracker; blue, DAPI; scale bar, 20 µm; (b) The protein levels of SLC25A10 in cardiomyocytes treated with FAC for indicated time was evaluated by WB; (c) Overexpression of SLC25A10 by pcDNA3.1 eukaryotic vector in cardiomyocytes; (d) Knockdown of SLC25A10 by siRNA in cardiomyocytes; (e) Cell death labeled with PI staining was detected in each group; (f) Cell death labeled with PI staining was detected in each group; (g) Fold changes of the fluorescence intensity of the C11-BO staining in mitochondria in each group; (h) Fold changes of the fluorescence intensity of the C11-BO staining in mitochondria in each group.
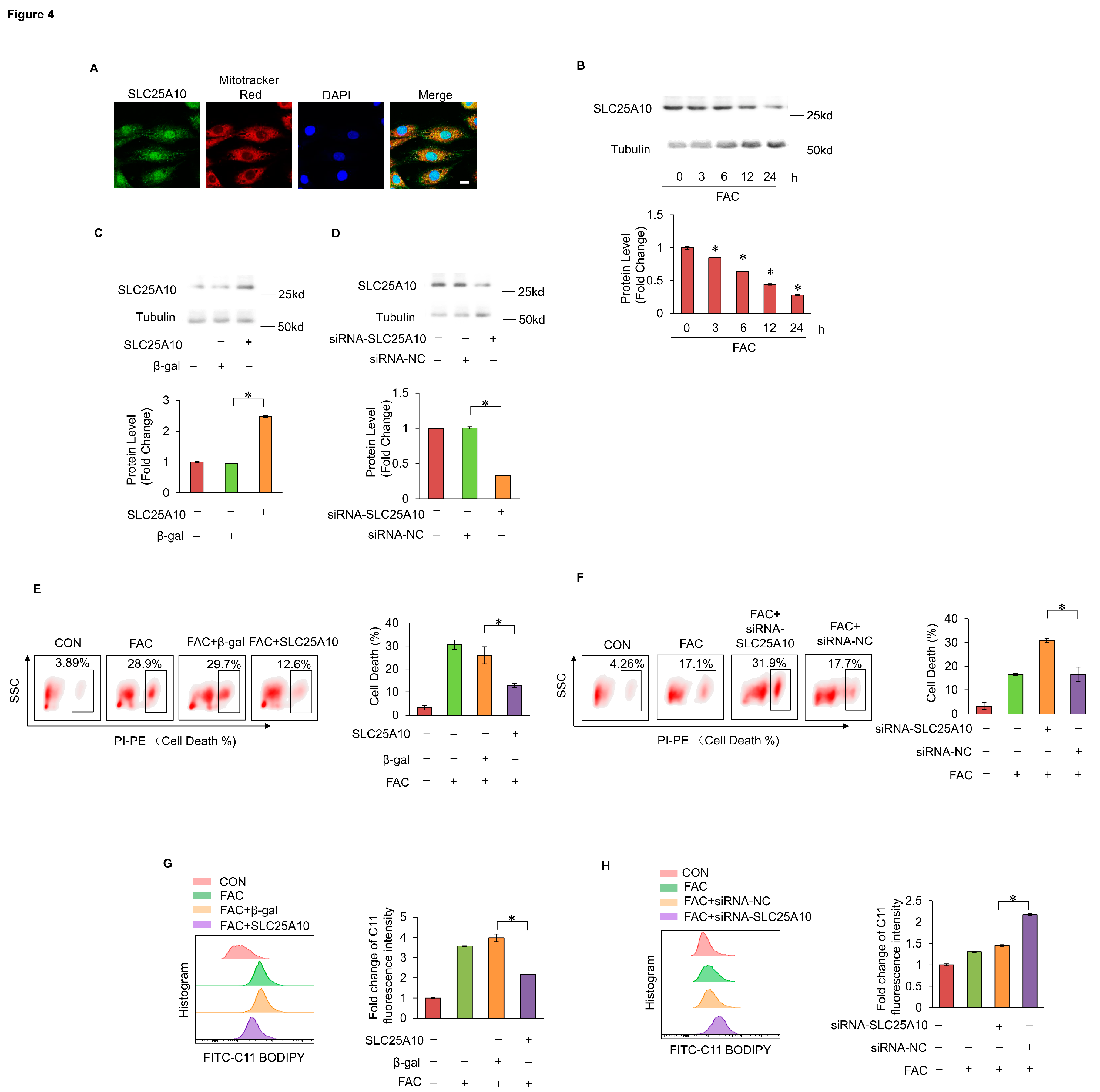
Figure 5.
(a) Colocalization of MPV17 and SLC25A10 detected by immunofluorescence and confocal imaging; green, MPV17; red, SLC25A10; blue, DAPI; scale bar, 20 µm; (b) Immunoprecipitation was performed to detect the interaction of endogenous MPV17 with SLC25A10 in cardiomyocytes; (c) The expression of SLC25A10 were evaluated by WB in each group; (d) WB was performed to detect the ubiquitination of SLC25A10; (e) The ratio of cell death labeled with PI staining in each group; (f) The ratio of cell death labeled with PI staining in each group; (g) Fold changes of the fluorescence intensity of the C11-BO staining in each group; (h) Fold changes of the fluorescence intensity of the C11-BO staining in each group.
Figure 5.
(a) Colocalization of MPV17 and SLC25A10 detected by immunofluorescence and confocal imaging; green, MPV17; red, SLC25A10; blue, DAPI; scale bar, 20 µm; (b) Immunoprecipitation was performed to detect the interaction of endogenous MPV17 with SLC25A10 in cardiomyocytes; (c) The expression of SLC25A10 were evaluated by WB in each group; (d) WB was performed to detect the ubiquitination of SLC25A10; (e) The ratio of cell death labeled with PI staining in each group; (f) The ratio of cell death labeled with PI staining in each group; (g) Fold changes of the fluorescence intensity of the C11-BO staining in each group; (h) Fold changes of the fluorescence intensity of the C11-BO staining in each group.
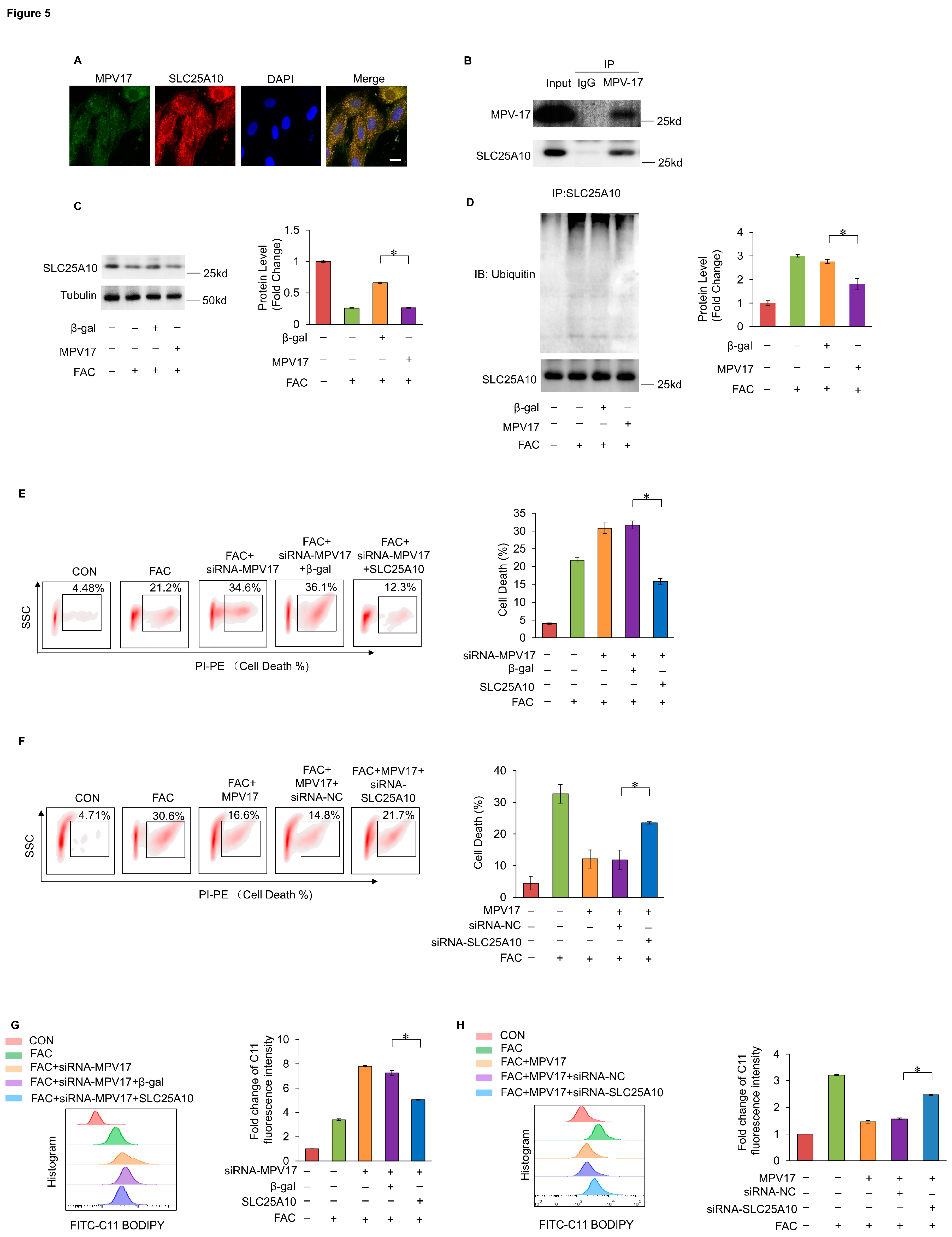
Figure 6.
(a) The expression levels of Nrf2 in cardiomyocytes treated with FAC for indicated time was evaluated by WB; (b) The 5’-flanking sequence of MPV17 contained a potential Nrf2 binding site; Chip assay was performed to examined the binding site of Nrf2 in MPV17 promotor region; (c) Luciferase activity was measured in HEK-293 cells transfected with pGL4-MPV17-WT or pGL4-MPV17-MU along with the Nrf2 overexpression constructs; (d) The expression of MPV17 in cardiomyocytes were evaluated by WB; (e) The percentage of cell death labeled with PI staining; (f) The expression of MPV17 in cardiomyocytes were evaluated by WB; (g) The percentage of cell death labeled with PI staining in each group; (h) Fold changes of the fluorescence intensity of the C11-BO staining in each group; (i) Fold changes of the fluorescence intensity of the C11-BO staining in each group.
Figure 6.
(a) The expression levels of Nrf2 in cardiomyocytes treated with FAC for indicated time was evaluated by WB; (b) The 5’-flanking sequence of MPV17 contained a potential Nrf2 binding site; Chip assay was performed to examined the binding site of Nrf2 in MPV17 promotor region; (c) Luciferase activity was measured in HEK-293 cells transfected with pGL4-MPV17-WT or pGL4-MPV17-MU along with the Nrf2 overexpression constructs; (d) The expression of MPV17 in cardiomyocytes were evaluated by WB; (e) The percentage of cell death labeled with PI staining; (f) The expression of MPV17 in cardiomyocytes were evaluated by WB; (g) The percentage of cell death labeled with PI staining in each group; (h) Fold changes of the fluorescence intensity of the C11-BO staining in each group; (i) Fold changes of the fluorescence intensity of the C11-BO staining in each group.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



