
Submitted:
24 September 2024
Posted:
24 September 2024
You are already at the latest version
Abstract
Fusarium solani KMZW-1, is recognized for its potential as a biocontrol agent against agricultural and forestry pests, particularly due to its compatibility with integrated pest management (IPM) strategies. This study aimed to investigate the complete genome of F. solani KMZW-1 and assess its pathogenicity against Bactrocera dorsalis. Whole genome sequencing revealed a genome size of 47,239,278 bp, comprising 27 contigs, with a GC content of 51.16% and fungus identified as F. solani KMZW-1. The genome completeness was assessed as 97.93% using BUSCO analysis, DFVF sequence identifier was Fusarium 0G092560.1, AntiSMASH analysis identified 29 gene clusters associated with secondary metabolite biosynthesis providing insights into the genetic basis of its pathogenic mechanisms and biocontrol potential. Comparative genomic analysis found 269 unique genes of F. solani KMZW-1 and the collinearity analysis exhibited a high degree of synteny with Fusarium solani-melongenae. The pathogenicity of F. solani KMZW-1 was assessed using concentrations ranging from 1×10^4 to 1×10^11 conidia/mL. Higher concentrations (1×10^10 to 1×10^11 conidia/mL), resulted in significantly increased cumulative mortality rates of B. dorsalis adults compared to the control group. Notably, the pathogenicity was higher in male adults than in females. Probit regression analysis yielded LogLC50 values as 5.662 for fe-male and 4.486 for male B. dorsalis adults. In summary, F. solani KMZW-1 exhibits strong insecti-cidal activity against B. dorsalis and shows potential as a biocontrol agent with IPM strategies. These findings provide robust genomic evidence supporting the use of F. solani KMZW-1 in managing against B. dorsalis populations.
Keywords:
Fusarium solani KMZW-1
; whole genome sequencing
; entomopathogenic fungus
; Bactrocera dorsa-lis
1. Introduction
Fusarium solani KMZW-1, is a well-recognized entomopathogenic fungus belonging to the family Tuberculariaceae; order Hypocreales. Recently, biological control has garnered increased attention for managing agricultural insect pests, due to its environmental friendliness. Biocontrol agents such as F. solani KMZW-1, have become a focal point for experts seeking sustainable pest management solutions. Bactrocera dorsalis (Diptera; Tephritidae), commonly known as the oriental fruit fly, is a significantly destructive and persistent pest affecting over 250 host plants as documented [1]. Effective control of B. dorsalis is crucial for minimizing crop damage and financial losses.
Previous researches have showed the insecticidal potential of biocontrol agents. For example Methoxyfenozide and Beauveria bassiana (Hypocreales; Cordycipitaceae) have demonstrated promising results in controlling B. dorsalis. Chandramohan et al., (2016) reported mortality rates of 67.5%, 65.0% and 63.5% for Methoxyfenozide and 60.0%, for B. bassiana [1]. Similarly, Li et al. (2024) screened six strains of B. bassiana and found mortality rate of 90.67% for adults of oriental fruit fly with most active strain B4 [2]. These studies provide a theoretical basis for exploring new biocontrol agents against B. dorsalis high-lighting the need for further research into the pathogenicity of F. solani KMZW-1.
In recent years, advancement in biotechnology have provided new insights into the success of biological control. Genomic sequencing and comparative genomics analyses have become focal points in studying biocontrol potential of entomopathogenic fungi, offering crucial insights about their biological characteristics and potential applications [3]. For instance, genetic data based fungal co-expression network analysis of the entomopathogenic fungus B. bassiana have revealed the infection associated modules and host pathogen interaction related to its biocontrol potential against insect pests [4]. These findings offers deep understanding of the genetic characteristics, pathogenic mechanisms, and biocontrol potential of entomopathogenic fungus.
Whole genome sequencing has emerged as a powerful tool for understanding the genetic basis of biological traits and behaviors in various organisms. The complete genome analysis of L. attenuatum Strain Lec8 revealed the genome size of 32.38 Mb, containing 9,531 genes, including those encoding proteins related to biosynthetic pathways which suggested strong entomopathogenic potential [5]. Similarly, The genome sequencing of F. solani-melongenae (CRI 24-3) revealed 49.6 Mb chromosome-level draft genome containing 15,374 putative coding genes, Contigs N50, 4,496,268 (bp) and GC content was 50.7%, it also showed relatively high numbers of virulence factors such as carbohydrate-active enzymes (CAZymes), pathogen host interaction (PHI) proteins, and terpene synthases (TSs) [6]. These findings provide significant insights on the molecular basis of fungus though further research is needed for exploring its pathogenic potential. Whole genome sequencing may explore the genetic basis of F. solani KMZW-1 as a biocontrol agent.
Fruit flies poses a major threat to a wide range of fruits and vegetables globally from tropical to subtropical environments due to their polyphagous nature [7,8]. B. dorsalis, in particular is a significant threat to horticultural crops in China and neighboring regions. Biological control of many insect pests including fruit flies, frequently employs entomopathogenic microorganisms [9,10,11,12]. This study aims to explore the entomopathogenic potential of F. solani KMZW-1 against B. dorsalis by utilizing complete genome sequencing and comparative genomics. Additionally, we compared its pathogenicity potential against different sex of B. dorsalis.
2. Materials and Methods
2.1. Fusarium Solani KMZW-1 Culture and Identification
Adult Boettcherisca peregrina flies infected with a wild strain of fungus were collected from a pig farm at Yunnan Agricultural University, Animal Science Institute, Kunming City. Newly emerged 4-day-old B. peregrina flies, disease free and of uniform size, were individually placed in small rearing cages and provided with ample water, and white sugar. They were maintained under controlled conditions of 25±0.5°C, and a (12L:12D) light cycle. The wild strain of fungus was isolated from naturally infected B. peregrina flies and cultured on Potato Dextrose Agar (PDA). The third strain was subsequently, reintroduced to the B. dorsalis flies. The strain was propagated and expanded on PDA medium at 26°C and a pH of 6.0 following Usman et al. (2021) with slight modifications [13].
The DNA of the fungal strain was extracted using the Ark DZ306-02 Fungal DNA Mini Kit (Tsingke Biotechnol-ogy Co., Ltd., Beijing, China). PCR amplification was conducted utilizing the universal primers ITS1 (5′-TCCGTAGGTGAACCTGCGG-3′) and ITS4 (5′-TCCTCCGCTTATTGATATGC-3′) which generated an approximately 700 bp. The amplified products were subsequently analyzed by agarose gel electrophoresis to assess band size accuracy, band singularity, and the presence of any smearing. Upon confirming the expected band size and quality of the PCR products, the target bands were excised and purified. The purified DNA was then subjected to Sanger sequencing. Sequencing results were assembled using ContigExpress software (Version 9.1), trimming off the inaccurate ends. The assembled sequences were subjected to batch BLASTn [14] (Basic Local Alignment Search Tool, version 2.13, available at https://blast.ncbi.nlm.nih.gov/Blast.cgi) to align against nucleotide database. This BLASTn search, conducted against the latest version of the of the version of the nucleotide (nt) database, facilitated the retrieval of accession numbers and the species identification and annotation of homologous sequences.
2.2. Insect Collection and Rearing
The population of B. dorsalis was collected from fallen fruits in a mango orchard located in the suburb of Kunming (latitude 23°36′ N, longitude 101°58′ E, altitude 460.97 meters). Infested fruits were placed on sandy soil until hatching and pupation. After pupation, the pupae were collected and moved indoors. Upon adult emergence, the flies were provided a diet consisting of 10% yeast extract, 5% peptone, 30% white sugar, and 15% agar, and plain water. For, egg collection mating females were allowed to oviposit in plastic bottles lined within stint fruit juices coating the inner walls. The eggs were then transferred to fresh keeping boxes containing larval feed composed of 10% yeast extract, 5% peptone, 30% white sugar, 10% agar, and 40% wheat bran. Larvae were allowed to mature and pupate in the soil. The emerged adults from the third generations were maintained in rearing cages (30cm × 30cm × 30cm) with 100 mesh nylon netting indoor environmental conditions were controlled at a temperature of 25±1°C, relative humidity of 60±10%, and photoperiod of (14L:10D).
2.3. Complete Genome Sequencing, Assembly and Annotation
2.3.1. Genome Sequencing
The experimental process followed the standard protocol provided by Oxford Nanopore Technologies (ONT), encompassing sample quality assessment, library construction, library quality detection and library sequencing by Biomarker Technologies Co., Ltd. (Beijing, China). The library construction process included the following steps as genomic DNA extraction and quality control, fragment recovery, library construction (SQK-LSK109 Sequencing kit) and sequencing. High quality genomic DNA was extracted, and its purity, concentration and integrity were checked using Nanodrop spectrophotometry, Qubit flurometry and 0.35% agarose gel electrophoresis. The BluePippin automatic nucleic acid recovery system was used to recovers the large DNA fragments.
- a)
- DNA damage repair and end repair were performed followed by magnetic bead purification
- b)
- DNA fragments were ligated with adapters followed by another round of, magnetic bead purification
- c)
- Qubit flurometry was used for library quantification
- d)
- Sequencing was done using a computer based system 2.4.2.
2.3.2. Genome Assembly
The filtered sub reads were assembled using NECAT software, and the initial assembly results were corrected using Racon software with the filtered sub reads. Subsequently, Pilon software [15] was utilized to further enhance the assembly accuracy by employing second generation data. The alignment rate was assessed using BWA software [16].
Mapped (%): The percentage of clean reads mapped to the reference genome out of all clean reads was calculated using the Samtools flagstat command.
Properly mapped (%): This metric showed the percentage of paired-end sequencing reads mapped to the reference genome with distances consistent with the expected length distribution of sequencing fragments, checked using the Samtools flagstat command.
Coverage (%): The coverage of the assembled genome, was determined using the Samtools depth command.
Depth (X): The depth of coverage across the assembled genome, was quantified using the Samtools depth command.
After completing the assembly of F. solani KMZW-1 genome, its completeness was done using BUSCO (Benchmarking Universal Single-Copy Orthologs, v3.0.2). The analysis employed the fungi_odb9 database [17], which includes 29,000 single-copy orthologous genes from 185 fungal species. The completeness of the genome assembly was determined by calculating the percentage of complete single-copy genes identified in the F. solani KMZW-1 genome sequence compared to the total number of single-copy genes in the database.
2.3.3. Genome Predictions
LTR_FINDER v1.05 [18], MiX-Hunter [19], RepeatScout v1.0.5 [20], PILER-DF v2.4 [21] were used to construct the repeat sequence database. The database was classified using PASTE Classifier [22], and merged with the Repbase [23] to form the final repeat database Repeat Masker v4.0.6 [24] was then used to predict the repeat sequence.
Gene structure prediction primarily involves ab initio prediction, homology-based protein prediction, and transcriptome data prediction, Ab initio prediction tools used were co Genscan [25], Augustus v2.4 [26], Glimmer HMM v3.0.4 [27], Gene ID v1.4 [28], and SNAP (version 2006-07-28). Homology-based protein prediction was performed using GeMoMa v1.3.1 [29]. Transcriptome based assembly was conducted using Hisat2 v2.0.4 and Stringtie v1.2.3, with TransDecoder v2.0 employed for Unigene sequence prediction. Predicted results were integrated using EVM v1.1.1 [30] and refined using PASA v2.0.2. tRNA genes were predicted using tRNAscan-SE v1.3.1 [31], and drRNA genes were pre-dicted using Infernal v1.1.1 [32].
Genomic scanning was performed using GenBlastA v1.0.1 [33] to filter predicted functional genes. Subsequently, GeneWise v2.2.0 [34] was utilized to identify immature and frame shift mutations, followed by analysis of presumed candidates. antiSMASH v6.0.0 [35] was employed to predict the secondary metabolite gene clusters.
2.3.4. Genome Protein Annotations
The predicted proteins were subjected to the BLAST [36] against various databases: Nr [37], Swiss-Prot [38], TrEMBL [38], KEGG [39], KOG [40] using e value threshold of 1e-5. GO annotations were performed using annotations using Blast2GO [41] based on the result of BLAST searches. Pfam domain were identified using HMMER [42]. In addition, pathogenicity related protein were determined using CAZy [43], TCDB [44], PHI [45,46] CYPED, DFVF [47] and others. Secreted proteins were detected using SignalP 4.0 [48] and transmembrane proteins were filtered by TMHMM [49] to isolate a in candidate secreted proteins. Effector proteins among the P secreted proteins were predicted utilizing [50].
2.4. Comparative Genome Analysis
Comparative genome analysis were conducted to comprehensively understand the distinctions between F. solani KMZW-1 other genomes (GenBank accession number: Fusarium solani-melongenae: GCA_023101225.1, Fusarium solani: GCA_023522795.1, Fusarium oxysporum: GCA_013085055.1, Fusarium sp. Ph1: GCA_025433565.1, Fusarium sp. LHS14.1: GCA_025433615.1). Phylogenetic and molecular evolutionary analyses were performed using MEGA version 11 [51]. OrthoFinder version (2.5.5) was employed to identify single-copy orthologous genes and classify gene families. The analysis was performed using the “-M msa” for the multiple sequence alignment method and “-S Diamond” for the sequence similarity search tool [52]. Genomic collinearity analysis were performed using TBtools v2.025 [53], with visualization of the results conducted through NGenomeSyn [54].
2.5. Pathogenicity Test of Fusarium solani KMZW-1 against Bactrocera Dorsalis
2.5.1. Conidial Suspension Preparation
The fungal strain F. solani KMZW-1, stored at -80° used recovered for fresh culture. A 100 μL aliquot of the fungal suspension was evenly spread onto the PDA solid medium containing kasugamycin using a disposable spreader. The plates were then incubated at a constant temperature of 26°C in an incubator for subsequent use. Initially, a sterile 0.05% (v/v) tween-80 solution was prepared by mixing 200 μL of tween-80 solution and 400 mL of deionized water, then it was sterilized by autoclaving at 115°C for 30 minutes. The conidial suspension derived from shaking flask cultures was filtered through a four-layer sterile gauze to eliminate the mycelial fragments. The resultant filtrate then distributed into 50 mL centrifuge tubes and centrifuged at 4°C at 12,000 rpm for 10 minutes. The supernatant was discarded, and the conidial pellet re-suspended in an adequate volume of the aforementioned 0.05% (v/v) Tween-80 solution. To ensure homogeneity, the suspension mixed vigorously on a vortex mixer for 3 minutes. Concentration determination of the conidia was done by using hemocytometer featuring a 25×16 grid pattern. Each sample undergone triplicate counts to enhance the precision. The targeted concentrations were as 1×104、1×105、1×106、1×107、1×108、1×109、1×1010 and 1×1011 conidia per milliliter. The calculation for the conidial suspension performed according to the following equation:
Conidial suspension = (Total count from five squares) × (Dilution factor) × 5 × 104 (conidia/mL).
2.5.2. Bioassays
The experiment utilized the hit and trial method. A small manual sprayer was employed to uniformly spray the conidial suspensions, adjusted to the distinct concentrations (see Table 1), onto the flies until their cuticles appeared to be visibly moistened. Mortality was monitored every 24 hours for 12 consecutive days. All treated according to Zhang et al. (2021) with slight modifications [55]. To ensure the equal conidial doses applied topically, we continuously shacked the sprayer and 5ml of the conidial suspension per replicate used for each concentration. For the control treatment, sterile 0.05% (v/v) tween-80 solution was sprayed. insects were placed in rearing chambers at a temperature of 26°C, relative humidity of 85%, and a 12:12h photoperiod. Treated insects were given with adult feed (liquid cultured medium) and water throughout the experiment. Each treatment group consisted of 30 insects and all experiments were performed in triplicate. Male and female adults were separated for the experimentation with no age specificity. The dead cadavers of flies were observed for fungal outgrowth (infection) under the light microscope (see Figure 11).
2.6. Data Analysis
The cumulative mortality rate of B. dorsalis was statistically analyzed using Microsoft Office Excel 2021. The LC50 values were calculated using Probit regression and the results were plotted on a logarithmic scale (base 10). Graphs were generated using GraphPad Prism, (USA, 8.3.0538).
3. Results
3.1. ITS-based identification of Fusarium solani KMZW-1
Through strain identification, the KMZW-1 strain is identified as F. solani with a homology of 569/572 (99.48%), the blast alignment score is 1038, and the e-value of the blast alignment result is 0.0. The annotation of the alignment result is F. solani genes for 18S rRNA, ITS1, 5.8S rRNA, ITS2, 28S rRNA, partial and complete sequence, isolate: FKI-6853. Based on these results, the strain is identified as F. solani KMZW-1 commonly known as Fusarium.
3.2. Genome Assembly
The genome assembly of F. solani KMZW-1 resulted in 27 contigs with a total length of 47,239,278 base pairs (bp). Key metrics include an N50 of 2,751,789 bp and an N90 values of 1,018,923 bp, indicating a higher proportion of larger contigs in the assembly and overall good contiguity. The GC content of the genome observed 51.16%, provided insights into its genetic composition (Table 2). Importantly the assembly process gained closure without any gaps showing the of the genome assembly. This achievement is crucial for subsequent analyses such as gene structure analysis, functional annotation, and evolutionary analysis. The statistical results of gene assembly were as follows:
3.3. BUSCO evaluation
We utilized the fungi_odb9 database in BUSCO [17] which contains 290 conserved core genes for fungi to evaluate the completeness of our fungal genome assembly using BUSCO (v2.0). A total of 284 complete BUSCO genes were identified in our assembly, resulting in a genome completeness evaluation of 97.93%. Among these, 281 genes were identified as single-copy, constituting 96.90% of the total BUSCO genes assessed. The presence of these genes in a single copy showed the high accuracy and completeness of the genome assembly. Additionally, 3 genes were identified as multi-copy, accounting for 1.03% of total genes. These genes likely exist in multiple copies or repetitive sequences influencing the assembly process. Four genes were predicted to be incomplete, accounting for 1.38% of total genes. These incomplete genes may result from assembly gaps or missing sequences in the genome as assembly process. Furthermore, 2 genes were not predicted at all, constituting 0.69% of total genes indicating that some conserved genes were not detected during the genome assembly process (Table 3).
3.4. General Database Annotations
Gene sets were annotated using different databases GO Annotation, KEGG Annotation, KOG Annotation, Pfam Annotation, Swissprot Annotation, TrEMBL Annotation, and nr Annotation (Table 4). A total number of 13,867 genes were annotated indicating that the majority of genes obtained at least one functional annotation, providing a solid foundation for subsequent gene function studies For example, annotations varied across data-bases; GO Annotation annotated 9,994 genes, KEGG Annotation annotated 3,775 genes, KOG Annotation annotated 7,059 genes, Pfam Annotation annotated 10,510 genes, Swis-sprot Annotation annotated 8,390 genes, TrEMBL Annotation annotated 13,862 genes, and nr Annotation annotated 13,864 genes. For genes with lengths in the range of 100≤length<300 bp, annotations varied from 913 (KEGG) to 3,733 (All Annotated), indi-cating that even shorter gene fragments can obtain a certain degree of annotation. Howev-er, the genes with lengths greater than or equal to 300 bp, generally received higher anno-tation quantities. For instance, the nr database annotated 9,993 genes, indicating that longer genes are more likely to receive functional annotations.
3.5. GO Annotations
Gene Ontology (GO) annotation classification for the genes of F. solani KMZW-1 given in Figure 1. Biological Process (BP) with 4,444 genes, Cellular Component (CC) with 2,279 genes, and Molecular Function (MF) with 7,603 genes were found. Among these categories, the ‘metabolic process’ category has the highest number of genes, with 1,401 genes, followed by the ‘binding’ category with 938 genes. Additionally, there were categories such as catalytic activity, structural molecule, cellular component, and others, with gene counts of 689, 462, 239, and 218, respectively.
3.6. KOG Annotations
Energy production and conversion (C) contains approximately 1500 genes, the highest among all categories. This suggests that F. solani KMZW-1 extensive energy metabolism pathways essential for adapting to variable environments (Figure 2). Amino acid transport and metabolism (E) included around 1000 genes indicating specialization in amino acid metabolism crucial for protein synthesis and cell growth. Nucleotide transport and metabolism (F) has a similar number of genes as the amino acid metabolism, ap-proximately 1000 genes, reflecting complexity in nucleotide metabolism, crucial for fundamental life activities such as DNA replication, repair, and RNA metabolism. Carbohydrate transport and metabolism (G) also comprises around 1000 genes, indicating F. solani KMZW-1′s ability to effectively utilize and regulate carbohydrate metabolism, crucial for energy production and carbon skeletons. Translation, ribosomal structure, and biogenesis (J) encompasses around 500 genes reflecting F. solani KMZW-1 activity in protein synthesis, vital for its growth and development. Replication, recombination, and repair (L) includes a moderate number of genes, indicating F. solani KMZW-1 mechanisms for maintaining genome stability, crucial for its long-term survival and adaptability. Secondary metabolites biosynthesis, transport, and catabolism (Q) involves relatively fewer genes, but is crucial for production of bioactive compounds in F. solani KMZW-1, potentially influencing pathogenic potential. Function unknown (S) encompasses genes whose functions are yet to be fully elucidated, its presence suggested un-explored functional areas in the genome of F. solani KMZW-1, which could include un-discovered novel functions or specific metabolic pathways.
3.7. KEGG Annotations
F. solani KMZW-1 has the complete metabolic capability to regulate its energy production and storage (see Figure 3). Additionally, the diagram illustrates pathways related to the sugar metabolism, such as Lactate fermentation and Pyruvate fermentation, which may function under anaerobic conditions or specific environmental condition.
3.8. Genetic Information Processing
Significant categories include “Ribosome” (101 genes, approximately 10%), “Spliceosome” (91 genes), and “mRNA surveillance pathway” (46 genes). These data indicate that F. solani KMZW-1 has complex protein synthesis and RNA processing mechanisms, which are crucial for its growth and adaptability (see Figure 4).
Cellular Processes: Processes like, “Endocytosis” (5%) and “Phagosome” (42 genes), which involve the uptake and processing of substances within the cell, likely play crucial role in F. solani KMZW-1 nutrient acquisition and defense mechanisms.
Metabolism: Categories such as “Biosynthesis of amino acids” (166 genes, approximately 16%) and “Purine metabolism” (96 genes) encompass a relatively large number of genes, indicating that F. solani KMZW-1 has complete pathways for amino acid synthesis, essential for cell growth and DNA synthesis.
3.9. DFVF Database Annotation
The Database of Known Fungal Virulence Factors (DFVF), available online at http://sysbio.unl.edu/DFVF, represents a comprehensive resource that aggregates data on the 2,058 genes associated with pathogenicity, derived from a diverse collection of 228 fungal strains encompassing 85 genera’s. In the present annotation endeavor, a total of 3,054 genes encoding factors pertinent to the fungal virulence have been meticulously annotated. Referencing to the Figure 5, the DFVF annotation bubble plot highlights that the gene designated by the sequence identifier Fusarium 0G092560.1 exhibited a strong virulence activity specifically relevant to the infection of insect hosts. It suggests its insecticidal potential against insect hosts.
3.10. Comparative Genomic analysis
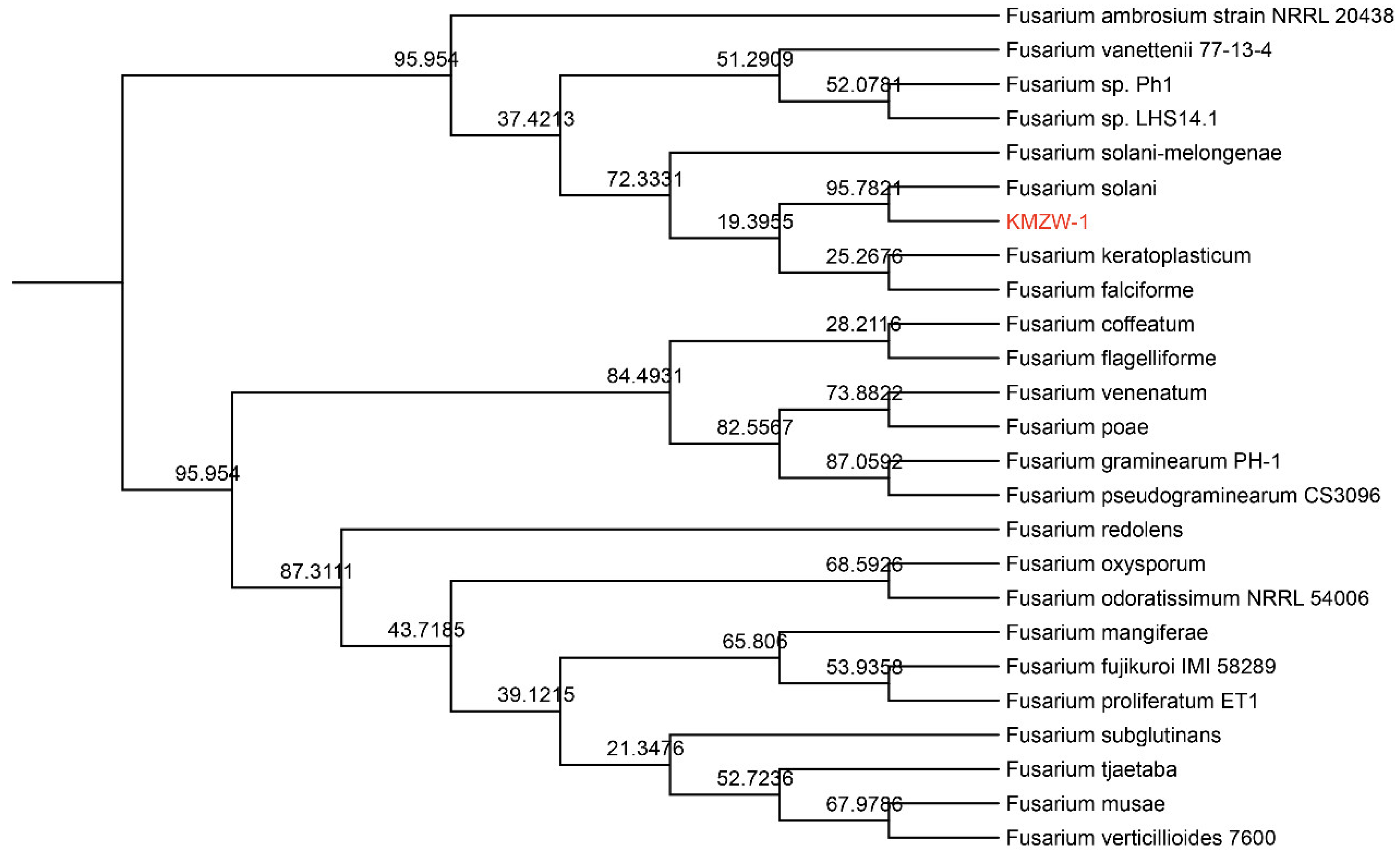
KMZW-1 has the closest phylogenetic relationship with F. solani in the phylogenetic tree, with a bootstrap value of 95.7824, indicating a high level of genetic similarity and the possibility that they share many evolutionary traits and ecological functions. It is also relatively close to the strain F. solani-melongenae, with a bootstrap value of 72.3331. The bootstrap value between F. solani KMZW-1 and F. keratoplasticum is 25.2676, suggesting that although they belong to the same Fusarium genus, they have significant genetic differences and evolutionary divergence. Furthermore, the bootstrap values between F. solani KMZW-1 and F. falciforme and F. coffeatum are 28.2116 and 19.3965, respectively, further indicating that these species have progressively diverged in the evolutionary process, and their phylogenetic relationship is relatively distant. In comparison, F. solani KMZW-1 is more distantly related to F. graminearum (bootstrap value 87.0592) and F. poae (bootstrap value 73.8822), showing greater genetic differences. The bootstrap value of 68.5926 with F. oxysporum indicates significant evolutionary divergence between F. solani KMZW-1 and this group of species. These data suggest that while KMZW-1 has a close evolutionary relationship with F. solani, it has shown different divergence pathways with other species during the evolutionary process, possibly reflecting its adaptive evolution in different ecological environments.
Figure 6.
The Phylogenetic tree of Fusarium solani KMZW-1.
The genomic comparison using OrthoFinder reveals distinct and shared genetic characteristics of F. solani KMZW-1 in relation to other Fusarium strains (Figure 7). Notably, F. solani KMZW-1 contains 269 unique genes, underscoring its genetic distinctiveness and potential for unique ecological adaptations. The strain shares 142 genes with F. solani, indicating a closer evolutionary relationship with this species. In contrast it shares fewer genes with F. solani-melongenae (77 genes), Fusarium sp. Ph1 (115 genes), Fusarium sp. LHS14.1 (82 genes), and the least with F. oxysporum (56 genes), suggesting varying degrees of genetic divergence among these species. The core genome shared across all analyzed strains consists of 8728 genes, likely represent essential functions conserved within the Fusarium species. These findings highlight the unique genetic profile of F. solani KMZW-1 and provides insights into its evolutionary relationships with other Fusarium strains.
The Figure 8 presents a synteny analysis of the genomes of F. solani KMZW-1, F. solani-melongenae, and F. solani. The blue lines depict shared syntenic regions among the three genomes, illustrating a high degree of genomic similarity, and conservation throughout their evolutionary histories. This conservation likely reflects shared biological functions or ecological adaptations among these species. Notably, the synteny between F. solani KMZW-1 and F. solani-melongenae is more extensive, indicating a higher degree of similarity across multiple genomic regions. This suggests that these strain may have been subjected to similar selective pressures or ecological adaptations. In contrast, the synteny with F. solani is relatively sparse, pointing to differences in their evolutionary trajectories. These differences may arise from divergent selective pressures, genome rearrangements, or specific functional evolutionary adaptations, further underscoring the genomic uniqueness of F. solani KMZW-1. These findings provide important genomic evidence for understanding the evolutionary relationships, as well as the ecological and functional characteristics, of these Fusarium strains.
3.11. Pathogenicity Test of Fusarium solani KMZW-1 at Different Concentrations
The cumulative mortality of B. dorsalis adults infected with varying concentrations of F. solani KMZW-1 conidial suspensions (female adults in Figure a, male adults in Figure b) over time are given in Figure 9. The lines represent different concentrations of F. solani KMZW-1 conidial suspensions ranging from 1×104 conidia/mL to 1×1011 conidia/mL, while control group represents the population not infected with the fungus. The cumulative mortality increases over time in all treatment groups, indicating that as time progresses, more B. dorsalis adults were infected. The difference between the control group and the treatment groups indicates that fungal infection was the main cause of increased mortality of B. dorsalis. Regarding the time effect, in the early stages (e.g., first 5 days), the differences in mortality rates among different concentration treatment groups were not significant. However, as the experiment progressed, the mortality rate in the high concentration treatment groups becomes significantly higher than that in the low concentration treatment groups. This indicates that F. solani KMZW-1 requires some time to proliferate within the host and cause death. By comparing the insecticidal rates of different high concentrations against female and male adults of B. dorsalis, it is evident that F. solani KMZW-1 has a significantly higher insecticidal effect against male adults. At each concentration gradient, the insecticidal rate for male adults is approximately 10% higher than that for female adults.
Based on the calculation, the LogLC50 of F. solani KMZW-1 against adult B. dorsalis females was determined as 5.662 and against male adults as 4.486 (Figure 10). A significant increase in the mortality observed as conidial concentration increased from LogC4 to LogC9. Particularly at LogC6 and higher concentrations, the mortality rate rap-idly reaches high levels, suggested a strong lethal effect of F. solani KMZW-1 against B. dorsalis.
Figure 10.
The calculation of lethal concentration for Bactrocera dorsalis adults over 12 days. Note: a: The calculation of lethal concentration for female Bactrocera dorsalis adults;b: The calculation of lethal concentration for male Bactrocera dorsalis adults. While error bars indicate significant differences among different replications of each treatment.
Figure 10.
The calculation of lethal concentration for Bactrocera dorsalis adults over 12 days. Note: a: The calculation of lethal concentration for female Bactrocera dorsalis adults;b: The calculation of lethal concentration for male Bactrocera dorsalis adults. While error bars indicate significant differences among different replications of each treatment.
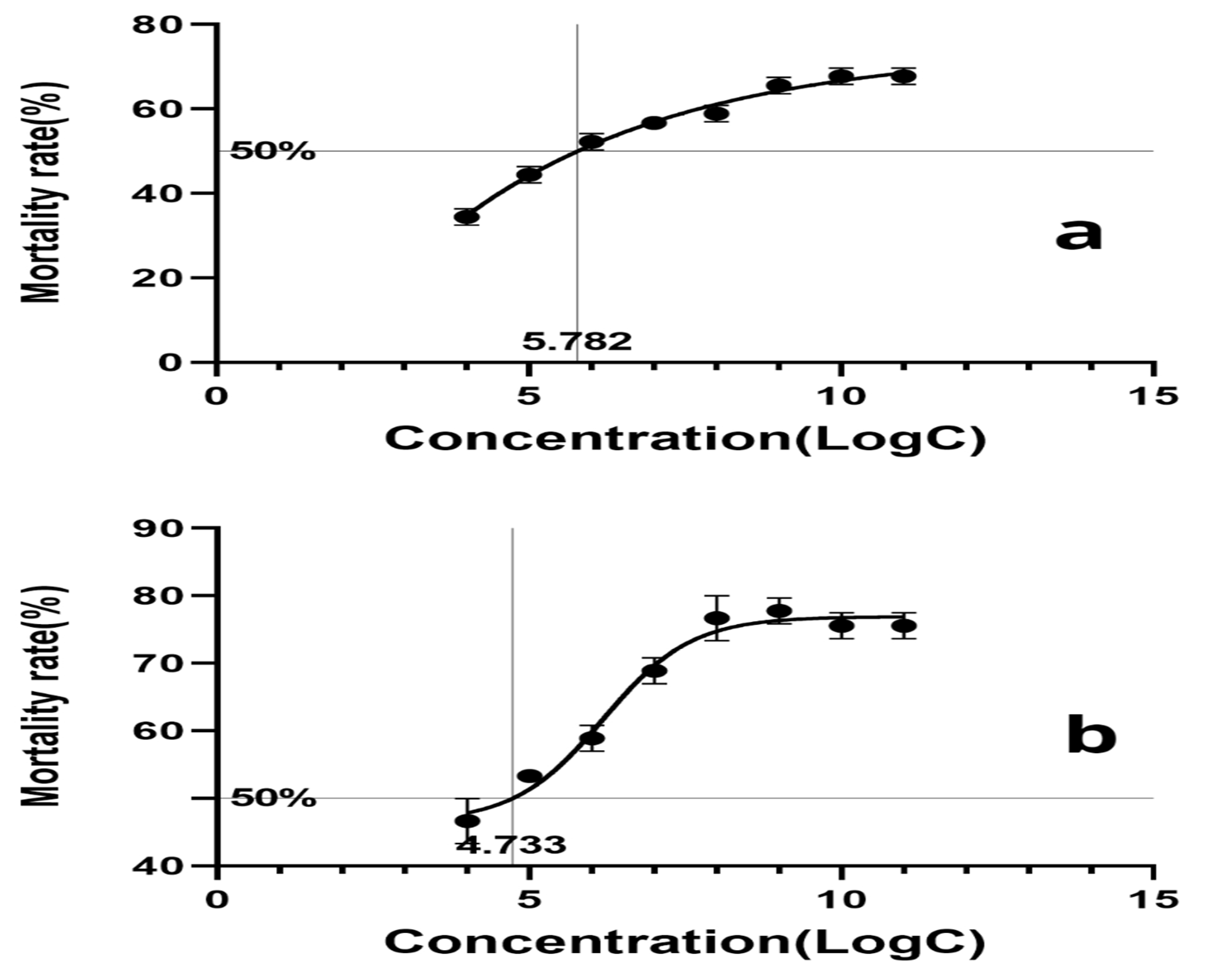
Figure 11.
Dead cadavers of Bactrocera dorsalis adults with fungal outgrowth. .

4. Discussion
In recent years, whole-genome sequencing emerged as important tool for elucidating the genetic characteristics and biological mechanisms of biocontrol fungal strains [56]. Comparative analyses have revealed distinct genomic features among these strains including variations in GC content, numbers of coding genes, and specific gene clusters. In our study, whole genome sequencing revealed a genome size of 47,239,278 bp, comprising 27 contigs, with a GC content of 51.16%. The genome completeness was assessed as 97.93% us-ing BUSCO analysis, DFVF sequence identifier was Fusarium 0G092560.1, AntiSMASH analysis identified 29 gene clusters associated with secondary metabolite biosynthesis providing insights into the genetic basis of its pathogenic mechanisms and biocontrol potential. Our findings align with previous whole genome sequencing studies on different organisms. For example, the whole genome sequence of entomopathogenic fungus B. bassiana JEF-350 revealed a total of 34,655,292 Illumina reads (5,232,949,092 bases), 49.61% G1C content, 8 assembled contigs and 22,202,500 filtered Illumina reads. Annotation of protein coding genes and functional annotation showed that there were 28,999 and 17,771 exons and introns, respectively; while a total of 11,225 protein coding genes were predicted. Total genes annotated for transport and metabolism related to amino acids, nucleotides, coenzymes, carbohydrates, lipids, and inorganic ions in the eggNOG classification were 1,593 [57]. A 49.6 mb chromosome-level draft genome containing 15,374 putatively coding genes was obtained with a GC content of 50.7% and 12 scaffolds by sequencing the pathogenic fungus F. solani-melongenae (CRI 24-3) using third-generation and next-generation sequencing techniques [5]. In another study by Lee et al. (2024), hybrid assembly approach was used to study the whole genome sequencing of B. bassiana strain KNU-101. The genome analysis revealed comprehensive insights into its genetic makeup. The genome sequencing of strain KNU-101 showed a maximum scaffold length 10,066,884 bp with a GC content of 49.49%, 26 contigs and 1,822,896 reads (9,827,426,196 bp) with an N50 read length of 13,949 bp through nanopore sequencing [58]. Similarly, the genome analysis of B. pseudobassiana strain RGM 2184 resulted in 114 genes encoding for extracellular enzymes, four biosynthetic gene clusters reported as producers of insecticidal and bactericidal factors such as oosporein, beauvericin, desmethyl-bassianin, and beau-veriolide. Comparative genomic analysis revealed that 65–95% of these genes are Beauveria genus-specific. Metabolic profiling of supernatant extracts from RGM 2184 cultures exhibited secondary metabolites such as beauveriolide, oosporein, inflatin C, and bassiatin, which were main factors involved in its insecticidal activity [59].
This study performed comprehensive functional annotation of the genome of F. solani KMZW-1, including annotations from GO, KEGG, KOG, and DVDF databases. The results showed 9994 GO annotations, 3775 KEGG annotations, 7059 KOG annotations, and 3054 DVDF annotations. In a similar study, Iwanicki et al., (2022) conducted genome sequencing of Metarhizium humberi (Hypocreales; Clavicipitaceae) in order to get its genomic signatures and insights into host niche adaptation. In M. humberi, 10633 genes were predicted by the genome annotation, of which 92.0% have putative functions assigned to them, and roughly 17% of the genome was marked as repetitive sequences. They discovered that 18.5% of the M. humberi genome resembles proteins linked to pathogen host interaction; while the M. humberi strain ESALQ1638 genome showed some distinct features that set it apart from the genomes of the other 8 Metarhizium species. These features included a greater number of genes functionally annotated as polyketide synthases (PKSs), over represended GO-terms linked to the transport of ions, organic matter, and amino ac-ids, a higher proportion of repetitive elements, and higher levels of RIP-induced point mutations [60]. Similarly, Binneck et al., (2019) studied whole genome sequencing of Metarhizium rileyi (Hypocreales; Clavicipitaceae), strain Cep018-CH2/ARSEF 7053. Results revealed as 31,808,756 bp was the total length of the final assembly, made up of 249 scaffolds, 240 of which were larger than 1,000 bp, and 1,044 contigs. The longest scaffold had a length of 2,535,063 bp while the N50 scaffold had a length of 815,204 bp while the L50 value was 10. There was 51.30% G+C content altogether. Gene prediction and annotation showed 8,945 protein-codings and 102 tRNA genes. antiSMASH analysis revealed 30 gene clusters that were involved in the biosynthesis of specialized metabolites and Functional annotation of the predicted proteins revealed key genes coding for peptidases, carbohydrate-active enzymes, secreted proteins, and transcription factors [61]. Gene annotation in the KOG, GO, KEGG and DVDF databases yielded 7059, 9994, 3775 and 3054 genes, respectively.
F. parceramosum and F. aff. solani with a bootstrap support of 48, and F. vanettenii with 32, indicating a close evolutionary relationship among these species. Additionally, F. solani KMZW-1 forms a clade with F. falciforme, and F. coffeatum are with a bootstrap support of 28.2116 and 19.3965 respectively, highlighting its phylogenetic associations. The similar findings revealed the taxonomic and evolutionary position of M. anisopliae var. anisopliae [62]. OrthoFinder analysis revealed that F. solani KMZW-1 possesses 269 unique genes and shares 142 genes with F. solani, while sharing fewer genes with other strains, indicating varying degrees of genetic divergence. All strains share a core genome of 8728 genes, representing fundamental conserved functions. These reports aligned with Guo et al., (2023), who reported the structure and specific gene categories in the P. herquei genome, and then comprehensively compared with those of the two well-studied Penicillium species, P. decumbens and P. chrysogenum, using OrthoFinder [63]. Additionally, the collinearity analysis between the genomes of F. solani KMZW-1 and F. solani-melongenae exhibited a high degree of synteny. Most of the contigs of F. solani KMZW-1 correspond to the contigs of F. solani-melongenae. These findings provide important evidence for understanding the evolutionary relationships and ecological characteristics of F. solani KMZW-1.
Due to high damaging potential of B. dorsalis, its control often involves multiple tactics including the use of etomopathogenic fungus. In this study, B. dorsalis adult females and males were treated with different conidial suspensions of F. solani KMZW-1 strain at different concentrations (1×107, 1×106, 1×105, 1×104 conidia/mL). After 12 days of treatment, the cumulative average mortality rates of B. dorsalis adults were recorded as follows: 56.67% (68.89%), 52.22% (58.89%), 44.44% (53.33%), and 34.44% (46.67%) for females (males), respectively, compared to a survival rate of 85.56% (80%) in the control group. The calculated LogLC50 for F. solani KMZW-1 against female and male B. dorsalis adults were 5.662 and 4.486, respectively. Furthermore, after treatment with high concentrations (1×1011, 1×1010, 1×109, 1×108 conidia/mL) of F. solani KMZW-1, the cumulative average mortality rates of B. dorsalis adults were significantly higher than the control 14.44% (20%) after 12 days, reaching 67.78% (75.56%), 67.78% (75.56%), 65.56% (77.78%), and 58.89% (76.67%), respectively.
These findings are consistent with the results of many other researchers assessing the pathogenicity of different entomopathogenic fungus against B. dorsalis. For example, Cherry and Moore, (2006) extensively observed the infection of M. anisopliae on B. dorsalis and found that approximately 70% of the treated fruit fly larvae were heavily infected, with a survival rate of only 30% [64]. These results indicated that M. anisopliae has high pathogenicity against B. dorsalis similar to the pathogenic potential observed with the F. solani KMZW-1 in this study. Similarly, seven different Fusarium species were tested against Helicoverpa armigera for their insecticidal potential. In field conditions, all of the collected fungi were insecticidal, however, some were severely lethal to H. armigera larvae. After nine days mortality reached to 91% at concentration 1×108 conidia/mL. While F. solani isolate displayed the highest toxicity against H. armigera larvae [65].
Jackson and Dunlap, (2012) found that Isaria fumosorosea (Hypocreales; Cordycipita-ceae) treatment significantly reduced the survival rate of B. dorsalis larvae upto 40% with an infection rate of 90% [66]. Similarly, Murtaza et al., (2022) tested the efficacy of three entompathogenic fungi including M. anisopliae, B. bassiana and V. lecanii, against Bactrocera zonata ((Diptera: Tephritidae) stages under different laboratory conditions. They found that B. bassiana and M. anisopliae were more pathogenic to all stages of the fruit fly as compared to V. lecanii. Following exposure to concentrations of 1 × 1010 conidia/ml of B. bassiana, the highest rates of mortality were observed for the 3rd larval instar and the pupal stage, with 68.67% and 89.67%, respectively; while adults were most susceptible to all three fungus. However, at 1 × 1010 conidial concentration, M. anisopliae was more virulent against adult B. zonata flies as compared to B. bassiana and V. lecanii [67]. In our studies, F. solani KMZW-1 exhibited a concentration-dependent effect on both male and female adults of B. dorsalis with a markedly higher impact on male adults. This suggests the strong potential of F. solani KMZW-1 for use in integrated pest management programs targeting B. dorsalis.
5. Conclusions
The fungus identified as F. solani KMZW-1 and its whole genome sequencing revealed a genome size of 47,239,278 bp, comprising 27 contigs, with a GC content of 51.16% and a completeness as 97.93%. Additionally, (DFVF) sequence identifier Fusarium 0G092560.1 and 29 gene clusters associated with secondary metabolite biosynthesis were identified. Comparative genomic analysis revealed that F. solani KMZW-1 possess 269 unique genes and the collinearity analysis of exhibited a high degree of synteny with F. solani-melongenae. These findings provide crucial insights into the genetic characteristics, pathogenic mechanisms, and bio-control potential of F. solani KMZW-1. Subsequently, pathogenicity of F. solani KMZW-1 against B. dorsalis demonstrated a concentration dependent increase in mortality rates. Notably the pathogenicity of F. solani KMZW-1 was significantly higher in male adults compared to females with LogLC50 values of 5.662 and 4.486 for female and male B. dorsalis adults, respectively. These findings provides robust genomic evidence supporting the potential of F. solani KMZW-1 for integrated pest management strategies against B. dorsalis.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Authors Contribution: J.Y. and M.H. contributed equally to this work. They jointly developed the research methodology, carried out the experiments, analyzed and interpreted the data, and drafted the initial manuscript. Both authors were actively involved in revisions and critical discussions throughout the manuscript preparation process. D.Q., M.W., C.S., L.S., S.H., and M.J. provided critical feedback on the experimental design, assisted in data interpretation, and helped in writing. G.W. and C.X. supervised the project, secured funding, and provided overall guidance and mentorship. All authors have read and agreed to the submitted version of the manuscript.
Funding
This work was supported by National Natural Science Foundation of China (32060639, 32060640, and 32260704), Reserve Talents Project for Yunnan Young and Middle-aged Academic and Technical Leaders (202105AC160037), and Yunnan Fundamental Research Projects (202401AT070240).
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Acknowledgments
We thank the editors and anonymous referees for their invaluable comments and suggestions. We would like to express our gratitude to Dr. Abid Ali, Associate Professor, Department of Entomology, University of Agriculture Faisalabad, Pakistan for commenting on earlier draft of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Meng, L.W.; Yuan, G.R.; Lu, X.P.; Jing, T.X.; Zheng, L.S.; Yong, H.X.; Wang, J.J. Two delta class glutathione S-transferases involved in the detoxification of malathion in Bactrocera dorsalis (Hendel). Pest Management Science. 2019, 75, 1527–1538. [CrossRef]
- Li XL, Zhang JJ, Li DD, Cai XY, Qi YX, Lu YY. Toxicity of Beauveria bassiana to Bactrocera dorsalis and effects on its natural predators. Frontiers in Microbiology. 2024 May 2;15:1362089. [CrossRef] [PubMed] [PubMed Central]
- Zhang L, Chen X, Hou Q, Et Al. 2018. Genome Sequencing And Comparative Genomics Reveal The Potential Pathogenic Mechanism Of Cercospora Sojina Hara On Soybean. Scientific Reports [J], 8: 1796.
- Huang S, Zhao X, Luo Z, Tang X, Zhou Y, Keyhani N, Zhang Y. Fungal co-expression network analyses identify pathogen gene modules associated with host insect invasion. Microbiology Spectrum. 2023 Sep 1;11(5):e0180923. [CrossRef] [PubMed] [PubMed Central]
- Cao Y, Wang Y, Feng M G 2020. Whole-Genome Sequence Of Lecanicillium Attenuatum Strain Lec8, An Entomopathogenic Fungus With High Potential As A Biocontrol Agent. *Microbiology Resource Announcements [J],9:E01480-01499. [CrossRef]
- Xie S Y, Ma T, Zhao N, Zhang X, Fang B, Huang L. Whole-Genome Sequencing and Comparative Genome Analysis of Fusarium solani-melongenae Causing Fusarium Root and Stem Rot in Sweet potatoes. Microbiology Spectrum. 2022 Aug 31;10(4):e0068322. [CrossRef] [PubMed] [PubMed Central]
- Weems, Jr., Howard V., John B. Heppner, Thomas R. Fasulo, and James L. Nation. 2004. “Caribbean Fruit Fly, Anastrepha Suspensa (Loew) (Insecta: Diptera: Tephritidae): EENY196/IN353, 3/2001”. EDIS 2004 (5). Gainesville, FL. [CrossRef]
- Vargas R.I., Piñero J.C., Leblanc L. An Overview Of Pest Species Of Bactrocera Fruit Flies (Diptera: Tephritidae) And The Integration Of Biopesticides With Other Biological Approaches For Their Management With A Focus On The Pacific Region. Insects. 2015;6:297–318. [CrossRef]
- Usman M., Gulzar S., Wakil W., Piñero J.C., Leskey T.C., Nixon L.J., Oliveira-Hofman C., Wu S., Shapiro-Ilan D. Potential Of Entomopathogenic Nematodes Against The Pupal Stage Of The Apple Maggot Rhagoletis Pomonella (Walsh) (Diptera:Tephritidae)Journal of Nematology. 2020 52:E2020–E2079. [CrossRef]
- Gulzar S., Wakil W., Shapiro-Ilan D.I. Combined Effect Of Entomopathogens Against Thrips Tabaci Lindeman (Thysanoptera: Thripidae): Laboratory, Greenhouse And Field Trials. Insects. 2021;12:456. [CrossRef]
- Wakil W., Tahir M., Al-Sadi A.M., Shapiro-Ilan D. Interactions Between Two Invertebrate Pathogens: An Endophytic Fungi And Externally Applied Bacterium. Frontiers in Microbiology. 2020;11:2624. [CrossRef]
- Tahir M., Wakil W., Ali A., Sahi S.T. Pathogenicity Of Beauveria Bassiana, And Metarhizium Anisopliae Isolates Against Larvae Of The Polyphagous Pest Helicoverpa Armigera. Entomologia Generalis. 2019;38:225–242. [CrossRef]
- Usman M, Wakil W, Piñero JC, Wu S, Toews MD, Shapiro-Ilan DI. Evaluation of Locally Isolated Entomopathogenic Fungi against Multiple Life Stages of Bactrocera zonata and Bactrocera dorsalis (Diptera: Tephritidae): Laboratory and Field Study. Microorganisms. 2021 Aug 23;9(8):1791. [CrossRef] [PubMed] [PubMed Central]
- Altschul S F, Gish W, Miller W, Myers EW, Lipman D J. Basic local alignment search tool. Journal of Molecular Biology. [J] 1990 Oct 5;215(3):403-10. [CrossRef] [PubMed]
- Walker B J, Abeel T, Shea T, Et Al. 2014. Pilon: An Integrated Tool For Comprehensive Microbial Variant Detection And Genome Assembly Improvement. Plos One [J], 9: E112963. [CrossRef]
- Li H, Durbin R 2009. Fast And Accurate Short Read Alignment With Burrows-Wheeler Transform. Bioinformatics [J], 25: 1754-1760. oi: 10.1093/bioinformatics/btp324.
- Simão F A, Waterhouse R M, Ioannidis P, Et Al. 2015. BUSCO: Assessing Genome Assembly And Annotation Completeness With Single-Copy Orthologs. Bioinformatics [J], 31: 3210-3212. [CrossRef]
- Xu Z, Wang H 2007. LTR_FINDER: An Efficient Tool For The Prediction Of Full-Length LTR Retrotransposons. Nucleic Acids Reserch [J], 35: W265-268. [CrossRef]
- Han Y, Wessler S R 2010. Mite-Hunter: A Program For Discovering Miniature Inverted-Repeat Transposable Elements From Genomic Sequences. Nucleic Acids Research [J], 38: E199. [CrossRef]
- Price A L, Jones N C, Pevzner P A 2005. De Novo Identification Of Repeat Families In Large Genomes. Bioinformatics [J], 21 Suppl 1: I351-358. [CrossRef]
- Edgar R C, Myers E W 2005. PILER: Identification And Classification Of Genomic Repeats. Bioinformatics [J], 21 Suppl 1: I152-158. [CrossRef]
- Wicker T, Sabot F, Hua-Van A, Et Al. 2009. Reply: A Unified Classification System For Eukaryotic Transposable Elements Should Reflect Their Phylogeny. Nature Review Genetics. [J], 10: 276-276. [CrossRef]
- Jurka J, Kapitonov V V, Pavlicek A, Et Al. 2005. Repbase Update, A Database Of Eukaryotic Repetitive Elements. Cytogenetic And Genome Research. 110: 462-467. [CrossRef]
- Tarailo-Graovac M, Chen N 2009. Using Repeatmasker To Identify Repetitive Elements In Genomic Sequences. Current Protocols in Bioinformatics [J], Chapter 4: 4.10.11-14.10.14. [CrossRef]
- Burge C, Karlin S 1997. Prediction Of Complete Gene Structures In Human Genomic DNA. Journal of Molecular Biology. [J], 268: 78-94. [CrossRef]
- Stanke M, Waack S 2003. Gene Prediction With A Hidden Markov Model And A New Intron Submodel. Bioinformatics [J], 19: Ii215-Ii225. [CrossRef]
- Majoros W H, Pertea M, Salzberg S L 2004. Tigrscan And Glimmerhmm: Two Open Source Ab Initio Eukaryotic Gene-Finders. Bioinformatics [J], 20: 2878-2879. [CrossRef]
- Alioto T, Blanco E, Parra G, Et Al. 2018. Using Geneid To Identify Genes. Current Protoc Bioinformatics [J], 64: E56. [CrossRef]
- Keilwagen J, Wenk M, Erickson J L, Et Al. 2016. H: Using Intron Position Conservation For Homology-Based Gene Prediction. Nucleic Acids Research [J], 44: E89-E89. [CrossRef]
- Haas B J, Salzberg S L, Zhu W, Et Al. 2008. Automated Eukaryotic Gene Structure Annotation Using Evidencemodeler And The Program To Assemble Spliced Alignments. Genome Biology [J], 9: R7. [CrossRef]
- Lowe T M, Eddy S R 1997. Trnascan-SE: A Program For Improved Detection Of Transfer RNA Genes In Genomic Sequence. Nucleic Acids Research. [J], 25: 955-964. [CrossRef]
- Nawrocki E P, Eddy S R 2013. Infernal 1.1: 100-Fold Faster RNA Homology Searches. Bioinformatics [J], 29: 2933-2935. [CrossRef]
- She R, Chu J S C, Wang K, Et Al. 2009. Genblasta: Enabling BLAST To Identify Homologous Gene Sequences. Genome Research. [J], 19: 143-149. [CrossRef]
- Birney E, Clamp M, Durbin R 2004. Genewise And Genomewise. Genome Research. [J], 14: 988-995. [CrossRef]
- Blin K, Shaw S, Kloosterman A M, Et Al. 2021. Antismash 6.0: Improving Cluster Detection And Comparison Capabilities. Nucleic Acids Research. [J], 49: W29-W35. [CrossRef]
- Altschul S F, Madden T L, Schäffer A A, Et Al. 1997. Gapped BLAST And PSI-BLAST: A New Generation Of Protein Database Search Programs. Nucleic Acids Research. [J], 25: 3389-3402. [CrossRef]
- Deng Y Y, Li J Q, Wu S F 2006. Integrated Nr Database In Protein Annotation System And Its Localization. J]. Comput Engineerig [J], 32: 71-74. [CrossRef]
- Boeckmann B, Bairoch A, Apweiler R, Et Al. 2003. The SWISS-PROT Protein Knowledgebase And Its Supplement Trembl In 2003. Nucleic Acids Research. [J], 31: 365-370. [CrossRef]
- Kanehisa M, Goto S, Kawashima S, Et Al. 2004. The KEGG Resource For Deciphering The Genome. Nucleic Acids Research. [J], 32: D277-280. [CrossRef]
- Tatusov R L, Galperin M Y, Natale D A, Et Al. 2000. The COG Database: A Tool For Genome-Scale Analysis Of Protein Functions And Evolution. Nucleic Acids Research. [J], 28: 33-36. [CrossRef]
- Conesa A, Götz S, García-Gómez J, Et Al. 2004. Blast2GO: A Universal Annotation And Visualization Tool For Functional Genomics Research. Bioinformatics. 15;21(18):3674-6. [CrossRef]
- Eddy S R 1998. Profile Hidden Markov Models. Bioinformatics [J]: 755-763. [CrossRef]
- Cantarel B L, Coutinho P M, Rancurel C, Et Al. 2009. The Carbohydrate-Active Enzymes Database (Cazy): An Expert Resource For Glycogenomics. Nucleic Acids Research [J]: D233-D238. [CrossRef]
- Saier M H, JR., Tran C V, Barabote R D 2006. TCDB: The Transporter Classification Database For Membrane Transport Protein Analyses And Information. Nucleic Acids Research. [J], 34: D181-186. [CrossRef]
- Winnenburg R 2006. PHI-Base: A New Database For Pathogen Host Interactions. Nucleic Acids Research. [J], 34: D459-D464. [CrossRef]
- Fischer M, Knoll M, Sirim D, Et Al. 2007. The Cytochrome P450 Engineering Database: A Navigation And Prediction Tool For The Cytochrome P450 Protein Family. Bioinformatics [J], 23: 2015-2017. [CrossRef]
- Lu T, Yao B, Zhang C 2012. DFVF: Database Of Fungal Virulence Factors. Database [J], 2012: Bas032-Bas032. [CrossRef]
- Petersen T N, Brunak S, Von Heijne G, Et Al. 2011. Signalp 4.0: Discriminating Signal Peptides From Transmembrane Regions. Nature Methods [J], 8: 785-786. [CrossRef]
- Krogh A, Larsson B, Von Heijne G, Et Al. 2001. Predicting Transmembrane Protein Topology With A Hidden Markov Model: Application To Complete Genomes. Journal of Molecular Biology. [J], 305: 567-580. [CrossRef]
- Sperschneider J, Gardiner D M, Dodds P N, Et Al. 2016. Effectorp: Predicting Fungal Effector Proteins From Secretomes Using Machine Learning. New Phytologist. [J], 210: 743-761. [CrossRef]
- Tamura K, Stecher G, Kumar S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Molecular Biology and Evolution. 2021 Jun 25;38(7):3022-3027. [CrossRef] [PubMed] [PubMed Central]
- Emms D M , Kelly S . OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy[J].Genome biology, 2015, 16(157):157. [CrossRef]
- Chen C., Wu Y., Li J., Wang X., Zeng Z., Xu J., Liu Y., Feng J., Chen H., He Y., and Xia R. (2023). TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Molecular Plant. 16,1733–1742. [CrossRef]
- He W, Yang J, Jing Y, Xu L, Yu K, Fang X. NGenomeSyn: an easy-to-use and flexible tool for publication-ready visualization of syntenic relationships across multiple genomes. Bioinformatics. 2023 Mar 1;39(3):btad121. [CrossRef] [PubMed] [PubMed Central]
- Zhang, Z.; Zheng, C.; Keyhani, N.O.; Gao, Y.; Wang, J. Infection of the Western Flower Thrips, Frankliniella occidentalis, by the Insect Pathogenic Fungus Beauveria bassiana. Agronomy 2021, 11, 1910. [CrossRef]
- Taylor J.W., Fisher M.C. Fungal multilocus sequence typing: It’s not just for bacteria. Current Opinion in Microbiology. 2003;6:351–356. [CrossRef]
- Kim JC, Park SE, Lee SJ, Kim JS. Whole-Genome Sequence of Beauveria bassiana JEF-350, a Strain with High Insecticidal Activity against Melon Thrips (Thrips palmi). Microbiology Resource Announcements. 2022 Sep 15;11(9):e0047022. [CrossRef]
- Lee G, Ibal JC, Park T, Kim M, Choi S, Shin J. 2024. Whole-genome sequencing of Beauveria bassiana KNU-101 using the hybrid assembly approach. Microbiology Resource Announcment. 13:e00681-23. [CrossRef]
- Altimira F, Arias-Aravena M, Jian L, Real N, Correa P, González C, Godoy S, Castro JF, Zamora O, Vergara C, et al. Genomic and Experimental Analysis of the Insecticidal Factors Secreted by the Entomopathogenic Fungus Beauveria pseudobassiana RGM 2184. Journal of Fungi. 2022; 8(3):253. [CrossRef]
- Iwanicki NSA, Botelho ABRZ, Klingen I, Júnior ID, Rossmann S, Lysøe E. Genomic signatures and insights into host niche adaptation of the entomopathogenic fungus Metarhizium humberi. G3 (Bethesda). 2022 Feb 4;12(2):jkab416. [CrossRef]
- Binneck E, Lastra CCL, Sosa-Gómez DR. Genome Sequence of Metarhizium rileyi, a Microbial Control Agent for Lepidoptera. Microbiology Resource Announcements. 2019 Sep 5;8(36):e00897-19. [CrossRef]
- Nam, J.-S., Lee, D.-H., Lee, K. H., Park, H.-M. in Bae, K. S. (1998). Cloning and phylogenetic analysis of chitin synthase genes from the insect pathogenic fungus, Metarhizium anisopliae var. anisopliae. FEMS Microbiology Letters, 159(1), 77-84. [CrossRef]
- Guo, W., Wang, W., Tang, J., Li, T. in Li, X. (2023). Genome analysis and genomic comparison of a fungal cultivar of the nonsocial weevil Euops chinensis reveals its plant decomposition and protective roles in fungus-farming mutualism. Frontiers in Microbiology, 14, 1048910. [CrossRef]
- Cherry A J, Moore D 2006. Infection Of The Non-Target Invertebrate Pests Bactrocera Dorsalis And Bactrocera Cucurbitae (Diptera: Tephritidae) With Metarhizium Anisopliae And Beauveria Bassiana. Biocontrol Science And Technology [J], 16: 701-713.
- Mantzoukas, S.; Kitsiou, F.; Lagogiannis, I.; Eliopoulos, P.A. Potential Use of Fusarium Isolates as Biological Control Agents: Helicoverpa armigera (Hübner) (Lepidoptera: Noctuidae) Case Study. Appl. Sci. 2022, 12, 8918. [CrossRef]
- Jackson M A, Dunlap C A 2012. Laboratory Evaluation Of Isaria Fumosorosea CCM 8367 For Controlling Bactrocera Dorsalis (Diptera: Tephritidae). Florida Entomologist [J], 95: 62-69. [CrossRef]
- Murtaza G, Naeem M, Manzoor S, Khan HA, Eed EM, Majeed W, Ahmed Makki H, Ramzan U, Ummara UE. Biological control potential of entomopathogenic fungal strains against peach Fruit fly, Bactrocera zonata (Saunders) (Diptera: Tephritidae). PeerJ. 2022 Apr 22;10:e13316. [CrossRef]
Figure 1.
Category statistics of GO annotations. Note: The horizontal coordinate is the content of GO classification, the left side of the vertical coordinate is the percentage of the number of genes, and the right side is the number of genes. This figure shows the gene enrichment of each secondary function of GO in the context of all genes, reflecting the status of each secondary function in this context.
Figure 1.
Category statistics of GO annotations. Note: The horizontal coordinate is the content of GO classification, the left side of the vertical coordinate is the percentage of the number of genes, and the right side is the number of genes. This figure shows the gene enrichment of each secondary function of GO in the context of all genes, reflecting the status of each secondary function in this context.
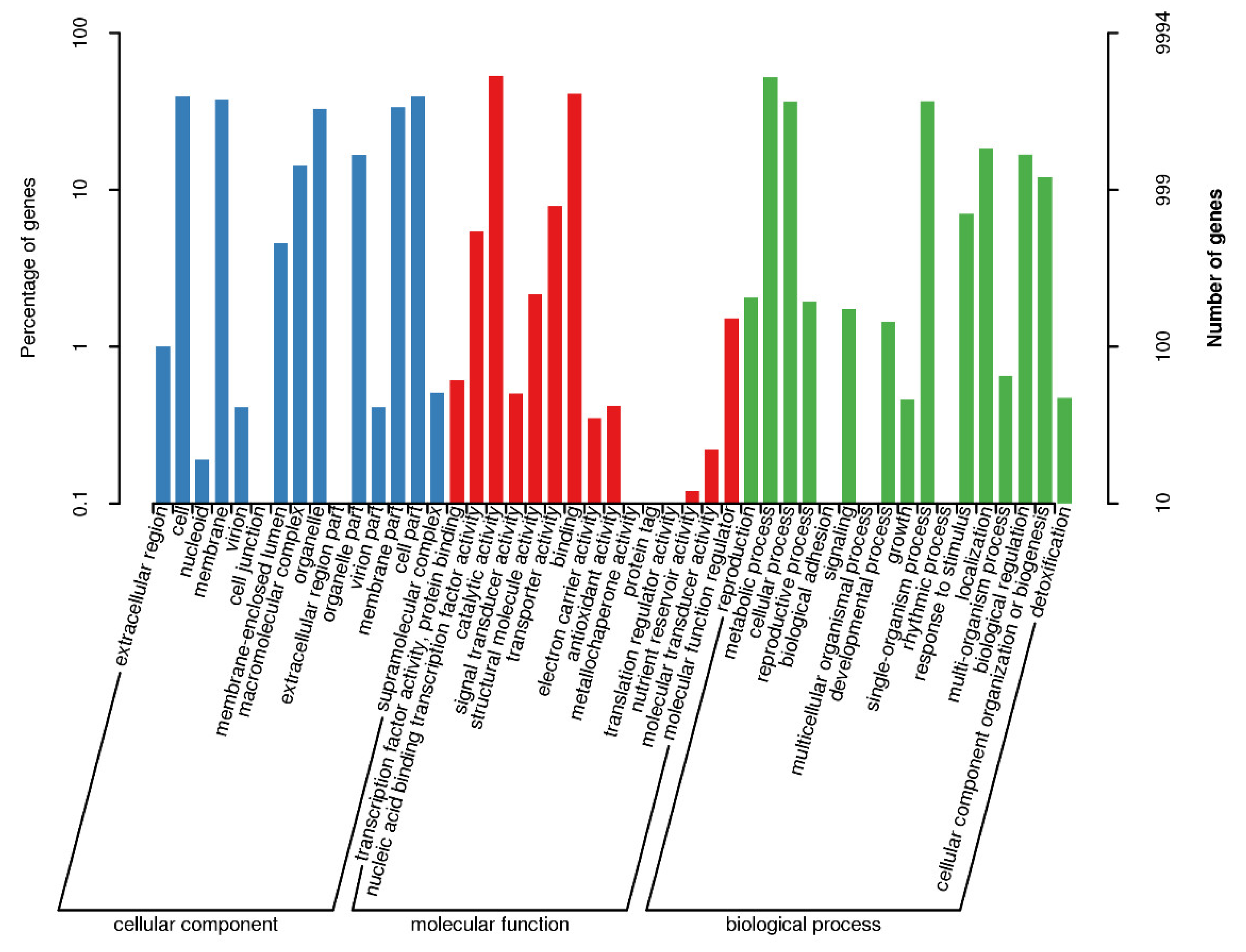
Figure 2.
Category statistics of KOG function annotations. Note: The horizontal coordinate is the classification content of KOG, and the vertical coordinate is the number of genes. In different functional categories, the proportion of genes reflects the metabolic or physiological bias in the corresponding period and environment, which can be scientifically explained in combination with the distribution of research objects in each functional category.
Figure 2.
Category statistics of KOG function annotations. Note: The horizontal coordinate is the classification content of KOG, and the vertical coordinate is the number of genes. In different functional categories, the proportion of genes reflects the metabolic or physiological bias in the corresponding period and environment, which can be scientifically explained in combination with the distribution of research objects in each functional category.
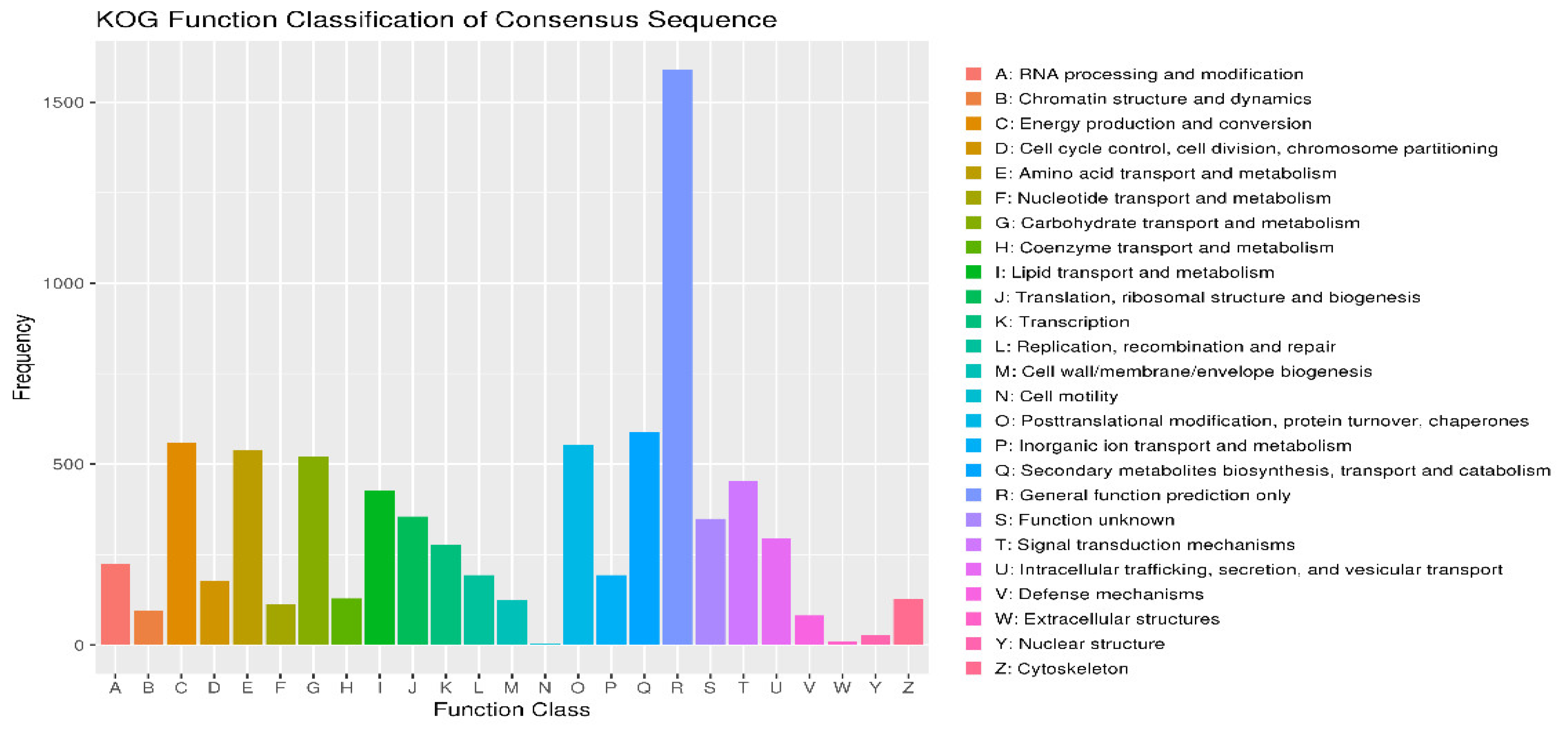
Figure 3.
Schematic diagram of KEGG metabolic pathway results. Note: The number in the box represents the EC number of the key enzyme in the metabolic pathway, and the blue box indicates the presence of the coding gene corresponding to the enzyme in the genome.
Figure 3.
Schematic diagram of KEGG metabolic pathway results. Note: The number in the box represents the EC number of the key enzyme in the metabolic pathway, and the blue box indicates the presence of the coding gene corresponding to the enzyme in the genome.
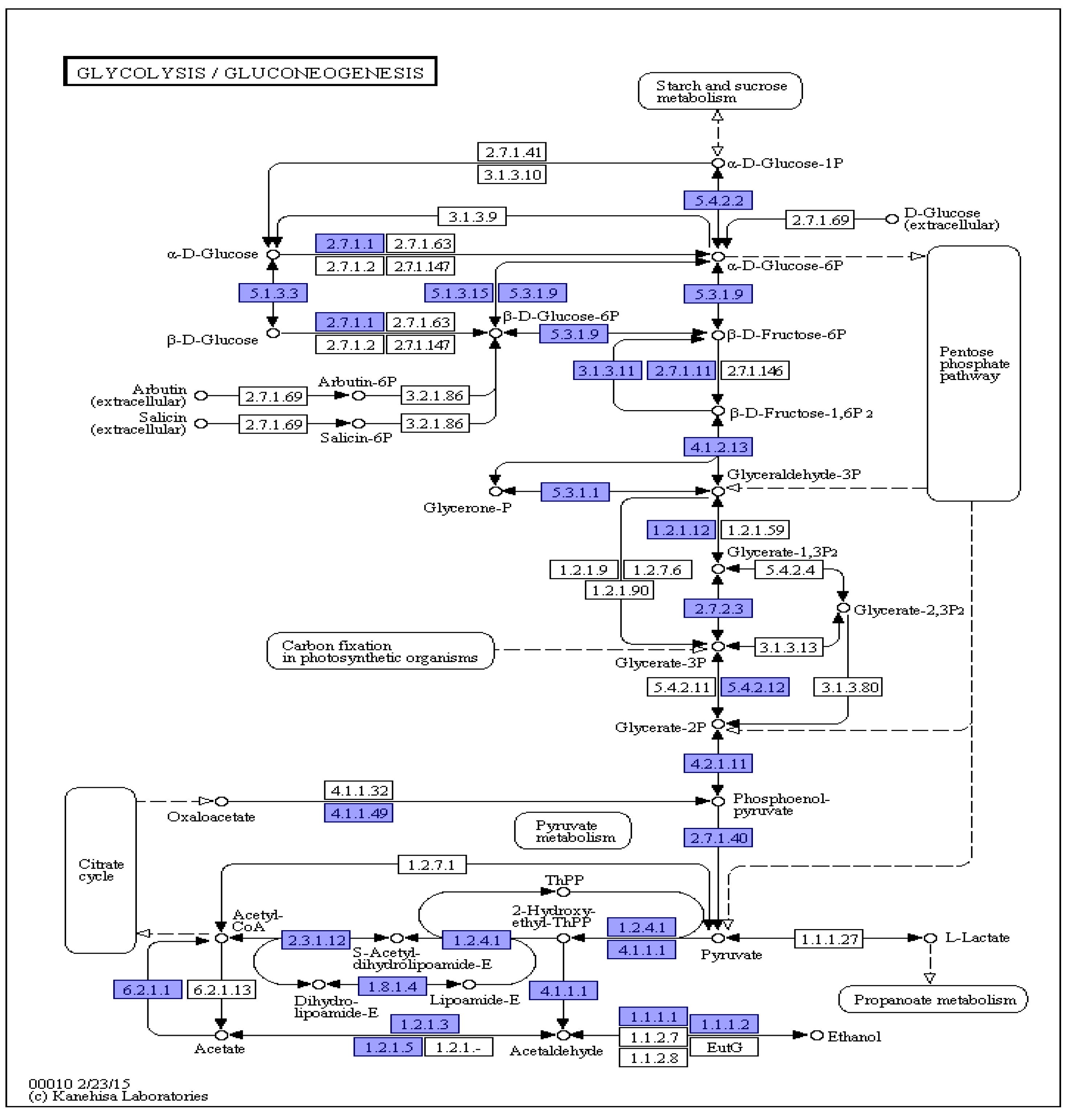
Figure 4.
Classification statistics of KEGG annotations. Note: The vertical coordinate is KEGG secondary classification, and the horizontal coordinate is percentage.
Figure 4.
Classification statistics of KEGG annotations. Note: The vertical coordinate is KEGG secondary classification, and the horizontal coordinate is percentage.
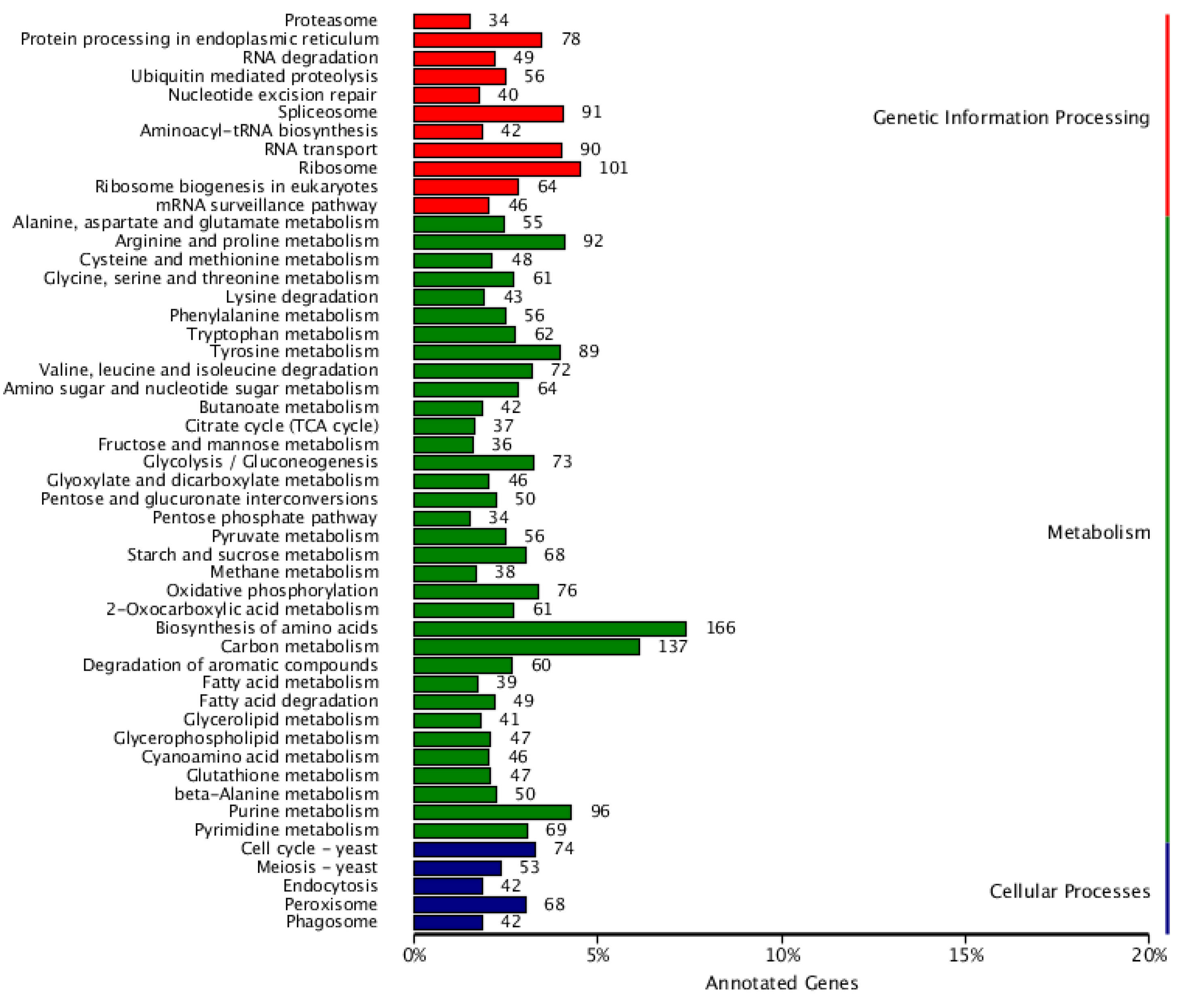
Figure 5.
DFVF Annotation Results of Fusarium solani KMZW-1.
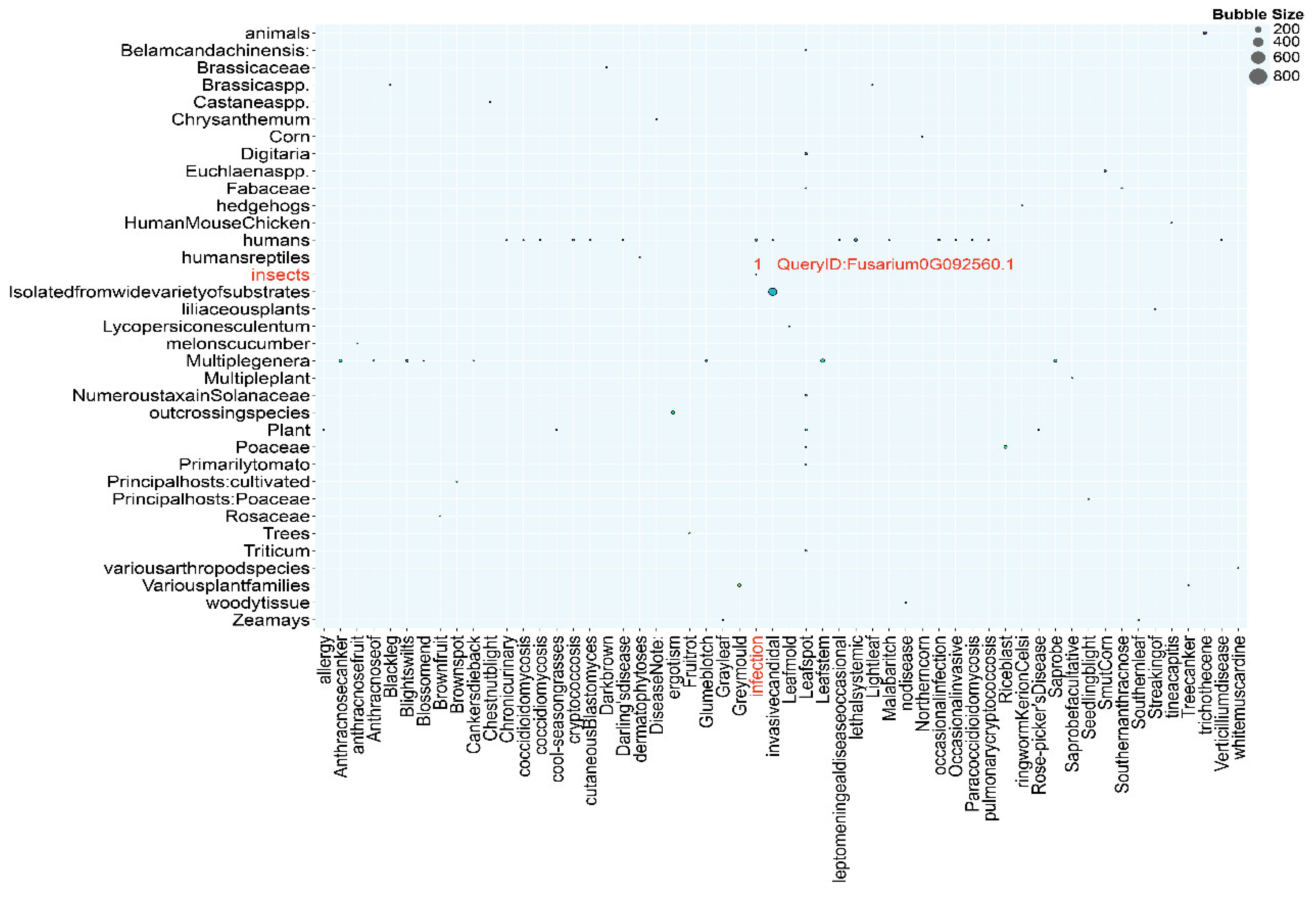
Figure 7.
Comparative Analysis of Homologous Genomes.
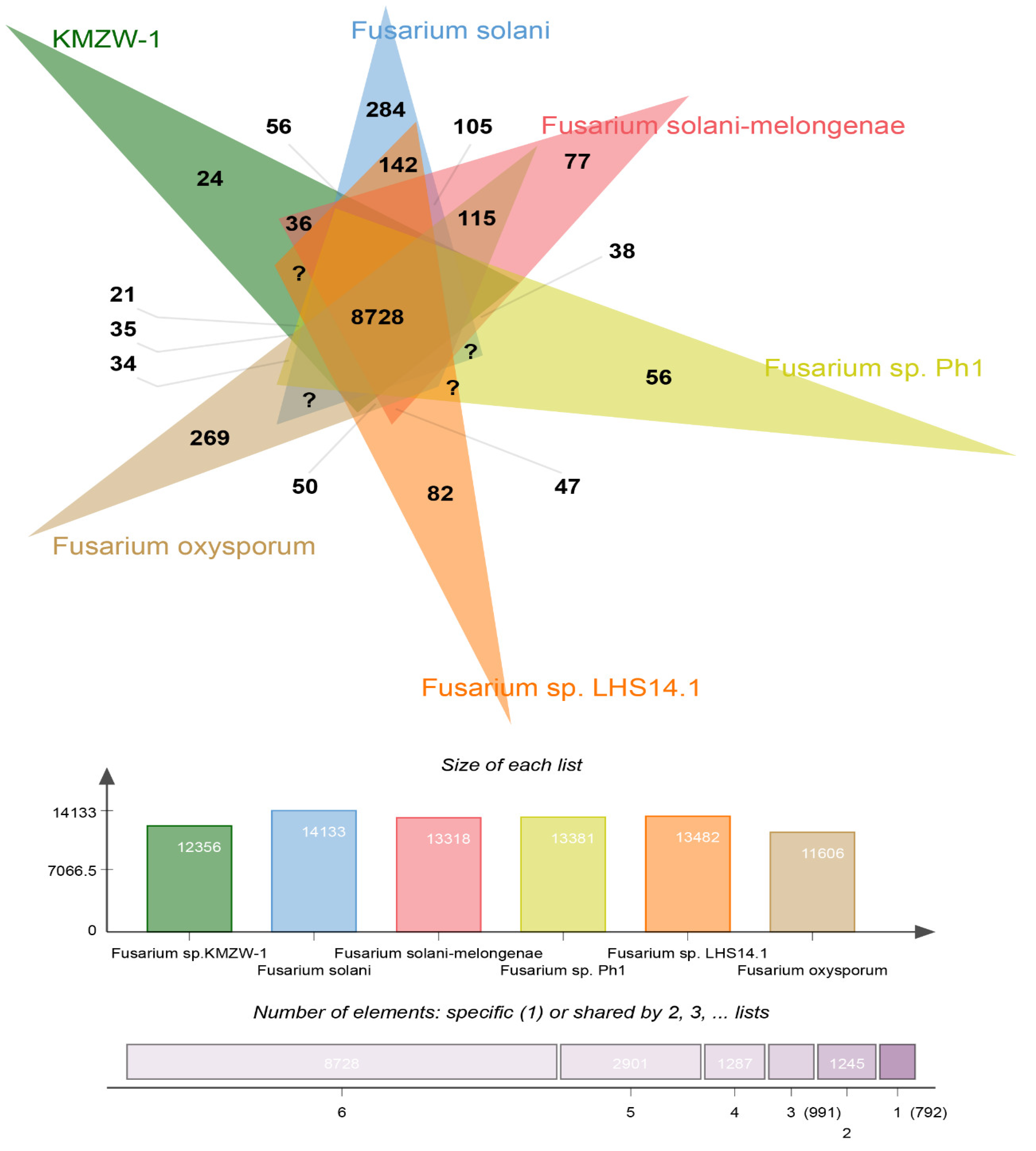
Figure 8.
Collinearity analysis between the genomes of Fusarium solani KMZW-1, Fusarium solani-melongenae and Fusarium solani.
Figure 8.
Collinearity analysis between the genomes of Fusarium solani KMZW-1, Fusarium solani-melongenae and Fusarium solani.

Figure 9.
The variation in cumulative mortality count under different treatments for Bactrocera dorsalis adults over 12 days. Note: Figure A shows the cumulative mortality of female Bactrocera dorsalis adults; Figure B shows the cumulative mortality of male Bactrocera dorsalis adults. Error Bars=SD, Data analyzed by Non-linear regression.
Figure 9.
The variation in cumulative mortality count under different treatments for Bactrocera dorsalis adults over 12 days. Note: Figure A shows the cumulative mortality of female Bactrocera dorsalis adults; Figure B shows the cumulative mortality of male Bactrocera dorsalis adults. Error Bars=SD, Data analyzed by Non-linear regression.
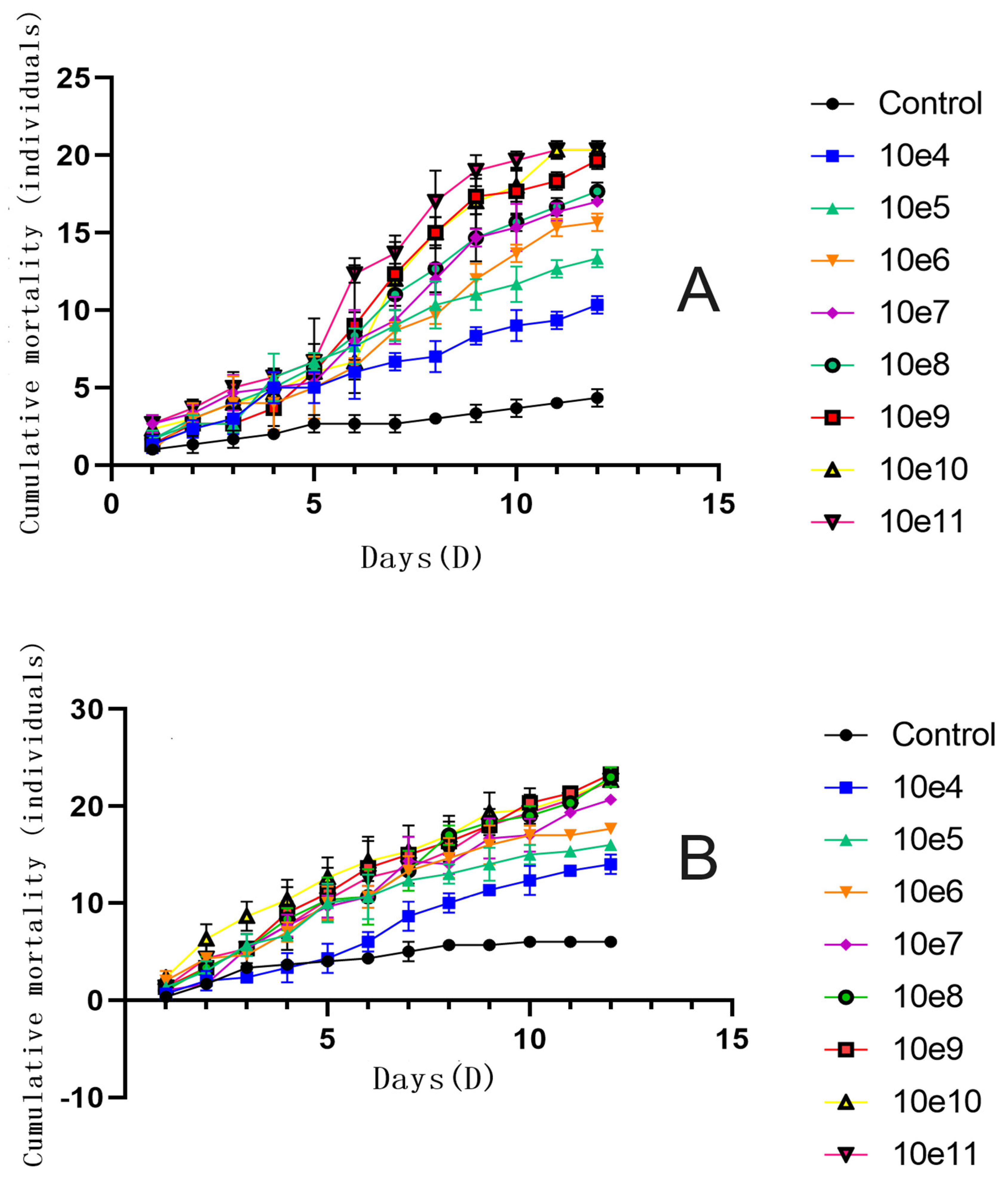

Table 1.
Concentration determination test concentration batches and groups.
| Concentration | Female Insect | Male Insect |
|---|---|---|
| 1×104 | 3*30 | 3*30 |
| 1×105 | 3*30 | 3*30 |
| 1×106 | 3*30 | 3*30 |
| 1×107 | 3*30 | 3*30 |
| 1×108 | 3*30 | 3*30 |
| 1×109 | 3*30 | 3*30 |
| 1×1010 | 3*30 | 3*30 |
| 1×1011 | 3*30 | 3*30 |
| Control | 3*30 | 3*30 |
| Total (Insect) | 810 | 810 |
Table 2.
Statistics of genome assembly.
| Contig Length (bp). | Contig Number | Contig N50 (bp) | Contig N90 (bp) | GC content (%) | Gaps Number |
|---|---|---|---|---|---|
| 47,239,278 | 27 | 2,751,789 | 1,018,923 | 51.16 | 0 |
Note: Contig Length (bp): indicates the length of contig that is more than 1 kb. Contig Number: Indicates the number of contig above 1 kb. contig N50 (bp): length of contig N50; contig N90 (bp): length of contig N90; GC Content (%): GC content; Gaps Number: The number of gaps.
Table 3.
BUSCO evaluation statistics.
| Complete BUSCOs(C) | Complete and single-copy BUSCOs(S) | Complete and duplicated BUSCOs(D) | Fragmented BUSCOs(F) | Missing BUSCOs(M) | Total Lineage BUSCOs |
|---|---|---|---|---|---|
| 284 (97.93%) | 281 (96.90%) | 3 (1.03%) | 4 (1.38%) | 2 (0.69%) | 290 |
Note: Complete BUSCOs: Find the complete gene number; Complete and single-copy BUSCOs: The number of single-copy genes; Complete and duplicated BUSCOs: number of duplicated genes; Fragmented BUSCOs: predicts incomplete gene number; Missing BUSCOs: No predicted number of genes; Total Lineage BUSCOs: The number of conserved gene sets.
Table 4.
Gene function annotation statistics for Fusarium solani KMZW-1.
| Database | Number | 100<=length<300 | length>=300 |
|---|---|---|---|
| GO_Annotation | 9,994 | 2,279 | 7,603 |
| KEGG_Annotation | 3,775 | 913 | 2,788 |
| KOG_Annotation | 7,059 | 1,422 | 5,575 |
| Pfam_Annotation | 10,510 | 2,346 | 8,076 |
| Swissprot_Annotation | 8,390 | 1,692 | 6,608 |
| TrEMBL_Annotation | 13,862 | 3,729 | 9,992 |
| nr_Annotation | 13,864 | 3,730 | 9,993 |
| All_Annotated | 13,867 | 3,733 | 9,993 |
Note: Database: Annotated functional database; Number: The number of genes on the annotation; 100<=length<300: the number of genes whose length is (100~300bp); length>=300: The number of genes with a length greater than 300 bp.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



