
Submitted:
25 September 2024
Posted:
25 September 2024
You are already at the latest version
Abstract
Ustekinumab is a fully human IgG1k monoclonal antibody that binds with high affinity and specificity to the p40 subunit of interleukins (IL-) 12 and 23, inhibiting their activity by preventing binding to their receptors. The European extension of the patent (Supplementary Protection Certificate) of ustekinumab expired on July 20, 2024. Biosimilar alternatives to ustekinumab are now an additional option for treating patients. The efficacy data for this drug in moderate-to-severe psoriasis, obtained both from clinical trials and indirect comparisons through meta-analyses, are superior to those of etanercept and adalimumab, and its safety profile is more favorable than that of tumor necrosis factor (TNF) inhibitors.
Several ustekinumab biosimilars have already been approved by regulatory agencies: between October 2023 and July 2024, Wezlana® (Amgen ABP 654), Uzpruvo® (Alvotech AVT04) and Pyzchiva® (Samsung/Bioepis SB17) have been approved by both the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA). SteQeyma® (Celltrion Healthcare CT-P43) has been approved by the EMA in August 2024. Several other potential biosimilar candidates are under development, including FYB202 (Formycon), BAT2206 (Bio-Thera), DMB-3115 (Dong-A ST), QX001S (Qyuns Therapeutic), BFI-751 (BioFactura), NeuLara (Neuclone), ONS3040 (Oncobiologics), and BOW090 (Epirus Biopharmaceuticals). In most cases, these monoclonal antibodies are expressed in cell lines (e.g. Chinese Hamster Ovary, CHO) different from those used for the originator (Sp2/0 spleen cell murine myeloma); of note, the cell line of origin is not a requirement for biosimilarity in the totality-of-evidence comparison exercise and may facilitate the production and reduce the immunogenicity of biosimilars originated in CHO cultures.
This narrative review will summarize the available data on characteristics of the full comparability exercises and comparative clinical trials of these drugs.
Keywords:
psoriasis
; biologics
; biosimilar
; ustekinumab
; systemic treatment
; IL‐23 inhibitors
1. Introduction
Psoriasis is a chronic immune-mediated inflammatory skin condition affecting approximately 1-3% of the global population [1]. The importance of the interleukin (IL) 12/23 pathway in the pathogenesis of psoriasis has been widely documented [2,3,4]. This inflammatory pathway is known to be overexpressed in psoriasis, driving the activation of Th1/Th17 lymphocytes, and promoting inflammation and keratinocyte proliferation [5].
Biologics have revolutionized the treatment of chronic inflammatory disorders like psoriasis. However, their high costs of acquisition have significantly increased healthcare expenditures and limited broader accessibility to biologic treatment [6]. To enhance access, regulatory agencies such as the US Food and Drug Administration (FDA), the European Medicines Agency (EMA), and others worldwide have established streamlined development and approval pathways for biosimilars—biologic therapies that are highly similar to an approved reference product (RP) [7]. US and EU guidelines advocate for a stepwise totality-of-evidence (TOE) approach, which involves compiling data from comparative analytical, nonclinical, and clinical studies [8]. The goal of this TOE approach is to ensure that there are no clinically meaningful differences in safety or efficacy between the biosimilar candidate and its RP. The abbreviated regulatory pathway for biosimilars minimizes the need for unnecessary animal and human testing. Instead, biosimilar development primarily emphasizes preclinical analytical evaluation and focuses on pharmacokinetic (PK) and pharmacodynamic studies. In order to be approved, biosimilars must undergo extensive physicochemical and biological characterization to demonstrate molecular similarity, including both in vivo and in vitro assays. This is followed by a phase I study to establish PK bioequivalence and a phase III study to confirm clinical biosimilarity [9,10].
Most biosimilars developed for the treatment of psoriasis and other immune mediated inflammatory diseases correspond to tumor necrosis factor (TNF) inhibitors [11,12,13]. While TNF inhibitors remain important therapies for psoriasis, other biologics with improved safety and efficacy profiles are currently available. Among the first of these newer treatments was the interleukin 12/23 inhibitor reference ustekinumab, which is approved for the treatment of psoriasis, psoriatic arthritis, and inflammatory bowel disease [14,15]. Ustekinumab offers several advantages over TNF inhibitors, including less frequent dosing and a more favorable safety profile [16].
The European extension of the Stelara® patent (Supplementary Protection Certificate) expired on July 20, 2024. As the exclusivity period for ustekinumab approached expiration, interest on ustekinumab biosimilars as safe and effective alternatives grew, and biosimilar alternatives to ustekinumab are now an additional option for treating patients.
This narrative review will summarize the available data on characteristics of the full comparability exercises and comparative clinical trials of these drugs.
2. Ustekinumab and Proposed Biosimilars
2.1. Ustekinumab
2.1.1. Generation
Ustekinumab is a human IgG1 kappa (κ) monoclonal antibody (mAb) originally developed by Centocor Research & Development, a division of Johnson & Johnson Pharmaceutical Research and Development, LLC. It was created using human immunoglobulin (hu-Ig) transgenic mice from GenPharm, a company later acquired by Medarex that is now part of Bristol-Myers Squibb, based in Princeton, New Jersey.
Four targeted genetic modifications replaced the murine immunoglobulin loci with human antibody transgenes, resulting in a mouse strain capable of producing human antibodies upon immunization with any antigen [17,18]. This human Ig transgenic mouse technology facilitated the generation of diverse, high-affinity, and specific monoclonal antibodies (mAbs). To develop human anti-human IL-12 therapeutic mAbs, the transgenic mice were immunized with human IL-12 antigen [19]. Mice with positive serum titers for anti-IL-12 antibodies were selected for hybridoma fusion, where splenocytes containing antibody-producing B cells were fused with an immortal cell line [19]. The resulting hybridoma cells were cultured, and IL-12-specific, growth-positive hybridomas were further subcloned [19]. Antibodies were screened through binding and functional assays using human T cells, selecting those that specifically bound IL-12 and inhibited IL-12-mediated responses [19]. A monoclonal hybridoma clone producing a human IgG1κ antibody with the ability to bind and neutralize human and nonhuman primate IL-12 was identified [19]. Initially called 12B75, then CNTO1275, and later ustekinumab, this antibody was chosen for further development due to its superior IL-12 binding and neutralization properties, but its IL-23 inhibitory action was later identified as much more relevant from a therapeutic perspective.
2.1.2. Mechanism of Action
Ustekinumab specifically targets the p40 subunit of IL-12 and IL-23 and inhibits their interaction with the IL-12Rβ1 receptor chain within the IL-12 (IL-12Rβ1/β2) and IL-23 (IL-12Rβ1/23R) receptor complexes on the surface of NK and T cells (Figure 1). By preventing IL-12 and IL-23 from binding to the IL-12Rβ1 receptor, ustekinumab effectively neutralizes IL-12- and IL-23-mediated cell signaling, activation, and cytokine production [19,20].
IL-12 is a heterodimeric cytokine composed of two protein subunits, p40 and p35, which were named according to their approximate molecular weights. IL-12 interacts with a heterodimeric receptor complex formed by the IL-12 receptor (IL-12R) β1 and IL-12Rβ2 chains, which are expressed on the surface of T cells and NK cells [21]. IL-12-mediated signaling involves intracellular phosphorylation of the signal transducers and activators of transcription (STAT)4 and STAT6 proteins, leading to functional responses such as the expression of cell surface molecules, enhancement of NK cell lytic activity, and IFNγ cytokine production[19].
The p40 protein subunit of IL-12 also combines with a p19 subunit to form IL-23 [22]. Since IL-23 shares the IL-12p40 subunit, ustekinumab is able to bind to and neutralize human IL-23. IL-23 also uses the IL-12Rβ1 chain to bind to the surface of effector cells. The association of the IL-23p19 subunit with the second component of the IL-23 receptor complex (IL-23R) triggers IL-23-specific intracellular signaling, including the phosphorylation of STAT3 and the activation of lymphocytes, leading to expression of the RORγT transcription factor and stabilization of a T17 phenotype with production of cytokines such as IL-17A [23].
2.1.3. Indications and Efficacy Data
Stelara® (ustekinumab RP) was first approved in 2009 for treatment of moderate-to-severe plaque psoriasis in adults and subsequently gained approval for psoriatic arthritis (2013), Crohn’s disease (2016), ulcerative colitis (2019), pediatric psoriasis (2020), and pediatric Crohn’s disease (2022) (Table 1).
The efficacy data of ustekinumab have been gathered from both clinical trials and indirect comparisons. The PHOENIX 1 and PHOENIX 2 clinical trials evaluated the efficacy and safety of ustekinumab in patients with moderate-to-severe plaque psoriasis and demonstrated significant improvement in psoriasis symptoms, with 66-76% of patients achieving at least a 75% improvement in Psoriasis Area and Severity Index (PASI 75) at week 12 [24,25]. Stable clinical response and safety profile were observed through 5 years of continuous treatment [26]. In real-world clinical practice, approximately 80% of patients maintain a PASI75 response at 2 years [27].
Compared to other biologics approved for psoriasis treatment, ustekinumab is less effective than newer anti-IL-17 and anti-IL-23 agents [28]. However, it has shown greater efficacy than etanercept in a phase III head-to-head study (ACCEPT trial) [29]. Indirect evidence from multiple meta-analyses suggests that ustekinumab demonstrates superior efficacy compared to etanercept and adalimumab with a more favorable safety profile than TNF inhibitors [30,31]. Ustekinumab is one of the biologic therapies with the lowest rate of adverse events, demonstrating a very low risk of serious infections and malignancies, with a safety profile similar to anti-IL-23 drugs [28].
Additionally, since its mechanism of action inhibits pathways involved in various inflammatory dermatoses, ustekinumab has been extensively used off-label with favorable results, for instance in patients with hidradenitis suppurativa or pityriasis rubra pilaris, among others (Table 1). There are currently two phase III trials evaluating the efficacy and safety of ustekinumab for the treatment of systemic lupus erythematosus (NCT03517722, NCT04060888).

| Ustekinumab approved indications | Adults and children 6 years and older with moderate to severe psoriasis who may benefit from taking injections or pills (systemic therapy) or phototherapy (treatment using ultraviolet light alone or with pills). Adults and children 6 years and older with active psoriatic arthritis. Adults 18 years and older with moderately to severely active Crohn’s disease. Adults 18 years and older with moderately to severely active ulcerative colitis. |
| Off-label reported uses of ustekinumab | Hidradenitis suppurativa Takayasu arteritis Giant cell arteritis Behçet disease Myelodysplastic syndrome Pyoderma grangrenosum Pityriasis rubra pilaris Synovitis, acne, pustulosis, hyperostosis, and osteitis (SAPHO) syndrome Lichen planus Atopic dermatitis Systemic lupus erythematosus |
2.2. ABP 654 (Wezlana® or Wezenla®)
Amgen’s ustekinumab biosimilar is called Wezenla® in Europe and Wezlana® in Canada and the US.
2.2.1. Pharmacology
ABP 654 is an ustekinumab biosimilar developed by Amgen, Thousand Oaks, California. While ustekinumab RP is produced using an Sp2/0 cell line system, ABP 654 is manufactured in a glyco-engineered Chinese hamster ovary (CHO) cell line, specifically designed to enhance its biosimilarity to ustekinumab RP [32]. ABP 654 has identical dosage forms and presentations as the RP [32]. Extensive analytical characterization has demonstrated that ABP 654 shares the same amino acid sequence as ustekinumab and is comparable in structure, function, purity, and potency [32,33].
Chow et al. conducted a randomized study in healthy subjects to determine the similarity of single-dose PKs, safety, and immunogenicity of ABP 654 compared with ustekinumab RP sourced from both the United States (US) and the European Union (EU) [33]. In total, 238 subjects from 3 centers in the US were included. In terms of PKs, the study demonstrated a high degree of similarity across all three treatment groups. Exposure levels were consistent among the groups, with peak concentrations occurring approximately 6-8 days post-treatment. Following peak levels, concentrations declined in a monophasic manner, with a half-life ranging from 21.9 to 23.9 days, consistent with the known half-life of ustekinumab [34]. The point estimates and 90% confidence intervals (CIs) for the geometric mean ratios of the primary PK endpoints (area under the concentration-time curve from time 0 extrapolated to infinity [AUCinf] and maximum observed serum concentration [Cmax]) were within the prespecified margin of 0.8–1.25 [33]. These findings confirm that there are no clinically meaningful differences in the PK profiles of ABP 654 and ustekinumab RP. Regarding immunogenicity, the potential for developing binding or neutralizing anti-drug antibodies (ADAs) was evaluated and resulted to be numerically lower in the ABP 654 group (15.4%) compared to both ustekinumab groups (36.3% in the EU ustekinumab group and 38.0% in the US ustekinumab group), with no significant impact on drug exposure across all treatments. Adverse events were reported by 28.2% of subjects in the ABP 654 group, 22.8% in the ustekinumab US group, and 36.3% in the ustekinumab EU group [33]. Most events were mild to moderate in severity (Common Terminology Criteria for Adverse Events, or CTCAE grade 1 or 2). The overall frequency, type, and severity of adverse events were similar across all treatment groups and aligned with the known safety profile of ustekinumab RP.
Similar results were reported in another randomized, double-blind study that compared the PKs, safety, tolerability, and immunogenicity of ABP 654 with US and EU ustekinumab [33]. A total of 238 healthy participants were randomized in a 1:1:1 ratio and stratified by gender and ethnicity (Japanese versus non-Japanese) to receive a single 90 mg subcutaneous injection of ABP 654, ustekinumab US, or ustekinumab EU. PK similarity was confirmed based on the 90% CIs for the primary endpoints—AUCinf and Cmax—falling within the predefined margin of 0.8–1.25. No clinically significant differences in immunogenicity were observed between the three products. Adverse events were comparable across treatment groups and consistent with the known safety profile of ustekinumab RP.
2.2.2. Clinical Efficacy and Safety
A phase III trial (NCT04607980) evaluated the efficacy and safety of ABP 654 compared to ustekinumab RP in adult patients with moderate-to-severe plaque psoriasis. The study was a multicenter, randomized, double-blinded, comparative clinical trial including 563 patients (281 patients in the ABP 654 group and 282 patients in the ustekinumab group). Clinical equivalence of the primary endpoint was evaluated by comparing the 2-sided 95% CIs of the mean difference in PASI percent improvement from baseline to week 12 between ABP 654 and ustekinumab with an equivalence margin of (-15, +15). Results of this trial confirmed the equivalence, with mean percent change in PASI score being 89.1% for both the ABP 654 and ustekinumab groups, and with CI 95% (-3.16 to 3.43) falling within the predefined equivalence margins. PASI75 response at week 12 was achieved by 69.8% and 70.2% of patients treated with ABP 654 and ustekinumab, respectively.
At week 28, patients in the ustekinumab group who achieved at least PASI75 response were re-randomized in a blinded fashion to either continue on ustekinumab or switch to ABP 654. Response was maintained to week 52, with PASI75 responses being held by 89.5%, 92.3% and 92.2% of participants in the ABP 654 / ABP 654 group, ustekinumab / ABP 654 group, and ustekinumab / ustekinumab group, respectively. PASI100 responses at week 52 were observed in 47%, 42.7% and 44.8% of patients in each group, respectively.
Treatment emergent adverse events (TEAEs) were found equally in the three groups after week 28 (34.4% in the ABP 654 / ABP 654 group, 37.6% in the ustekinumab / ABP 654 group, and 34.5% in the ustekinumab / ustekinumab group), mostly infections. Only 2.6% of patients in the ustekinumab / ustekinumab group presented with serious TEAEs, compared to less than 1% in the other groups.
In terms of immunogenicity, binding ADAs were identified in 18.6% of patients receiving ABP 654 and 37.1% of those receiving ustekinumab RP. Among ADA-positive individuals, neutralizing ADAs were found in 8.6% of the ABP 654 group and 17.9% of the ustekinumab group.
2.2.3. Switching from ustekinumab RP
Results from another phase III, multicenter, randomized, double-blinded study evaluating the PKs, efficacy and safety of multiple switches between ustekinumab and ABP 654 compared with continued use of ustekinumab in subjects with moderate-to-severe plaque psoriasis (NCT04761627) have been recently made available. In total, 494 participants were included. Initially, all participants received ustekinumab RP 45 mg (baseline body weight ≤ 100 kg) or 90 mg (baseline body weight > 100 kg) on day 1 (week 0), week 4, and week 16. At week 28, eligible participants with a PASI 50 response or better were randomized either to receive ustekinumab RP (continued-use group) or to receive ABP 654 at week 28, ustekinumab RP at week 40, and ABP 654 at week 52 (switching group). Primary outcome measures included Area Under the Curve over the dosing interval (AUCtau) between week 52 and week 64, and Cmax between week 52 and week 64. Again, PK similarity was confirmed based on the 90% CIs for the primary endpoints falling within the predefined margin of 0.8–1.25.
On October 31, 2023, Wezlana® (ustekinumab-auub) was approved by the FDA as a biosimilar to and interchangeable with Stelara®, with the same indications as ustekinumab RP; this was the first approval for a ustekinumab biosimilar. Subsequently, on April 2024, the Committee for Medicinal Products for Human Use of the EMA recommended marketing authorization for Wezenla®.
2.3. AVT04 (Uzpruvo® or Selarsdi®)
Alvotech’s ustekinumab biosimilar is called Uzpruvo® in Europe, Japan and Canada, and Selarsdi® (ustekinumab-aekn) in the US.
2.3.1. Pharmacology
AVT04 is produced in a murine myeloma Sp2/0 line. The primary amino acid sequences of AVT04 are identical to those of ustekinumab RP. AVT04 exhibits comparable physicochemical and functional properties to reference ustekinumab. PK equivalence between AVT04 and both EU and US sourced ustekinumab was established in a randomized, multicenter, double-blind, phase I trial involving 298 healthy adults [35]. The 90% CIs for the geometric mean ratios of key PK parameters, including AUCinf and Cmax, as well as the secondary endpoint of AUC from time zero to the last quantifiable concentration (AUClast), fell within the predefined equivalence range of 0.8-1.25 after normalization for protein content. Other PK measures, such as median time to Cmax, clearance, volume of distribution, and terminal half-life, were also similar between AVT04 and the ustekinumab RP groups.
Moreover, in another phase III trial comparing AVT04 and ustekinumab RP in patients with moderate-to-severe chronic plaque psoriasis, no clinically significant differences in mean serum trough concentrations were observed between AVT04 and ustekinumab through week 52 [36].
2.3.2. Clinical Efficacy and Safety
A randomized, double-blind, multicenter phase III trial evaluated efficacy, safety, tolerability, and immunogenicity between AVT04 and ustekinumab RP in adult patients with moderate-to-severe plaque psoriasis (NCT04930042) [36]. The study included 581 patients and was conducted in two distinct phases: the first phase (weeks 1–15) focused on evaluating primary efficacy, while the second phase (weeks 16–52) aimed to assess long-term efficacy and safety. The primary endpoint was the percentage improvement in PASI from baseline to week 12. Patients were randomized to receive subcutaneous injections of either AVT04 (n = 194) or reference ustekinumab (n = 387). At week 16, those receiving AVT04 continued their treatment with AVT04, while patients initially treated with reference ustekinumab were re-randomized to either switch to AVT04 (n = 192) or continue on ustekinumab RP (n = 189).
AVT04 demonstrated comparable efficacy to ustekinumab RP in terms of percentage improvement in PASI at week 12. The percent PASI improvement for AVT04 vs ustekinumab RP was 87.3% vs 86.8%, respectively, and the least squares mean (LSM) difference in PASI improvement between the two products was 0.4%, with both 90% CI (−2.14% to 3.01%) and 95% CI (−2.63% to 3.50%) falling within the predefined equivalence margins, confirming therapeutic equivalence [36]. Secondary endpoints such as proportions of patients achieving PASI50, 75, 90 and 100 responses, the proportion of patients achieving a static Physician’s Global Assessment (sPGA) response of ‘clear’ or ‘almost clear’, the change from baseline in Dermatology Life Quality Index (DLQI) scores, and the change in percent body surface area (BSA), were also similar in both groups. Efficacy in terms of PASI improvement rates and secondary endpoints were maintained throughout the switching period (weeks 16–52), irrespective of whether patients continued on AVT04, transitioned from ustekinumab RP to AVT04, or remained on ustekinumab RP [36].
AVT04 showed a tolerability and safety profile similar to that of ustekinumab RP, with TEAEs being reported in 34.5% and 33.6% of the participants, respectively. Most TEAEs were mild or moderate and the most frequently reported TEAEs were infections and infestations. Safety profiles were also comparable across all three treatment groups during the switching period (weeks 16–52).
In terms of immunogenicity, binding ADAs were identified in 25.4% of patients receiving AVT04 and 48.2% of those receiving ustekinumab RP. Among ADA-positive individuals, neutralizing ADAs were found in 26.5% of the AVT04 group and 31.0% of the ustekinumab group. The presence of ADAs did not seem to affect the therapeutic efficacy of AVT04.
Based on these results, Uzpruvo® became the first ustekinumab biosimilar approved in Europe after the European Medicines Agency (EMA) granted marketing authorization (to STADA) on January 2024, with the same indications as Stelara®. It has also been approved in Japan, Canada and more recently in the US; FDA-approval was received in April 2024 under the name of Selarsdi® (ustekinumab-aekn), marketed by Teva Pharmaceuticals, although the FDA has only approved Selarsdi® for the treatment of psoriasis and psoriatic arthritis; at the time of writing the only presentations available in the American market are 45 mg/0.5 mL or 90 mg/mL solution in single-dose prefilled syringes.
2.4. SB17 (Pyzchiva®, Eksunbi®)
2.4.1. Pharmacology
SB17 is a fully human IgG1κ ustekinumab biosimilar developed by Samsung Bioepis, produced in a recombinant Chinese Hamster Ovary (CHO) cell line cultured by continuous perfusion, and physicochemically similar to ustekinumab RP.
A randomized, double-blind, three-arm, single-dose phase I study (NCT04772274) compared PK, safety, tolerability, and immunogenicity of SB17 with US and EU ustekinumab RP in 201 healthy subjects [37]. The primary PK endpoints were AUCinf and Cmax. Equivalence for the primary PK endpoints was concluded since the 90% CIs for the geometric mean ratios of the compared groups were within the predefined equivalence margin of 0.80-1.25. Regarding immunogenicity, overall incidences of post-dose ADAs were comparable (26.9% in the SB17 group and 34.3% in both the EU-ustekinumab and US-ustekinumab groups). The proportions of subjects experiencing TEAEs were also similar across the groups, with rates of 68.7% for SB17, 58.2% for EU-ustekinumab, and 65.7% for US-ustekinumab. Importantly, no deaths, serious adverse events, severe TEAEs, or discontinuations due to TEAEs related to the study products occurred. All three ustekinumab products were generally well tolerated and exhibited similar safety profiles.
2.4.2. Clinical Efficacy and Safety
A randomized, double-blind, multicenter phase III study was conducted to evaluate efficacy, safety, PKs, and immunogenicity of SB17 compared to ustekinumab RP in adult subjects with moderate-to-severe plaque psoriasis (NCT04967508) [38]. Patients were randomized to receive 45 mg of SB17 or ustekinumab RP subcutaneously at week 0, 4, and every 12 weeks. The primary endpoint was the percent change from baseline in PASI at week 12 with an equivalence margin of [-15%, 15%]. Other secondary endpoints were measured through week 28.
A total of 503 subjects were included (249 subjects were randomized to SB17 and 254 subjects to ustekinumab). The adjusted difference in PASI change from baseline at week 12 was 0.6% (95% CI: −3.780 to 2.579), remaining within the predefined equivalence margin. Comparable outcomes were also observed for the PGA and DLQI. TEAEs occurred at similar rates across both groups (48.2% and 48.8% in the SB17 and ustekinumab RP group, respectively). Most were mild to moderate and considered not related to the investigational products. The overall incidence of ADAs up to week 28 was 13.3% for SB17, lower than for ustekinumab (39.4%). Similarly, neutralizing antibodies up to week 28 were found in 13.7% of subjects in the SB17 group, a lower rate than in the ustekinumab group (35.4%).
These results indicate that SB17 shows comparable efficacy, safety and PK to reference ustekinumab, as well as lower immunogenicity, up to week 28 in patients with moderate-to-severe psoriasis.
In April 2024, Samsung Bioepis announced that the European Commission granted marketing authorization for Pyzchiva®, two months after the EMA’s Committee for Medicinal Products for Human Use adopted a positive opinion for SB17 for the treatment of plaque psoriasis, psoriatic arthritis, ulcerative colitis and Crohn’s disease. On July 2024, Pyzchiva® (ustekinumab-ttwe) was approved by the USA FDA as an ustekinumab biosimilar with interchageability designation. Pyzchiva® is marketed by Sandoz. Eksunbi® (Samsung Bioepis) received a positive opinion from the Committee for Medicinal Products for Human Use of the EMA in July 2024.
2.5. CT-P43 (SteQeyma®)
CT-P43 is a fully human IgG1κ ustekinumab biosimilar developed by Celltrion. It is produced in a recombinant Chinese Hamster Ovary (CHO) cell line cultured by continuous perfusion and is physicochemically similar to ustekinumab RP.
A phase I, randomized, double-blind, single-dose study evaluated the PK, safety and immunogenicity of CT-P43 compared to US and EU ustekinumab in 271 male subjects (NCT04428814). Equivalent efficacy was subsequently demonstrated in a randomized, active-controlled, double-blind, phase III trial including 509 adult patients with moderate-to-severe psoriasis (NCT04673786) [39]. Subjects were initially randomized to receive either CT-P43 (n=256) or ustekinumab RP (n=254) at week 0 and week 4. At week 16, patients receiving ustekinumab RP were re-randomized to continue ustekinumab or switch to CT-P43. The primary endpoint was mean per cent improvement from baseline in PASI score at week 12. Equivalence was established if the CIs for the estimated treatment difference fell within the predefined equivalence margins: ±10% [90% CI; modified intent-to-treat set; FDA approach] or ±15% [95% CI; full analysis set; EMA approach]. Additional efficacy, safety, PK, and immunogenicity endpoints were evaluated through week 52.
The mean percentage improvement in PASI score at week 12 was 77.93% for CT-P43 and 75.89% for ustekinumab RP under the FDA criteria. According to the EMA criteria, the respective values were 78.26% and 77.33%. Equivalence was demonstrated under both sets of assumptions. Additionally, other efficacy parameters, as well as safety, PK, and immunogenicity outcomes, were similar across treatment groups, even after switching from originator ustekinumab to CT-P43.
After receiving a positive opinion from the EMA’s Committee for Medicinal Products for Human Use in June 2024, the European Commission has recently approved the use of SteQeyma® as an ustekinumab biosimilar in August 2024.
As a final note, Rani Therapeutics has recently announced a phase I clinical trial to evaluate the safety and tolerability of RT-111 (RaniPill®), an orally administered capsule containing CT-P43 (NCT05890118). RT-111 has already shown comparable bioavailability to subcutaneous injection in preclinical testing in animal models.
2.6. BAT2206
BAT2206 is a proposed ustekinumab biosimilar developed by Bio-Thera Solutions, Ltd. (Guangzhou, China). Biosimilarity of BAT2206 and ustekinumab RP in PK profiles, safety, and immunogenicity was evaluated in a double-blinded, randomized, single-dose, parallel-group, phase I trial (NCT04371185) [40]. In total, 270 healthy male subjects were included and received a single subcutaneous injection of either BAT2206 or ustekinumab RP (US or EU).
The results showed that the 90% CIs of the geometric mean ratios for primary PK parameters (AUCinf and Cmax) among BAT2206 and ustekinumab groups were all within the predefined equivalent interval of 0.8-1.25, supporting the bioequivalence between the groups. Incidence of TEAEs was similar across the groups, with most cases being mild or moderate in severity grading.
ADAs were detected in 26.7% of subjects in the BAT2206 group, a higher rate than in the EU and US ustekinumab groups (14.8% and 18.9%, respectively), although the incidence of neutralizing ADAs was similar among the three groups.
A multicenter, randomized, double-blind, parallel-arm, phase 3 trial was subsequently designed to compare efficacy, safety, immunogenicity, and PK of BAT2206 with ustekinumab in 556 patients with moderate-to-severe plaque psoriasis (NCT04728360). Bio-Thera has already announced positive outcomes from this phase III trial, although results are not yet posted on ClinicalTrials.gov.
The FDA has accepted Bio-Thera’s Biologics License Application for BAT2206 as an interchangeable biosimilar to the reference product Stelara®, while the EMA has accepted the Marketing Authorization Application for BAT2206 as a biosimilar to reference product Stelara®, but the respective final approvals are still pending. Furthermore, in June 2024, BAT2206 received approval in Brazil as a monotherapy or along with methotrexate to treat active psoriatic arthritis in adults.
2.7. DMB-3115
DMB-3115 is a proposed ustekinumab biosimilar developed by Dong-A Socio Holdings and Meiji Seika Pharma. On May 2021, the company announced positive results of a randomized, double-blind, three-arm, single-dose phase I study comparing the PK, safety and tolerability of DMB-3115 with ustekinumab RP (US and EU sourced Stelara®) in 296 healthy volunteers. After that, a randomized, double-blind, multicentric, parallel group phase III study was conducted to compare the efficacy, safety and immunogenicity of DMB-3115 and ustekinumab RP in patients with moderate-to-severe plaque psoriasis (Opportuniti, NCT04785326). In total, 598 subjects were included. Patients were initially randomized to receive either DMB-3115 or ustekinumab RP. At week 28, patients who received ustekinumab RP at the beginning of the study were re-randomized in a 1:1 ratio to either continue on ustekinumab RP or transition to DMB-3115. The primary endpoint was the percentage improvement in PASI from baseline. The results showed comparable efficacy between DMB-3115 and ustekinumab RP. Specifically, the least squares mean percent change in PASI from baseline to week 12 was 87.59% for the DMB-3115 group and 87.89% for the ustekinumab RP group. The difference in LSM percent change between the two treatments was within the predefined equivalence margin of ±10%. The overall incidence of TEAEs was similar between the two groups. The most common TEAEs included nasopharyngitis, upper respiratory tract infection, and headache, occurring at comparable rates between the two treatments.
Overall, DMB-3115 demonstrated equivalent efficacy, safety, and immunogenicity to ustekinumab RP, supporting its potential as a biosimilar treatment for psoriasis. In January 2024, the FDA accepted the Biologics License Application for the proposed biosimilar DMB-3115; similarly, the EMA has recently accepted the Marketing Authorization Application in July 2024, with final approval pending at the time of writing.
2.8. FYB202 (Fymskina®, Otulfi®)
FYB202 is a proposed ustekinumab biosimilar developed by Formycon AG. After successful conclusion of the extended phase I clinical study comparing the PK of FYB202 and ustekinumab RP, a randomized, double-blind, multicenter phase III trial (VESPUCCI study; NCT04595409) achieved the primary endpoint of percent improvement of PASI with respect to baseline at 12 weeks, demonstrating the comparable efficacy, safety and immunogenicity of FYB202 and the reference drug in patients with moderate-to-severe plaque psoriasis.
In February 2023, Formycon concluded a license agreement with Fresenius Kabi AG for the global commercialization of FYB202. The EMA accepted the Marketing Authorization Application for FYB202 in September 2023 and a positive opinion from the Committee for Medicinal Products for Human Use (CHMP) of the EMA was announced in July 2024, while the FDA accepted the Biologics License Application for FYB202 in November 2023; the respective final approvals are still pending.
2.9. QX001S
QX001S is a proposed ustekinumab biosimilar developed by Qyuns Therapeutics. A randomized, double-blind, single-dose phase I trial was conducted to assess the PK, tolerance and variability of QX001S in a cohort of healthy Chinese men [41]. In total, 178 participants were included and received either 45 mg of QX001S or ustekinumab RP.
The results demonstrated that QX001S exhibited a PK profile closely aligned with ustekinumab RP. The 90% CIs for the geometric mean ratios of key PK parameters (Cmax, AUClast, and AUCinf) fell within the predefined bioequivalence range of 0.8-1.25. Despite notable inter-subject variability (32.0% to 33.5%), the biosimilarity between the two drugs was affirmed. ADAs were detected in 19.1% of QX001S recipients and 40.9% of ustekinumab recipients, though these did not impact the bioequivalence outcomes. Adverse reactions were comparable between the groups, with upper respiratory infections and elevated alanine aminotransferase levels being the most frequently reported. Overall, this study confirmed that QX001S is pharmacokinetically equivalent to ustekinumab, despite the high variability observed among subjects.
Qyuns Therapeutics has reported positive outcomes of a phase III clinical trial evaluating clinical equivalence of QX001S to ustekinumab in terms of efficacy, safety, immunogenicity and PK profile in psoriasis patients, although results have not been published. Zhongmei Huadong, a subsidiary of Huadong Medicine and commercialization partner for QX001S, submitted a Biologics License Application in China in July 2023, which was accepted by the National Medical Products Administration in August 2023 and is currently under review.
2.10. BFI-751
BFI-751 is a proposed biosimilar to ustekinumab developed by BioFactura. A a multicenter, randomized, double-blind, phase I study has assessed the PK, safety, and immunogenicity of BFI-751 compared to EU and US ustekinumab RP (NCT04843631) [42]. The primary PK endpoints were AUCinf, AUClast and Cmax. Results showed that BFI-751 met bioequivalence criteria when compared to both reference products, with 90% CIs of the primary endpoints within the accepted range of 0.8-1.25. No significant safety or tolerability differences were noted, and treatment-emergent adverse events were mild to moderate across all groups. ADAs were detected in 16% of BFI-751 recipients, compared to 44% of EU ustekinumab recipients and 49% of US ustekinumab recipients.
2.11. Other Proposed Ustekinumab Biosimilars
Other molecules are currently under evaluation as potential ustekinumab biosimilars, including NeuLara, which has already shown positive results in phase I trial (although results are not published), or ONS3040 and BOW090, which are in pre-clinical development stages (Table 2).
3. Discussion
Ustekinumab, an interleukin-12/23 inhibitor, has been a cornerstone in the management of chronic inflammatory conditions such as psoriasis, psoriatic arthritis, and inflammatory bowel disease. With expiration of the patent exclusivity for ustekinumab, the development and approval of biosimilars have gained momentum. Ustekinumab biosimilars, designed to be highly similar in structure, efficacy, and safety to the reference product (Stelara®), offer the potential for broader patient access and reduced healthcare costs.
At the moment of writing, four ustekinumab biosimilars have been approved by the FDA and/or the EMA: ABP 654 (Wezenla®/Wezlana® in US), AVT04 (Uzpruvo®/Selarsdi® in US), SB17 (Pyzchiva®/EpyztekTM in Korea) and CT-P43 (SteQeyma®). At least three more candidates (FYB202, BAT2206, DMB-3115) are expected to be approved in the upcoming months, after the acceptance of the FDA’s Biologics License Application and the EMA’s Marketing Authorization Application. While the acceptance of these applications by the FDA and EMA does not guarantee approval, it is a significant step forward. The regulatory agencies will still need to conduct a thorough review before deciding whether to grant final approval for the biosimilars’ use in different clinical settings. Other molecules are being evaluated at pre-clinical and clinical level (Table 2).
The phase I and phase III studies required for the approval of ustekinumab biosimilars have consistently demonstrated PK equivalence, with key metrics such as Cmax and AUC falling within the predefined bioequivalence margins, as well as comparable clinical efficacy and safety. These findings align with the rigorous standards set by regulatory agencies such as the FDA and EMA for the approval of biosimilars.
Immunogenicity remains a relevant consideration, with ustekinumab biosimilars generally showing comparable or slightly lower rates of ADA formation compared to the RP (Table 3), which do not impinge on the TOE-determined biosimilarity.
The lower immunogenicity of ustekinumab biosimilars has been attributed to the differences in manufacturing processes compared to the RP. The most common mammalian cell lines used for biopharmaceutical manufacturing include Chinese hamster ovary (CHO) cells, baby hamster kidney cells (BHK21) and murine myeloma cells (NS0 and Sp2/0) [43]. Ustekinumab RP is produced using Sp2/0 cells, while most ustekinumab biosimilars are produced using CHO cell lines, which provide manufacturing advantages. Murine cell lines produce post-translational modifications that are not expressed in humans and are potentially immunogenic, such as galactose-a1,3-galactose (a-gal) and N-glycolylneuraminic acid (NGNA), but these modifications in recombinant CHO cells are apparently closer to those in humans [44]. Thus, different cell lines may lead to variations in glycosylation profiles, which could potentially influence the immunogenicity of biosimilars. Interestingly, lower immunogenicity has been observed even in biosimilars that utilize the same cell line as the RP, like AVT04 [36]. The underlying cause of the reduced immunogenicity of ustekinumab biosimilars remains unclear.
However, the presence of ADAs does not appear to compromise the clinical efficacy of these biosimilars. In the case of SB17, for instance, the primary efficacy endpoint—measured by the percentage change in PASI scores at week 12—was comparable between ADA-positive and ADA-negative patients. This suggests that ADAs may not significantly affect the therapeutic outcome in patients treated with these biosimilars. In psoriasis, the pooled ADA rate to ustekinumab RP is 4.1% across nine studies with 4044 patients [45], but sensitivity of current tests for ADA has greatly increased. The presence of ADA decreases the serum concentrations of ustekinumab and slightly attenuates the clinical response, especially after switching in patients with prior ADAs to adalimumab [46,47,48]. Interestingly, in patients with psoriatic arthritis, ADA formation is not associated with impairments in ustekinumab RP safety, efficacy or trough levels, and concomitant methotrexate has no effect in ustekinumab RP immunogenicity [49]. On the other hand, development of ustekinumab ADAs is associated with loss of response to ustekinumab RP in patients with inflammatory bowel disease [50]. Further investigation in real-world settings to fully understand the long-term impact of ustekinumab biosimilars with lower immunogenicity on clinical outcomes in different settings.
All approved and proposed ustekinumab biosimilars have undergone clinical trials in patients with psoriasis. Extrapolation allows regulatory authorities to extend the approved indications of the biosimilar to other conditions treated by the RP, such as psoriatic arthritis or Crohn’s disease, without the need for conducting separate clinical trials for each condition. This process is based on the principle that the ustekinumab mechanism of action is the same across all indications of the RP.
A recent systematic review of 14 randomized clinical trials and 3 cohort studies assessed the efficacy and safety of biosimilars of adalimumab, etanercept, infliximab, and ustekinumab compared to originator biologics in treating psoriasis [51]. The findings showed no clinically or statistically significant differences in efficacy (in terms of achieving PASI75 response) or in the risk of adverse events between biosimilars and originators at weeks 16 and 52. This results confirm the remarkable equivalence between biosimilars and originators, and support the adoption of biosimilars in the treatment of psoriasis and other inflammatory diseases pathogenetically mediated by IL-23.
Despite their potential cost benefits, several barriers hinder the widespread adoption of biosimilars [52]. Public perception often favors brand-name biologics over biosimilars, viewing the latter as inferior “copies.” Additionally, the unique development process of biosimilars, which differs from traditional drug development, has contributed to the hesitancy of some prescribers. Long-term efficacy and safety assessments for biosimilars in different indications also remain partially uncertain. However, biosimilars offer significant societal benefits, including broader access to treatments and potential cost savings. As an important and expanding class of biologics, their widespread adoption should lead to improved patient outcomes and more sustainable healthcare systems.
4. Conclusions
Ustekinumab biosimilars represent a promising advancement in dermatologic therapy, potentially offering cost-effective alternatives to the reference product. Continued research and pharmacovigilance are essential to fully establish their role in clinical practice, ensuring that patients receive the same high standards of care as with the original biologic.
Authorship declaration
All authors declare to have contributed to the conception and design of the article, the writing of the draft, the critical review of the intellectual content and the final approval of the version presented.
Conflicts of interest
ECR has perceived speaker’s honoraria from Boehringer-Ingelheim and Amgen. LP has perceived consultancy/speaker’s honoraria from and/or participated in clinical trials sponsored by Abbvie, Almirall, Amgen, Biogen, Bristol Myers Squibb, Boehringer Ingelheim, Celgene, Fresenius-Kabi, Gebro, Janssen, JS BIOCAD, Leo-Pharma, Lilly, Novartis, Pfizer, Samsung-Bioepis, and UCB.
References
- Michalek, I.M.; Loring, B.; John, S.M. A Systematic Review of Worldwide Epidemiology of Psoriasis. Journal of the European Academy of Dermatology and Venereology 2017, 31, 205–212. [CrossRef]
- Rendon, A.; Schäkel, K. Psoriasis Pathogenesis and Treatment. Int J Mol Sci 2019, 20, 1475. [CrossRef]
- Blauvelt, A.; Chiricozzi, A. The Immunologic Role of IL-17 in Psoriasis and Psoriatic Arthritis Pathogenesis. Clin Rev Allergy Immunol 2018, 55, 379–390. [CrossRef]
- Girolomoni, G.; Strohal, R.; Puig, L.; Bachelez, H.; Barker, J.; Boehncke, W.H.; Prinz, J.C. The Role of IL-23 and the IL-23/TH17 Immune Axis in the Pathogenesis and Treatment of Psoriasis. Journal of the European Academy of Dermatology and Venereology 2017, 31, 1616–1626. [CrossRef]
- Nussbaum, L.; Chen, Y.L.; Ogg, G.S. Role of Regulatory T Cells in Psoriasis Pathogenesis and Treatment. British Journal of Dermatology 2021, 184, 14–24. [CrossRef]
- Baumgart, D.C.; Misery, L.; Naeyaert, S.; Taylor, P.C. Biological Therapies in Immune-Mediated Inflammatory Diseases: Can Biosimilars Reduce Access Inequities? Front Pharmacol 2019, 10, 279. [CrossRef]
- Kabir, E.R.; Moreino, S.S.; Sharif Siam, M.K. The Breakthrough of Biosimilars: A Twist in the Narrative of Biological Therapy. Biomolecules 2019, 9. [CrossRef]
- Markus, R.; Liu, J.; Ramchandani, M.; Landa, D.; Born, T.; Kaur, P. Developing the Totality of Evidence for Biosimilars: Regulatory Considerations and Building Confidence for the Healthcare Community. BioDrugs 2017, 31, 175–187,. [CrossRef]
- EMA. Guideline on Similar Biological Medicinal Products Containing Monoclonal AntibodiesdNonclinical and Clinical Issues. EMA; 2012.
- FDA. Scientific Considerations in Demonstrating Biosimilarity to a Reference Product. FDA; 2015.
- Griffiths, C.E.M.; Thaçi, D.; Gerdes, S.; Arenberger, P.; Pulka, G.; Kingo, K.; Weglowska, J.; EGALITY study group; Hattebuhr, N.; Poetzl, J.; et al. The EGALITY Study: A Confirmatory, Randomized, Double-Blind Study Comparing the Efficacy, Safety and Immunogenicity of GP2015, a Proposed Etanercept Biosimilar, vs. the Originator Product in Patients with Moderate-to-Severe Chronic Plaque-Type Psoriasis. Br J Dermatol 2017, 176, 928–938. [CrossRef]
- Hercogová, J.; Papp, K.A.; Chyrok, V.; Ullmann, M.; Vlachos, P.; Edwards, C.J. AURIEL-PsO: A Randomized, Double-Blind Phase III Equivalence Trial to Demonstrate the Clinical Similarity of the Proposed Biosimilar MSB11022 to Reference Adalimumab in Patients with Moderate-to-Severe Chronic Plaque-Type Psoriasis. Br J Dermatol 2020, 182, 316–326. [CrossRef]
- Papp, K.; Bachelez, H.; Costanzo, A.; Foley, P.; Gooderham, M.; Kaur, P.; Narbutt, J.; Philipp, S.; Spelman, L.; Weglowska, J.; et al. Clinical Similarity of Biosimilar ABP 501 to Adalimumab in the Treatment of Patients with Moderate to Severe Plaque Psoriasis: A Randomized, Double-Blind, Multicenter, Phase III Study. J Am Acad Dermatol 2017, 76, 1093–1102. [CrossRef]
- EMA. Stelara®: EPARdProduct Information; Annex I, Summary of Product Characteristics. EMA; 2023.
- FDA. Stelara®: Prescribing Information. FDA; 2023.
- Kalb, R.E.; Fiorentino, D.F.; Lebwohl, M.G.; Toole, J.; Poulin, Y.; Cohen, A.D.; Goyal, K.; Fakharzadeh, S.; Calabro, S.; Chevrier, M.; et al. Risk of Serious Infection With Biologic and Systemic Treatment of Psoriasis: Results From the Psoriasis Longitudinal Assessment and Registry (PSOLAR). JAMA Dermatol 2015, 151, 961–969. [CrossRef]
- Fishwild, D.M.; O’Donnell, S.L.; Bengoechea, T.; Hudson, D. V; Harding, F.; Bernhard, S.L.; Jones, D.; Kay, R.M.; Higgins, K.M.; Schramm, S.R.; et al. High-Avidity Human IgG Kappa Monoclonal Antibodies from a Novel Strain of Minilocus Transgenic Mice. Nat Biotechnol 1996, 14, 845–851. [CrossRef]
- Lonberg, N. Human Antibodies from Transgenic Animals. Nat Biotechnol 2005, 23, 1117–1125. [CrossRef]
- Benson, J.M.; Peritt, D.; Scallon, B.J.; Heavner, G.A.; Shealy, D.J.; Giles-Komar, J.M.; Mascelli, M.A. Discovery and Mechanism of Ustekinumab: A Human Monoclonal Antibody Targeting Interleukin-12 and Interleukin-23 for Treatment of Immune-Mediated Disorders. MAbs 2011, 3.
- Luo, J.; Wu, S.-J.; Lacy, E.R.; Orlovsky, Y.; Baker, A.; Teplyakov, A.; Obmolova, G.; Heavner, G.A.; Richter, H.-T.; Benson, J. Structural Basis for the Dual Recognition of IL-12 and IL-23 by Ustekinumab. J Mol Biol 2010, 402, 797–812. [CrossRef]
- Presky, D.H.; Yang, H.; Minetti, L.J.; Chua, A.O.; Nabavi, N.; Wu, C.Y.; Gately, M.K.; Gubler, U. A Functional Interleukin 12 Receptor Complex Is Composed of Two Beta-Type Cytokine Receptor Subunits. Proc Natl Acad Sci U S A 1996, 93, 14002–14007. [CrossRef]
- Oppmann, B.; Lesley, R.; Blom, B.; Timans, J.C.; Xu, Y.; Hunte, B.; Vega, F.; Yu, N.; Wang, J.; Singh, K.; et al. Novel P19 Protein Engages IL-12p40 to Form a Cytokine, IL-23, with Biological Activities Similar as Well as Distinct from IL-12. Immunity 2000, 13, 715–725. [CrossRef]
- Parham, C.; Chirica, M.; Timans, J.; Vaisberg, E.; Travis, M.; Cheung, J.; Pflanz, S.; Zhang, R.; Singh, K.P.; Vega, F.; et al. A Receptor for the Heterodimeric Cytokine IL-23 Is Composed of IL-12Rbeta1 and a Novel Cytokine Receptor Subunit, IL-23R. J Immunol 2002, 168, 5699–5708. [CrossRef]
- Leonardi, C.L.; Kimball, A.B.; Papp, K.A.; Yeilding, N.; Guzzo, C.; Wang, Y.; Li, S.; Dooley, L.T.; Gordon, K.B.; PHOENIX 1 study investigators Efficacy and Safety of Ustekinumab, a Human Interleukin-12/23 Monoclonal Antibody, in Patients with Psoriasis: 76-Week Results from a Randomised, Double-Blind, Placebo-Controlled Trial (PHOENIX 1). Lancet 2008, 371, 1665–1674. [CrossRef]
- Papp, K.A.; Langley, R.G.; Lebwohl, M.; Krueger, G.G.; Szapary, P.; Yeilding, N.; Guzzo, C.; Hsu, M.-C.; Wang, Y.; Li, S.; et al. Efficacy and Safety of Ustekinumab, a Human Interleukin-12/23 Monoclonal Antibody, in Patients with Psoriasis: 52-Week Results from a Randomised, Double-Blind, Placebo-Controlled Trial (PHOENIX 2). Lancet 2008, 371, 1675–1684. [CrossRef]
- Kimball, A.B.; Papp, K.A.; Wasfi, Y.; Chan, D.; Bissonnette, R.; Sofen, H.; Yeilding, N.; Li, S.; Szapary, P.; Gordon, K.B.; et al. Long-Term Efficacy of Ustekinumab in Patients with Moderate-to-Severe Psoriasis Treated for up to 5 Years in the PHOENIX 1 Study. J Eur Acad Dermatol Venereol 2013, 27, 1535–1545. [CrossRef]
- Gerdes, S.; Hoffmann, M.; Asadullah, K.; Korge, B.; Mortazawi, D.; Krüger, N.; Personke, Y.; Tabori, S.; Gomez, M.; Wegner, S.; et al. Effectiveness, Safety and Quality-of-Life Effects of Guselkumab and Ustekinumab in Patients with Psoriasis: Week 104 Results from the Non-Interventional, Prospective, German Multicentre PERSIST Study. J Eur Acad Dermatol Venereol 2023. [CrossRef]
- Yiu, Z.Z.N.; Becher, G.; Kirby, B.; Laws, P.; Reynolds, N.J.; Smith, C.H.; Warren, R.B.; Griffiths, C.E.M.; BADBIR Study Group Drug Survival Associated With Effectiveness and Safety of Treatment With Guselkumab, Ixekizumab, Secukinumab, Ustekinumab, and Adalimumab in Patients With Psoriasis. JAMA Dermatol 2022, 158, 1131–1141. [CrossRef]
- Griffiths, C.E.M.; Strober, B.E.; van de Kerkhof, P.; Ho, V.; Fidelus-Gort, R.; Yeilding, N.; Guzzo, C.; Xia, Y.; Zhou, B.; Li, S.; et al. Comparison of Ustekinumab and Etanercept for Moderate-to-Severe Psoriasis. N Engl J Med 2010, 362, 118–128. [CrossRef]
- Armstrong, A.W.; Soliman, A.M.; Betts, K.A.; Wang, Y.; Gao, Y.; Puig, L.; Augustin, M. Comparative Efficacy and Relative Ranking of Biologics and Oral Therapies for Moderate-to-Severe Plaque Psoriasis: A Network Meta-Analysis. Dermatol Ther (Heidelb) 2021, 11, 885–905. [CrossRef]
- Sbidian, E.; Chaimani, A.; Garcia-Doval, I.; Doney, L.; Dressler, C.; Hua, C.; Hughes, C.; Naldi, L.; Afach, S.; Le Cleach, L. Systemic Pharmacological Treatments for Chronic Plaque Psoriasis: A Network Meta-Analysis. Cochrane Database Syst Rev 2022, 5, CD011535. [CrossRef]
- Cantin, G.; Liu, Q.; Shah, B.; Kuhns, S.; Wikström, M.; Cao, S.; Liu, J. Analytical and Functional Similarity of the Biosimilar Candidate ABP 654 to Ustekinumab Reference Product. Drugs in R and D 2023, 23, 421–438. [CrossRef]
- Chow, V.; Mytych, D.T.; Das, S.; Franklin, J. Pharmacokinetic Similarity of ABP 654, an Ustekinumab Biosimilar Candidate: Results from a Randomized, Double-Blind Study in Healthy Subjects. Clin Pharmacol Drug Dev 2023, 12, 863–873. [CrossRef]
- Zhu, Y.; Wang, Q.; Frederick, B.; Bouman-Thio, E.; Marini, J.C.; Keen, M.; Petty, K.J.; Davis, H.M.; Zhou, H. Comparison of the Pharmacokinetics of Subcutaneous Ustekinumab between Chinese and Non-Chinese Healthy Male Subjects across Two Phase 1 Studies. Clin Drug Investig 2013, 33, 291–301. [CrossRef]
- Wynne, C.; Hamilton, P.; McLendon, K.; Stroissnig, H.; Smith, M.; Duijzings, P.; Ruffieux, R.; Otto, H.; Sattar, A.; Haliduola, H.N.; et al. A Randomized, Double-Blind, 3-Arm, Parallel Study Assessing the Pharmacokinetics, Safety, Tolerability and Immunogenicity of AVT04, an Ustekinumab Candidate Biosimilar, in Healthy Adults. Expert Opin Investig Drugs 2023, 32, 417–427. [CrossRef]
- Feldman, S.R.; Reznichenko, N.; Berti, F.; Duijzings, P.; Ruffieux, R.; Otto, H.; Haliduola, H.N.; Leutz, S.; Stroissnig, H. Randomized, Double-Blind, Multicenter Study to Evaluate Efficacy, Safety, Tolerability, and Immunogenicity between AVT04 and the Reference Product Ustekinumab in Patients with Moderate-to-Severe Chronic Plaque Psoriasis. Expert Opin Biol Ther 2023, 23, 759–771,. [CrossRef]
- Jeong, H.; Kang, T.; Lee, J.; Im, S. Comparison of SB17 and Reference Ustekinumab in Healthy Adults: A Randomized, Double-Blind, Single-Dose, Phase I Study. Int J Clin Pharmacol Ther 2024, 62, 231–240. [CrossRef]
- Feldman, S.R.; Narbutt, J.; Girolomoni, G.; Brzezicki, J.; Reznichenko, N.; Zegadło-Mylik, M.A.; Pulka, G.; Dmowska-Stecewicz, M.; Kłujszo, E.; Rekalov, D.; et al. A Randomized, Double-Blind, Phase III Study Assessing Clinical Similarity of SB17 (Proposed Ustekinumab Biosimilar) to Reference Ustekinumab in Subjects with Moderate-to-Severe Plaque Psoriasis. J Am Acad Dermatol 2024. [CrossRef]
- Papp, K.A.; Lebwohl, M.G.; Thaçi, D.; Jaworski, J.; Kwiek, B.; Trefler, J.; Dudek, A.; Szepietowski, J.C.; Reznichenko, N.; Narbutt, J.; et al. Efficacy and Safety of Candidate Biosimilar CT-P43 Versus Originator Ustekinumab in Moderate to Severe Plaque Psoriasis: 28-Week Results of a Randomised, Active-Controlled, Double-Blind, Phase III Study. BioDrugs 2024, 38, 121–131. [CrossRef]
- Wu, M.; Li, X.; Yang, D.; Wang, M.; Zhang, H.; Li, C.; Mai, J.; Yang, L.; Qi, Y.; Yu, J.C.; et al. Comparison of Pharmacokinetic Similarity, Immunogenicity, and Safety of Ustekinumab and BAT2206 in Healthy Chinese Male Subjects in a Double-Blind, Randomized, Single-Dose, Parallel-Group Phase I Trial. BioDrugs 2023, 37, 89–98. [CrossRef]
- Gao, L.; Li, Q.; Zhang, H.; Wu, M.; Fang, M.; Yang, L.; Li, X.; Liu, J.; Li, C.; Chen, H.; et al. A Biosimilarity Study Between QX001S and Ustekinumab in Healthy Chinese Male Subjects. Front Pharmacol 2021, 12. [CrossRef]
- Hausfeld, J.N.; Challand, R.; McLendon, K.; Macapagal, N.; Bruce-Staskal, P.; Fiaschetti, C.; Sampey, D.B. Pharmacokinetic Profiles of a Proposed Biosimilar Ustekinumab (BFI-751): Results From a Randomized Phase 1 Trial. Clin Pharmacol Drug Dev 2023, 12, 1001–1012,. [CrossRef]
- Estes, S.; Melville, M. Mammalian Cell Line Developments in Speed and Efficiency. In; 2013; pp. 11–33.
- Dumont, J.; Euwart, D.; Mei, B.; Estes, S.; Kshirsagar, R. Human Cell Lines for Biopharmaceutical Manufacturing: History, Status, and Future Perspectives. Crit Rev Biotechnol 2016, 36, 1110–1122. [CrossRef]
- Sun, X.; Cui, Z.; Wang, Q.; Liu, L.; Ding, X.; Wang, J.; Cai, X.; Li, B.; Li, X. Formation and Clinical Effects of Anti-Drug Antibodies against Biologics in Psoriasis Treatment: An Analysis of Current Evidence. Autoimmun Rev 2024, 23, 103530. [CrossRef]
- Loeff, F.C.; Tsakok, T.; Dijk, L.; Hart, M.H.; Duckworth, M.; Baudry, D.; Russell, A.; Dand, N.; van Leeuwen, A.; Griffiths, C.E.M.; et al. Clinical Impact of Antibodies against Ustekinumab in Psoriasis: An Observational, Cross-Sectional, Multicenter Study. J Invest Dermatol 2020, 140, 2129–2137. [CrossRef]
- Tsai, T.-F.; Ho, J.-C.; Song, M.; Szapary, P.; Guzzo, C.; Shen, Y.-K.; Li, S.; Kim, K.-J.; Kim, T.-Y.; Choi, J.-H.; et al. Efficacy and Safety of Ustekinumab for the Treatment of Moderate-to-Severe Psoriasis: A Phase III, Randomized, Placebo-Controlled Trial in Taiwanese and Korean Patients (PEARL). J Dermatol Sci 2011, 63, 154–163,. [CrossRef]
- Chiu, H.-Y.; Chu, T.W.; Cheng, Y.-P.; Tsai, T.-F. The Association between Clinical Response to Ustekinumab and Immunogenicity to Ustekinumab and Prior Adalimumab. PLoS One 2015, 10, e0142930. [CrossRef]
- Mojtahed Poor, S.; Henke, M.; Ulshöfer, T.; Köhm, M.; Behrens, F.; Burkhardt, H.; Schiffmann, S. The Role of Antidrug Antibodies in Ustekinumab Therapy and the Impact of Methotrexate. Rheumatology (Oxford) 2023, 62, 3993–3999. [CrossRef]
- Roblin, X.; Duru, G.; Papamichael, K.; Cheifetz, A.S.; Kwiatek, S.; Berger, A.-E.; Barrau, M.; Waeckel, L.; Nancey, S.; Paul, S. Development of Antibodies to Ustekinumab Is Associated with Loss of Response in Patients with Inflammatory Bowel Disease. J Clin Med 2023, 12. [CrossRef]
- Phan, D.B.; Elyoussfi, S.; Stevenson, M.; Lunt, M.; Warren, R.B.; Yiu, Z.Z.N. Biosimilars for the Treatment of Psoriasis. JAMA Dermatol 2023, 159, 763. [CrossRef]
- Naldi, L.; Addis, A. Biosimilars for the Treatment of Psoriasis-A Systematic Review of Clinical Trials and Observational Studies Looking Beyond Single-Trial Evidence for a Valuable Choice. Photography in Clinical Medicine 2014, 2020, 491–499,.
- Ustekinumab Prescribing Information. Www.Stelarainfo.Com. August 2024.
- Colquhoun M, Kemp AK. Ustekinumab. [Updated 2023 Mar 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: Https://Www.Ncbi.Nlm.Nih.Gov/Books/NBK570645/.
- Masson, R.; Seivright, J.; Grogan, T.; Atluri, S.; Hamzavi, I.; Hogeling, M.; Shi, V.Y.; Hsiao, J.L. Ustekinumab in Hidradenitis Suppurativa: A Systematic Review and Meta-Analysis. Dermatol Ther (Heidelb) 2024, 14, 1901–1916. [CrossRef]
- Ye, L.; Wu, Z.; Li, C.; Zhao, X.; Wan, M.; Wang, L. Off-Label Uses of Ustekinumab. Dermatol Ther 2022, 35, e15910. [CrossRef]
Figure 1.
Ustekinumab mechanism of action. Ustekinumab binds to the p40 subunit of interleukin (IL)-12 and IL-23, blocking their interaction with the IL-12Rβ1 receptor on the cell surface of natural killer cells and T cells. This inhibits IL-12- and IL-23-driven cell signaling, activation, and cytokine production. Adapted from Benson et al. [19]. Created in BioRender. Puig sanz, L. (2024) BioRender.com/b86x734.
Figure 1.
Ustekinumab mechanism of action. Ustekinumab binds to the p40 subunit of interleukin (IL)-12 and IL-23, blocking their interaction with the IL-12Rβ1 receptor on the cell surface of natural killer cells and T cells. This inhibits IL-12- and IL-23-driven cell signaling, activation, and cytokine production. Adapted from Benson et al. [19]. Created in BioRender. Puig sanz, L. (2024) BioRender.com/b86x734.
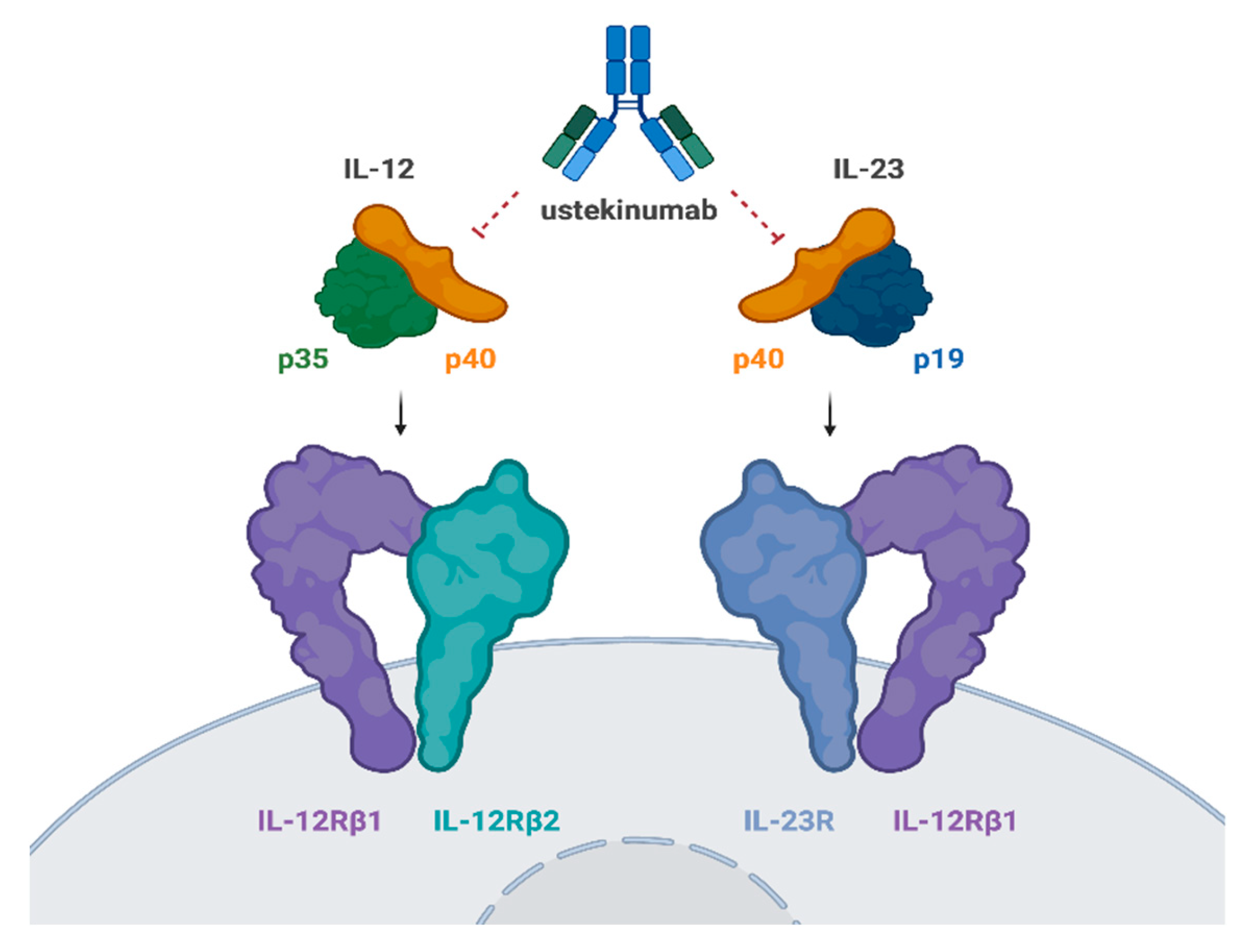
Table 2.
Ustekinumab and biosimilars of ustekinumab (approved, proposed, or under development).
| Product name | Company name | Production system | Stage of development |
|---|---|---|---|
| Ustekinumab (Stelara®) | Janssen Biotech | Sp2/0 cell line | Reference product |
| ABP 654 (Wezlana® in US and Canada; Wezenla® in Europe) | Amgen | CHO cell line | FDA-approved* in October 2023 Australian Therapeutic Goods Administration approval in January 2024 Health Canada approval in March 2024 EMA-approved in April 2024 |
| AVT04 (Uzpruvo® in Europe and Japan; Serlarsdi® in US; Jamteki™ in Canada) | Alvotech / Stada / Teva | Sp2/0 cell line | Japan approval in September 2023 Health Canada approval in November 2023 EMA-approved in January 2024 FDA-approved** in April 2024 |
| SB17 (Pyzchiva® in US and Europe; Eksunbi® in Europe; EpyztekTM in Korea) | Samsung Bioepis | CHO cell line |
EMA-approved in April 2024 FDA-approved* in July 2024 Korea approval in April 2024 |
| CT-P43 (SteQeyma®) | Celltrion Healthcare | CHO cell line |
Approved In South Korea (South Korean Ministry of Food and Drug Safety) in June-2024 Approved In Canada in July-2024 EMA-approved in August 2024 BLA submitted to FDA, pending acceptance |
| RT-111 (oral CT-P43; RaniPill®) | Rani Therapeutics | Unknown | Phase I study ongoing (NCT05890118) |
| FYB202 (Fymskina®, Otulfi®) | Formycon – Fresenius Kabi | CHO cell line |
Phase I and phase III trials completed with positive results BLA accepted by FDA MAA accepted by EMA with positive opinion from the CHMP in July 2024 |
| BAT2206 | Bio-Thera / Hikma | CHO cell line |
Phase I and phase III trials completed with positive results BLA accepted by FDA MAA accepted by EMA Approved in Brazil for psoriatic arthritis |
| DMB-3115 |
Dong-A ST / Accord BioPharma |
Sp2/0 cells |
Phase I and phase III trials completed with positive results BLA accepted by FDA MAA accepted by EMA |
| QX001S | Qyuns Therapeutics | CHO cell line |
Phase I and phase III trials completed with positive results BLA accepted by China National Medical Products Administration |
| BFI-751 | BioFactura / CuraTeQ | Murine myeloma, NS0 | Phase 1 completed with positive results (NCT04843631) |
| NeuLara | Neuclone / Serum Institute of India | Unknown | Phase 1 completed in April 2020 with positive results |
| ONS3040 | Oncobiologics | Unknown | Pre-clinical development |
| BOW090 | Epirus Biopharmaceuticals / Bioceros Holding | Unknown | Pre-clinical development |
Abbreviations: CHO, Chinese hamster ovary; BLA, Biologic License Application; MAA, Marketing Authorization Application; CHMP, Committee for Medicinal Products for Human Use *Approved as interchangeable biosimilar, meaning it can be substituted for the reference product at the pharmacy level, without the intervention of the healthcare provider who prescribed the original product. **Only for psoriasis and psoriatic arthritis.
Table 3.
Immunogenicity of ustekinumab RP and approved biosimilars.
| Product | ADA incidence in healthy individuals | ADA incidence in psoriasis patients* | NADA incidence in psoriasis patients** |
|---|---|---|---|
| UST RP [15] | 5.6% | 4.1% | 67% |
| ABP 654 [32,33] | ABP 654: 15.4% EU UST: 36.3% US UST: 38.0% |
ABP 654: 18.6% UST: 37.1% |
ABP 654: 8.6% UST: 17.9% |
| AVT04 [35,36] | AVT04: 36.7% EU UST: 59.6% US UST: 53.6% |
AVT04: 21.2% UST: 26.2%% |
AVT04: 33.3% UST: 22.9% |
| SB17 [37,38] | SB17: 26.9% EU UST: 34.3% US UST: 34.3% |
SB17: 13.3% UST: 39.4% |
SB17: 13.7% UST: 35.4% |
| CT-P43 [39] | N/A | CT-P43: 10.2% UST: 17.0% |
CT-P43: 5.9% UST: 7.9% |
*ADA incidences in psoriasis patients were obtained from Phase II studies with UST RP (Week 52) and from Phase III studies with ABP 654 (Week 28), AVT04 (Week 52), SB17 (Week 28), and CT-P43 (Week 28). **NADA were evaluated in patients with positive results for ADAs. Abbreviations: ADA, anti-drug antibody; NADA, neutralizing anti-drug antibody; EU, European Union; US, United States; N/A, not available; UST, ustekinumab; RP, reference product.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



