
Submitted:
25 September 2024
Posted:
26 September 2024
You are already at the latest version
Abstract
Obesity is one of the world's major public health challenges. Its pathogenesis and comorbid metabolic disorders share common mechanisms, such as mitochondrial or endoplasmic reticulum dysfunction or oxidative stress, gut dysbiosis, chronic in-flammation and altered autophagy. Numerous pro-autophagy dietary interventions are being investigated for their potential obesity-preventing or therapeutic effects. In this review, we summarize current data on the relationship between autophagy and obesity and discuss various non-pharmacological dietary interventions as regu-lators of autophagy-related genes in the prevention and ultimately treatment of obe-sity in humans, available in scientific databases and published through July 2024. Lifestyle modification (such as calorie restriction, intermittent fasting, physical exer-cise), a diet rich in flavonoids, antioxidants, specific fatty acids, specific amino acids and others have shown a beneficial role in the induction of this process. Induction of autophagy by heterogeneous interventions elicits a relatively homogeneous response characterized by activation of specific kinases (AMPK, IKK, JNK1, TAK1, ULK1, VPS34), inhibition of others (such as mTORC1), protein deacetylation reactions (at least in part providing SIRT1 activation and/or EP300 inhibition), and reversal of in-hibitory interactions such as those between BECN1 and members of the Bcl-2 family.
Keywords:
nutrients
; dietary intake
; autophagy
; overweight
; obesity
; humans
1. Introduction
Obesity is one of the world's major public health challenges. The estimates for global levels of high Body Mass Index (BMI > 25 suggest that nearly 3.3 billion adults living 2035 will be obese, compared with 2.2 billion in 2020. This reflects an increase from 42% of adults in 2020 to over 54% by 2035 [1]. In Poland, more than 35 percent of adult men (aged 20 years or older) and more than 25 percent of adult women will be struggling with obesity in 2035. Also, obesity among children and adolescents will increase year after year. On present trends, by 2035 more than 750 million children (age 5-19 years) are expected to be living with overweight and obesity as measured by body mass. That is equivalent to two children in every five globally, and most of these children will be living in middle-income countries [2].
Obesity is defined as excessive accumulation or abnormal distribution of body fat (globally, regionally, and in organs as ectopic lipids) that poses health risks [3]. The World Obesity Federation has recognized obesity as a chronic, recurrent and progressive disease [4]. Several factors can play a role in gaining and retaining excess weight. These include: Genetics/physiology (for example, metabolism, appetite, satiety and body fat distribution),health inequalities, environmental factors, commercial determinants (for example, media and advertising, retail environments) [4,5].
Overweight and obesity are the key risk factors for numerous Non- communicable diseases (NCDs) such as hypertension, type 2 diabetes mellitus, cardiovascular disease, metabolic syndrome, musculoskeletal disorders and 13 types of cancer. Of the 41 million adult deaths each year due to NCDs, 5 million are driven by high BMI (≥ 25 kg/m2). Nearly 4 million of these are from diabetes, stroke, coronary heart disease and cancer alone [1,6,7].
Metabolic syndrome (MetS) originally described by Reaven in 1988 as "syndrome X" or "insulin resistance syndrome", is a cluster of common abnormalities, including insulin resistance, impaired glucose tolerance, abdominal obesity, reduced high-density lipoprotein (HDL)-cholesterol levels, elevated triglycerides, and hypertension [8,9].
The coexistence of these metabolic disorders impairs liver function, manifesting as Metabolic Dysfunction–Associated Liver Disease (MASLD) –[10].
The pathogenesis of obesity and concomitant metabolic disorders shares common mechanisms such as mitochondrial dysfunction or oxidative and endoplasmic reticulum stress, chronic inflammation, gut dysbiosis, and altered autophagy [11].
According to recent studies, it is clear that the common denominator of such metabolic diseases is autophagy. The researchers have discovered that autophagy may play an important role in adipose tissue differentiation and function [12,13,14,15].
Autophagy is a highly conservative mechanism of self-digestion, responsible for the removal of damaged proteins and organelles as well as proteins distorted during biosynthesis. The function of the autophagy process is to maintain homeostasis in the body by removing damaged proteins, fatty acids and cellular organelles, which are degraded by lysosomal enzymes [16]. Autophagy can be selective or non-selective, as it can target the degradation of a specific organelle (i.e. mitophagy or lipophagy). It is induced by nutrient deficiency and stress factors to increase the number of autophagosomes. Autophagy begins with the formation and elongation of a membrane, the phagophore, which in turn develops into a vesicle surrounded by two membranes, called an autophagosome. Autophagy-related gene and protein (ATG) products are involved in this process. Autophagosomes surround abnormally folded proteins or damaged organelles and then fuse with lysosomes to form autophagolysosomes, where proteins to be degraded are digested [17,18]—Figure 1.
ATG1 and microtubule-associated protein light chain 3 (LC3) are widely regarded as important autophagy-initiating genes. Conversion of LC3-I to LC3-II results in the formation of autophagosomes. P62/secretosome (SQSTM) is a ubiquitin-binding protein that recognises ubiquitinated cargo and binds to autophagosomes through direct interaction with LC3-II [19,20]. Because both LC3-II and p62 are degraded in the autolysosome, the accumulation of LC3-II and p62 is considered a robust marker of impaired autophagy [21]. Lysosome-associated membrane proteins (LAMPs) are essential for autophagosome-lysosome fusion and are responsible for the proteolytic activity of lysosomes. While LAMP1 and LAMP2 are inhibited, autophagosome-lysosome fusion is also inhibited, which is associated with the accumulation of LC3-II and p62, resulting in reduced autophagy activity [22].
The regulatory mechanism of autophagy is closely linked to nutritional status. When nutrients are supplied to the body or under the influence of insulin, phosphatidylinositol 3-kinase (PI3K) class I activates mTOR and mTOR complex 1 (mTORC1), thereby inhibiting ATG1 activation. In contrast, the PI3K-beclin1 class III complex is triggered in the presence of nutrient deficiency, which promotes the formation of the ATG12-ATG5-ATG16L and ATG8/LC3 complex and subsequently stimulates autophagosome formation [23].
Following phagophore formation via both the mTOR and PI3K pathways, the phagophore complex and ATG5-ATG12-ATG16 bind to caveolin-1 (CAV-1) and then interact with LC3 to promote autophagosome formation and CAV-1 degradation [24]. During CAV-1 deficiency, increased levels of autophagy-related gene 7 (ATG7), beclin1 and LC3-II were demonstrated, indicating increased autophagy activity and protection against atherosclerosis [25].
Components of MetS, including elevated glucose [26] and dyslipidemia [27], inhibited autophagosome formation through CAV-1 activation.
Liver and adipose tissue are rich in lysosomes and have high levels of cellular stress-induced autophagy. In fact, reactive oxygen species (ROS) cause lysosome dysfunction and impair autophagy, preventing the degradation of damaged cellular components [28]. In addition, increased endoplasmic reticulum (ER) stress can deregulate lysosome acidification, thereby blocking autophagy in hepatocytes. This leads to hepatotoxicity, cell death and changes in liver function. Obesity, autophagy and metabolic syndrome are mutually dependent.
Based on studies, human adipose tissue and adipocytes contain autophagosomes and show increased expression of autophagy genes, such as autophagy-related genes (ATG5, ATG7 and ATG12), Beclin1, LC3I(A) and LC3II(B), LC3-II and p62 [29,30]. Interestingly, increased autophagy in obese individuals is more pronounced in visceral than in subcutaneous adipose tissue, which is also associated with greater metabolic and cardiovascular risk. The rate of fat accumulation and fat cell hypertrophy are significantly associated with autophagy gene expression, and changes in autophagy are accompanied by obesity-associated IR, preceding metabolic and cardiovascular dysfunction [29].
The growing number of studies show altered (increased or decreased) autophagy in genetically modified or diet-induced animal models of obesity [31,32,33,34,35,36]. Simultaneously, a limited number of studies in obese humans have examined whether there is a possible link between dietary nutrient intake and autophagy related genes expression.
Nutritional interventions that induce of autophagy can be used to manipulate metabolism in vivo. New insights into the functions of dietary ingredients in relation to autophagy modulation may provide useful tools in planning diet modifications during obesity treatment and its prevention in humans. In this review, we discuss the importance of various nutritional interventions as induction regulators of autophagy related genes in the prevention and/or treatment of obesity in humans.
2. Methods
This narrative review was designed and reported in accordance with the guidelines of the preferred reporting items for systematic reviews and meta-analyses (PRISMA). The following databases: PubMed, Springer, ScienceDirect and Scopus were search using the terms: “autophagy -related genes”, “overweight”, “obesity”, “ obesity diseases”, “ human studies”, “”, “metabolic syndrome” and in combination with “nutrition”, “dietary nutrition intake”, “natural compounds”, “Calorie restriction”, „Intermittent fasting” and secondary searches were conducted, adding the terms “in vitro,” “in vivo,” “review,” or “clinical trial” to the previous terms. Due to the extensive literature, only the most relevant articles were selected, taking into account primarily the quality of the study, the most recent years of publication, and the variety of mechanisms and models studied. The inclusion criteria were: 1/peer-reviewed articles in English , 2/ full-text publications, 3/use of a clear study design (cross-sectional or observational studies, etc.), 4/ studies published in the period January 1,2010 – July,1 2024. Exclusion criteria were: non-English articles, opinion pieces, scientific dissertations, abstracts. We also excluded studies of short duration (<2 weeks) and studies that focused on intercurrent medical conditions. Two independent reviewers conducted the search and selected the legal acts and list of qualified articles, which we described. Reference lists from all selected articles were also examined for additional relevant studies.
Ethical Approval: This study did not require ethical approval as it is a narrative review of previously published studies.
3. Results
3.1. Relationship between Autophagy and Obesity
Adipose tissue is a major lipid store and also plays a key role in energy metabolism. Adipose tissue differentiation involves extensive remodeling of progenitor cells, where the removal of cytoplasmic contents, particularly mitochondria, is one of the main changes that occur during adipocyte maturation. In the early stages of adipose tissue differentiation and adipogenesis, there is a huge increase in the number of mitochondria and mitochondrial proteins [37]. In mature adipocytes, on the other hand, the number of mitochondria is significantly lower compared to preadipocytes. This condition is caused by mitophagy (a type of autophagy during which mitochondria are degraded), which is strongly activated during adipocyte maturation. In addition to reducing the number of mitochondria during adipose tissue maturation, mitophagy is also involved in maintaining proper mitochondrial function in mature adipocytes [38]. Although autophagy is crucial for proper adipocyte function and differentiation, defective regulation associated with obesity results in metabolic abnormalities, leading to the development of Mets [39]. Meanwhile, the exact regulatory mechanisms of autophagy in adipogenesis are unknown. Presumably, autophagosomes facilitate the reorganization of cytoplasmic components by mobilizing membranes in the cell, thereby contributing to adipogenesis [40]. In addition, autophagy has been shown to increase the stability of peroxisome proliferator-activated receptor (PPAR) γ, a master regulator of adipogenesis and adipocyte differentiation. Studies confirm that inhibition of autophagy decreases PPARγ activity and directly attenuates adipocyte differentiation [41]. PPARγ is the rate-limiting enzyme for adipogenesis and fat accumulation in excessive adipose tissue [42]. Thus, PPARγ activation by autophagy may be the mechanism by which autophagy induces obesity and may become a future target for preventing obesity-associated autophagy during MetS [43]. In addition, PPARγ activation during obesity depends on a number of other factors, including polyunsaturated fatty acids (PUFAs) and prostaglandins (e.g. J2 or D2) [44]. Therefore, additional studies are needed to determine exactly whether the activated PPARγ pathway induces or inhibits autophagy during obesity. Adipogenesis is a two-step process in which multipotent adipose tissue-derived mesenchymal stem cells (ASCs) transform into mature adipocytes, which in turn are involved in energy storage in the form of fat. In obesity, an increase in the size and number of these cells leads to adipose tissue proliferation, which is closely linked to IR [45].
Obesity is characterized by a significant increase in fat mass and is a major risk factor for the development of IR. Autophagy plays a key role in adipogenesis. Adipocyte differentiation is associated with increased levels of autophagy, while inhibition of autophagy inhibits adipogenesis [46]. The pathogenesis of obesity underlies a significant accumulation of potential autophagic substrates, such as lipid droplets, protein aggregates and damaged mitochondria. Therefore, inhibition of autophagy can be expected to accelerate the development of obesity and its associated pathologies. The pathogenesis of obesity underlies a significant accumulation of potential autophagic substrates, such as lipid droplets, protein aggregates and damaged mitochondria [47]. Thus, inhibition of autophagy might be expected to accelerate the development of obesity and its associated pathologies. However, recent studies indicate that a myriad of intracellular and extracellular factors are involved in the etiogenesis and development of obesity. Therefore, it is imperative that the autophagy process in obese patients is thoroughly investigated in order to fully exploit its therapeutic potential in the prevention and treatment of obesity. To date, changes in autophagy, both its increase and decrease, have been shown to be involved in the pathogenesis of various diseases, including cancer, neurological, cardiovascular and metabolic diseases [48].
Activation of autophagy facilitates adipocyte differentiation, induces adipogenesis and increases fat accumulation in adipose tissue. A clinical study by Kovsan et al.[49] involving non-obese, obese and severely obese patients (with and without diabetes) confirmed a possible link between induced autophagy activity and fat accumulation.
Studies performed on adipose tissue from obese subjects showed higher expression of autophagy-related genes ATG5-12 and autophagosome membrane-binding proteins LC3I(A) and LC3II(B). The expression of LC3-II, the ATG5-12 protein complex, mTOR and ATG was analyzed by Western blot. It was confirmed that autophagy was increased in both visceral and subcutaneous adipose tissue in obese patients relative to control tissues. The protein Beclin1, which is a master regulator of autophagy, was also elevated in obese patients. Furthermore, ATG12 mRNA expression was positively associated with the degree of obesity, the presence of visceral adipose tissue and adipocyte hypertrophy, confirming increased autophagy [50].
In addition, ER stress, inflammation or hypoxia, which are processes that are active in adipose tissue during obesity, promote autophagy through suppression of mTORC1 [51]. Overall, autophagy may be a protective mechanism against the increased inflammation associated with obesity or serve as a compensatory response to the excessive accumulation of nutrients and damaged organelles in hypertrophied adipocytes [52].
Insulin is an anabolic hormone that acts as a potent inhibitor of autophagy [53]. It can prevent autophagy by activating mTORC1, resulting in suppression of FoxO and ULK1 factors. The PI3K-Akt pathway is a key component of the insulin signaling pathway that contributes to the hormone's inhibition of autophagy. Akt inhibits FoxO 1/3 and induces mTORC1 activity, thus revealing a major link between insulin signaling and autophagy [54]. However, targeted deletion of ATG7 causes deleterious white adipose tissue (WAT) differentiation and browning, resulting in improved insulin sensitivity and glucose consumption, as well as increased β-oxidation of fatty acids [52]. These metabolic changes through inhibition of autophagy play a key role in IR [38]. It has indeed been shown that specific deficiency of ATG3 and ATG16L1 in adipocytes caused IR. In particular, inhibition of autophagy in adipocytes interfered with insulin signaling to Akt in adipose tissue, liver and skeletal muscle. Based on the studies performed to date, innate inhibition of autophagy impairs adipogenesis and leads to insulin sensitivity, whereas selective inhibition of this pathway in mature adipocytes results in IR [38,55,56]—Figure 2 and Figure 3 show the insulin signaling pathway in adipocytes and liver.
An increase in the transcription factor E2F1 in adipose tissue of obese individuals was found to be associated with the expression of ATG genes, mainly those involved in the later stages of autophagy, such as ATG12, LC3-II and DRAM1[57,58]. E2F1-deficient adipocytes under the influence of inflammatory cytokines showed less activation of autophagy. Interestingly, E2F1 induction in adipose tissue occurs simultaneously with inflammatory activation. This fact suggests that activation of autophagy by E2F1 may act as a protective mechanism against obesity-related inflammation [59,60]. These results indicate that there is a concomitant correlation between the regulation of autophagy and inflammation. Many cytokines or adipokines released during mild inflammation induce autophagy, which is an important mechanism for the clearance of invading pathogens.
The action of autophagy in the liver differs significantly from its behavior in adipose tissue during MetS. In obesity, autophagy in hepatocytes is significantly reduced (in contrast, an increase is observed in adipose tissue) because impaired metabolism is observed in the liver along with deformed mitochondria [61,62]. In contrast to the role of autophagy in adipose tissue, inhibition of autophagy promotes lipid accumulation in hepatocytes through lipolysis of lipid droplets accumulated in TG [63]. Furthermore, a constantly positive energy balance favors mTORC1 activity at the expense of AMPK, achieving consequent inhibition of autophagy [64]. In a study in mice, it was shown that long-term use of HFD induced induction of mTORC1 activity and decreased ATG5 and ATG7 expression in the liver, where autophagy is markedly activated during starvation [65,66,67]. Yang et al. [65] showed lower protein expression of ATG7, beclin1 (ATG6), LC3, ATG5, and elevated expression of p62 in livers of obese mice. Moreover, higher levels of ER and IR stress were observed in these mice due to impaired autophagy activity in hepatocytes. Furthermore, reduced autophagy in the liver was observed in both diet-induced obesity and genetic obesity models, which could be explained by obesity-associated hyperinsulinaemia (insulin inhibits autophagy). Yet, insulin is not the main cause of reduced autophagy in the liver in obese individuals. It is likely that other mechanisms co-exist here. One is the action of calpain 2, a Ca2+-dependent protease whose higher levels in hepatocytes reduce autophagy in obese patients[68,69], and whose inhibition increases autophagy [65]. Another possible mechanism that reduces autophagy in the liver is the transcription factor FoxO, which acts as a key regulator of the Vps34 and ATG12 proteins responsible for the initiation of autophagy [70]. Elevated insulin levels and activated Akt suppress FoxO activity, thereby reducing the rate of autophagy in MetS [71]. Thus, long-term inhibition of autophagy due to IR and hyperinsulinaemia in MetS can be explained by reduced FoxO activity in hepatocytes [70]. Similarly, genetic or pharmacological inhibition of autophagy counteracts starvation-induced weight loss while contributing to obesity and T2DM [72]. Therefore, chronic HDF use is thought to alter the intracellular ion balance in hepatocytes, ultimately impeding autophagosome-lysosome fusion [73,74]. In addition to the altered autophagy in the adipose tissue of obese mice, other tissues such as the hypothalamus and kidney also show lower levels of autophagy [75,76] suggesting the involvement of systemic factors. In addition, mitochondrial and ER oxidative stress and the accumulation of toxic substances may be responsible for, among other things, inducing IR [77]. Soussi et al. described a reduction in autophagic flow in adipocytes of subcutaneous tissue in obese subjects [14]. In contrast, other authors have reported a relationship between nutrient restriction in obese subjects and an increase in autophagy activity associated with improved insulin sensitivity [78].
In contrast, subsequent studies have shown that HFD-induced hepatic steatosis and obesity-related ER stress essentially activate autophagy as a protective mechanism against cellular damage [79]. Autophagy protects hepatocytes from lipotoxicity-related ER stress, as well as from SFA (palmitic acid) induced apoptosis [74,80], and this may be the reason for the observed induction of autophagy in the early stages of obesity. Nevertheless, studies have shown that the effects of HDF-induced autophagy persist for the first few weeks, with autophagy activity eventually declining due to the ongoing cellular stress that occurs in chronic obesity [65]. Furthermore, a mouse study showed that mRNA and protein levels of beclin1 and LC3 were significantly higher in severely obese mice compared to controls. In contrast, the same obese mice showed significantly reduced LC3-II levels and LC3-II/LC3-I ratios compared to control mice, indicating impaired autophagy [81]. In summary, some of these results indicate increased expression of autophagy markers and number of autophagosomes in obesity. However, without adequate measurement of autophagic flow (total autophagosome synthesis, substrate delivery and lysosome degradation), it cannot really be said that these results indicate increased autophagy activity. Hence, there is a need for further research to clearly understand and describe the existing relationships. Regarding human studies [82,83,84,85], the general depicted trend is an increased White Adipose Tissue (WAT) autophagy in obese and/or diabetic humans. It was found that higher mRNA and/or protein levels of several autophagic markers: Beclin-1, ATG5, ATG12, ATG7, LC3A and B, LC3-II, p62 and decreased mTOR expression in subcutaneous WAT and/or visceral WAT from obese compared with lean individuals.
3.2. Nutritional Interwentions for Autophagy Activation in Overweigth/obese Humans
Lifestyle modification (such as calorie restriction, intermittent fasting, sleep, stress control, various diets, exercise), nutritional interventions and pharmacological modulation of autophagy have been proved to be beneficial in preventing and treating obesity and its complications by improving metabolic health [11]. Nutrition interventions are defined as deliberately planned actions that aim to positively change a nutrition-related behaviors, environmental conditions, or an aspect of the health status of an individual, a target group or an entire community [87].
Data from epidemiological, experimental and clinical studies have shown that nutritional strategies are well-known methods for inducing autophagy [31,88,89,90]—Table 1.
3.2.1. Calorie Restriction; Intermittent Fasting
According to number of studies [91,92,93,94,95] Calorie Restriction - CR (usually 20%–40% intake) and Intermittent Fasting - IF (various short- and long-term diet programs with regular cycles between eating and fasting times) stimulate autophagy mainly by decreasing mTOR signaling through decreased insulin and IGF-1 levels and increased AMP/ATP ratio, leading to activation of AMPK as well as several other products involved in stimulating this process (ATG6, ATG7, ATG8, LC3-II, Beclin1, p62, SIRT1, LAMP2, ULK1 and ATG101). Calorie restriction also alters the level and/or activity of CoA (the sole donor of acetyl groups), acetyltransferases and/or deacetylases, leading to the induction of autophagy through deacetylation of cellular proteins. Deep activation of autophagy occurs after 72 hours of fasting, but 3-days periods without food intake are challenging for individuals. Therefore, various less restrictive dietary models have been developed to help induce autophagy [96].
In study by Yang et al [97] serum cortisol, molecular chaperones and autophagy proteins were measured in skeletal muscle of subjects on CR diet for 3–15 years and in control volunteers. The authors found that CR significantly upregulated multitude autophagy genes, including ULK1, ATG101, Beclin-1, APG12, microtubule-associated protein 1 light chain 3 (LC3), GAPRAP/GATE-16, and autophagin-1. At the same time, the expression levels of Beclin-1 and LC3 proteins were significantly higher in the skeletal muscles of the CR volunteers compared to the control group. Chaudhary and co-workers [98] reported, that skeletal muscle autophagy may be suppressed in obese woman in response to weight loss. In our study, fifty women (51 ± 2 years; BMI 31.8 ± 4.3 kg/m2) were randomly assigned to one of two IF protocols (24-hours fasting, 3 non-consecutive days per week) and fed at 70% (IF70) or 100% (IF100) of energy requirements for 8 weeks. Vastus lateralis muscle was sampled after 12- and 24-h fasting. The 24-h fast increased mRNA levels of SQSTM1, BECLIN1, SQSTM1 and LAMP2, which were reduced in IF70 after a 12-h overnight fast. The benefits of food and energy restriction in promoting optimal health were supported by a 2017 study that found that IF (60% calorie restriction for 2 days per week or every other day) delays pathological processes through adaptive stress signaling cascades to improve mitochondrial health, DNA repair and autophagy [99,100]. Kim and Li conclude that: these findings are consistent with the hypothesis that humans and other animals evolved survival mechanisms in food-deficient environments and developed adaptations to improve both physical and cognitive functions [101].
In report by Kitada, Kume et al. [102] four overweight male participants were enrolled and treated with 25% CR of their basal energy requirements for 7 weeks, allowing the researchers to demonstrate the effects of human serum taken from CR participants on AMPK and SIRT1 activation and mitochondrial biogenesis in cultured human skeletal muscle cells. AMPK and SIRT1 activation was assessed by the deacetylation of H2O2-induced increase in acetylated-p53 expression and was shown to be significantly increased in human skeletal muscle cells cultured with serum after CR. The authors observed a correlation between SIRT1 gene expression and lower serum levels of insulin, free fatty acids and interleukin 6.
The interplay between Sirtuins and autophagy, both potential longevity-promoting factors, has gained more attention in recent years. SIRT1 is one of the main mammalian sirtuins that is upregulated in response to CR [103].
Other studies [104,105] have shown that SIRT1 is expressed in visceral adipose tissue and is reduced by obesity. Sirtuin 1 (SIRT1) increased basal autophagic activity, and SIRT1 knock-out cells and mice demonstrated unusually high levels of acetylation of essential autophagic proteins like Autophagy Protein 5 (ATG5), ATG7, and Light Chain 3B (LC3B) [102,106].
It has been suggested that suppression of mTOR signaling and increased autophagy contribute to many of the beneficial adaptations observed with IF protocols in the subjects analyzed. Time-restricted eating (TRE) is a popular form of IF that limits caloric intake, without changing the quantity and quality of diet, to a period of 6–10 h. Overweight adults (6 Females, 3 Males; aged 65 years and older) were advised to fast for approximately 16 h per day for four weeks with the daily target range set for 14–18 h. Participants were asked to abstain from caloric intake during the target fasting window of 16 continuous hours. There were no dietary restrictions on the amount or types of food consumed during the 8 h eating window, and participants could choose the time frame that best fit their lifestyle [107]. Blood was collected from all participants before and after the TRE regimen in the morning, and the expression of 2083 human miRNAs was quantified using HTG molecular whole transcriptome miRNA assay, and ultimately fourteen miRNAs were differentially expressed before and after TRE regimen. Notably, downregulated miRNA targets suggested increased expression of transcripts, including PTEN, TSC1, and ULK1. The serine/threonine protein kinase ULK1 (unc-51-like kinase 1), which functions in a complex with at least three protein partners: FIP200 (focal adhesion kinase family interacting protein of 200 kDa), ATG (autophagy-related protein) 13 (ATG13), and ATG10 to regulate the formation of autophagophores, the precursors of autophagosomes . It also acts as both as a downstream effector and a negative regulator of the mammalian target of rapamycin complex 1 (mTORC1) [108]. It has been suggested that suppression of mTOR signaling and increased autophagy contribute to many of the beneficial adaptations observed in TRE protocols in clinical populations.
The beneficial effects on autophagy of both CR and IF appear to be associated with increases in fat mobilization, oxidation, metabolic flexibility, insulin sensitivity and redox imbalance along with a reduction in systemic inflammation, cardiovascular risks and body weight.
Many nutrients with significant health/translational properties (nutraceuticals), may affect the chronic disease risk through various mechanisms that include the activation or inhibition of autophagy [31,88,109]. These include, for exemple: amino acids (i.e., leucine), fatty acids (i.e., omega 3 polyunsaturated fatty acids), vitamins (carotenoids and retinoids, ascorbic acid, calciferol, tocopherols, and tocotrienols), coenzyme Q10, bioactive compounds (i.e., mainly polyphenols like curcumin, caffeine, EGCG, resveratrol, allicin), minerals ( zinc or iron), ergothioneine, lipoic acid, N acetylcysteine and spermidine [110].
3.2.2. Mediterranean Diet (Met Diet)
In 2010, UNESCO acknowledged Mediterranean Diet as an Intangible Cultural Heritage of Humanity and developed the food pyramid model in order to communicate the “Me Diet” model to people and health professionals [110].
The Mediterranean diet is characterized by a high intake of plants and is rich in dietary fiber, vitamins, polyunsaturated fatty acids, oligoelements, polyphenols, and others [111]. One of the dietary ingredients common in plant-based diets are polyphenols, which are particularly abundant in fruits, vegetables, whole grains, and legumes but also in cocoa, tea, coffee, and red wine [112]. Polyphenols are classified as flavonoids and seven described subclasses (flavonols, flavones, flavanones, flavanonols, flavanols, anthocyanidins, and isoflavones) and non-flavonoid molecules (phenolic acids, hydroxycinnamic acids, lignans, stilbenes, and tannins) [113,114]. Flavonoids are polyphenolic secondary metabolites that are commonly found in most plants. These compounds can occur as glycosides or aglycones. Flavonoids exhibit a broad spectrum of biological activities, such as neuroprotective, anti-inflammatory, antibacterial, hepatoprotective, anti-mutagenic, anticancer, cardiovascular protective, antifungal, antiviral, and anti-allergic effects [115].
The high content of protective phenolic compounds in the MedDiet ingredients, especially those present in vegetables and fruits, may also help explain their multiple benefits [116]. Some well-known polyphenols include: resveratrol, quercetin, curcumin, epigallocatechin gallate, catechin, hesperetin, cyanidin, procyanidin, caffeic acid, and genistein [117].
A study by Osorio – Conles et co-workers [118] evaluated the short-term effects of a dietary intervention based on the Mediterranean diet (MedDiet ) supplemented with almonds (MDSA) on the main features of obesity-related white adipose tissue (WAT) dysfunction. A total of 38 obese women (aged 18–68 years, with a BMI of 40–50 kg/m2 ) were randomly assigned to a 3-month intervention with MDSA (19 women) vs. maintaining their usual diet ( 17 women). Biopsies of subcutaneous (SAT) and visceral adipose tissue (VAT) were obtained before and after the dietary intervention. The expression of angiogenesis-related genes PDGFRB, VEGFA, VEGFR1 and VEGFR2 was significantly increased after MDSA intervention compared to controls. In VAT the expression of genes associated with adipogenesis, angiogenesis, autophagy and fatty acid usage was increased. Among other things, the authors found increased expression of autophagy-related ATG 7 and ATG12 in VAT from the MDSA group, while ATG5 showed a non-significant trend (p = 0.054).
3.2.3. Dietary Polyphenols
Dietary polyphenols have beneficial effects on adipose tissue mass in humans. Many autophagic pathways, including cAMP, AMPK, MAPK, AKT, SIRT1, PI3K, Nrf2/HO-1, PINK1/Parkin, PPARδ, and miRNAs have been implicated in the improvement of glucolipid metabolic diseases by polyphenols [119]. Epigallocatechin-3-gallate and resveratrol are dietary polyphenols abundantly available in green tea and in grapes, respectively.
In a randomized, placebo-controlled study of Most et al. [120] 25 (10 women) overweight and obese humans received a combination of the polyphenols epigallocatechin-gallate (EGCG) and resveratrol (RES) (282 mg/d, 80 mg/d, respectively, EGCG+RES, n = 11) or placebo (PLA, n = 14) for 12 weeks. Abdominal subcutaneous adipose tissue (SAT) biopsies were taken for assessment of adipocyte morphology and micro-array analysis. EGCG+RES supplementation had no significant effect on mean adipocyte size or surface area in abdominal subcutaneous AT and illustrated that it can induce a suppression of gene sets related to adipocyte turnover (adipogenesis and apoptosis/autophagy), inflammation and the immune system in AT in overweight and obese humans. Increased gene expression: ATP6V1A, ATP6V1H, CD68, HSL/LIPE, LAMP2, PI4K2A, UCP2, GAPDH.
In placebo-controlled, double-blind cross-over study [121] 11 healthy obese patients (52.5 ± 2.1 years) received placebo followed by 150 mg of resveratrol once daily for 30 days after a 4-week wash-out period. After muscle biopsy, the authors also found increased AMPK phosphorylation and SIRT-1 and PGC-1α expression. Konings E, Timmers S et al. [122], indicated that in the obese men analyzed previously, it reduced the size of subcutaneous adipocyte in the abdominal region and enhanced and improved adipogenesis, probably through modulating gene expression.. At the same time, RES supplementation induced, among other things, the expression of TFEB (transcriptional factor EB) in the subcutaneous AT. TFEB controls multiple crucial steps in the autophagic pathway [123]. Moskot M, Montefusco S et al. [124] discovered a regulatory network linking phytoestrogen genistein-mediated control of EB transcription factor (TFEB) gene expression, TFEB nuclear translocation, and activation of TFEB-dependent lysosome biogenesis to lysosomal metabolism. Resveratrol suppresses mTOR activity, which should mediate resveratrol-induced autophagy. Upstream regulators including PI3K, AMPK, and SIRT1, have been shown to be involved in mechanism of inhibition [125,126,127]. Park et al. [128] suggested that resveratrol stimulates autophagy through convergent modalities that include the activation of the AMPK–SIRT1–PGC-1α axis and the inhibition of the mTOR-ULK1 pathway.
Forty-eight healthy participants (24 women) aged 55-65years, BMI <29.9 kg/m2 were randomized to 30 days of resveratrol administration (500mg/day) or caloric restriction (1000cal/day) in study by Mansur et co-workers [129]. Plasma SIRT1 concentrations and gene expression were examined by real-time PCR. Resveratrol and caloric restriction increased serum concentrations of SIRT1, from 1.06±0.71 to 5.75±2.98ng/mL; p<0.0001, and from 1.65±1.81 to 5.80±2.23ng/mL; p<0.0001, respectively.
3.2.4. Dietary Fatty Acids
Modulation of autophagy of subcutaneous visceral adispose tissue by dietary intervention has also been reported in obese patients. Evidence suggests that this modulation depends on the type of dietary fat, protein and carbohydrates consumed [82]. A high-fat diet is one of the factors causing obesity. Dietary fatty acids profile is also an important variable in the development of obesity and the role of autophagy in this process [130]. Saturated fatty acids (SFAs) have been shown to contribute more to obesity than monounsaturated fatty acids (MUFAs) and polyunsaturated fatty acids (PUFAs) [131]. As the most common monosaturated fatty acid (MUFA) in the daily diet, oleic acid could induce autophagy, which is responsible for regulating lipids metabolism in hepatocytes. ω-3 and ω-6 polyunsaturated fatty acids (PUFA) are essential for normal physiology and metabolism and play a role in the occurrence and development of several diseases [132]. O’Rourke data showed that ω-6 PUFA supplementation activates autophagy in human epithelial cells [133].
According Ciesielska K, Gajewska M [134] SFAs induce autophagy, which appears to be directly correlated with the activation of the diabetogenic stress kinase JNK1, as well as increased ER stress observed in hypertrophic adipocytes. PUFAs exert an opposite effect, being able to alleviate SFA-induced mitochondrial dysfunction, reduce oxidative stress, and modulate the inflammatory response of the adipose tissue. Yang B and co-workers [135] concluded that ω-6 PUFAs (linoleic acid) activate both autophagy and antioxidation in a synergistic feedback loop through TOR-dependent and TOR-independent signaling pathways. Camargo et al.[136] conducted a randomized, controlled trial involving 39 obese volunteers with metabolic syndrome, receiving one of four diets for 12 weeks: a high-saturated fatty acid diet (HSFA), a high-monounsaturated fatty acid diet (HMUFA), and two low-fat, high-complex carbohydrate diets supplemented with long-chain n-3 polyunsaturated fatty acids (LFHCC n-3) or placebo (LFHCC). After the dietary intervention period, adipose tissue samples were collected from the superficial abdominal subcutaneous adipose tissue. The authors noted significantly increased expression of autophagy-related genes (BECN1 and ATG7) after long-term consumption of the HMUFA diet, and increased expression of the apoptosis-related CASP3 gene after the long-term consumption of the LFHCC and LFHCC n-3 diets. Expression of the other autophagy markers analyzed (LC3, LAMP2, and ULK1), tended to increase after consumption of the LFHCC n-3 diet. The number of autophagy-related genes tended to increase after long-term use of the LFHCC n-3 diet, consistent with previous descriptions of apoptosis and autophagy being related.The authors concluded that enhanced autophagy may contribute to the maintenance of adipose tissue homeostasis.
The studies described above indicate that PUFAs may be involved in the regulation of autophagy directly, but also indirectly via their small bioactive lipid mediators.
3.2.5. Diet Modifications
The 648 participants in the European multicenter NUGENOB (Nutrient-Gene Interaction in Human Obesity) study [137,138] were randomly assigned to 10-weeks dietary intervention of two hypoenergetic diets: low-fat, high-carbohydrate diet (LF) or a moderate-fat, low-carbohydrate diet (MF). Both diets were designed to provide 600 kcal/d less than the individual’s estimated energy requirement. Fat provided 20–25 and 40–45%, carbohydrates provided 60–65 and 40–45% of total energy in the LF and the MF diets respectively. Both diets provided 15% of total energy from protein. Gene expression in adipose tissue before and after 10 weeks of diet was assessed in two sets 47 obese subjects selected within each dietary group. The expression of five genes: FABP4, SIRT3, NR3C1, GABARAPL2, and FNTA was 15–65% higher in the MF than the LF. In this study, energy restriction had a more pronounced effect on changes in human adipose tissue gene expression than macronutrient composition. Macronutrient-sensitive regulation of a subset of genes may influence adipose tissue function and metabolic response.
3.2.6. Protein Intake
Protein intake is one of the strongest dietary regulators of circulating levels of IGF-1, a potent growth factor that activates the Akt/mTOR pathway [139]. According Liu Ch, Ji L et al. [106] regulation of autophagy by amino acids is still a burgeoning field of research. In particular, functional amino acids (FAAs) (e.g., arginine, leucine, glutamine, and methionine) are involved in protein synthesis and homeostasis and regulate autophagy through sensor-mediated activation of mTORC1 kinase. In clinical study by Xu C. et al. [140] 19 morbidly obese participants (BMI approximately 45 kg/m2 ) undergoing bariatric surgery were analyzed in two hypocaloric (1500-1600 kcal/day) dietary groups: low protein (10E% protein) and high protein (30E% protein), for three weeks prior to surgery. Serum levels of intrahepatic lipids (IHL) and fibroblast growth factor 21 (FGF21) were measured before and after the dietary intervention. Autophagy flux, histology, mitochondrial activity and gene expression analyses were performed in liver samples collected during surgery. From dynamic analyses of autophagy flux after 3 weeks of intervention, this study confirmed that the LP group displayed significantly elevated autophagy flux and FGF21 levels in the liver and circulation compared to HP, but the HP diet more was more effective in reducing intrahepatic fat. Expression of autophagy-related genes (LC3A, LC3B and Atg5) was positively correlated with ER-stress genes (BiP, XBP1s, XBP1, ATF4 and DDIT3).
4. Conclusions
Numerous studies have shown that autophagy plays an important role in adipose tissue - in its differentiation and function in states of physiology and pathology. Autophagy is crucial for the proper functioning and differentiation of adipocytes, and its defective regulation associated with obesity results in metabolic abnormalities, leading to the development of metabolic syndrome. Increased expression of autophagy-related genes correlates with the degree of visceral fat mass obesity, and adipocyte hypertrophy and autophagy in adipose tissue are associated with impaired glucose tolerance in a manner independent of BMI and insulin. Many nutrients with significant health/translational properties (nutraceuticals) can affect chronic disease risk through various mechanisms that include activation or inhibition of autophagy. Nutritional intervention in the regulation of autophagy in obesity and its comorbidities is not yet clear, especially in obese individuals.
This is due to the fact that most experimental studies on this topic are conducted on cell lines or animal models. Most human studies mainly evaluate anthropometric measurements, selected biochemical indicators before, during and after the designed diet. It does not, for example, address changes in autophagy gene expression in adipose tissue (visceral or subcutaneous), which is mainly due to the invasiveness of this procedure. So many questions about nutrient bioavailability, optimal dosage and overall efficacy remain unanswered.
At the same time, a limited number of studies in obese people have examined whether there is a possible link between dietary nutrient intake and the expression of autophagy-related genes. There are also few clinical studies, with a sufficiently large number of participants (patients, volunteers) emphasizing the importance of dietary and lifestyle strategies in health maintenance and secondary and tertiary prevention. Clinical trials of autophagy-inducing nutrients are strongly encouraged, while emphasizing the dose, duration and possible synergistic effects of various compounds.
Future research should focus on longitudinal studies, and clinical trials will provide a comprehensive understanding of how to harness the potential of nutrition to regulate specific signaling pathways that maintain efficient autophagy in obese humans. Nutrient-sensitive pathways influenced by nutrient compounds and dietary components offer an important untapped perspective for treating obesity by affecting inflammation, oxidative stress and nutrient metabolism, which seems reasonable and needed.
Author Contributions
Conceptualization, M.M-W. and M.B.; methodology, M.B.,N.D-S.; D.L.and M.M-W; resources, M.B.;N.D-S.;D.L; S.D-G. and D.W.; writing—original draft preparation M.B.; M.M-W;S.D-G;D.W; writing—review and editing, M.M-W.;S.D-G;D.W.; visualization, N.D-S.;D.L.; supervision, M.B. and M.M-W.; All authors have read and agreed to the published version of the manuscript.
Funding
The grant number BN-W-1-056/K/3/Z Medical University of Silesia in Katowice.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- World Obesity Federation. World Obesity Atlas 2024. London: World Obesity Federation, 2024. https://data.worldobesity.org/publications/.
- World Health Organization. Obesity and overweight. Available from: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed: 29 June 2024).
- Purnell,J. Definitions, Classification, and Epidemiology of Obesity. Endotext 2023, https://www.ncbi.nlm.nih.gov/books/NBK279167/.
- https://www.worldobesity.org/what-we-do/our-policy-priorities/obesity-as-a-disease (accessed: 28 August 2024).
- Li, X. Qi, L. Gene-Environment Interactions on Body Fat Distribution. Int J Mol Sci. 2019 Jul 27;20(15):3690. [CrossRef] [PubMed]
- World Health Organization. Noncommunicable diseases. 2023. https://www.who.int/news-room/fact-sheets/detail/noncommunicable-diseases#%7Etext=Noncommunicable.20diseases.20(NCDs).20kill.2041-.20and.20middle-income.
- Abarca-Gómez L., Abdeen Z. A., Hamid Z. A., et al. Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: a pooled analysis of 2416 population-based measurement studies in 9 million children, adolescents, and adults. The Lancet . 2017;390(10113):2627–2642. [CrossRef]
- Lemieux I, Després, J.P. Metabolic syndrome: past, present and future. Nutrients. 2020; 12(11):3501.
- Fathi Dizaji B. The investigations of genetic determinants of the metabolic syndrome. Diabetes Metab Syndr. 2018;12(5):783-789. [CrossRef]
- Pal, SC, Méndez-Sánchez N. Screening for MAFLD: who, when and how? Ther Adv Endocrinol Metab. 2023;14. [CrossRef]
- Jakubek, P.; Pakula, B.; Rossmeisl, M.; et al. Autophagy alterations in obesity, type 2 diabetes, and metabolic dysfunction-associated steatotic liver disease: the evidence from human studies. Intern Emerg Med 2024. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J; Petroni, G; Amaravadi,R.K; Baehrecke, E.H; Ballabio, A; Boya, P. et al. Autophagy in major human diseases. EMBO J. 2021 ;40(19):e108863. [CrossRef]
- Hassanpour, M; Rahbarghazi, R; Nouri, M; Aghamohammadzadeh, N; Safaei, N; Ahmadi, M. Role of autophagy in atherosclerosis: foe or friend? J Inflamm (Lond). 2019;16:8. [CrossRef]
- Soussi, H.; Clément, K.; Dugail, I. Adipose tissue autophagy status in obesity: Expression and flux--two faces of the picture. Autophagy. 2016;12(3):588-589. [CrossRef]
- Xu, Q.; Mariman, E.; Roumans, N.; Vink, R.G.; Goossens, G.H.; Blaak, E.E. et al. Adipose tissue autophagy related gene expression is associated with glucometabolic status in human obesity. Adipocyte. 2018;7(1):12-19. [CrossRef]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P. et al. Autophagy in major human diseases. EMBO J. 2021; 40(19):e108863. [CrossRef]
- Gatica, D.; Chiong, M.; Lavandero, S.; Klionsky, D.J. Molecular mechanisms of autophagy in the cardiovascular system. Circ Res. 2015;116(3):456-467. [CrossRef]
- Nishimura, T.; Tooze, S.A. Emerging roles of ATG proteins and membrane lipids in autophagosome formation. Cell Discov. 2020; 6(1):32. [CrossRef]
- Nussenzweig, S.C.; Verma, S.; Finkel, T. The role of autophagy in vascular biology. Circ Res. 2015;116(3):480-488. [CrossRef]
- Hassanpour, M.; Rahbarghazi, R.; Nouri, M.; Aghamohammadzadeh, N.; Safaei, N.; Ahmadi, M. Role of autophagy in atherosclerosis: foe or friend? J Inflamm (Lond). 2019;16:8. [CrossRef]
- Klionsky, D.J,; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.; Adams, P.D.; Adeli, K.; Adhihetty, P.J.; Adler, S.G.; Agam, G.; Agarwal, R.; Aghi, M.K.; Agnello, M. et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12:1–222.
- Terasawa, K.; Tomabechi ,Y.; Ikeda, M.; Ehara, H.; Kukimoto-Niino ,M.; Wakiyama, M.; Podyma-Inoue, K.A.; Rajapakshe, A.R.; Watabe, T.; Shirouzu, M.; Hara-Yokoyama, M. Lysosome-associated membrane proteins-1 and -2 (LAMP-1 and LAMP-2) assemble via distinct modes. Biochem Biophys Res Commun. 2016 Oct 21;479(3):489-495. Epub 2016 Sep 20. [CrossRef] [PubMed]
- Shao, B.Z.; Han, B.Z.; Zeng, Y.X.; Su, D.F.; Liu, C. The roles of macrophage autophagy in atherosclerosis. Acta Pharmacol Sin. 2016;37(2):150-156. [CrossRef]
- Hou, K.; Li, S.; Zhang, M.; Qin, X. Caveolin-1 in autophagy: A potential therapeutic target in atherosclerosis. Clin Chim Acta. 2021;513:25-33. [CrossRef]
- Wu, Z.; Huang. C.; Xu, C.; Xie, L., Liang, J.J.; Liu. L, et al. Caveolin-1 regulates human trabecular meshwork cell adhesion, endocytosis, and autophagy. J Cell Biochem. 2019;120(8):13382-13391. [CrossRef]
- Bai, X.; Yang, X.; Jia, X.; Rong, Y.; Chen, L.; Zeng, T. et al. CAV1-CAVIN1-LC3B-mediated autophagy regulates high glucose-stimulated LDL transcytosis. Autophagy. 2020;16(6):1111-1129. [CrossRef]
- Chen, Z.; Nie, S.D.; Qu, M.L.; Zhou, D.; Wu, L.Y.; Shi, X.J. et al. The autophagic degradation of Cav-1 contributes to PA-induced apoptosis and inflammation of astrocytes. Cell Death Dis. 2018;9(7). [CrossRef]
- Chen, X.; Chan, H.; Zhang, L.; Liu, X.; Ho, IHT.; Zhang, X. et al. The phytochemical polydatin ameliorates non-alcoholic steatohepatitis by restoring lysosomal function and autophagic flux. J Cell Mol Med. 2019;23(6):4290-4300. [CrossRef]
- Clemente-Postigo, M.; Tinahones, A.; Bekay, R.; Malagón, M.M.; Tinahones, F.J. The Role of Autophagy in White Adipose Tissue Function: Implications for Metabolic Health. Metabolites. 2020;10(5). [CrossRef]
- Soussi, H.; Clément, K.; Dugail, I. Adipose tissue autophagy status in obesity: Expression and flux--two faces of the picture. Autophagy. 2016;12(3):588-589. [CrossRef]
- Zhang, Y.; Sowers, J.R.; Ren, J. Targeting autophagy in obesity: from pathophysiology to management. Nat Rev Endocrinol. 2018 Jun;14(6):356-376. [CrossRef] [PubMed]
- Lopez-Vicario, C.; Alcaraz-Quiles, J.; García-Alonso, V.; Rius, B.; Hwang, S.H., Titos, E.; Lopategi, A.; Hammock, B.D.; Arroyo, V.; Clària J. Inhibition of soluble epoxide hydrolase modulates inflammation and autophagy in obese adipose tissue and liver: Role for omega-3 epoxides. Proc. Natl. Acad. Sci. USA. 2015;112:536–541. [CrossRef]
- Yasuda-Yamahara, M.; Kume, S.; Yamahara, K.; Nakazawa, J.; Chin-Kanasaki, M.; Araki, H.; Araki S.; Koya D.; Haneda, M.; Ugi, S. et al. Lamp-2 deficiency prevents high-fat diet-induced obese diabetes via enhancing energy expenditure. Biochem. Biophys. Res. Commun. 2015;465:249–255. [CrossRef]
- Sekar, M.; Thirumurugan, K. Autophagic Regulation of Adipogenesis Through TP53INP2: Insights from In Silico and In Vitro Analysis. Mol Biotechnol. 2024 May;66(5):1188-1205. Epub 2024 Jan 18. [CrossRef] [PubMed]
- Wang, Y.; Chen, G.; Xu, M.; Cui, Y.; He, W.; Zeng ,H.; Zeng, T.; Cheng, R.; Li, X. Caspase-1 Deficiency Modulates Adipogenesis through Atg7-Mediated Autophagy: An Inflammatory-Independent Mechanism. Biomolecules. 2024 Apr 20;14(4):501. PMCID: PMC11048440. [CrossRef] [PubMed]
- García-Barrado, M.J., Iglesias-Osma, M.C.; Pérez-García, E.; Carrero, S.; Blanco, E.J.; Carretero-Hernández, M.; Carretero, J. Role of Flavonoids in The Interactions among Obesity, Inflammation, and Autophagy. Pharmaceuticals (Basel). 2020 Oct 26;13(11):342. PMCID: PMC7692407. [CrossRef] [PubMed]
- Ambele, M.A.; Dhanraj, P.; Giles,R.; Pepper, M.S. Adipogenesis: A Complex Interplay of Multiple Molecular Determinants and Pathways. Int J Mol Sci. 2020 Jun 16;21(12):4283. PMCID: PMC7349855. [CrossRef] [PubMed]
- Cai, J.; Pires, K.M.; Ferhat, M.; Chaurasia, B.; Buffolo, M.A.; Smalling, R. et al. Autophagy Ablation in Adipocytes Induces Insulin Resistance and Reveals Roles for Lipid Peroxide and Nrf2 Signaling in Adipose-Liver Crosstalk. Cell Rep. 2018;25(7):1708-1717.e5. [CrossRef]
- Kosacka, J.; Kern, M.; Klöting, N.; Paeschke, S; Rudich, A.; Haim, Y. et al. Autophagy in adipose tissue of patients with obesity and type 2 diabetes. Mol Cell Endocrinol. 2015;409:21-32. [CrossRef]
- Zhang, C.; He, Y.; Okutsu, M.; Ong, L.C.; Jin, Y. Zheng, L. et al. Autophagy is involved in adipogenic differentiation by repressesing proteasome-dependent PPARγ2 degradation. Am J Physiol Endocrinol Metab. 2013;305(4). [CrossRef]
- Ma, X.; Wang, D.; Zhao, W.; Xu, L. Deciphering the Roles of PPARγ in Adipocytes via Dynamic Change of Transcription Complex. Front Endocrinol (Lausanne). 2018 Aug 21;9:473. PMCID: PMC6110914. [CrossRef] [PubMed]
- Vegiopoulos A, Rohm M, Herzig S. Adipose tissue: between the extremes. EMBO J. 2017. 36:1999–2017.
- Wang, S.; Lin, Y.; Gao, L.; Yang, Z.; Lin, J.; Ren, S.; Li, F.; Chen, J.; Wang, Z.; Dong, Z.; Sun, P.; Wu, B. PPAR-γ integrates obesity and adipocyte clock through epigenetic regulation of Bmal1. Theranostics. 2022 Jan 16;12(4):1589-1606. PMCID: PMC8825584. [CrossRef] [PubMed]
- Fujimori, Ko. Prostaglandin D2 and F2α as Regulators of Adipogenesis and Obesity. Biol and Pharmacol. 2022;45:8.
- Armani A, Berry A, Cirulli F, Caprio M. Molecular mechanisms underlying metabolic syndrome: the expanding role of the adipocyte. FASEB J. (2017) 31:4240–55.
- Ferhat, M.; Funai, K.; Boudina, S. Autophagy in Adipose Tissue Physiology and Pathophysiology. Antioxid Redox Signal. 2019;31(6):487-501. [CrossRef]
- Pietrocola, F.; Bravo-San Pedro, J.M. Targeting Autophagy to Counteract Obesity-Associated Oxidative Stress. Antioxidants (Basel). 2021 Jan 12;10(1):102. [CrossRef] [PubMed]
- Ichimiya, T.; Yamakawa, T.; Hirano, T.; Yokoyama, Y.; Hayashi, Y.; Hirayama, D.; Wagatsuma, K.; Itoi, T.; Nakase, H. Autophagy and Autophagy-Related Diseases: A Review. Int J Mol Sci. 2020 Nov 26;21(23):8974. [CrossRef] [PubMed]
- Kovsan, J.; Blüher, M.; Tarnovscki, T.; Klöting, N.; Kirshtein, B.; Madar, L. et al. Altered autophagy in human adipose tissues in obesity. J Clin Endocrinol Metab. 2011;96(2). [CrossRef]
- Xu Q, Mariman ECM, Roumans NJT, Vink RG, Goossens GH, Blaak EE, et al. Adipose tissue autophagy related gene expression is associated with glucometabolic status in human obesity. Adipocyte. 2018;7(1):12-19. [CrossRef]
- Chipurupalli, S.; Samavedam, U.; Robinson, N. Crosstalk Between ER Stress, Autophagy and Inflammation. Front Med (Lausanne). 2021 Nov 5;8:758311. PMCID: PMC8602556. [CrossRef] [PubMed]
- Singh, R.; Cuervo, A.M. Lipophagy: connecting autophagy and lipid metabolism. Int J Cell Biol. 2012;2012:282041. [CrossRef]
- Tokarz, V. L.; MacDonald, P. E.; Klip, A. The cell biology of systemic insulin function. J. Cell Biol. 2018:217, 1–17. [CrossRef]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol. 2012;4(9). [CrossRef]
- Kang, Y.H.; Cho, M.H.; Kim, J.Y.; Kwon, M.S.; Peak, J.J.; Kang, S.W. et al. Impaired macrophage autophagy induces systemic insulin resistance in obesity. Oncotarget. 2016;7(24):35577-35591. [CrossRef]
- Frendo-Cumbo, S.; Tokarz, V.L.; Bilan, P.J.; Brumell, J.H.; Klip, A. Communication Between Autophagy and Insulin Action: At the Crux of Insulin Action-Insulin Resistance? Front Cell Dev Biol. 2021;9. [CrossRef]
- Maixner, N.; Bechor, S.; Vershinin, Z.; Pecht, T.; Goldstein, N.; Haim, Y. et al. Transcriptional Dysregulation of Adipose Tissue Autophagy in Obesity. Physiology (Bethesda). 2016;31(4):270-282. [CrossRef]
- Maixner, N.; Pecht, T.; Haim, Y.; Chalifa-Caspi, V.; Goldstein, N.; Tarnovscki ,T. et al. A TRAIL-TL1A Paracrine Network Involving Adipocytes, Macrophages, and Lymphocytes Induces Adipose Tissue Dysfunction Downstream of E2F1 in Human Obesity. Diabetes. 2020;69(11):2310-2323. [CrossRef]
- Haim, Y.; Blüuher, M.; Slutsky, N.; Goldstein, N.; Klöting, N.; Harman-Boehm, I. et al. Elevated autophagy gene expression in adipose tissue of obese humans: A potential non-cell-cycle-dependent function of E2F1. Autophagy. 2015;11(11):2074-2088. [CrossRef]
- Böni-Schnetzler, M.; Häuselmann, S.P.; Dalmas, E.; Meier, D.T.; Thienel, C.; Traub, S. et al. β Cell-Specific Deletion of the IL-1 Receptor Antagonist Impairs β Cell Proliferation and Insulin Secretion. Cell Rep. 2018;22(7):1774-1786. [CrossRef]
- Ward, C.; Martinez-Lopez, N.; Otten, E.G.; Carroll, B.; Maetzel, D.; Singh, R. et al. Autophagy, lipophagy and lysosomal lipid storage disorders. Biochim Biophys Acta. 2016;1861(4):269-284. [CrossRef]
- Qian,Q.; Zhang, Z.; Orwig, A.; Chen, S.; Ding, W.X.; Xu, Y. et al. S-Nitrosoglutathione Reductase Dysfunction Contributes to Obesity-Associated Hepatic Insulin Resistance via Regulating Autophagy. Diabetes. 2018;67(2):193-207. [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M. et al. Autophagy regulates lipid metabolism. Nature. 2009;458(7242):1131-1135. [CrossRef]
- Dann, S.G.; Selvaraj, A.; Thomas, G. mTOR Complex1-S6K1 signaling: at the crossroads of obesity, diabetes and cancer. Trends Mol Med. 2007;13(6):252-259. [CrossRef]
- Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11(6):467-478. [CrossRef]
- Yoshizaki,T.; Kusunoki, C.; Kondo, M.; Yasuda, M.; Kume, S.; Morino, K. et al. Autophagy regulates inflammation in adipocytes. Biochem Biophys Res Commun. 2012;417(1):352-357. [CrossRef]
- Ezaki, J.; Matsumoto, N.; Takeda-Ezaki, M.; Komatsu, M.; Takahashi, K.; Hiraoka, Y. et al. Liver autophagy contributes to the maintenance of blood glucose and amino acid levels. Autophagy. 2011;7(7):727-736. [CrossRef]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffner, T.; Scapozza, L. et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8(10):1124-1132. [CrossRef]
- Kim, K.H.; Jeong, Y.T.; Oh, H.; Kim,S.H.; Cho, J.M.; Kim, Y.N. et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med. 2013;19(1):83-92. [CrossRef]
- Liu, H.Y.; Han, J.; Cao, S.Y.; Hong, T.; Zhuo, D.; Shi, J. et al. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J Biol Chem. 2009;284(45):31484-31492. [CrossRef]
- Martins, R.; Lithgow, G.J.; Link, W. Long live FOXO: unraveling the role of FOXO proteins in aging and longevity. Aging Cell. 2016;15(2):196-207. [CrossRef]
- Fernández ÁF, Bárcena C, Martínez-García GG, Tamargo-Gómez I, Suárez MF, Pietrocola F, et al. Autophagy couteracts weight gain, lipotoxicity and pancreatic β-cell death upon hypercaloric pro-diabetic regimens. Cell Death Dis. 2017;8(8). [CrossRef]
- Park HW, Park H, Semple IA, Jang I, Ro SH, Kim M, et al. Pharmacological correction of obesity-induced autophagy arrest using calcium channel blockers. Nat Commun. 2014;5. [CrossRef]
- González-Rodríguez A, Mayoral R, Agra N, Valdecantos MP, Pardo V, Miquilena-Colina ME, et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014;5(4):e1179. [CrossRef]
- Meng Q, Cai D. Defective hypothalamic autophagy directs the central pathogenesis of obesity via the IkappaB kinase beta (IKKbeta)/NF-kappaB pathway. J Biol Chem. 2011;286(37):32324-32332. [CrossRef]
- Yamahara K, Kume S, Koya D, Tanaka Y, Morita Y, Chin-Kanasaki M, et al. Obesity-mediated autophagy insufficiency exacerbates proteinuria-induced tubulointerstitial lesions. J Am Soc Nephrol. 2013;24(11):1769-1781. [CrossRef]
- Öst A, Svensson K, Ruishalme I, Brännmark C, Franck N, Krook H, et al. Attenuated mTOR signaling and enhanced autophagy in adipocytes from obese patients with type 2 diabetes. Mol Med. 2010;16(7-8):235-246.
- Bagherniya M, Butler AE, Barreto GE, Sahebkar A. The effect of fasting or calorie restriction on autophagy induction: A review of the literature. Ageing Res Rev. 2018;47:183-197. [CrossRef]
- Mei S, Ni HM, Manley S, Bockus A, Kassel KM, Luyendyk JP, et al. Differential roles of unsaturated and saturated fatty acids on autophagy and apoptosis in hepatocytes. J Pharmacol Exp Ther. 2011;339(2):487-498. [CrossRef]
- Li S, Dou X, Ning H, Song Q, Wei W, Zhang X, et al. Sirtuin 3 acts as a negative regulator of autophagy dictating hepatocyte susceptibility to lipotoxicity. Hepatology. 2017;66(3):936-952. [CrossRef]
- de Luxán-Delgado B, Potes Y, Rubio-González A, Caballero B, Solano JJ, Fernández-Fernández M, et al. Melatonin reduces endoplasmic reticulum stress and autophagy in liver of leptin-deficient mice. J Pineal Res. 2016;61(1):108-123. [CrossRef]
- Clemente-Postigo, M.; Tinahones ,A.; El Bekay, R.; Malagón, M.M,; Tinahones, F.J. The Role of Autophagy in White Adipose Tissue Function: Implications for Metabolic Health. Metabolites. 2020 Apr 30;10(5):179. [CrossRef] [PubMed]
- Q, Mariman ECM, Roumans NJT, Vink RG, Goossens GH, Blaak EE, Jocken JWE. Adipose tissue autophagy related gene expression is associated with glucometabolic status in human obesity. Adipocyte. 2018 Jan 2;7(1):12-19. Epub 2018 Feb 5. PMCID: PMC5915036. [CrossRef] [PubMed]
- Nuñez CE, Rodrigues VS, Gomes FS, Moura RF, Victorio SC, Bombassaro B, Chaim EA, Pareja JC, Geloneze B, Velloso LA, Araujo EP. Defective regulation of adipose tissue autophagy in obesity. Int J Obes (Lond). 2013 Nov;37(11):1473-80. Epub 2013 Mar 12. [CrossRef] [PubMed]
- Xu Q, Mariman ECM, Roumans NJT, Vink RG, Goossens GH, Blaak EE, Jocken JWE. Adipose tissue autophagy related gene expression is associated with glucometabolic status in human obesity. Adipocyte. 2018 Jan 2;7(1):12-19. Epub 2018 Feb 5. [CrossRef] [PubMed]
- Chung KW, Chung HY. The Effects of Calorie Restriction on Autophagy: Role on Aging Intervention. Nutrients. 2019 Dec 2;11(12):2923. PMCID: PMC6950580. [CrossRef] [PubMed]
- International Dietetics and Nutrition Terminology (IDNT) reference manual : standardized language for the nutrition care proces. 2011.
- He, C. et al. Exercise- induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012; 481, 511–515.
- Alrushud, A. S., Rushton, A. B., Kanavaki, A. M. & Greig, C. A. Effect of physical activity and dietary restriction interventions on weight loss and the musculoskeletal function of overweight and obese older adults with knee osteoarthritis: a systematic review and mixed method data synthesis. BMJ Open 2017; 7, e014537.
- Halling, J. F. & Pilegaard, H. Autophagy- dependent beneficial effects of exercise. Cold Spring Harb. Perspect. Med. 2017; 7, a029777.
- Shabkhizan R, Haiaty S, Moslehian MS, Bazmani A, Sadeghsoltani F, Saghaei Bagheri H, Rahbarghazi R, Sakhinia E. The Beneficial and Adverse Effects of Autophagic Response to Caloric Restriction and Fasting. Adv Nutr. 2023 Sep;14(5):1211-1225. Epub 2023 Jul 30. PMCID: PMC10509423. [CrossRef] [PubMed]
- Sun L., Xiong H., Chen L., Dai X., Yan X., Wu Y., et al. Deacetylation of ATG4B promotes autophagy initiation under starvation. Sci. Adv. 2022;8(31):eabo0412. [CrossRef]
- Chaudhary R., Liu B., Bensalem J., Sargeant T.J., Page A.J., Wittert G.A., et al. Intermittent fasting activates markers of autophagy in mouse liver, but not muscle from mouse or humans. Nutrition. 2022;101:111662. [CrossRef]
- Noda N.N., Fujioka Y. Atg1 family kinases in autophagy initiation. Cell. Mol. Life Sci. 2015;72(16):3083–3096. [CrossRef]
- Bagherniya M, Butler AE, Barreto GE, Sahebkar A. The effect of fasting or calorie restriction on autophagy induction: A review of the literature. Ageing Res Rev. 2018 Nov;47:183-197. Epub 2018 Aug 30. [CrossRef] [PubMed]
- Kocot AM, Wróblewska B. Nutritional strategies for autophagy activation and health consequences of autophagy impairment. Nutrition. 2022 Nov-Dec;103-104:111686. Epub 2022 Apr 11. [CrossRef] [PubMed]
- Yang L, Licastro D, Cava E, Veronese N, Spelta F, Rizza W, Bertozzi B, Villareal DT, Hotamisligil GS, Holloszy JO, Fontana L. Long-Term Calorie Restriction Enhances Cellular Quality-Control Processes in Human Skeletal Muscle. Cell Rep. 2016 Jan 26;14(3):422-428. 4; Epub 2016 Jun 7. [CrossRef] [PubMed]
- Chaudhary R, Liu B, Bensalem J, Sargeant TJ, Page AJ, Wittert GA, Hutchison AT, Heilbronn LK. Intermittent fasting activates markers of autophagy in mouse liver, but not muscle from mouse or humans. Nutrition. 2022 Sep;101:111662. Epub 2022 Mar 26. [CrossRef] [PubMed]
- Zhang Y, Sowers JR, Ren J. Targeting autophagy in obesity: from pathophysiology to management. Nat Rev Endocrinol. 2018 Jun;14(6):356-376. [CrossRef] [PubMed]
- Song DK, Kim YW. Beneficial effects of intermittent fasting: a narrative review. Journal of Yeungnam Medical Science. 2023;40(1):4–11.
- Kim KH, Lee MS. Autophagy--a key player in cellular and body metabolism. Nat Rev Endocrinol. 2014 Jun;10(6):322-37. Epub 2014 Mar 25. [CrossRef] [PubMed]
- Kitada M, Kume S, Takeda-Watanabe A, Tsuda S, Kanasaki K, Koya D. Calorie restriction in overweight males ameliorates obesity-related metabolic alterations and cellular adaptations through anti-aging effects, possibly including AMPK and SIRT1 activation. Biochim Biophys Acta. 2013 Oct;1830(10):4820-7.
- Hanningan, A.; Gorski,S. Macroautophagy. Autophagy.2009, 5; 140-151.
- Mariani S, di Giorgio MR, Martini P, Persichetti A, Barbaro G, Basciani S, Contini S, Poggiogalle E, Sarnicola A, Genco A, Lubrano C, Rosano A, Donini LM, Lenzi A, Gnessi L. Inverse Association of Circulating SIRT1 and Adiposity: A Study on Underweight, Normal Weight, and Obese Patients. Front Endocrinol (Lausanne). 2018 Aug 7;9:449. [CrossRef] [PubMed]
- Baeken MW. Sirtuins and their influence on autophagy. J Cell Biochem. 2023 Feb 6. Epub ahead of print. [CrossRef] [PubMed]
- Liu C, Ji L, Hu J, Zhao Y, Johnston LJ, Zhang X, Ma X. Functional Amino Acids and Autophagy: Diverse Signal Transduction and Application. Int J Mol Sci. 2021 Oct 22;22(21):11427. PMCID: PMC8592284. [CrossRef] [PubMed]
- Saini, S.K.; Singh, A.; Saini, M.; Gonzalez-Freire, M.; Leeuwenburgh, C.; Anton, S.D. Time-Restricted Eating Regimen Differentially Affects Circulatory miRNA Expression in Older Overweight Adults. Nutrients 2022, 14, 1843. [CrossRef]
- Zachari M, Ganley IG. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017 Dec 12;61(6):585-596. [CrossRef] [PubMed]
- Ortega MA, Fraile-Martinez O, de Leon-Oliva D, Boaru DL, Lopez-Gonzalez L, García-Montero C, Alvarez-Mon MA, Guijarro LG, Torres-Carranza D, Saez MA, Diaz-Pedrero R, Albillos A, Alvarez-Mon M. Autophagy in Its (Proper) Context: Molecular Basis, Biological Relevance, Pharmacological Modulation, and Lifestyle Medicine. Int J Biol Sci. 2024 Apr 22;20(7):2532-2554. [CrossRef] [PubMed]
- Sbai O, Torrisi F, Fabrizio FP, Rabbeni G, Perrone L. Effect of the Mediterranean Diet (MeDi) on the Progression of Retinal Disease: A Narrative Review. Nutrients. 2024; 16(18):3169. [CrossRef]
- García-Montero C, Fraile-Martínez O, Gómez-Lahoz AM, Pekarek L, Castellanos AJ, Noguerales-Fraguas F, et al. Nutritional components in western diet versus mediterranean diet at the gut microbiota-immune system interplay. implications for health and disease. Nutrients. 2021;13(2):1–53.
- Franco GA, Interdonato L, Cordaro M, Cuzzocrea S, Di Paola R. Bioactive Compounds of the Mediterranean Diet as Nutritional Support to Fight Neurodegenerative Disease. Int J Mol Sci. 2023 Apr 15;24(8):7318. [CrossRef] [PubMed]
- Di Lorenzo C, Colombo F, Biella S, Stockley C, Restani P. Polyphenols and Human Health: The Role of Bioavailability. Nutrients. 2021 Jan 19;13(1):273. [CrossRef] [PubMed]
- Pandey KB, Rizvi SI. Plant polyphenols as dietary antioxidants in human health and disease. Oxid Med Cell Longev. 2009 Nov-Dec;2(5):270-8. [CrossRef] [PubMed]
- Sokal, A; Stocerz K, ; Olczyk, P.; Kadela-Tomanek, M. Therapeutic potential of flavonoids used in traditional Chinese medicine – a comparative study of galangin, kaempferol, chrysin and fisetin. AAMS. 2024,78.
- Domonese N, Di Bella G, Cusumano C, Parisi A, Tagliaferri F, Ciriminna S, Barbagallo M. Mediterranean diet in the management and prevention of obesity. Exp Gerontol. 2023 Apr;174:112121. Epub 2023 Feb 17. [CrossRef] [PubMed]
- Brimson JM, Prasanth MI, Malar DS, Thitilertdecha P, Kabra A, Tencomnao T, Prasansuklab A. Plant Polyphenols for Aging Health: Implication from Their Autophagy Modulating Properties in Age-Associated Diseases. Pharmaceuticals (Basel). 2021 Sep 27;14(10):982. [PubMed]
- Osorio-Conles Ó, Olbeyra R, Moizé V, Ibarzabal A, Giró O, Viaplana J, Jiménez A, Vidal J, de Hollanda A. Positive Effects of a Mediterranean Diet Supplemented with Almonds on Female Adipose Tissue Biology in Severe Obesity. Nutrients. 2022 Jun 24;14(13):2617. [PubMed]
- Wang, Z.; Quan, W., Zeng, M., Wang, Z., Chen, Q., Chen, J., Christian, M.; He, Z. . Regulation of autophagy by plant-based polyphenols: A critical review of current advances in glucolipid metabolic diseases and food industry applications. Food Frontiers.2023 4, 1039–1067. [CrossRef]
- Most J, Warnke I, Boekschoten MV, Jocken JWE, de Groot P, Friedel A, Bendik I, Goossens GH, Blaak EE. The effects of polyphenol supplementation on adipose tissue morphology and gene expression in overweight and obese humans. Adipocyte. 2018;7(3):190-196. Epub 2018 May 22. [PubMed]
- Timmers S., Konings E., Bilet L., Houtkooper R.H., van de Weijer T., Goossens G.H., Hoeks J., van der Krieken S., Ryu D., Kersten S., et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 2011;14:612–622. [CrossRef]
- Konings E, Timmers S, Boekschoten MV, Goossens GH, Jocken JW, Afman LA, Müller M, Schrauwen P, Mariman EC, Blaak EE. The effects of 30 days resveratrol supplementation on adipose tissue morphology and gene expression patterns in obese men. Int J Obes (Lond). 2014 Mar;38(3):470-3. Epub 2013 Aug 20. [PubMed]
- ettembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin Set al.TFEB links autophagy to lysosomal biogenesis.Science 2011;332: 1429–1433.
- Moskot M, Montefusco S, Jakóbkiewicz-Banecka J, Mozolewski P, Węgrzyn A, Di Bernardo D, Węgrzyn G, Medina DL, Ballabio A, Gabig-Cimińska M. The phytoestrogen genistein modulates lysosomal metabolism and transcription factor EB (TFEB) activation. J Biol Chem. 2014 Jun 13;289(24):17054-69. Epub 2014 Apr 25. [CrossRef] [PubMed]
- Widlund, A. L.; Baur, J.; A.; Vang O. mTOR: more targets of resveratrol? Expert Rev Mol Med 2013;15, e10.
- Zhang J, Chiu JF, Zhang HW, et al. Autophagic cell death induced by resveratrol depends on the Ca2+/AMPK/mTOR pathway in A549 cells. Biochem Pharmacol. 2013;86(2):317–328. [CrossRef]
- Tian Y, Song W, Li D, Cai L, Zhao Y. Resveratrol As A Natural Regulator Of Autophagy For Prevention And Treatment Of Cancer. Onco Targets Ther. 2019 Oct 17;12:8601-8609. [CrossRef] [PubMed]
- Park D, Jeong H, Lee MN, Koh A, Kwon O, Yang YR, Noh J, Suh PG, Park H, Ryu SH. Resveratrol induces autophagy by directly inhibiting mTOR through ATP competition. Sci Rep. 2016 Feb 23;6:21772. [CrossRef] [PubMed]
- Mansur AP, Roggerio A, Goes MFS, Avakian SD, Leal DP, Maranhão RC, Strunz CMC. Serum concentrations and gene expression of sirtuin 1 in healthy and slightly overweight subjects after caloric restriction or resveratrol supplementation: A randomized trial. Int J Cardiol. 2017 Jan 15;227:788-794. Epub 2016 Oct 27. [CrossRef] [PubMed]
- Phillips, C.M.; Kesse-Guyot, E.; McManus, R.; Hercberg, S.; Lairon, D.; Planells, R.; Roche, H.M. High dietary saturated fat intake accentuates obesity risk associated with the fat mass and obesity-associated gene in adults. J. Nutr. 2012, 142, 824–831.
- Liu W, Zhu M, Gong M, Zheng W, Zeng X, Zheng Q, Li X, Fu F, Chen Y, Cheng J, Rao Z, Lu Y, Chen Y. Comparison of the Effects of Monounsaturated Fatty Acids and Polyunsaturated Fatty Acids on Liver Lipid Disorders in Obese Mice. Nutrients. 2023 Jul 19;15(14):3200. PMCID: PMC10386220. [CrossRef] [PubMed]
- Wang,J.; Nie, D.Modulation autophagy by Fatty acids. In: Cell Death - Autophagy, Apoptosis and Necrosis. IntechOpen 2015.
- O'Rourke EJ, Kuballa P, Xavier R, Ruvkun G. ω-6 Polyunsaturated fatty acids extend life span through the activation of autophagy. Genes Dev. 2013 Feb 15;27(4):429-40. Epub 2013 Feb 7. [CrossRef] [PubMed]
- Ciesielska K, Gajewska M. Fatty Acids as Potent Modulators of Autophagy Activity in White Adipose Tissue. Biomolecules. 2023 Jan 30;13(2):255. [CrossRef] [PubMed]
- Yang B, Zhou Y, Wu M, Li X, Mai K, Ai Q. ω-6 Polyunsaturated fatty acids (linoleic acid) activate both autophagy and antioxidation in a synergistic feedback loop via TOR-dependent and TOR-independent signaling pathways. Cell Death Dis. 2020 Jul 30;11(7):607. [CrossRef] [PubMed]
- Camargo A, Rangel-Zúñiga OA, Alcalá-Díaz J, Gomez-Delgado F, Delgado-Lista J, García-Carpintero S, Marín C, Almadén Y, Yubero-Serrano EM, López-Moreno J, Tinahones FJ, Pérez-Martínez P, Roche HM, López-Miranda J. Dietary fat may modulate adipose tissue homeostasis through the processes of autophagy and apoptosis. Eur J Nutr. 2017 Jun;56(4):1621-1628. Epub 2016 Mar 30. [CrossRef] [PubMed]
- Viguerie N, Vidal H, Arner P, Holst C, Verdich C, Avizou S, Astrup A, Saris WH, Macdonald IA, Klimcakova E, Clément K, Martinez A, Hoffstedt J, Sørensen TI, Langin D; Nutrient-Gene Interactions in Human Obesity--Implications for Dietary Guideline (NUGENOB) project. Adipose tissue gene expression in obese subjects during low-fat and high-fat hypocaloric diets. Diabetologia. 2005 Jan;48(1):123-31. Epub 2004 Dec 29. [CrossRef] [PubMed]
- Capel F, Viguerie N, Vega N, Dejean S, Arner P, Klimcakova E, Martinez JA, Saris WH, Holst C, Taylor M, Oppert JM, Sørensen TI, Clément K, Vidal H, Langin D. Contribution of energy restriction and macronutrient composition to changes in adipose tissue gene expression during dietary weight-loss programs in obese women. J Clin Endocrinol Metab. 2008 Nov;93(11):4315-22. Epub 2008 Sep 9. [CrossRef] [PubMed]
- Hanjani NA, Vafa M. Protein Restriction, Epigenetic Diet, Intermittent Fasting as New Approaches for Preventing Age-associated Diseases. Int J Prev Med. 2018 Jun 29;9:58. [CrossRef] [PubMed]
- Xu C, Markova M, Seebeck N, Loft A, Hornemann S, Gantert T, Kabisch S, Herz K, Loske J, Ost M, Coleman V, Klauschen F, Rosenthal A, Lange V, Machann J, Klaus S, Grune T, Herzig S, Pivovarova-Ramich O, Pfeiffer AFH. High-protein diet more effectively reduces hepatic fat than low-protein diet despite lower autophagy and FGF21 levels. Liver Int. 2020 Dec;40(12):2982-2997. Epub 2020 Jul 21. [CrossRef] [PubMed]
- Soussi H, Reggio S, Alili R, Prado C, Mutel S, Pini M, Rouault C, Clément K, Dugail I. DAPK2 Downregulation Associates With Attenuated Adipocyte Autophagic Clearance in Human Obesity. Diabetes. 2015 Oct;64(10):3452-63. Epub 2015 Jun 2. [CrossRef] [PubMed]
Figure 1.
Mechanism regulating autophagy.
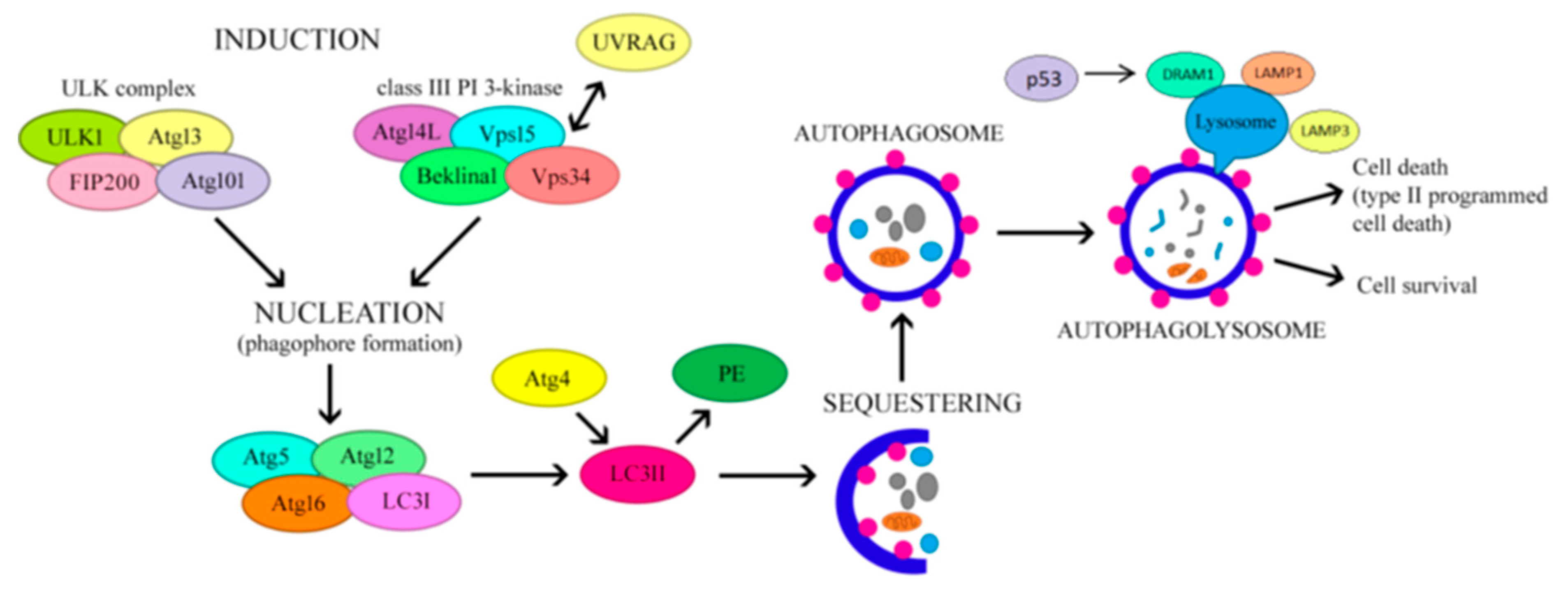
Figure 2.
Regulation of autophagy in lean (normal) and obese (pathologic) subjects. In the former case, induction of mTORC1 by insulin results in inhibition of autophagy. Inhibition of autophagy induces a “browning” phenotype in adipocytes. During obesity, ER stress, hypoxia and inflammation stimulate insulin resistance, resulting in inhibition of mTORC1, followed by induction of autophagy. Autophagy improves adipocyte function by eliminating damaged organelles and misfolded proteins and preventing pro-inflammatory responses. In addition, excessive stimulation of autophagy can increase energy storage by adipocytes and promote cell death.
Figure 2.
Regulation of autophagy in lean (normal) and obese (pathologic) subjects. In the former case, induction of mTORC1 by insulin results in inhibition of autophagy. Inhibition of autophagy induces a “browning” phenotype in adipocytes. During obesity, ER stress, hypoxia and inflammation stimulate insulin resistance, resulting in inhibition of mTORC1, followed by induction of autophagy. Autophagy improves adipocyte function by eliminating damaged organelles and misfolded proteins and preventing pro-inflammatory responses. In addition, excessive stimulation of autophagy can increase energy storage by adipocytes and promote cell death.
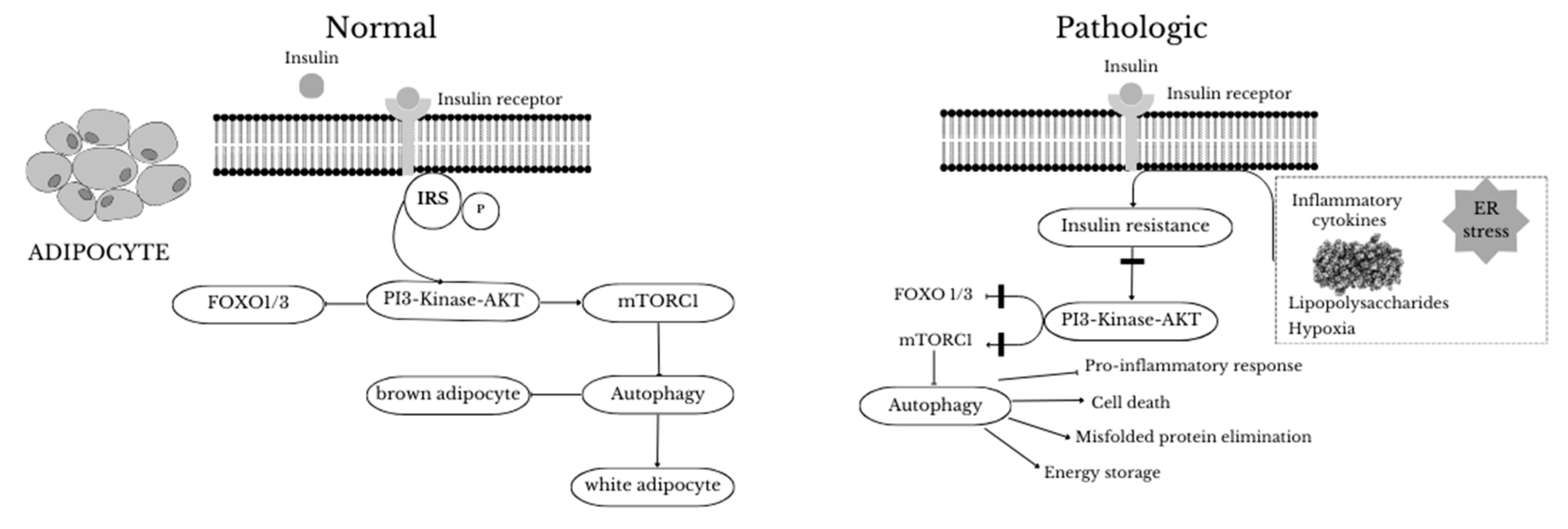
Figure 3.
In the physiological (normal) state, after insulin binds to its receptor, Akt is activated and leads to inhibition of glycogen synthase kinase 3 (GSK3) and forkhead box O (FOXO) 1 in the liver. Inhibition of GSK3 leads to increased glycogen synthase activity and therefore increased glycogen synthesis. When FOXO1 is inhibited, the expression of glucose-6 phosphatase and phosphoenolpyruvate carboxykinase genes decreases, resulting in decreased hepatic glucose production. In addition, insulin inhibits autophagy by activating Akt and inhibiting FOXO1. Akt increases lipid synthesis in hepatocytes through activation of SREBP1c. The increase in lipid content by SREBP1c results in impaired lipophagy and hepatic steatosis. In the insulin-resistant (pathologic) state, GSK3 is activated and inhibits glycogen synthase (GS). FOXO1 activity increases, resulting in activation of the gluconeogenesis pathway, lipid and very low-density lipoprotein (VLDL) synthesis and increased autophagy. SREBP1c increases its activity through ER stress or through IRS-1.
Figure 3.
In the physiological (normal) state, after insulin binds to its receptor, Akt is activated and leads to inhibition of glycogen synthase kinase 3 (GSK3) and forkhead box O (FOXO) 1 in the liver. Inhibition of GSK3 leads to increased glycogen synthase activity and therefore increased glycogen synthesis. When FOXO1 is inhibited, the expression of glucose-6 phosphatase and phosphoenolpyruvate carboxykinase genes decreases, resulting in decreased hepatic glucose production. In addition, insulin inhibits autophagy by activating Akt and inhibiting FOXO1. Akt increases lipid synthesis in hepatocytes through activation of SREBP1c. The increase in lipid content by SREBP1c results in impaired lipophagy and hepatic steatosis. In the insulin-resistant (pathologic) state, GSK3 is activated and inhibits glycogen synthase (GS). FOXO1 activity increases, resulting in activation of the gluconeogenesis pathway, lipid and very low-density lipoprotein (VLDL) synthesis and increased autophagy. SREBP1c increases its activity through ER stress or through IRS-1.
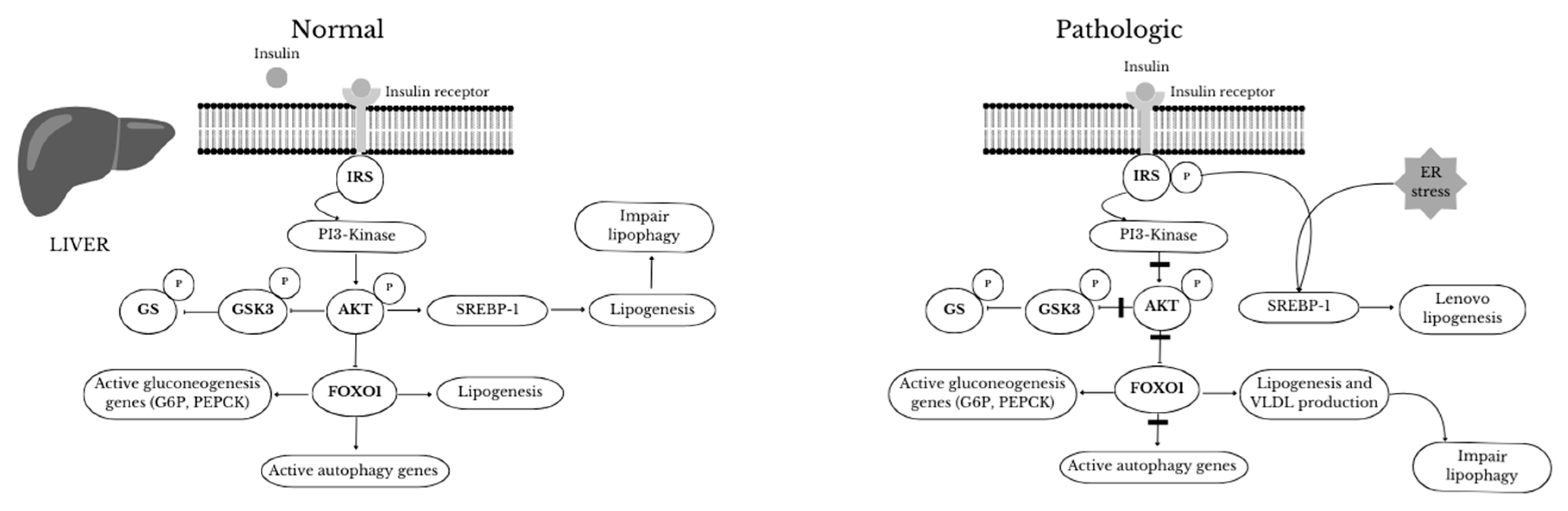

Table 1.
Modulation of autophagy by dietary strategy in overweight/obese humans.
| Dietary strategies | Model of Obesity | Parametr studies | Effect on Autophagy | Ref. |
|---|---|---|---|---|
| Calorie restriction or Intermittent fasting |
Sceletal muscle of body fat-matched endurance athletes ; sceletal muscle of obese women; Obese human (subcutaneous, white adipose tissue |
Decreased mTOR signaling through reducing insulin and IGF-1 levels and increased the AMP/ATP ratio, which leads to the activation of AMPK as well as several other products involved in the stimulation of this process (ATG 5, ATG6, ATG7, ATG8, LC3-II, Beclin1, p62, SIRT1, LAMP2, ULK1 and ATG101) | Enhanced | [96,97,98,99,100] |
| Calorie restriction 25% for 7 weeks | Peripheral blood mononuclear cells (PBMNCs) of overweight male | Activated of AMPK and SIRT1 | Enhanced | [106] |
| Mediterranean Diet vs Mediterranean Diet with almonds |
Obese human (subcutaneous, white adipose tissue |
Increased expression of autophagy-related ATG 7 and ATG12 in VAT from the MDSA group, while ATG5 show non-significant trend (p=0.054) | Enhanced | [117] |
| Dietary polyphenols | Obese human (subcutaneous, white adipose tissue |
Activated cAMP, AMPK, MAPK, increased AKT, SIRT1, PI3K, Nrf2/HO-1, PINK1/Parkin, PPARδ genes expression | Enhanced | [118] |
| Epigallocatechin-3-gallate + resveratrol (280 mg +80 mg/d) vs placebo – 12 weeks | Obese human (subcutaneous, white adipose tissue |
Activated genes expression of ATP6V1A, ATP6V1H, CD68, HSL/LIPE, LAMP2, PI4K2A, UCP2, GAPDH | Enhanced | [119] |
| Resveratrol 150 mg once daily for 30 days | Obese men (sceletal muscle) | Activated AMPK, increased SIRT1 and PGC-1α protein levels | Enhanced | [120] |
| Resveratrol 150 mg once daily for 30 days | Obese men (sceletal muscle) | Activated TFEB (transcriptional factor EB) expression Inhibited mTOR activity |
Enhanced | [121] |
| Resveratrol (500 mg/d) vs Calorie restriction (1000 kcal/d) | Overweight human (blood) | Resveratrol and caloric restriction increased significantly serum concentrations of SIRT1 proteins | Enhanced | [128] |
| Four diets for 12 weeks: a high-saturated fatty acid diet (HSFA), a high-monounsaturated fatty acid diet (HMUFA), and two low-fat, high-complex carbohydrate diets supplemented with long-chain n-3 polyunsaturated fatty acids (LFHCC n-3) or placebo (LFHCC). | Obese human (subcutaneous) white adipose tissue |
Significantly increased expression of autophagy-related BECN1 and ATG7 genes after the HMUFA diet; increased the expression of the apoptosis-related CASP3 gene after the LFHCC and LFHCC n-3 diets. Expression of other autophagy markers, LC3, LAMP2, and ULK1, tended to increase after the consumption of the LFHCC n-3 diet. | Enhanced | [135] |
| Low-fat, high-carbohydrate diet (LF) vs moderate-fat, low-carbohydrate diet (MF) for 10 weeks | Obese human (subcutaneous) white adipose tissue |
Expression FABP4, SIRT3, NR3C1, GABARAPL2, and FNTA genes was 15–65% higher in the MF than the LF. | Enhanced in MF diet vs LF | [136,137] |
| Hypocaloric diet (1500-1600 kcal/day) and low protein (10%) vs Hypocaloric (1500-1600 kcal/day) and high protein (30E%) for 3 weeks prior bariatric surgery | Liver sample collected during surgery | Significantly elevated autophagy flux and FGF21 levels in liver in patients in LP diet versus HP | Enhanced in LP diet vs HP | [138] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



