Submitted:
26 September 2024
Posted:
27 September 2024
You are already at the latest version
Abstract
Inherited heart diseases (IHDs) are caused by genetic mutations that disrupt the physiological structure and function of the heart. Understanding the mechanisms behind these diseases is crucial for developing personalised interventions in cardiovascular medicine. Development of induced pluripotent stem cells, which can then be differentiated to any nucleated adult cell type, has enabled the creation of personalised single-cell and multicellular models, providing unprecedented insights into the pathophysiology of IHDs. This review provides a comprehensive overview of recent advancements in human iPSC models used to dissect the molecular and genetic underpinnings of common IHDs. We examine multicellular models and tissue engineering approaches, such as cardiac organoids, engineered heart tissue, and multicellular co-culture systems, which simulate complex intercellular interactions within heart tissue. Recent advancements in stem cell models offer a more physiologically relevant platform to study disease mechanisms, enabling researchers to observe cellular interactions, study disease progression, and identify therapeutic strategies. By leveraging these innovative models, we can gain deeper insights into the molecular and cellular mechanisms underlying IHDs, ultimately paving the way for more effective diagnostic and therapeutic strategies.
Keywords:
Inherited heart disease
; stem cell
; disease modelling
; arrhythmia
; tissue engineering
1. Introduction
1.1. Global Health Burden of Inherited Heart Diseases
Inherited Heart Diseases (IHDs) pose a significant and growing burden on global public health. These conditions, which include hypertrophic cardiomyopathy (HCM), arrhythmogenic right ventricular cardiomyopathy (ARVC), long QT syndrome (LQTS), and others, often lead to severe cardiac complications, including heart failure, arrhythmias, and sudden cardiac death. IHDs not only impact individual patients but also place considerable strain on healthcare systems due to the need for long-term management, specialised care, and in some cases, heart transplants. The emotional and economic toll on affected families and communities is profound, particularly in regions with limited access to advanced diagnostic and therapeutic options. Addressing the global health burden of IHDs requires a comprehensive approach that includes raising awareness, improving diagnostic accuracy, expanding access to genetic testing, and advancing research into effective treatments.
1.2. Genetic Basis and Pathophysiology of IHDs
The genetic basis of IHDs is complex and involves mutations in various genes that encode proteins crucial for cardiac function. These mutations can lead to structural abnormalities, impaired cardiac muscle function, or electrical disturbances within the heart. For instance, HCM is often caused by mutations in genes that encode sarcomeric proteins, leading to abnormal thickening of the heart muscle [1]. Similarly, LQTS, a condition that predisposes individuals to life-threatening arrhythmias, is typically linked to mutations in genes responsible for ion channel function [2]. The pathophysiology of IHDs varies depending on the specific genetic mutation and its impact on cardiac tissue. In many cases, the disease process begins at the molecular level, where defective proteins disrupt normal cardiac function, leading to progressive structural changes, impaired contractility, or altered electrical activity [3,4,5]. Understanding these genetic and pathophysiological mechanisms is crucial for developing targeted therapies and improving outcomes for patients with IHDs.
1.3. Challenges in Understanding IHD Mechanisms
Despite significant advances in the study of IHDs, numerous challenges remain in fully understanding their underlying mechanisms. One major challenge is the genetic heterogeneity of these diseases; mutations in different genes can result in similar clinical phenotypes, complicating diagnosis, and treatment. Additionally, the penetrance and expressivity of genetic mutations that lead to IHDs can vary widely, meaning that individuals with the same mutation may experience vastly different symptoms, making it difficult to predict disease course and outcomes [6]. Another challenge lies in the complexity of the heart as an organ, where multiple cell types, signalling pathways, and structural components interact in ways that are not yet fully understood. Traditional animal models often fail to capture the full spectrum of human IHDs, and while advances in genetic technology have improved our ability to study these diseases, translating these findings into effective therapies remains a formidable task [7]. Moreover, ethical and practical limitations in obtaining human heart tissue for study hinder progress [8]. As a result, there is an ongoing need for innovative research approaches, including the use of stem cells and advanced imaging techniques, to better understand the mechanisms driving IHDs and develop more effective treatments.
1.4. Advances in Stem Cell Technology for IHD Research
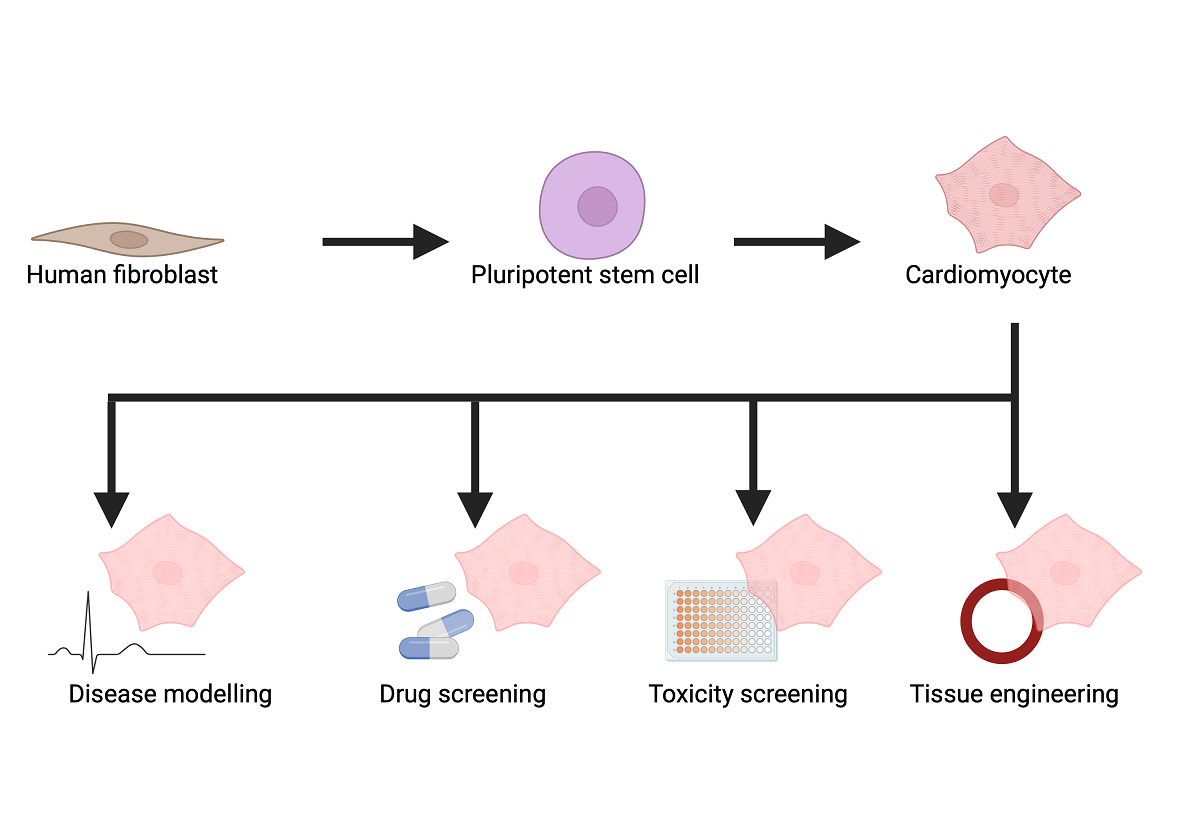
Stem cell technology has revolutionised research into IHDs, offering new insights into disease mechanisms and potential therapeutic avenues. One of the most promising developments is the use of induced pluripotent stem cells (iPSCs), which can be generated from a patient’s own cells and differentiated into cardiomyocytes or other cell types that make up heart muscle. These patient-specific iPSCs allow researchers to model IHDs in vitro, studying the effects of genetic mutations on heart cells and identifying potential drug targets. Furthermore, human iPSC-derived cardiomyocytes (iPSC-CMs) can be used to screen for drug efficacy and toxicity, paving the way for personalised medicine approaches in the treatment of IHDs. Recent advances have also seen the development of three-dimensional cardiac tissue models, or “heart-on-a-chip” systems, which more accurately replicate the complex structure and function of the human heart [9]. These models are invaluable for studying the pathophysiology of IHDs and testing new therapeutic interventions in a controlled environment. As stem cell technology continues to evolve, it holds great promise not only for understanding the molecular mechanisms of IHDs but also for developing innovative treatments that could one day repair or replace damaged heart tissue in affected patients.
2. Stem Cell-Derived Models
2.1. Historical Overview: From hESCs to iPSCs
Unicellular stem cell models enable a focused and controlled environment to study the intricacies of cellular function and dysfunction in IHDs. These models are particularly useful in investigating the molecular and genetic underpinnings of IHDs at a cellular level, offering insights that are often difficult to obtain from more complex multicellular models. The first human stem cell-derived cell types were developed from human embryonic stem cells (ESCs) over 25 years ago [10,11,12]. The genes that encode transcription factors pivotal in the maintenance of pluripotency are Oct4, Sox2, and Nanog [13,14]. However, unless the embryo for the human ESCs carried a specific, highly penetrant disease mutation, human ESC-derived cell types had poor predictive value. Furthermore, the sourcing of these cells had major ethical issues, limiting their use [15]. The field then moved to exploring alternative patient-specific stem cells.
2.2. Human iPSCs
2.2.1. Generation of Patient-Specific iPSCs
A more recent approach for producing stem cell-derived cell types, including cardiomyocytes, came following the production of iPSCs [16]. These cells are created by reprogramming somatic cells back to a pluripotent state. High expression of four specific transcription factors, Oct4/Sox2/Klf4/c-Myc or Oct4/Sox2/Nanog/LIN28 by virus, can reprogram fibroblast cells into an embryonic-like state [16,17]. Since then, alternative methods of reprogramming have been shown, such as generating iPSCs from fibroblasts by viral integration of Oct4/Sox2/Klf4 without c-Myc [18]. These cells are then capable of differentiating into any cell type, including cardiomyocytes. Since the first production of iPSCs, they have been differentiated into various cell types found in the adult healthy myocardium (Table 1).
2.2.2. Differentiation of iPSCs into Cardiomyocytes
Various protocols have been established to promote differentiation of iPSCs into cardiomyocytes. These protocols are discussed in detail elsewhere [42]. The most efficient method to generated iPSC-CMs has a two-stage approach, in which iPSCs are treated with a Glycogen Synthase Kinase (GSK) inhibitor to induce the beta-catenin pathway which drives conversion of iPSCs into cardiac progenitor cells [43]. These cells are then cultured in the presence of broad-spectrum pharmacological agents which target Wnt pathways, producing a phenotype that can contract and respond to physiological and pharmacological stimuli in a manner that is comparable to the adult, human cardiomyocyte.
2.2.3. Advantages of Human iPSC Models for IHD Research
The use of human iPSC models to investigate IHDs allows a personalised model that reflects the patient’s unique genetic background. To date, human iPSC models have been generated on a wide range of channelopathies, cardiomyopathies and vascular diseases [44]. Furthermore, they allow researchers to observe the development and progression of IHD from the early stages, providing valuable insight into the initial cellular and molecular events critical to the development of IHDs. Virtually all models used for drug screening and disease modelling rely on the use of cardiomyocytes from animal models or isolated human adult cardiomyocytes [45,46]. There are many challenges with the use of current human and animal models, including high costs, difficulties in manipulation, and ethical issues [45,46]. Importantly, human iPSCs hold significant value in drug screening, as differentiated cells developed from iPSCs can be used to test the efficacy and safety of novel drugs, enabling personalised medicine approaches.
2.3. Studying Cellular Function and Dysfunction
2.3.1. Electrophysiology Studies
Researchers can use human iPSC-CMs to investigate how genetic mutations associated with IHDs affect cellular function. From identifying the first genetic mutation causing an arrhythmia in 1995, there are now over 28 genes implicated in inherited cardiac arrhythmic disorders [47,48]. Electrophysiology studies allow researchers to investigate how mutations alter the electrical properties of cardiomyocytes, leading to arrhythmias and other electrical disturbances associated with IHDs. LQTS type 1 (LQTS1) was the first ion channelopathy modelled using human iPSC-CMs. In a study by Moretti and colleagues, human iPSCs were derived from two LQT1 patients with heterozygous missense mutation R190Q in the gene KCNQ1 [49]. The human iPSC-CMs created from these cells demonstrated the reduction in action potential repolarising current IKs identified in adults with this condition. The resulting prolongation of the action potential duration puts these patients at risk of arrhythmic events when exposed to isoproterenol, a beta-adrenergic agonist. Homozygous mutations in KCNQ1 has been shown to cause Jervell and Lange-Nielsen Syndrome (JLNS), an autosomal recessive disorder characterised by a prominent prolongation of the QT interval and deafness [50]. Human iPSC-CMs derived from patients with JLNS or genetically engineered to have a homozygous mutation in KCNQ1 demonstrate the expected pronounced action potential duration prolongation and susceptibility to drug-induced arrhythmia [51].
2.3.2. Contractility Studies
Contractility studies involving the analysis of how genetic defects impact the contractile function of cardiomyocytes are important for investigations into conditions like HCM and dilated cardiomyopathy (DCM). Mutations that truncate the sarcomere protein titin are the most common genetic cause for DCM [52]. Hinson and colleagues used human iPSC-CMs to identify missense mutations that cause truncation in the A-band domain of titin causes DCM. Interestingly, truncation in the I-band is better tolerated [53].
2.3.3. Calcium Handling Studies
Calcium handling studies investigate how mutations affect the cycling of calcium within cardiomyocytes, a process that is crucial for physiological contraction and relaxation of heart muscle [54]. El-Brattrawy and colleagues investigated the pathogenesis of patients with Short QT-Syndrome (SQTS) [55]. They generated human iPSC-CMs from the skin fibroblasts of a patient with SQTS type 1 carrying a mutation (N588K) in KCNH2 and two healthy control subjects. This mutation increased the expression of HERG channels and altered calcium homeostasis in the human iPSC-CMs, contributing to a greater frequency of irregular, delayed afterdepolarisation-like and early afterdepolarisation-like events than control.
2.4. Genome Editing in iPSC-CMs
2.4.1. CRISPR/Cas9 Technology
The advent of CRISPR/Cas9 genome editing has revolutionised the ability to study genetic diseases. This technology allows precise modification of specific genes within iPSCs, enabling researchers to introduce or correct genetic mutations associated with IHDs [56,57]. This system introduces double-strand breaks (DSBs) at specific sites in the genome, which are then repaired endogenously by the cell. This is often by error-prone nonhomologous end joining (NHEJ), but if a DNA template with homology to the sequence around the DSB is present, this repair can also be driven by homology-directed repair [58]. By using CRISPR/Cas9, researchers can generate, within 2-6 months, isogenic cell lines where only the gene of interest differs between the experimental and control groups [59]. This approach eliminates background genetic variability, allowing for a clearer understanding of the specific effects of a mutation.
2.4.2. Knockout and Knock-in Models
Gene editing technologies have been utilised in studying the function of genes involved in the pathophysiology of IHDs. Knockout models disable specific genes in iPSC-CMs to study their role in heart function and disease. Knock-in models introduce specific mutations associated with IHDs to replicate the disease phenotype and study its mechanisms. Human iPSCs have been derived from a patient with LQTS and confirmed to carry a novel variant of uncertain significance (VUS) in KCNH2 [60]. Upon correcting the VUS with a knockout model, the electrophysiological abnormalities observed in the patient human iPSC-CMs were corrected and introducing the homozygous variant into a control human iPSC line by knock-in enabled researchers to confirm its contribution to electrophysiological dysfunction.
3. Multicellular Models and Tissue Engineering
Multicellular models and tissue engineering approaches offer a more comprehensive and physiologically relevant platform than single cell models to study the mechanisms of IHDs. These models simulate the complex interactions between different cell types within the heart, providing deeper insights into disease pathophysiology [61,62,63]. Notable multicellular models and tissue engineering models used in investigating mechanisms of IHDs include cardiac organoids, engineered heart tissue, and co-culture systems.
3.1. Cardiac Organoids
3.1.1. Structure and Function of Cardiac Organoids
The human heart is primarily composed of cardiomyocytes (60%), with the predominant non-myocyte cell types being cardiac fibroblasts (24%) and endothelial cells (14%). The rest of the heart consists of a small number of smooth muscle cells, epicardial cells, conductance cells as well as immune cells [64,65]. Cardiac organoids are three-dimensional structures that replicate aspects of heart tissue architecture and function. These organoids are derived from stem cells, typically iPSCs, which are induced to differentiate into various cardiac cell types, including cardiomyocytes, fibroblasts, and endothelial cells. Organoids can be generated through self-organisation or by guiding the differentiation process using specific growth factors and signalling molecules [66,67]. By studying how mutations affect the development and function of cardiac organoids, researchers can gain insights into the early events in the pathogenesis of IHDs. Furthermore, cardiac organoids offer a platform for high-throughput screening of potential drugs and therapeutic agents, allowing researchers to assess their efficacy and safety in a more physiologically relevant context.
3.1.2. Applications in IHD Research
Researchers have been able to perform mutagenic studies by using iPSCs derived from patient tissue to create cardiac organoids physiologically relevant to accurately model familial cardiomyopathy in vitro. Yang and colleagues created cardiac organoid constructs using human iPSCs and human marrow stromal cells from patients with a myosin heavy-chain 7 mutation E848G [68]. The resulting organoid structures demonstrated decreased alignment, reduced systolic function with little impact on diastolic function, recapitulating the contractile dysfunction in these patients. They were able to demonstrate that the E848G allele disrupts the protein-protein interaction between the MYH7 and cardiac myosin binding protein C, presenting a potential mechanism of action [68].
3.1.3. Case Studies: Modelling Cardiomyopathies and DMD
Similarly, Buono and colleagues used human iPSC-CMs derived from a HCM patient with an R719Q mutation in MYH7, a single point mutation that has been known to play a role in the onset of HCM causing sudden cardiac death [69,70]. They combined these human iPSC-CMs with human cardiac endothelial cells and cardiac fibroblasts to create an organoid that exhibited an arrhythmogenic phenotype [70]. This can then play a critical role in drug screening for therapeutic strategies against sudden cardiac death in HCM patients.
Cardiac organoids have also been constructed using cardiomyocytes derived from patients with Duchenne Muscular Dystrophy (DMD) [71,72]. This disease affects approximately 1 in 5000 males and is caused by mutations in the X-linked dystrophin gene (DMD) [73]. The dystrophin protein links the cytoskeleton and extracellular matrix of muscle cells, as well as maintaining the integrity of the plasma membrane [74]. In DMD, the dystrophin protein is absent or dysfunctional, causing muscle degeneration and often DCM. By creating organoids where the DMD mutation was corrected using CRISPR/Cas9 technology, the researchers were able to restore dystrophin expression and improve the contractile ability of the tissue. These studies highlight the value of human cardiac organoids for modelling IHDs and demonstrate the functional impact that gene editing can have on human cardiac tissue.
3.2. Engineered Heart Tissue (EHT)
3.2.1. Construction and Mechanical Properties
EHTs involve the creation of three-dimensional structures that replicate the mechanical and electrical characteristics of native heart tissue. These tissues are constructed using cardiomyocytes derived from iPSCs, along with other cardiac cell types, seeded onto biodegradable scaffolds or hydrogels [75]. These materials provide structural support and facilitate tissue organisation. The mechanical properties of EHT can be customised to mimic the stiffness and elasticity of native heart tissue, which is essential for studying how genetic mutations impact the heart’s mechanical function. EHT can also undergo mechanical stimulation, such as stretching and contraction, to further analyse the biomechanical effects of mutations [76].
3.2.2. Functional Studies in EHT Models
By incorporating sensors and electrodes into EHT, researchers can measure electrical activity and contractile force, offering valuable insights into how mutations influence the electrophysiological and mechanical functions of the heart [76]. A recent study by Wang and colleagues utilised a human EHT model of HCM to replicate key features of the disease [77]. They generated EHTs using healthy human cardiac fibroblasts alongside iPSC-CMs carrying a beta-myosin mutation (MYH7-R403Q) or its isogenic control. Although both R403Q and control EHTs exhibited mature cardiac phenotypes, the R403Q EHTs showed increased tissue size, cell volume, shortened sarcomere length, and disorganised sarcomere structures, mirroring those seen in mature myocardium. Additionally, the R403Q EHTs exhibited increased twitch amplitude, slower contractile kinetics, prolonged action potential durations, and slower calcium transient decay time—typical findings in myocardial tissue from HCM patients.
3.2.3. Drug Screening and Therapeutic Testing
EHTs serve as a powerful platform for screening potential drugs and therapeutic agents, enabling researchers to evaluate their effects on heart tissue function in a controlled and physiologically relevant environment. In the study by Wang and colleagues, the impact of chronic mavacamten treatment—a first-in-class cardiac myosin inhibitor for HCM patients—was investigated [77]. After five weeks of mavacamten treatment, the R403Q EHTs exhibited shortened relaxation time, reduced action potential prolongation, and decreased expression of B-type natriuretic peptide (BNP) mRNA and protein. Additionally, mavacamten treatment led to an increase in sarcomere length and improved sarcomere organisation. Mavacamten has since been identified as a potentially crucial therapy for obstructive HCM patients as an alternative to myomectomy surgery [78].
4. Insights into Cellular and Molecular Mechanisms
4.1. Paracrine Signalling Between Cardiac Cells
Cells in the heart communicate through the secretion of signalling molecules, which can influence the behaviour of neighbouring cells. In co-culture systems, researchers can study how mutations affect paracrine signalling pathways, such as those involving growth factors, cytokines, and extracellular vesicles. A study by Kane and colleagues investigated the effect of cardiac fibroblasts on human iPSC-CMs Ca2+ cycling. It identified that when human iPSC-CMs are cultured in cardiac fibroblast-conditioned media or in non-contact co-cultures with cardiac fibroblasts, they demonstrated slower Ca2+ cycling [79]. This study demonstrates the importance of understanding the key paracrine mediators of cardiac fibroblast crosstalk if we are to better understand the pathological fibrosis that occurs in many IHDs.
4.2. Mechanical and Electrical Cell-Cell Interactions
Direct interactions between cardiac cell types, such as the interactions mediated by gap junctions and adherens junctions, are critical for maintaining the structural and functional integrity of the heart and dysfunction of these interactions have been implicated in IHDs [80]. Co-culture systems allow for the examination of how these interactions are disrupted by genetic mutations. For example, Giacomelli and colleagues identified that human iPSC-CMs in tricellular combinations with human iPSC-derived cardiac stromal cells and cardiac fibroblasts have improved contractility and mitochondrial respiration than organoids in the absence of cardiac fibroblasts [81]. They identified that these improvements are through connexin 43 (CX43) gap channels and increased intracellular cyclic AMP. Interestingly, they also identified that organoids containing cardiac fibroblasts derived from patients with arrhythmogenic cardiomyopathy recapitulate features of the disease.
4.3. ECM Remodelling
The ECM provides structural support to cardiac cells and regulates their behaviour. In co-culture systems, researchers can investigate how mutations in genes that regulate ECM production or degradation contribute to pathological remodelling, as seen in conditions like myocardial fibrosis. It is well known that ECM proteins interact with cardiomyocytes through via the integrin receptors found on the cardiomyocytes [82]. A study investigating the role that ECM proteins play in cardiac remodelling identified that the integrin-binding sequence of arginine-glycine-aspartic acid induces recruitment of the sarcoplasmic reticulum for Ca2+ cycling in excitation-contraction coupling in cardiomyocytes [83]. The flanking sequence around this tripeptide integrin-binding sequence determines affinity for integrins. Integrin ligand-receptor interactions in the in vitro setting, reflecting the interactions between ECM proteins and cardiomyocytes, has been found to modulate cardiomyocyte Ca2+ cycling [83]. This has important implications in our understanding of pathophysiology of IHDs and therapeutic strategies that can correct the Ca2+ cycling dysfunction often found in cardiomyopathies.
4.4. Metabolic Coupling in the Heart
The heart is a highly metabolically active organ, and the metabolic interactions between different cell types are crucial for maintaining cardiac function. Barth syndrome, an x-linked cardiac and skeletal myopathy, results from a mutation in the gene tafazzin [84]. Tafazzin is responsible for the production of the enzyme acyltransferase. Reduced acyltranferase function leads to decreased acetylation of cardiolipin—the major phospholipid of the mitochondrial inner membrane [85]. By using cas9-mediated genome editing of cardiomyocytes derived from patients, it has been shown that a mutation in tafazzin can recapitulate the patient disease phenotype in wild type cells [86]. Tafazzin deficiency increased the production of reactive oxygen species (ROS), and suppression of ROS ameliorates the metabolic dysfunction in Barth syndrome human iPSC-CMs, demonstrating the critical role of metabolic regulation in the disease.
5. Limitations and Future Directions
5.1. Limitations and Challenges
The potential applications of human iPSC-CMs in both research and therapy are significant. However, it is essential to address the immaturity of human iPSC-CM structures [87]. These cells exhibit structural characteristics more like neonatal or embryonic cardiomyocytes than to those of adult cardiomyocytes (Table 2). Typically, human iPSC-CMs are large, flat, round, and possess a single nucleus, though they can adapt considerably to their culture environment [88,89]. They generally mimic the phenotype of human cardiomyocytes during the early stages of cardiogenesis. At this developmental phase, the myocardium has not yet transitioned from hyperplastic to physiological hypertrophic growth. In physiological hypertrophic growth, elongated, rod-like cardiomyocytes align anisotropically and connect electrically via unidirectional gap junctions, facilitating efficient contraction of the cardiomyocyte syncytium in the developing heart [90]. However, this directional propagation of cardiomyocyte excitation is not observed in spontaneously beating human iPSC-CMs in vitro, likely due to the absence of extracellular stimuli found in the developing heart [91].
One significant limitation of human iPSC-CMs is their restricted contractility, which is attributed to the immaturity of their sarcomere structure [92,93]. Sarcomeres, defined as the distance between two Z-lines, are crucial for force production. In adult cardiomyocytes, the optimal sarcomere length for maximum contractile force is 2.2 μm under loaded conditions and 1.8 μm under unloaded conditions [94]. In contrast, human iPSC-CMs possess shorter (1.6 μm) and disorganised sarcomeres, akin to those in fetal cardiomyocytes or in the pathological state [95]. Studies have shown that human iPSC-CMs primarily exhibit Z-disk and I-band structures, although these characteristics can vary significantly depending on the seeding conditions and extracellular interactions [96]. Despite variations in human iPSC-CMs phenotypes across different studies, it is generally agreed that these cells produce much less force than healthy adult cardiomyocytes [97].
Additionally, human iPSC-CMs exhibit suboptimal mitochondrial maturation. In adult cardiomyocytes, mitochondrial biogenesis significantly increases mitochondrial content to meet higher energetic demands [98]. In adult ventricular myocytes, mitochondria occupy about 30% of the total cell volume, compared to just 2% in human iPSC-CMs [99]. Moreover, mitochondria in human iPSC-CMs have lower activity due to immature cristae on their inner membrane [99,100]. Unlike healthy adult cardiomyocytes, which primarily rely on fatty acid oxidation for energy, human iPSC-CMs, like the fetal heart, heavily depend on glycolysis [101].
It has been observed that many ultrastructural features of human iPSC-CMs maturation develop with prolonged culture. Increases in nucleation, myofibril density, and expression of contractile proteins such as β-myosin heavy chain have been reported in cultures 120-360 days post-differentiation [96,102]. However, it remains unclear whether these developments are due to genuine maturation over time or result from senescence caused by the culture conditions.
5.2. Future Directions in IHD Research
The use of human iPSCs in investigating IHDs has significantly advanced our understanding of the molecular and cellular mechanisms underlying these conditions. However, there are still several key areas where future research could further enhance the field. One promising direction is the integration of human iPSC technology with advanced gene-editing tools such as CRISPR-Cas9. This combination allows for the precise modelling of genetic mutations associated with IHDs, enabling researchers to study not only the impact of specific mutations but also the potential for correcting them at the genetic level.
Another important area of future research is the development of more complex and physiologically relevant models, such as organ-on-a-chip systems and bioengineered heart tissue. These models could incorporate multiple cell types, including those involved in the immune response and vasculature, to better mimic the in vivo environment. This will provide a more comprehensive understanding of IHD pathophysiology, particularly in the context of how these diseases develop and progress over time.
Furthermore, the integration of human iPSC-derived cardiac models with high-throughput screening technologies holds significant potential. This approach could enable large-scale screening of genetic variants and environmental factors that contribute to IHDs, as well as the identification of novel biomarkers and therapeutic targets. As our ability to generate and analyse large datasets improves, future research may also focus on leveraging artificial intelligence and machine learning to identify patterns and predict disease outcomes based on human iPSC-derived data.
Finally, the exploration of patient-specific human iPSC models will continue to be a critical focus. By generating human iPSCs from diverse populations, including those with rare IHD mutations, researchers can better understand the variability in disease presentation and response to treatments. This personalised approach to IHD research has the potential to revolutionise the way we diagnose, study, and ultimately treat inherited heart diseases.
6. Conclusion
The development of stem cell-derived models has revolutionised the study of inherited heart diseases, providing unprecedented insights into the molecular and cellular mechanisms underlying these conditions. Single-cell models using human iPSC-CMs allow researchers to study the effects of genetic mutations at a cellular level, offering valuable information on the roles of specific genes and proteins in disease pathogenesis. Genome editing technologies, such as CRISPR/Cas9, further enhance the ability to investigate the genetic underpinnings of IHDs, enabling precise manipulation of specific genes and mutations. In addition, multicellular models and tissue engineering approaches, such as cardiac organoids, engineered heart tissue, and co-culture systems, provide a more comprehensive and physiologically relevant platform to study disease mechanisms. These models allow researchers to investigate the complex interactions between different cell types within the heart, providing deeper insights into the pathophysiology of IHDs and enabling the development of more effective diagnostic and therapeutic strategies. By leveraging these innovative models, researchers can gain a deeper understanding of the molecular and cellular mechanisms underlying IHDs, ultimately paving the way for more effective diagnostic and therapeutic strategies.
Author Contributions
Conceptualization, B.X.W; writing—B.X.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ALDH-2 | Aldehyde dehydrogenase 2 |
| ARVC | Arrhythmogenic right ventricular cardiomyopathy |
| BNP | B-type natriuretic peptide |
| CX43 | Connexin 43 |
| DCM | dilated cardiomyopathy |
| DMD | Duchenne Muscular Dystrophy |
| DSB | Double-strand break |
| EC | Endothelial cell |
| EHT | Engineered Heart Tissue |
| ELN | Elastin |
| ESC | Embryonic stem cells |
| GSK | Glycogen Synthase Kinase |
| HCM | Hypertrophic cardiomyopathy |
| IHD | Inherited heart disease |
| iPSC | Induced pluripotent stem cells |
| iPSC-CM | induced pluripotent stem cell-derived cardiomyocyte |
| LQTS | Long QT syndrome |
| NHEJ | Nonhomologous end joining |
| RBC | Red blood cell |
| ROS | Reactive oxygen species |
| RYR2 | Ryanodine receptor 2 |
| SMC | Smooth muscle cell |
| SQTS | Short QT syndrome |
| VUS | Variant of uncertain significance |
References
- Marian AJ, Braunwald E (2017) Hypertrophic cardiomyopathy: Genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res 121:749–770. [CrossRef]
- Wallace E, Howard L, Liu M, et al. (2019) Long QT Syndrome: Genetics and Future Perspective. Pediatr Cardiol 40:1419–1430.
- Wang S, Zhang Z, He J, et al. (2023) Comprehensive review on gene mutations contributing to dilated cardiomyopathy. Front Cardiovasc Med 10. [CrossRef]
- Badura K, Buławska D, Dąbek B, et al. (2024) Primary Electrical Heart Disease—Principles of Pathophysiology and Genetics. Int J Mol Sci 25. [CrossRef]
- Garfinkel AC, Seidman JG, Seidman CE (2018) Genetic Pathogenesis of Hypertrophic and Dilated Cardiomyopathy. Heart Fail Clin 14:139–146.
- Kathiresan S, Srivastava D (2012) Genetics of human cardiovascular disease. Cell 148:1242–1257.
- Bracken MB (2009) Why animal studies are often poor predictors of human reactions to exposure. J R Soc Med 102:120–122. [CrossRef]
- Kiani AK, Pheby D, Henehan G, et al. (2022) Ethical considerations regarding animal experimentation. J Prev Med Hyg 63:E255–E266. [CrossRef]
- King O, Cruz-Moreira D, Worrapong Kit-Anan S, et al. (2020) Vascularized Myocardium-On-A-Chip: Excitation-Contraction Coupling in Perfused Cardiac Co-Cultures. Biophys J 118:410a. [CrossRef]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, et al. (1998) Embryonic stem cell lines derived from human blastocysts. Science (1979) 282:1145–1147. [CrossRef]
- Takahashi K, Tanabe K, Ohnuki M, et al. (2007) /SUPP/ Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131:861–72. [CrossRef]
- Shelton M, Kocharyan A, Liu J, et al. (2016) Robust generation and expansion of skeletal muscle progenitors and myocytes from human pluripotent stem cells. Methods 101:73–84. [CrossRef]
- Boyer LA, Lee TI, Cole MF, et al. (2005) Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122:947–956. [CrossRef]
- Loh Y-H, Wu Q, Chew J-L, et al. (2006) The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet 38:431–440. [CrossRef]
- Deb K, Sarda K (2008) Human embryonic stem cells: Preclinical perspectives. J Transl Med 6:1–8. [CrossRef]
- Takahashi K, Yamanaka S (2006) Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 126:663–676. [CrossRef]
- Yu J, Vodyanik MA, Smuga-Otto K, et al. Induced Pluripotent Stem Cell Lines Derived from Human Somatic Cells.
- Nakagawa M, Koyanagi M, Tanabe K, et al. (2008) Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol 26:101–106. [CrossRef]
- Dudek J, Cheng IF, Balleininger M, et al. (2013) Cardiolipin deficiency affects respiratory chain function and organization in an induced pluripotent stem cell model of Barth syndrome. Stem Cell Res 11:806–819. [CrossRef]
- Nasilli G, Verkerk AO, O’Reilly M, et al. (2024) Chronic Mexiletine Administration Increases Sodium Current in Non-Diseased Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Biomedicines 12:. [CrossRef]
- Nasilli G, Yiangou L, Palandri C, et al. (2023) Beneficial effects of chronic mexiletine treatment in a human model of SCN5A overlap syndrome. Europace 25:. [CrossRef]
- Novak A, Barad L, Zeevi-Levin N, et al. (2012) Cardiomyocytes generated from CPVT D307H patients are arrhythmogenic in response to β-adrenergic stimulation. J Cell Mol Med 16:468–482. [CrossRef]
- Sasaki K, Makiyama T, Yoshida Y, et al. (2016) Patient-specific human induced pluripotent stem cell model assessed with electrical pacing validates S107 as a potential therapeutic agent for catecholaminergic polymorphic ventricular tachycardia. PLoS One 11:. [CrossRef]
- Lan F, Lee ASS, Liang P, et al. (2013) Abnormal Calcium Handling Properties Underlie Familial Hypertrophic Cardiomyopathy Pathology in Patient-Specific Induced Pluripotent Stem Cells. Cell Stem Cell 12:101–113. [CrossRef]
- Jiang Y, Habibollah S, Tilgner K, et al. (2014) An Induced Pluripotent Stem Cell Model of Hypoplastic Left Heart Syndrome (HLHS) Reveals Multiple Expression and Functional Differences in HLHS-Derived Cardiac Myocytes. Stem Cells Transl Med 3:416–423. [CrossRef]
- Ebert AD, Kodo K, Liang P, et al. (2014) Characterization of the molecular mechanisms underlying increased ischemic damage in the aldehyde dehydrogenase 2 genetic polymorphism using a human induced pluripotent stem cell model system. Sci Transl Med 6:255ra130-255ra130. [CrossRef]
- Lauriol J, Kontaridis MI (2011) PTPN11-Associated Mutations in the Heart: Has LEOPARD Changed Its RASpots? Trends Cardiovasc Med 21:97–104.
- Moretti A, Bellin M, Welling A, et al. (2010) Patient-Specific Induced Pluripotent Stem-Cell Models for Long-QT Syndrome. New England Journal of Medicine 363:1397–1409. [CrossRef]
- Yazawa M, Hsueh B, Jia X, et al. (2011) Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature 471:230–236. [CrossRef]
- Davis RP, Casini S, Van Den Berg CW, et al. (2012) Cardiomyocytes derived from pluripotent stem cells recapitulate electrophysiological characteristics of an overlap syndrome of cardiac sodium channel disease. Circulation 125:3079–3091. [CrossRef]
- Lahti AL, Kujala VJ, Chapman H, et al. (2012) Model for long QT syndrome type 2 using human iPS cells demonstrates arrhythmogenic characteristics in cell culture. Dis Model Mech 5:220–230. [CrossRef]
- Limpitikul WB, Dick IE, Tester DJ, et al. (2017) A Precision Medicine Approach to the Rescue of Function on Malignant Calmodulinopathic Long-QT Syndrome. Circ Res 120:39–48. [CrossRef]
- Rocchetti M, Sala L, Dreizehnter L, et al. (2017) Elucidating arrhythmogenic mechanisms of long-QT syndrome CALM1-F142L mutation in patient-specific induced pluripotent stem cell-derived cardiomyocytes. Cardiovasc Res 113:531–541. [CrossRef]
- Adams WJ, Zhang Y, Cloutier J, et al. (2013) Functional vascular endothelium derived from human induced pluripotent stem cells. Stem Cell Reports 1:105–113. [CrossRef]
- Liu C, Shen M, Tan WLW, et al. (2023) Statins improve endothelial function via suppression of epigenetic-driven EndMT. Nature Cardiovascular Research 2:467–485. [CrossRef]
- Matrone G, Thandavarayan RA, Walther BK, et al. (2019) Dysfunction of iPSC-derived endothelial cells in human Hutchinson–Gilford progeria syndrome. Cell Cycle 18:2495–2508. [CrossRef]
- Richardson SE, Ghazanfari R, Chhetri J, et al. (2021) In vitro differentiation of human pluripotent stem cells into the B lineage using OP9-MS5 co-culture. STAR Protoc 2:. [CrossRef]
- Fan F, Yu Y, Sun L, et al. (2018) Induction of Pluripotent Stem Cell-Derived Cardiomyocyte Toxicity by Supernatant of Long Term-Stored Red Blood Cells in Vitro. Cellular Physiology and Biochemistry 46:1230–1240. [CrossRef]
- Haro-Mora JJ, Uchida N, Demirci S, et al. (2020) Biallelic correction of sickle cell disease-derived induced pluripotent stem cells (iPSCs) confirmed at the protein level through serum-free iPS-sac/erythroid differentiation. Stem Cells Transl Med 9:590–602. [CrossRef]
- Ge X, Ren Y, Bartulos O, et al. (2012) Modeling supravalvular aortic stenosis syndrome with human induced pluripotent stem cells. Circulation 126:1695–1704. [CrossRef]
- Granata A, Serrano F, Bernard WG, et al. (2017) An iPSC-derived vascular model of Marfan syndrome identifies key mediators of smooth muscle cell death. Nat Genet 49:97–109. [CrossRef]
- Lyra-Leite DM, Gutiérrez-Gutiérrez Ó, Wang M, et al. (2022) A review of protocols for human iPSC culture, cardiac differentiation, subtype-specification, maturation, and direct reprogramming. STAR Protoc 3.
- Burridge PW, Matsa E, Shukla P, et al. (2014) Chemically defned generation of human cardiomyocytes. Nat Methods 11:855–860. [CrossRef]
- Moretti A, Laugwitz KL, Dorn T, et al. (2013) Pluripotent stem cell models of human heart disease. Cold Spring Harb Perspect Med 3:. [CrossRef]
- Parameswaran S, Kumar S, Verma RS, Sharma RK (2013) Cardiomyocyte culture — an update on the in vitro cardiovascular model and future challenges. Can J Physiol Pharmacol 91:985–998. [CrossRef]
- Li D, Wu J, Bai Y, et al. (2014) Isolation and Culture of Adult Mouse Cardiomyocytes for Cell Signaling and <em>in vitro</em> Cardiac Hypertrophy. Journal of Visualized Experiments 2–9. [CrossRef]
- Wang Q, Shen J, Splawski I, et al. (1995) SCN5A Mutations Associated with an Inherited Cardiac Arrhythmia, Long QT Syndrome.
- Funke BH (2016) Development of a Comprehensive Sequencing Assay for Inherited Cardiac Condition Genes. J Cardiovasc Transl Res 9:1–2.
- Moretti A, Bellin M, Welling A, et al. (2010) Patient-Specific Induced Pluripotent Stem-Cell Models for Long-QT Syndrome. New England Journal of Medicine 363:1397–1409. [CrossRef]
- Skinner JR, Winbo A, Abrams D, et al. (2019) Channelopathies That Lead to Sudden Cardiac Death: Clinical and Genetic Aspects. Heart Lung Circ 28:22–30.
- Zhang M, D’Aniello C, Verkerk AO, et al. (2014) Recessive cardiac phenotypes in induced pluripotent stem cell models of Jervell and Lange-Nielsen syndrome: Disease mechanisms and pharmacological rescue. Proc Natl Acad Sci U S A 111:E5383–E5392. [CrossRef]
- Tabish AM, Azzimato V, Alexiadis A, et al. (2017) Genetic epidemiology of titin-truncating variants in the etiology of dilated cardiomyopathy. Biophys Rev 9:207–223.
- Hinson JT, Chopra A, Nafissi N, et al. (2015) Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science (1979) 349:982–986. [CrossRef]
- Eisner DA, Caldwell JL, Kistamás K, Trafford AW (2017) Calcium and excitation-contraction coupling in the heart. Circ Res 121:181–195. [CrossRef]
- El-Battrawy I, Lan H, Cyganek L, et al. (2018) Modeling Short QT syndrome using human-induced pluripotent stem cell-derived cardiomyocytes. J Am Heart Assoc 7:. [CrossRef]
- Kolanu ND (2024) CRISPR–Cas9 Gene Editing: Curing Genetic Diseases by Inherited Epigenetic Modifications. Glob Med Genet 11:113–122. [CrossRef]
- Asif M, Khan WJ, Aslam S, et al. (2024) The Use of CRISPR-Cas9 Genetic Technology in Cardiovascular Disease: A Comprehensive Review of Current Progress and Future Prospective. Cureus. [CrossRef]
- Ben-Tov D, Mafessoni F, Cucuy A, et al. (2024) Uncovering the dynamics of precise repair at CRISPR/Cas9-induced double-strand breaks. Nat Commun 15:. [CrossRef]
- Jacinto F, V. Jacinto F V., Link W, Ferreira BI (2020) CRISPR/Cas9-mediated genome editing: From basic research to translational medicine. J Cell Mol Med 24:3766–3778.
- Garg P, Oikonomopoulos A, Chen H, et al. (2018) Genome Editing of Induced Pluripotent Stem Cells to Decipher Cardiac Channelopathy Variant. J Am Coll Cardiol 72:62–75. [CrossRef]
- King O, Kermani F, Wang B, et al. (2019) Endothelial Cell Regulation of Excitation-Contraction Coupling in Induced Pluripotent Stem Cell Derived Myocardium. Biophys J 116:153a. [CrossRef]
- Wang BX, Nicastro L, Couch L, et al. (2022) Extracellular Vesicles from Human Cardiac Fibroblasts Modulate Calcium Cycling in Human Stem Cell-Derived Cardiomyocytes. Cells 11:. [CrossRef]
- Zwi-Dantsis L, Winter CW, Kauscher U, et al. (2020) Highly purified extracellular vesicles from human cardiomyocytes demonstrate preferential uptake by human endothelial cells. Nanoscale 12:19844–19854. [CrossRef]
- Pinto AR, Ilinykh A, Ivey MJ, et al. (2016) Revisiting cardiac cellular composition. Circ Res 118:400–409. [CrossRef]
- Lewis-Israeli YR, Wasserman AH, Aguirre A (2021) Heart organoids and engineered heart tissues: Novel tools for modeling human cardiac biology and disease. Biomolecules 11.
- Brassard JA, Lutolf MP (2019) Engineering Stem Cell Self-organization to Build Better Organoids. Cell Stem Cell 24:860–876.
- Lancaster MA, Knoblich JA (2014) Organogenesisin a dish: Modeling development and disease using organoid technologies. Science (1979) 345.
- Yang KC, Breitbart A, De Lange WJ, et al. (2018) Novel Adult-Onset Systolic Cardiomyopathy Due to MYH7 E848G Mutation in Patient-Derived Induced Pluripotent Stem Cells. JACC Basic Transl Sci 3:728–740. [CrossRef]
- Li X, Fu W, Guo G, et al. (2021) A heterozygous MYH7 (c. 2156G > A) mutant human induced pluripotent stem cell line (ZZUNEUi020-A) generated from a patient with hypertrophic cardiomyopathy. Stem Cell Res 51:. [CrossRef]
- Buono MF, Boehmer L von, Strang J, et al. (2020) Human cardiac organoids for modeling genetic cardiomyopathy. Cells 9:1–19. [CrossRef]
- Long C, Li H, Tiburcy M, et al. (2018) Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing.
- Marini V, Marino F, Aliberti F, et al. (2022) Long-term culture of patient-derived cardiac organoids recapitulated Duchenne muscular dystrophy cardiomyopathy and disease progression. Front Cell Dev Biol 10:. [CrossRef]
- Duan D, Goemans N, Takeda S, et al. (2021) Duchenne muscular dystrophy. Nat Rev Dis Primers 7.
- Gao QQ, McNally EM (2015) The dystrophin complex: Structure, function, and implications for therapy. Compr Physiol 5:1223–1239. [CrossRef]
- Jabbour R, Owen T, Reinsch M, et al. (2019) Development and preclinical testing of upscaled engineered heart tissue for use in translational studies. Eur Heart J 40:0345–0345.
- Stoppel WL, Kaplan DL, Black LD (2016) Electrical and mechanical stimulation of cardiac cells and tissue constructs. Adv Drug Deliv Rev 96:135–155.
- Wang K, Schriver BJ, Aschar-Sobbi R, et al. (2023) Human engineered cardiac tissue model of hypertrophic cardiomyopathy recapitulates key hallmarks of the disease and the effect of chronic mavacamten treatment. Front Bioeng Biotechnol 11:. [CrossRef]
- Braunwald E, Saberi S, Abraham TP, et al. (2023) Mavacamten: a first-in-class myosin inhibitor for obstructive hypertrophic cardiomyopathy. Eur Heart J 44:4622–4633.
- Kane C, Terracciano CM (2018) Human cardiac fibroblasts engage the sarcoplasmic reticulum in induced pluripotent stem cell-derived cardiomyocyte excitation–contraction coupling. J Am Coll Cardiol 72:1061–1063. [CrossRef]
- Sheikh F, Ross RS, Chen J (2009) Cell-Cell Connection to Cardiac Disease. Trends Cardiovasc Med 19:182–190.
- Giacomelli E, Meraviglia V, Campostrini G, et al. (2020) Human-iPSC-Derived Cardiac Stromal Cells Enhance Maturation in 3D Cardiac Microtissues and Reveal Non-cardiomyocyte Contributions to Heart Disease. Cell Stem Cell 26:862-879.e11. [CrossRef]
- Israeli-Rosenberg S, Manso AM, Okada H, Ross RS (2014) Integrins and integrin-associated proteins in the cardiac myocyte. Circ Res 114:572–586.
- Wang BX, Kane C, Nicastro L, et al. (2022) Integrins Increase Sarcoplasmic Reticulum Activity for Excitation—Contraction Coupling in Human Stem Cell-Derived Cardiomyocytes. Int J Mol Sci 23:. [CrossRef]
- Jefferies JL (2013) Barth syndrome. Am J Med Genet C Semin Med Genet 163:198–205. [CrossRef]
- Ross Pennington E, Funai K, Brown DA, et al. (2019) The role of cardiolipin concentration and acyl chain composition on mitochondrial inner membrane molecular organization and function HHS Public Access Author manuscript. Biochim Biophys Acta Mol Cell Biol Lipids 1039–1052. [CrossRef]
- Wang G, McCain ML, Yang L, et al. (2014) Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat Med 20:616–623. [CrossRef]
- Karakikes I, Ameen M, Termglinchan V, Wu JC (2015) Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes: Insights into Molecular, Cellular, and Functional Phenotypes. Circ Res 117:80–88. [CrossRef]
- Gherghiceanu M, Barad L, Novak A, et al. (2011) Cardiomyocytes derived from human embryonic and induced pluripotent stem cells: Comparative ultrastructure. J Cell Mol Med 15:2539–2551. [CrossRef]
- Du DTM, Hellen N, Kane C, Terracciano CMN (2015) Action potential morphology of human induced pluripotent stem cell-derived cardiomyocytes does not predict cardiac chamber specificity and is dependent on cell density. Biophys J 108:1–4. [CrossRef]
- Bernardo BC, Weeks KL, Pretorius L, McMullen JR (2010) Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol Ther 128:191–227. [CrossRef]
- Wang B, Kit-Anan W, Terracciano C (2018) Many Cells Make Life Work—Multicellularity in Stem Cell-Based Cardiac Disease Modelling. Int J Mol Sci 19:3361. [CrossRef]
- Zhang J, Wilson GF, Soerens AG, et al. (2009) Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ Res 104:e30-41. [CrossRef]
- Zwi L, Caspi O, Arbel G, et al. (2009) Cardiomyocyte differentiation of human induced pluripotent stem cells. Circulation 120:1513–1523. [CrossRef]
- Hanft LM, McDonald KS (2010) Length dependence of force generation exhibit similarities between rat cardiac myocytes and skeletal muscle fibres. J Physiol 588:2891–2903. [CrossRef]
- Wheelwright M, Win Z, Mikkila JL, et al. (2018) Investigation of human iPSC-derived cardiac myocyte functional maturation by single cell traction force microscopy. PLoS One 13:e0194909. [CrossRef]
- Kamakura T, Makiyama T, Sasaki K, et al. (2013) Ultrastructural maturation of human-induced pluripotent stem cell-derived cardiomyocytes in a long-term culture. Circ J 77:1307–1314.
- Schick R, Mekies L, Shemer Y, et al. (2017) P3496Functional abnormalities in induced pluripotent stem cell-derived cardiomyocytes generated from titin-mutated dilated cardiomyopathy patients. Eur Heart J 38:1–25. [CrossRef]
- Dorn GW, Vega RB, Kelly DP (2015) Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev 29:1981–1991. [CrossRef]
- Dai D-F, Danoviz ME, Wiczer B, et al. (2017) Mitochondrial Maturation in Human Pluripotent Stem Cell Derived Cardiomyocytes. Stem Cells Int 2017:5153625. [CrossRef]
- Schaper J, Meiser E, Stammler G (1985) Ultrastructural morphometric analysis of myocardium from dogs, rats, hamsters, mice, and from human hearts. Circ Res 56:377–391.
- Lopaschuk GD, Spafford MA, Marsh DR (1991) Glycolysis is predominant source of myocardial ATP production immediately after birth. Am J Physiol 261:H1698-705. [CrossRef]
- Lundy SD, Zhu W-Z, Regnier M, Laflamme MA (2013) Structural and Functional Maturation of Cardiomyocytes Derived from Human Pluripotent Stem Cells. Stem Cells Dev 22:1991–2002. [CrossRef]

Table 1.
Available human induced pluripotent stem cell-derived cells used in disease modelling. CM = cardiomyocyte, EC = endothelial cell, RBC = red blood cell, SMC = smooth muscle cell.
Table 1.
Available human induced pluripotent stem cell-derived cells used in disease modelling. CM = cardiomyocyte, EC = endothelial cell, RBC = red blood cell, SMC = smooth muscle cell.
| Pathology | Cell type involved | Mutation | (Drug/Treatment) Test | Ref. |
|---|---|---|---|---|
| Cardiomyocytes | ||||
| Barth syndrome | CM | Tafazzin | Genetic rescue | [19] |
| Brugada syndrome | CM | SCN5A-1795insD mutation | Mexiletine | [20,21] |
| Catecholaminergic polymorphic ventricular tachycardia type 1 | CM | Ryanodine receptor 2 (RYR2) | Isoproterenol | [22,23] |
| Familial hypertrophic cardiomyopathy | CM | MYH7 Arg663His | Verapamil, diltiazem, mexiletine among 15 drugs | [24] |
| Hypoplastic left heart syndrome | CM | Patient-derived (GM12601) | Isoproterenol | [23,25] |
| Ischaemic heart damage | CM | Aldehyde dehydrogenase 2 (ALDH-2) deficiency | siRNA knockdown | [26] |
| LEOPARD syndrome | CM and 3 germ layers | PTPN11 | Rapamycin | [27] |
| LQT1,2,3,5,8,14 | CM | Patient-derived | Common drugs | [28,29,30,31,32,33] |
| Endothelial | ||||
| Healthy | EC | N/A | Flow-induced disease and simvastatin | [34,35] |
| Hutchison-Gilford Progeria Syndrome | EC | Patient-derived | N/A | [36] |
| Lymphocytes | ||||
| Healthy | B-cell lymphoid lineage | N/A | N/A | [37] |
| Red blood cells | ||||
| Health | CM and RBC | N/A | Toxicity of RBC | [38] |
| Sickle | RBC | Patient-derived | Biallelic correction | [39] |
| Smooth muscle cells | ||||
| Supravalvular aortic stenosis | SMC | Elastin (ELN) | Elastin recombinant protein | [40] |
| Marfan syndrome | SMC | FBN1 | Gene editing | [41] |
Table 2.
Structural, electrophysiological, and molecular marker differences between immature and mature cardiomyocytes.
Table 2.
Structural, electrophysiological, and molecular marker differences between immature and mature cardiomyocytes.
| Immature CMs | Mature CMs | ||
|---|---|---|---|
| Structure | |||
| Shape | Irregular | Rod-shaped | |
| Area | ∼ 480 µm2 | ∼ 1700 µm2 | |
| Volume | Small | Large | |
| Sarcomere organisation | Disorganised and less developed | Organised and M-line developed | |
| Mitochondrial population | Few | Abundant | |
| T-tubule organisation | Absent | Scarce in atrial, abundant in ventricular | |
| Glucose metabolism | High | Low | |
| Nucleus Morphology | Mononuclear | Mononuclear, binuclear, multinuclear | |
| Electrophysiology | |||
| Spontaneous activity | Very frequent | Absent | |
| Maximum diastolic potential | -60 mV | -70mV (atrial) to -80 mV (ventricular) | |
| Maximum upstroke velocity | 44–50 V/s | 200 V/s | |
| Action potential amplitude | 94–113 mV | 80–130 mV | |
| *Action potential duration at 50% | 60–130 ms | 200 ms (atrial), 200–300 ms (ventricular) | |
| *Action potential duration at 90% | 80–160 ms | 200–400 ms | |
| Force Generation | 100–150 Pa for a single cell | Myocardium tensile force ≈ 56 kPa | |
| Elastic modulus | 466 Pa | 22–55 kPa | |
| Molecular markers | |||
| Gap junction | Cx40 | - | + (atrial), − (ventricular) |
| Cx43 | + | + | |
| Cx45 | + | - | |
| Ion channel | KCNA5 | + | + (atrial), − (ventricular) |
| NCX1 | + | + | |
| SERCA2a | + | + | |
| RYR2 | + | + | |
| Cav 1.2 | + | + | |
| Kir 2.1 | + | + | |
| Kv 4.3 | + | + | |
| KChip 2 | + | + | |
| KCNH2 (HERG) | + | + | |
| Structural protein | TNNT2 | + | + |
| ACTN2 | + | + | |
| MLC2A | + | + | |
| MLC2V | + | - (atrial), + (ventricular) | |
| MYL2 | + | + | |
| MYH6 | + | + | |
| Master gene | NKX2.5 | + | ± |
* Action potential duration for human iPSC-CMs depends on seeding conditions [89].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



