
Submitted:
27 September 2024
Posted:
30 September 2024
You are already at the latest version
Abstract
Epilepsy is a brain disease characterized by a persistent predisposition to epileptic seizures. The current issue is the development of new methods of epilepsy therapy, In particular, using gene therapy methods. Promising areas of research in this field are the modulation of the expression of a number of neuropeptides, ion channels, transcription factors, neurotrophic factors, etc., as well as the use of antisense oligonucleotides. One of the optimal vectors for the delivery of therapeutic transgenes is the adenoassociated virus (AAV), which is most often used in the gene therapy of neurological disorders due to the high transduction ability of neuronal tissues and low immunogenicity/cytotoxicity. In this study, we analyze the existing achievements in the use of AAV vectors in the treatment of epilepsy of various etiologies.
Keywords:
adeno-associated virus
; epilepsy
; gene therapy
; neuropeptides
; ion channels
; receptors
; transcription factors
; neurotrophic factors
; antisense oligonucleotides
1. Introduction
Epilepsy is a brain disease, characterized by a persistent predisposition to the occurrence of seizures. It is diagnosed by at least two unprovoked seizures with an interval of > 24 hours [1]. Epilepsy syndrome is defined as a characteristic set of clinical and electroencephalographic (EEG) signs, often supported by specific etiological data (structural, genetic, metabolic, immune and infectious) [2]. According to the International League against Epilepsy (ILAE), epilepsy can be classified into the following types: structural, genetic, infectious, metabolic and immune [3]. However, epilepsy often falls into several categories. The area of origin of the attack in the brain is the basis for classifying epilepsy into focal, generalized and epilepsy with unspecified onset [4].
Regardless of the etiology and type of seizures, epilepsy is characterized by a constant predisposition to unprovoked seizures caused by an imbalance between mediators of excitation and inhibition of neuronal activity [5]. Studies of the genetic forms of epilepsy have revealed several pathogenic variants in various genes [6,7,8], most of which encode ion channel subunits, which helped to better study the pathogenesis of the disease, as well as identify potential targets for gene therapy. However, in some cases, especially for focal epilepsy, the presence of pathogenic variants is uncharacteristic [9,10]. Nevertheless, proteomic and/or transcriptomic profiling demonstrates aberrant expression of a number of gene transcripts and their products involved in the regulation of excitatory and inhibitory activity [11,12,13].
A characteristic feature, especially for structural forms of epilepsy, is that epileptic seizures are insufficiently stopped or not stopped at all with the help of antiepileptic drugs (AEDs). In general, the formation of pharmacoresistance to AEDs is typical in one-third of all patients [14,15]. These facts indicate the need to develop new therapies for epileptic disorders, in particular, advanced therapy medicinal products (ATMPs).
ATMP drugs are innovative, complex therapeutic agents that can potentially be used to treat various diseases, including cancer, neurodegenerative diseases (such as Huntington's and Parkinson's diseases), hereditary diseases and autoimmune diseases [16].
ATMPs are usually divided into three categories: 1) gene therapy (drugs for gene therapy); 2) somatic cell therapy [17]; 3) tissue engineering products, as well as any combination of the above. A detailed review of the methods of advanced therapy in the treatment of epilepsy is described in the work of Shaimardanova et al. [18,19].
To date, there are many studies devoted to the development of vector viral systems potentially used in the treatment of diseases of the central nervous system (CNS) [20]. Most of the efforts were aimed at developing vectors based on the following viruses: adenovirus, lentivirus, adenoassociated virus, herpes simplex virus [21]. Adenoviral vectors, despite their ability to transduce neuronal and glial cells, which is the main target in the treatment of CNS diseases [22], as well as the ability to carry a large transgenic insert [23], re less suitable for clinical use due to high levels of cellular toxicity [24,25]. According to research, the herpes simplex virus is also potentially toxic to cells [26], in addition, the production of viral particles based on it is a rather laborious process [27]. Lentivirus is a representative of the retrovirus class [28], and therefore has the ability to integrate into the genome, which can lead to an increase in the mutational load of the genome in cases of inappropriate embedding of genetic material [29,30].
Adenoassociated virus (AAV) is one of the most appealing viruses for developing vector systems, it is most often used in gene therapy for neurological disorders [31] due to the high transduction ability of neuronal tissues for a variety of serotypes and the absence of toxicity to cells [32]. Attempts to use AAV vectors in the treatment of neurological disorders have been made for a relatively long time [33]. In particular, for a number of disorders classified as lysosomal storage diseases [34], clinical trials were conducted using viral vectors [35] such as: Canavan disease [36,37]— a pediatric neurodegenerative disease associated with pathogenic variants in the aspartoacylase gene (ASPA); late infantile neuronal ceroid lipofuscinosis (LINCL) [38] and a number of other diseases, a detailed analysis of these materials can be found in a number of publications [39,40,41,42].
The introduction of a targeted gene using AAV to significantly reduce the frequency of seizures and relieve symptoms may be an effective strategy for the treatment of epilepsy. In this study, we analyze the existing achievements in the use of AAV vectors in the treatment of epilepsy of various etiologies.
2. Results
Cap ORF: encodes overlapping sequences of three capsid proteins, VP1, VP2 and VP3, and two accessory proteins, membrane-associated accessory protein (MAAP) and associated accessory protein (AAP), which start from the same p40 promoter [50]. In total, the icosahedral capsid includes about 60 monomers of VP proteins (VP1, VP2, VP3), in a ratio of 1:1:10, respectively [55].
All three VP proteins are translated from a single mRNA, which can be spliced in three ways: by excision of a long or short intron. The CAP gene also produces an additional nonstructural protein, AAP, whose precise function in capsid assembly is not yet clear [56]. The AAV-based expression vector must be co-transfected with a helper plasmid that mediates the expression of the Rep and Cap genes. In addition, specific adenovirus genes required for AAV replication must be delivered in trans [57].
2.1. Tropism and Transduction Capacity of AAV Vectors
A variety of naturally occurring AAV serotypes have been described in the literature [58], which, despite high sequence homology, differ in the surface properties of the capsid [43,59,60]. Combining AAV-based genomic constructs with Cap genes derived from different AAV serotypes leads to the possibility of creating so-called pseudotyped recombinant AAV (rAVV). Packaging systems for approximately 10 different serotypes are available for vector construction [61].
Efficient transduction depends on the interaction of capsid proteins with cellular receptors, which ensures endocytosis, transportation and nuclear internalization of viral particles [62]. Thus, a number of studies have described significant differences in the transduction efficiency of different AAV serotypes for specific tissues and cell types [63,64,65,66]. Therefore, efficient expression of the target gene in certain cells directly depends on the serotype of the virus used. In addition to tropism due to the AAV serotype, efficient expression of the target gene using AAV vectors can vary significantly, also depending on the type of expression cassette promoter used [67]. The effect of the promoter on tissue specificity and expression capacity was demonstrated in studies using the myelin basic protein promoter [68] and the glial fibrillary acidic protein (GFAP) promoter [69]. Often, cytomegalovirus (CMV), chicken β-actin (CBA), and cytomegalovirus/chicken beta-actin (CBh) promoters are used in the vector cassette [70]. In particular, AAV9 under the control of CBA demonstrated dominant neuronal transduction, while AAV under the control of a truncated CBA - CBh promoter shifted transduction towards oligodendrocytes [71]. In the work of Jackson et al. [72] a more specific neuronal transduction capacity of an AAV vector using the synapsin promoter was demonstrated, compared to the hybrid CBh promoter, which, despite higher expression, has tissue specificity outside the CNS.
Despite their low immunogenicity profile, AAV vectors can stimulate antiviral immune responses directed against the capsid and/or transgene, especially when delivered systemically or at higher vector doses [73]. Neutralizing antibodies (NAbs) against AAV can be produced following exposure to naturally occurring AAVs. An anti-capsid humoral immune response is triggered in patients receiving gene therapy, which subsequently leads to the neutralization of cross-reactive injected AAV [74,75]. As a result, NAb screening is a prerequisite before inclusion in AAV-based clinical trials, with high pre-existing titers being an exclusion parameter. This criterion immediately limits the widespread use of AAV for the treatment of diseases until further optimization creates the opportunity to evade NAb [76]. Immunomodulatory methods are currently being actively developed to reduce the immunogenicity of the AAV vector and ensure safe and repeatable interventions. Corticosteroids are often used in trials and help modulate immune-mediated toxicity and achieve long-term transgene expression [77].
To date, the most studied AAV serotype is AAV serotype 2 (AAV2). Vectors based on AAV serotype 2 are well suited for transferring target genes to the CNS, which has been demonstrated in the studies of Parkinson's disease [78] and Alzheimer's disease [79]. According to studies, AAV2 is capable of transducing postmitotic cells [80]. Also, according to Du et al., neurons are highly susceptible to this serotype [81]. Pathogenicity for humans has not been identified using AAV2, in addition, the virus is unable to replicate without the auxiliary functions provided by another virus, such as adenovirus or herpes simplex virus. rAAV2 vectors are less immunogenic than other viral vectors, since they do not express any viral genes and are not able to effectively transduce antigen-presenting cells after vector administration in vivo [82]. In animal models, rAAV2 vectors provide long-term stable expression of encoded transgenes in both the brain and retina [83,84].
Preclinical studies using rAAV2 vectors show that following intraparenchymal injection into the brain of animals, transduction occurs predominantly in neurons, with transduction of other cell types such as astrocytes and microglia being rare [63,69,83,85]. Using larger animals to assess transduction capacity, such as Felis catus [86] and primates [87,88], have also demonstrated predominantly neuronal transduction of AAV2. Resected human hippocampal tissue that had undergone a surgery for refractory temporal lobe epilepsy also showed a strictly neuronal tropism for this serotype [89]. However, one of the problems with using this serotype is that transgene expression following direct injection of the rAAV2 vector into a specific brain region is usually limited to the region around the injection needle site and, accordingly, the virus has a poor ability to spread to more distal regions [86,90]. The efficiency of transduction and expression capacity can be increased by additional administration of agents that facilitate binding of the viral capsid to the cellular receptor. For example, methods have been investigated for additional administration of heparin, which binds to heparan sulfate proteoglycan (HSPG), thereby increasing the efficiency of viral vector binding to CNS cells, promoting more successful transduction [91,92], fibroblast growth factor (FGF) has a high affinity for HSPG and can interact with heparin-like glycosaminoglycans (HLGAG) of the extracellular matrix (ECM), affecting the efficiency of viral vector binding and penetration [93], or mannitol [94,95], an osmotic agent that increases the permeability of the blood-brain barrier (BBB), thereby enhancing the distribution of AAV in the CNS [96], [97]. In particular, the delivery of AAV vectors to the brain of mice with prior administration of mannitol to increase BBB permeability was reported [98], which led to greater accumulation of the transgene. However, the use of additional agents is associated with a number of difficulties, for example, a potential risk of bleeding associated with heparin sulfate injection has been described [90]. Another problem with the use of AAV2 is the fact that 18–70% of the population already have neutralizing antibodies to AAV2 [99,100], which may reduce the efficiency of transduction and expression of this serotype vector rAAV2 in the CNS [101,102].
Other AAV serotypes have also been evaluated for their ability to transfer genes to the CNS. In particular, the work by Davidson et al. reported that rAAV vectors derived from serotypes 4 and 5 exhibited increased gene transfer efficiency and broader distribution of transduced cells throughout the brain following direct injection into the mouse striatum compared to serotype 2. Higher transduction capacity for AAV4 and AAV5 was indicated following intraventricular injection compared to AAV2. Intrastriatal injection showed the highest efficiency for AAV5, its expression, as well as the AAV4 expression, was stable, but lost 15 weeks after administration [103]. These findings were also supported by another study [104] showing that injection of AAV-5 into the cerebellar cortex of mice resulted in extensive dissemination of the virus beyond the injection site. The transduction profile of rAAV1 vectors has also been assessed following direct injection into the CNS of mice [63,105]and Felis catus [86], demonstrating higher transduction efficiency with this serotype than with rAAV2, as well as pronounced neurotropism. However, transduction profiles may differ depending on the target brain region [63]. Nabs that reduce AAV2 CNS transduction may be overcome using rAAV vectors based on AAV5, as it is the most non-homologous to AAV2 of all existing serotypes [101].
Evaluation of the in vivo transduction profiles of rAAV7 and rAAV8 vectors revealed higher transduction efficiency compared to rAAV2 vectors in many tissues, including liver and muscle tissues [106,107,108,109]. More recently, Harding et al. examined recombinant AAV vectors pseudotyped with viral capsids derived from AAV serotypes 7 and 8 and assessed their transduction capacity into mouse striatal cells in comparison with vectors pseudotyped with AAV serotypes 2, 5, and 6. The study showed that pseudotyped AAV7 and AAV8 vectors had increased transduction efficiency into the mouse CNS, in the rank order rAAV7 > rAAV8 > rAAV5 > rAAV2 = rAAV6, with all vectors demonstrating strong tropism for neuronal transduction. The obtained data demonstrate that rAAV vectors pseudotyped with capsids derived from AAV serotypes 7 and 8 can provide more efficient gene transfer into the CNS [110].
AAV9 is known to transduce approximately twice as many neurons as astrocytes throughout the adult rodent CNS. Co-administration of mannitol had only a moderate effect on CNS transduction, suggesting that AAV9 crosses the BBB via an active transport mechanism. However, when this approach was applied to young primates at the mid-dose tested in mice, it showed reduced transduction of peripheral organs and brain compared to mice, a clear shift towards predominantly glial transduction, and the presence of low levels of pre-existing NAbs largely blocked CNS and peripheral transduction [111].
Studies have shown differences in the transduction profiles of six commonly used AAV serotypes (AAV1, AAV2, AAV5, AAV6, AAV8, AAV9) in different regions of the mouse brain. Despite a common ability to transduce the main brain cell types, expression levels varied for different serotypes and cell types. Notably, rAAV8 was found to be particularly effective in transducing astrocytes, whereas rAAV9 was most suitable for transducing cortical neurons [111,112].
Epilepsy is characterized not only by aberrant functioning of neurons, but also by the extensive involvement of glial cells in the formation and persistence of the pathological process [113]. Therefore, one possible strategy may be to target not only neurons of the brain, but also glial tissues [114].
The route and site of vector administration play an important role in determining its transduction and expression capabilities. AAV9-egfp and AAV9-fLuc delivery via intrastriatal (IST), intracisternal (ICM) and lumbar intrathecal (LIT) routes was evaluated in adult rats. Results showed that IST administration provided robust transgene expression in the striatum, thalamus and cortex with lower transduction in peripheral tissues compared to ICM or LIT administration. ICM administration provided strong expression in more brain regions and similar expression in the spinal cord compared to LIT administration. LIT administration showed lower expression in the brain compared to ICM administration. A similar study was conducted for AAV5. Results showed that IST administration provided centralized localized vector distribution and expression in the frontal part of the brain. Intrathalamic injection demonstrated transduction and gradient expression from the rostral brain to the lumbar spinal cord. Intracerebroventricular administration resulted in more uniform, albeit relatively superficially distributed, transduction and expression throughout the central nervous system. Thus, the choice of route and site of vector administration significantly influences its efficacy and expression distribution, which must be taken into account when developing gene therapies and other biomedical applications [115].
A study by Foust et al. described intravenous delivery of AAV9 to mice, which effectively affects the brain, dorsal root ganglia, and spinal motor neurons in neonatal animals and astrocytes of the brain and spinal cord of adult mice. This study demonstrated a direct dependence of AAV9 tropism on the age of the animal injected with the vector: widespread transduction of neurons, motor neurons in neonatal mice in particular, and extensive transduction of astrocytes in adult mice after intravenous delivery [116]. The ability of AAV9 to penetrate the BBB has also been demonstrated in a number of other studies [111,117]. Directed evolution of this serotype led to the emergence of AAV-PHP.B, which also penetrates the BBB [118]. High potency of the recombinant serotype was demonstrated in preclinical studies in a synucleinopathy model [119]. A drug was also approved in the European Union and the UK in 2022 for the correction of aromatic L-amino acid decarboxylase (AADC) deficiency using AAV vector therapy [120]. To date, several AAV gene therapies have been approved worldwide. The first AAV2-based drug is Glybera, approved by the European Commission in 2012 for the treatment of lipoprotein lipase deficiency [121]. One of the commercially successful drugs is a drug for the treatment of spinal muscular atrophy (SMA). It is a vector based on the AAV9 virus. Intravenous administration of the drug has been shown to improve motor skills in patients and reduce clinical manifestations of the disease [122]. There is also a gene therapy product Hemgenix, which was approved in 2022 by the Food and Drug Administration (FDA) and is a recombinant AAV5 containing a highly active Paduan variant of the human factor 9 (F9) gene with a codon-optimized nucleotide sequence [123].
Thus, the development of AAV vectors for delivery of genetic material to the epileptic brain must take into account a number of factors, including the type of cells and regions of the brain, the serotype of the virus used, the selection of an appropriate promoter, and the route of administration. Although the data on efficient transduction of brain tissue are encouraging, the tropism and efficiency of the vector may be significantly reduced when used in animals evolutionarily closer to humans. Vector tropism may vary depending on the type and site of administration, as well as the physiological characteristics of the recipient. Although there is no data on the pathogenicity of AAV, its efficiency may be reduced by the presence of antibodies, as well as by improper capsid construction or promoter use.
The ability of AAV to transfer and induce expression of the lacZ marker gene in brain slices from patients undergoing temporal lobectomy was assessed. The AAV-lacZ vector was introduced into brain slices and incubated for 24 hours. Expression of the lacZ gene was observed after 5 hours and was maintained until the end of the experiment, predominantly in neurons, without signs of cytotoxicity. The results confirm the efficiency of AAV in gene transfer in the human CNS. Replacement of the lacZ gene with a functional gene may allow localized genetic intervention in focal seizures using stereotactic or endovascular delivery [89].
Development of a suitable AAV vector for the treatment of epilepsy is a complex and multi-step task that requires consideration of all the above factors. These components are essential for efficient transduction of brain tissue and successful treatment of epilepsy using AAV gene therapy. The components for the effective use of AAV vectors are summarized in Figure 1.
2.2. Pathogenesis of Epilepsy
The mechanism of epilepsy development consists of the formation of an epileptic focus, the formation of epileptic systems in the brain, and brain epileptization. Seizures occur due to under- or over-activation of neurons, and this results in the inability of the brain to coordinate the rest of the human body. Epilepsy can affect one area of the body (focal epilepsy) or the entire body (generalized epilepsy), depending on the location of the affected neurons in the brain [124]. Epilepsy occurs due to genetic susceptibility, development of the nervous system, cerebrovascular factors, and other acquired factors that irritate nervous tissue. In addition, it can occur due to neoplasms, metabolic or neurodegenerative disorders, especially among the elderly.
The molecular mechanisms of epileptogenesis are complex and not fully understood, but are thought to involve an imbalance between excitatory (glutamate, aspartate) and inhibitory (gamma-aminobutyric acid (GABA), taurine, glycine, norepinephrine, dopamine, serotonin) neurotransmitters. A seizure occurs when there is a decrease in inhibitory signaling such as GABA or an increase in excitatory signaling such as glutamate [125,126]. The accumulation of glutamate leads to the degeneration of glutamate receptors, activation of Na+ and Ca2+ channels, accumulation of Na+ and Ca2+ ions inside the cell, and K+ ions in the extracellular fluid. This in turn promotes the release of Ca2+ from the intracellular depot and the activation of enzymes (phospholipase, protease, etc.), the accumulation of arachidonic acid, increased lipid peroxidation and the destruction of cell membranes [127]. In contrast, GABA-A receptors (ligand-dependent cl- ion channels) mediate fast inhibitory presynaptic potentials by increasing chloride influx, and GABA-B receptors (G-protein-coupled receptors) mediate slow inhibitory presynaptic potentials by increasing potassium conductivity and decreasing calcium entry [128]. It is assumed that a decrease or loss of GABAergic inhibition may increase the probability of generating excitatory postsynaptic potentials and synchronization volley discharges, and, consequently, cause epileptogenesis [129].
Other causes of epileptic seizures are changes in ion concentrations and dysfunction of ion channels - channelopathies. Ion channels are involved in the generation of electrical currents through ion charges, cation channels mainly generate action potentials and contribute to neuronal excitability, on the other hand anion channels are involved in the mechanism of inhibition of the neuronal excitatory process, and thus ion imbalance can cause epileptogenesis [130]. Mutations in genes expressing potassium, sodium, chloride, calcium channels and acetylcholine and GABA receptors have been reported in epilepsy [131,132]. In addition, channelopathies can also be the pathogenesis of acquired epilepsy due to secondary changes in ion channels through transcriptional and post-translational mechanisms [132].
Inflammation and impaired immune regulation may also play a role in triggering an epileptic seizure. Inflammatory cells release molecules that can alter neuronal signaling, which may lead to seizures. Following seizures, cytokines such as interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF-α) are released, modulating inflammatory responses in the brain. Studies show that these cytokines influence N-Methyl-D-aspartic acid (NMDA) receptors, synaptic plasticity, GABAergic neurotransmission, and neuronal excitability, contributing to seizure development and recurrence [133]. During the epileptic phase, studies of synaptic protein expression, brain inflammation, and hippocampal neurogenesis in adult synapsin 2 null mice showed elevated levels of IL-6 and TNF-α [134]. Currently, CNS inflammation caused by BBB permeability is associated with the induction and progression of epilepsy.
2.3. Gain of Function Using an AAV Vector in the Treatment of Epileptic Disorders
One of the key directions in the treatment of epilepsy is gene therapy, which is a promising method for correcting the deficiency or absence of a gene in the body, as well as enhancing its expression to increase the production of the necessary protein. As part of gene therapy, researchers are exploring various strategies for delivering genetic material aimed at restoring normal cell function and reducing the frequency of epileptic seizures. This can be accomplished through the introduction of specific vectors aimed at modulating the operation of ion channels or other molecular components responsible for regulating neuronal excitability. In addition, methods for delivering neuropeptides and neurotrophic factors that can modulate neuronal activity and promote neuronal survival are being actively studied, as well as antisense oligonucleotides, leading to the repression of target genes.
One approach to reducing the frequency and severity of epileptic seizures is to deliver genetic material capable of correcting the lost and/or reduced function of a particular protein product to a cell. The genetic material delivered by the vector construct can encode various functional products, in particular: neuropeptides, ion channel subunits, cellular receptors, transcription factors, etc.
2.3.1. Neuropeptides
Most of the studies are aimed at modulating seizure activity by modifying the level of neuropeptides in neuronal tissue. Neuropeptides are messenger molecules that mediate neuronal communication and intercellular signaling in association with neurotransmitters [135]. Indeed, it is known that neuropeptides can regulate inhibition and excitatory processes by repressing the release of glutamate induced by membrane depolarization, which provides an antiepileptic effect [136,137,138].
In existing studies aimed at modulating seizure activity using the AAV vector cassette, neuropeptide Y (NPY) was most often delivered. A study using rAAV2-NPY was conducted on rats exposed to kainic acid. The vector was injected into the rat hippocampus in a volume of 3 μl at a concentration of 1.06 × 10¹² particles/ml. NPY overexpression was restricted to the hippocampus and was observed only in neurons. Peak expression was observed at 2 weeks and persisted for at least 3 months. Immunohistochemical staining revealed NPY immunoreactivity in the molecular layer of the hippocampus, but not in CA1 and CA3 pyramidal cells or granule neurons. NPY mRNA expression was also observed in interneurons and granule cells, as well as in CA1-CA3 pyramidal cells. Applying this viral vector resulted in a reduction in seizure frequency, meanwhile transduction with the AAV1/2 serotype showed a greater reduction in discrete seizures [139]. In the work of Noe et al. A chimeric rAAV1/2 vector (expressing both serotype 1 and 2 capsid proteins, with an AAV1, AAV2 ratio of 1:1) expressing the NPY gene was applied to a model of progressive and spontaneous seizures of temporal lobe epilepsy induced by electrical stimulation of the temporal pole of the hippocampus. 3 μl of rAAV1/2-NPY with a concentration of 5.2 × 10¹² particles/ml were injected into 4 areas of the hippocampus of rats with early epilepsy, additionally 0.5 μl of heparin was administered. As a result, the introduction of rAAV1/2-NPY into the rat brain led to a significant decrease in seizure progression compared to the control groups. The effect of reducing seizure progression correlated with an increase in NPY expression in the hippocampus. The frequency of spontaneous seizures was significantly reduced in 40% of experimental animals compared to the baseline level before injection [140]. Furthermore, with intrahippocampal administration of rAAV1-NPY with the CBA promoter, a significantly better anticonvulsant effect was shown compared to AAV1/2 [141].
Studies have also shown that in addition to NPY, its receptors (Y2) play an important role in the antiepileptic action [142,143,144]. In addition, it was noted that in resected epileptic tissues of patients with epilepsy [145] and rodents [146], a decrease in the expression of Y1 receptors is observed, while the expression of Y2 receptors increases. These data suggest an increase in the effectiveness of the antiepileptic effect of NPY, which is an agonist of the Y2 receptor. In fact, intracerebral delivery of the agonist NPY13-36 with affinity for the Y2 receptor reduces the predisposition to epileptic seizures after systemic administration of kainic acid [147]. The truncated neuropeptide fragment - NPY13-36 is the C-terminal peptide fragment of NPY, which primarily activates the Y2 receptor, which mediates anticonvulsant activity. When AAV2-NPY or AAV2-NPY13-3 was injected into the piriform cortex of rats, no seizures were observed after kainic acid administration, only control animals developed seizures for 90 minutes. 3/7 rats receiving AAV2-NPY and 1/7 rats receiving AAV2-NPY13-36 developed seizures during the entire observation period [148].
Additionally, studies in rats using rAAV vectors aimed at overexpression of the Y2 receptor in the hippocampus have demonstrated seizure suppression effects. Combined overexpression of Y2 and NPY showed a more pronounced effect in reducing seizure activity [149].
Gene therapy with AAV1/2-NPY/Y2 in a post-intrahippocampal rat model of chronic epilepsy induced by kainic acid demonstrated not only the prevention of the progressive increase in seizure frequency in treated animals compared to controls, but even a 45% reduction in seizure frequency in 80% of epileptic animals [150].
It was also tested whether delivery of the human NPY gene to the thalamus or somatosensory cortex of rats using chimeric AAV1/2 could induce sustained seizure suppression in rats with genetic absence epilepsy. Three cohorts of rats were injected with rAAV-NPY bilaterally into the thalamus or somatosensory cortex. Vector-mediated overexpression of NPY in the thalamus and somatosensory cortex of the genetic absence epilepsy rat from Strasbourg (GAERS) led to a significant reduction in time of convulsive activity and number of seizures, however, a reduction in seizure duration was only seen when the vector was administered into the thalamus. Expression of both human and rat NPY receptor was significantly increased in the somatosensory cortex [151].
More recent studies have also explored the seizure suppression potential of combinatorial gene therapy using AAV1-based vector carrying the NPY gene and the Y2 receptor in rats. A preliminary study of dose-effect correlation was conducted in a model of systemic acute seizure induced by kainic acid, which revealed the optimal vector titer of 1 μl in 3 hippocampal sites at a concentration of 1012 particles/ml. The vector construct was then administered intrahippocampally in a model of spontaneous recurrent seizures (SRS). The response rate to therapy was 31% (more than 50% reduction in SRS frequency) and the seizure freedom rate was 13%, while no such effects were observed in control animals. An increase in the intervals between SRS and a decrease in the duration of SRS themselves were also observed in the experimental group [152]. Wickham et al. infused AAV-NPY vector into resected hippocampal tissue slices in vitro for 48 h. Whole-cell patch-clamp analysis of the dentate gyrus (DG) preparation of the hippocampus demonstrated a strong inhibitory effect of NPY on epileptiform activity. In particular, a decrease in the number of paroxysmal depolarizing shifts and action potentials was recorded, which was mediated by Y2 receptors, since the use of a selective Y2 antagonist blocked the action of NPY [153].
The internal ribosome entry site (IRES) sequence separating two gene sequences, such as those for the NPY and Y2 receptor, reduces translation of the downstream gene, which may also have implications for the potential anticonvulsant effect. Thus, a comparative analysis of three serotypes (AAV1, AAV2, and AAV8) and two transgenes, NPY-IRES-Y2 and Y2-IRES-NPY, in a rat model of acutely induced seizures showed that AAV1-NPY-IRES-Y2 was more effective than other serotypes or transgene sequences, considering both transgene expression and the ability to suppress induced seizures in rats. The vector also demonstrated a transgene-induced decrease in glutamate release from excitatory neuron terminals and significantly increased NPY and Y2 expression in resected human hippocampal tissue from patients with drug-resistant temporal lobe epilepsy [154].
In particular, the combined induction of neuropeptides and their receptors can influence the influx of Ca2+, through a decrease in the opening of voltage-dependent calcium channels [155]. Galanin (GAL), for example, is a modulator of neurotransmitter levels in the CNS and peripheral nervous system (PNS) [156] and has been shown to hyperpolarize brain neurons through the opening of K+ channels [157,158], as well as through inhibition of adenylate cyclase activity [159], inhibition of voltage-dependent Ca2+ channels [160], and regulation of the release of dopamine [161], acetylcholine [162], and glutamate [163].
In the case of NPY, there is evidence that prolonged and persistent epileptic activity can lead to depletion of neuropeptide-storing vesicles, which contributes to a decrease in the release of this neuropeptide [164]. The seizure threshold is reduced in preprodynorphin (pDyn) knockout mice, leading to an increased susceptibility to developing epilepsy [165]. Similarly, low dynorphin levels in humans are correlated with increased vulnerability to this disease [166,167]. Therefore, the introduction of transgene copies is a relevant therapeutic strategy for reducing seizure activity.
The excitatory inhibitory effect of neuropeptides has been demonstrated in a number of studies, in particular: GAL is able to inhibit the release of acetylcholine in the striatum of the brain and hypothalamus [168], and GAL is also able to repress the release of glutamate and aspartate in the hippocampus [163]. Ultimately, efforts to use neuropeptides in the treatment of epilepsy are aimed at reducing the excitatory activity of brain neurons.
In the work of Haberman et al., cells were transduced in vitro and in vivo with an AAV vector carrying the gene for the galanin neuropeptide GAL (AAV-GAL) and also carrying the GAL gene and the fibronectin secretory signal sequence FIB (AAV-FIB-GAL), which promotes the secretion of galanin from the cells. In vitro, HEK293 cells transfected with AAV-FIB-GAL demonstrated significant galanin content in the medium 24 h after transfection (32 ng/ml), whereas cells transfected with AAV-GAL showed no evidence of secreted galanin [169]. For the in vivo study, a rat model of focal seizures in the lower collicular cortex induced by brief electrical stimulation was used. As a result of the introduction of AAV-FIB-GAL, an increase in the seizure stimulation threshold was observed after 4 weeks. A protective effect was also found after injection of AAV-FIB-GAL into the hilar region of the hippocampus in the kainic model of epilepsy, resulting in the preservation of neurons at a distance of up to 500 μm from the infusion site. Real-time polymerase chain reaction (RT-PCR) demonstrated the stability of galanin expression 4 months after injection of AAV-FIB-GAL into the hippocampus. The work of Lin et al. confirms these results. Administration of an AAV2-GAL-based vector also led to a decrease in the duration of seizure activity in the rats’ injected hippocampus, where galanin was detected in granule cells and DG interneurons of the hippocampus [170]. AAV-FIB-GAL was also infused laterally into the piriform cortex of rats (2 μl, 8 × 1012 particles/ml). Following the infusion, experimental rats received a 10 mg/kg dose of kainic acid. Bilateral infusion of AAV-FIB-GAL significantly attenuated kainic acid-induced seizures, so that 11 of 12 rats did not exhibit limbic seizures and one rat exhibited only a brief, single class III seizure. AAV-FIB-GAL infusion prevented electrographic seizure activity. In contrast, bilateral infusion of AAV-FIB-GFP did not alter either behavioral or electrographic seizure activity. Since prior seizure exposure could affect the efficiency of the transporter, another group of rats received daily electrical stimulation of the piriform cortex until three consecutive class V seizures occurred. AAV-FIB-GAL or AAV-FIB-GFP (3 μl/30 min) were then injected into the electrode site. After one week, AAV-FIB-GAL rats showed a significant increase in the stimulation current required to elicit limbic seizure activity, while AAV-FIB-GFP did not alter the seizure threshold. Thus, AAV-mediated galanin expression and secretion significantly suppresses limbic seizure activity in vivo [171].
Gene therapy with AAV encoding preprodynorphin (pDyn) has been shown to suppress seizures for several months in mice and rats with an epileptogenic model of temporal lobe epilepsy. Preprodynorphin is a precursor of dynorphins. Dynorphins are a family of endogenous opioids stored in vesicles that are recognized as natural anticonvulsants [172]. AAV1-pDyn and AAV1-GFP at a dose of 2 × 109 genomic particles (gp) were administered into the epileptogenic lesion approximately 1 month after KA injection, when focal epilepsy developed. Delivery of AAV1-pDyn caused a gradual decrease in the frequency of generalized seizures. Generalized seizures completely disappeared within 1 week, and no further seizures were observed during the entire observation period (3 months). In contrast, animals treated with AAV1-GFP continued to have seizures throughout the observation period. To demonstrate that Dyn’s action is mediated by kappa opioid receptors, mice were injected with the KOR antagonist norBNI (20 mg/kg) 30 days after AAV1-pDyn delivery, at which time seizures had disappeared. Drug treatment resulted in a transient resumption of seizures, which disappeared when the antagonist was washed out.
2.3.2. Ion Channels, Receptors and Membrane Proteins
Another direction is the delivery of ion channel subunits or membrane receptors designed to compensate for insufficient function or reduce excessive function. Dysfunction of ion channels can promote the propagation of excitation and/or the disruption of inhibitory processes, which ultimately lead to the occurrence of seizure activity [173]. Work in this orientation can go both in the direction of enhancing the function and in the direction of decreasing it.
Ion channels are transmembrane proteins that are selective pores for ions that regulates neuronal excitability [173]. Induction of ion channel function, as well as specific cellular receptors, has also been one of the goals in a number of studies. In particular, a decrease in the function of the local NMDA receptor can reduce sensitivity to epileptic seizures [174]. Oral vaccines are taken up by intestinal M cells, are rapidly taken up by antigen-presenting cells (APCs) in Peyer's patches and lamina propria, and can induce potent humoral immune responses. Use of an oral AAV vaccine targeting the NR1 subunit of the NMDA receptor resulted in the production of an autoantibody and a potent humoral response to NR1, which contributed to potent antiepileptic and neuroprotective activity in both the KA model of epilepsy and the stroke model of rats [175].
The SCN1A gene encodes the alpha subunit of the voltage-gated sodium channel type I (NaV1.1) [176]. NaV1.1 is predominantly expressed in the axon initial segment of GABAergic inhibitory interneurons, where it generates and propagates action potentials [177]. Genetic reduction of NaV1.1 significantly reduces the frequency and amplitude of action potentials generated by GABAergic inhibitory interneurons, thereby impairing their inhibitory function [178,179,180].
In an analysis of the therapeutic efficacy of SCN1A gene regulation using AAV9, AAV9-REGABA-eTFSCN1A was designed to activate the SCN1A gene from the endogenous genome since the SCN1A gene exceeds the packaging capacity of AAV, which expresses an engineered transcription factor (eTF SCN1A) designed to upregulate the SCN1A gene from the endogenous genome. A cell-selective regulatory element (RE GABA) was included to target transgene expression specifically to GABAergic inhibitory interneurons. A single bilateral intracerebroventricular administration of AAV9-REGABA-eTFSCN1A to experimental mice at postnatal day 1 resulted in an increase in SCN1A mRNA transcripts, especially in GABAergic inhibitory interneurons, and NaV1.1 protein levels in the brain. A significant reduction in spontaneous and hyperthermic seizure frequency, as well as an increase in survival, was demonstrated. In non-human primates (NHPs), delivery of AAV9-REGABA-eTFSCN1A via unilateral intracerebral injection resulted in widespread biodistribution of the vector and transgene expression throughout the brain, including key structures involved in epilepsy such as the hippocampus and cerebral cortex, with no adverse effects, no noticeable changes in clinical observations, no adverse histopathological findings, and no dorsal root ganglia-related toxicity [181].
At the same time, enhancing the function of certain types of receptors and ion channels can also lead to a reduction in seizure activity. For example, the endocannabinoid system is considered a therapeutic target in epilepsy [182], and therefore effective treatment strategies that exploit CB1 receptor regulation require a detailed understanding of the effects of the CB1 receptor in neuronal subtypes. To this end, analysis of conditional mutant mice lacking CB1 receptors on different neuronal subtypes subjected to kainic acid (KA)-induced seizures showed that CB1 receptors on glutamatergic, but not GABAergic, hippocampal neurons are required for protection against excitotoxic seizures [183]. In line with these preclinical data, specific reductions in CB1 receptor protein and mRNA levels at glutamatergic but not GABAergic axon terminals have been reported in human epileptic hippocampal tissue [184]. However, these conditional loss-of-function studies have not yet been complemented by a corresponding gain-of-function approach involving CB1 overexpression, thereby precluding a full picture of CB1-mediated control of hyperexcitability. Also, potassium channels are considered promising tools for gene therapy due to their remarkable ability to hyperpolarize neurons and thus suppress their activity [185]. Most genetic approaches to suppress seizures utilize the expression of potassium channels or other potassium conductance-related proteins [186,187,188,189]. Moreover, many forms of epilepsy are known to be correlated with loss of potassium channel function [190,191,192,193,194]. Voltage-gated Kv1 channels are involved in the generation of action potentials (APs) [195,196].. The duration of APs controls the amplitude of evoked postsynaptic potentials [197,198], which affects downstream signal transmission.
Studies in humans with temporal lobe epilepsy (TLE) and in rodent models of TLE have found decreased expression of the GABA receptor GABR1 subunit and increased expression of GABR4 subunits in the DG of humans with epilepsy [199]. These changes begin within 24 h of pilocarpine-induced status epilepticus (SE) in adult rats and persist for several months as these animals gradually develop epilepsy [200]. Rats that experienced pilocarpine-induced SE on postnatal day 10 had increased GABR1 expression in the DG and enhanced benzodiazepine-induced increases in GABA type I currents, and none developed epilepsy later in life [201].
Raol et al. used AAV5 containing GABRA4 promoter to transduce the DG after induced status epilepticus (SE). They detected an increase in GABRA4 subunit mRNA and protein expression in the DG 1–2 weeks after SE. Increased GABRA4 expression in the DG resulted in a three-fold increase in the mean seizure-free time after induced SE and a 60% decrease in the number of rats that developed epilepsy (recurrent spontaneous seizures) in the first 4 weeks after SE [202].
Nikitin et al. [203] reported an AAV vector designed to reduce epileptiform activity by expressing the human CA2 +- gated K+ channel KCNN4 gene, a calcium-activated potassium channel, KCa3.1, to target virus expression to glutamatergic excitatory pyramidal neurons, which make up the majority of the cerebral cortex. AAV2-KCNN4-Venus, driven by the CaMKII promoter, was injected into adult mouse brains (2 × 1010 vg/μl). Results showed that KCNN4-transduced cells exhibited Ca2+-dependent slow afterhyperpolarization, which significantly reduced the ability of KCNN4-positive neurons to generate high-frequency spike trains without affecting their low-frequency coding capacity and action potential shape. Antiepileptic activity tests showed potent suppression of pharmacologically induced seizures in vitro at both single cell and local field levels, with a reduction in peaks during ictal discharges.
AAV1/2-CB1 was injected into the hippocampus of adult mice to evaluate the cannabinoid receptor CB1, the most abundant G protein-coupled receptor in the brain and a key regulator of neuronal excitability, and to investigate the effects of increasing the CB1 gene dosage in the hippocampus on the development of epileptiform seizures and neuronal injury in the kaine epilepsy model. Transgene expression was restricted exclusively to Cre-recombinase-expressing principal neurons using an AAV expression cassette with a transcription stop cassette flanked by loxP sites preceding the transgene. Collectively, it has been demonstrated that ectopic CB1 receptor is highly expressed by cell type, particularly in hippocampal pyramidal neurons, localized to presynaptic sites and coupled to G proteins. The severity and mortality of KA-induced seizures were reduced in CB1 receptor overexpressors compared to AAV-treated controls. Neuronal damage in the CA3 region of the hippocampus is notably absent in AAV-treated Cre transgenic animals but is evident in all cortical regions of both treatment groups [204].
Neuroligins 2 (NL2) is selectively found at inhibitory synapses. the interaction between presynaptic neurexin and postsynaptic NL2 enhances inhibitory synaptic transmission. NL2 binds GABA receptors in the scaffold in the postsynaptic neuron through molecular interactions with gephyrin and collybistin, and global induction of NL2 may alleviate generalized seizures. Application of AAV9 to express NL2 resulted in a significant reduction in seizure duration, severity, and frequency in an epilepsy mouse model. Exogenous NL2 was expressed widely in the brain and co-localized with the postsynaptic inhibitory molecule gephyrin [205].
Collectively, these studies demonstrate the high potential of gene therapy using different vectors and genes to effectively control epileptic seizures. These approaches open new horizons in the development of personalized and targeted therapies for epilepsy, providing significant improvement in the quality of life of patients.
2.3.3. Neurotrophic and Transcription Factors
Neurotrophic factors are peptides or small proteins involved in the growth, differentiation, development and survival of neurons. Brain-derived neurotrophic factors are involved in the pathogenesis of epilepsy and may have a neuroprotective effect [206]. Overexpression of rAAV2-based glial cell-derived neurotrophic factor (GDNF) in the rat hippocampus has been shown to suppress seizures in two models of temporal lobe epilepsy [207]. In the case of GDNF, upregulation of GDNF messenger RNA and protein have been demonstrated in the granule cell layer (GC) of the DG and hilus, as well as in the pyramidal layer of the hippocampal cornu ammonis (CA1–CA3) regions. After induction of status epilepticus with kainic acid [208,209] and lithium-pilocarpine [210], suggesting the involvement of GDNF in the epileptogenic process. Most importantly, KA-induced generalized tonic-clonic seizures [211] and kindling-induced epileptogenesis are suppressed by intraventricular infusion of GDNF [212]. Based on these data, it could be hypothesized that GDNF gene transfer to the epileptogenic focus would have an inhibitory effect on both epileptogenesis and the occurrence of generalized seizures. In line with the latter concept, adenoviral vector-mediated overexpression of GDNF in the hippocampus has been shown to suppress KA-induced generalized tonic-clonic seizures [213].
Also, the delivery of transcription factors seems promising. For example, Nrf2 regulates the expression of a number of genes encoding detoxifying, antioxidant, anti-inflammatory mediators, calcium homeostasis regulators, and signaling molecules, which leads to an organized protective response. Genes predominantly regulated by Nrf2 include those encoding heme oxygenase-1 (HO-1), NAD(P)H quinone oxidoreductase 1 (NQO1), microsomal glutathione S-transferase (MGST), glutathione peroxidase, and superoxide dismutase [214,215,216], which may provide a cascade of protective events.
A study by Mazzuferi demonstrated that overexpression of the transcription factor Nrf2, which promotes the expression of numerous antioxidant, anti-inflammatory, and neuroprotective proteins, using AAV can reduce the incidence of spontaneous recurrent seizures (SRS) and preserve neurons in the hippocampus. Following administration of AAV-Nrf2 into the hippocampus of mice, mRNA levels of Nrf2 and related genes (HO-1, NQO1, and MGST) increased, peaked 72 hours after the seizure episode, and then decreased. Mice receiving AAV-Nrf2 showed a significant reduction in the number of generalized seizures and decreased microglial activation, while the number of neurons was preserved and the number of astrocytes was not affected [217].
2.3.4. Other Transgenes for Delivery
In addition to neuropeptides and channels, other genes have been investigated for targeting delivery to the CNS. A study by Klugmann et al. examined the role of overexpressing different domains of the Homer 1 protein in the hippocampus of adult rats using rAAV. Homer 1c has both a ligand-binding domain and a coiled-coil domain for self-multimerization, but Homer 1a lacks the coiled-coil domain. In this study, a new isoform, 1g, which lacks the ligand-binding domain of Homer, was analyzed. AAV vectors were generated expressing Homer 1a, 1c, and 1g under the control of the CBA promoter. The viral vectors were injected into the rat hippocampus. Histological analysis revealed no gross brain abnormalities 4 weeks after vector injection. To evaluate the effect of AAV-Homer 1a overexpression, a model of self-sustaining limbic status epilepticus (SSLSE) induced by focal hippocampal stimulation was used. In rats treated with AAV-Homer 1a, SSLSE was strongly suppressed, in contrast to Homer 1c, Homer 1g, and control groups, where SSLSE was successfully induced. During SSLSE, control and Homer 1c and 1g rats exhibited intense motor seizure activity, including clonic seizures and forelimb raising [218].
Another way to reduce excitatory activity and excitotoxicity is to deliver genes responsible for glutamate metabolism. Glutamate transporters are a family of neurotransmitter carrier proteins that transfer glutamate, the major excitatory neurotransmitter, across the membrane. Transporters play an important role in regulating the concentration of glutamate in the extracellular space by transporting it along with other ions across cell membranes [219]. Changes in the expression of excitatory amino acid transporter 2 (EAAT2) and glutamine synthetase (GS) have been detected in sclerotic hippocampal tissue removed during epilepsy surgery [220,221,222,223], and the finding that elevated extracellular glutamate levels appear to trigger spontaneous seizures in humans with TLE [224], have provided some impetus to determine whether these proteins would be suitable targets for gene therapy. However, whether glutamate transporters are altered in human epilepsy remains controversial; two studies found no changes in transporter expression in resected tissue from TLE patients [225,226], whereas region-specific changes in EAAT1-3 expression have also been reported [221,223,225].
GS in the brain is involved in metabolic regulation of glutamate, brain ammonia detoxification, ammonia assimilation, neurotransmitter recycling, and neurotransmitter signaling termination [227,228]. GS in the brain is found predominantly in astrocytes [229]. Astrocytes protect neurons from excitotoxicity by scavenging excess ammonia and glutamate [228]. GS expression in astrocytes is also reduced in resected epileptic tissue [223,225].
In another study, AAV-mediated overexpression of GS, EAAT2, or expression of adenosine kinase-targeting microRNA (miR-ADK) under the control of the glial GFAP promoter modulated susceptibility to kainate-induced seizures and neuronal cell loss in rat hippocampal astrocytes. After injection of AAV9 vectors into the rat hippocampus, transgene expression was detected after 3 weeks, predominantly in astrocytes. ADK expression in miR-ADK vector-loaded rats was reduced by 94–96%, which was accompanied by a ~50% reduction in kainate-induced seizure duration and better protection of dentate neurons, but not CA3 neurons, compared with controls. In contrast, infusion of AAV9-GS and AAV9-EAAT2 vectors did not provide protection against seizures or neuronal injury due to low transcriptional activity of the GFAP promoter, which did not result in a significant increase in transgenic GS or EAAT2 levels [230].
Data on AVV-mediated delivery of therapeutic genes are compiled in Table 1.
2.4. Loss of Function Using AAV Vectors
A less used, but nonetheless effective approach in gene therapy is the delivery or expression of antisense sequences. Traditionally, antisense oligonucleotides have been employed, but specific genes can also be targeted using antisense RNAs inserted into an AAV vector cassette [231]. Antisense oligonucleotides are synthetic single-stranded nucleic acid sequence that bind to RNA and thereby alter or reduce the expression of the target RNA. They can not only reduce the expression of mutant proteins by cleaving the target transcript, but also restore protein expression or modify proteins by interfering with pre-mRNA splicing [232].
GABA is the major inhibitory neurotransmitter in the brain. In the inferior colliculus and other brain regions, GABA receptors directly modulate seizure susceptibility. Increased GABA-A receptor activity attenuates seizure genesis, while blockade of GABA-A receptor function increases seizure susceptibility. Thus, an AAV vector with a CMV promoter and truncated human GABA-A-α1 cDNA in both sense and antisense orientations was constructed. Following microinjection of AAV-GABA-A-α1 sense vectors at a dose of 3 × 109 particles/μl, neurons exhibited GABA-A-α-like immunoreactivity significantly exceeding endogenous concentrations after 7 days. Infusion of antisense vector (3 × 108 particles/μl) resulted in increased seizure duration and decreased [3H]zolpidem binding, which may affect seizure susceptibility in vivo [233].
In contrast, reduction of local N-methyl-d-aspartic acid (NMDA) receptor function may attenuate seizure susceptibility. The work of Haberman et al. was one of the first to explore the approach of blocking specific molecules by delivering antisense sequences using AAV vectors. An NMDA receptor cDNA fragment in the antisense orientation was cloned into AAV vectors (AAV-NR1A) where expression was driven by either tetracycline-off regulatable promoter (AAV-tTAK-NR1A) or a cytomegalovirus promoter (AAV-CMV-NR1A). In vitro infection of neurons with AAV-tTAK-NR1A resulted in decreased NMDA-evoked currents and a decrease in the number of NMDA receptors, and intracortical administration of AAV-tTAK-NR1A to rats reduced NMDAR1 subunit protein levels in vivo. However, administration of AAV-CMV-NR1A caused a significant reduction in seizure sensitivity. Additional administration of AAV-tTAK-GFP and AAV-CMV-LacZ transduced different neuronal populations. The results indicate that promoters can significantly influence the physiological outcome of NMDA receptor-based gene therapy [234].
Adenosine kinase (ADK) is a negative regulator of the endogenous brain anticonvulsant adenosine in astrocytes. ADK-transgenic mice and wild-type mice were injected with the AAV8-Adk vector in sense or antisense orientation under the control of gfaABC1D promoter to overexpress or downregulate ADK in astrocytes. It was shown that in wild-type mice, overexpression of ADK in astrocytes was sufficient to trigger spontaneous recurrent seizures in the absence of any other epileptogenic events, whereas downregulation of ADK with antisense AAV8-Adk almost completely abolished spontaneous recurrent seizures in ADK-transgenic mice [235].
Tubulin β-III (TUBB3) is the most dynamic β-tubulin isoform expressed in neurons and is highly expressed in the CNS. TUBB3 expression has been found to be increased in human and rat epileptic tissues. Moreover, TUBB3 expression is associated with inhibitory GABAergic neurons and the inhibitory postsynaptic scaffold protein gephyrin. TUBB3 downregulation attenuated seizure behavioral phenotypes during pilocarpine-induced chronic seizure phase and pentylenetetrazole kindling, whereas TUBB3 overexpression had the opposite effect. Importantly, TUBB3 interacted with GABA-A receptor-associated protein, which is known to be involved in GABA-A receptor trafficking. These results indicate that TUBB3 plays a critical role in the regulation of epileptic seizures through the trafficking of GABA-A receptors, suggesting a molecular mechanism for new therapeutic strategies by AAV-delivery of this ion channel subunits [236].
A promising approach is the delivery of antisense nucleotides to block the expression of potentially epileptogenic proteins. In particular, blocking the expression of long non-coding RNA (lncRNA) H19 can prevent its induced apoptosis [237]. LncRNAs are RNA molecules longer than 200 nucleotides that do not have the ability to encode a protein. Individual lncRNAs demonstrate cell-, tissue-, and line-specific expression patterns, also indicating the involvement of lncRNAs in the regulation of gene expression at both the transcriptional and translational levels. In addition, lncRNAs are likely to contribute to the development of various diseases, including nervous system disorders. In the study of Han et al., lncRNAs H19 was analyzed, which is reactivated in the latent period of epilepsy. Vectors carrying H19 (AAV9-H19) or short hairpin RNA targeting H19 (AAV9-shRNA) were delivered in a KA rat model of epilepsy. According to the functional annotation of genes, many genes inhibited or stimulated by H19 are involved in myelin assembly, immune responses, apoptosis, and MAPK activation. Demyelination of nerve fibers, hyperactivation of MAPK, and apoptosis of hippocampal neurons are key processes in epileptogenesis. It is indicated that H19 can induce epileptogenesis by inhibiting myelin assembly and inducing demyelination of nerve fibers. In addition, H19 can both enhance and suppress MAPK activity, which is also associated with epileptogenesis. Functional and pathway analysis reveals that H19 has diverse functions in epileptogenesis, representing potential targets for future studies of H19-modulated mechanisms [238].
Wong et al. used small hairpin RNA (shRNA) directed against the Scn8a gene, which resulted in selective downregulation of Scn8a expression in the hippocampus. SCN8A encodes the voltage-gated sodium channel (VGSC) Nav1.6, which is widely expressed in the CNS and PNS, where it strongly modulates neuronal excitability by setting the action potential initiation threshold and generating subthreshold depolarizing currents in the soma and dendrites. AAV expressing shRNA against Scn8a (shAAV-Scn8a) was injected acutely into the hippocampus of mesial temporal lobe epilepsy (MTLE) mice 24 h after KA administration, and was found to arrest the development of spontaneous seizures, attenuate KA-induced hyperactivity, and reduce reactive gliosis. Thus, downregulation of Scn8a expression can prevent the occurrence of spontaneous seizures in a mouse model of MTLE [239].
In temporal lobe epilepsy, recurrent mossy fibers sprouting from Dentate Gyrus Granule Cells (DGGCs) create an aberrant epileptogenic network between DGGCs that acts through ectopically expressed GluK2/GluK5-containing kainite acid receptors (KARs). Pharmacological inhibition of KARs or genetic deletion of GluK2-containing KARs is known to reduce interictal epileptiform discharges [240]. Therefore, an AAV9 vector expressing anti-grik2 microRNA was designed to specifically downregulate GluK2 expression. Hippocampal delivery of AAV9-anti grik2 miRNA was shown to markedly reduce chronic seizure activity in mice with temporal lobe epilepsy [241].
The data on the repression of some genes using AAV vectors are compiled in Table 2.
Thus, there are different approaches to using AAV vectors for the treatment of epileptic disorders. Enhancement of function of a particular target protein is the most commonly used, however, data suggest that AAV-induced gene knockout may also be quite effective. Unfortunately, most of the data were obtained in animals that are quite evolutionarily distant from humans, so the question of the prospects for clinical trials remains open. Further research in this area would help to select the most effective serotype and promoter for sufficient transduction of human cells without the occurrence of undesirable effects.
3. Materials and Methods
The following scientific journal aggregators were used in writing the systematic review: MDPI, PubMed, ScienceDirect, Springer, and Web of Science. The search for scientific articles was carried out using the following keywords: adenoassociated viral vector, AAV, epilepsy, gene therapy. The review included studies from 2000 to the present day (Figure 2).
3.1. Inclusion Criteria
The review includes studies on the delivery of AAV-based viral vectors for the treatment of epileptic disorders of various etiologies. No restrictions were imposed on the serotype of the AAV vector, the method of delivery of the viral vector, the place of delivery of the viral vector, and the experimental model of epilepsy. The study included both AAV vectors carrying the target gene and its functional fragments, as well as antisense sequences of the targeted gene.
3.2. Exclusion Criteria
The absence of using an AAV-based vector was the main criterion for excluding studies from the review. Methodologies using additional modifications of AAV delivery, such as: AAV vectors using the CRISPR-Cas system, were also excluded.
4. Conclusions
In this article, we reviewed the use of AAV in gene therapy for epilepsy, examining their mechanisms of action, advantages, and disadvantages. Different AAV serotypes differ in their surface properties, which affects their tropism, ability to penetrate the BBB, and the efficiency of cell transduction. This diversity allows for the selection of the optimal serotype for a specific therapy. Combining AAV genomic constructs with capsid proteins of different serotypes, creates pseudotyped recombinant AAVs, and expands their application to various cell types and tissues. The use of different promoters in vector constructs allows for tissue specificity and maximum efficiency of gene therapy [61].
Studies using AAV vectors in gene therapy for epilepsy demonstrate significant potential in modulating seizure activity by delivering various genes and neuropeptides. In particular, delivery of the NPY gene and its receptors to the hippocampus has been shown to be effective in reducing seizure frequency. Pseudotyped vectors such as rAAV1/2-NPY have also been shown to be effective in progressive and spontaneous seizure models. In addition to NPY, other neuropeptides such as galanin have been studied to modulate the activity of neurotransmitter systems and reduce seizure frequency and severity [154,169].
Studies have shown that ion channels are promising targets in gene therapy for epilepsy. Introduction of genes encoding various ion channels and receptors such as GABRA4, SCN1A, and KCNN4 using AAV vectors resulted in significant reductions in seizure frequency and severity in preclinical models. Gene therapy targeting the expression of neurotrophic factors and other genes in the central nervous system also represents a promising approach for the treatment of epilepsy. Factors such as GDNF and transcription factors such as Nrf2 have been shown to reduce seizure frequency and protect neurons [202,203,205]. Incorporation of antisense RNAs into vector cassettes allows for targeted manipulation of the expression of specific genes such as GABA and NMDA receptors, as well as ADK, which significantly impacts seizure activity [234,235].
Thus, modulating the expression of various neuropeptides, ion channels, transcription factors, etc. can regulate seizure activity and, consequently, reduce the severity of epileptic disorders. Targeting both neuronal and glial proteins may be a promising strategy in treatment. Another important strategy is gene knockout, since the cDNA transcript is shorter than the functional gene sequence, which simplifies the use of AAV, since their packaging capacity is small. Additionally, delivering transcription factors required for inducing target gene transcription appears to be a promising strategy, since a number of functionally proteins can be under the control of the same transcription factor, which would provide the possibility of multiplex targeting of a number of genes. Study of proteomic and transcriptomic expression profiles of epileptic tissues and animal models can help in finding potential targets for knockout or enhancement of expression by transgene delivery.
Author Contributions
Conceptualization, E.T., A.Y. and D.D.; writing – original draft preparation, A.M. and V.S.; writing – review and editing, V.S., A.S. and G.S.; visualization, D.D. and AI; supervision, A.R. All authors have read and agreed to the published version of the manuscript.
Funding
The work was carried out at the expense of the subsidy allocated to Kazan Federal University to fulfill the state task in the field of scientific activity (PROJECT No. FZSM-2023-0011).
Data Availability Statement
Data sharing applicable.
Acknowledgments
This paper has been supported by the Kazan Federal University Strategic Academic Leadership Program (PRIORITY-2030).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fisher, R.S.; et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia, 2014, 55, 475–82. [Google Scholar] [CrossRef] [PubMed]
- Riney, K.; et al. International League Against Epilepsy classification and definition of epilepsy syndromes with onset at a variable age: position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia, 2022, 63, 1443–1474. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia, 2017, 58, 512–521. [Google Scholar] [CrossRef]
- Fisher, R.S.; et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia, 2017, 58, 522–530. [Google Scholar] [CrossRef]
- Dos Santos, R.R.; et al. Neurochemical abnormalities in the hippocampus of male rats displaying audiogenic seizures, a genetic model of epilepsy. Neurosci Lett 2021, 761, 136123. [Google Scholar] [CrossRef] [PubMed]
- Numis, A.L.; et al. Whole-exome sequencing with targeted analysis and epilepsy after acute symptomatic neonatal seizures. Pediatr Res, 2022, 91, 896–902. [Google Scholar] [CrossRef]
- Møller, R.S., H.A. Dahl. and I. Helbig, The contribution of next generation sequencing to epilepsy genetics. Expert Rev Mol Diagn, 2015, 15, 1531–8. [Google Scholar] [CrossRef]
- Jiang, Y.L.; et al. Clinical Utility of Exome Sequencing and Reinterpreting Genetic Test Results in Children and Adults With Epilepsy. Front Genet, 2020, 11, 591434. [Google Scholar] [CrossRef] [PubMed]
- Salzmann, A. and A. Malafosse, Genetics of temporal lobe epilepsy: a review. Epilepsy Res Treat, 2012, 2012: 863702.
- Cavalleri, G.L.; et al. Failure to replicate previously reported genetic associations with sporadic temporal lobe epilepsy: where to from here? Brain, 2005, 128 Pt 8, 1832-40.
- Pfisterer, U.; et al. Identification of epilepsy-associated neuronal subtypes and gene expression underlying epileptogenesis. Nat Commun, 2020, 11, 5038. [Google Scholar] [CrossRef]
- Pires, G.; et al. Proteomic differences in the hippocampus and cortex of epilepsy brain tissue. Brain Commun, 2021, 3, fcab021. [Google Scholar] [CrossRef]
- Bando, S.Y.; et al. Correction: Complex Network Analysis of CA3 Transcriptome Reveals Pathogenic and Compensatory Pathways in Refractory Temporal Lobe Epilepsy. PLoS One. 2014, 9. [Google Scholar] [CrossRef]
- Löscher, W.; et al. Drug Resistance in Epilepsy: Clinical Impact, Potential Mechanisms, and New Innovative Treatment Options. Pharmacol Rev, 2020, 72, 606–638. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D. and W. Löscher. Drug resistance in epilepsy: putative neurobiologic and clinical mechanisms. Epilepsia, 2005, 46, 858–77. [Google Scholar] [CrossRef] [PubMed]
- Meij, P.; et al. Advanced therapy medicinal products. Brief Pap, 2019, 3, 2020. [Google Scholar]
- 320/12, D.o.t.G.C.o.A.J.C.N.I.A., “Bolar Exemption–Poland” Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community Code Relating to Medicinal Products for Human Use, Art. 10 (6); Law on Industrial Property, Arts. 61 (1)(4), 63 (1). 2013, Springer.
- Shaimardanova, A.A.; et al., Gene and Cell Therapy for Epilepsy: A Mini Review. Front Mol Neurosci, 2022, 15: 868531.
- Detela, G.; Lodge, A. EU Regulatory Pathways for ATMPs: Standard, Accelerated and Adaptive Pathways to Marketing Authorisation. Mol Ther Methods Clin Dev, 2019, 13, 205–232. [Google Scholar] [CrossRef]
- Dulak, J., J. Uchto. 50 years of gene therapy--a contribution of Wacław Szybalski to science and humanity. Gene, 2013, 525, 149–50. [Google Scholar] [CrossRef]
- Lentz, T.B., S.J. Gray. Viral vectors for gene delivery to the central nervous system. Neurobiol Dis, 2012, 48, 179–88. [Google Scholar] [CrossRef]
- Candolfi, M.; et al. Optimization of adenoviral vector-mediated transgene expression in the canine brain in vivo, and in canine glioma cells in vitro. Neuro Oncol, 2007, 9, 245–58. [Google Scholar] [CrossRef]
- Volpers, C. and S. Kochanek, Adenoviral vectors for gene transfer and therapy. J Gene Med, 2004, 6 Suppl 1: S164-71.
- Thomas, C.E.; et al. Acute direct adenoviral vector cytotoxicity and chronic, but not acute, inflammatory responses correlate with decreased vector-mediated transgene expression in the brain. Mol Ther, 2001, 3, 36–46. [Google Scholar] [CrossRef]
- Lozier, J.N.; et al. Toxicity of a first-generation adenoviral vector in rhesus macaques. Hum Gene Ther, 2002, 13, 113–24. [Google Scholar] [CrossRef]
- Markert, J.M.; et al. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol Ther, 2009, 17, 199–207. [Google Scholar] [CrossRef]
- Saeki, Y., X.O. Breakefield. Improved HSV-1 amplicon packaging system using ICP27-deleted, oversized HSV-1 BAC DNA. Methods Mol Med, 2003, 76, 51–60. [Google Scholar]
- Goff, S.P. Retroviridae: The Retroviruses and Their Replication, in Fields Virology, D.M. Knipe and P.M. Howley, Editors. 2007, Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia. 2000–2069.
- Bokhoven, M.; et al. Insertional gene activation by lentiviral and gammaretroviral vectors. J Virol, 2009, 83, 283–94. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science, 2003, 302, 415–9. [Google Scholar] [CrossRef]
- Thomas, C.E., A. Ehrhardt. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet, 2003, 4, 346–58. [Google Scholar] [CrossRef]
- Grimm, D. and M.A. Kay. From virus evolution to vector revolution: use of naturally occurring serotypes of adeno-associated virus (AAV) as novel vectors for human gene therapy. Curr Gene Ther, 2003, 3, 281–304. [Google Scholar] [CrossRef]
- Pena, S.A.; et al. Gene therapy for neurological disorders: challenges and recent advancements. J Drug Target, 2020, 28, 111–128. [Google Scholar] [CrossRef]
- Mullagulova, A.; et al. Safety and Efficacy of Intravenous and Intrathecal Delivery of AAV9-Mediated ARSA in Minipigs. Int J Mol Sci, 2023, 24(11).
- Massaro, G.; et al. Gene Therapy for Lysosomal Storage Disorders: Ongoing Studies and Clinical Development. Biomolecules, 2021, 11(4).
- Janson, C.; et al. Clinical protocol. Gene therapy of Canavan disease: AAV-2 vector for neurosurgical delivery of aspartoacylase gene (ASPA) to the human brain. Hum Gene Ther, 2002, 13, 1391–412. [Google Scholar] [CrossRef]
- Corti, M.; et al. Adeno-associated virus-mediated gene therapy in a patient with Canavan disease using dual routes of administration and immune modulation. Mol Ther Methods Clin Dev, 2023, 30: 303-314.
- Worgall, S.; et al. Treatment of late infantile neuronal ceroid lipofuscinosis by CNS administration of a serotype 2 adeno-associated virus expressing CLN2 cDNA. Hum Gene Ther, 2008, 19, 463–74. [Google Scholar] [CrossRef]
- Tardieu, M.; et al. Intracerebral administration of adeno-associated viral vector serotype rh.10 carrying human SGSH and SUMF1 cDNAs in children with mucopolysaccharidosis type IIIA disease: results of a phase I/II trial. Hum Gene Ther, 2014, 25, 506–16. [Google Scholar] [CrossRef]
- Hocquemiller, M.; et al. AAVrh10 Vector Corrects Disease Pathology in MPS IIIA Mice and Achieves Widespread Distribution of SGSH in Large Animal Brains. Mol Ther Methods Clin Dev, 2020, 17: 174-187.
- Mingozzi, F. and K.A. High. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat Rev Genet, 2011, 12, 341–55. [Google Scholar] [CrossRef]
- Kido, J., K. Sugawara. Gene therapy for lysosomal storage diseases: Current clinical trial prospects. Front Genet, 2023, 14, 1064924. [Google Scholar] [CrossRef]
- Wu, Z., A. Asokan. Adeno-associated virus serotypes: vector toolkit for human gene therapy. Mol Ther, 2006, 14, 316–27. [Google Scholar] [CrossRef]
- Zengel, J.; Carette, J.E. Structural and cellular biology of adeno-associated virus attachment and entry. Adv Virus Res, 2020, 106, 39–84. [Google Scholar]
- Naso, M.F.; et al. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs, 2017, 31, 317–334. [Google Scholar] [CrossRef]
- Samulski, R.J., L.S. Chang. Helper-free stocks of recombinant adeno-associated viruses: normal integration does not require viral gene expressio. J Virol, 1989, 63, 3822–8. [Google Scholar] [CrossRef]
- Samulski, R.J.; et al. Rescue of adeno-associated virus from recombinant plasmids: gene correction within the terminal repeats of AAV. Cell, 1983, 33, 135–43. [Google Scholar] [CrossRef]
- Kyöstiö, S.R.; et al. Analysis of adeno-associated virus (AAV) wild-type and mutant Rep proteins for their abilities to negatively regulate AAV p5 and p19 mRNA levels. J Virol, 1994, 68, 2947–57. [Google Scholar] [CrossRef]
- Henckaerts, E. and R.M. Linden. Adeno-associated virus: a key to the human genome? Future Virol, 2010, 5, 555–574. [Google Scholar] [CrossRef]
- Qiu, J.; et al. Parvovirus RNA processing strategies. 2006: Hodder Arnold, London, UK.
- James, J.A.; et al. Crystal structure of the SF3 helicase from adeno-associated virus type 2. Structure, 2003, 11, 1025–35. [Google Scholar] [CrossRef]
- Hickman, A.B.; et al. The nuclease domain of adeno-associated virus rep coordinates replication initiation using two distinct DNA recognition interfaces. Mol Cell, 2004, 13, 403–14. [Google Scholar] [CrossRef]
- Surosky, R.T.; et al. Adeno-associated virus Rep proteins target DNA sequences to a unique locus in the human genome. J Virol, 1997, 71, 7951–9. [Google Scholar] [CrossRef]
- Yoon-Robarts, M.; et al. Residues within the B' motif are critical for DNA binding by the superfamily 3 helicase Rep40 of adeno-associated virus type 2. J Biol Chem, 2004, 279, 50472–81. [Google Scholar] [CrossRef]
- Chapman, M.S. and M. Agbandje-McKenna, Atomic structure of viral particles. Parvoviruses, 2006: 107-23.
- Sonntag, F.; et al. The assembly-activating protein promotes capsid assembly of different adeno-associated virus serotypes. J Virol, 2011, 85, 12686–97. [Google Scholar] [CrossRef]
- de Backer, M.W.; et al. Recombinant adeno-associated viral vectors. Methods Mol Biol, 2011, 789, 357–76. [Google Scholar]
- Issa, S.S.; et al. Various AAV Serotypes and Their Applications in Gene Therapy: An Overview. Cells, 2023, 12(5).
- Zincarelli, C.; et al. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol Ther, 2008, 16, 1073–80. [Google Scholar] [CrossRef]
- Cearley, C.N. and J.H. Wolfe. Transduction characteristics of adeno-associated virus vectors expressing cap serotypes 7, 8, 9, and Rh10 in the mouse brain. Mol Ther, 2006, 13, 528–37. [Google Scholar] [CrossRef]
- Gao, G., L.H. Vandenberghe. New recombinant serotypes of AAV vectors. Curr Gene Ther, 2005, 5, 285–97. [Google Scholar] [CrossRef]
- Harbison, C.E., J.A. Chiorini. The parvovirus capsid odyssey: from the cell surface to the nucleus. Trends Microbiol, 2008, 16, 208–14. [Google Scholar] [CrossRef]
- Burger, C.; et al. Recombinant AAV viral vectors pseudotyped with viral capsids from serotypes 1, 2, and 5 display differential efficiency and cell tropism after delivery to different regions of the central nervous system. Mol Ther, 2004, 10, 302–17. [Google Scholar] [CrossRef]
- Taymans, J.M.; et al. Comparative analysis of adeno-associated viral vector serotypes 1, 2, 5, 7, and 8 in mouse brain. Hum Gene Ther, 2007, 18, 195–206. [Google Scholar] [CrossRef]
- Inagaki, K.; et al. Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol Ther, 2006, 14, 45–53. [Google Scholar] [CrossRef]
- Zabner, J.; et al. Adeno-associated virus type 5 (AAV5) but not AAV2 binds to the apical surfaces of airway epithelia and facilitates gene transfer. J Virol, 2000, 74, 3852–8. [Google Scholar] [CrossRef]
- Srivastava, A. In vivo tissue-tropism of adeno-associated viral vectors. Curr Opin Virol, 2016, 21, 75–80. [Google Scholar] [CrossRef]
- Chen, H.; et al. Gene transfer and expression in oligodendrocytes under the control of myelin basic protein transcriptional control region mediated by adeno-associated virus. Gene Ther, 1998, 5, 50–8. [Google Scholar] [CrossRef]
- Xu, R.; et al. Quantitative comparison of expression with adeno-associated virus (AAV-2) brain-specific gene cassettes. Gene Ther, 2001, 8, 1323–32. [Google Scholar] [CrossRef]
- Gray, S.J.; et al. Optimizing promoters for recombinant adeno-associated virus-mediated gene expression in the peripheral and central nervous system using self-complementary vectors. Hum Gene Ther, 2011, 22, 1143–53. [Google Scholar] [CrossRef]
- Powell, S.K., R.J. Samulski. AAV Capsid-Promoter Interactions Determine CNS Cell-Selective Gene Expression In Vivo. Mol Ther, 2020, 28, 1373–1380. [Google Scholar] [CrossRef]
- Jackson, K.L.; et al. Better Targeting, Better Efficiency for Wide-Scale Neuronal Transduction with the Synapsin Promoter and AAV-PHP.B. Front Mol Neurosci, 2016, 9: 116.
- Verdera, H.C., K. Kuranda. AAV Vector Immunogenicity in Humans: A Long Journey to Successful Gene Transfer. Mol Ther, 2020, 28, 723–746. [Google Scholar] [CrossRef]
- Au, H.K.E., M. Isalan, and M. Mielcarek, Gene Therapy Advances: A Meta-Analysis of AAV Usage in Clinical Settings. Front Med (Lausanne), 2021, 8: 809118.
- Dhungel, B.P.; et al. Understanding AAV vector immunogenicity: from particle to patient. Theranostics, 2024, 14, 1260–1288. [Google Scholar] [CrossRef]
- Salganik, M., M. L. Hirsch, and R.J. Samulski, Adeno-associated Virus as a Mammalian DNA Vector. Microbiol Spectr, 2015, 3(4).
- Arjomandnejad, M.; et al. Immunogenicity of Recombinant Adeno-Associated Virus (AAV) Vectors for Gene Transfer. BioDrugs, 2023, 37, 311–329. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; et al. Subthalamic GAD gene therapy in a Parkinson's disease rat model. Science, 2002, 298, 425–9. [Google Scholar] [CrossRef] [PubMed]
- andel, R.J. and C. Burger. Clinical trials in neurological disorders using AAV vectors: promises and challenges. Curr Opin Mol Ther, 2004, 6, 482–90. [Google Scholar]
- Podsakoff, G., K.K. Wong. Efficient gene transfer into nondividing cells by adeno-associated virus-based vectors. J Virol, 1994, 68, 5656–66. [Google Scholar] [CrossRef] [PubMed]
- Du, B.; et al. Efficient transduction of human neurons with an adeno-associated virus vector. Gene Ther, 1996, 3, 254–61. [Google Scholar]
- Jooss, K.; et al. Transduction of dendritic cells by DNA viral vectors directs the immune response to transgene products in muscle fibers. J Virol, 1998, 72, 4212–23. [Google Scholar] [CrossRef]
- Kaplitt, M.G.; et al. Long-term gene expression and phenotypic correction using adeno-associated virus vectors in the mammalian brain. Nat Genet, 1994, 8, 148–54. [Google Scholar] [CrossRef]
- Ali, R.R.; et al. Gene transfer into the mouse retina mediated by an adeno-associated viral vector. Hum Mol Genet, 1996, 5, 591–4. [Google Scholar] [CrossRef]
- Bartlett, J.S., R.J. Samulski. Selective and rapid uptake of adeno-associated virus type 2 in brain. Hum Gene Ther, 1998, 9, 1181–6. [Google Scholar] [CrossRef]
- Vite, C.H.; et al. Adeno-associated virus vector-mediated transduction in the cat brain. Gene Ther, 2003, 10, 1874–81. [Google Scholar] [CrossRef]
- During, M.J.; et al. In vivo expression of therapeutic human genes for dopamine production in the caudates of MPTP-treated monkeys using an AAV vector. Gene Ther, 1998, 5, 820–7. [Google Scholar] [CrossRef] [PubMed]
- Bankiewicz, K.S.; et al. Convection-enhanced delivery of AAV vector in parkinsonian monkeys; in vivo detection of gene expression and restoration of dopaminergic function using pro-drug approach. Exp Neurol, 2000, 164, 2–14. [Google Scholar] [CrossRef]
- Freese, A.; et al. Direct gene transfer into human epileptogenic hippocampal tissue with an adeno-associated virus vector: implications for a gene therapy approach to epilepsy. Epilepsia, 1997, 38, 759–66. [Google Scholar] [CrossRef]
- Mastakov, M.Y.; et al. Recombinant adeno-associated virus serotypes 2- and 5-mediated gene transfer in the mammalian brain: quantitative analysis of heparin co-infusion. Mol Ther, 2002, 5, 371–80. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.B.; et al. Convection-enhanced delivery of AAV-2 combined with heparin increases TK gene transfer in the rat brain. Neuroreport, 2001, 12, 1961–4. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, J.; et al. Extensive Transduction and Enhanced Spread of a Modified AAV2 Capsid in the Non-human Primate CNS. Mol Ther, 2018, 26, 2418–2430. [Google Scholar] [CrossRef]
- Hoganson, D.K.; et al. Uptake of adenoviral vectors via fibroblast growth factor receptors involves intracellular pathways that differ from the targeting ligand. Mol Ther, 2001, 3, 105–12. [Google Scholar] [CrossRef] [PubMed]
- Mastakov, M.Y.; et al. Combined injection of rAAV with mannitol enhances gene expression in the rat brain. Mol Ther, 2001, 3, 225–32. [Google Scholar] [CrossRef]
- Fu, H.; et al. Self-complementary adeno-associated virus serotype 2 vector: global distribution and broad dispersion of AAV-mediated transgene expression in mouse brain. Mol Ther, 2003, 8, 911–7. [Google Scholar] [CrossRef]
- Tajiri, N.; et al. Breaking the Blood-Brain Barrier With Mannitol to Aid Stem Cell Therapeutics in the Chronic Stroke Brain. Cell Transplant, 2016, 25, 1453–60. [Google Scholar] [CrossRef]
- Burger, C.; et al. Systemic mannitol-induced hyperosmolality amplifies rAAV2-mediated striatal transduction to a greater extent than local co-infusion. Mol Ther, 2005, 11, 327–31. [Google Scholar] [CrossRef] [PubMed]
- Foley, C.P.; et al. Intra-arterial delivery of AAV vectors to the mouse brain after mannitol mediated blood brain barrier disruption. J Control Release, 2014, 196: 71-78.
- Chirmule, N.; et al. Immune responses to adenovirus and adeno-associated virus in humans. Gene Ther, 1999, 6, 1574–83. [Google Scholar] [CrossRef] [PubMed]
- Moskalenko, M.; et al. Epitope mapping of human anti-adeno-associated virus type 2 neutralizing antibodies: implications for gene therapy and virus structure. J Virol, 2000, 74, 1761–6. [Google Scholar] [CrossRef] [PubMed]
- Peden, C.S.; et al. Circulating anti-wild-type adeno-associated virus type 2 (AAV2) antibodies inhibit recombinant AAV2 (rAAV2)-mediated, but not rAAV5-mediated, gene transfer in the brain. J Virol, 2004, 78, 6344–59. [Google Scholar] [CrossRef]
- Sanftner, L.M.; et al. Striatal delivery of rAAV-hAADC to rats with preexisting immunity to AAV. Mol Ther, 2004, 9, 403–9. [Google Scholar] [CrossRef]
- Davidson, B.L.; et al. Recombinant adeno-associated virus type 2, 4, and 5 vectors: transduction of variant cell types and regions in the mammalian central nervous system. Proc Natl Acad Sci U S A, 2000, 97, 3428–32. [Google Scholar] [CrossRef]
- Alisky, J.M.; et al. Transduction of murine cerebellar neurons with recombinant FIV and AAV5 vectors. Neuroreport, 2000, 11, 2669–73. [Google Scholar] [CrossRef]
- Passini, M.A.; et al. Intraventricular brain injection of adeno-associated virus type 1 (AAV1) in neonatal mice results in complementary patterns of neuronal transduction to AAV2 and total long-term correction of storage lesions in the brains of beta-glucuronidase-deficient mice. J Virol, 2003, 77, 7034–40. [Google Scholar]
- Gao, G.P.; et al. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci U S A, 2002, 99, 11854–9. [Google Scholar] [CrossRef]
- Sarkar, R.; et al. Total correction of hemophilia A mice with canine FVIII using an AAV 8 serotype. Blood, 2004, 103, 1253–60. [Google Scholar] [CrossRef]
- Nakai, H.; et al. Unrestricted hepatocyte transduction with adeno-associated virus serotype 8 vectors in mice. J Virol, 2005, 79, 214–24. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; et al. Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat Biotechnol, 2005, 23, 321–8. [Google Scholar] [CrossRef]
- Harding, T.C.; et al. Enhanced gene transfer efficiency in the murine striatum and an orthotopic glioblastoma tumor model, using AAV-7- and AAV-8-pseudotyped vectors. Hum Gene Ther, 2006, 17, 807–20. [Google Scholar] [CrossRef]
- Gray, S.J.; et al. Preclinical differences of intravascular AAV9 delivery to neurons and glia: a comparative study of adult mice and nonhuman primates. Mol Ther, 2011, 19, 1058–69. [Google Scholar] [CrossRef]
- Brown, D.; et al., Deep Parallel Characterization of AAV Tropism and AAV-Mediated Transcriptional Changes via Single-Cell RNA Sequencing. Front Immunol, 2021, 12: 730825.
- Devinsky, O.; et al. Glia and epilepsy: excitability and inflammation. Trends Neurosci, 2013, 36, 174–84. [Google Scholar] [CrossRef] [PubMed]
- O'Carroll, S.J., W.H. Cook, and D. Young, AAV Targeting of Glial Cell Types in the Central and Peripheral Nervous System and Relevance to Human Gene Therapy. Front Mol Neurosci, 2020, 13: 618020.
- Chandran, J.; et al. Assessment of AAV9 distribution and transduction in rats after administration through Intrastriatal, Intracisterna magna and Lumbar Intrathecal routes. Gene Ther, 2023, 30(1-2): 132-141.
- Foust, K.D.; et al. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol, 2009, 27, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, J., R. J. Nobre, and L. Pereira de Almeida, Gene therapy for the CNS using AAVs: The impact of systemic delivery by AAV9. J Control Release, 2016, 241: 94-109.
- Deverman, B.E.; et al. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat Biotechnol, 2016, 34, 204–9. [Google Scholar] [CrossRef]
- Morabito, G.; et al. AAV-PHP.B-Mediated Global-Scale Expression in the Mouse Nervous System Enables GBA1 Gene Therapy for Wide Protection from Synucleinopathy. Mol Ther, 2017, 25, 2727–2742. [Google Scholar] [CrossRef]
- Das, S., S. Huang. Acceleration of rare disease therapeutic development: a case study of AGIL-AADC. Drug Discov Today, 2019, 24, 678–684. [Google Scholar] [CrossRef]
- Keeler, A.M. and T.R. Flotte. Recombinant Adeno-Associated Virus Gene Therapy in Light of Luxturna (and Zolgensma and Glybera): Where Are We, and How Did We Get Here? Annu Rev Virol, 2019, 6, 601–621. [Google Scholar] [CrossRef]
- Blair, H.A. Onasemnogene Abeparvovec: A Review in Spinal Muscular Atrophy. CNS Drugs, 2022, 36, 995–1005. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.A. Etranacogene Dezaparvovec: First Approval. Drugs, 2023, 83, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Shariff, S.; et al. Advances in understanding the pathogenesis of epilepsy: Unraveling the molecular mechanisms: A cross-sectional study. Health Sci Rep, 2024, 7, e1896. [Google Scholar] [CrossRef]
- Staley, K. Molecular mechanisms of epilepsy. Nat Neurosci, 2015, 18, 367–72. [Google Scholar] [CrossRef]
- Yi, J.H. and A.S. Hazell. Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem Int, 2006, 48, 394–403. [Google Scholar] [CrossRef] [PubMed]
- DeLorenzo, R.J., D.A. Sun. Erratum to "Cellular mechanisms underlying acquired epilepsy: the calcium hypothesis of the induction and maintenance of epilepsy." [Pharmacol. Ther. 105(3) (2005) 229-266]. Pharmacol Ther, 2006, 111, 288–325. [Google Scholar] [CrossRef]
- Treiman, D.M. GABAergic mechanisms in epilepsy. Epilepsia, 2001, 42 Suppl 3: 8-12.
- Aroniadou-Anderjaska, V.; et al. Pathology and pathophysiology of the amygdala in epileptogenesis and epilepsy. Epilepsy Res, 2008, 78(2-3): 102-16.
- Hirose, S.; et al. Are some idiopathic epilepsies disorders of ion channels?: A working hypothesis. Epilepsy Res, 2000, 41, 191–204. [Google Scholar] [CrossRef]
- Graves, T.D. Ion channels and epilepsy. QJM, 2006, 99, 201–17. [Google Scholar] [CrossRef]
- Berkovic, S.F.; et al. Human epilepsies: interaction of genetic and acquired factors. Trends Neurosci, 2006, 29, 391–397. [Google Scholar] [CrossRef]
- Boleti, A.P.A.; et al. Pathophysiology to Risk Factor and Therapeutics to Treatment Strategies on Epilepsy. Brain Sci, 2024, 14(1).
- Chugh, D.; et al. Alterations in Brain Inflammation, Synaptic Proteins, and Adult Hippocampal Neurogenesis during Epileptogenesis in Mice Lacking Synapsin2. PLoS One, 2015, 10, e0132366. [Google Scholar] [CrossRef]
- Clynen, E.; et al. Neuropeptides as targets for the development of anticonvulsant drugs. Mol Neurobiol, 2014, 50, 626–46. [Google Scholar] [CrossRef] [PubMed]
- Laing, B.T.; et al. AgRP/NPY Neuron Excitability Is Modulated by Metabotropic Glutamate Receptor 1 During Fasting. Front Cell Neurosci, 2018, 12: 276.
- Colmers, W.F. and D. Bleakman, Effects of neuropeptide Y on the electrical properties of neurons. Trends Neurosci, 1994, 17, 373–9. [Google Scholar] [PubMed]
- Greber, S., C. Schwarzer. Neuropeptide Y inhibits potassium-stimulated glutamate release through Y2 receptors in rat hippocampal slices in vitro. Br J Pharmacol, 1994, 113, 737–40. [Google Scholar] [CrossRef] [PubMed]
- Richichi, C.; et al. Anticonvulsant and antiepileptogenic effects mediated by adeno-associated virus vector neuropeptide Y expression in the rat hippocampus. J Neurosci, 2004, 24, 3051–9. [Google Scholar] [CrossRef] [PubMed]
- Noè, F.; et al. Neuropeptide Y gene therapy decreases chronic spontaneous seizures in a rat model of temporal lobe epilepsy. Brain, 2008, 131(Pt 6): 1506-15.
- Noe, F.; et al. Anticonvulsant effects and behavioural outcomes of rAAV serotype 1 vector-mediated neuropeptide Y overexpression in rat hippocampus. Gene Ther, 2010, 17, 643–52. [Google Scholar] [CrossRef]
- Dumont, Y.; et al. Potent and selective tools to investigate neuropeptide Y receptors in the central and peripheral nervous systems: BIB03304 (Y1) and CGP71683A (Y5). Can J Physiol Pharmacol, 2000, 78, 116–25. [Google Scholar] [CrossRef]
- Lin, E.J.; et al. Differential actions of NPY on seizure modulation via Y1 and Y2 receptors: evidence from receptor knockout mice. Epilepsia, 2006, 47, 773–80. [Google Scholar]
- El Bahh, B.; et al. The anti-epileptic actions of neuropeptide Y in the hippocampus are mediated by Y and not Y receptors. Eur J Neurosci, 2005, 22, 1417–30. [Google Scholar] [CrossRef]
- Furtinger, S.; et al. Plasticity of Y1 and Y2 receptors and neuropeptide Y fibers in patients with temporal lobe epilepsy. J Neurosci, 2001, 21, 5804–12. [Google Scholar] [CrossRef]
- Schwarzer, C., N. Kofler. Up-regulation of neuropeptide Y-Y2 receptors in an animal model of temporal lobe epilepsy. Mol Pharmacol, 1998, 53, 6–13. [Google Scholar] [CrossRef]
- Vezzani, A.; et al. Plastic changes in neuropeptide Y receptor subtypes in experimental models of limbic seizures. Epilepsia, 2000, 41 Suppl 6: S115-21.
- Foti, S.; et al. Adeno-associated virus-mediated expression and constitutive secretion of NPY or NPY13-36 suppresses seizure activity in vivo. Gene Ther, 2007, 14, 1534–6. [Google Scholar] [CrossRef] [PubMed]
- Woldbye, D.P.; et al. Adeno-associated viral vector-induced overexpression of neuropeptide Y Y2 receptors in the hippocampus suppresses seizures. Brain, 2010, 133, 2778–88. [Google Scholar] [CrossRef] [PubMed]
- Ledri, L.N.; et al. Translational approach for gene therapy in epilepsy: Model system and unilateral overexpression of neuropeptide Y and Y2 receptors. Neurobiol Dis, 2016, 86: 52-61.
- Powell, K.L.; et al. Gene therapy mediated seizure suppression in Genetic Generalised Epilepsy: Neuropeptide Y overexpression in a rat model. Neurobiol Dis, 2018, 113: 23-32.
- Melin, E.; et al. Disease Modification by Combinatorial Single Vector Gene Therapy: A Preclinical Translational Study in Epilepsy. Mol Ther Methods Clin Dev, 2019, 15: 179-193.
- Wickham, J.; et al. Inhibition of epileptiform activity by neuropeptide Y in brain tissue from drug-resistant temporal lobe epilepsy patients. Sci Rep, 2019, 9, 19393. [Google Scholar] [CrossRef]
- Melin, E.; et al. Combinatorial gene therapy for epilepsy: Gene sequence positioning and AAV serotype influence expression and inhibitory effect on seizures. Gene Ther, 2023, 30(7-8): 649-658.
- Qian, J., W.F. Colmers. Inhibition of synaptic transmission by neuropeptide Y in rat hippocampal area CA1: modulation of presynaptic Ca2+ entry. J Neurosci, 1997, 17, 8169–77. [Google Scholar] [CrossRef] [PubMed]
- Webling, K.; et al. Ala(5)-galanin (2-11) is a GAL(2)R specific galanin analogue. Neuropeptides, 2016, 60: 75-82.
- Konopka, L.M., T. W. McKeon, and R.L. Parsons, Galanin-induced hyperpolarization and decreased membrane excitability of neurones in mudpuppy cardiac ganglia. J Physiol, 1989, 410: 107-22.
- Papas, S. and C.W. Bourque. Galanin inhibits continuous and phasic firing in rat hypothalamic magnocellular neurosecretory cells. J Neurosci, 1997, 17, 6048–56. [Google Scholar] [CrossRef]
- Nishibori, M.; et al. Galanin inhibits noradrenaline-induced accumulation of cyclic AMP in the rat cerebral cortex. J Neurochem, 1988, 51, 1953–5. [Google Scholar] [CrossRef]
- Merriam, L.A. and R.L. Parsons. Neuropeptide galanin inhibits omega-conotoxin GVIA-sensitive calcium channels in parasympathetic neurons. J Neurophysiol, 1995, 73, 1374–82. [Google Scholar] [CrossRef]
- Nordstrom, O.; et al. Evidence for an inhibitory effect of the peptide galanin on dopamine release from the rat median eminence. Neurosci Lett, 1987, 73, 21–6. [Google Scholar] [CrossRef]
- Ogren, S.O.; et al. Differential effects of the putative galanin receptor antagonists M15 and M35 on striatal acetylcholine release. Eur J Pharmacol, 1993, 242, 59–64. [Google Scholar] [CrossRef]
- Zini, S.; et al. Galanin reduces release of endogenous excitatory amino acids in the rat hippocampus. Eur J Pharmacol, 1993, 245, 1–7. [Google Scholar] [CrossRef]
- Bellmann, R.; et al. Enhanced rate of expression and biosynthesis of neuropeptide Y after kainic acid-induced seizures. J Neurochem, 1991, 56, 525–30. [Google Scholar] [CrossRef] [PubMed]
- Loacker, S.; et al. Endogenous dynorphin in epileptogenesis and epilepsy: anticonvulsant net effect via kappa opioid receptors. Brain, 2007, 130(Pt 4): 1017-28.
- Stogmann, E.; et al. A functional polymorphism in the prodynorphin gene promotor is associated with temporal lobe epilepsy. Ann Neurol, 2002, 51, 260–3. [Google Scholar] [CrossRef] [PubMed]
- de Lanerolle, N.C.; et al. Dynorphin and the kappa 1 ligand [3H]U69,593 binding in the human epileptogenic hippocampus. Epilepsy Res, 1997, 28, 189–205. [Google Scholar] [CrossRef] [PubMed]
- Bartfai, T.; et al. M-15: high-affinity chimeric peptide that blocks the neuronal actions of galanin in the hippocampus, locus coeruleus, and spinal cord. Proc Natl Acad Sci U S A, 1991, 88, 10961–5. [Google Scholar] [CrossRef] [PubMed]
- Haberman, R.P., R.J. Samulski. Attenuation of seizures and neuronal death by adeno-associated virus vector galanin expression and secretion. Nat Med, 2003, 9, 1076–80. [Google Scholar] [CrossRef]
- Lin, E.J.; et al. Recombinant AAV-mediated expression of galanin in rat hippocampus suppresses seizure development. Eur J Neurosci, 2003, 18, 2087–92. [Google Scholar] [CrossRef]
- McCown, T.J. Adeno-associated virus-mediated expression and constitutive secretion of galanin suppresses limbic seizure activity in vivo. Mol Ther, 2006, 14, 63–8. [Google Scholar] [CrossRef]
- Agostinho, A.S.; et al. Dynorphin-based "release on demand" gene therapy for drug-resistant temporal lobe epilepsy. EMBO Mol Med, 2019, 11, e9963. [Google Scholar] [CrossRef]
- Armijo, J.A.; et al. Ion channels and epilepsy. Curr Pharm Des, 2005, 11, 1975–2003. [Google Scholar] [CrossRef]
- Bradford, H.F. Glutamate, GABA and epilepsy. Prog Neurobiol, 1995, 47, 477–511. [Google Scholar] [CrossRef]
- During, M.J.; et al. An oral vaccine against NMDAR1 with efficacy in experimental stroke and epilepsy. Science, 2000, 287, 1453–60. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.W.; et al. Incidence of Dravet Syndrome in a US Population. Pediatrics, 2015, 136, e1310–5. [Google Scholar] [CrossRef] [PubMed]
- Ogiwara, I.; et al. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci, 2007, 27, 5903–14. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.H.; et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci, 2006, 9, 1142–9. [Google Scholar] [CrossRef]
- Han, S.; et al. Autistic-like behaviour in Scn1a+/- mice and rescue by enhanced GABA-mediated neurotransmission. Nature, 2012, 489, 385–90. [Google Scholar] [CrossRef]
- Tai, C.; et al. Impaired excitability of somatostatin- and parvalbumin-expressing cortical interneurons in a mouse model of Dravet syndrome. Proc Natl Acad Sci U S A, 2014, 111, E3139–48. [Google Scholar] [CrossRef]
- Tanenhaus, A.; et al. Cell-Selective Adeno-Associated Virus-Mediated SCN1A Gene Regulation Therapy Rescues Mortality and Seizure Phenotypes in a Dravet Syndrome Mouse Model and Is Well Tolerated in Nonhuman Primates. Hum Gene Ther, 2022, 33(11-12): 579-597.
- van der Stelt, M.; et al. Acute neuronal injury, excitotoxicity, and the endocannabinoid system. Mol Neurobiol, 2002, 26(2-3): 317-46.
- Monory, K.; et al. The endocannabinoid system controls key epileptogenic circuits in the hippocampus. Neuron, 2006, 51, 455–66. [Google Scholar] [CrossRef]
- Ludanyi, A.; et al. Downregulation of the CB1 cannabinoid receptor and related molecular elements of the endocannabinoid system in epileptic human hippocampus. J Neurosci, 2008, 28, 2976–90. [Google Scholar] [CrossRef]
- Nikitin, E.S. and L.V. Vinogradova. Potassium channels as prominent targets and tools for the treatment of epilepsy. Expert Opin Ther Targets, 2021, 25, 223–235. [Google Scholar] [CrossRef]
- Wykes, R.C.; et al. Optogenetic and potassium channel gene therapy in a rodent model of focal neocortical epilepsy. Sci Transl Med, 2012, 4, 161ra152. [Google Scholar] [CrossRef]
- Snowball, A.; et al. Epilepsy Gene Therapy Using an Engineered Potassium Channel. J Neurosci, 2019, 39, 3159–3169. [Google Scholar] [CrossRef] [PubMed]
- Magloire, V.; et al. KCC2 overexpression prevents the paradoxical seizure-promoting action of somatic inhibition. Nat Commun, 2019, 10, 1225. [Google Scholar] [CrossRef]
- Qiu, Y.; et al. On-demand cell-autonomous gene therapy for brain circuit disorders. Science, 2022, 378, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Kohling, R. and J. Wolfart, Potassium Channels in Epilepsy. Cold Spring Harb Perspect Med, 2016, 6(5).
- Masnada, S.; et al. Clinical spectrum and genotype-phenotype associations of KCNA2-related encephalopathies. Brain, 2017, 140, 2337–2354. [Google Scholar] [CrossRef]
- Muona, M.; et al. A recurrent de novo mutation in KCNC1 causes progressive myoclonus epilepsy. Nat Genet, 2015, 47, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Corbett, M.A.; et al. Dominant KCNA2 mutation causes episodic ataxia and pharmacoresponsive epilepsy. Neurology, 2016, 87, 1975–1984. [Google Scholar] [CrossRef]
- Simons, C.; et al. Mutations in the voltage-gated potassium channel gene KCNH1 cause Temple-Baraitser syndrome and epilepsy. Nat Genet, 2015, 47, 73–7. [Google Scholar] [CrossRef]
- Kole, M.H., J.J. Letzkus. Axon initial segment Kv1 channels control axonal action potential waveform and synaptic efficacy. Neuron, 2007, 55, 633–47. [Google Scholar] [CrossRef]
- Foust, A.J.; et al. Somatic membrane potential and Kv1 channels control spike repolarization in cortical axon collaterals and presynaptic boutons. J Neurosci, 2011, 31, 15490–8. [Google Scholar] [CrossRef]
- Shu, Y.; et al. Modulation of intracortical synaptic potentials by presynaptic somatic membrane potential. Nature, 2006, 441, 761–5. [Google Scholar] [CrossRef]
- Roshchin, M.V.; et al. A BK channel-mediated feedback pathway links single-synapse activity with action potential sharpening in repetitive firing. Sci Adv, 2018, 4, eaat1357. [Google Scholar] [CrossRef] [PubMed]
- Brooks-Kayal, A.R.; et al. Human neuronal gamma-aminobutyric acid(A) receptors: coordinated subunit mRNA expression and functional correlates in individual dentate granule cells. J Neurosci, 1999, 19, 8312–8. [Google Scholar] [CrossRef] [PubMed]
- Brooks-Kayal, A.R.; et al. Selective changes in single cell GABA(A) receptor subunit expression and function in temporal lobe epilepsy. Nat Med, 1998, 4, 1166–72. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; et al. Effects of status epilepticus on hippocampal GABAA receptors are age-dependent. Neuroscience, 2004, 125, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Raol, Y.H.; et al. Enhancing GABA(A) receptor alpha 1 subunit levels in hippocampal dentate gyrus inhibits epilepsy development in an animal model of temporal lobe epilepsy. J Neurosci, 2006, 26, 11342–6. [Google Scholar] [CrossRef]
- Nikitin, E.S.; et al. Overexpression of KCNN4 channels in principal neurons produces an anti-seizure effect without reducing their coding ability. Gene Ther, 2024, 31(3-4): 144-153.
- Guggenhuber, S.; et al. AAV vector-mediated overexpression of CB1 cannabinoid receptor in pyramidal neurons of the hippocampus protects against seizure-induced excitoxicity. PLoS One, 2010, 5, e15707. [Google Scholar] [CrossRef]
- Oguro, K.; et al. Global brain delivery of neuroligin 2 gene ameliorates seizures in a mouse model of epilepsy. J Gene Med, 2022, 24, e3402. [Google Scholar] [CrossRef]
- Deister, C. and C.E. Schmidt. Optimizing neurotrophic factor combinations for neurite outgrowth. J Neural Eng, 2006, 3, 172–9. [Google Scholar] [CrossRef]
- Kanter-Schlifke, I.; et al. Seizure suppression by GDNF gene therapy in animal models of epilepsy. Mol Ther, 2007, 15, 1106–13. [Google Scholar] [CrossRef]
- Humpel, C.; et al. Neurons of the hippocampal formation express glial cell line-derived neurotrophic factor messenger RNA in response to kainate-induced excitation. Neuroscience, 1994, 59, 791–5. [Google Scholar] [CrossRef]
- Mikuni, N.; et al. Time course of transient expression of GDNF protein in rat granule cells of the bilateral dentate gyri after unilateral intrahippocampal kainic acid injection. Neurosci Lett, 1999, 262, 215–8. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Kastner, R.; et al. Glial cell-line derived neurotrophic factor (GDNF) mRNA upregulation in striatum and cortical areas after pilocarpine-induced status epilepticus in rats. Brain Res Mol Brain Res, 1994, 26(1-2): 325-30.
- Martin, D.; et al. Potent inhibitory effects of glial derived neurotrophic factor against kainic acid mediated seizures in the rat. Brain Res, 1995, 683, 172–8. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; et al. Glial cell line-derived neurotrophic factor modulates kindling and activation-induced sprouting in hippocampus of adult rats. Exp Neurol, 2002, 178, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Yoo, Y.M.; et al. Neuroprotection of adenoviral-vector-mediated GDNF expression against kainic-acid-induced excitotoxicity in the rat hippocampus. Exp Neurol, 2006, 200, 407–17. [Google Scholar] [CrossRef]
- Lee, J.M. and J.A. Johnson. An important role of Nrf2-ARE pathway in the cellular defense mechanism. J Biochem Mol Biol, 2004, 37, 139–43. [Google Scholar] [CrossRef]
- Nguyen, T., P. J. Sherratt, and C.B. Pickett, Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol, 2003, 43: 233-60.
- Hybertson, B.M.; et al. Oxidative stress in health and disease: the therapeutic potential of Nrf2 activation. Mol Aspects Med, 2011, 32(4-6): 234-46.
- Mazzuferi, M.; et al. Nrf2 defense pathway: Experimental evidence for its protective role in epilepsy. Ann Neurol, 2013, 74, 560–8. [Google Scholar] [CrossRef]
- Klugmann, M.; et al. AAV-mediated hippocampal expression of short and long Homer 1 proteins differentially affect cognition and seizure activity in adult rats. Mol Cell Neurosci, 2005, 28, 347–60. [Google Scholar] [CrossRef]
- Zerangue, N. and M.Kavanaugh. Flux coupling in a neuronal glutamate transporter. Nature, 1996, 383, 634–7. [Google Scholar] [CrossRef]
- Rothstein, J.D.; et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron, 1996, 16, 675–86. [Google Scholar] [CrossRef]
- Mathern, G.W.; et al. Hippocampal GABA and glutamate transporter immunoreactivity in patients with temporal lobe epilepsy. Neurology, 1999, 52, 453–72. [Google Scholar] [CrossRef]
- Proper, E.A.; et al. Distribution of glutamate transporters in the hippocampus of patients with pharmaco-resistant temporal lobe epilepsy. Brain, 2002, 125(Pt 1): 32-43.
- van der Hel, W.S.; et al. Reduced glutamine synthetase in hippocampal areas with neuron loss in temporal lobe epilepsy. Neurology, 2005, 64, 326–33. [Google Scholar] [CrossRef] [PubMed]
- During, M.J. and D.D. Spencer. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet, 1993, 341, 1607–10. [Google Scholar] [CrossRef] [PubMed]
- Eid, T.; et al. Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet, 2004, 363, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Tessler, S.; et al. Expression of the glutamate transporters in human temporal lobe epilepsy. Neuroscience, 1999, 88, 1083–91. [Google Scholar] [CrossRef]
- Liaw, S.H., I. Kuo. and D. Eisenberg, Discovery of the ammonium substrate site on glutamine synthetase, a third cation binding site. Protein Sci, 1995, 4, 2358–65. [Google Scholar] [CrossRef] [PubMed]
- Suarez, I., G. Bodega, and B. Fernandez, Glutamine synthetase in brain: effect of ammonia. Neurochem Int, 2002, 41(2-3): 123-42.
- Venkatesh, K.; et al. In vitro differentiation of cultured human CD34+ cells into astrocytes. Neurol India, 2013, 61, 383–8. [Google Scholar]
- Young, D.; et al. Adenosine kinase, glutamine synthetase and EAAT2 as gene therapy targets for temporal lobe epilepsy. Gene Ther, 2014, 21, 1029–40. [Google Scholar] [CrossRef]
- Phillips, M.I.; et al. Prolonged reduction of high blood pressure with an in vivo, nonpathogenic, adeno-associated viral vector delivery of AT1-R mRNA antisense. Hypertension, 1997, 29(1 Pt 2): 374-80.
- Evers, M.M., L. J. Toonen, and W.M. van Roon-Mom, Antisense oligonucleotides in therapy for neurodegenerative disorders. Adv Drug Deliv Rev, 2015, 87: 90-103.
- Xiao, X.; et al. Adeno-associated virus (AAV) vector antisense gene transfer in vivo decreases GABA(A) alpha1 containing receptors and increases inferior collicular seizure sensitivity. Brain Res, 1997, 756(1-2): 76-83.
- Haberman, R.; et al. Therapeutic liabilities of in vivo viral vector tropism: adeno-associated virus vectors, NMDAR1 antisense, and focal seizure sensitivity. Mol Ther, 2002, 6, 495–500. [Google Scholar] [CrossRef]
- Theofilas, P.; et al. Adenosine kinase as a target for therapeutic antisense strategies in epilepsy. Epilepsia, 2011, 52, 589–601. [Google Scholar] [CrossRef]
- Xu, X.; et al. Tubulin β-III modulates seizure activity in epilepsy. J Pathol, 2017, 242, 297–308. [Google Scholar] [CrossRef]
- Han, C.L.; et al. Long non-coding RNA H19 contributes to apoptosis of hippocampal neurons by inhibiting let-7b in a rat model of temporal lobe epilepsy. Cell Death Dis, 2018, 9, 617. [Google Scholar] [CrossRef] [PubMed]
- Han, C.L.; et al. Whole-transcriptome screening reveals the regulatory targets and functions of long non-coding RNA H19 in epileptic rats. Biochem Biophys Res Commun, 2017, 489, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.C.; et al. Selective targeting of Scn8a prevents seizure development in a mouse model of mesial temporal lobe epilepsy. Sci Rep, 2018, 8, 126. [Google Scholar] [CrossRef] [PubMed]
- Peret, A.; et al. Contribution of aberrant GluK2-containing kainate receptors to chronic seizures in temporal lobe epilepsy. Cell Rep, 2014, 8, 347–54. [Google Scholar] [CrossRef]
- Boileau, C.; et al. GluK2 Is a Target for Gene Therapy in Drug-Resistant Temporal Lobe Epilepsy. Ann Neurol, 2023, 94, 745–761. [Google Scholar] [CrossRef]
Figure 1.
Components of the effective use of AAV vectors in the treatment of CNS disorders.
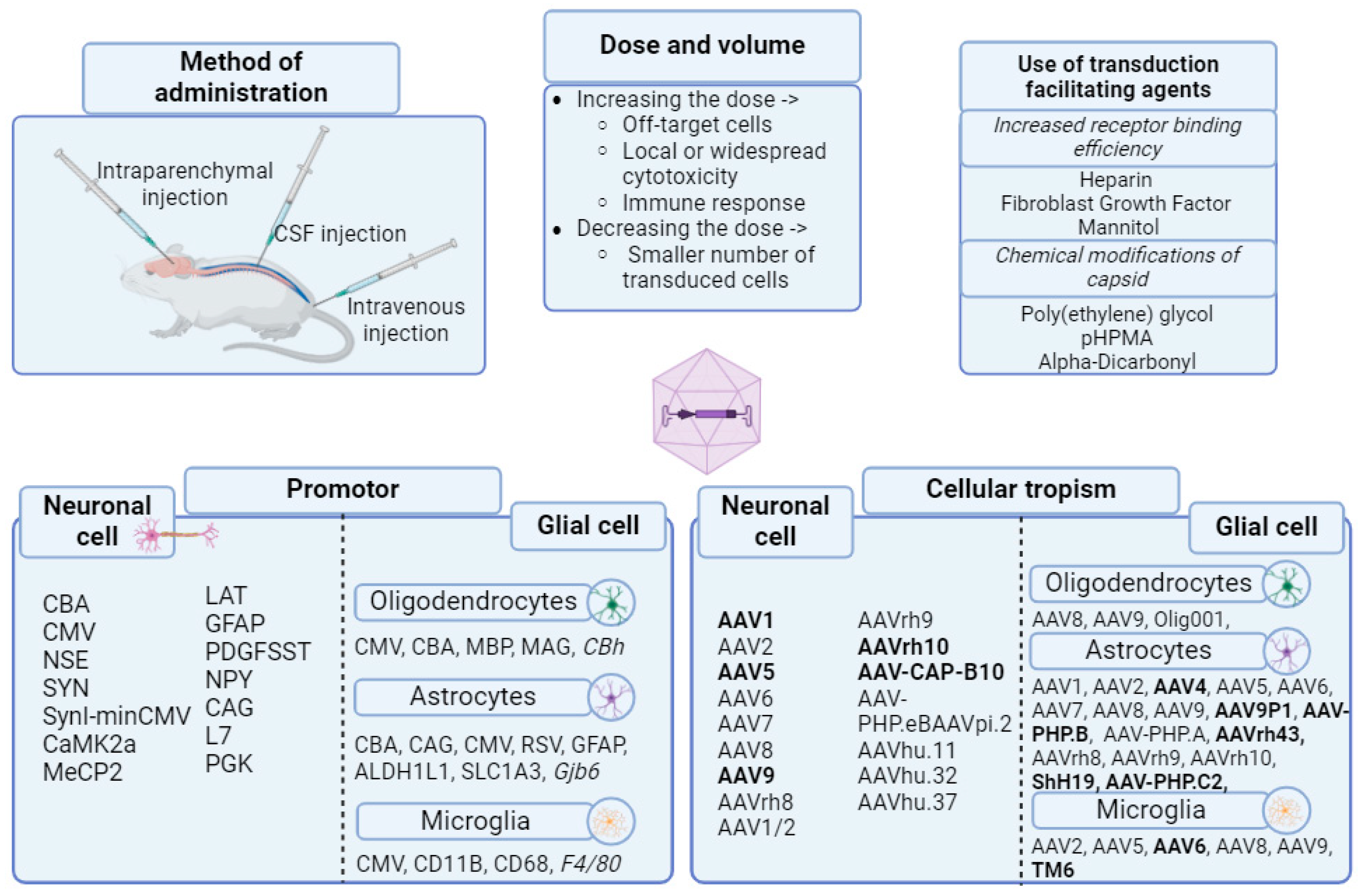
Figure 2.
PRISMA chart. The PRISMA flow diagram follows the PRISMA checklist (http://prisma-statement.org/PRISMAStatement/Checklist).
Figure 2.
PRISMA chart. The PRISMA flow diagram follows the PRISMA checklist (http://prisma-statement.org/PRISMAStatement/Checklist).
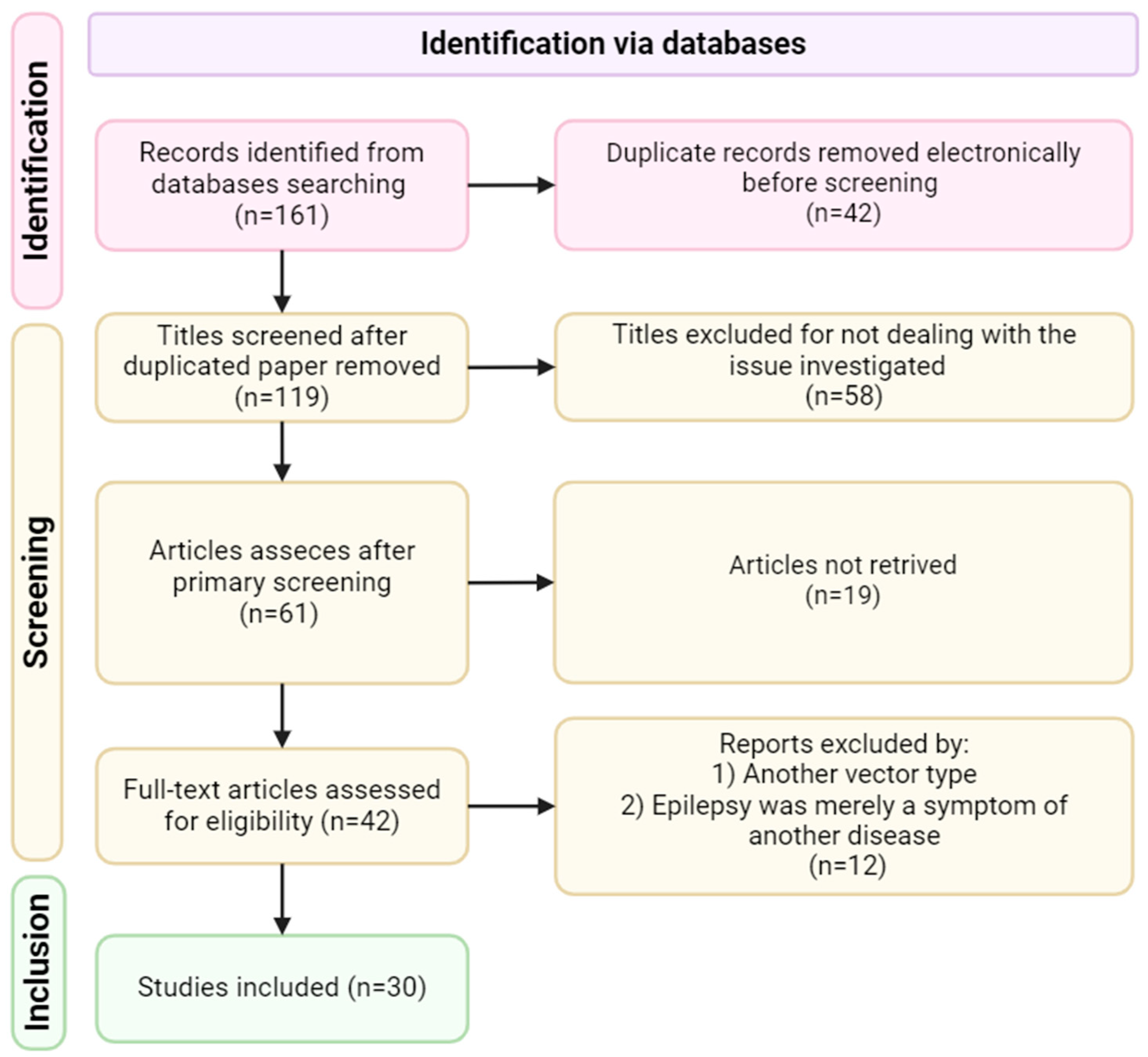

Table 1.
Data on AVV-mediated delivery of therapeutic genes.
| Serotype | Delivery gene | Model | Antiepileptic effect | Reference |
|---|---|---|---|---|
| AAV2; AAV1/2 | NPY | КА, rats | Reducing the number of seizures. Reducing the duration of seizure activity | [139] |
| AAV2 | КА, rats | Increase in the latent period of limbic convulsive activity | [148] | |
| AVV1/2 | Electrical stimulation, rats | Reducing the frequency of seizures. Decrease in the progression of seizures by 80% | [140] | |
| AAV1 | КА, rats | Reducing the number of seizures by 55%. Reducing the duration of ictal activity | [141] | |
| AAV1/2 | КА, rats | Preventing the progression of the frequency of seizures by 57.5 ± 19.3%. Reducing the duration of seizure activity | [150] | |
| AAV1/2 | GAERS GGE, rats | Reducing the duration of seizure activity, reducing the number of seizures | [151] | |
| AAV1 | КА, rats | Reducing the severity of seizures. Increase in the latent time. Decrease in the frequency and total time of seizure activity. SRS suppression | [152] | |
| AAV1, AAV2, AAV8 | КА, rats + resected human hippocampus | AAV1 reduces the time spent on motor seizures and increases the latency period to SE | [154] | |
| AAV2 | Y2 | Kindling, КА, rats | Reduction of the cumulative degree of seizures. Reducing the number of severe grade 4-5 seizures and increasing the amount of stimulation needed to achieve grade 3 or 4-5 seizures | [149] |
| AAV2 | GAL | КА, rats, HEK 293 cells | Decrease of in vivo sensitivity to focal seizures and prevention of hilar cell death caused by kainic acid | [169] |
| AAV2 | КА, rats, HEK 293 cells | Increase in the threshold of excitability. Reduction of neuronal death | [170] | |
| AAV2 | КА, Kindling, rats | Suppression of limbic seizures. Increase in the stimulation current required to induce limbic convulsive activity | [171] | |
| AAV1/2 | HOMER1 | Electrical stimulation, rats | Suppression of SSLSE | [218] |
| AAV2 | GABRA4 | Electrical stimulation, rats | Suppression of SSLSE | [202] |
| AAV2 | GDNF | Kindling, rats | Suppression of SSLSE. Increased survival after SE | [207] |
| AAV1/2 | CB1 | КА, mice | Reducing the severity of seizures. Protection from excitotoxicity and neuronal death | [204] |
| AAV2 | Nrf2 | Lithium-pilocarpine model, mice | Decrease in microglia activation. The ratio between astrocytes/neurons and activated microglia/neurons was significantly lower | [217] |
| AAV9 | EAAT2 | КА, rats | There were no differences in the total duration of seizures between the animals that were injected with the EAAT2, GS vector, and the control vector | [230] |
| GS | ||||
| AAV1 | Dyn | КА, electrical stimulation, rats, mice | Suppression of seizures. The disappearance of generalized seizures | [172] |
| AAV2 | NL2 | Models of polygenic epilepsy, mice | Significant decrease in the duration, strength and frequency of seizures was observed over a 14-week period | [205] |
| AAV9 | SCN1A | Scn1a+/-, mice, primates | Reducing the frequency, duration and severity of spontaneous seizures and reduces the sensitivity of HTS in Dravet mice | [181] |
| AAV2 | KCNN4 | 4-aminopyridine model, mice | Powerful suppression of pharmacologically induced seizures in vitro both at the level of single cells and at the level of the local field with a decrease in peaks during ictal discharges | [203] |
Table 2.
The data on the repression of some genes using AAV vectors.
| Serotype | Gene for blocking | Model | Effect | Reference |
|---|---|---|---|---|
| AAV2 | GABA(A) | КА, Kindling, rats | Modulation of colliculi inferiores convultions | [233] |
| AAV2 | NMDAR1 | КА, rats | Significant decrease in sensitivity to focal seizures within 4 weeks. The design of the pTet promoter caused the opposite effect — a significant increase in sensitivity to seizures. It has been shown that in the brain, NMDA receptor excitation can cause GABA inhibition [21], therefore, removing NMDA receptor excitation from these inhibitory interneurons can create a state of hyperexcitability. | [234] |
| AAV8 | ADK | Transgenic mice with spontaneous epilepsy | Preventing of convulsive activity | [235] |
| AAV9 | lnRNA H19 | КА, rats | The CA3 neurons were preserved | [238] |
| AAV10 | Scn8a | КА, mice | Protection from spontaneous seizures. Reduction of gliosis | [238] |
| AAV9 | GluK2 | Pilocarpine, mice | Reduction of chronic convulsive activity | [241] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



