
Submitted:
30 September 2024
Posted:
01 October 2024
You are already at the latest version
Abstract
Canine Multiple System Degeneration (CMSD) is an early onset, progressive movement disorder affecting Kerry Blue Terriers and Chinese Crested dogs. The associated pathologic lesions include degeneration of the cerebellum, caudate nucleus and substantia nigra. CMSD is inherited as an autosomal recessive trait in both dog breeds. Previous linkage mapping localized the CMSD locus to a 15 MB region on canine chromosome 1 (CFA1). Next generation sequencing was used to generate whole genome sequences from DNA of an affected dog from each breed. The resulting sequence reads were aligned to the NCBI canine reference genome (build 3.1). Among the homozygous sequence variants within the CFA1 target region, a nonsense variant in exon 15 of SERAC1 was identified in the affected Kerry Blue Terrier, while in the Chinese Crested dog, a 4 bp deletion in the SERAC1 exon 4 acceptor splice site was found. RT-PCR showed that this deletion resulted in exon 4 skipping. Genotyping of large cohorts of Kerry Blue Terriers and Chinese Crested dogs for the respective breed-specific SERAC1 variants showed complete concordance between genotype and disease phenotype. Genotype-phenotype concordance was also observed in offspring generated by cross breeding between SERAC1-heterozygous Kerry Blue Terrier and Chinese Crested dogs, with only the compound heterozygotes exhibiting the disease phenotype, further confirming the recessive inheritance of CMSD. Variants in human SERAC1 are associated with disorders with a range of ages of disease onset and patterns of clinical signs, but that are all characterized by movement abnormalities similar to those of the dogs with CMSD. Canine CMSD could serve as a valuable model to elucidate the mechanisms underlying SERAC1-deficiency disorders and to evaluate potential therapeutic interventions.

Keywords:
movement disorder
; dog
; mitochondria
; whole genome sequencing
; neurodegeneration
1. Introduction
Canine multiple system degeneration (CMSD) is a progressive early onset movement disorder that occurs in Kerry Blue Terrier and Chinese Crested dogs [1,2]. Disease onset is characterized by cranial intention tremor and cerebellar ataxia that first become apparent at 3 to 6 months of age. The affected dogs exhibit a goose-stepping gait and infrequent falls during this early stage. By 6 to 8 months of age, the falling episodes become more frequent and the gait changes to festination with dysmetria. As the disease progresses, affected dogs develop akinesia and severe postural instability and frequent falls. Magnetic resonance imaging of the brains demonstrated disease-related cerebellar atrophy. Due to the progressive severity of neurologic signs, affected dogs are typically euthanized humanely by 13 to 18 months of age. Postmortem examination of the brains revealed Purkinje cell loss in the cerebellum and neuronal loss from the substantia nigra, putamen and caudate nucleus [1]. The patterns of inheritance in both breeds indicated that CMSD is an autosomal recessive disorder.
Previous microsatellite-based linkage mapping localized the CMSD disease loci to chromosome 1 in both breeds [1]. Further haplotype analysis using 11 additional microsatellite markers on CFA1 narrowed the target region to a 15 MB region containing 89 positional candidate genes. Among the genes in this region is PARK2, a gene associated with autosomal recessive juvenile parkinsonism, which presents with similar signs to CMSD including akinesia, gait abnormalities and postural instability. [3]. Degeneration of neurons in the substantia nigra, loss of Purkinje cells, and cerebellar ataxia are also reported in some human patients with Parkinson-plus syndromes [4,5,6,7]. Given these similarities, it was hypothesized that variants in PARK2 may be responsible for CMSD. However, this has not yet been directly tested. To identify the underlying genetic variants underlying CMSD, whole genome sequence analyses were performed on affected dogs from both Kerry Blue Terrier and Chinese Crested breeds.
2. Materials and Methods
2.1. Interbreed Mating Study
Because the disease phenotypes in the Chinese Crested and Kerry Blue Terrier dogs were identical and the causal variant mapped to the same region of CFA1 in both breeds, we hypothesized that the disease in both breeds resulted from variants in the same gene. To test this hypothesis, we evaluated the pattern of inheritance of the disease phenotype in offspring by crossing obligate carriers from each breed (Chinese Crested X Kerry Blue Terrier). The offspring of this cross were monitored for the development of the characteristic CMSD phenotype.
2.2. Whole Genome Sequence Analyses
Genomic DNA was isolated from blood leukocytes from an affected Kerry Blue Terrier and an affected Chinese Crested dog as described previously [8]. DNA samples were submitted to the University of Missouri Genomics Technology Core Facility for library construction and 2 x 150 base pair paired-end sequencing on an Illumina HiSeq 2000, with a target coverage of 20x. Raw sequence reads were aligned to the canine reference genome (build 3.1) by a modified Burrows-Wheeler Transform (BMT) alignment method with NextGENe v2.3.3 software which takes into account parameters including mapping quality, Phred quality for each base, read depth, repetitiveness and minor allele frequency. Sequence variant calling and reference sequence annotation were performed by integrating the NCBI canine genome reference sequence annotation with NextGENe software.
To identify candidate pathogenic mutations, we generated a sequence variants report table for each dog that listed all the homozygous variants detected in the coding sequences relative to the reference genome. This included homozygous splice site variants, nonsynonymous missense variants, indel variants, stop to read-through variants, and premature stop codon variants. The sequence variants were further filtered to exclude homozygous variants that were present in the whole genome sequences of 102 dogs that had not exhibited signs of movement disorders. These whole genome sequences had been generated previously by us using approaches similar to those used in this study. By focusing only on the sequence variants within the CFA1 disease target region identified by our previous linkage analysis, we generated final variant reports for each of the CMSD-affected dogs.
Among these sequence variants were a nonsense variant caused by a G to A substitution in exon 15 of SERAC1 in the affected Kerry Blue Terrier and a 4 bp deletion in SERAC1 exon 4 acceptor splice site in the Chinese Crested dog. Individual canine DNA samples were genotyped with respect to the 4 bp deletion by direct re-sequencing DNA with an automated Sanger sequencer (3730xl; Applied Biosystems). The forward primer was 5′-GGAAATATAATAAAGTTTACTGG-3′ and reverse primer was 5′- CAAAATTTATACATATTTGCCAC-3′. The resulting amplicon was 140 bp in length. Individual canine DNA samples were genotyped with respect to the G to A nucleotide change which produced a premature stop codon by RFLP-PCR. The PCR forward primer was 5′- CCCAATAAAAGCTCTTGCCT-3′ and reverse primer was 5′- GGCCAGAATTAAGTGAACCA-3′. Restriction enzyme BstXI enzyme was used to digest the PCR product by using the following reaction conditions: 3U BstXI enzyme and 1X NEBuffer 3 incubated at 37oC for 2 hours. For the ancestral allele, the 299 bp PCR product remains intact; whereas, the digestion cuts the mutant allele into 182 bp and 117 bp oligonucleotides. Restriction fragment sizes were determined with a microcapillary electrophoresis system (QIAxel, Qiagen).
TRIzol Reagent (Invitrogen) was used to extract total RNA from the brains of one affected and two unaffected Chinese Crested dogs. The affected dog was euthanized due to progression of the movement disorder signs and the unaffected dogs were euthanized due to unrelated health issues. RT-PCR amplifications were performed with a GeneAmp®EZ rTth RNA PCR kit (Applied Biosystems) using the following primer pairs: 5′-TGCAGAAGAATAGGAACCTCA-3′ and 3′-TTGCCTGGTAGGTGATTCCAT-5′. The resulting amplicons were evaluated with a microcapillary electrophoresis system (QIAxcel, Qiagen).
3. Results
Litters of both Kerry Blue Terriers and Chinese Crested dogs from unaffected parents included both CMSD-affected and unaffected offspring (Figure 1), indicating that the disorder is a recessive trait in both breeds. The cross breeding of obligate carrier Kerry Blue Terrier and Chinese Crested dogs produced a litter that included both CMSD-affected and unaffected offspring (Figure 1). This finding indicated that the casual variants likely to reside in the same gene in the two breeds.
The whole-genome sequence from a CMSD-affected Chinese Crested dog consisted of 167,497,918 reads, of which 120,222 were duplicate reads and 167,377,696 were unique. The whole-genome sequence from an affected Kerry Blue Terrier consisted of 384,505,652 reads, of which only 360 were duplicate reads and 384,505,292 were unique. The unique reads generated from both dogs were aligned separately to the canine reference genome using NextGENe software, resulting in an aligned sequence with average 18-fold coverage for the Chinese Crested dog and average 22.3-fold coverage for the Kerry Blue Terrier. For each dog, the sequence variants were filtered to include only those that were homozygous in the probands. Variants found to be homozygous in the whole genome sequences of any of 102 other dogs in our internal database that did not suffer from CMSD were also excluded. The remaining homozygous variants were further filtered to include only those located within the CFA1 region to which we previously mapped the causal variant [1].
Two variants in the Kerry Blue Terrier met the filtering criteria: a nonsense variant in SERAC1 (Figure 2) and a missense variant in SLC22A2. The SERAC1 variant, which introduces a premature stop codon (p.W512X) in exon 15, was considered more likely to be causal due to its predicted impact on the encoded protein and the associations of variants in this gene with neurological disorders [9].
The whole genome sequence of the CMSD-affected Chinese Crested dog contained a 4bp (GTAA) deletion in the acceptor splice site of SERAC1 exon 4 (Figure 3). SplicePort (http://spliceport.cbcb.umd.edu/SplicingAnalyser.html), an interactive splice site analysis tool, has predicted that the mutation would cause exon skipping. This prediction was confirmed by the RT-PCR amplification of SERAC1 transcripts between exon 3 and exon 5, which showed a reduced transcript length in RNA from an affected Chinese Crested dog corresponding to the omission of exon 4 (Figure 4). Automated Sanger sequencing of the RT-PCR product confirmed exon 4 skipping in the transcript from the affected dog (Figure 5).
A PCR-RFLP assay was used to genotype individual Kerry Blue Terriers for the SERAC1 nonsense variant (Figure 6). Genotyping was performed on a cohort of 228 Kerry Blue Terriers with known clinical status. All CMSD-affected dogs were homozygous for the variant allele, while clinically unaffected dogs were either homozygous for the reference allele or heterozygous. Homozygosity for the mutant allele was highly significantly associated with the CMSD phenotype (p < 1.0 x 10-9, Fisher’s exact test 2×2). Table 1 summarizes the genotype distribution. 90 dogs from 25 other breeds that did not exhibit CMSD signs were all homozygous for the reference allele.
A cohort of 183 Chinese Crested dogs with known clinical status was genotyped to screen for the SERAC1 4bp deletion using automated Sanger sequencing of PCR amplicons generated with primers spanning the deletion site (Figure 7). Table 2 summarizes the genotype and phenotype distributions. All 41 CMSD-affected dogs were homozygous for the mutant allele, while clinically unaffected dogs were either homozygous for the reference allele or heterozygous. Homozygosity for the mutant allele was highly significantly associated with the CMSD phenotype (p < 1.0 x 10-21, Fisher’s exact test 2×2). 131 dogs from 11 other breeds, none of which exhibited CMSD signs, were homozygous for the reference allele.
4. Discussion
Hereditary CMSD was first described in Kerry Blue Terriers as early as 1946 [2,10] and subsequently in Chinese Crested dogs [1]. Linkage analyses using microsatellite markers localized the genetic variants underlying the disease to a 15-Mb region of CFA1 in both breeds, but the specific variants underlying the disorder in these breeds were not identified. Because the clinical signs of CMSD resemble those in some forms of human Parkinson’s disease associated with variants in PARK2, and because the canine ortholog of PARK2 is located within the mapped CFA1 region, it was hypothesized that that variants in PARK2 might underlie CMSD in both breeds. However, the whole genome sequences of CMSD-affected dogs from both breeds did not contain homozygous variants in PARK2 relative to the reference sequence.
With advances in technology and improved annotation of the canine genome, we were able to identify the causal variants in SERAC1 in both breeds. The SEARAC1 protein is localized to the outer mitochondrial membrane where it facilitates serine transport from the cytosol to the mitochondria [11]. SERAC1 has also been shown to play a role in modulating mitochondrial membrane phospholipid composition and intracellular cholesterol trafficking [12]. Lack of functional SERAC1 protein results in depletion of mitochondrial DNA leading to mitochondrial dysfunction [11]. Thus, it appears likely that the signs of CMSD are the result of impaired mitochondrial function. Evidence suggests that the mitochondrial dysfunction resulting from SERAC1 deficiency is due at least in part to insufficient supply of nucleotides to the mitochondria, and that supplementation with nucleosides or nucleotides can restore mitochondrial function [11].
Variants in human SERAC1 are associated with a spectrum of disorders, including MEGDHEL syndrome (3-methylglutaconic aciduria with deafness-dystonia, hepatopathy, encephalopathy, and Leigh-like syndrome) [9,13,14,15], juvenile-onset complicated spastic paraplegia [16], and adult-onset generalized dystonia [9,17]. Age of onset and the spectrum and severity of signs in human SERAC1-associated disorders varies widely. MEGDHEL, the most severe form, typically presents with neonatal or infantile onset with signs that include transient hypoglycemia, feeding problems, failure to thrive, optic atrophy, developmental delay followed by motor and cognitive regression, progressive sensorineural hearing loss, progressive dystonia, and progressive spasticity [9,14,18]. Most children with this form of SERAC1 deficiency are completely dependent on care for all activities of daily living, and in some cases, the disease is fatal. Over 40 different SERAC1 variants, including missense, splice site, frame shift and nonsense variants, have been found to be associated with the early-onset MEGDHEL [13,14,15,19,20]. A juvenile-onset disorder associated with SERAC1 deficiency is characterized by cognitive delay, and slowly progressive lower limb spasticity beginning in adolescence [16]. This disorder was associated with a splice variant that resulted in the absence of full-length SERAC1 protein. The adult-onset disease associated with SERAC1 deficiency is characterized by cognitive regression and progressive dystonia beginning in early adulthood [17,21,22].
All of the human SERAC1 deficiency disorders are characterized by dystonia, which is the most prominent sign in the canine disease. Although cognitive function and hearing loss were not assessed in the affected dogs, the canine disorder appears to closely resemble early-onset MEGDHEL syndrome. Because dogs with SERAC1-related CMSD could serve as a valuable model to test therapeutic interventions for the human disorders, further phenotypic characterization of the canine disease is warranted. Evidence suggests that nucleotide supplementation corrects the SERAC1 deficiency-related mitochondrial dysfunction and therefore may have therapeutic benefits [11]. This could be tested in dogs with CMSD.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, G.S.J; methodology, R.Z., G.S.J., G.B., and J.G..; software, G.B.; validation, R.Z., G.B. and J.G..; formal analysis, R.Z., G.B., and M.L.K..; investigation, R.Z. and J.G.; resources, G.S.J. and M.L.K. data curation, G.B..; writing—original draft preparation, R.Z. and M.L.K.; writing—review and editing, R.Z., J.G., G.B., S.C., and M.L.K..; supervision, M.L.K.; project administration, M.L.K.; funding acquisition, G.S.J. and M.L.K. All authors other than G.S.J. who was deceased prior to final manuscript preparation have read and agreed to the submitted version of the manuscript.
Funding
This research was supported by funding provided by the Orthopedic Foundation for Animals.
Institutional Review Board Statement
This study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board of the University of Missouri (protocol 20520, approved 22 December 2021).
Informed Consent Statement
Not applicable.
Data Availability Statement
DNA sequence data for the dogs included in this study have been archived and deposited in the NCBI Sequence Read Archive as BioSamples SAMN03580379 and SAMN03580387.
Acknowledgments
Our thanks to Robert Schnabel for assistance with whole genome sequence analyses.
Conflicts of Interest
The Canine Genetics Laboratory at the University of Missouri provides fee-for-service genetic testing for dogs. The authors disclose no other conflict of interest.
References
- O’Brien, D.P.; Johnson, G.S.; Schnabel, R.D.; Khan, S.; Coates, J.R.; Johnson, G.C.; Taylor, J.F. Genetic Mapping of Canine Multiple System Degeneration and Ectodermal Dysplasia Loci. J. Hered. 2005, 96, 727–734. [Google Scholar] [CrossRef] [PubMed]
- deLahunta, A.; Averill, D.R.J. Hereditary Cerebellar Cortical and Extrapyramidal Nuclear Abiotrophy in Kerry Blue Terriers. J Am Vet Med Assoc 1976, 168, 1119–1124. [Google Scholar] [PubMed]
- Shimura, H.; Hattori, N.; Kubo, S. i; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K.; et al. Familial Parkinson Disease Gene Product, Parkin, Is a Ubiquitin-Protein Ligase. Nat Genet 2000, 25, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.-W.; Hong, S.-W.; Youn, Y.C. Clinical Aspects of the Differential Diagnosis of Parkinson’s Disease and Parkinsonism. J Clin Neurol 2022, 18, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; McFarland, N. Recognizing and Treating Atypical Parkinson Disorders. Handb Clin Neurol 2019, 167, 301–320. [Google Scholar] [CrossRef]
- Glasmacher, S.A.; Leigh, P.N.; Saha, R.A. Predictors of Survival in Progressive Supranuclear Palsy and Multiple System Atrophy: A Systematic Review and Meta-Analysis. J Neurol Neurosurg Psychiatry 2017, 88, 402–411. [Google Scholar] [CrossRef]
- Armstrong, M.J.; McFarland, N. Recognizing and Treating Atypical Parkinson Disorders. Handb Clin Neurol 2019, 167, 301–320. [Google Scholar] [CrossRef]
- Katz, M.L.; Khan, S.; Awano, T.; Shahid, S.A.; Siakotos, A.N.; Johnson, G.S. A Mutation in the CLN8 Gene in English Setter Dogs with Neuronal Ceroid-Lipofuscinosis. Biochem Biophys Res Commun 2005, 327. [Google Scholar] [CrossRef]
- Wortmann, S.B.; de Brouwer, A.P.M.; Wevers, R.A.; Morava, E. SERAC1 Deficiency. 1993.
- Metler, F.; Goss, L. Canine Chorea Due to Striatocerebellar Degeneration of Unknown Etiology. J Am Vet Med Assoc 1946, 108, 377–384. [Google Scholar]
- Fang, H.; Xie, A.; Du, M.; Li, X.; Yang, K.; Fu, Y.; Yuan, X.; Fan, R.; Yu, W.; Zhou, Z.; et al. SERAC1 Is a Component of the Mitochondrial Serine Transporter Complex Required for the Maintenance of Mitochondrial DNA. Sci Transl Med 2022, 14, eabl6992. [Google Scholar] [CrossRef]
- Wortmann, S.B.; Vaz, F.M.; Gardeitchik, T.; Vissers, L.E.L.M.; Renkema, G.H.; Schuurs-Hoeijmakers, J.H.M.; Kulik, W.; Lammens, M.; Christin, C.; Kluijtmans, L.A.J.; et al. Mutations in the Phospholipid Remodeling Gene SERAC1 Impair Mitochondrial Function and Intracellular Cholesterol Trafficking and Cause Dystonia and Deafness. Nat Genet 2012, 44, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Unal, O.; Ozgul, R.K.; Yucel, D.; Yalnizoglu, D.; Tokatli, A.; Sivri, H.S.; Hismi, B.; Coskun, T.; Dursun, A. Two Turkish Siblings with MEGDEL Syndrome Due to Novel SERAC1 Gene Mutation. Turk J Pediatr 2015, 57, 388–393. [Google Scholar] [PubMed]
- Snanoudj, S.; Mordel, P.; Dupas, Q.; Schanen, C.; Arion, A.; Gerard, M.; Read, M.-H.; Nait Rabah, D.; Goux, D.; Chapon, F.; et al. Identification of a Novel Splice Site Mutation in the SERAC1 Gene Responsible for the MEGDHEL Syndrome. Mol Genet Genomic Med 2019, 7, e815. [Google Scholar] [CrossRef] [PubMed]
- Alagoz, M.; Kherad, N.; Turkmen, S.; Bulut, H.; Yuksel, A. A Novel Mutation in the SERAC1 Gene Correlates with the Severe Manifestation of the MEGDEL Phenotype, as Revealed by Whole-Exome Sequencing. Exp Ther Med 2020, 19, 3505–3512. [Google Scholar] [CrossRef]
- Roeben, B.; Schule, R.; Ruf, S.; Bender, B.; Alhaddad, B.; Benkert, T.; Meitinger, T.; Reich, S.; Bohringer, J.; Langhans, C.-D.; et al. SERAC1 Deficiency Causes Complicated HSP: Evidence from a Novel Splice Mutation in a Large Family. J Med Genet 2018, 55, 39–47. [Google Scholar] [CrossRef]
- Martins, E.; Duraes, J.; Nogueira, C.; Gomes, J.; Vilarinho, L.; Macario, C. SERAC1 Deficiency- A New Phenotype. Endocr Metab Immune Disord Drug Targets 2023. [Google Scholar] [CrossRef]
- Finsterer, J.; Scorza, F.A.; Fiorini, A.C.; Scorza, C.A. MEGDEL Syndrome. Pediatr Neurol 2020, 110, 25–29. [Google Scholar] [CrossRef]
- Maas, R.R.; Iwanicka-Pronicka, K.; Kalkan Ucar, S.; Alhaddad, B.; AlSayed, M.; Al-Owain, M.A.; Al-Zaidan, H.I.; Balasubramaniam, S.; Baric, I.; Bubshait, D.K.; et al. Progressive Deafness-Dystonia Due to SERAC1 Mutations: A Study of 67 Cases. Ann Neurol 2017, 82, 1004–1015. [Google Scholar] [CrossRef]
- Radha Rama Devi, A.; Lingappa, L. Novel Mutations in SERAC1 Gene in Two Indian Patients Presenting with Dystonia and Intellectual Disability. Eur J Med Genet 2018, 61, 100–103. [Google Scholar] [CrossRef]
- Giron, C.; Roze, E.; Degos, B.; Meneret, A.; Jardel, C.; Lannuzel, A.; Mochel, F. Adult-Onset Generalized Dystonia as the Main Manifestation of MEGDEL Syndrome. Tremor Other Hyperkinet Mov (N Y) 2018, 8, 554. [Google Scholar] [CrossRef]
- Ashton, C.; Davis, M.; Laing, N.; Ravenscroft, G.; Lamont, P. Novel SERAC1 Variant Presenting With Adult-Onset Extrapyramidal Dystonia-Parkinsonism Phenotype: A Case Report. Neurol Genet 2023, 9, e200067. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Pedigree showing segregation consistent with an autosomal recessive inheritance for CMSD and that the causal variants likely occurred in the same gene in the two breeds. The background is blue-shaded behind the Kerry Blue Terrier family, yellow-shaded behind the Chinese Crested dogs, and green-shaded behind the cross-bred litter. (Adapted from [1]).
Figure 1.
Pedigree showing segregation consistent with an autosomal recessive inheritance for CMSD and that the causal variants likely occurred in the same gene in the two breeds. The background is blue-shaded behind the Kerry Blue Terrier family, yellow-shaded behind the Chinese Crested dogs, and green-shaded behind the cross-bred litter. (Adapted from [1]).
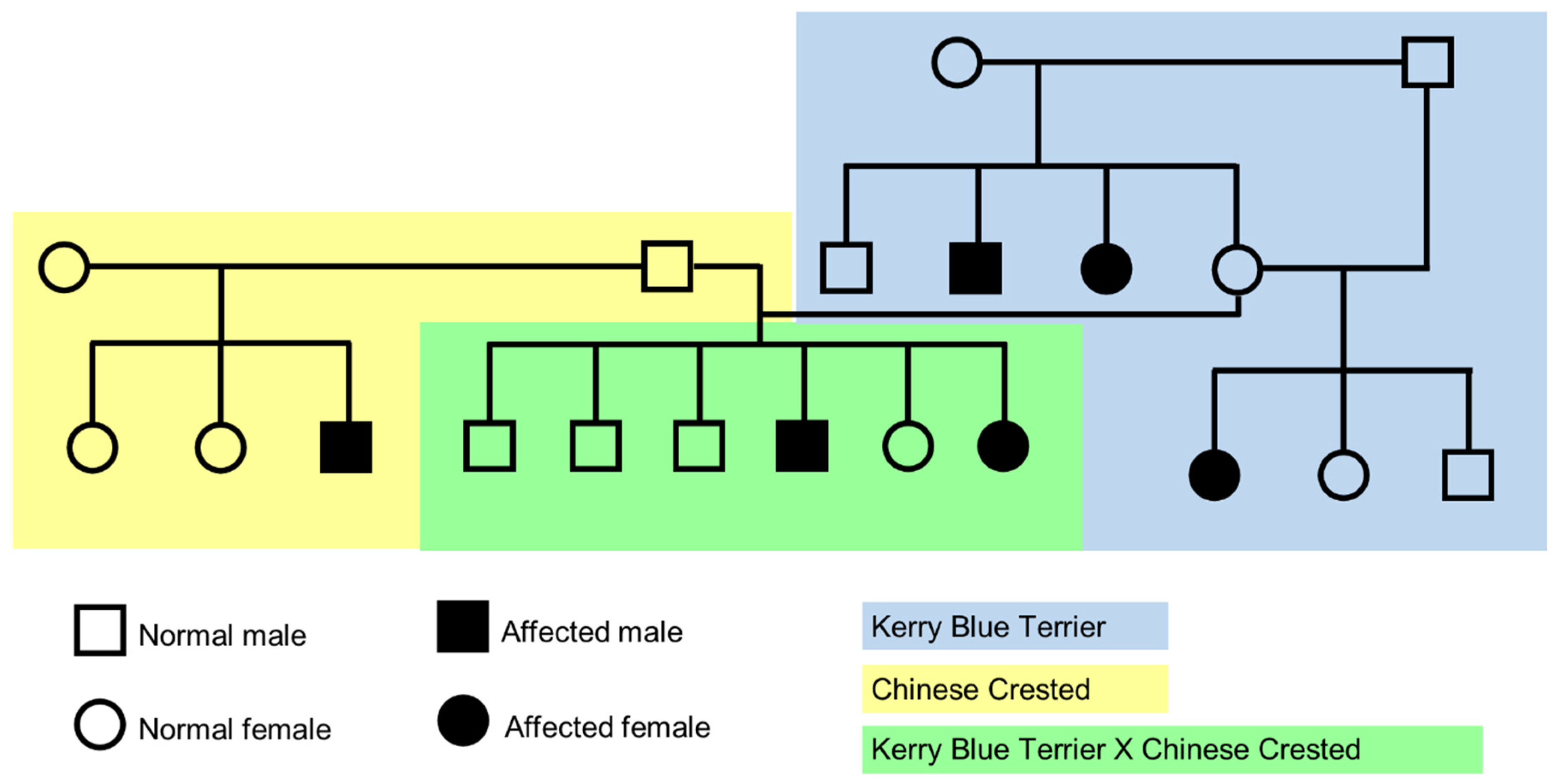
Figure 2.
NextGENe software viewer shows an alignment of whole genome sequence reads DNA from a CMSD-affected Kerry Blue Terrier in the antisense orientation for SERAC1. This region contains a homozygous a C>T which converts a tyrosine codon (TGG on the sense strand) to a premature stop codon (TGA on the sense strand). Sequence variants are highlighted in blue.
Figure 2.
NextGENe software viewer shows an alignment of whole genome sequence reads DNA from a CMSD-affected Kerry Blue Terrier in the antisense orientation for SERAC1. This region contains a homozygous a C>T which converts a tyrosine codon (TGG on the sense strand) to a premature stop codon (TGA on the sense strand). Sequence variants are highlighted in blue.
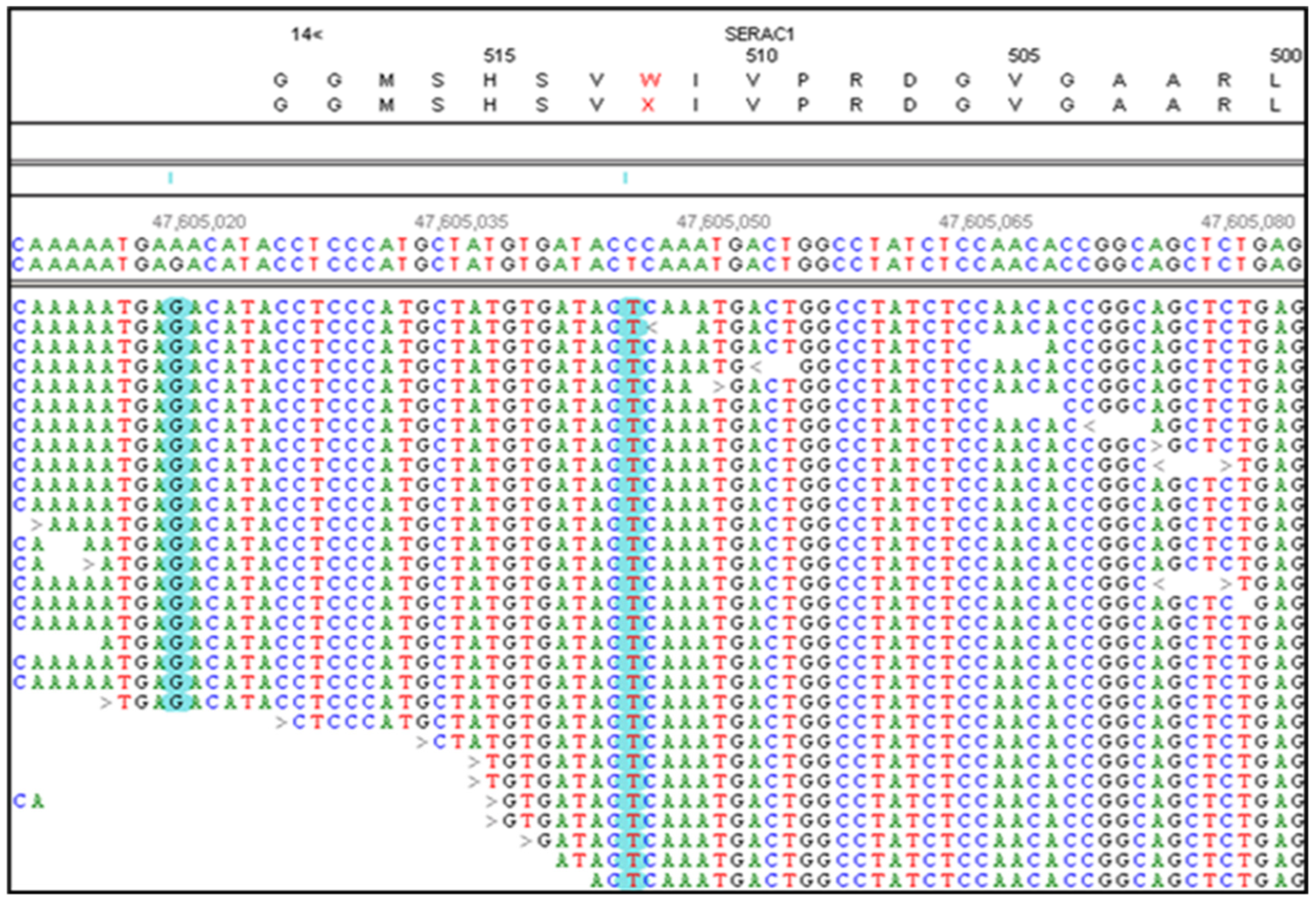
Figure 3.
NextGENe software viewer shows an alignment of whole genome sequence reads generated from DNA from a CMSD-affected CCD in the antisense orientation for SERAC1. This region contains a homozygous a 4bp deletion (GTAA in the antisense strand) in the exon 15 acceptor splice site. The deleted nucleotides are highlighted in blue. The SERAC1 exon 15 coding region is highlighted in red.
Figure 3.
NextGENe software viewer shows an alignment of whole genome sequence reads generated from DNA from a CMSD-affected CCD in the antisense orientation for SERAC1. This region contains a homozygous a 4bp deletion (GTAA in the antisense strand) in the exon 15 acceptor splice site. The deleted nucleotides are highlighted in blue. The SERAC1 exon 15 coding region is highlighted in red.
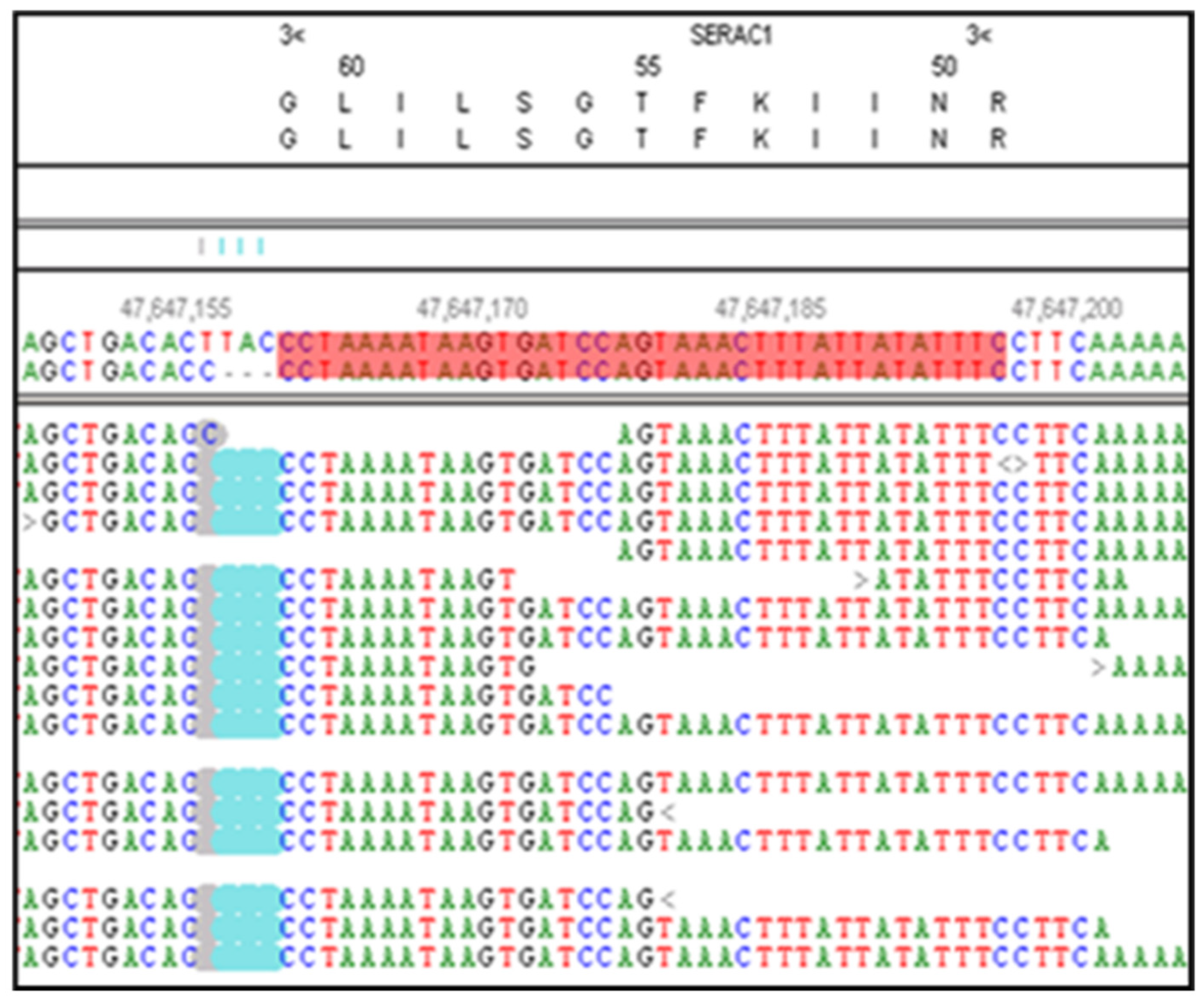
Figure 4.
Microcapillary electrophoretograms of RT-PCR amplicons produced with primers designed to anneal to exon 3 and exon 5 of SERAC1. The RNA from a CMSD-affected (lane 4) and two unaffected (Lanes 2 and 3) Chinese Crested dogs. A band at 253 bp was expected from the consecutive splicing of exon 3 to exon 4 to exon 5. A band at 216 bp was expected from the splicing of exon 3 to exon 5. Lane 1 was a no-template control (lane 1). The band at 500 bp is from an internal molecular weight marker.
Figure 4.
Microcapillary electrophoretograms of RT-PCR amplicons produced with primers designed to anneal to exon 3 and exon 5 of SERAC1. The RNA from a CMSD-affected (lane 4) and two unaffected (Lanes 2 and 3) Chinese Crested dogs. A band at 253 bp was expected from the consecutive splicing of exon 3 to exon 4 to exon 5. A band at 216 bp was expected from the splicing of exon 3 to exon 5. Lane 1 was a no-template control (lane 1). The band at 500 bp is from an internal molecular weight marker.
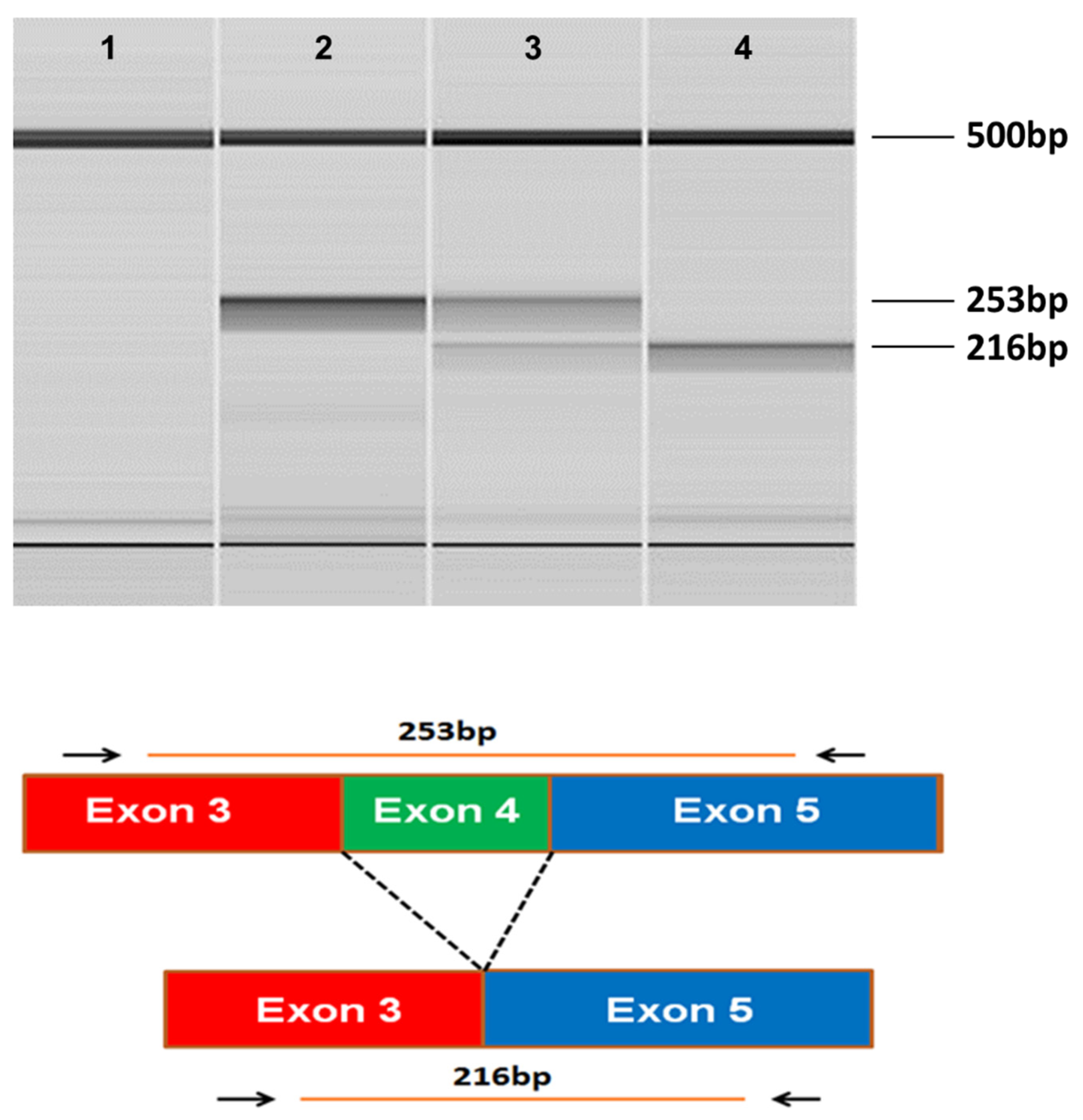
Figure 5.
Automated Sanger sequence electrophoretograms of the RT-PCR amplicon produced with RNA from a CMSD-affected Chinese Crested dog confirming the splicing of SERAC1 exon 3 to exon 5.
Figure 5.
Automated Sanger sequence electrophoretograms of the RT-PCR amplicon produced with RNA from a CMSD-affected Chinese Crested dog confirming the splicing of SERAC1 exon 3 to exon 5.
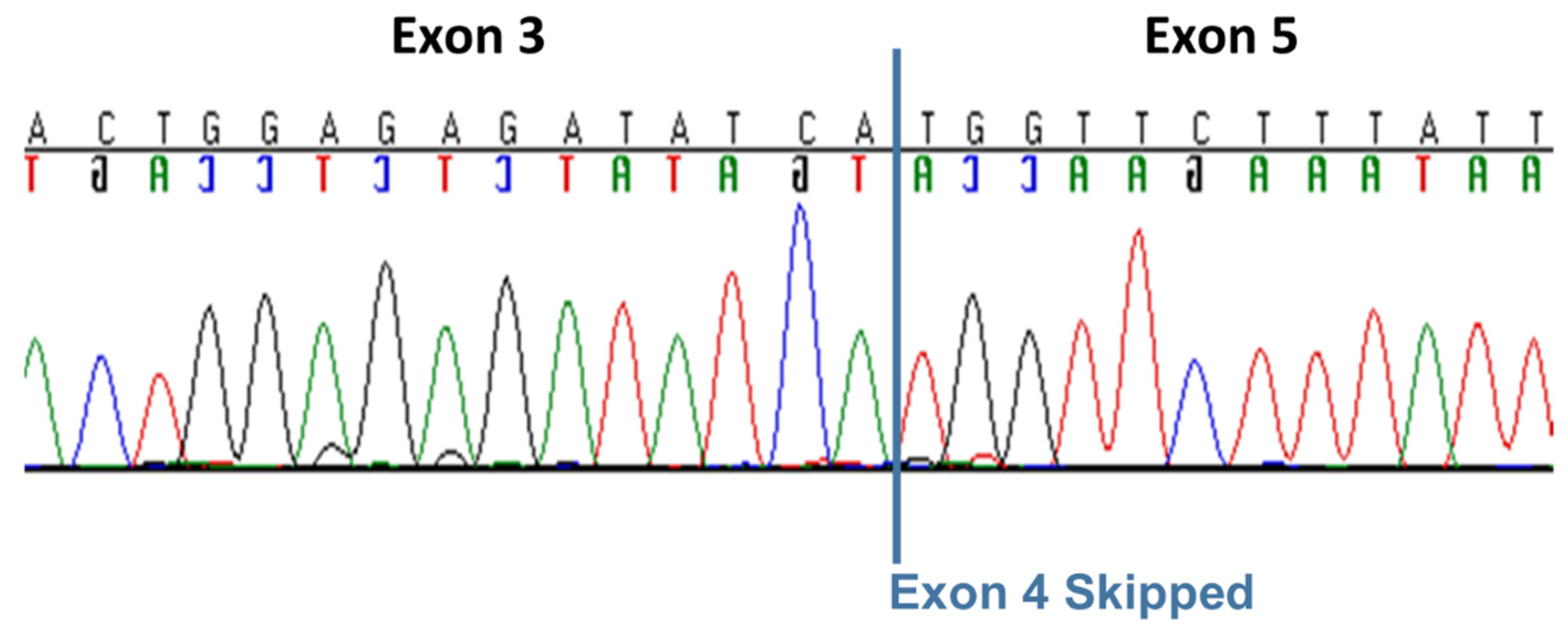
Figure 6.
Microcapillary electrophoretograms of amplicons produced with the RFLP-PCR genotyping assay with DNA from two CMSD-affected KBTs, (lanes 1 and 2), DNA from a clinically normal KBT with a “carrier” (heterozygous) test result (lane 3) and two other clinically normal KBTs with a homozygous normal test result (lanes 4 and 5). The bands at 15 bp and 500 bp are from internal molecular weight markers.
Figure 6.
Microcapillary electrophoretograms of amplicons produced with the RFLP-PCR genotyping assay with DNA from two CMSD-affected KBTs, (lanes 1 and 2), DNA from a clinically normal KBT with a “carrier” (heterozygous) test result (lane 3) and two other clinically normal KBTs with a homozygous normal test result (lanes 4 and 5). The bands at 15 bp and 500 bp are from internal molecular weight markers.

Figure 7.
Automated Sanger sequence electrophoretograms of PCR amplicons produced with DNA from unaffected (Top), CMSD-affected (Middle), and heterozygous unaffected Chinese Crested dogs (Bottom).
Figure 7.
Automated Sanger sequence electrophoretograms of PCR amplicons produced with DNA from unaffected (Top), CMSD-affected (Middle), and heterozygous unaffected Chinese Crested dogs (Bottom).
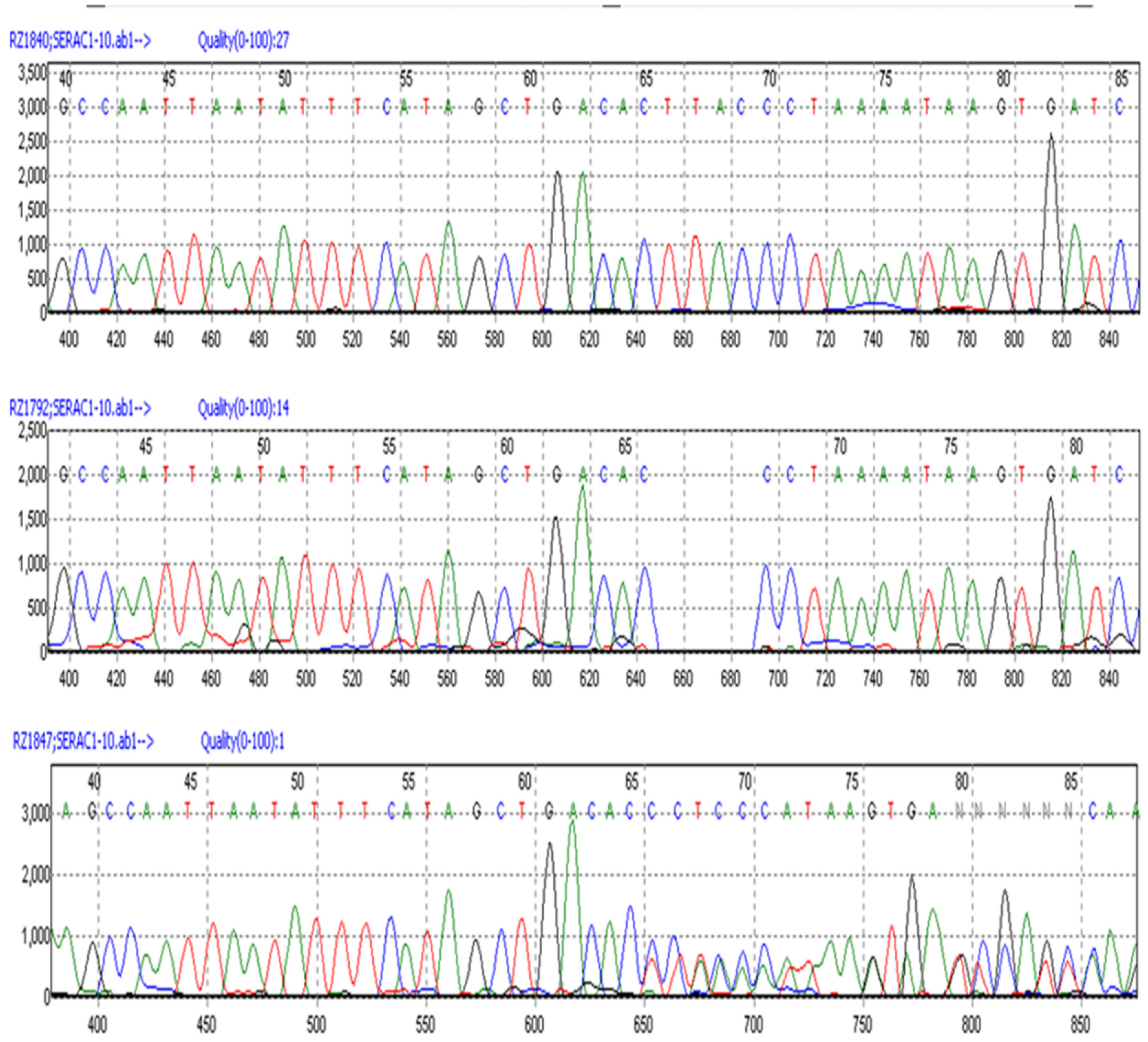

Table 1.
Distribution of genotypes and phenotypes among affected and unaffected Kerry Blue Terriers.
Table 1.
Distribution of genotypes and phenotypes among affected and unaffected Kerry Blue Terriers.
| Phenotype | Genotype | Total | ||
|---|---|---|---|---|
| Mut/Mut | Mut/WT | WT/WT | ||
| Affected | 5 | 0 | 0 | 5 |
| Unaffected | 0 | 11 | 212 | 223 |
| Total | 5 | 11 | 212 | 228 |
Table 2.
Distribution of genotypes and phenotypes among affected and unaffected Chinese Crested dogs.
Table 2.
Distribution of genotypes and phenotypes among affected and unaffected Chinese Crested dogs.
| Phenotype | Genotype | Total | ||
|---|---|---|---|---|
| Mut/Mut | Mut/WT | WT/WT | ||
| Affected | 41 | 0 | 0 | 41 |
| Unaffected | 0 | 55 | 87 | 142 |
| Total | 41 | 55 | 87 | 183 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



