
Submitted:
30 September 2024
Posted:
03 October 2024
You are already at the latest version
Abstract
Flashes of superoxide anion (SA) in mitochondria are generated spontaneously or during the opening of the permeability transition pore (PTP) and a sudden change in the metabolic state of a cell. Under certain conditions, SA can leave the mitochondrial matrix and perform signaling functions beyond mitochondria. In this work, we studied the kinetics of the release of SA and H2O2 from isolated mitochondria upon PTP opening and the modulation of the metabolic state of mitochondria by the substrates of respiration and oxidative phosphorylation. It was found that PTP opening leads to suppression of H2O2 emission and activation of SA burst. When the induction of PTP was blocked by its antagonists (cyclosporine A, ruthenium red, EGTA), the level of substrates of respiration and oxidative phosphorylation and the selective inhibitors of complexes I and V determined the type of ROS emitted by mitochondria. It was concluded that upon complete and partial reduction and complete oxidation of redox centers of the respiratory chain, mitochondria emit H2O2, SA, and nothing, respectively. The results indicate that the PTP- and substrate-dependent switching of the type of ROS leaving mitochondria may be the basis for SA- and H2O2-selective redox signaling in a cell.
Keywords:
mitochondria
; superoxide anion
; hydrogen peroxide
; kinetics
; redox signaling
; OXPHOS substrates
1. Introduction
In recent years, the area of research associated with redox signaling in cells in normal and pathological states has been actively developing [1,2]. The data obtained indicate the complexity and ramification of the system of redox signaling, which includes subsystems of production and transformation of reactive oxygen species (ROS), amplification/weakening of a redox signal, redox sensors, and redox-dependent effectors that provide a local or generalized cellular response [3,4]. Presently, more than 200 mammalian proteins are known whose activity can be regulated by the redox state of thiol groups or the level of ROS. It is proposed to call this community the “redoxome” [5].
Mitochondria are one of the main sources of ROS in the majority of cell types except specialized ROS producers: leukocytes and macrophages [6]. ROS produced by mitochondria contribute to the development of many pathologic states including cancer, cardiovascular diseases, diabetes, neurological disorders, muscular dystrophy, etc. [7]. One of the most interesting phenomena associated with ROS generated by mitochondria is the so-called superoxide anion (SA) flashes discovered about 15 years ago [8,9,10,11,12]. An SA flash is a sharp short-term acceleration of SA production by individual mitochondria in the cell [10,12]. As a rule, the time to achieve the maximum SA production in a single mitochondrion does not exceed a few seconds, while the decay time is about 20 seconds [10,11,12,13,14]. SA flashes can occur spontaneously, but their frequency can increase, and their generation can be synchronized under certain conditions. In particular, oxidants, Ca2+ and other inducers of the mitochondrial permeability transition pore (PTP), and respiratory substrates increase the frequency of flashes, while antioxidants, uncouplers and inhibitors of the electron transport chain (ETC), and PTP blockers reduce the frequency of flashes or completely abolish them [10,11,12,13,14]. Moreover, flashes can propagate from mitochondria to mitochondria, causing the permanent or temporary depolarization, i.e., a “wave of dysfunction” [11,15,16]. Though flashes are often associated with transient or long-term PTP opening, this condition is not obligatory [17,18]. It was demonstrated that flashes can appear in intact mitochondria [11,14] and, presumably, play an important role in cell physiology and pathophysiology. Flashes participate in cell excitation [21,22] and regulation of cell differentiation, proliferation [23,24,25,26], muscular development [14], and animal longevity [27], as well as in the progress of muscular diseases [28], ischemia-reperfusion-dependent injury [9], amyotrophic lateral sclerosis [29], and oxidant-induced apoptosis [30]. However, in recent years, the flow of works devoted to the study of SA flashes is gradually drying up. It can be assumed that this is due to a serious criticism of the methods used to detect SA flashes, on the one hand, and the paradoxical mechanism of their generation, on the other.
The data on SA flashes in mitochondria were obtained predominantly using the SA-sensing matrix-targeted circularly permuted yellow fluorescent protein (mt-cpYFP) [8,31] and the cationic derivative of hydroethidine Mito-SOX red [11,13,32]. Another matrix-targeted protein, the circularly permuted green fluorescent protein (pericam), was applied for the recording of SA flashes in mouse skeletal muscle mitochondria [11]. Besides, uncharged and relatively hydrophilic 2, 7-dichlorodihydrofluorescein diacetate was used for detecting H2O2 flashes in the cell cytosole [12].
However, the ability of the mt-cpYFP protein to detect SA was questioned in several studies. In particular, it was shown that an increase in the fluorescence of the detector can reflect the alkalization of the matrix, but not the generation of SA [33,34,35]. In addition, another SA probe, Mito-SOX red, being sensitive to the mitochondrial membrane potential (ΔΨm), relatively non-specific, and reactive toward DNA, also cannot be a reliable detector in mitochondria, when their functional state changes and, especially, when the PTP opens [36,37].
The proposed mechanism for the emergence of SA flashes also raises a number of questions. First, it is widely believed that “mitoflashes are quantal bursts of ROS production accompanied by the modest matrix alkalinization and depolarization of the mitochondrial membrane potential” [36]. However, mitochondrial depolarization by definition should cause the acidification of the matrix, especially if the PTP is irreversibly opened, since in this case the protons pumped out by the pumps of the respiratory chain immediately return to the matrix. Theoretically, ΔΨm dissipation and the alkalization of the matrix may co-exist during the transient PTP opening or stochastic short-term drops in ΔΨm [39] in accordance with the mechanism proposed by Schwarzlander et al.: stochastic depolarization causes the activation of proton pumps, which, in turn, induces matrix alkalization and a burst of ROS [40]. However, it was shown in a model of UCP-3 knockout that the burst of SA and alkalization can be separated [31]. Moreover, this mechanism cannot explain the generation of SA flashes upon permanent PTP opening, since a large pore will prevent the formation of the pH gradient across the inner mitochondrial membrane (IMM). Second, flashes require the presence of respiratory substrates, i.e., redox centers must be reduced. However, both inhibitors, which cause the complete reduction of certain segments of the ETC, and uncouplers, which induce the full oxidation of electron carriers, inhibit SA flashes [8]. The question arises: How is the degree of reduction of redox centers related to the intensity of SA production? Third, experiments with isolated mitochondria demonstrated that the rates of ROS production are maximum upon good mitochondrial coupling and at high ΔΨm [41,42,43]. Thus, the mechanism of SA flashes upon mitochondrial depolarization is unclear.
Trying to resolve the paradox of the mechanism of SA flashes, we have previously shown that the SA burst can be induced in mitochondrial suspension upon permeabilization of the IMM due to the opening of PTP or the incorporation of a pore-forming peptide [19,20]. Using uncharged and relatively hydrophilic 3,7-dihydro-2-methyl-6-(4-methoxyphenyl)imidazol [1,2-a]pyrazine-3-one (MCLA), whose accumulation in intact mitochondria is limited but which can pass through the large pores in the IMM, we demonstrated that added NADH and, especially, NADPH strongly stimulated SA bursts, which occur only after considerable oxidation of pyridine nucleotides [20]. However, we were unable to reliably compare the kinetics of SA and H2O2 generation since pyridine nucleotides (which were added to permeabilized mitochondria at high concentrations) can interfere with the oxidation of fluorogenic substrates by horseradish peroxidase (HRP) [44].
In the present work, we compared the kinetics of the release of SA and H2O2 from isolated mitochondria in the presence of endogenous and exogenous substrates of different mitochondrial dehydrogenases. We assessed the effect of the concentration of added substrates and PTP opening on these processes. The data obtained indicate that the opening of the PTP and the presence of substrates cause a bidirectional switch of the type of the ROS signal (SA or H2O2) emitted by mitochondria to the medium. The possible mechanism of this phenomenon and its probable physiological significance are discussed.
2. Materials and Methods
Materials
ADP (sodium salt) (A2754), ATP (disodium salt hydrate) (A7699), bovine serum albumin (BSA) (A7030), FCCP (C2920), CATR (C4992), DMSO (276855), DNP (D198501), 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) (H3375), sucrose (S7903), succinate (S3674), Trizma Base (93352), Ampliflu™ Red (AR), ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA), glutamate, 2-oxoglutarate, malate, 3-hydroxybutyrate, mannitol, MCLA, myxothiazol, NADH, NAD, potassium peroxide, pyruvate, rotenone (R8875), and superoxide dismutase (SOD) were obtained from the Sigma-Aldrich Corporation (St. Louis, MO; USA). Other chemicals were of analytical grade and were purchased from local suppliers.
Preparation of mitochondria
All manipulations with animals before the isolation of the liver were performed in accordance with the Helsinki Declaration of 1975 (revised in 1983), the national requirements for the care and use of laboratory animals, and protocol 26/2024 of 18.03.2024 approved by the Commission on Biological Safety and Bioethics of the Institute of Theoretical and Experimental Biophysics, Russian Academy of Sciences (ITEB RAS). Rat liver mitochondria (RLM) were isolated by a standard procedure [45] with minor modifications [46] Adult male Wistar rats (200-250 g) were decapitated after anesthesia with CO2. The liver was homogenized in ice-cold isolation buffer containing 220 mM mannitol, 70 mM sucrose, 1 mM EGTA, 0.3% BSA, and 10 mM HEPES-Tris (pH 7.4). The homogenate was centrifuged at 600×g for 10 min at 4°C, and the supernatant fraction was then centrifuged at 7,000×g for 15 min to sediment mitochondria. The RLM were washed three times (7,000×g for 20 min) in the above medium without EGTA and BSA (1× washing medium). The final mitochondrial pellet was suspended in the washing medium to yield 70-80 mg protein/ml and kept on ice until used. The total mitochondrial protein was determined by the Biuret method using BSA as a standard [47]. All measurements unless otherwise stated were performed at 37°C using the standard KCl-based medium (KCl-BM): 125 mM KCl, 20 mM mannitol, 10 mM HEPES (pH 7.3), 2 mM KH2PO4, and 2 mM MgCl2.
Measurement of oxygen uptake
Isolated RLM (1 mg protein/ml) were incubated at 25°C in the standard KCl-BM supplemented with substrates of complex I (5 mM glutamate and 5 mM malate) (GM) or complex II (5 mM succinate) in the presence of 2 μM rotenone (SR). Oxygen uptake was measured with a Clark-type electrode using an Oroboros Oxygraph-2 k respirometer (Austria). In order to assess V3 and V4 respiration rates, 500 μM ADP was added to resting RLM (V2). The respiratory control coefficient (V3/V4) for RLM taken for experiments was ≥ 6 for GM and ≥ 5 for SR.
Recording of mitochondrial swelling
The opening of PTP in isolated RLM was assessed from the initiation of EGTA- and CsA-sensitive high-amplitude swelling. Mitochondrial swelling was determined by measuring a decrease in absorbance at 550 (A550) or 535 nm (A535) in suspension using the plate readers Infinite 200 Tecan and Infinite 200 Tecan Pro (Groedig, Austria), respectively, and 96-well plates. Other details are given in the figures and figure legends.
Assessment of ROS production
RLM were placed in standard KCl-BM without substrates, and the suspension was immediately poured into two tubes with either MCLA at indicated concentrations or with 40 µM AR and HRP (3 U/ml). The suspensions were then distributed into the wells of plates for luminescence and fluorescence measurements, which contained substrates and other additions, as specified in figure legends, and analyzed in parallel using two plate readers.
H2O2 release
Resorufin accumulation (which linearly depends on H2O2 production) was traced at excitation and emission wavelengths of 530 and 595 nm. For the quantitative measurement of H2O2, fluorescence was calibrated by the addition of an excess of H2O2 (100 μM final concentration) at the beginning and at the end of the recording to several wells. This was done to assess the rate of the conversion of AR/resorufin to non-fluorescent product [48] and thus to deduce the fluorescence of 40 µM resorufin at zero time. The rate of H2O2 release from RLM was calculated for the each point of the record except the first as an increment in resorufin concentration per minute per mg of mitochondrial protein: R = Increment in fluorescence (ΔAU) · 40 · Fluorescence of 40 μM resorufin-1 (AU)·nmol·ml-1 · mg protein-1.
SA release
Since SA flashes usually occur during a spontaneous drop in ΔΨm, we used MCLA for SA detection. MCLA is a highly sensitive SA probe that requires single-electron transfer to some oxidant for activation [49]. The resulting radical reacts with SA with high rate and selectivity. The rate constant is about 2.54 ·108 M-1 s-1 [50], which is only an order of magnitude lower than that of SOD [51]. The relative chemiluminescence intensity of MCLA in reaction with SA, H2O2, 1O2, and NO was determined to be 230000, 0.4, 1400, and 17000, respectively [52]. The product of the reaction is an unstable anionic dioxetanone, which is immediately decarboxylated to form oxy-MCLA in an excited state emitting a quantum of blue chemiluminescence upon transition to the ground state. A quantum yield of the MCLA chemiluminescence in aqueous solutions is in the range from 0.0079 to 0.066 [53]. Both SA-sensing MCLA radical and oxy-MCLA are uncharged [49,54], which makes MCLA-derived chemiluminescence (MDCL) independent of ΔΨm.
In the present study, MDCL was recorded approximately once a minute. Each value on the curve is the mean ± S.D. of three integrations of luminescence for 900 ms expressed in arbitrary units. In order to separate relatively bright spontaneous SA-insensitive MDCL [54] from SA-sensitive one, some wells contained SOD at indicated concentrations. Since MDCL in solution is effectively quenched by sulfur-containing compounds [55], we excluded sulfur-containing antioxidants from the experimental protocol. It should be stressed that MDCL reflects the quantity of photons emitted by the excited oxy-MCLA within 900 ms-period of luminescence accumulation at each experimental point (AU accumulated per 900 ms). Thus, MDCL is the measure of the rate of SA production. As the rate of SA production by mitochondria and, consequently, the intensity of MDCL were not constant during long incubation, in order to assess the net SA production for long period, we integrated MDCL values within initial 60 min interval of incubation (Σ(MDCL)0-60 min, AU).
Statistical analysis
Representative data from 3 to 20 independent experiments are given. The values on all swelling/shrinkage and ΔΨm curves are the means ± SEM for three wells (n = 3). Statistical significance (P) was determined using the Student’s t-test.
3. Results
3.1. Effect of Respiratory Substrates and Permeability Transition Pore Inhibitors on the Release of SA and H2O2 from Mitochondria
Initially, we studied the effect of respiratory substrates at saturating concentrations on the kinetics of the release of SA and H2O2 from RLM (Figure 1A and 1B, respectively). The release of SA was assessed as SOD-sensitive MDCL. The rate of H2O2 release was determined by measuring the increment in resorufin fluorescence per mg protein per minute. A parallel study of SA and H2O2 release revealed several interesting features. First, the highest rate of SA release occurred when mitochondria oxidized endogenous substrates (Endo) (Figure 1A). In contrast, the rate of H2O2 release was the least and decreased with time (Figure 1B). Second, the highest rate of H2O2 release was recorded in the first minutes of incubation (presumably, when mitochondrial coupling was maximum (Supplementary Figure S1)), while the maximum in MDCL was postponed. Third, respiratory substrates, namely 3-hydroxybutyrate (3-HB), 2-oxoglutarate (2-OG), and succinate (Suc) (all at a concentration of 5 mM) (Figure 1A–F), as well as GM, pyruvate (Pyr), and SR (Supplementary Figure S2) suppressed the release of SA and stimulated the release of H2O2. Since SA more poorly penetrates through lipid membranes than H2O2, the strong SA release from RLM, which oxidize endogenous substrates, may be connected with a faster PTP opening (either transient or permanent).
The data presented in Figure 1C–F suggest that this assumption may be correct. Indeed, the antagonists of PTP opening, namely the Ca2+ chelator EGTA and the inhibitors of cyclophilin D isomerase cyclosporine A (CsA) and of the mitochondrial Ca2+ uniporter ruthenium red (RR) suppressed the SA release and delayed its maximum (Figure 1C,E). In contrast, the inhibitors increased the rate of H2O2 release and made it last longer. These data can be explained by the improved mitochondrial coupling and a higher degree of reduction of redox centers of the ETC (Supplementary Figure S1), as well as by a worse permeability of the IMM to SA upon the inhibition of PTP opening. In all of the above cases, the rates of the release of SA and H2O2 mirrored each other (corrected for the effect of RR on MDCL due to the intense staining of the inhibitor), indicating that the redox signal leaving the mitochondria switches from SA to H2O2 and vice versa.
3.2. Composition of an MDCL Signal in a Mitochondrial Suspension
In order to reveal the relationship between the level of MDCL and the rate of SA generation, we studied the effect of the SA scavenger SOD and the SA generator xanthine oxidase (XO) on MDCL in solution (Figure 2). Figure 2A shows that spontaneous SA generation in solution may be responsible for the minor, SOD-sensitive, portion of MDCL. The rest of MDCL is SOD-insensitive and can be referred to as basal (MDCLb). Nevertheless, XO at increasing concentrations caused a linear enhancement in MDCL (Figure 2B), indicating the linear dependence of MDCL on the SA concentration. In solutions with XO, the maximum MDCL was highly sensitive to added SOD (Figure 2C). However, the integrated MDCL (Figure 2C, insert) was affected to a minor extent, demonstrating that SOD effectively competes with MCLA for SA and thus protects the probe from oxidation. In a mitochondrial suspension, the recorded total MDCL might comprise several constituents: MDCLb (SOD-insensitive and SA-independent), SOD-sensitive SA-dependent MDCL in solution between mitochondria (MDCLExt), SOD-insensitive SA-dependent MDCL in the intermembrane space (MDCLIMS), and, probably, SOD-insensitive SA-dependent MDCL in the matrix (MDCLMtx). (In all cases, we are talking about the added SOD.) Figure 2D shows that the microbial peptide alamethicin capable of forming large pores (permeable to ~2.2 kDa substances) in mitochondrial membranes [19] and ensuring free passage of MCLA and SA through the IMM caused a sharp increase in MDCL in the mitochondrial suspension. The addition of SOD suppressed MDCL in both intact and permeabilized mitochondria to the same level. Thus, the contribution of MDCLMtx to total MDCL in the suspension of intact RLM seems negligible, presumably, due to a slow penetration of MCLA through the intact IMM.
3.3. PTP Opening Switches the Type of a ROS Signal Emitted by Mitochondria
In order to demonstrate unequivocally that PTP opening can switch the type of a ROS signal from mitochondria, we conducted a series of experiments in which MDCL and mitochondrial swelling (a consequence of PTP opening, assessed as a decrease in A535 or A550) were recorded in the same wells, and H2O2 production was measured in parallel (Figure 3). In this case, the points on the luminescence curves lagged behind the points on the absorption curves by approximately 1 min. In the presence of the added respiratory substrates Pyr, 2-OG, and Suc, (all at a concentration of 5 mM) (Figure 3C, 3E, and 3G, respectively) and in RLM oxidizing Endo substrates in the absence of 1 mM EGTA (Figure 3A), SA bursts followed the PTP opening. Again, the intensity of the bursts was a mirror image of the intensity of H2O2 release. The inhibition of PTP opening and mitochondrial swelling at a high EGTA concentration strongly suppressed SA bursts in the presence of all respiratory substrates but Endo. In the latter case, EGTA suppressed the swelling, but SA bursts occurred approximately at the same time as in its absence. In the presence of all substrates but Endo, the inhibition of PTP opening increased the time of H2O2 production. Thus, both PTP opening and, probably, respiratory substrates regulate the SA release from mitochondria.
3.4. PTP Antagonists Do Not Prevent SA Flashes in the Absence of Added Substrates
In order to discriminate the role of Ca2+ and of PTP opening per se in the persistence of SA bursts in RLM that oxidize Endo substrates in the presence of EGTA (Figure 3B), we studied the effect of the other PTP antagonists (CsA and RR) on the kinetics of swelling and SA/H2O2 release (Figure 4). In all cases, the highest rate of H2O2 release was noted in the first minutes of incubation followed by the inhibition of H2O2 production. Since PTP inhibitors did not prevent the suppression of H2O2 release, the suppression might be a consequence of the exhaustion of Endo substrates and/or the mild uncoupling (Supplementary Figure S1), which declined the extent of the reduction of ETC redox centers. The decline in H2O2 generation preceded the activation of SA release. SA bursts could occur before high-amplitude mitochondrial swelling when PTP opening was blocked by EGTA, CsA, and RR (Figure 4B–D). Therefore, respiratory substrates can limit the SA release from mitochondria independently of PTP opening. Hence, the presence/absence of a respiratory substrate can be another trigger of the switching of the type of a ROS signal emitted by mitochondria (when the PTP is closed).
3.5. Modulation of the Type of a ROS Signal by Different Respiratory Substrates
Different respiratory substrates are transported into the matrix by appropriate carriers via the symport with H+ or electrically neutral exchange with an anion and are oxidized by specific dehydrogenases (Figure 5). The resulting set of reduced electron donors can donate electrons to several ROS-generating sites, depending on the combination of substrates. The switching of the type of the emitted ROS signal can be associated either with a change in the rate of the production of SA/H2O2 in the same or different mitochondrial redox centers or with an alteration in the efficiency of their release. For H2O2, it is known that conditions promoting its formation in mitochondria also contribute to the maintenance of the systems of its scavenging in the active state [56]. Therefore, it can be assumed that the rate of the release of H2O2 is approximately proportional to the rate of its production. The mechanism of SA release from the matrix of intact mitochondria is poorly understood. Acidic pH can dramatically increase the capability of SA for penetration through phospholipid membranes [57,58]. Indirect evidence indicates that, at slightly alkaline pH of the mitochondrial matrix, the release of SA may require the operation of carriers of anionic substrates [59,60,61].
Therefore, we studied the effect of various respiratory substrates at different concentrations and their combinations on the emission of SA/H2O2 from mitochondria (Figure 6, Supplementary Figure S3). In order to distinguish the own effects of substrates on ROS emission from the effect on PTP opening, the incubation medium contained the PTP inhibitor 1 μM CsA, and membrane intactness was monitored as indicated in the legend to Figure 3 and Figure 4. Figure 6A–C and Supplementary Figure S3 show that the addition of any exogenous substrates, especially Suc, at near-physiologic concentrations (250 μM) was sufficient to sharply increase the release of H2O2 from mitochondria. The most intense production was observed in the presence of combinations of the OXPHOS substrates 3-HB/2-OG/Suc (A) and 3-HB/2-OG/Suc/Pyr/GM (C). A rise in the substrate concentrations to 5 mM (saturation) caused an increase in H2O2 generation in the presence of all substrates except the combination of six substrates (C). Thus, the mixture of six substrates at near-physiologic concentrations caused the reduction of redox centers sufficient for maximum production of H2O2. The decrease in H2O2 release with the mixture of 5 mM substrates is obviously due to the competition between the substrates for coenzymes, the bidirectional transport of substrates and intermediates, and the different activities of transporters and dehydrogenases of different substrates (Figure 5). In all cases, the SA release decreased with increasing substrate concentration regardless of whether the substrate was symported with H+ or not. The decrease in MDCL contrasted with the increase in H2O2 production. A noteworthy exception was the presence of the six-substrate combination, where the maximum boost and the slight decrease in H2O2 production at 250 μM and 5 mM substrate concentrations, respectively, coincided with a moderate and an almost complete suppression of SA liberation (Figure 6C). These data indicate that the carriers of anionic substrates may contribute to the SA release from the matrix and that the transport of substrates competes with this process.
The mechanism of suppression of the SA efflux by respiratory substrates may involve a switch in the redox centers and an alkalization of the matrix. Therefore, we studied the effect of the CI inhibitor rotenone and the protonophore FCCP on the rate of SA and H2O2 release in the presence of substrates of CI and CII. The oxidation of Endo substrates in the forward electron transport (FET) mode (CI-CIII-CIV segment of ETC) yielded maximum SA and minimum H2O2 (Figure 6D,E). An additional reduction of CI (and, subsequently, CIII) with 3-HB accelerated the generation of H2O2 and diminished the SA release. Rotenone, which induces the complete reduction of the flavin site of CI and the oxidation of the Q-CIII-CIV segment of the ETC accompanied with the disruption of the H+ gradient across the IMM, moderately stimulated the SA release and slightly suppressed H2O2 production, indicating the major contribution of the CI flavin site to the H2O2 production (Figure 6D). In the case that the addition of Suc activated both FET (CII-CIII-CIV segment) and reversed electron transport (RET) (CII-CI segment), the release of H2O2 was strongly enhanced, while the SA release was suppressed even more strongly than upon FET (Figure 6E). Rotenone, which stops RET without affecting the FET and the H+ gradient, strongly suppressed the H2O2 efflux and had a minor effect on the SA emission. FCCP, which causes a complete oxidation of ETC downstream the rotenone block and the dissipation of H+ gradient across the IMM, restrained the generation of SA/H2O2 on both substrates, though, to a different extent. These results indicate that the switching of the type of ROS signal is associated primarily with a change in the extent of the reduction of ETC redox centers, but not with the H+ availability in the matrix. The type of signal emitted by mitochondria corresponds to a pattern of ROS production as if fully reduced redox centers generated H2O2, partially reduced ones generated SA, and oxidized ones generated nothing.
3.6. Effect of Respiratory Substrates on the Spontaneous and Xanthine Oxidase-Dependent Generation of SA
Another possible mechanism for the suppression of SA release from mitochondria by respiratory substrates is the direct antioxidant effect of the latter. Figure 7 shows the effect of different OXPHOS substrates (all at a concentration of 5 mM except ATP (1 mM)) on the spontaneous and xanthine/xanthine oxidase-dependent SA generation (SOD-sensitive MDCL) in aqueous solution. At a near-physiologic concentration (250 μM), neither individual substrates nor their combinations affected the MDCL (not shown). As follows from the data presented, Pyr and, to a lesser extent, 2-OG decreased the SA level in the xanthine/xanthine oxidase system (Figure 7A,C,E,G). Spontaneously generated SA was scavenged by 2-OG, and, to a lesser extent, ATP and Pyr (Figure 7B,D,F,H). In contrast, 3-HB, Suc, and GM as well as fumarate and malate added separately (not shown) had a minimal effect on the SA level. These data indicate that alpha-keto acids can operate as the weak SA scavengers (Supplementary Table S1), while other substrates tested cannot.
3.7. Effect of Adenine Nucleotides on the SA/H2O2 Release from RLM
Then, we explored the effect of added adenine nucleotides (AN) on the SA/H2O2 release from RLM (Figure 8). AN are known to strongly inhibit the opening of PTP [46]. In our preparations, 1 mM ADP and 1 mM ATP increased the calcium retention capacity of RLM oxidizing Endo substrates from 45 to 195 and 240 nmol Ca2+ per mg protein, respectively. In order to discriminate the effect of AN on the SA/H2O2 release from the effect on the PTP opening, the medium contained 1 μM CsA, and the IMM intactness was monitored as indicated in the legends to Figure 3 and Figure 4. As follows from the figure, AN considerably suppressed the mitochondrial swelling and SA release but prolonged the H2O2 emission, probably, due to the support of mitochondrial coupling and the reduced state of mitochondrial redox centers (Figure 8A–C,F, Supplementary Figure S1). The inhibitor of FOF1-ATP synthase oligomycin slightly enhanced the AN-dependent suppression of SA release. Simultaneously, it stabilized the H2O2 production at a lower level than in the presence of AN alone (Figure 8D–F). This may be connected with the allosteric effect of AN on the ETC complexes. Thus, changes in mitochondrial coupling without PTP opening and changes in the levels of substrates may contribute to a switch of the ROS-type signal emitted from mitochondria.
4. Discussion
In the vast majority of studies devoted to the role of ROS in redox signaling and various physiological and pathological processes, the authors do not discriminate between the effects of SA and H2O2, considering SA exclusively as a relatively short-lived precursor of stable H2O2. Even in cases that the production of both species or even SA alone was measured, the authors prefer to use the umbrella term “ROS” [8]. A feature of the experimental approach in this study was the parallel recording of the long-term kinetics of SA and H2O2 release in a mitochondrial suspension with the simultaneous assessment of the permeability of the IMM to low-molecular-weight compounds (Figure 3, Figure 4, Figure 6 and Figure 8). This allowed exploring the SA/H2O2 release in the context of redox signaling. The SA probe MCLA is well suited for the long-term registration of SA production in a mitochondrial suspension [20,55,62,63,64]. It is very sensitive to SA and can also recognize singlet oxygen [52,54,65]. Since SA flashes are usually accompanied by fluctuations in ΔΨm, the lack of charge in both MCLA radical and its SA adduct [49,54] is a great advantage compared to lucigenin [66] and the oxidized products of hydroethidine (Mito-SOX), ethidium and 2-hydroxyethidium [67]. MCLA detects SA in suspension between mitochondria (SOD-sensitive MDCL, MDCLExt) (Figure 1, Figure 3, Figure 4, Figure 6 and Figure 8) and in the intermembrane space (SOD-insensitive, OXPHOS substrate-sensitive MDCL, MDCLIMS) (Supplementary Figure S4) but not in the matrix of intact mitochondria (Figure 2). The production of H2O2 using AR/HRP is usually measured in the initial period of the process [43,68,69], but this technique also allows recording the long-term kinetics of H2O2 production by mitochondria (Figure 1, Figure 3, Figure 4 and Figure 8). Though resorufin accumulation interfered with the recording of both swelling and MDCL (not shown), the use of two plate readers in parallel allowed us to solve this problem.
The main finding of this study is that changes in the physiological state of mitochondria lead to changes not only in the rate of ROS release from mitochondria, but also in the type of the ROS signal (or more broadly, the redox signal) emerging from the organelles (Figure 1, Figure 3, Figure 6 and Supplementary Figure S3). This conclusion, important in the context of redox signaling, raises two questions. First, what is the mechanism for the switching of the type (SA or H2O2) of the outgoing redox signal? Second, what is the possible physiological role and the pathological effect of this switch? It is obvious that two main mechanisms of the observed switching of the type of ROS signal are possible: a real switching in the redox centers of mitochondria and an apparent switching due to the facilitation of the release of SA from mitochondria (PTP/pore-dependent or -independent mechanism).
According to modern concepts, the majority of mitochondrial ROS-generating enzymes and complexes produce ROS as a result of one-electron leaks in flavin- and Q-binding sites with the formation of SA (CI, CII, and CIII, dihydroorotate dehydrogenase, glycerol-3-phosphate dehydrogenase, electron-transporting flavoprotein) [70,71,72,73,74], which subsequently dismutate to H2O2. These views were based on the data of early studies of ROS in mitochondria. The studies conducted using complex III inhibitors, which stabilize semiubiquinone at one of the binding sites, led to the conclusion that the only precursor of all ROS in mitochondria is SA [75,76,77]. In this case, a necessary condition for the maximum generation of SA and, as a consequence, H2O2, is the maximum reduction of the ETC segment that ends with the semiquinone form of flavin or of ubiquinone [75,77].
If this model is the only correct one, then the only possibility of switching the type of ROS signal exiting the mitochondria would be a disproportional facilitation of the exit of one of the ROS types (SA) through PTP (or another pore) or by another mechanism. However, there is evidence indicating that this model is not universal. In inside-out submitochondrial particles, in which there are no restrictions on the release of ROS, it was shown that 1 mM NADH suppresses the generation of SA in both FET and RET (10 mM Suc) in the presence of rotenone [68]. Moreover, the dependence of the SA production rate on the NADH concentration is bell-shaped with the maximum rate at a concentration of about 50 μM, while the maximum rate of H2O2 generation (mainly in CI) occurs at a NAD(H) concentration of ~100–500 μM and the maximum NADH/NAD+ ratio [43,69]. The same pattern of H2O2 production was observed for isolated dihydrolipoamide dehydrogenase [43]. It is important that, at low NADH concentrations, the only type of ROS generated in submitochondrial particles and isolated CI is SA, while at high NADH levels, the main type of ROS becomes H2O2, which, according to the authors, should be the main type of ROS at physiological substrate concentrations [68]. We previously demonstrated that, in mitochondria with the IMM permeabilized due to PTP opening or the incorporation of a pore-forming peptide, an SA burst occurs after significant oxidation of the added substrates NADH or NADPH (positive shift in ENAD(P)H)[20]. These data indicate the possibility of a real switch in the type of ROS generated, at least in mitochondrial complex I.
It should be mentioned that initially we suggested that NADPH-dependent SA bursts are associated with the activity of adrenodoxin-adrenodoxin reductase complex [20]. However, since SA bursts are not detected in mitochondria lacking functional ETC complexes [10], and CI can oxidize NADPH at a low rate [80], it can be assumed that CI is also responsible for the NADPH-dependent generation of SA. At the same time, in intact mitochondria, succinate, which is devoid of antioxidant properties (Figure 7, Supplementary Table S1), potently suppressed SA generation (Figure 1, Figure 3, Figure 6 and Supplementary Figure S3), and rotenone had a minor effect on this process (Supplementary Figure S4). This suggests the participation of other, non-CI, partially reduced electron carriers of the CII-CIII-CIV segment of ETC in the generation of SA.
In this work, we confirm the data of other groups [42,43,69] indicating that, in intact mitochondria, the rate of H2O2 generation is maximum at the maximum coupling (Figure 1 and Figure 8) and the maximum concentration of respiratory substrates (Figure 1, Figure 3, Figure 6 and Supplementary Figure S3). This mirrors the situation with the release of SA, which maximum rate was observed at low (near-physiological) substrate concentrations, on Endo substrates and upon reduced coupling (Figure 4, Figure 6, Figure 8 and Supplementary Figure S1) [78,79]. Thus, these data confirm that the switching of the ROS signal type during changes in the functional state of intact mitochondria occurs in redox centers. PTP induction (or creation of any other pore in the IMM) not only opens the way for the free release of SA (Figure 3), but also causes oxidation of the mitochondrial redox centers, the degree of which depends on the concentration of available substrate (Supplementary Figure S1) [20].
The mechanism of switching the type of ROS signal may be different in redox centers of different complexes. As for complex I, its electron transfer pathway consists of terminal two-electron carriers FMN and ubiquinone and intermediate one-electron-transferring FeS clusters (N3, N1b, N4, N5, N6a, N6b, and N2). The chain is organized in such a way that a) electron transport is slow, b) electrons can move in both directions, and c) the chain is reduced in coupled mitochondria [80]. In addition to the main chain of FeS clusters, there are two additional ones, N7 and the highly conserved N1a, which is adjacent to FMN and is thought to play an important role in preventing ROS generation [81]. Since FeS clusters can be inactivated by SA [82,83,84], to prevent self-inactivation of the complex, mechanisms for the elimination of SA or its additional reduction to H2O2 could have been evolutionarily developed. Although the presence of SA in the membrane is extremely thermodynamically unfavorable [85], the membrane can be a buffer for the reactive hydroperoxyl radical [57,58,86,87]. Therefore, additional reduction of salvated SA to H2O2 is not only thermodynamically beneficial (Gibbs energy of formation of SA and H2O2 in aqueous solutions (ΔG0) are +7 and -134 kJ/mol; standard electrode potentials (E0) are -0.18 and + 0.36 V, respectively [88,89]), but also rational from the point of view of preserving the activity of the complex. It can be assumed that in coupled mitochondria at a high NADH/NAD+ ratio, the probability of electron return to partially reduced FMN (or ubiquinone) and two-electron reduction of oxygen to H2O2 increases. In contrast, partial oxidation of electron carriers (Figure 6D,E) may decrease the probability of SA reduction by second electron and thus promote the production of SA by both intact and permeabilized mitochondria.
The question arises whether there is any contribution of the facilitated SA exit via PTP or alternative mechanism to the switching of the type of emitted signal? In the presence of Endo substrates, the decrease in the level of H2O2 generation is accompanied by near the same activation of SA generation, regardless of whether the decline in H2O2 generation was accompanied by PTP opening or not (Figure 4). Thus, the contribution of PTP opening per se to the activation of SA output appears to be small.
Theoretically, there may be at least two PTP-independent mechanism of the facilitated SA release from the mitochondrial matrix. First should involve a protonation of SA (anion) to neutral hydroperoxyl radical (pKa = 4.8) [88,89], which penetrates through phospholipid membranes even more effectively than H2O2 [57,58]. The efficient protonation requires the decline in the matrix pH from the physiological values of 7.8–8.0 to 7.1–7.3 (common to the pH of cytosol) or even lower. Second mechanism implied by a body of indirect evidence [59,60,61] should include the extrusion of SA by the transporters of anionic substrates.
Since neither protonophore FCCP nor substrates transported to the matrix via the symport with protons (in comparison with other substrates) increased the release of SA produced by the whole ETC or its segments either before or after the rotenone block (Figure 5 and Figure 6), SA protonation, presumably, has a minor effect on the activation of the SA efflux in our experimental model. At variance, the dose-dependent effect of the combination of substrates on the SA/H2O2 release (Figure 6C) supports the possibility of SA extrusion by the carriers of anionic substrates. Though, this question requires a more detailed study.
It should be stressed that dose-dependent decline in the level of the SA by all respiratory substrates tested but alpha-keto acids (Pyr and 2-OG) (Figure 3 and Figure 6) is not related to their SA-scavenging ability (Figure 7, Supplementary Table S1). Moreover, though alpha-keto acids can operate as the weak SA scavengers, one still can observe SA bursts in their presence under certain conditions (Figure 3), which indicates that their scavenging effect cannot cancel strong SA generation in redox centers.
The physiological or pathophysiological significance of the switching of the type of ROS signal emitted by mitochondria becomes more understandable when considering the modern data on the organization of the redox signaling system in a cell [3,90]. This system includes the subsystems of ROS generation (more than 40 enzymes and complexes), transformation of ROS/redox signal (SOD, GSH-peroxidase, nitric oxide synthase, etc.), of signal attenuation/amplification (cytochrome c, catalase, GSSG reductase, enzymes supporting ROS-induced ROS release), ROS/redox-dependent adapters (thioredoxins, glutaredoxins, peroxiredoxins, etc.), and ROS/adapter-dependent effectors (protein kinases, phosphatases, ion channels, transcription factors) [3,90,91,92,93,94,95]. Among ROS generated in mitochondria, the hydroxyl radical is the most reactive and short-lived (10-9 s) [96], which strongly limits the range of its activity. By contrast, H2O2, stable and capable of penetrating through membranes, is considered to be the main signaling ROS species, which links the subsystems of redox signaling in the cell [3,90]. Not much is known about the own, H2O2-independent, role of SA in the redox signaling. On the one hand, being an anionic reductant scarcely penetrating through membranes [86], it should act locally: inside or in close proximity to mitochondria. In particular, SA can reversibly inhibit FeS-containing proteins like aconitase, CI and CII [82,83,84], and, to some extent, adenine nucleotide translocase [97]. This, in turn, can modify ionic homeostasis and cristae morphology [98]. On the other hand, protonated SA (hydroperoxyl radical) can easily escape from mitochondria or propagate within membranes [57,58,86,87]. This should increase the range of its effectiveness as a redox signal transmitter, although it is not clear to what extent. It is also unclear whether SA- and hydroperoxyl radical-specific adapters and effectors exist beyond mitochondria. At the same time, a body of indirect evidence indicates that an SA/hydroperoxyl radical-dependent subsystem of redox signaling may exist. Indeed, SA directly or through a local intermediary can affect many intracellular processes of both physiologic and pathologic nature [9,14,21,22,23,24,25,26,27,28,29,30]. It is important that, in some cases, SA-mediated redox signaling was clearly distinguished from the H2O2-mediated one on the level of a whole cell [99,100]. The results of our study indicate that the level of substrates, the degree of mitochondrial coupling and PTP induction can switch the type of the ROS signal emitted by mitochondria, which makes it possible to specifically activate SA- or H2O2-dependent signaling cascades.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Dynamics of membrane potential and redox state of pyridine nucleotides (PN) in mitochondria during prolonged incubation in the presence of PTP modulator, Figure S2: Effect of respiratory substrates and PTP inhibitors on the release of SA (A, C, and E) and H2O2 (B, D, and F) from RLM, Figure S3: Dose-response effect of dicarboxylates on the release of SA and H2O2 from mitochondria, Figure S4: Effect of Suc, Rot, and CsA on the kinetics of SA release from intact RLM ; Table S1: Suppression of spontaneous and xanthine oxidase-dependent SA generation by respiratory substrates that are alpha-keto acids.
Author Contributions
Conceptualization K.A.G.; methodology K.A.G. and N.A.B.; software, K.A.G. and N.A.B.; formal analysis, K.A.G. and N.A.B.; investigation, K.A.G. and N.A.B.; data curation, K.A.G.; writing—original draft preparation, K.A.G.; writing—review and editing, K.A.G.; visualization, K.A.G. and N.A.B.; supervision, K.A.G.; project administration, K.A.G.; funding acquisition, K.A.G. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by a grant to Kruglov A.G. from the Russian Science Foundation (project no. 24-24-00522).
Institutional Review Board Statement
All experiments involving animals were carried out in accordance with the European Convention for the Protection of Vertebrates for experimental and other scientific purposes (European Community Council Directive 86/609/EEC, Strasbourg, 19 June 2003) and the principles of the Helsinki Declaration (2000). The study with laboratory animals was approved by the Institute of Theoretical and Experimental Biophysics RAS Ethics Committee (Protocol No 26/2024 of 18.03. 2024).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122(6), 877–902. [Google Scholar] [CrossRef] [PubMed]
- Lennicke, C.; Cochemé, H.M. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell. 2021, 81(18), 3691–3707. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 7, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Li, S.; Hudlikar, R.; Wang, L.; Shannar, A.; Peter, R.; Chou, P.J.; Kuo, H.D.; Liu, Z.; Kong, A.N. Redox signaling, mitochondrial metabolism, epigenetics and redox active phytochemicals. Free Radic Biol. Med. 2022, 179, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Held, J.M. Redox Systems Biology: Harnessing the Sentinels of the Cysteine Redoxome. Antioxid. Redox Signal. 2020, 32(10), 659–676. [Google Scholar] [CrossRef]
- Magnani, N.D.; Marchini, T.; Calabró, V.; Alvarez, S.; Evelson, P. Role of Mitochondria in the Redox Signaling Network and Its Outcomes in High Impact Inflammatory Syndromes. Front. Endocrinol. (Lausanne) 2020, 11, 568305. [Google Scholar] [CrossRef]
- Jomova, K.; Raptova, R.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Valko, M. Reactive oxygen species, toxicity, oxidative stress, and antioxidants: chronic diseases and aging. Arch. Toxicol. 2023, 97(10), 2499–2574. [Google Scholar] [CrossRef]
- Wang, W.; Gong, G.; Wang, X.; Wei-LaPierre, L.; Cheng, H.; Dirksen, R.; Sheu, S.S. Mitochondrial Flash: Integrative Reactive Oxygen Species and pH Signals in Cell and Organelle Biology. Antioxid. Redox Signal. 2016, 25(9), 534–549. [Google Scholar] [CrossRef]
- Sheu, S.S.; Wang, W.; Cheng, H.; Dirksen, R.T. Superoxide flashes: illuminating new insights into cardiac ischemia/ reperfusion injury. Future Cardiol. 2008, 4, 551–554. [Google Scholar] [CrossRef]
- Wang, W.; Fang, H.; Groom, L.; Cheng, A.; Zhang, W.; Liu, J.; Wang, X.; Li, K.; Han, P.; Zheng, M.; et al. Superoxide flashes in single mitochondria. Cell 2008, 134, 279–290. [Google Scholar] [CrossRef]
- Pouvreau, S. Superoxide flashes in mouse skeletal muscle are produced by discrete arrays of active mitochondria operating coherently. PLoS One 2010, 5, 13035. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Huang, Z.; Hou, T.; Xu, J.; Wang, Y.; Shang, W.; Ye, T.; Cheng, H.; Gao, F.; Wang, X. Superoxide constitutes a major signal of mitochondrial superoxide flash. Life Sci. 2013, 93, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Wei-LaPierre, L.; Gong, G.; Gerstner, B.J.; Ducreux, S.; Yule, D.I.; Pouvreau, S.; Wang, X.; Sheu, S.S.; Cheng, H.; Dirksen, R.T.; et al. Respective contribution of mitochondrial superoxide and pH to mitochondria-targeted circularly permuted yellow fluorescent protein (mt-cpYFP) flash activity. J. Biol. Chem. 2013, 288, 10567–10577. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Salahura, G.; Boncompagni, S.; Kasischke, K.A.; Protasi, F.; Sheu, S.S.; Dirksen, R.T. Mitochondrial superoxide flashes: metabolic biomarkers of skeletal muscle activity and disease. FASEB J. 2011, 25, 3068–3078. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Javadov, S.; Saks, V.; Margreiter, R.; Grimm, M. Synchronism in mitochondrial ROS flashes, membrane depolarization and calcium sparks in human carcinoma cells. Biochim. Biophys. Acta Bioenerg. 2017, 1858(6), 418–431. [Google Scholar] [CrossRef]
- Zhou, L.; Aon, M.A.; Almas, T.; Cortassa, S.; Winslow, R.L.; O’Rourke, B. A reaction-diffusion model of ROS-induced ROS release in a mitochondrial network. PLoS Comput. Biol. 2010, 6(1), 1000657. [Google Scholar] [CrossRef]
- Hou, T.; Wang, X.; Ma, Q.; Cheng, H. Mitochondrial flashes: new insights into mitochondrial ROS signalling and beyond. J. Physiol. 2014, 592(17), 3703–3713. [Google Scholar] [CrossRef]
- Fang, H.; Chen, M.; Ding, Y.; Shang, W.; Xu, J.; Zhang, X.; Zhang, W.; Li, K.; Xiao, Y.; Gao, F.; et al. Imaging superoxide flash and metabolism-coupled mitochondrial permeability transition in living animals. Cell Res. 2011, 21(9), 1295–1304. [Google Scholar] [CrossRef]
- Mikkola, R.; Andersson, M.; Kharechkina, E.; Kruglova, S.; Kruglov, A. Fusaricidin-Type Compounds Create Pores in Mitochondrial and Plasma Membranes of Mammalian Cells. Biomolecules 2019, 9(9), 433. [Google Scholar] [CrossRef]
- Kharechkina, E.S.; Nikiforova, A.B.; Kruglov, A.G. Pyridine nucleotides regulate the superoxide anion flash upon permeabilization of mitochondrial membranes: An MCLA-based study. Free Radic Biol. Med. 2018, 124, 473–483. [Google Scholar] [CrossRef]
- Li, K.; Zhang, W.; Fang, H.; Xie, W.; Liu, J.; Zheng, M.; Wang, X.; Wang, W.; Tan, W.; Cheng, H. Superoxide flashes reveal novel properties of mitochondrial reactive oxygen species excitability in cardiomyocytes. Biophys. J. 2012, 102(5), 1011–1021. [Google Scholar] [CrossRef] [PubMed]
- Gong, G.; Liu, X.; Wang, W. Regulation of metabolism in individual mitochondria during excitation-contraction coupling. J. Mol. Cell Cardiol. 2014, 76, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Ghosh, P.; Wan, R.; Ouyang, X.; Cheng, H.; Mattson, M.P.; Cheng, A. Permeability transition pore-mediated mitochondrial superoxide flashes mediate an early inhibitory effect of amyloid beta1-42 on neural progenitor cell proliferation. Neurobiol. Aging 2014, 35(5), 975–989. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Mattson, M.P.; Cheng, A. Permeability transition pore-mediated mitochondrial superoxide flashes regulate cortical neural progenitor differentiation. PLoS One 2013, 8(10), 76721. [Google Scholar] [CrossRef]
- Hou, Y.; Ouyang, X.; Wan, R.; Cheng, H.; Mattson, M.P.; Cheng, A. Mitochondrial superoxide production negatively regulates neural progenitor proliferation and cerebral cortical development. Stem Cells 2012, 30(11), 2535–2547. [Google Scholar] [CrossRef]
- Ying, Z.; Chen, K.; Zheng, L.; Wu, Y.; Li, L.; Wang, R.; Long, Q.; Yang, L.; Guo, J.; Yao, D.; et al. Transient activation of mitoflashes modulates nanog at the early phase of somatic cell reprogramming. Cell Metab. 2016, 23, 220–226. [Google Scholar] [CrossRef]
- Zhang, M.; Sun, T.; Jian, C.; Lei, L.; Han, P.; Lv, Q.; Yang, R.; Zhou, X.; Xu, J.; Hu, Y.; et al. Remodeling of Mitochondrial Flashes in Muscular Development and Dystrophy in Zebrafish. PLoS One 2016, 10(7), 0132567. [Google Scholar] [CrossRef]
- Xiao, Y.; Karam, C.; Yi, J.; Zhang, L.; Li, X.; Yoon, D.; Wang, H.; Dhakal, K.; Ramlow, P.; Yu, T.; et al. ROS-related mitochondrial dysfunction in skeletal muscle of an ALS mouse model during the disease progression. Pharmacol. Res. 2018, 138, 25–36. [Google Scholar] [CrossRef]
- Ma, Q.; Fang, H.; Shang, W.; Liu, L.; Xu, Z.; Ye, T.; Wang, X.; Zheng, M.; Chen, Q.; Cheng, H. Superoxide flashes: early mitochondrial signals for oxidative stress-induced apoptosis. J. Biol. Chem. 2011, 286(31), 27573–27581. [Google Scholar] [CrossRef]
- Shen, E.Z.; Song, C.Q.; Lin, Y.; Zhang, W.H.; Su, P.F.; Liu, W.Y.; Zhang, P.; Xu, J.; Lin, N.; Zhan, C.; et al. Mitoflash frequency in early adulthood predicts lifespan in Caenorhabditis elegans. Nature 2014, 508(7494), 128–132. [Google Scholar] [CrossRef]
- McBride, S.; Wei-LaPierre, L.; McMurray, F.; MacFarlane, M.; Qiu, X.; Patten, D.A.; Dirksen, R.T.; Harper, M.E. Skeletal muscle mitoflashes, pH, and the role of uncoupling protein-3. Arch. Biochem. Biophys. 2019, 663, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Azarias, G.; Chatton, J.Y. Selective ion changes during spontaneous mitochondrial transients in intact astrocytes. PLoS One 2011, 6, 28505. [Google Scholar] [CrossRef] [PubMed]
- Schwarzlander, M.; Logan, D.C.; Fricker, M.D.; Sweetlove, L.J. The circularly permuted yellow fluorescent protein cpYFP that has been used as a superoxide probe is highly responsive to pH but not superoxide in mitochondria: implications for the existence of superoxide ‘flashes’. Biochem. J. 2011, 437, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Schwarzlander, M.; Murphy, M.P.; Duchen, M.R.; Logan, D.C.; Fricker, M.D.; Halestrap, A.P.; Muller, F.L.; Rizzuto, R.; Dick, T.P.; Meyer, A.J.; et al. Mitochondrial ‘flashes’: a radical concept repHined. Trends Cell Biol. 2012, 22, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Schwarzlander, M.; Wagner, S.; Ermakova, Y.G.; Belousov, V.V.; Radi, R.; Beckman, J.S.; Buettner, G.R.; Demaurex, N.; Duchen, M.R.; Forman, H.J.; et al. The ‘mitoflash’ probe cpYFP does not respond to superoxide. Nature 2014, 514, E12–E14. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, J.; Kalyanaraman, B. Hydroethidine- and MitoSOX-derived red fluorescence is not a reliable indicator of intracellular superoxide formation: another inconvenient truth. Free Radic Biol. Med. 2010, 48(8), 983–1001. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Dirksen, R.T. Perspectives on: SGP symposium on mitochondrial physiology and medicine: mitochondrial superoxide flashes: from discovery to new controversies. J. Gen. Physiol. 2012, 139(6), 425–434. [Google Scholar] [CrossRef]
- Wei-LaPierre, L.; Ainbinder, A.; Tylock, K.M.; Dirksen, R.T. Substrate-dependent and cyclophilin D-independent regulation of mitochondrial flashes in skeletal and cardiac muscle. Arch. Biochem. Biophys. 2019, 665, 122–131. [Google Scholar] [CrossRef]
- Rosselin, M.; Santo-Domingo, J.; Bermont, F.; Giacomello, M.; Demaurex, N. L-OPA1 regulates mitoflash biogenesis independently from membrane fusion. EMBO Rep. 2017, 18(3), 451–463. [Google Scholar] [CrossRef]
- Schwarzlander, M.; Logan, D.C.; Johnston, I.G.; Jones, N.S.; Meyer, A.J.; Fricker, M.D.; Sweetlove, L.J. Pulsing of membrane potential in individual mitochondria: a stressinduced mechanism to regulate respiratory bioenergetics in Arabidopsis. Plant Cell 2012, 24, 1188–1201. [Google Scholar] [CrossRef]
- Loschen, G.; Flohé, L.; Chance, B. Respiratory chain linked H2O2 production in pigeon heart mitochondria. FEBS Lett. 1971, 18, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Kareyeva, A.V.; Grivennikova, V.G.; Vinogradov, A.D. Mitochondrial hydrogen peroxide production as determined by the pyridine nucleotide pool and its redox state. Biochim. Biophys. Acta 2012, 1817(10), 1879–1885. [Google Scholar] [CrossRef] [PubMed]
- Votyakova, T.V.; Reynolds, I.J. Detection of hydrogen peroxide with Amplex Red: interference by NADH and reduced glutathione auto-oxidation. Arch. Biochem. Biophys. 2004, 431(1), 138–144. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.; Lardy, H.A. Isolation of liver or kidney mitochondria. Methods Enzymol. 1967, 10, 94–96. [Google Scholar]
- Kharechkina, E.S.; Nikiforova, A.B.; Teplova, V.V.; Odinokova, I.V.; Krestinina, O.V.; Baburina, Y.L.; Kruglova, S.A.; Kruglov, A.G. Regulation of permeability transition pore opening in mitochondria by external NAD(H). Biochim. Biophys. Acta Gen. Subj. 2019, 1863(5), 771–783. [Google Scholar] [CrossRef]
- Gornall, A.G.; Bardawill, C.J.; David, M.M. Determination of serum proteins by means of the biuret reaction. J. Biol. Chem. 1949, 177, 751–766. [Google Scholar] [CrossRef]
- Towne, V.; Will, M.; Oswald, B.; Zhao, Q. Complexities in horseradish peroxidase-catalyzed oxidation of dihydroxyphenoxazine derivatives: Appropriate ranges for pH values and hydrogen peroxide concentrations in quantitative analysis. Analytical Biochemistry 2004, 334(2), 290–296. [Google Scholar] [CrossRef]
- Hirano, T.; Takahashi, Y.; Kondo, H.; Maki, S.; Kojima, S.; Ikeda, H.; Niwa, H. The reaction mechanism for the high quantum yield of Cypridina (Vargula) bioluminescence supported by the chemiluminescence of 6-aryl-2-methylimidazo [1,2-a]pyrazin-3(7H)-ones (Cypridina luciferin analogues). Photochem. Photobiol. Sci. 2008, 7(2), 197–207. [Google Scholar] [CrossRef]
- Suzuki, N.; Suetsuna, K.; Mashiko, S.; Yoda, B.; Nomoto, T.; Toya, Y. Reaction rates for the chemiluminescence of Cypridina luciferin analogs with superoxide: a quenching experiment with superoxide dismutase. Agric. Biol. Chem. 1991, 55(1), 157–160. [Google Scholar] [CrossRef]
- Forman, H.J.; Fridovich, I. Superoxide dismutase: a comparison of rate constants. Arch. Biochem. Biophys. 1973, 158(1), 396–400. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, S.; Kishikawa, N.; Ohyama, K.; Ohba, Y.; Kohno, M.; Masuda, T.; Takadate, A.; Nakashima, K.; Kuroda, N. Evaluation of chemiluminescence reagents for selective detection of reactive oxygen species. Anal. Chim. Acta 2010, 665(1), 74–78. [Google Scholar] [CrossRef] [PubMed]
- Teranishi, K.; Hisamatsu, M.; Yamada, T. Chemiluminescence of 2-methyl-6-arylimidazo-[1,2-a]pyrazin-3(7H)-one in protic solvents: electron-donating substituent effect on the formation of the neutral singlet excited-state molecule. Luminescence 1999, 14(6), 297–302. [Google Scholar] [CrossRef]
- Kambayashi, Y.; Ogino, K. Reestimation of Cypridina luciferin analogs (MCLA) as a chemiluminescence probe to detect active oxygen species--cautionary note for use of MCLA. J. Toxicol. Sci. 2003, 28(3), 139–148. [Google Scholar] [CrossRef] [PubMed]
- Kruglov, A.G.; Nikiforova, A.B.; Shatalin, Y.V.; Shubina, V.V.; Fisyuk, A.S.; Akatov, V.S. Sulfur-containing compounds quench 3,7-dihydro-2-methyl-6-(4-methoxyphenyl)imidazol [1,2-a]pyrazine-3-one chemiluminescence: Discrimination between true antioxidants and quenchers using xanthine oxidase. Anal. Biochem. 2010, 406(2), 230–232. [Google Scholar] [CrossRef]
- Yin, F.; Sancheti, H.; Cadenas, E. Mitochondrial thiols in the regulation of cell death pathways. Antioxid. Redox Signal. 2012, 17(12), 1714–1727. [Google Scholar] [CrossRef]
- Gus’kova, R.A.; Ivanov, I.I.; Kol’tover, V.K.; Akhobadze, V.V.; Rubin, A.B. Permeability of bilayer lipid membranes for superoxide (O2-.) radicals. Biochim. Biophys. Acta 1984, 778(3), 579–585. [Google Scholar] [CrossRef]
- Cordeiro, R.M. Reactive oxygen species at phospholipid bilayers: distribution, mobility and permeation. Biochim. Biophys. Acta 2014, 1838 1 Pt B, 438–444. [Google Scholar] [CrossRef]
- Lustgarten, M.S.; Bhattacharya, A.; Muller, F.L.; Jang, Y.C.; Shimizu, T.; Shirasawa, T.; Richardson, A.; Van Remmen, H. Complex I generated, mitochondrial matrix-directed superoxide is released from the mitochondria through voltage dependent anion channels. Biochem. Biophys. Res. Commun. 2012, 422(3), 515–521. [Google Scholar] [CrossRef]
- Lançar-Benba, J.; Foucher, B.; Saint-Macary, M. Characterization, purification and properties of the yeast mitochondrial dicarboxylate carrier (Saccharomyces cerevisiae). Biochimie 1996, 78(3), 195–200. [Google Scholar] [CrossRef]
- Dierks, T.; Stappen, R.; Salentin, A.; Krämer, R. Probing the active site of the reconstituted aspartate/glutamate carrier from bovine heart mitochondria: carbodiimide-catalyzed acylation of a functional lysine residue. Biochim. Biophys. Acta 1992, 1103(1), 13–24. [Google Scholar] [CrossRef] [PubMed]
- Kruglov, A.G.; Teplova, V.V.; Saris, N.E. The effect of the lipophilic cation lucigenin on mitochondria depends on the site of its reduction. Biochem. Pharmacol. 2007, 74(4), 545–556. [Google Scholar] [CrossRef] [PubMed]
- Kruglov, A.G.; Subbotina, K.B.; Saris, N.E. Redox-cycling compounds can cause the permeabilization of mitochondrial membranes by mechanisms other than ROS production. Free Radic Biol. Med. 2008, 44(4), 646–656. [Google Scholar] [CrossRef] [PubMed]
- Nikiforova, A.B.; Saris, N.E.; Kruglov, A.G. External mitochondrial NADH-dependent reductase of redox cyclers: VDAC1 or Cyb5R3? Free Radic Biol. Med. 2014, 74, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Oosthuizen, M.M.; Engelbrecht, M.E.; Lambrechts, H.; Greyling, D.; Levy, R.D. The effect of pH on chemiluminescence of different probes exposed to superoxide and singlet oxygen generators. J. Biolumin. Chemilumin. 1997, 12(6), 277–284. [Google Scholar] [CrossRef]
- Kruglov, A.G.; Yurkov, I.S.; Teplova, V.V.; Evtodienko, Y.V. Lucigenin-derived chemiluminescence in intact isolated mitochondria. Biochemistry (Moscow) 2002, 67(11), 1262–1270. [Google Scholar] [CrossRef]
- Budd, S.L.; Castilho, R.F.; Nicholls, D.G. Mitochondrial membrane potential and hydroethidine-monitored superoxide generation in cultured cerebellar granule cells. FEBS Lett. 1997, 415(1), 21–24. [Google Scholar] [CrossRef]
- Grivennikova, V.G.; Vinogradov, A.D. Generation of superoxide by the mitochondrial Complex I. Biochim. Biophys. Acta 2006, 1757(5-6), 553–561. [Google Scholar] [CrossRef]
- Grivennikova, V.G.; Vinogradov, A.D. Partitioning of superoxide and hydrogen peroxide production by mitochondrial respiratory complex I. Biochim. Biophys. Acta 2013, 1827(3), 446–454. [Google Scholar] [CrossRef]
- Bleier, L.; Dröse, S. Superoxide generation by complex III: from mechanistic rationales to functional consequences. Biochim. Biophys. Acta 2013, 1827(11-12), 1320–1331. [Google Scholar] [CrossRef]
- Speijer, D. Can All Major ROS Forming Sites of the Respiratory Chain Be Activated By High FADH2/NADH Ratios?: Ancient evolutionary constraints determine mitochondrial ROS formation. Bioessays 2019, 41(1), 1800180. [Google Scholar] [CrossRef] [PubMed]
- Tretter, L.; Takacs, K.; Hegedus, V.; Adam-Vizi, V. Characteristics of alpha-glycerophosphate-evoked H2O2 generation in brain mitochondria. J. Neurochem. 2007, 100(3), 650–663. [Google Scholar] [CrossRef] [PubMed]
- Koufos, O.; Mailloux, R.J. Protein S-glutathionylation and sex dimorphic effects on hydrogen peroxide production by dihydroorotate dehydrogenase in liver mitochondria. Free Radic Biol. Med. 2023, 194, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Henriques, B.J.; Katrine, J.; Olsen, R.; Gomes, C.M.; Bross, P. Electron transfer flavoprotein and its role in mitochondrial energy metabolism in health and disease. Gene 2021, 776, 145407. [Google Scholar] [CrossRef]
- Loschen, G.; Azzi, A.; Richter, C.; Flohé, L. Superoxide radicals as precursors of mitochondrial hydrogen peroxide. FEBS Lett。 1974, 42(1), 68–72. [Google Scholar] [CrossRef]
- Dionisi, O.; Galeotti, T.; Terranova, T.; Azzi, A. Superoxide radicals and hydrogen peroxide formation in mitochondria from normal and neoplastic tissues. Biochim. Biophys. Acta 1975, 403(2), 292–300. [Google Scholar] [CrossRef]
- Boveris, A. Mitochondrial production of superoxide radical and hydrogen peroxide. Adv Exp Med Biol 1977, 78, 67–82. [Google Scholar] [CrossRef]
- Arce-Molina, R.; Cortés-Molina, F.; Sandoval, P.Y.; Galaz, A.; Alegría, K.; Schirmeier, S.; Barros, L.F.; San Martín, A. A highly responsive pyruvate sensor reveals pathway-regulatory role of the mitochondrial pyruvate carrier MPC. Elife 2020, 9, 53917. [Google Scholar] [CrossRef]
- Stern, A.; Fokra, M.; Sarvin, B.; Alrahem, A.A.; Lee, W.D.; Aizenshtein, E.; Sarvin, N.; Shlomi, T. Inferring mitochondrial and cytosolic metabolism by coupling isotope tracing and deconvolution. Nat. Commun. 2023, 14(1), 7525. [Google Scholar] [CrossRef]
- Sazanov, L.A. A giant molecular proton pump: structure and mechanism of respiratory complex I. Nat. Rev. Mol. Cell Biol. 2015, 16(6), 375–388. [Google Scholar] [CrossRef]
- Sazanov, L.A.; Hinchliffe, P. Structure of the hydrophilic domain of respiratory complex I from Thermus thermophilus. Science 2006, 311, 1430–1436. [Google Scholar] [PubMed]
- Vasquez-Vivar, J.; Kalyanaraman, B.; Kennedy, M.C. Mitochondrial aconitase is a source of hydroxyl radical. An electron spin resonance investigation. J. Biol. Chem. 2000, 275(19), 14064–14069. [Google Scholar] [CrossRef] [PubMed]
- Sadek, H.A.; Szweda, P.A.; Szweda, L.I. Modulation of mitochondrial complex I activity by reversible Ca2+ and NADH mediated superoxide anion dependent inhibition. Biochemistry 2004, 43(26), 8494–8502. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.S.; Mezera, V.; Dighe, P.; Melov, S.; Gerencser, A.A.; Sweis, R.F.; Pliushchev, M.; Wang, Z.; Esbenshade, T.; McKibben, B.; et al. Superoxide produced by mitochondrial site IQ inactivates cardiac succinate dehydrogenase and induces hepatic steatosis in Sod2 knockout mice. Free Radic Biol. Med. 2021, 164, 223–232. [Google Scholar] [CrossRef]
- Sawyer, D.T.; Joan, S. Valentine How super is superoxide? Accounts of Chemical Research 1981, 14(12), 393–400. [Google Scholar] [CrossRef]
- Wardman, P. Molecular mechanisms of oxygen and “electron-affinic” radiosensitizers. Int. J. Radiat. Oncol. Biol. Phys. 1989, 16(1), 286–288. [Google Scholar] [CrossRef]
- Benon Bielski, H.J.; Diane, E.C.; Ravindra, L.A.; Alberta, B.R. Reactivity of HO2/O−2 Radicals in Aqueous Solution. J. Phys. Chem. Ref. Data 1985, 14(4), 1041–1100. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Stanbury, D.M.; Bounds, P.L. Electrode potentials of partially reduced oxygen species, from dioxygen to water. Free Radic Biol. Med. 2010, 49(3), 317–322. [Google Scholar] [CrossRef]
- Wood, P.M. The potential diagram for oxygen at pH 7. Biochem. J. 1988, 253(1), 287–289. [Google Scholar] [CrossRef]
- Bindoli, A.; Fukuto, J.M.; Forman, H.J. Thiol chemistry in peroxidase catalysis and redox signaling. Antioxid. Redox Signal. 2008, 10(9), 1549–1564. [Google Scholar] [CrossRef]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol. Med. 2002, 33(3), 337–349. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef] [PubMed]
- Veal, E.A.; Underwood, Z.E.; Tomalin, L.E.; Morgan, B.A.; Pillay, C.S. Hyperoxidation of Peroxiredoxins: Gain or Loss of Function? Antioxid. Redox Signal. 2018, 28(7), 574–590. [Google Scholar] [CrossRef] [PubMed]
- Glorieux, C.; Calderon, P.B. Catalase, a remarkable enzyme: targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017, 398(10), 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Filburn, C.R.; Klotz, L.O.; Zweier, J.L.; Sollott, S.J. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med 2000, 192, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Strategies of antioxidant defense. Eur. J. Biochem. 1993, 215(2), 213–219. [Google Scholar] [CrossRef] [PubMed]
- Zini, R.; Berdeaux, A.; Morin, D. The differential effects of superoxide anion, hydrogen peroxide and hydroxyl radical on cardiac mitochondrial oxidative phosphorylation. Free Radic. Res. 2007, 41(10), 1159–1166. [Google Scholar] [CrossRef]
- Ježek, P.; Jabůrek, M.; Holendová, B.; Engstová, H.; Dlasková, A. Mitochondrial Cristae Morphology Reflecting Metabolism, Superoxide Formation, Redox Homeostasis, and Pathology. Antioxid. Redox Signal. 2023, 39(10-12), 635–683. [Google Scholar] [CrossRef]
- Thorpe, G.W.; Reodica, M.; Davies, M.J.; Heeren, G.; Jarolim, S.; Pillay, B.; Breitenbach, M.; Higgins, V.J.; Dawes, I.W. Superoxide radicals have a protective role during H2O2 stress. Mol. Biol. Cell 2013, 24(18), 2876–2884. [Google Scholar] [CrossRef]
- Hao, S.; Cai, D.; Gou, S.; Li, Y.; Liu, L.; Tang, X.; Chen, Y.; Zhao, Y.; Shen, J.; Wu, X.; et al. Does each Component of Reactive Oxygen Species have a Dual Role in the Tumor Microenvironment? Curr Med Chem. 2023. [Google Scholar] [CrossRef]
Figure 1.
Effect of respiratory substrates and PTP inhibitors on the release of SA (A, C, and E) and H2O2 (B, D, and F) from RLM. RLM (0.75 mg protein/ml) were placed in the standard KCl-BM without substrates and EGTA (the concentration of free Ca2+ was ~15 µM, as indicated by titration with EGTA), and the suspension was immediately poured into two tubes with either 15 µM MCLA or 40 µM AR and HRP (3 U/ml). The suspensions were then distributed into the wells of plates for luminescence and fluorescence measurements and analyzed in parallel using two plate readers. Where indicated, the wells contained 5 mM 3-HB, 5 mM 2-OG, 5 mM Suc, 1 mM EGTA, 2 µM RR, 1 µM CsA, and SOD (100 U/ml). Panels A, C and E show the MDCL accumulated over 900 ms at each point on the curve and expressed in AU. Points on the curves are the means ± standard deviation (n = 3) of three technical replicates. Panels B, D, and F show the rate of H2O2 production (pmol·min-1·mg protein-1). Points on the curves are the means of three technical replicates (n = 3). Inserts in the panels are the data of cumulative MDCL (Σ(MDCL)0-60 min, AU) and H2O2 production per hour. The figure shows one representative experiment of at least five similar.
Figure 1.
Effect of respiratory substrates and PTP inhibitors on the release of SA (A, C, and E) and H2O2 (B, D, and F) from RLM. RLM (0.75 mg protein/ml) were placed in the standard KCl-BM without substrates and EGTA (the concentration of free Ca2+ was ~15 µM, as indicated by titration with EGTA), and the suspension was immediately poured into two tubes with either 15 µM MCLA or 40 µM AR and HRP (3 U/ml). The suspensions were then distributed into the wells of plates for luminescence and fluorescence measurements and analyzed in parallel using two plate readers. Where indicated, the wells contained 5 mM 3-HB, 5 mM 2-OG, 5 mM Suc, 1 mM EGTA, 2 µM RR, 1 µM CsA, and SOD (100 U/ml). Panels A, C and E show the MDCL accumulated over 900 ms at each point on the curve and expressed in AU. Points on the curves are the means ± standard deviation (n = 3) of three technical replicates. Panels B, D, and F show the rate of H2O2 production (pmol·min-1·mg protein-1). Points on the curves are the means of three technical replicates (n = 3). Inserts in the panels are the data of cumulative MDCL (Σ(MDCL)0-60 min, AU) and H2O2 production per hour. The figure shows one representative experiment of at least five similar.
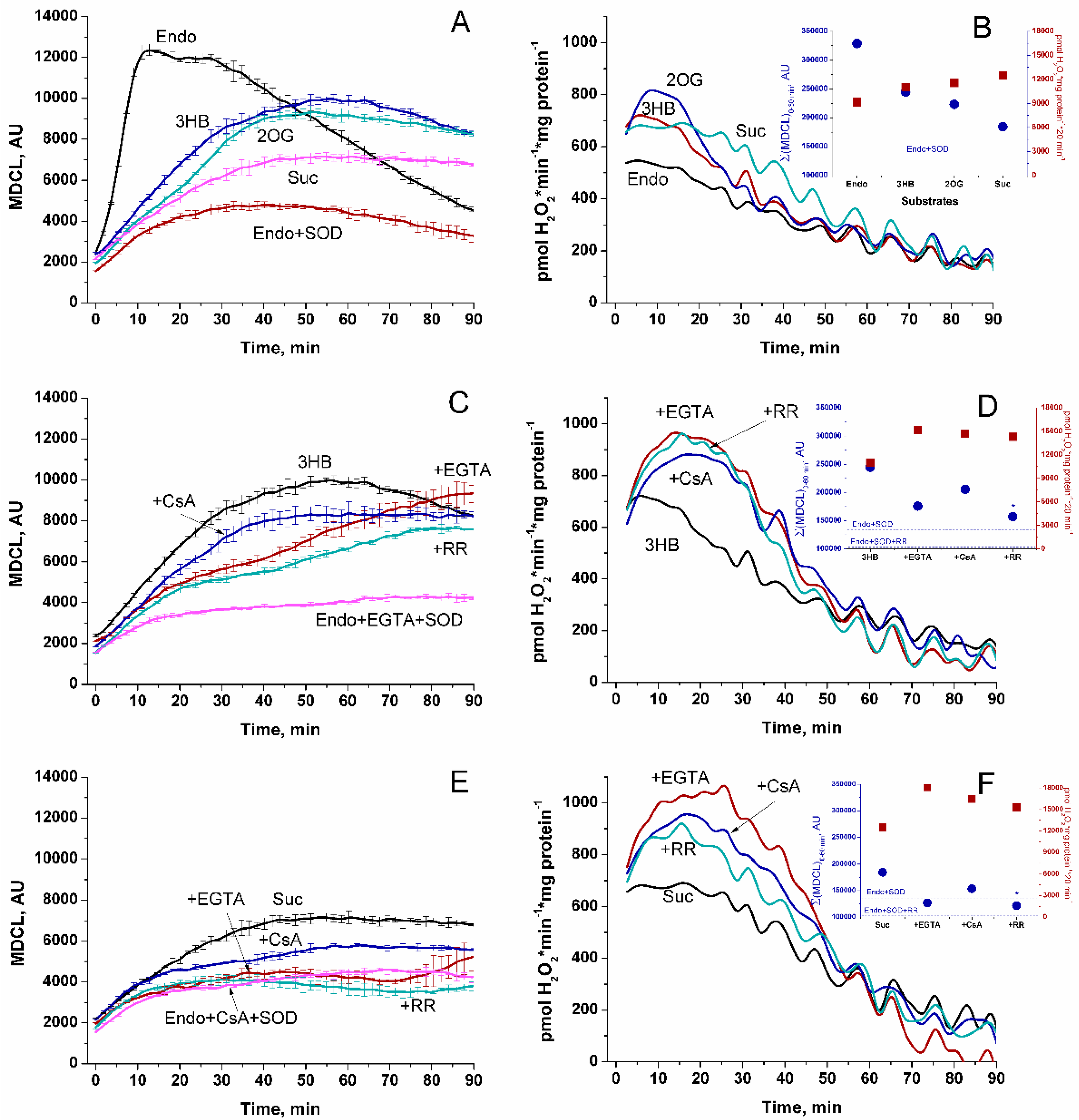
Figure 2.
Modulation of an MDCL signal by SA and SOD in solution and mitochondrial suspension. A. Standard KCl-BM contained 10 μM MCLA and, where indicated, SOD 100 U/ml. B. The medium contained 20 μM MCLA, 400 μM xanthine, and 0–0.083 U/ml of XO. The insert shows the dependence of maximum MDCL on the concentration of XO. C. Medium contained 15 μM MCLA, 400 μM xanthine, 0.15 U/ml of XO, and SOD at an indicated concentration (0–50 U/ml). The insert shows the dependence of the maximum and the integrated (for 105 min) MDCL on the concentration of SOD. D. The medium contained 5 mM Suc, 1 mM EGTA, 15 μM MCLA, rotenone (2 μg/ml), and, where shown, SOD (100 U/ml). RLM (0.5 mg/ml) were added just before measurements. The arrow shows the addition of alamethicin (40 μg/mg protein). Points on the curves are the means ± standard deviation (n = 3) of three technical replicates. Panels show the representative data of at least three similar experiments.
Figure 2.
Modulation of an MDCL signal by SA and SOD in solution and mitochondrial suspension. A. Standard KCl-BM contained 10 μM MCLA and, where indicated, SOD 100 U/ml. B. The medium contained 20 μM MCLA, 400 μM xanthine, and 0–0.083 U/ml of XO. The insert shows the dependence of maximum MDCL on the concentration of XO. C. Medium contained 15 μM MCLA, 400 μM xanthine, 0.15 U/ml of XO, and SOD at an indicated concentration (0–50 U/ml). The insert shows the dependence of the maximum and the integrated (for 105 min) MDCL on the concentration of SOD. D. The medium contained 5 mM Suc, 1 mM EGTA, 15 μM MCLA, rotenone (2 μg/ml), and, where shown, SOD (100 U/ml). RLM (0.5 mg/ml) were added just before measurements. The arrow shows the addition of alamethicin (40 μg/mg protein). Points on the curves are the means ± standard deviation (n = 3) of three technical replicates. Panels show the representative data of at least three similar experiments.
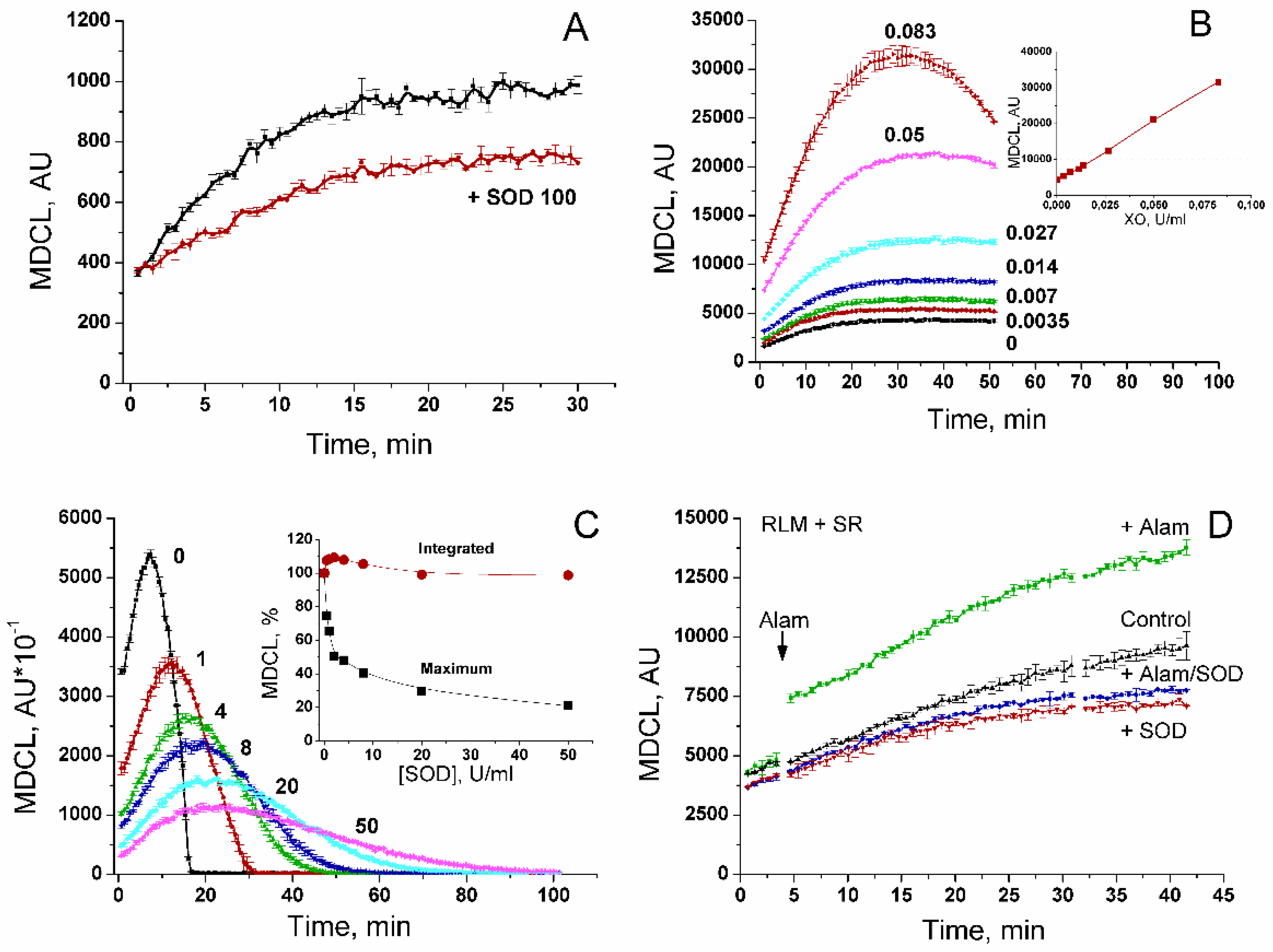
Figure 3.
PTP opening switches the SA/H2O2 redox signal coming from mitochondria. RLM (0.75 mg protein/ml) were placed in the standard KCl-BM without added respiratory substrates but supplemented with 10 μM EGTA (the concentration of free Ca2+ was ~5 µM, as indicated by titration with EGTA). The suspension was immediately poured into two tubes with either 15 µM MCLA or 40 µM AR plus HRP (3 U/ml), transferred to the wells of plates for absorbance and fluorescence measurements, and analyzed as in Figure 1. Were indicated, the wells contained 5 mM Pyr, 5 mM 2-OG, 5 mM Suc, 1 mM EGTA, and SOD (100 U/ml). In all panels, black, red, and blue lines indicate changes in absorbance, H2O2 production, and MDCL, respectively. In panels B, D, F, and H, cyan lines are MDCL in the presence of SOD. Points on the curves are the means (n = 3) of three technical replicates. The figure shows one representative experiment of at least five similar.
Figure 3.
PTP opening switches the SA/H2O2 redox signal coming from mitochondria. RLM (0.75 mg protein/ml) were placed in the standard KCl-BM without added respiratory substrates but supplemented with 10 μM EGTA (the concentration of free Ca2+ was ~5 µM, as indicated by titration with EGTA). The suspension was immediately poured into two tubes with either 15 µM MCLA or 40 µM AR plus HRP (3 U/ml), transferred to the wells of plates for absorbance and fluorescence measurements, and analyzed as in Figure 1. Were indicated, the wells contained 5 mM Pyr, 5 mM 2-OG, 5 mM Suc, 1 mM EGTA, and SOD (100 U/ml). In all panels, black, red, and blue lines indicate changes in absorbance, H2O2 production, and MDCL, respectively. In panels B, D, F, and H, cyan lines are MDCL in the presence of SOD. Points on the curves are the means (n = 3) of three technical replicates. The figure shows one representative experiment of at least five similar.
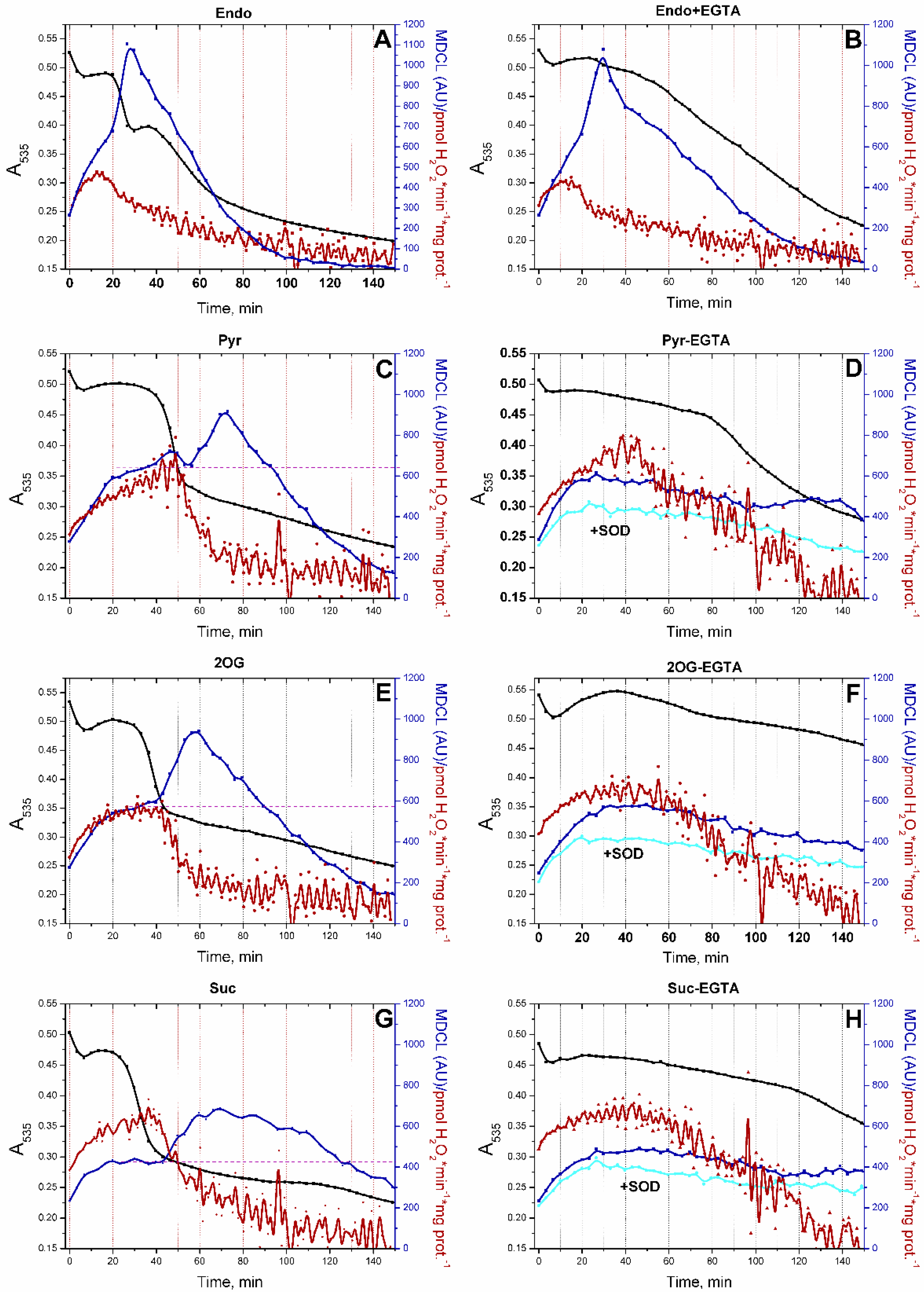
Figure 4.
Effect of PTP inhibitors on the SA/H2O2 release from RLM oxidizing Endo substrates. RLM (0.75 mg protein/ml) were treated exactly as described in Figure 2. Were indicated, the wells contained 1 mM EGTA, 1 μM CsA, and 2 μM RR. In all panels, black, red, and blue lines indicate changes in absorbance, H2O2 production, and MDCL, respectively. Points on the curves are the means (n = 3) of three technical replicates. The figure shows one representative experiment of at least five similar.
Figure 4.
Effect of PTP inhibitors on the SA/H2O2 release from RLM oxidizing Endo substrates. RLM (0.75 mg protein/ml) were treated exactly as described in Figure 2. Were indicated, the wells contained 1 mM EGTA, 1 μM CsA, and 2 μM RR. In all panels, black, red, and blue lines indicate changes in absorbance, H2O2 production, and MDCL, respectively. Points on the curves are the means (n = 3) of three technical replicates. The figure shows one representative experiment of at least five similar.
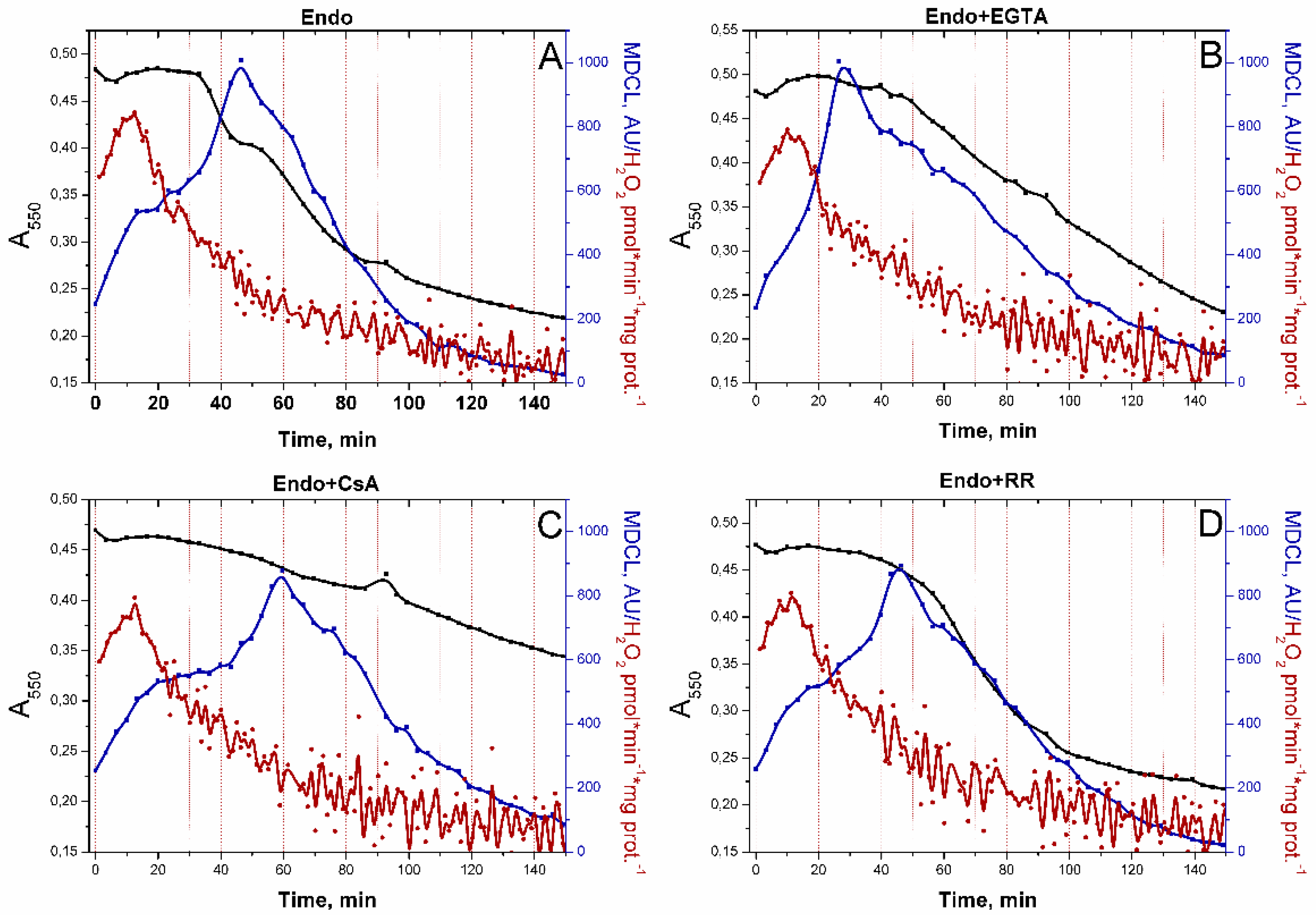
Figure 5.
ROS producers in mitochondria supplemented with different respiratory substrates. Carriers: MPC, mitochondrial pyruvate carrier; GC, mitochondrial glutamate carrier; OGCP, mitochondrial 2-oxoglutarate/malate carrier protein; DIC, mitochondrial dicarboxylate carrier; and ANT, adenine nucleotide translocator. Dehydrogenases: BHBDH, 3-hydroxybutyrate dehydrogenase; PDH, pyruvate dehydrogenase; OGDH, oxoglutarate dehydrogenase; SDH, succinate dehydrogenase; MDH, malate dehydrogenase; and GDH, glutamate dehydrogenase. ETC complexes: RCI, RCII, RCIII, and RCIV. Electron donor: LPAH2, reduced lipoamide. The asterisk shows indirect modulators of ROS production.
Figure 5.
ROS producers in mitochondria supplemented with different respiratory substrates. Carriers: MPC, mitochondrial pyruvate carrier; GC, mitochondrial glutamate carrier; OGCP, mitochondrial 2-oxoglutarate/malate carrier protein; DIC, mitochondrial dicarboxylate carrier; and ANT, adenine nucleotide translocator. Dehydrogenases: BHBDH, 3-hydroxybutyrate dehydrogenase; PDH, pyruvate dehydrogenase; OGDH, oxoglutarate dehydrogenase; SDH, succinate dehydrogenase; MDH, malate dehydrogenase; and GDH, glutamate dehydrogenase. ETC complexes: RCI, RCII, RCIII, and RCIV. Electron donor: LPAH2, reduced lipoamide. The asterisk shows indirect modulators of ROS production.
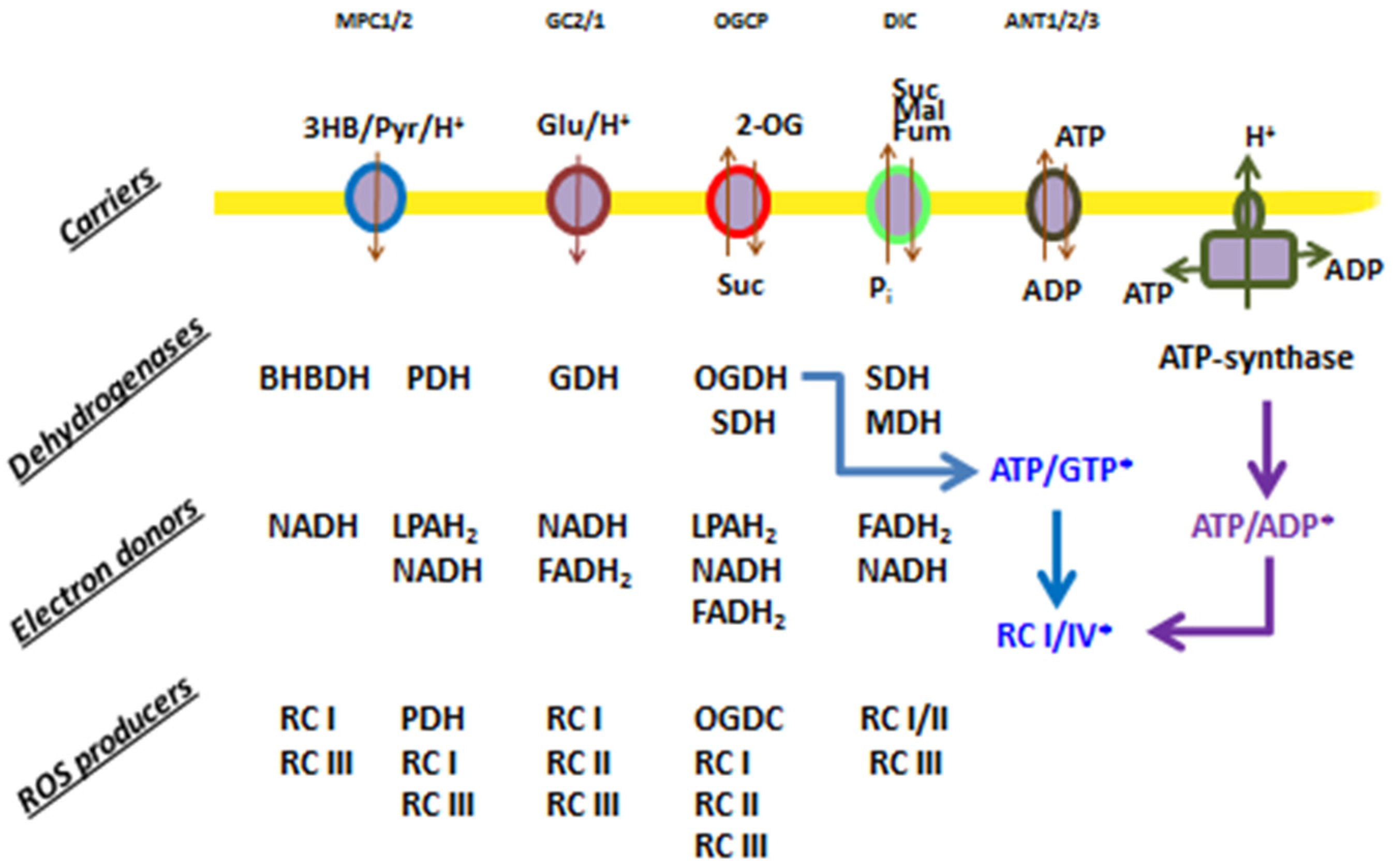
Figure 6.
The opposite dose-response effect of respiratory substrates on the release of SA and H2O2 from mitochondria. RLM (0.4 mg protein/ml) were placed in the standard KCl-BM without added respiratory substrates but supplemented with 1 μM CsA, and the suspension was processed as described in the legend to Figure 3. Were indicated, the wells contained 1 mM ATP, SOD (100 U/ml), 2 μM rotenone, 500 nM FCCP, 3-HB, 2-OG, Suc, Pyr, GM and their combinations (all substrates at either 250 μM or 5 mM concentrations). In all panels, blue and red symbols show the cumulative MDCL (Σ(MDCL)0-60 min, AU) and H2O2 production in the first hour of incubation, respectively. Blue doted lines show MDCL in the presence of SOD and indicated substrates. The left ordinate axis on all panels starts with the value of MDCL in the presence of SOD and all substrates at a concentration of 5 mM. Experimental points are the means ± S.D. (n = 3) of three technical replicates. The figure shows one representative experiment of at least three similar.
Figure 6.
The opposite dose-response effect of respiratory substrates on the release of SA and H2O2 from mitochondria. RLM (0.4 mg protein/ml) were placed in the standard KCl-BM without added respiratory substrates but supplemented with 1 μM CsA, and the suspension was processed as described in the legend to Figure 3. Were indicated, the wells contained 1 mM ATP, SOD (100 U/ml), 2 μM rotenone, 500 nM FCCP, 3-HB, 2-OG, Suc, Pyr, GM and their combinations (all substrates at either 250 μM or 5 mM concentrations). In all panels, blue and red symbols show the cumulative MDCL (Σ(MDCL)0-60 min, AU) and H2O2 production in the first hour of incubation, respectively. Blue doted lines show MDCL in the presence of SOD and indicated substrates. The left ordinate axis on all panels starts with the value of MDCL in the presence of SOD and all substrates at a concentration of 5 mM. Experimental points are the means ± S.D. (n = 3) of three technical replicates. The figure shows one representative experiment of at least three similar.
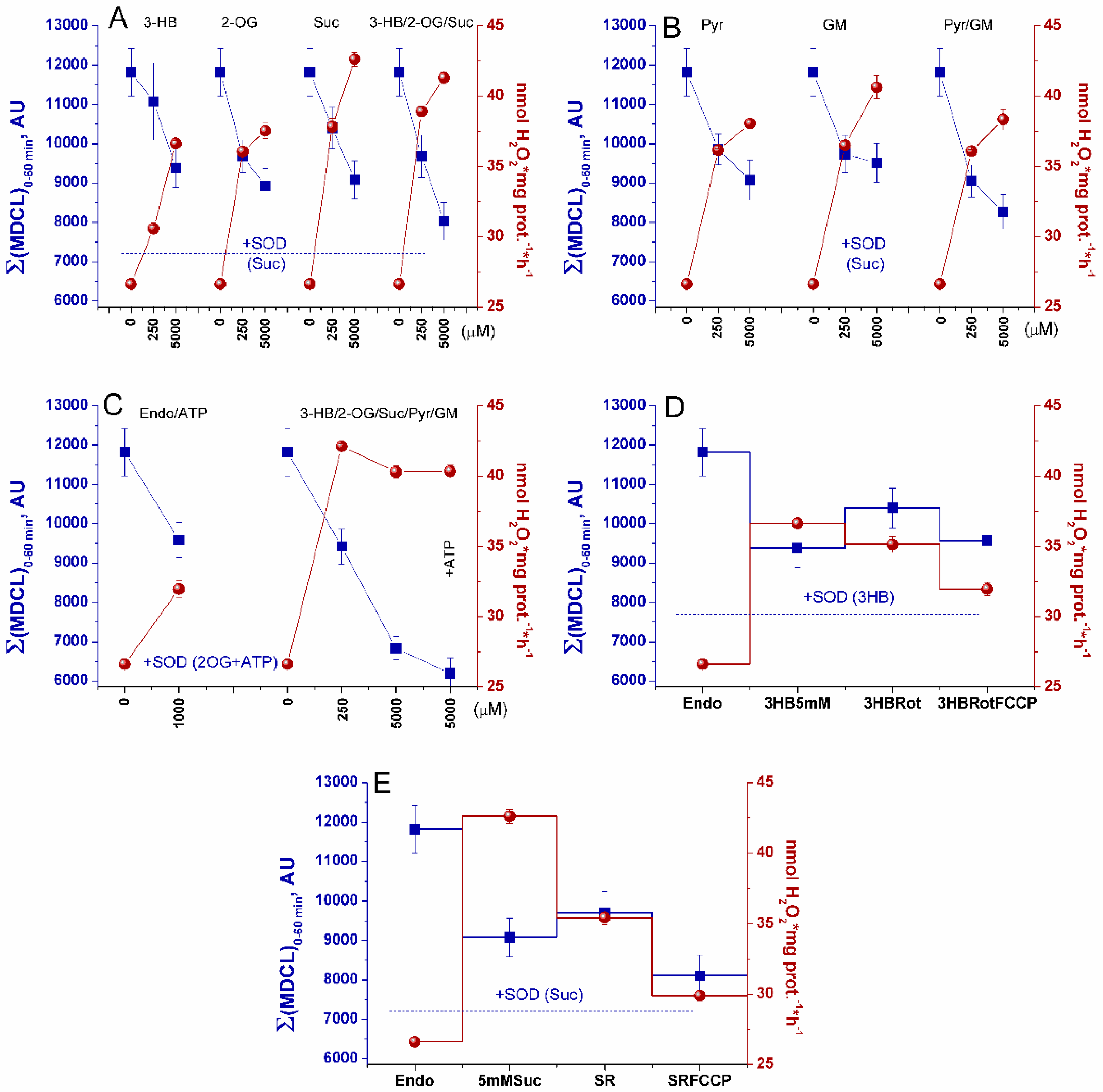
Figure 7.
Effect of respiratory substrates on the spontaneous and XO-dependent SA generation. Standard incubation medium was supplemented with 15 μM MCLA (A-H) and 400 μM xanthine plus XO (0.005 U/ml) (XXO) (A,C,E, and G), extensively mixed, and immediately added to the wells of a 96-well plate containing, where shown, 1 mM ATP, Pyr, GM, Suc, 3-HB, 2-OG (all at a concentration of 5 mM)), and SOD (200 U/ml). A-F. Original traces of one representative experiment of three similar. Points on the curves are the means ± S.D. (n = 3) for three technical replicates. G and H. Cumulative MDCL (± cumulative S.D.) within 60 min of incubation for the curves presented in panels A-F. Asterisks show the significant difference with the control (* ̶ P < 0.05, ** ̶ P < 0.01).
Figure 7.
Effect of respiratory substrates on the spontaneous and XO-dependent SA generation. Standard incubation medium was supplemented with 15 μM MCLA (A-H) and 400 μM xanthine plus XO (0.005 U/ml) (XXO) (A,C,E, and G), extensively mixed, and immediately added to the wells of a 96-well plate containing, where shown, 1 mM ATP, Pyr, GM, Suc, 3-HB, 2-OG (all at a concentration of 5 mM)), and SOD (200 U/ml). A-F. Original traces of one representative experiment of three similar. Points on the curves are the means ± S.D. (n = 3) for three technical replicates. G and H. Cumulative MDCL (± cumulative S.D.) within 60 min of incubation for the curves presented in panels A-F. Asterisks show the significant difference with the control (* ̶ P < 0.05, ** ̶ P < 0.01).
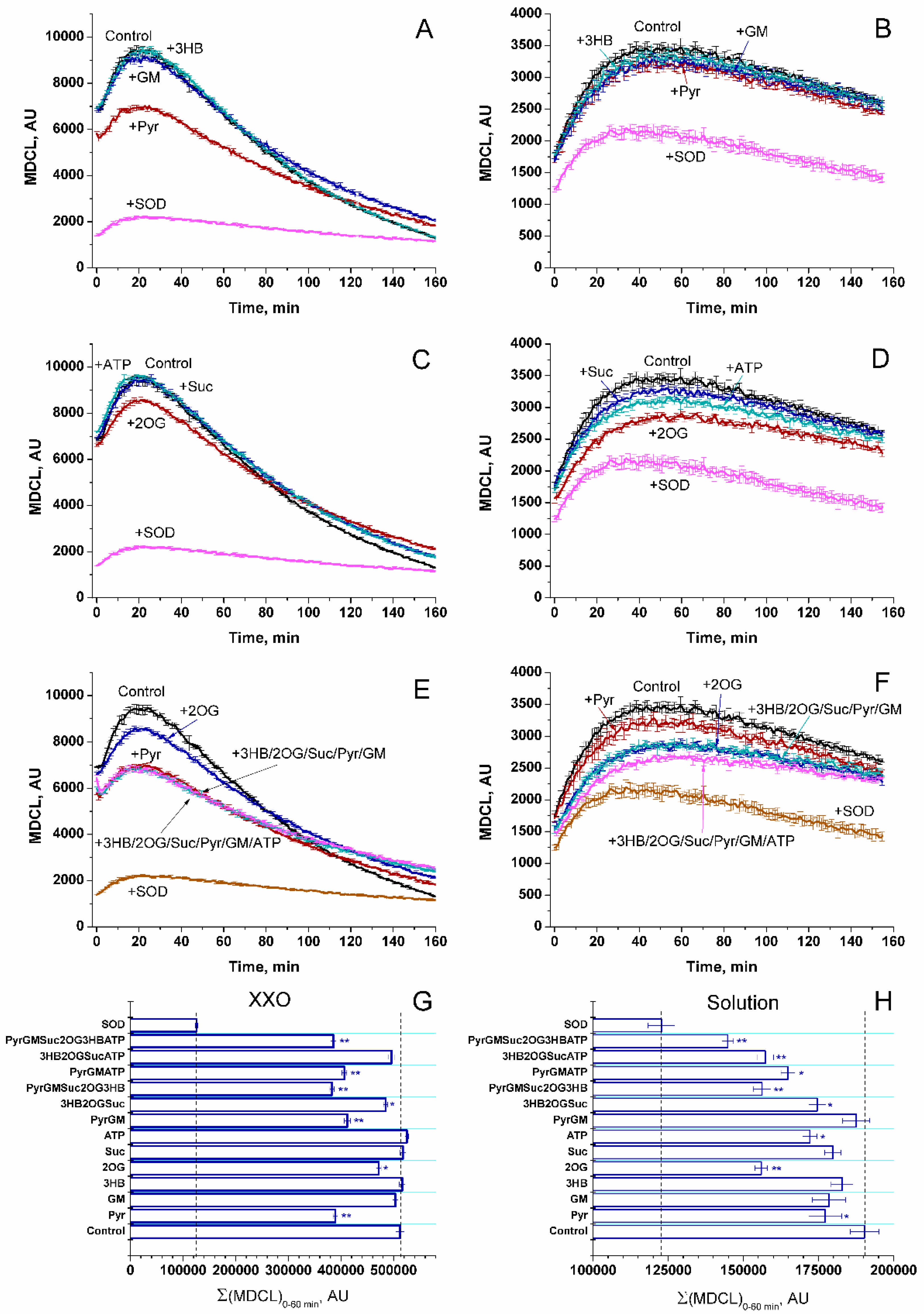
Figure 8.
Effect of AN on the SA/H2O2 release from RLM oxidizing Endo substrates. RLM (0.4 mg protein/ml) were treated exactly as indicated in the legend to Figure 3 except that 1 μM CsA was added to the incubation medium. Were indicated, the wells also contained 1 mM ATP, 1 mM ADP, 2.5 μM oligomycin, and SOD (100 U/ml). In all panels, black, red, and blue lines indicate the changes in absorbance, H2O2 production, and MDCL, respectively. In panels A and B, cyan lines show the MDCL in the presence of SOD. Panel F shows the data on cumulative MDCL and H2O2 production per hour. Points on the curves are the means (n = 3) of three technical replicates. The figure shows one representative experiment of at least three similar.
Figure 8.
Effect of AN on the SA/H2O2 release from RLM oxidizing Endo substrates. RLM (0.4 mg protein/ml) were treated exactly as indicated in the legend to Figure 3 except that 1 μM CsA was added to the incubation medium. Were indicated, the wells also contained 1 mM ATP, 1 mM ADP, 2.5 μM oligomycin, and SOD (100 U/ml). In all panels, black, red, and blue lines indicate the changes in absorbance, H2O2 production, and MDCL, respectively. In panels A and B, cyan lines show the MDCL in the presence of SOD. Panel F shows the data on cumulative MDCL and H2O2 production per hour. Points on the curves are the means (n = 3) of three technical replicates. The figure shows one representative experiment of at least three similar.
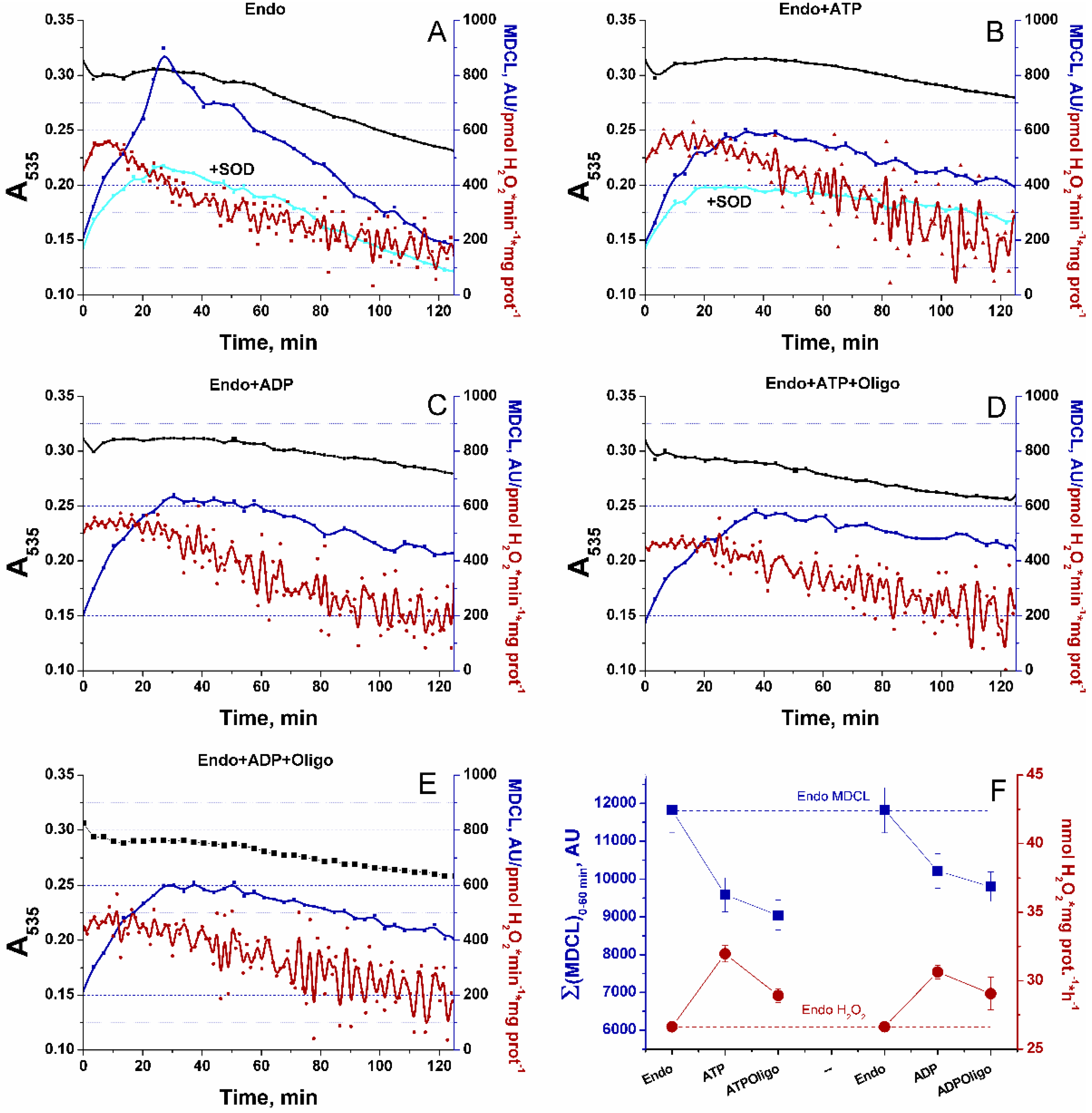
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



