
Submitted:
02 October 2024
Posted:
03 October 2024
You are already at the latest version
Abstract
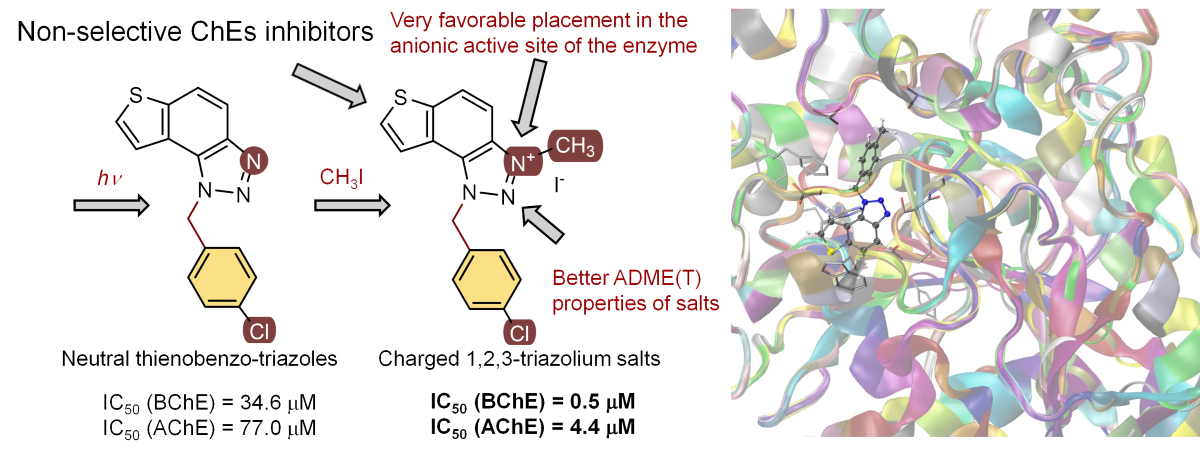
In previous research, 1,2,3-triazolium salts showed significant biological activity as potential in-hibitors of the enzymes cholinesterases (ChEs), crucial for neurotransmission. In this research, pairs of uncharged thienobenzo-triazoles and their charged salts were prepared in order to ex-amine further the role of the positive charge on the nitrogen of the triazole ring on the interac-tions in the active site of the enzymes and to compare the selectivity of 1,2,3-triazolium salts in relation to their uncharged analogs obtained by photochemical cyclization. Neutral thienoben-zo-triazoles showed very good selective activity toward butyrylcholinesterase (BChE), while their salts showed excellent non-selective inhibition toward both BChE (the most active 23: IC50 0.47 mikroM) and acetylcholinesterase (AChE) enzymes (the most active 23: IC50 4.4 mikroM). These new structures with incorporated 1,2,3-triazolium salts present the new scaffold for drug development as it is known that the current therapy in Alzheimer's disease (AD) are selective AChE inhibitors, while in Parkinson's and all stages of AD, non-selective inhibitors of ChEs are preferred. Molecular docking of the selected compounds and their corresponding salts into the active sites of ChEs was conducted to identify the interactions responsible for the stability of the non-covalent cholinesterase-ligand complexes. As genotoxicity studies are crucial when devel-oping new active substances and finished drug forms, in silico studies for all the synthesized compounds have shown that compound 18 is the most promising candidate for genotoxic safety.
Keywords:
cholinesterase inhibition
; docking
; genotoxicity
; 1
; 2
; 3-triazolium salts
1. Introduction
1,2,3-Triazolium salts can be directly synthesized in several steps, including CuAAC, N-alkylation, and salt metathesis. These methods can achieve a wide range of 1,2,3-triazole derivatives with different substituents and substitution patterns [1]. In addition to their impressive applications as ionic liquids, catalysts, and metal ligands, 1,2,3-triazolium salts exhibit a wide range of biological activities, such as antibacterial [2,3,4], antifungal [5], anticancer [6], and antileishmanial [7] properties. They are valuable in developing pesticides and pharmaceutical products [1]. Incorporating 1,2,3-triazole groups into polymeric materials via simple synthetic methods can generate antibacterial properties. The use of such materials can solve the problem of microbial contamination in medical devices or specialized antibacterial environments. The incorporation of 1,2,3-triazolium salts into anticancer drug carriers may bring additional opportunities. Modifying special nanotubes with triazolium salts can result in positively charged carriers of anticancer drugs with improved encapsulation efficiency, controlled release ability, increased water solubility, and thus improved anticancer activity. Based on the structure-activity relationship of the 1,2,3-triazolium salt derivatives, the substituents at the N-1 and N-3 positions are believed to play key roles in biological activities. The lipophilicity/hydrophilicity balance of 1,2,3-triazolium salts is crucial for their high biological activity and selectivity. In addition, 1,2,3-triazole derivatives can have multiple modes of action, and electrostatic interactions with anionic components on the cell membrane are among the most important.
Compared with neutral triazoles, 1,2,3-triazolium salts have superior biological activities and water solubility and are promising in developing new drugs for drug-resistant pathogens. 1,2,3-triazolium salts possess unique chemical properties, such as high stability, tunable reactivity, and electronic properties [8]. They are valuable building blocks in organic synthesis and have also been used to develop new catalysts and fluorescent dyes [9]. These salts have three active sites, including the N-1, N-3, and C-4 positions, which can bind to different functional groups and thus regulate their biological activities and cytotoxicity.
Numerous examples of 1,2,3,-triazole derivatives that act as inhibitors of cholinesterase enzymes (ChEs), inspired by tacrine and acridones, can be found in the literature [10,11,12,13,14,15]. These are molecules with a substituted benzyl group on the 1,2,3-triazole ring. The structure-activity relationship (SAR) showed that by replacing hydrogen with chlorine in the para-position of the benzyl ring, the inhibitory activity against acetylcholinesterase (AChE) is improved [11]. Similarly, the structure-activity relationship showed that the methoxy group in the para-position of the benzyl ring has the best inhibitory activity, while chlorine, fluorine, methyl, and the unsubstituted benzyl ring decrease the activity [12]. Since it has a large dipole moment and can form hydrogen bonds with active sites, benzyl-1,2,3-triazole was also used for molecular hybridization with methylindolinone [15]. The synthesized compound showed good to moderate activity towards butyrylcholinesterase (BChE) but weak inhibition of AChE. The IC50 value for BChE was better than that of AChE inhibitor donepezil.
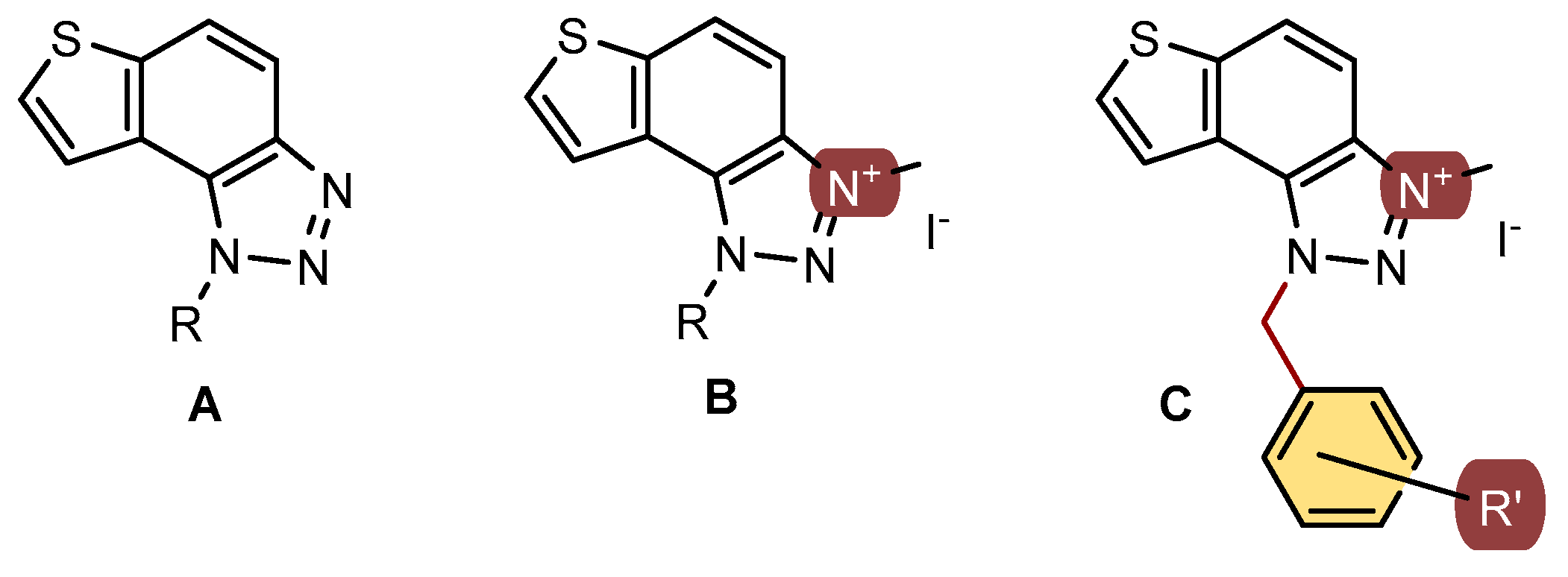
In our previous study, different thienobenzo-triazoles A and triazolium salts B possessing various substituents on the triazole ring (Figure 1) were synthesized [16,17,18,19,20] and spectroscopically and biologically evaluated. They exhibited the capability to bind to proteins, displaying diverse inhibition properties towards AChE and BChE. Triazolium salts B exhibited excellent inhibition and demonstrated very good non-selective inhibition of both BChE and AChE enzymes. In contrast, neutral thienobenzo-triazoles A were shown to be selective towards BChE, even with very good inhibition potential.
Additionally, the representatives of those salts remained stable in an aqueous solution and displayed intriguing fluorimetric properties characterized by strong Stokes shifts exceeding 160 nm [20]. The molecular docking of the best-performing candidates to cholinesterases’ active site identified cation–π and π-π interactions as responsible for the stability of the enzyme–ligand complexes. Regarding the safety of triazolium salts B, no structural alerts were found by the Derek Nexus considering potential genotoxicity, so they could be considered the safest choices for further development into drug formulations, particularly given their high inhibitory potential against AChE and BChE.
In this study, the second series of triazolium salts C (Figure 1) with the substituted benzyl group on the charged 1,2,3-triazole ring were designed and biologically evaluated as cholinesterase inhibitors. These structures present the new scaffold of 1,2,3-triazolium salts for drug development as the current therapy in Alzheimer’s disease (AD) are selective AChE inhibitors, while in Parkinson’s and all stages of AD, non-selective inhibitors of ChE are preferred.
2. Results and Discussion
2.1. Synthesis and Spectroscopic Characterization of New 1,2,3-Triazolium Salts
New targeted triazolium salts with the substituted benzyl group on the charged 1,2,3-triazole ring were synthesized to investigate their inhibition potential toward AChE and BChE based on the previous encouraging results. To obtain the same, a series of four consecutive reactions was carried out (Scheme 1). The 1,4-disubstituted triazole aldehydes 1–5 were prepared by the reaction of 1-(4-nitrophenyl)-1H-1,2,3-triazole-4-carbaldehyde and the corresponding amine (isolated yields 23-82%, Scheme 1) [21].
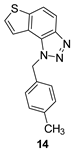
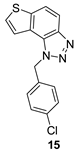
The obtained aldehydes 1–5 entered into the Wittig reaction, where they react with the corresponding phosphorus ylides containing the thiophene ring, thus obtaining triazole-thienostilbenes 6–13 as mixtures of cis- and trans-isomers (isolated yields for derivatives 6–8 80-91% and isolated yields for derivatives 9–13 13-32%, Scheme 1). Mixtures of the geometrical isomers of triazole-thienostilbenes 6–13 were not separated, and due to the presence of the resulting conjugated system, they were subjected to photochemical cyclization leading to thienobenzo-triazoles 14–21 (isolated yields 15-73%, Scheme 1). The photoproducts 14–21 were transferred to triazolium salts 22–29 as targeted products using methyl iodide (isolated yields 15-42%, except 29 obtained only in traces according to NMR analysis, not isolated and evaluated, Scheme 1). Two additional triazolium salts, 30 and 31, were also prepared from the previously obtained and characterized neutral thienobenzo-triazoles (See Materials and Methods) [20].
All isolated triazole aldehydes 1–5, triazole-thienostilbenes 6–13, thienobenzo-triazoles 14–21, and triazolium salts 22–31 were fully spectroscopically characterized (except 29 obtained only in traces, See Materials and Methods and ESI). In the 1H NMR spectra of 6–13, it can be seen the resolved patterns for ethylenic protons of both geometrical isomers between 6.5 and 7.0 ppm (cis-isomers) and between 6.8 and 7.7 ppm (trans-isomers) with the characteristic coupling constants, as well as signals for the protons on various substituents and singlets for the protons on the triazole rings (6.9 and 7.9 ppm). Preparative irradiations of 6–13 in toluene solutions under aerobic conditions gave the thienobenzo-triazoles 14–21. They were isolated in high yields (Scheme 1) and characterized by NMR spectroscopy (See Materials and Methods and ESI). The formation of the electrocyclization photoproducts 14–21 was generally accompanied by the formation of some high-molecular-weight products, which were not investigated. In their 1H NMR spectra, it can be seen the disappearance of the ethylenic protons and the singlets for the protons on the 1,2,3-triazole rings in comparison with the starting 6–13 (Figure 2). In the 1H NMR spectra of triazolium salts 22–31, a new signal of the methyl group on the triazole nitrogen is visible around 4.8 ppm. All signals in the triazolium salts 22–31 are shifted to a less shaded area compared to their neutral analogues 14–21.
2.2. Inhibitory Activity of Thienobenzo-Triazoles 14–21 and triazolium Salts 22–31 Toward Enzymes Cholinesterases
Newly prepared thienobenzo-triazoles 14–21 and triazolium salts 22–31 were tested for inhibitory activity toward AChE and BChE in various concentrations, depending on solubility. Spectrophotometric Ellman assay was applied [22], and the results obtained were compared to those of commercially available Galantamine as the reference standard. The IC50 values of triazole derivatives 14–31 for AChE and BChE inhibition are presented in Table 1.
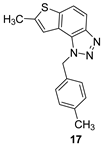
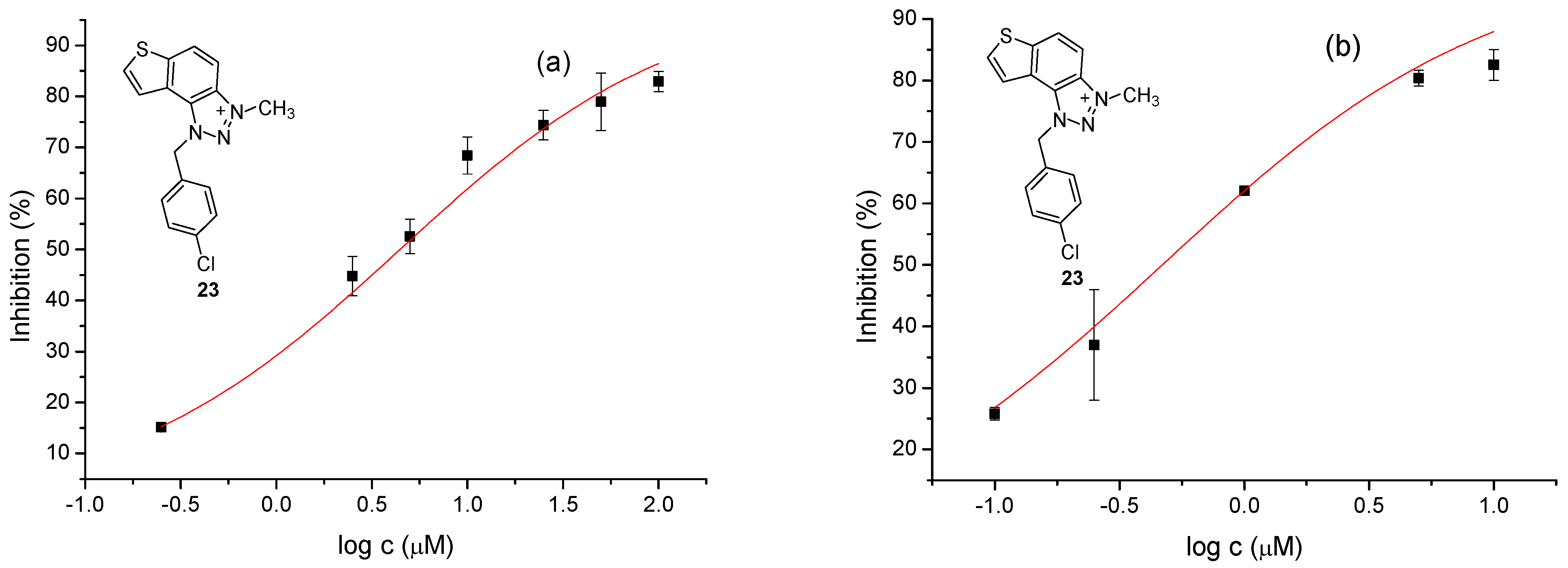
The tested compounds were divided into two main groups: thienobenzo-triazoles, presented on the left side of Table 1, and triazolium salts, presented on the right. Analogs can be found between these two groups that differ only in charge, while the type and position of the substituents are the same. According to structural features, the following structure-activity relationship can be noted. The presence of a charge on the triazole ring favored more potent inhibition toward both enzymes, AChE and BChE. Leading compounds were 22, 23, 26, 27, and 30, which inhibited both enzymes in the excellent to very good range of concentrations compared to the standard (Table 1, Figure 3 and Figure 4). The strongest inhibitor of both enzymes was derivative 23, with a value for BChE in the nanomolar range that was better than the standard (Table 1) and a slightly higher but still excellent value for AChE. The exception among the salts was 25, whose IC50 values were not in such a good range of concentrations. The presence of chlorine on the benzyl ring supports the activity since three salts with excellent results, 23, 26, and 30, possess this substituent. Introduction of methyl substituent on thiophene ring, from 22 to 25, significantly reduced activity toward enzymes. The same structural change from 23 to 26 resulted similarly. Derivatives 24 and 27 were an exception, but only toward AChE, because the methylated charged derivative 27 was a better inhibitor of this enzyme than the unmethylated 24.
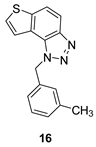
Uncharged thienobenzo-triazoles 14–21 prefer inhibition of BChE over AChE. This observation is consistent with the conclusions of our previous research, which indicated that neutral photoproducts tend to be weaker inhibitors and show selectivity toward BChE [20]. It is supported additionally by the fact that the uncharged analog of 30 analyzed in the previous research [20] showed weaker inhibition of both enzymes. For herein tested thienobenzo-triazoles, conversion to salt significantly increased inhibition of AChE and, to a good extent, also of BChE. Exceptions were 16 and 17, which achieved better IC50 values for BChE than the corresponding salts 24 and 25.
It can be concluded that the most important structural feature for thienobenzo-triazoles is the presence/absence of charge. Triazolium salts were shown to be dual inihibitors whose potency of inhibition varies depending on the substituent of thiophene and triazole heterocycles.

Table 1.
Calculated IC50 values for the inhibition of AChE and BChE by compounds 14–31.
| Compound | IC50 (AChE/BChE) /μM | Compound | IC50 (AChE/BChE)/μM |
|---|---|---|---|
 |
117.2 / 24.3 | 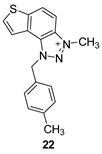 |
39.2 / 1.00 |
 |
77.0 / 34.6 | 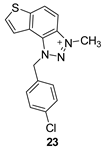 |
4.4 / 0.47 |
 |
77.1 / 2.9 | 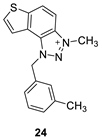 |
55.1 / 7.0 |
 |
> 250 / 57.6 | 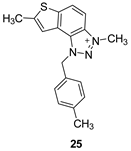 |
211.1 / 102.3 |
 |
> 100 / 59.0 | 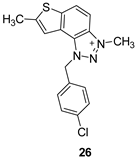 |
8.1 / 13.4 |
 |
> 250 / 70.3 | 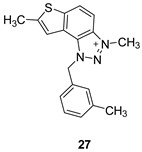 |
8.3 / 9.7 |
 |
185.8 / 90.2 | 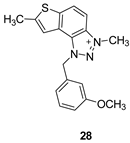 |
54.8 / 10.6 |
 |
464.7 / 171.9 | 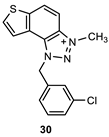 |
12.2 / 2.7 |
| Galantamine | 0.15 / 7.9 | 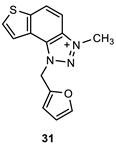 |
157.0 / 16.1 |
Figure 3.
Dose-response curve for the inhibition of AChE (a) and BChE (b) by 22.
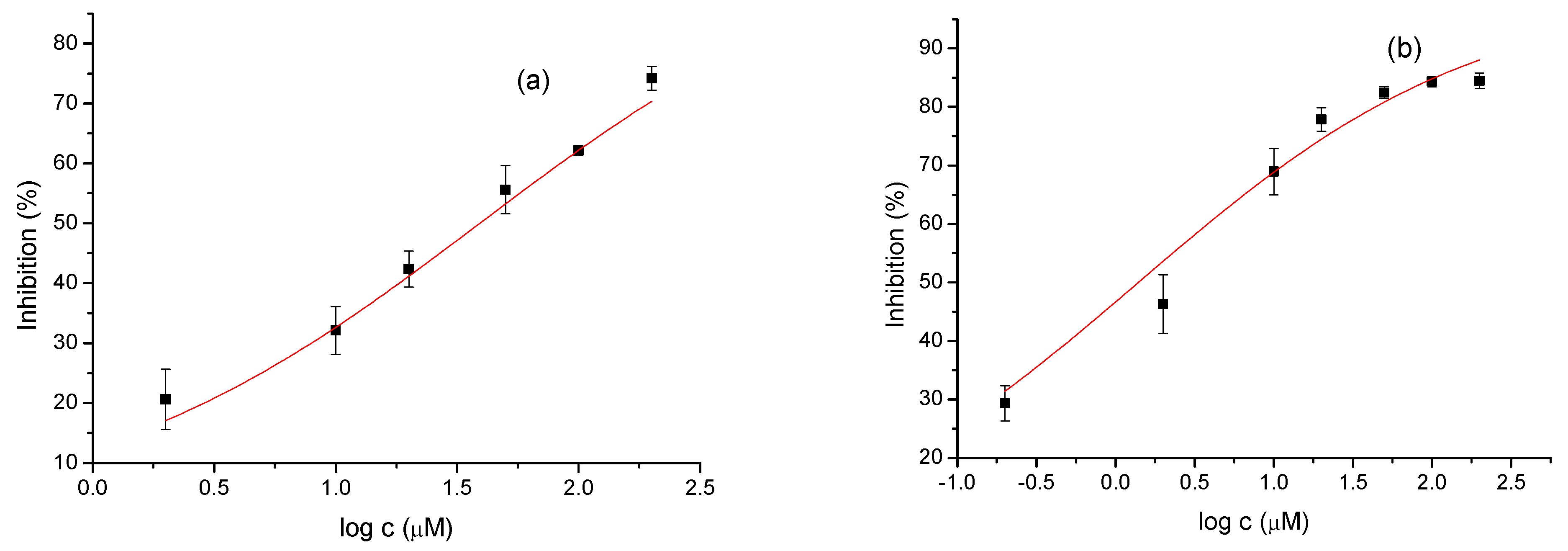
Figure 4.
Dose-response curve for the inhibition of AChE (a) and BChE (b) by 23.
2.3. ADMET Properties of Biologically Active Thienobenzo-Triazoles and Triazolium Salts
Absorption, distribution, metabolism, excretion, and toxicity (ADME(T)) studies are critical in drug development. They give insights into whether the compound exhibits drug-like pharmacokinetic properties and whether it has properties that will cause safety concerns in people [23]. ADMET properties are investigated in silico, in vitro, and in vivo. In early drug discovery, in silico tools are indispensable. For this study, free in silico tools [24,25] were employed to screen the candidate compounds.
Table 2.
In silico study for more indicators about the absorption, distribution, metabolism, excretion, and toxicity (ADME(T)) in the human body of some pairs of thienobenzo-triazoles 14, 15, and 17 and their biologically active charged triazolium salt 22, 23, 25 and 30.
Table 2.
In silico study for more indicators about the absorption, distribution, metabolism, excretion, and toxicity (ADME(T)) in the human body of some pairs of thienobenzo-triazoles 14, 15, and 17 and their biologically active charged triazolium salt 22, 23, 25 and 30.
| Property Model Name | Compound | Unit | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 14 | 22 | 15 | 23 | 17 | 25 | 30 | |||
| Absorption | Water solubility | -6,53 | -4,782 | -6,30 | -5,18 | -6,2 | -4,62 | -4,69 | log mol/L |
| Absorption | Caco2 permeability | 0.82 | 0.852 | 0,892 | 1.209 | 0,794 | 0,78 | 0,761 | log Papp in 10-6 cm/s |
| Absorption | Intestinal absorption (human) | 87,71 | 95,798 | 76,09 | 95,903 | 76,1 | 76,2 | 76,1 | % Absorbed |
| Distribution | BBB permeability | 0,6378 | 0,484 | 0,563 | 0,457 | 0,63 | 0,453 | 0,396 | log BB |
| Metabolism | CYP2D6 substrate | No | No | No | No | No | No | No | |
| Metabolism | CYP3A4 substrate | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Metabolism | CYP2C9 inhibitior | No | No | No | No | No | No | No | |
| Metabolism | CYP2D6 inhibitior | Yes | Yes | Yes | No | No | No | No | |
| Metabolism | CYP3A4 inhibitior | No | No | No | No | No | No | No | |
| Excretion | Total Clearance | 0,96 | 0,874 | 0,901 | 0,855 | 0,873 | 0,882 | 0,925 | log ml/min/kg |
| Excretion | Renal OCT2 substrate | No | No | No | No | No | No | No | |
| Toxicity | AMES toxicity | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Toxicity | hERG I inhibitor | No | No | No | No | No | No | No | |
| Toxicity | hERG II inhibitor | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Toxicity | Oral Rat Acute Toxicity (LD50) | 2,31 | 2,251 | 2,47 | 2,322 | 2,25 | 2,25 | 2,24 | mol/kg |
| Toxicity | Oral Rat Chronic Toxicity (LOAEL) | - | 0.286 | - | 1.444 | - | - | - | log mg/kg_bw/day |
| Toxicity | Hepatotoxicity | No | No | No | Yes | Yes | Yes | Yes | |
| Toxicity | Skin Sensitisation | No | No | No | No | No | No | No | |
As the compounds in the form of salts (22–31) are developed mainly to enhance the ADmet part of the properties, it is important to compare the neutral molecule against its salt counterpart. It can be seen (Table 2) that the water solubility, although higher, remains very low, and all other properties are alike. To determine whether the in silico models could differentiate between the neutral molecule and the salt, multiple software programs were utilized, revealing variations in the absorption estimates. As other parts of the adMET are concerned, the results are similar. The CNS bioavailability is somewhat reduced, as can be expected, as this is a delicate balance between water solubility and lipophilicity.
2.4. Molecular Docking of Biologically Active Thienobenzo-Triazoles and Triazolium Salts into Cholinesterases
To reveal the structure of non-covalent complexes formed between the tested compounds and enzymes and identify the interactions responsible for their stabilization, molecular docking of two thienobenzo-triazoles and their triazolium counterparts into the active sites of acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) was performed.
The most promising inhibitory activity was observed for triazolium salts 22 and 23. Their corresponding neutral thienobenzo-triazoles 14 and 15, which contain the same substituents, also performed very well (Table 1). Figure 5 presents the structures of the complexes resulting from the docking of compounds 14 (left panel) and 15 (right panel) into the active site of AChE, while the results of docking of cations 22 and 23 into the AChE are shown in Figure 6.
Both compounds occupy a similar position within the peripheral anionic subsite (PAS), allowing for π-π stacking between the thienobenzo moiety and Trp286 and similar interactions with Tyr341. The phenyl substituent in both cases is favorably oriented towards Phe338, also engaging in π-π stacking. In compound 14, the methyl group at the para position of the phenyl substituent participates in an alkyl-π interaction with Phe338. In the protein-ligand complex with 15, the chlorine atom is situated 4.2 Å from the centroid of Phe338, suggesting the presence of halogen-π dispersive attraction. The attractive interactions between the chlorine of the ligand and the π-system of the aromatic protein residues can occur in two distinct geometries, depending on the positioning of the Cl atom relative to the aromatic ring: “edge-on” and “face-on” [26]. When the difference between the distance from the chlorine to the center of the aromatic ring and the distance from Cl to the closest carbon of the aromatic ring exceeds 0.3 Å, the “edge-on” geometry is observed. In this case, the distances are 4.2 Å and 3.8 Å, indicating that the interaction can be classified as “edge-on.”
The structures of the complexes formed between acetylcholinesterase and triazolium cations 22 and 23 (Figure 6) reveal that, in both cases, the quaternary nitrogen of the triazole engages in electrostatic interaction with the anionic residues of the active site, specifically aspartate in the peripheral anionic subdomain (Asp74). The distance between the quaternary nitrogen and the oxygen of Asp74 is 3.9 Å, forming a salt bridge that significantly contributes to the stability of the complex. The conformations of the docked cations 22 and 23 are nearly identical (Figure 6), facilitating alkyl-π interactions between the methyl group on the triazole and Tyr332, as well as parallel π-π stacking of the triazolo-benzo fragment with Tyr341 and the thiophene core with Phe338. For cation 23, the preferred conformation of the ligand is unfavorable for establishing a chlorine-π interaction, as the Cl atom is positioned at distances exceeding 4.5 Å from the closest aromatic residues.
Figure 6.
The AChE’s active site structure docked with triazolium salts 22 (left) and 23 (right panel). Distances in angstroms; hydrogen atoms of the residues omitted for clarity.
Figure 6.
The AChE’s active site structure docked with triazolium salts 22 (left) and 23 (right panel). Distances in angstroms; hydrogen atoms of the residues omitted for clarity.
The docking of compounds 14 and 15 into butyrylcholinesterase (BChE) resulted in the structures shown in Figure 7. In both cases, the ligands adopt similar orientations within the active site. The thienobenzo moiety engages in π-π stacking interactions with Trp82, which belongs to the anionic subdomain of the enzyme’s active site. Simultaneously, the triazole ring interacts similarly with the histidine residue in the esteratic site. The phenyl substituent is oriented to facilitate π-π stacking with residue Phe329.
In compound 14, the methyl group on the phenyl substituent is conveniently positioned within the acyl pocket of BChE, interacting with residues Leu286 and Val288 to achieve alkyl-alkyl dispersive interaction. In molecule 15, the chlorine atom behaves similarly, establishing hydrophobic contacts with the aliphatic carbons of the residues in the acyl pocket.
Finally, the structures of the complexes between 22 and 23 and BChE, shown in Figure 8, again reveal the presence of the electrostatic attraction between the positively charged ligands’ region and the anionic residue of the enzyme.
As noted previously, the anion involved in AChE is aspartate from the peripheral anionic subsite (PAS); however, in BChE, the anionic residue engaged in this charge-charge interaction is Glu197, which is part of the active site’s anionic subdomain. Similar to the structures of the complexes with ligands 14 and 15, the thienobenzo moiety establishes parallel π-π stacking with the tryptophan in the anionic subsite, Trp82. In both cations, the phenyl substituent participates in stacking with Phe329. The methyl group on the phenyl of 22 is engaged in alkyl-alkyl dispersion with the acyl pocket. The chlorine atom in 23 is positioned too far from the nearest aromatic residue to form halogen-π dispersion effectively; however, it achieves hydrophobic interaction with residues of the acyl pocket.
2.5. Potential Genotoxicity of Thienobenzo-Triazoles 14–21 and Triazolium Salts 22–31
The pharmaceutical development process necessitates the evaluation of all involved compounds (including drug substances, impurities, intermediates, etc.) for their potential mutagenic or carcinogenic effects. The ICH M7 Guideline governs the regulation of such compounds, requiring that levels present in the drug substance or drug product be calculated based on their acceptable daily intake (ADI) and the drug’s maximum daily dose (MDD). Experimental data for new drug substances and products are often unavailable in the literature databases. The quantitative structure-activity relationship (QSAR) approach is essential in these cases. QSAR models predict biological activity based on structural components [27]. This approach is particularly useful during the early stages of identifying potentially active drug substances, as it helps eliminate compounds that exhibit biological activity but may also have mutagenic potential. The most commonly utilized tool for this purpose is Lhasa software (Nexus v.2.5.2 (Build 5, Jul 2022), Derek Nexus v.6.2.1, Meteor Nexus v.3.1.0, Sarah Nexus v.3.2.1, Vitic Link v.2.4.2), which incorporates two complementary models—one rule-based and the other statistical. Predictions from these models are subsequently reviewed by an expert to ensure accuracy.
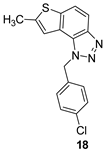
In the case of compounds 14–31 that are investigated for their potential biological activity with the emphasis on the inhibition of enzymes cholinesterases, the software, as it only recognizes special functionalities, cannot determine the potential for salts, so the results are inconclusive. It does not treat them as their neutral counterparts, as the Derek Nexus will give them a negative, but with less assurance. As for the neutral molecules 14–21, the best candidate is compound 18. Here the Sarah prediction is inconclusive, but as Derek is strongly negative, the overall prediction comes out negative. For the rest of the neutral compounds, Sarah prediction has a positive result, and if they are to be tested further, it would be wise to do an experimental mutagenicity AMES test.
Table 3.
The mutagenic potential of thienobenzo-triazoles 14–21 and triazolium salts 22–31 by Lhasa M7 evaluation (green square – negative, red square – positive, white square – no data available); grey highlight – negative, orange highlight – positive, white – strongly negative).
Table 3.
The mutagenic potential of thienobenzo-triazoles 14–21 and triazolium salts 22–31 by Lhasa M7 evaluation (green square – negative, red square – positive, white square – no data available); grey highlight – negative, orange highlight – positive, white – strongly negative).
| Structure | ICH M7 Class | DerekPrediction | Sarah Prediction |
Overall In Silico |
|---|---|---|---|---|
| 14 | Class 3 |  |
 |
Positive |
| 15 | Class 3 | |
|
Positive |
| 16 | Class 3 | |
|
Positive |
| 17 | Class 3 | |
|
Positive |
| 18 | Inconclusive |  |
 |
Negative |
| 19 | Class 3 | |
|
Positive |
| 20 | Class 3 | |
|
Positive |
| 21 | Class 3 | |
|
Positive |
| 22 | Inconclusive |  |
 |
Inconclusive |
| 23 | Inconclusive | |
|
Inconclusive |
| 24 | Inconclusive | |
|
Inconclusive |
| 25 | Inconclusive | |
|
Inconclusive |
| 26 | Inconclusive | |
|
Inconclusive |
| 27 | Inconclusive | |
|
Inconclusive |
| 28 | Inconclusive | |
|
Inconclusive |
| 29 | Inconclusive | |
|
Inconclusive |
| 30 | Inconclusive | |
|
Inconclusive |
| 31 | Inconclusive | |
|
Inconclusive |
3. Materials and Methods
3.1. General Procedure
1H NMR and 13C NMR techniques were used to confirm the structure of the synthesized compounds, and the spectra were recorded on a Bruker Avance instrument at 300 and 600 MHz for 1H NMR and at 75 and 150 MHz for 13C NMR. Chemical shifts (δ) are expressed in parts per million values (ppm) and coupling constants (J) in Hz. As a standard, tetramethylsilane, TMS, was used. The following signals in the 1H NMR spectra were specific and did not correspond to compounds: the signal for water in chloroform at about 1.50 ppm, the signal for chloroform at 7.24 ppm, the signal for dichloromethane in chloroform at 5.26 ppm, and the signal for acetone in chloroform at 2.17 ppm. Each 13C NMR spectrum contained one specific signal (group of three peak lines) at 77 ppm corresponding to the used solvent – deuterated chloroform. High-resolution mass spectrometry (HRMS) analyses were performed on a MALDI TOF/TOF analyzer mass spectrometer fitted with an Nd:YAG laser at 355 nm (fitting rate of 200 Hz). Photochemical reactions were carried out in a 50.0 mL solution in quartz cuvettes that transmitted light. For this purpose, a Rayonet photochemical reactor equipped with UV lamps (10) with a wavelength of 313 nm was used. All solvents used in this work were purified by distillation and were commercially available. The phosphonium salts were synthesized in our laboratory, and 1-(4-nitrophenyl)-1H-1,2,3-triazole-4-carbaldehyde used was previously synthesized in our laboratory [21]. After each Wittig reaction, an extraction was performed three times, separating the organic and aqueous layers.
The organic layer was dried over anhydrous magnesium sulfate, MgSO4. Thin-layer chromatography was performed on plates coated with silica gel (0.2 mm, 60/Kieselguhr F254) immersed in 10 mL of the dissolution system. Column chromatography was performed in glass columns of different diameters. The columns were filled with silica gel (60 Å, technical grade) of different heights. Abbreviations used in the experimental part of the work are the following: ACN – acetonitrile, DCM – dichloromethane, E – diethyl ether, EtOAc – ethyl acetate, PE – petroleum ether, NMR – nuclear magnetic resonance, UV – ultraviolet spectroscopy, NaOEt –sodium ethoxide, s – singlet, d – doublet, t – triplet, m – multiplet, dd – doublet of doublets, and q – quartet. UV/Vis spectra were recorded on a Varian Cary 100 Bio spectrometer and fluorescence spectra on a Varian Cary Eclipse fluorimeter in appropriate quartz cuvettes (path length = 1 cm).
3.2. Synthesis of Phosphonium Salts 1’ and 2’
In a 250 mL three-necked flask, 7.9 g (0.063 mol) of 2-thiophene methanol was dissolved in 72.3 mL of dry solvent E and added to the flask. Then, 5.71 g (0.022 mol) of phosphorus tribromide, PBr3 was dissolved in 7.23 mL of dry solvent E and added dropwise to the reaction flask over 40 minutes. The reaction mixture was stirred at room temperature on a magnetic stirrer for 1 hour. Subsequently, 15 mL of methanol and 100 mL of distilled water were added, and the mixture was extracted with a total of 300 mL of solvent E. The ether layer was dried overnight with anhydrous magnesium sulfate, MgSO4, filtered, and evaporated to dryness using a rotary evaporator, leaving 2-thiophene bromide as a yellow oil in the flask. This oil was then dissolved in 20 mL of toluene, while 16.52 g (0.037 mol) of triphenylphosphine, PPh3 was dissolved in 50 mL of toluene, and both solutions were added to the reaction flask. The mixture was stirred on a magnetic stirrer for three days and then filtered through a Büchner funnel under reduced pressure. The resulting light-yellow salt was dried in a desiccator for 12 hours. The dry thiophene-phosphonium salt 1’ obtained was used in all subsequent experiments for synthesizing compounds 6–8.

In a 250 mL round flask, 4.925 g (0.0440 mol) of dimethylthiophene was dissolved in 133 mL of carbon tetrachloride, CCl4. Then, 7.83 g (1 equivalent) of N-bromosuccinimide, NBS, was added to the mixture. The chemicals were stirred in an oil bath at a reflux temperature (150 ºC). Once reflux started, a small amount of α,α-azobisisobutyronitrile, AIBN catalyst was added. The oil bath temperature was then reduced to approximately 100 ºC due to excessive reflux. With reflux established, the reaction mixture was illuminated with a lamp to initiate the reaction. After 1.5 hours, bromide formation began, accompanied by the appearance of white, fluffy succinimide crystals on the surface. After another hour, a second dose of AIBN was added, and the mixture was refluxed for an additional 3 hours. The succinimide was filtered out using pleated filter paper, and the clear orange liquid was evaporated using a rotary evaporator, leaving a brown, oily precipitate of thiophene bromide in the flask. In the second step, the thiophene bromide was dissolved in 10 mL of benzene with added PPh3. The mixture was heated to reflux (80 ºC, with the mixer set to 110 ºC) and stirred at that temperature for 1 hour. The heating was then turned off, and the mixture was stirred at room temperature for the next 24 hours. The resulting salt was filtered using a Büchner funnel, and the crystallizer was weighed before and after adding the salt. The 2-methylthiophene salt 2’ was dried in a desiccator under vacuum for 4-5 hours and then used for synthesizing compounds 9–13.
3.3. Synthesis of Triazole Aldehydes 1–5
Triazole aldehydes 1–5 were synthesized in small glass vials from 200 mg of 1-(4-nitrophenyl)-1H-1,2,3-triazole-4-carbaldehyde [21] dissolved in 2 mL of dry 1,4-dioxane. The 1.2 eq of appropriate benzylamine was then added to the reaction mixture, and the mixture was briefly purged with argon. Depending on the applied benzylamine, the reaction took place for a certain period of time (24 – 48 h). The course of the reaction was monitored by thin-layer chromatography. At the end of the reaction, the solvent was removed by evaporation on a rotary vacuum evaporator. The solid product was purified by column chromatography using an appropriate solvent system, mostly DCM/EtOAc.

1-(4-methylbenzyl)-1H-1,2,3-triazole-4-carbaldehyde (1): 46 mg (isolated 23%), yellow oil; Rf (DCM) = 0.15; 1H NMR (CDCl3, 600 MHz) δ/ppm: 10.11 (s, 1H), 7.95 (s, 1H), 7.22 (d, J = 8.7 Hz, 2H), 7.20 (d, J = 8.7 Hz, 2H), 5.54 (s, 2H), 2.37 (s, 3H).
1-(4-chlorobenzyl)-1H-1,2,3-triazole-4-carbaldehyde (2): 131 mg (isolated 65%), yellow oil; Rf (DCM) = 0.40; 1H NMR (CDCl3, 300 MHz) δ/ppm: 10.13 (s, 1H), 8.01 (s, 1H), 7.39 (d, J = 7.4 Hz, 2H), 7.25 (d, J = 8.9 Hz, 2H), 5.57 (s, 2H).

1-(3-methylbenzyl)-1H-1,2,3-triazole-4-carbaldehyde (3): 130 mg (isolated 65%), yellow oil; Rf (DCM) = 0.22; 1H NMR (CDCl3, 600 MHz) δ/ppm: 10.12 (s, 1H), 7.98 (s, 1H), 7.29 (t, J = 7.3 Hz, 1H), 7.21 (d, J = 8.1 Hz, 1H), 7.10 (d, J = 6.5 Hz, 2H), 5.54 (s, 2H), 2.36 (s, 3H).
1-(3-methoxybenzyl)-1H-1,2,3-triazole-4-carbaldehyde (4): 121 mg (isolated 61%), yellow oil; Rf (DCM) = 0.30; 1H NMR (CDCl3, 300 MHz) δ/ppm: 10.13 (s, 1H), 7.98 (s, 1H), 7.32 (t, J = 7.8 Hz, 1H), 6.93 (dd, J = 8.7, 2.6 Hz, 1H), 6.88 (d, J = 7.8 Hz, 1H), 6.82 (s, 1H), 5.55 (s, 2H), 3.80 (s, 3H).

1-(3-fluorobenzyl)-1H-1,2,3-triazole-4-carbaldehyde (5): 163 mg (isolated 82%), yellow oil; Rf (DCM) = 0.42; 1H NMR (CDCl3, 600 MHz) δ/ppm: 10.14 (s, 1H), 8.03 (s, 1H), 7.41 – 7.37 (m, 1H), 7.11 (dd, J = 8.0, 2.4 Hz, 1H), 7.08 (d, J = 8.0 Hz, 1H), 7.00 (d, J = 8.8 Hz, 1H), 5.59 (s, 2H).
3.4. Synthesis of Triazolostilbenes 6–13 by Wittig Reaction
The apparatus, in a three-necked flask, a dropping funnel, a chlorine-calcium tube, and a balloon filled with nitrogen, was blown with argon for 5 min. A magnet was placed in the flask, the addition funnel was closed, and 30 mL of absolute ethanol was poured in (depending on the amount of starting reactants). A portion of absolute ethanol (10 mL) was poured into the flask, and the required amount of triphenylphosphonium salt was added. Sodium previously weighed in PE on an analytical balance with a precision of 0.0001 g was added to the remaining amount of absolute ethanol. After all the sodium had reacted in the ethanol with the evolution of hydrogen, a little portion of NaOEt was added to the flask. The aldehyde was dissolved in ethanol and transferred to a flask, then the rest of the NaOEt from the funnel was added dropwise. The flask was closed with a glass stopper, and the reaction mixture was left to stir in a magnetic stirrer at room temperature (6 – 108 h, 7 – 72 h, 8 – 48 h, 9 – 96 h, 10 – 96 h, 11 – 96 h, 12 – 168 h and 13 – 120 h). Compounds 6–13 were synthesized as mixtures of isomers and their pure isomers were not isolated due to the next reaction step.

1-(4-methylbenzyl)-4-(2-(thiophen-2-yl)vinyl)-1H-1,2,3-triazole (6): 37 mg (mixture of isomers, cis : trans = 1 : 1, 80%), white oil; Rf (PE/E = 80%) = 0.48.
1-(4-chlorobenzyl)-4-(2-(thiophen-2-yl)vinyl)-1H-1,2,3-triazole (7): 145 mg (mixture of isomers, cis : trans = 1 : 0.6, 90%), white oil; Rf (PE/E = 60%) = 0.25. MS (ESI) m/z (%, fragment): 302 (100); HRMS (m/z) for C15H12ClN3S: [M + H]+calcd = 301.0441, and [M + H]+measured = 301.0440.

1-(3-methylbenzyl)-4-(2-(thiophen-2-yl)vinyl)-1H-1,2,3-triazole (8): 108 mg (mixture of isomers, cis : trans = 1 : 0.8, 91%), white oil; Rf (PE/E = 80%) = 0.67.
1-(4-methylbenzyl)-4-(2-(5-methylthiophen-2-yl)vinyl)-1H-1,2,3-triazole (9): 13 mg (mixture of isomers, cis : trans = 1 : 0.9, 13%), white oil; Rf (PE/E = 50%) = 0.60. MS (ESI) m/z (%, fragment): 296 (100); HRMS (m/z) for C17H17N3S: [M + H]+calcd = 295.1143, and [M + H]+measured = 295.1147.

1-(4-chlorobenzyl)-4-(2-(5-methylthiophen-2-yl)vinyl)-1H-1,2,3-triazole (10): 50 mg (mixture of isomers, cis : trans = 1 : 0.8, 32%), white oil; Rf (PE/E = 50%) = 0.50. MS (ESI) m/z (%, fragment): 316 (100); HRMS (m/z) for C16H14ClN3S: [M + H]+calcd = 315.0597, and [M + H]+measured = 315.0599.
1-(3-methylbenzyl)-4-(2-(5-methylthiophen-2-yl)vinyl)-1H-1,2,3-triazole (11): 12 mg (mixture of isomers, cis : trans = 1 : 0.7, 10%), white oil; Rf (PE/E = 90%) = 0.58. MS (ESI) m/z (%, fragment): 296 (100); HRMS (m/z) for C17H17N3S: [M + H]+calcd = 295.1143, and [M + H]+measured = 295.1139.

1-(3-methoxybenzyl)-4-(2-(5-methylthiophen-2-yl)vinyl)-1H-1,2,3-triazole (12): 35 mg (mixture of isomers, cis : trans = 1 : 0.7, 24%), white oil; Rf (PE/E = 50 %) = 0.50. MS (ESI) m/z (%, fragment): 312 (100); HRMS (m/z) for C17H17N3OS: [M + H]+calcd = 311.1092, and [M + H]+measured = 311.1098.
1-(3-fluorobenzyl)-4-(2-(5-methylthiophen-2-yl)vinyl)-1H-1,2,3-triazole (13): 16 mg (mixture of isomers, cis : trans = 1 : 0.7, 23%), white oil; Rf (PE/E = 50%) = 0.54.
3.5. Synthesis of Photoproducts 14–21
The corresponding 1,2,3-triazolostilbenes 6–13, as mixtures of isomers, were dissolved in 50 mL of toluene. These mixtures were then transferred to quartz tubes, which allowed light to pass through. A small amount of iodine was added as an oxidizing agent. The solutions, with a concentration of approximately 10−3 mol L−1, were exposed to light for 1–4 hours using ten lamps emitting at a wavelength of about 313 nm in a Rayonet photoreactor. The resulting products, 14–21, were purified through column chromatography. Spectroscopic data for photoproducts 14–21 are provided below.

1-(4-methylbenzyl)-1H-thieno [3’,2’:3,4]benzo[1,2-d][1,2,3]triazole (14): 27 mg (isolated 73%), yellow oil; Rf (PE/E (40 %)) = 0.34; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.99 (d, J = 9.0 Hz, 1H), 7.80 (d, J = 9.0 Hz, 1H), 7.55 (d, J = 6.0 Hz, 1H), 7.51 (d, J = 5.6 Hz, 1H), 7.12 (d, J = 8.2 Hz, 2H), 7.09 (d, J = 8.2 Hz, 2H), 6.08 (s, 2H), 2.29 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 144.7, 140.0, 138.1, 132.1, 129.8, 128.7, 127.7, 126.5, 122.7, 120.3, 119.2, 116.1, 52.9, 21.1; MS (ESI) m/z (%, fragment): 280 (100); HRMS (m/z) for C16H13N3S: [M + H]+calcd = 279.0830, and [M + H]+measured = 279.0832.
1-(4-chlorobenzyl)-1H-thieno[3’,2’:3,4]benzo[1,2-d][1,2,3]triazole (15): 36 mg (isolated 25%), yellow oil; Rf (PE/E (80%)) = 0.25; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.00 (d, J = 8.6 Hz, 1H), 7.83 (d, J = 8.1 Hz, 1H), 7.58 (d, J = 5.4 Hz, 1H), 7.46 (d, J = 5.4 Hz, 1H), 7.29 (d, J = 8.6 Hz, 2H), 7.13 (d, J = 9.2 Hz, 2H), 6.09 (s, 2H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 144.7, 140.2, 134.3, 133.6, 129.4, 128.6, 128.0, 127.9, 122.5, 119.9, 119.4, 116.1, 52.4; MS (ESI) m/z (%, fragment): 300 (100); HRMS (m/z) for C15H10ClN3S: [M + H]+calcd = 299.0284, and [M + H]+measured = 299.0283.
1-(3-methylbenzyl)-1H-thieno[3’,2’:3,4]benzo[1,2-d][1,2,3]triazole (16): 26 mg (isolated 24%), yellow oil; Rf (PE/E (40%)) = 0.35; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.00 (d, J = 8.9 Hz, 1H), 7.81 (d, J = 9.1 Hz, 1H), 7.55 (d, J = 5.9 Hz, 1H), 7.51 (d, J = 5.7 Hz, 1H), 7.20 (t, J = 8.0 Hz, 1H), 7.09 (d, J = 8.0 Hz, 1H), 6.99 (d, J = 8.4 Hz, 2H), 6.09 (s, 2H), 2.27 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 144.7, 140.0, 139.0, 135.1, 129.1, 128.9, 127.7, 127.2, 123.6, 122.8, 120.3, 119.2, 116.1, 53.1, 21.4; MS (ESI) m/z (%, fragment): 280 (100); HRMS (m/z) for C16H13N3S: [M + H]+calcd = 279.0830, and [M + H]+measured = 279.0828.

7-methyl-1-(4-methylbenzyl)-1H-thieno[3’,2’:3,4]benzo[1,2-d][1,2,3]triazole (17): 12 mg (isolated 15%), yellow oil; Rf (PE/E (30%)) = 0.46; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.91 (d, J = 8.6 Hz, 1H), 7.68 (d, J = 8.7 Hz, 1H), 7.18 (s, 1H), 7.12 (d, J = 8.3 Hz, 2H), 7.06 (d, J = 8.3 Hz, 2H), 6.04 (s, 2H), 2.60 (s, 3H), 2.30 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 142.6, 138.0, 132.3, 129.7, 126.6, 123.0, 118.9, 118.1, 115.0, 52.8, 21.1, 16.1; MS (ESI) m/z (%, fragment): 294 (100); HRMS (m/z) for C17H15N3S: [M + H]+calcd = 293.0987, and [M + H]+measured = 293.0988.
1-(4-chlorobenzyl)-7-methyl-1H-thieno[3’,2’:3,4]benzo[1,2-d][1,2,3]triazole (18): 14 mg (isolated 28%), colourless oil; Rf (PE/E = 50%) = 0.51; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.91 (d, J = 9.0 Hz, 1H), 7.70 (d, J = 9.0 Hz, 1H), 7.30 (d, J = 8.4 Hz, 2H), 7.15 – 7.09 (m, 3H), 6.05 (s, 2H), 2.61 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 144.7, 143.0, 139.5, 134.3, 133.9, 129.3, 128.0, 122.8, 119.1, 117.7, 115.1, 52.3, 16.2; MS (ESI) m/z (%, fragment): 314 (100); HRMS (m/z) for C16H12ClN3S: [M + H]+calcd = 313.0440, and [M + H]+measured = 313.0441.
7-methyl-1-(3-methylbenzyl)-1H-thieno[3’,2’:3,4]benzo[1,2-d][1,2,3]triazole (19): 13 mg (isolated 29%), yellow oil; Rf (PE/E (40%)) = 0.46; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.92 (d, J = 8.9 Hz, 1H), 7.65 (d, J = 8.8 Hz, 1H), 7.23 – 7.18 (m, 2H), 7.09 (d, J = 7.5 Hz, 1H), 7.03 – 6.96 (m, 2H), 6.04 (s, 2H), 2.60 (s, 3H), 2.28 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 142.6, 139.3, 138.9, 135.3, 129.1, 128.9, 127.3, 123.6, 123.0, 118.9, 118.1, 115.1, 52.9, 21.4, 16.2; MS (ESI) m/z (%, fragment): 294 (100); HRMS (m/z) for C17H15N3S: [M + H]+calcd = 293.0987, and [M + H]+measured = 293.0981.
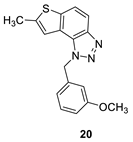
1-(3-methoxybenzyl)-7-methyl-1H-thieno[3’,2’:3,4]benzo[1,2-d][1,2,3]triazole (20): 14 mg (isolated 35%), yellow oil; Rf (PE/E (40%)) = 0.38; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.91 (d, J = 8.9 Hz, 1H), 7.68 (d, J = 8.9 Hz, 1H), 7.22 (d, J = 7.9 Hz, 1H), 7.17 (s, 1H), 6.85 – 6.75 (m, 2H), 6.70 (s, 1H), 6.05 (s, 2H), 3.71 (s, 3H), 2.60 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 160.1, 144.7, 142.6, 139.3, 136.9, 130.2, 128.3, 123.0, 118.9, 118.8, 118.1, 115.0, 113.6, 112.3, 55.2, 52.8, 16.2; MS (ESI) m/z (%, fragment): 310 (100); HRMS (m/z) for C17H15N3OS: [M + H]+calcd = 309.0936, and [M + H]+measured = 309.0939.
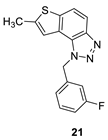
1-(3-fluorobenzyl)-7-methyl-1H-thieno[3’,2’:3,4]benzo[1,2-d][1,2,3]triazole (21): 8 mg (isolated 16%), colourless oil; Rf (PE/E (40%)) = 0.44; 1H NMR (CDCl3, 300 MHz) δ/ppm: 7.85 (d, J = 8.9 Hz, 1H), 7.63 (d, J = 8.9 Hz, 1H), 7.27 – 7.19 (m, 1H), 7.05 (s, 1H), 6.96 – 6.87 (m, 2H), 6.80 (d, J = 9.5 Hz, 1H), 6.01 (s, 2H), 2.51 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 161.9 (d, JCF = 247.8 Hz), 143.7, 141.9, 138.5, 136.8 (d, JCF = 7.4 Hz), 129.8 (d, JCF = 8.5 Hz), 127.1, 121.8, 121.1 (d, JCF = 3.1 Hz), 118.1, 116.7, 114.3 (d, JCF = 20.9 Hz), 114.1, 112.7 (d, JCF = 22.4 Hz), 51.4, 15.3; MS (ESI) m/z (%, fragment): 298 (100); HRMS (m/z) for C16H12FN3S: [M + H]+calcd = 297.0736, and [M + H]+measured = 297.0734.
3.6. Synthesis of Triazolium Salts 22–31
In a glass reaction tube with a magnetic stirring bar, the corresponding thienobenzo-triazoles 14–21 (0.2 mmol, 1 equivalent), dry DCM (0.4 mL), and iodomethane (2 mmol, 10 equivalents) were added. The reaction mixtures were purged with argon and then stirred at 60 °C for 24 hours. After this period, the mixtures were cooled to 0 °C and diluted with 5 mL of solvent E. Following precipitation, the tubes were centrifuged three times for 10 minutes at 3000 rpm. The solvent was decanted, and the precipitate was washed three to five times with solvent E. The crude powders were then dried under high vacuum, yielding the pure triazolium salts 22–29. The solubility of triazolium salts 22–29 is low in CDCl3, which is reflected in the signals, especially in the 13C NMR spectra.

3-methyl-1-(4-methylbenzyl)-1H-thieno[3’,2’:3,4]benzo[1,2-d][1,2,3]triazol-3-ium (22): 8 mg (isolated 30%), yellow powder; Rf (DCM (100%)) = 0.19; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.99 (d, J = 9.1 Hz, 1H), 7.80 (dd, J = 9.1, 0.7 Hz, 1H), 7.55 (d, J = 5.5 Hz, 1H), 7.51 (dd, J = 5.5, 0.7 Hz, 1H), 7.10 (q, J = 17.3, 8.7 Hz, 4H), 2.29 (s, 3H), 1.59 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 143.1, 139.3, 135.1, 132.8, 130.9, 130.4, 127.8, 127.5, 127.2, 126.5, 120.6, 109.0, 57.2, 40.2, 21.2; MS (ESI) m/z (%, fragment): 294 (100); HRMS (m/z) for C17H16N3S+: [M + H]+calcd = 293.0987, and [M + H]+measured = 293.0985.
1-(4-chlorobenzyl)-3-methyl-1H-thieno[3’,2’:3,4]benzo[1,2-d][1,2,3]triazol-3-ium (23): 15 mg (isolated 42%), yellow powder; Rf (DCM (100%)) = 0.15; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.34 (d, J = 8.6 Hz, 1H), 8.10 (d, J = 8.8 Hz, 1H), 7.98 (d, J = 4.9 Hz, 1H), 7.85 (d, J = 5.7 Hz, 1H), 7.43 – 7.40 (m, 4H), 6.45 (s, 2H), 4.86 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 150.9, 130.9, 130.1, 129.5, 129.4, 128.9, 122.6, 121.3, 57.3, 34.6 (signals for 4 quaternary carbons are missing); MS (ESI) m/z (%, fragment): 314 (100); HRMS (m/z) for C16H13ClN3S+: [M + H]+calcd = 313.0440, and [M + H]+measured = 313.0436.
3-methyl-1-(3-methylbenzyl)-1H-thieno[3’,2’:3,4]benzo[1,2-d][1,2,3]triazol-3-ium (24): 4 mg (isolated 15%), yellow powder; Rf (DCM (100%)) = 0.20; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.34 (d, J = 11.1 Hz, 1H), 8.25 (d, J = 11.1 Hz, 1H), 7.94 (d, J = 6.3 Hz, 1H), 7.78 (d, J = 6.3 Hz, 1H), 7.31 (t, J = 7.4 Hz, 1H), 7.22 (d, J = 7.4 Hz, 1H), 7.18 – 7.16 (m, 2H), 6.34 (s, 2H), 4.92 (s, 3H), 2.35 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 142.7, 132.8, 130.7, 127.6, 127.1, 124.5, 122.7, 120.5, 116.0, 53.4, 30.9, 21.5 (signals for 5 quaternary carbons are missing); MS (ESI) m/z (%, fragment): 294 (100); HRMS (m/z) for C17H16N3S+: [M + H]+calcd = 293.0987, and [M + H]+measured = 293.0991.

3,7-dimethyl-1-(4-methylbenzyl)-1H-thieno[3’,2’:3,4]benzo[1,2-d][1,2,3]triazol-3-ium (25): 6 mg (isolated 27%), white powder; Rf (DCM (100%)) = 0.22; 1H NMR (CDCl3, 300 MHz) δ/ppm: 8.21 (d, J = 6.6 Hz, 1H), 8.07 – 8.04 (m, 1H), 7.52 (s, 1H), 7.30 – 7.28 (m, 1H), 7.28 – 7.27 (m, 1H), 7.24 – 7.23 (m, 1H), 7.23 – 7.21 (m, 1H), 6.31 (s, 2H), 4.84 (s, 3H), 2.74 (s, 3H), 2.36 (s, 3H); MS (ESI) m/z (%, fragment): 308 (100); HRMS (m/z) for C18H18N3S+: [M + H]+calcd = 307.1216, and [M + H]+measured = 307.1218.
1-(4-chlorobenzyl)-3,7-dimethyl-1H-thieno[3’,2’:3,4]benzo[1,2-d][1,2,3]triazol-3-ium (26): 5 mg, (isolated 22%), white powder; Ryf (DCM (100%)) = 0.21; 1H NMR (CDCl3, 300 MHz) δ/ppm: 8.20 (d, J = 6.2 Hz, 1H), 7.98 – 7.90 (m, 1H), 7.58 (s, 1H), 7.44 – 7.38 (m, 4H), 6.41 (s, 2H), 4.79 (s, 3H), 2.75 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 156.2, 149.1, 136.0, 129.9, 129.4, 127.3, 126.9, 123.4, 118.4, 65.9, 16.6, 15.3 (signals for 3 quaternary carbons are missing); MS (ESI) m/z (%, fragment): 328 (100); HRMS (m/z) for C17H15ClN3S+: [M + H]+calcd = 327.0122, and [M + H]+measured = 327.0120.
3,7-dimethyl-1-(3-methylbenzyl)-1H-thieno[3’,2’:3,4]benzo[1,2-d][1,2,3]triazol-3-ium (27): 2 mg (isolated 15%), white powder; Rf (DCM (100%)) = 0.24; 1H NMR (CDCl3, 300 MHz) δ/ppm: 8.21 (d, J = 9.3 Hz), 8.11 – 8.09 (m, 1H), 7.49 (s, 1H), 7.31 (t, J = 7.6 Hz, 1H), 7.23 (d, J = 7.1 Hz, 1H), 7.19 – 7.16 (m, 2H), 6.31 (s, 2H), 4.86 (s, 3H), 2.73 (s, 3H), 2.36 (s, 3H); MS (ESI) m/z (%, fragment): MS (ESI) m/z (%, fragment): 308 (100); HRMS (m/z) for C18H18N3S+: [M + H]+calcd = 307.1216, and [M + H]+measured = 307.1218.
1-(3-methoxybenzyl)-3,7-dimethyl-1H-thieno[3’,2’:3,4]benzo[1,2-d][1,2,3]triazol-3-ium (28): 4 mg, (isolated 26%), yellow powder; Rf (DCM (100%)) = 0.18; 1H NMR (CDCl3, 300 MHz) δ/ppm: 8.19 (br s, 1H), 8.00 (br s, 1H), 7.53 (br s, 1H), 7.30 (d, J = 8.4 Hz, 1H), 7.13 – 7.10 (m, 1H), 6.95 – 6.91 (m, 2H), 6.35 (br s, 2H), 4.81 (br s, 3H), 3.81 (s, 3H), 2.73 (s, 3H); MS (ESI) m/z (%, fragment): 324 (100).
1-(3-fluorobenzyl)-3,7-dimethyl-1H-thieno[3’,2’:3,4]benzo[1,2-d][1,2,3]triazol-3-ium (29): obtained only in traces according to NMR analysis, not isolated and evaluated. Its synthesis should be optimized in the following investigations.
Two additional triazolium salts, 30 and 31, were also prepared from the previously obtained and characterized neutral thienobenzo-triazole [20] for comparison of the non-charged and charged analogues.

1-(3-chlorobenzyl)-3-methyl-1H-thieno[3’,2’:3,4]benzo[1,2-d][1,2,3]triazol-3-ium (30): 10 mg (isolated 43%), yellow powder; Rf (DCM (100%)) = 0.20; 1H NMR (CDCl3, 300 MHz) δ/ppm: 8.39 – 8.31 (m, 2H), 8.13 – 8.07 (m, 2H), 8.02 – 7.96 (m, 2H), 7.86 – 7.82 (m, 2H), 6.47 (s, 2H), 4.87 (s, 3H); MS (ESI) m/z (%, fragment): 314 (100); HRMS (m/z) for C16H13ClN3S+: [M + H]+calcd = 313.0440, and [M + H]+measured = 313.0436.
1-(furan-2-ylmethyl)-3-methyl-1H-thieno[3’,2’:3,4]benzo[1,2-d][1,2,3]triazol-3-ium (31): 6 mg (isolated 29%), yellow powder; Rf (DCM (100%)) = 0.17; 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.34 (d, J = 9.0 Hz, 1H), 8.11 (d, J = 9.0 Hz, 1H), 7.99 (d, J = 5.8 Hz, 1H), 7.86 (d, J = 5.8 Hz, 1H), 7.50 – 7.47 (m, 2H), 7.13 (d, J = 3.8 Hz, 2H), 6.43 (s, 2H), 4.82 (s, 3H); MS (ESI) m/z (%, fragment): 270 (100); HRMS (m/z) for C14H12N3OS+: [M + H]+calcd = 269.0623, and [M + H]+measured = 269.0627.
3.7. Inhibition Activity
The modified Ellman’s method was used to determine the synthesized triazole compounds’ cholinesterase inhibitory potential [22]. Acetylcholinesterase from electric eel (eeAChE, type VI-S), butyrylcholinesterase from equine serum (eqBChE, E.C. 3.1.1.8), acetylthiocholine iodide (ATChI), S-butyrylthiocholine iodide (BTChI), Trisma base and reference standard Galantamine were purchased from Sigma Aldrich (ST. Louis, MO). Ellman’s reagent 5,50-dithiobis-(2-nitrobenzoic acid) (DTNB) was purchased from Zwijndrecht (Belgium). All tested samples were dissolved in ethanol in concentrations range from 0.1 to 500 μM (final concentration in reaction mixture). Reaction mixtures were prepared as follows: 10 μL of tested solution of different concentrations, 10 μL of enzyme (AChE and BChE final concentrations 0.03 U/mL, prepared in 20 mM Tris buffer, pH 7.5), and 10 μL of DTNB (final concentration 0.3 mM, prepared in 50 mM Tris buffer, pH 8) were added to 180 μL Tris buffer (50 mM, pH 8.0). The reaction started by adding 10 μL of substrate ATChI/BTChI (final concentration 0.5 mM, prepared in Tris buffer, pH 8.0). The tested sample was replaced by 10 μL of buffer solution in the control measurement. A blank test for each sample contained the same solutions only without enzymes; its volume was replaced by adding 10 μL of buffer. The absorbance of the reaction mixture was measured at 405 nm over 6 min at room temperature using a 96-well microplate reader (Bio Tek 800TSUV Absorbance Reader, Agilent). Experiments were run in triplicate. The percentage of enzyme inhibition was calculated according to the equation: Inhibition (%) = [(AC – AT) / AC] × 100 where AC is the control enzyme activity, without the test sample, and AT is the enzyme activity with the test sample, and represented as mean value ± standard deviation. Data of mean inhibition values were used for IC50 value calculation by nonlinear curve fit of compound concentrations (log) vs. response. Ethanol contribution to inhibition was measured and subtracted from calculations.
3.8. Computational Study
The geometries of examined ligands were optimized using the B3LYP/6-31G(d) theoretical model with the Gaussian16 program package [28]. The crystallographic data of AChE (PDB ID: 4EY7) and BChE (PDB ID: 7AIY) are taken from the Protein Data Bank [29,30]. The molecular docking was performed employing the Lamarckian Genetic Algorithm, yielding a set of 25 genetic algorithm dockings, i.e., 25 binding poses for each of the examined ligands, with residues of the enzymes kept rigid during the docking process.
4. Conclusions
In this research, pairs of uncharged thienobenzo-triazoles and their charged salts were prepared in order to examine further the role of the positive charge on the nitrogen of the triazole ring on the interactions in the active site of the enzymes and to compare the selectivity of 1,2,3-triazolium salts in relation to their uncharged analogs obtained by photochemical cyclization. Uncharged thienobenzo-triazoles 14–21 prefer inhibition of BChE over AChE. This is consistent with our previous findings that neutral photoproducts are generally weaker inhibitors than their charged salts and show selectivity toward BChE. For herein tested thienobenzo-triazoles, conversion to salt form (compounds 22–31) significantly increased inhibition of AChE (the most active 23: IC50 4.4 μM), and to a good extent also of BChE (the most active 23: IC50 0.47 μM). It can be concluded that the most important structural feature for thienobenzo-triazoles is the presence/absence of charge. Triazolium salts 22–31 were shown to be dual inhibitors whose potency of inhibition varies depending on the substituent on thiophene and triazole heterocycles. These compounds present a novel scaffold for drug development, as current therapies for Alzheimer’s disease (AD) primarily use selective AChE inhibitors, while non-selective inhibitors of cholinesterases (ChEs) are favored in Parkinson’s and all stages of AD. Molecular docking of the two most promising salts and their neutral counterparts revealed that, in addition to π-π stacking and hydrophobic interactions, positively charged ligands can form salt bridges with anionic residues in the active sites of ChEs, strongly stabilizing the non-covalent complexes. Since genotoxicity studies are critical in the development of new active substances and final drug forms, in silico assessments of all synthesized compounds were conducted. These studies indicated that compound 18 is the most promising candidate concerning genotoxic safety.
Supplementary Materials
The following supporting information can be downloaded at website of this paper posted on Preprints.org, Figure S1-S80: 1H and 13C NMR spectra of compounds 1–31, Figures S81-S101: Mass spectra and HRMS analyses of compounds 1–31; Cartesian coordinates of docked ligands; Tables S1 and S2, free energies of binding obtained by docking.
Author Contributions
Conceptualization, I.Š.; methodology, Ana R., and I.S.; formal analysis, Z.L. and A.R.; investigation, M.M., M.R., I.O., D.B.; resources, I.Š., D.B., I.O. and I.S.; writing—original draft preparation, I.Š., D.B., I.O. and I.S.; writing—review and editing, all; supervision, I.Š. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the University of Zagreb for short-term scientific support for 2023 under the title Novel styryl-heterocyclic systems: synthesis, biological activity and computational studies. We thank the University of Zagreb (Croatia) Computing Centre (SRCE) for granting computational time on the Supercomputer Supek.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to privacy.
Acknowledgments
The authors acknowledge the NMR Centre at RBI for recording the NMR spectra. We thank the University of Zagreb (Croatia) Computing Centre (SRCE) for granting computational time on the Supercomputer Supek.
Conflicts of Interest
Author Ana Ratković was employed by the company Selvita Ltd. and Zlata Lasić by the company Pliva Hrvatska d.o.o. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Song, J.; Lv, J.; Jin, J.; Jin, Z.; Li, T.; Wu, J. Research Advances on the Bioactivity of 1,2,3-Triazolium Salts. Int. J. Mol. Sci. 2023, 24, 10694. [Google Scholar] [CrossRef] [PubMed]
- Tejero, R.; López, D.; López-Fabal, F.; Gómez-Garcés, J.L.; Fernández-García, M. Antimicrobial polymethacrylates based on quaternized 1,3-thiazole and 1,2,3-triazole side-chain groups. Polym. Chem. 2015, 6, 3449–3459. [Google Scholar] [CrossRef]
- Burujeny, S.B.; Yeganeh, H.; Atai, M.; Gholami, H.; Sorayya, M. Bactericidal dental nanocomposites containing 1,2,3-triazoliumfunctionalized POSS additive prepared through thiol-ene click polymerization. Dent. Mater. 2017, 33, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.T.; Sobczyk, J.M.; Gwazdacz, S.C.; Blanck, A.J. Antimicrobial 1,3,4-trisubstituted-1,2,3-triazolium salts. Bioorg. Med. Chem. Lett. 2018, 28, 3320–3323. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Qiu, L.; Tan, W.; Gu, G.; Guo, Z. Novel 1,2,3-triazolium-functionalized inulin derivatives: Synthesis, free radical-scavenging activity, and antifungal activity. RSC Adv. 2017, 7, 42225–42232. [Google Scholar] [CrossRef]
- Steiner, I.; Stojanovic, N.; Bolje, A.; Brozovic, A.; Polancec, D.; Ambriovic-Ristov, A.; Stojkovic, M.R.; Piantanida, I.; Eljuga, D.; Kosmrlj, J.; et al. Discovery of ‘click’ 1,2,3-triazolium salts as potential anticancer drugs. Radiol. Oncol. 2016, 50, 280–288. [Google Scholar] [CrossRef]
- Almeida, A.C.; Meinel, R.S.; Leal, Y.L.; Silva, T.P.; Glanzmann, N.; Mendonca, D.V.C.; Perin, L.; Cunha-Júnior, E.F.; Coelho, E.A.F.; Melo, R.C.N.; et al. Functionalized 1,2,3-triazolium salts as potential agents against visceral leishmaniasis. Parasitol. Res. 2022, 121, 1389–1406. [Google Scholar] [CrossRef]
- Yacob, Z.; Liebscher, J. 1,2,3-Triazolium salts as a versatile new class of ionic liquids. In Ionic Liquids; Handy, S.T., Ed.; IntechOpen: Rijeka, Germany, 2011; pp. 3–23. [Google Scholar]
- Yacob, Z.; Liebscher, J. Chemistry of 1,2,3-triazolium salts. In Chemistry of 1,2,3-Triazoles; Dehaen, W., Bakulev, V.A., Eds.; Springer International Publishing: Cham, Germany, 2015; pp. 167–210. [Google Scholar]
- Mohammadi-Khanaposhtani, M.; Saeedi, M.; Zafarghandi, N.S.; Mahdavi, M.; Sabourian, R.; Razkenari, E.K.; Alinezhad, H.; Khanavi, M.; Foroumadi, A.; Shafiee, A.; Akbarzadeh, T. Potent acetylcholinesterase inhibitors: design, synthesis, biological evaluation, and docking study of acridone linked to 1,2,3-triazole derivatives. Eur. J. Med. Chem. 2015, 92, 799–806. [Google Scholar] [CrossRef]
- Mohammadi-Khanaposhtani, M.; Mahdavi, M.; Saeedi, M.; Sabourian, R.; Safavi, M.; Khanavi, M.; Foroumadi, A.; Shafiee, A.; Akbarzadeh, T. Design, synthesis, biological evaluation, and docking study of acetylcholinesterase inhibitors: New acridone-1,2,4-oxadiazole-1,2,3-triazole hybrids. Chem. Biol. Drug Des. 2015, 86, 1425–1432. [Google Scholar] [CrossRef]
- Najafi, Z.; Mahdavi, M.; Saeedi, M.; Karimpour-Razkenari, E.; Asatouri, R.; Vafadarnejad, F.; Moghadam, F.H.; Khanavi, M.; Sharifzadeh, M.; Akbarzadeh, T. Novel tacrine-1,2,3-triazole hybrids: In vitro, in vivo biological evaluation and docking study of cholinesterase inhibitors. Eur. J. Med. Chem. 2017, 125, 1200–1212. [Google Scholar] [CrossRef]
- Sugimoto, H.; Yamanish, Y.; Iimura, Y.; Kawakami, Y. Donepezil hydrochloride (E2020) and other acetylcholinesterase inhibitors. Curr. Med. Chem. 2000, 7, 303–339. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.T.; Anh, D.T.; Dung, D.T.M.; Huong, L.T.T.; Park, E.J.; Jeon, H.W.; Kang, J.S.; Thuan, N.T.; Han, S.B.; Nam, N.H. Design, synthesis, and bioevaluation of novel oxoindolin-2-one derivatives incorporating 1-benzyl-1H-1,2,3-triazole. Med. Chem. Res. 2020, 29, 396–408. [Google Scholar] [CrossRef]
- Saeedi, M.; Maleki, A.; Iraji, A.; Hariri, R.; Akbarzadeh, T.; Edraki, N.; Firuzi, O.; Mirfazli, S.S. Synthesis and bio-evaluation of new multifunctional methylindolinone-1,2,3-triazole hybrids as anti-Alzheimer’s agents. J. Mol. Struct 2021, 1229, 129828. [Google Scholar] [CrossRef]
- Mlakić, M.; Faraho, I.; Odak, I.; Kovačević, B.; Raspudić, A.; Šagud, I.; Bosnar, M.; Škorić, I.; Barić, D. Cholinesterase inhibitory and anti-inflammatory activity of the naphtho- and thienobenzo-triazole photoproducts: Experimental and computational study. Int. J. Mol. Sci. 2023, 24, 14676. [Google Scholar] [CrossRef] [PubMed]
- Mlakić, M.; Odak, I.; Faraho, I.; Talić, S.; Bosnar, M.; Lasić, K.; Barić, D.; Škorić, I. New naphtho/thienobenzo-triazoles with interconnected anti-inflammatory and cholinesterase inhibitory activity. Eur. J. Med. Chem. 2022, 241, 114616. [Google Scholar] [CrossRef] [PubMed]
- Mlakić, M.; Selec, I.; Ćaleta, I.; Odak, I.; Barić, D.; Ratković, A.; Molčanov, K.; Škorić, I. New thienobenzo/naphtho-triazoles as butyrylcholinesterase inhibitors: Design, synthesis and computational study. Int. J. Mol. Sci. 2023, 24, 5879. [Google Scholar] [CrossRef]
- Mlakić, Milena; Faraho, Ivan; Odak, Ilijana; Talić, Stanislava; Vukovinski, Ana; Raspudić, Anamarija; Bosnar, Martina; Zadravec, Rahela; Ratković, Ana; Lasić, Kornelija; Marinić, Željko; Barić, Danijela; Škorić, Irena. Synthesis, photochemistry and computational study of novel 1,2,3-triazole heterostilbenes: expressed biological activity of their electrocyclization photoproducts. Bioorg. Chem. 2022, 121, 105701. [Google Scholar] [CrossRef]
- Mlakić, M.; Barić, D.; Ratković, A.; Šagud, I.; Čipor, I.; Piantanida, I.; Odak, I.; Škorić, I. New Charged Cholinesterase Inhibitors: Design, Synthesis, and Characterization. Molecules 2024, 29, 1622. [Google Scholar] [CrossRef]
- Ratković, A.; Mlakić, M.; Dehaen, W.; Opsomer, T.; Barić, D.; Škorić, I. Synthesis and photochemistry of novel 1,2,3-triazole di-heterostilbenes. An experimental and computational study. Spectr. Acta A Mol. Biomol. Spectr. 2021, 261, 120056. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtnex, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Vrbanac, J.; Slauter, R. ADME in Drug Discovery, In A Comprehensive Guide to Toxicology in Nonclinical Drug Development (2nd Edition); Faqi, A.S., Ed.; Academic Press, Cambridge, USA, 2017; pp. 39-67.
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: predicting small-molecule pharmacokinetic properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- https://ai-druglab.smu.edu/admetresult.
- Imai, Y.N.; Inoue, Y.; Nakanishi, I.; Kitaura, K. Cl–π interactions in protein-ligand complexes, Protein Sci. 2008, 27, 1129-1137. [CrossRef]
- Hasselgren, C.; Bercu, J.; Cayley, A.; Cross, K.; Glowienke, S.; Kruhlak, N.; Muster, W.; Nicolette, J.; Reddy, M.V.; Saiakhov, R.; Dobo, K. Management of pharmaceutical ICH M7 (Q)SAR predictions - The impact of model updates. Regul. Toxicol. Pharmacol. 2020, 118, 104807. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision, C01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Cheung, J., Rudolph, M., Burshteyn, F., Cassidy, M., Gary, E., Love, J., Height, J., Franklin, M. Crystal Structure of Recombinant Human Acetylcholinesterase in Complex with Donepezil, PDB. [CrossRef]
- Coquelle, N., Colletier, J.P. Crystal structure of human butyrylcholinesterase in complex with 2-{1-[4-(12-Amino-3-chloro-6,7,10,11-tetrahydro-7,11-methanocycloocta[b]quinolin-9-yl)butyl]-1H-1,2,3-triazol-4-yl}-N-[4-hydroxy-3-methoxybenzyl]acetamide, PDB. [CrossRef]
Figure 1.
Structures of previously evaluated thienobenzo-triazoles A and triazolium salts B, and new triazolium salts C with the substituted benzyl group on the charged 1,2,3-triazole ring in this study.
Figure 1.
Structures of previously evaluated thienobenzo-triazoles A and triazolium salts B, and new triazolium salts C with the substituted benzyl group on the charged 1,2,3-triazole ring in this study.
Scheme 1.
Reaction pathway for the synthesis of thienobenzo-triazoles 14–21 and their 1,2,3-triazolium salts 22–31.
Scheme 1.
Reaction pathway for the synthesis of thienobenzo-triazoles 14–21 and their 1,2,3-triazolium salts 22–31.
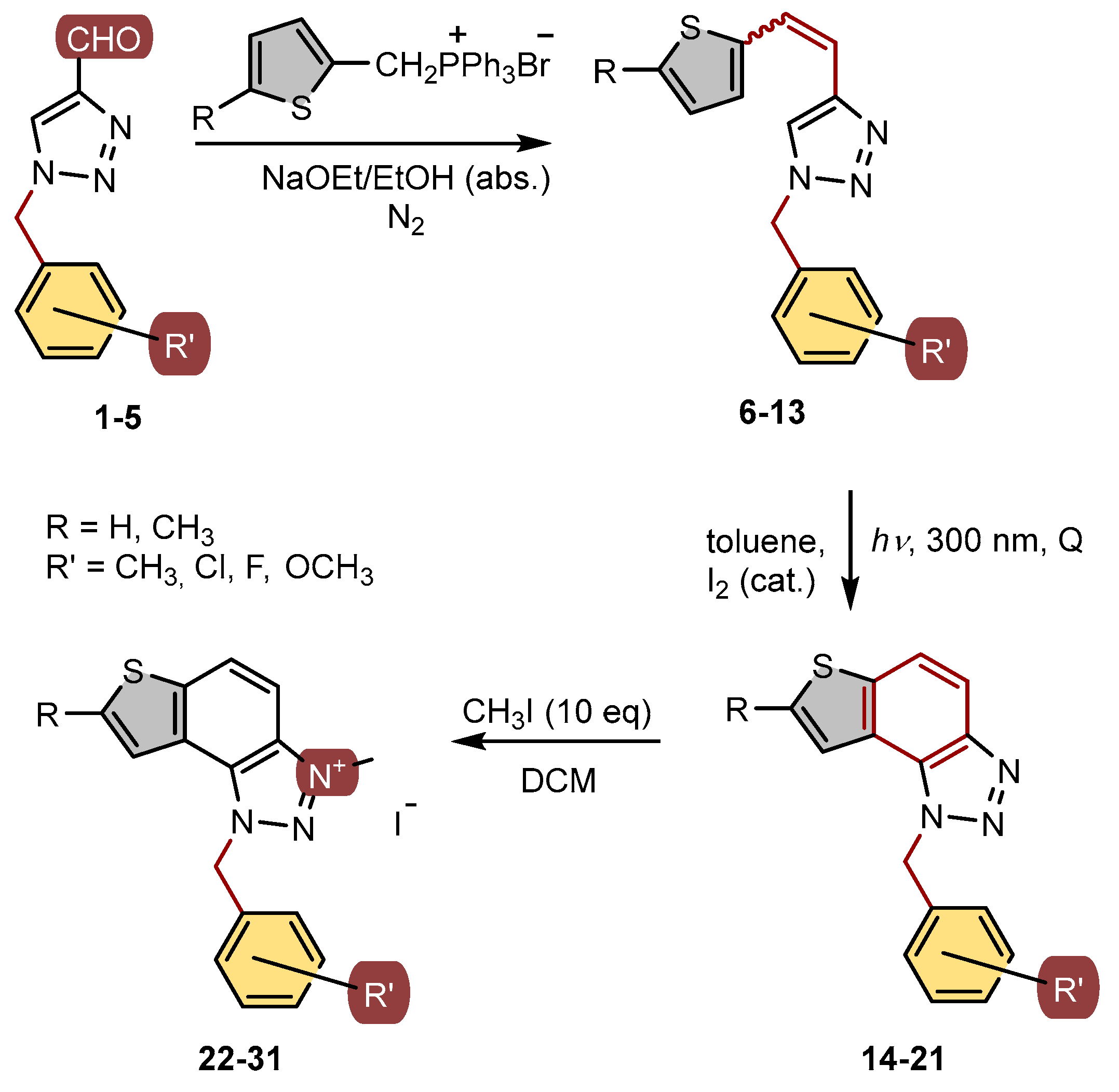
Figure 2.
1H NMR spectra (CDCl3) of the thienobenzo-triazole 16 (a) and its triazolium salt 24 (b).
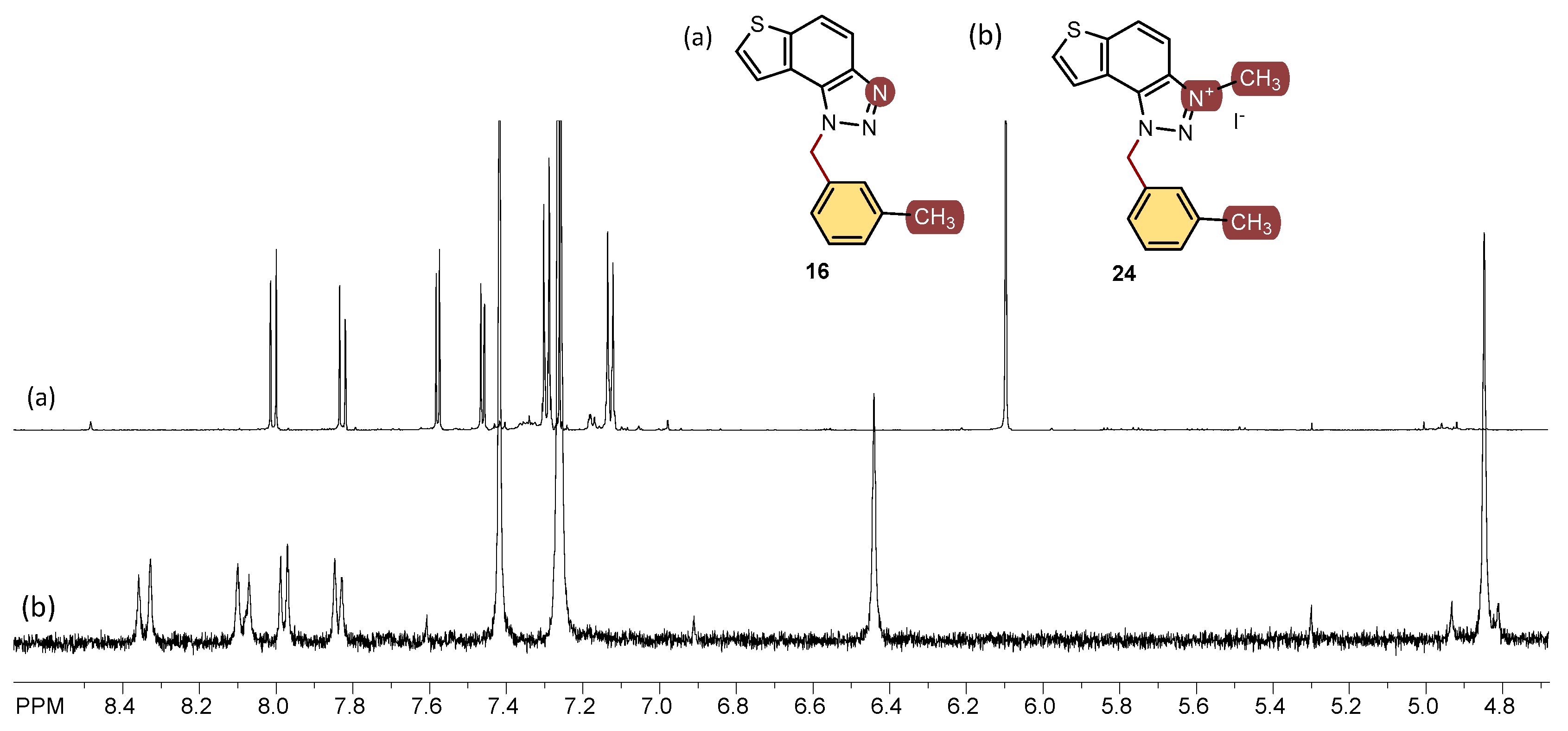
Figure 5.
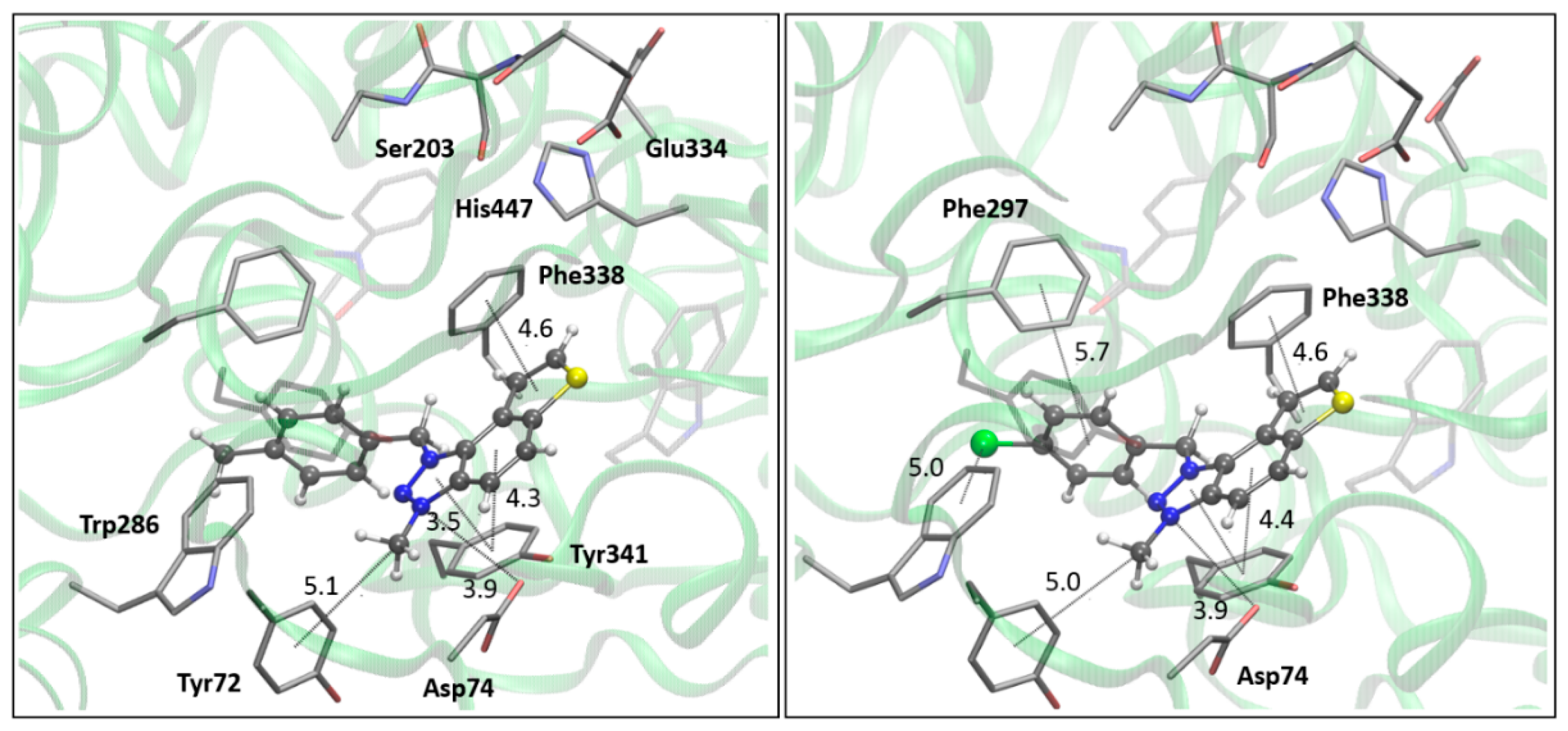
The AChE’s active site structure docked with compounds 14 (left) and 15 (right panel). Distances in angstroms; hydrogen atoms of the residues omitted for clarity.
Figure 5.
The AChE’s active site structure docked with compounds 14 (left) and 15 (right panel). Distances in angstroms; hydrogen atoms of the residues omitted for clarity.
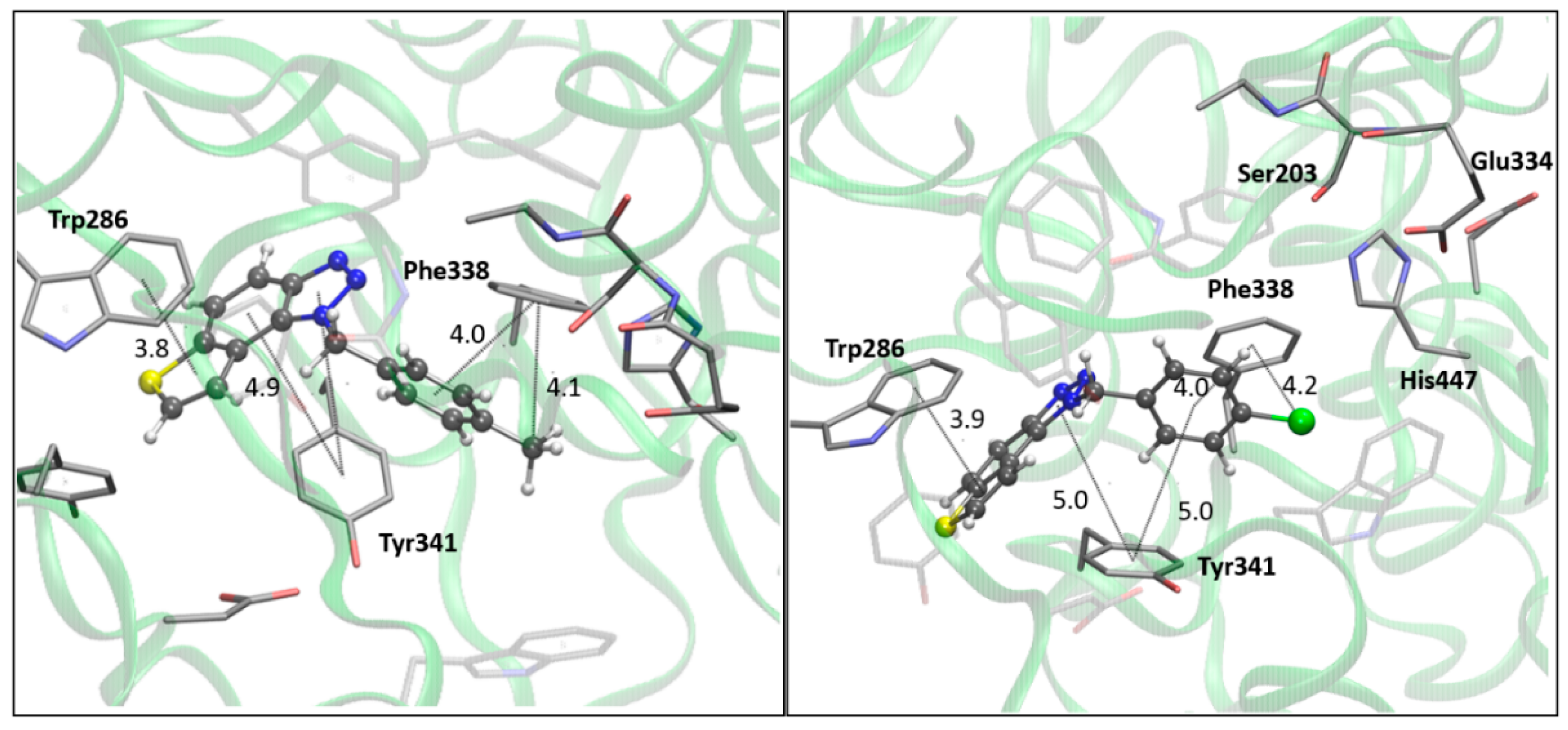
Figure 7.
The BChE’s active site structure docked with compounds 14 (left) and 15 (right). Distances in angstroms; hydrogen atoms of the residues omitted for clarity.
Figure 7.
The BChE’s active site structure docked with compounds 14 (left) and 15 (right). Distances in angstroms; hydrogen atoms of the residues omitted for clarity.
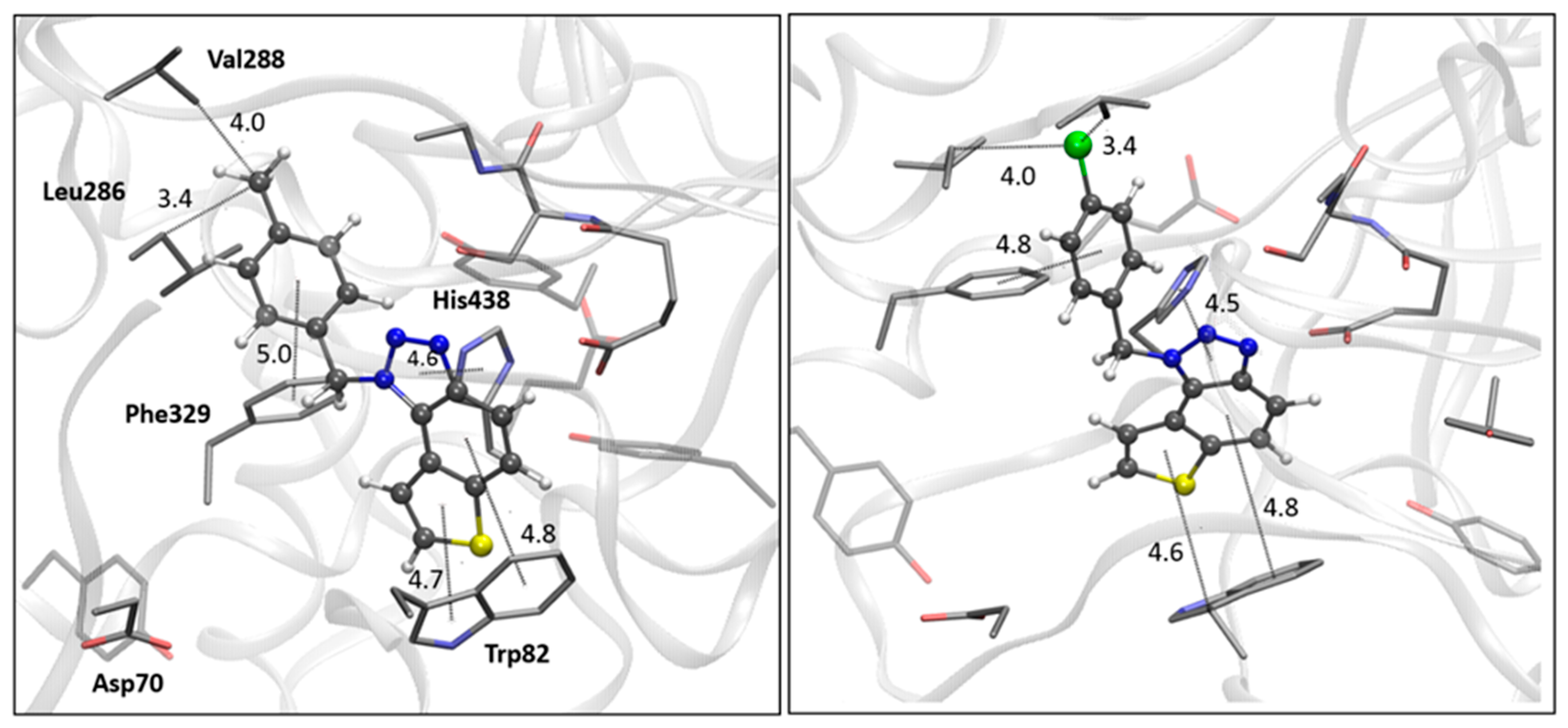
Figure 8.
The BChE’s active site structure docked with triazolium salts 22 (left) and 23 (right panel). Distances in angstroms; hydrogen atoms of the residues omitted for clarity.
Figure 8.
The BChE’s active site structure docked with triazolium salts 22 (left) and 23 (right panel). Distances in angstroms; hydrogen atoms of the residues omitted for clarity.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



