
Submitted:
02 October 2024
Posted:
04 October 2024
You are already at the latest version
Abstract
The persistent challenge of IPF, characterized by disease progression and high mortality, underscores the urgent need for innovative therapeutic strategies. We have developed a novel small molecule – catechin derivative ABI-171 – selectively targeting DYRK1A and PIM1 kinases, crucial in the pathogenesis of fibrotic processes. We employed the Bleomycin-induced (intratracheal) mouse model of pulmonary fibrosis (PF) to evaluate the therapeutic efficacy of ABI-171. Mice with induced PF were treated QD with ABI-171 either prophylactically or therapeutically using oral and intranasal routes. Pirfenidone (100mg/kg, TID) and Epigallocatechin gallate (EGCG, 100 mg/kg, QD), a natural catechin currently in a Phase 1 clinical trial, were used as reference compounds. ABI-171, administered prophylactically, led to a significant reduction in hydroxyproline levels and fibrotic tissue formation compared to the control group. Treatment with ABI-171 improved body weight, indicating mitigation of disease-related weight loss. Additionally, ABI-171 demonstrated anti-inflammatory activity, reducing lymphocyte and neutrophil infiltration. In the therapeutic setting, ABI-171, administered 7 days post-induction, reduced mortality rates (P = 0.04) compared with the bleomycin and EGCG control groups. ABI-171 also ameliorated the severity of lung injuries assessed by improved Masson’s trichrome scores when administered both orally and intranasally. ABI-171 significantly decreases bleomycin-induced PF and improves survival in mice, showcasing promising therapeutic potential beyond current medications like Pirfenidone and EGCG for patients with IPF. Based on these results, further studies with ABI-171 are ongoing in preclinical studies.
Keywords:
EGCG
; green tea
; IPF
; Bleomycin
1. Introduction
Idiopathic Pulmonary Fibrosis (IPF) is a severe, progressive interstitial lung disease characterized by the abnormal and excessive deposition of fibrotic tissue within the lungs.[1] This fibrotic tissue disrupts the normal lung architecture, leading to increased stiffness and reduced elasticity, which impairs the lungs’ ability to facilitate efficient gas exchange. Clinically, IPF manifests as chronic, worsening dyspnea (shortness of breath), a persistent dry cough, and fatigue, significantly diminishing the quality of life and functional capacity of affected individuals. The prognosis for IPF is poor, with a median survival time of 3 to 5 years from diagnosis.[2]
The pathogenesis of IPF is complex and not fully understood. It is believed to involve a combination of genetic predisposition and environmental exposures that lead to repeated injury and aberrant repair of alveolar epithelial cells (AECs). This results in the activation and proliferation of myofibroblasts, which produce and deposit extracellular matrix (ECM) proteins such as collagen, contributing to the progressive scarring (fibrosis) of lung tissue.[3] Transforming Growth Factor-beta (TGF-β), a key mediator of fibrosis, is released from injured AECs and other cells, promoting ECM production, deposition, and inhibiting ECM-degrading enzymes. Additional signaling molecules, such as connective tissue growth factor (CTGF), platelet-derived growth factor (PDGF), and fibroblast growth factor (FGF), also contribute to the fibrotic process.[4]
Currently, the treatment options for IPF are limited and primarily focused on slowing disease progression and managing symptoms. The two FDA-approved drugs for IPF, Pirfenidone and Nintedanib,[5] have been shown to slow the decline in lung function modestly. However, these treatments have significant limitations, including side effects that can impact patient adherence and their inability to improve overall survival or quality of life significantly. There remains an urgent need for more effective therapies that can halt or reverse the fibrotic process and provide meaningful clinical benefits for patients with IPF.
Epigallocatechin gallate (EGCG), a natural catechin derived from green tea, has emerged as a potential therapeutic agent for IPF due to its anti-fibrotic and anti-inflammatory properties.[6] EGCG has been shown to inhibit several key pathways involved in fibrosis, including the TGF-β signaling pathway, which is critical for activating fibroblasts and producing ECM proteins.[7] By blocking TGF-β1 responses and reducing the expression of pro-fibrotic markers such as Snail and collagen, EGCG can mitigate the fibrotic process.[8] Additionally, EGCG has demonstrated the ability to reduce oxidative stress and inflammation, which are significant contributors to the pathogenesis of IPF.[9]
Preclinical studies have shown that EGCG can reduce fibrosis in animal models of IPF, leading to improved lung function and reduced fibrotic markers. In a small human trial with IPF patients, EGCG was found to be effective in lessening fibrotic indicators.[10] Despite its promising anti-fibrotic effects, EGCG faces several challenges that limit its therapeutic potential. One of the primary limitations is its poor bioavailability, which restricts the amount of compound that can reach the target tissues in the lungs.[11] Furthermore, EGCG’s selectivity profile and safety concerns, particularly related to liver toxicity,[12] pose additional hurdles to its development as a therapeutic agent for IPF.
To address these limitations, we have explored the development of novel derivatives of EGCG that retain their beneficial properties while improving bioavailability and safety.[13] ABI-171, a novel fluoro-catechin derivative, has emerged as a promising candidate for the treatment of IPF. ABI-171 is a potent dual inhibitor of dual-specificity tyrosine-phosphorylation-regulated kinase 1A (DYRK1A) and proviral integration site for Moloney murine leukemia virus 1 (PIM1) kinases (this study). Both DYRK1A and PIM1 are serine/threonine kinases implicated in various cellular processes, including cell proliferation, survival, and differentiation.
The mechanism of action of ABI-171 potentially involves several key pathways and processes that contribute to its potential efficacy in treating IPF. DYRK1A plays a significant role in regulating neuronal development, cell cycle regulation, and stress response,[14] while also being involved in the regulation of inflammation and fibroblast activation in IPF.[15] ABI-171’s inhibition of DYRK1A leads to the attenuation of inflammatory responses and a reduction in fibroblast activation—critical steps in the development of pulmonary fibrosis. By targeting DYRK1A, ABI-171 helps to modulate the inflammatory environment within the lungs, thereby reducing the recruitment and activation of fibroblasts and myofibroblasts, the primary cells responsible for the production and deposition of ECM proteins.
PIM1 kinase is another important target of ABI-171. PIM1 is involved in controlling cell survival, proliferation, and differentiation.[16] In IPF, PIM1 is upregulated and contributes to epithelial-to-mesenchymal transition (EMT), a process where epithelial cells lose their characteristics and gain mesenchymal, fibrogenic properties.[17],[18]This is a crucial event in IPF, as it leads to the generation of myofibroblasts, which are responsible for excessive collagen deposition. By inhibiting PIM1, ABI-171 may disrupt this process, thereby reducing the generation of myofibroblasts and the subsequent fibrosis. Inhibition of PIM1 has also been shown to alleviate lung fibrosis in a bleomycin-induced mouse model. Additionally, PIM1 inhibition decreases phosphorylation and activation of nuclear factor of activated T cells (NFATc1),[19] a transcription factor promoting cell proliferation and survival, further contributing to ABI-171’s anti-fibrotic effects by reducing fibroblast proliferation and survival.
ABI-171, as a trihydroxyphenolic compound, could serve as a potential inhibitor of lysyl oxidase-like 2 (LOXL2) and, indirectly, impede TGF-β signaling. Polyphenolic compounds inhibit LOXL2 activity by inducing auto-oxidation of specific lysine residues in LOXL2, preventing the enzyme from facilitating the conversion of lysine residues in collagen to aldehydes, reducing collagen cross-linking and tissue stiffening, both hallmarks of fibrosis.[20]
Polyphenolic compounds also inhibit TGF-β signaling; however, the inhibition requires the presence of LOXL2. Apparently, inhibition of TGF-β signaling is mediated through the putative amino-catechol metabolites of polyphenolic compounds resulted from the interaction with LOXL2.7 Both LOXL2 and TGF-β signaling contribute to the elevation of Snail1, a transcription factor key to EMT. By inhibiting LOXL2 and TGF-β signaling, ABI-171 could reduce Snail1 expression and prevent EMT progression, thus mitigating fibrotic tissue development and the pathological collagen deposition seen in IPF.
This dual inhibition of LOXL2 and TGF-β signaling, combined with its inhibition of DYRK1A and PIM1 kinases, makes ABI-171 a promising candidate for therapeutic intervention in fibrotic diseases. Based on these mechanisms, we hypothesize that ABI-171 will significantly reduce fibrosis and improve lung function in preclinical models of IPF, providing superior efficacy compared to current treatments. This multifaceted mechanism of action highlights ABI-171’s potential as a disease-modifying agent for IPF, offering a promising therapeutic approach for this devastating disease.
This study aims to comprehensively evaluate the therapeutic potential of ABI-171 for IPF. The objectives include characterizing its pharmacokinetic properties, assessing its therapeutic potential in preventive and therapeutic animal models, and elucidating its effects on key molecular pathways involved in IPF pathogenesis. The findings from this research will support the development of ABI-171 as a novel therapeutic agent for IPF and provide a foundation for subsequent clinical trials.
2. Materials and Methods
2.1. Material and Reagents
Bleomycin (Selleck Chemicals, Cat: S1214) was used as the fibrosis-inducing agent. CMC-Na (carboxymethyl cellulose sodium) was sourced from Sigma-Aldrich (Cat: C4888-500G) as a vehicle solution. Pirfenidone was obtained from Bidepharm (Cat: BD28958-5g) for positive control treatment. (2-Hydroxypropyl)-β-cyclodextrin (HP-β-CD) was supplied by MedChemExpress (Cat: HY-101103), and polyethylene glycol 400 (PEG400) from Sigma-Aldrich (Cat: 81172) was used as a solvent. Na2EDTA (disodium ethylenediaminetetraacetic acid) from Sigma-Aldrich (Cat: E5134-50G) was used in reagent preparations. ABI-171 (purity 98%) was synthesized as described in our previous publication (Araldi GL, Hwang YW., Pharmaceuticals 2023), and its solutions were prepared in PEG400 and HP-β-CD mixtures. Hematoxylin for histological analysis was purchased from ZSGB-BIO (Cat: ZLI-9610).
2.2. ABI-171 Pharmacokinetics and Lung-Tissue Distribution
Female C57BL/6 mice with body weights of 17-24 g were purchased and maintained in air-conditioned quarters with 12-hr light/dark cycles. They were given a commercial mice chow and water ad libitum. The experiments started after acclimation for at least 1 week. Dosing solutions were made fresh. Fasted mice were administered the test drug by oral (po), intranasal (IN), and intravenous (iv) route. The vehicle for IN and po routes was composed of 12% PEG400, 10% HP-β-CD, and saline q.b.; for iv, water was used. Post-dosing, ~500 µL of blood samples will be collected (cardiac puncture) from three mice at each time point at 0.5, 1, 2, 4, 6, 8, and 24 h under isoflurane anesthesia. Blood is collected into K2EDTA microtubes and kept on ice all the time (must be centrifuged within 30 minutes of collection). Blood in EDTA vacuum tubes are centrifuged at 4 °C at 15,000 g for 4 min; ~250 µL of plasma are collected into polyethylene tubes containing 50 µL of the ascorbic acid (AA)/TCEP stabilizing solution (20 mM AA and 13 mM TCEP in 50 mM K2HPO4 buffer. The pH of the solution is adjusted to 6.5 using 2 M NaOH). The plasma samples right after collection are immediately snap frozen in isopropanol/carbon-dioxide dry ice and stored at -80 °C until analysis.
Test catechin is extracted from the above-collected plasma through 3 successive additions of 350 µL of acetonitrile. The plasma and solvent mixture are vortex-mixed for 2 min then snap-frozen with isopropanol/carbon-dioxide dry ice and the extraction solvent is removed. The solvent fractions are pooled into a polyethylene tube kept on ice. The combined fractions are evaporated under a gentle stream of nitrogen at ambient temperature. The residue obtained after evaporation is reconstituted in 200 µL of 75 mM citric acid//25 mM ammonium acetate: acetonitrile (75:25, by vol), vortexed vigorously for 5 min, and 20 µL of the resulting solution is injected into the HPLC column. Lung tissue was collected and treated immediately to avoid product degradation. Within 5 minutes from extraction, about ~0.4 g of the tissue is homogenized with 1 mL of ice-cold 0.4 M sodium phosphate buffer containing 6 mg of ascorbic acid and 0.5 mg of Na2EDTA (final pH of 6.5). After centrifugation at 4 °C at 15,000 g for 4 min; the supernatant is collected into polyethylene tubes containing 50 µL of the AA/TCEP stabilizing solution (20 mM AA and 13 mM TCEP in 50 mM K2HPO4 buffer. The pH of the solution is adjusted to 6.5 using 2 M NaOH) and immediately snap frozen in isopropanol/carbon-dioxide dry ice and stored at -80 °C until analysis. Extraction is performed as described above for plasma.
2.3. Efficacy Studies: Bleomycin-Induced Animal Models of IPF
To evaluate the therapeutic potential of ABI-154 and ABI-171, we utilized two well-established bleomycin-induced animal models of IPF: a preventive model and a therapeutic model. These models are widely recognized for their ability to mimic the fibrotic processes observed in human IPF, including inflammation, fibroblast proliferation, and excessive deposition of extracellular matrix (ECM) proteins.
2.3.1. Preventive Model
In the preventive model, male C57BL/6 mice, aged 8-10 weeks, were used to simulate the early intervention scenario where treatment begins immediately following the induction of lung injury. The animals were randomly assigned to different groups (n = 6 per group): Control (normal), BLM (vehicle), Pirfenidone (100 mg/kg orally, thrice daily), ABI-154 (60 mg/kg intranasally, once daily), and ABI-171 (60 mg/kg intranasally, once daily). On day 0, pulmonary fibrosis was induced by administering a single intratracheal infusion of bleomycin at a dose of 3 U/kg body weight, dissolved in 50 μL of sterile saline. Treatment with the test compounds commenced immediately after bleomycin administration and continued daily for 21 days. Body weight, survival rate, and clinical signs were monitored throughout the study.
At the end of the study period, the mice were sacrificed, and their lungs were harvested for histological and biochemical analyses. Lung tissues were fixed in 10% formalin and embedded in paraffin for histological examination using hematoxylin and eosin (H&E) staining to assess tissue morphology and Masson’s trichrome staining to evaluate collagen deposition. Additionally, lung hydroxyproline content, a marker of collagen accumulation, was quantified using a colorimetric assay. Inflammatory cell infiltration was assessed by counting lymphocytes and neutrophils in bronchoalveolar lavage fluid (BALF).
2.3.2. Therapeutic Model
In the therapeutic model, the efficacy of ABI-171 was assessed in a delayed treatment scenario, where the intervention begins after the establishment of lung fibrosis. Male C57BL/6 mice, aged 8-10 weeks, were randomly assigned to the following groups (n = 6 per group): Control (normal), BLM (vehicle), EGCG (100 mg/kg orally, once daily), and ABI-171 (intranasally at 10 mg/kg and 50 mg/kg once daily or orally at 100 mg/kg once daily). Pulmonary fibrosis was induced by a single intratracheal infusion of bleomycin at a dose of 3 U/kg body weight. Seven days post-bleomycin administration, when fibrosis was established, treatment with the test compounds commenced and continued daily for an additional 21 days.
Throughout the treatment period, body weight, survival rate, and clinical signs were monitored. At the study’s conclusion, mice were sacrificed, and their lungs were harvested for comprehensive histological and biochemical analyses. Lung tissues were processed for H&E and Masson’s trichrome staining to evaluate fibrosis and collagen deposition. Lung hydroxyproline content was measured to quantify collagen accumulation. Inflammatory cell infiltration in BALF was assessed by counting lymphocytes and neutrophils. Additionally, the expression levels of fibrosis-related markers, including PIM1, E-cadherin, p-Smad3, α-SMA, and Snail, were analyzed using Western blotting and immunohistochemistry to elucidate the molecular mechanisms underlying the anti-fibrotic effects of ABI-171.
These two complementary bleomycin-induced models allowed us to assess the preventive and therapeutic efficacy of ABI-154 and ABI-171, providing a robust framework for evaluating their potential as treatments for IPF.
2.4. Statistical Analysis
Results are expressed as mean ± SD. All data were analyzed using GraphPad Prism Version 10.0 software. Statistical comparisons were performed using Student t-test for two-group comparison and one or two-way ANOVA with a Tukey’s post hoc test for multiple comparisons. p < 0.05 was considered to be statistically significant.
3. Results
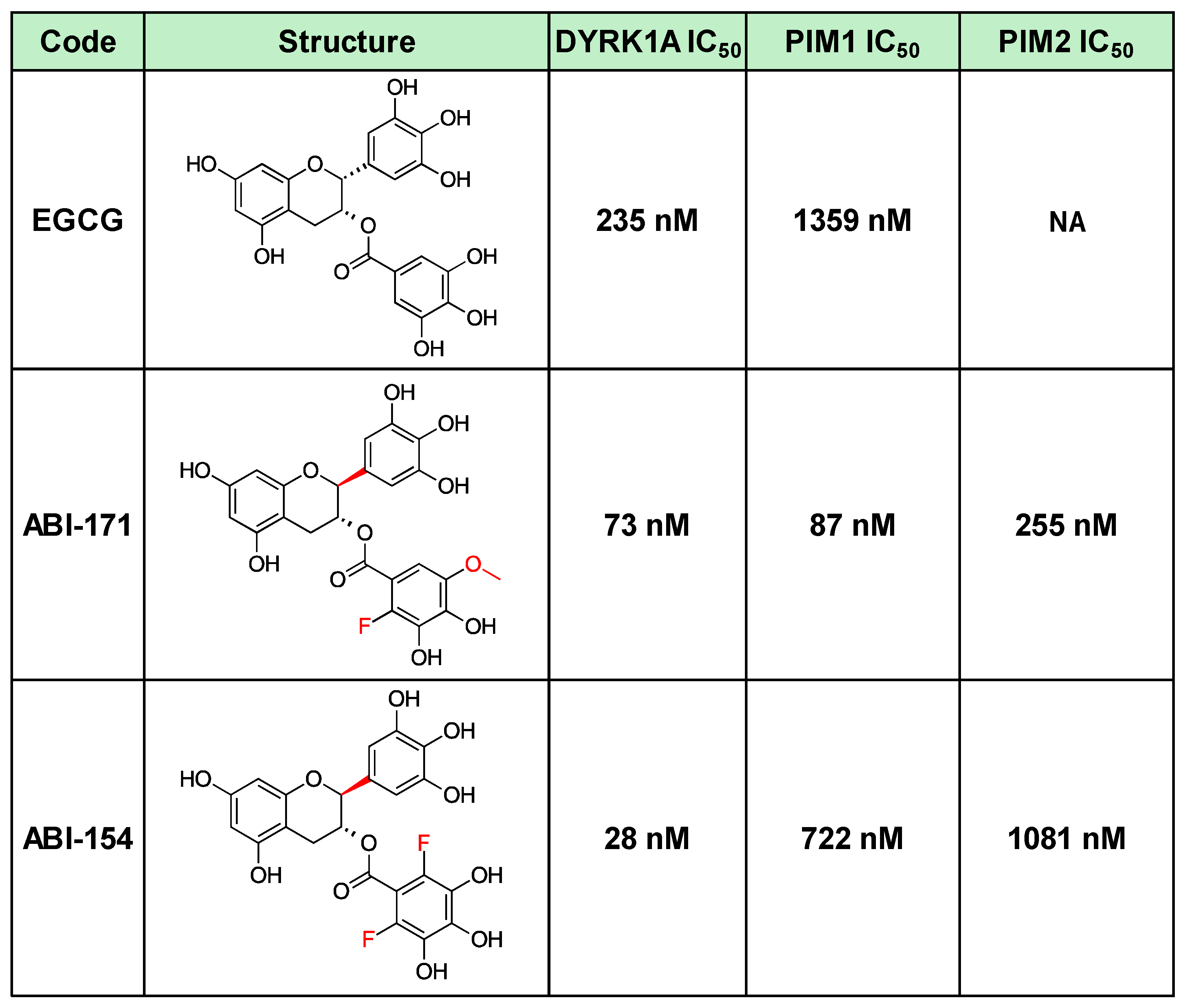
In the study, two compounds, ABI-154 and ABI-171, were initially evaluated for their therapeutic potential in treating IPF (Table 1). ABI-154, a potent inhibitor of DYRK1A, exhibited strong efficacy in initial preclinical tests due to its ability to modulate key pathways involved in inflammation and fibrosis. Despite its high potency, ABI-154 faced challenges related to pharmacokinetics and bioavailability (ref 13 and Table 2), which limited its overall therapeutic impact. ABI-171, a fluoro-methyl catechin derivative, was further developed to address these limitations. ABI-171 not only targets DYRK1A but also inhibits PIM1 kinase, broadening its anti-fibrotic activity by disrupting multiple pathways involved in fibrosis progression, including EMT and fibroblast activation. This dual inhibitory action, coupled with improved pharmacokinetic properties, such as better bioavailability and a favorable safety profile, positions ABI-171 as a more promising candidate for IPF treatment. The comparative evaluation of these compounds in this study aims to highlight the superior efficacy of ABI-171 over ABI-154, supporting its potential as a disease-modifying therapy for IPF

Table 1.
Best DYRK1A inhibitors.
 |
3.1. ABI-171 Pharmacokinetics and Lung-Tissue Distribution
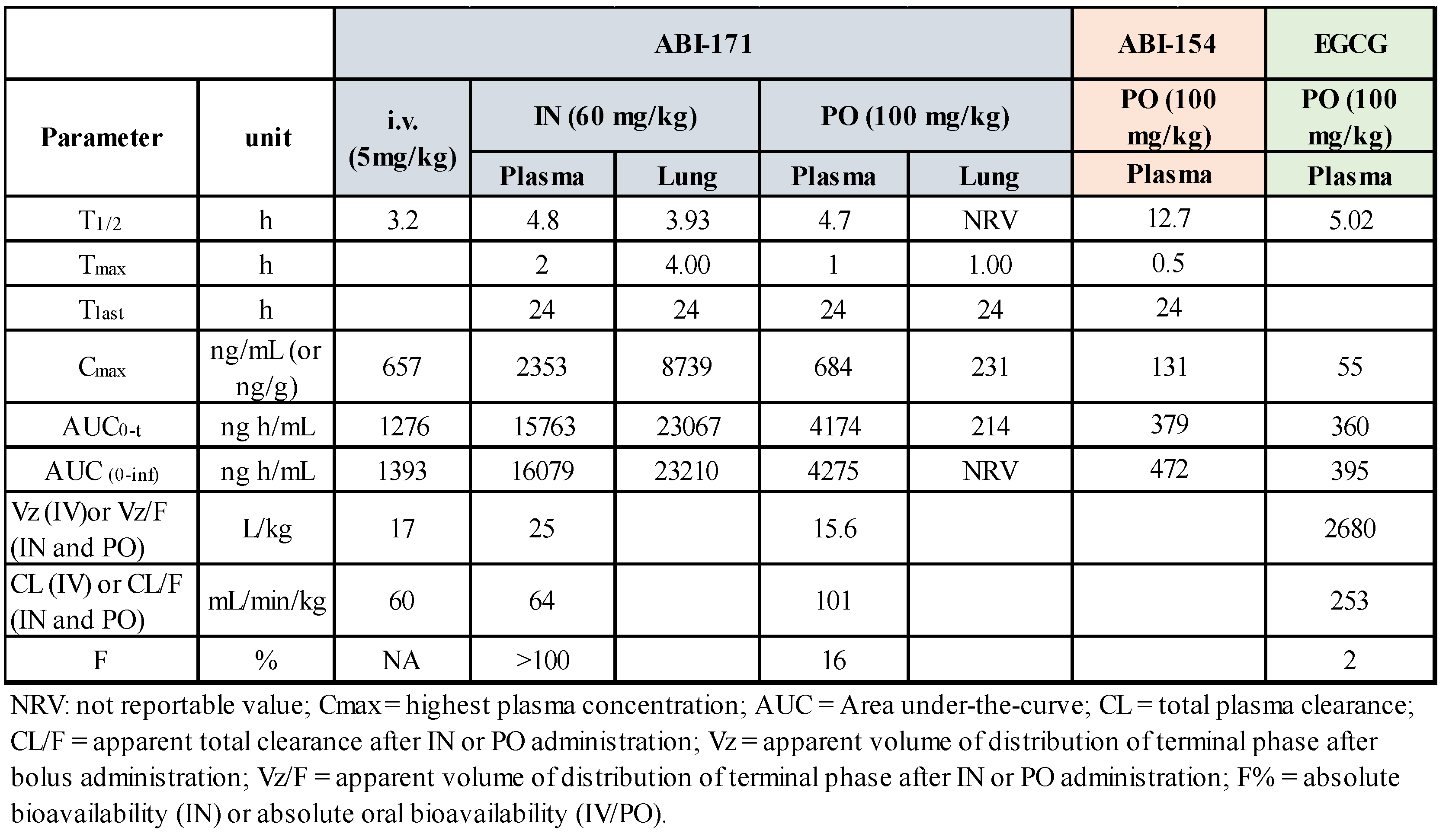
The pharmacokinetics (PK) of ABI-171 and ABI-154 were evaluated in male C57BL/6 mice following single doses administered via intranasal (IN 60 mg/Kg), oral (PO 100 mg/Kg), and intravenous (iv 5 mg/Kg) routes. Plasma concentrations were measured using LC-MS/MS, and PK parameters were calculated with WinNonlin. To facilitate drug delivery, we have developed a proprietary liquid formulation that allows the delivery of these drugs in high concentrations for oral or intranasal use (ref). The formulation is based on the solubilizer and penetration enhancer (2-hydroxypropyl)-β-cyclodextrin (HP-β-CD), PEG-400, and water. This vehicle allows us to achieve high drug concentration in dosing solution (up to 20% w/w) when needed. PK studies for these compounds in plasma and the brain have been reported in an earlier publication.13 Here we report the concentration of the drug in the lungs. As shown previously (ref 13 also in Table 2), despite the absolute total bioavailability of ABI-171 in plasma was higher when the drug was delivered via IN than via PO (100% vs. 16%), the IN route is less efficient than the PO route in delivering drug into tissues such as the brain and liver (ref). In the lungs, however, the outcome is opposite with the IN route shown to be highly superior to the PO route in delivering ABI-171 to the target (Table 2). Using the IN route, we can achieve a high and stable exposure in the lungs with a theoretical cellular exposure far surpassing the IC50 of inhibiting DYRK1A and PIM1. Considering the Cmax and the T1/2, it is possible that a single IN dose may still sustain a concentration of ABI-171 in the lungs above the IC50 of inhibiting DYRK1A and PIM1 after 24 hours. To further confirm the PK results, we selected both ABI-154 and ABI-171 using the IN routes for the BLM efficacy studies.
3.2. Efficacy in Animal Models
3.2.1. Preventative IPF Animal Model (Treatment Start @ Day 0 after BLM Instillation)
PK studies using the IN route showed significantly elevated lung drug concentrations. Consequently, we opted for intranasal delivery in our first IPF animal model, employing a validated BLM mouse model. In this study, 30 male C57BL/6 mice were randomly assigned to five groups: Control (Normal), BLM (vehicle), Pirfenidone (100 mg/kg, TD, oral), ABI-154 (60 mg/kg, QD, intranasal), and ABI-171 (60 mg/kg, QD, intranasal). The experiment considered the day of bleomycin injection as Day 0. The IPF model was successfully induced through a single intratracheal infusion of bleomycin. The experiment was completed after 21 days. Body weight loss induced by bleomycin was observed in the BLM group (*** P<0.001), while the ABI-171 group showed a significant increase in body weight from Day 14 to 21 (** P<0.01; *** P<0.001) and an improved delta body weight (* P<0.05) compared to the model group (Figure 1 A). The collagen content in lung tissues was assessed by measuring the hydroxyproline content.
Figure 1.
Preventative treatent study. A) BLM animal model scheme; B) body weight; C) Hydroxyproline level; D, E) Lymphocytes and neutrophile assy; F) H&E score; G) Masson score; H) Presentative histology pictures with blue arrow indicating fibrosis with lung structural destruction. *P<0.05; **P<0.01; ***P<0.001;****P<0.0001 one-way ANOVA. B-F data are presented as the mean ± standard error of the mean (n=6).
Figure 1.
Preventative treatent study. A) BLM animal model scheme; B) body weight; C) Hydroxyproline level; D, E) Lymphocytes and neutrophile assy; F) H&E score; G) Masson score; H) Presentative histology pictures with blue arrow indicating fibrosis with lung structural destruction. *P<0.05; **P<0.01; ***P<0.001;****P<0.0001 one-way ANOVA. B-F data are presented as the mean ± standard error of the mean (n=6).
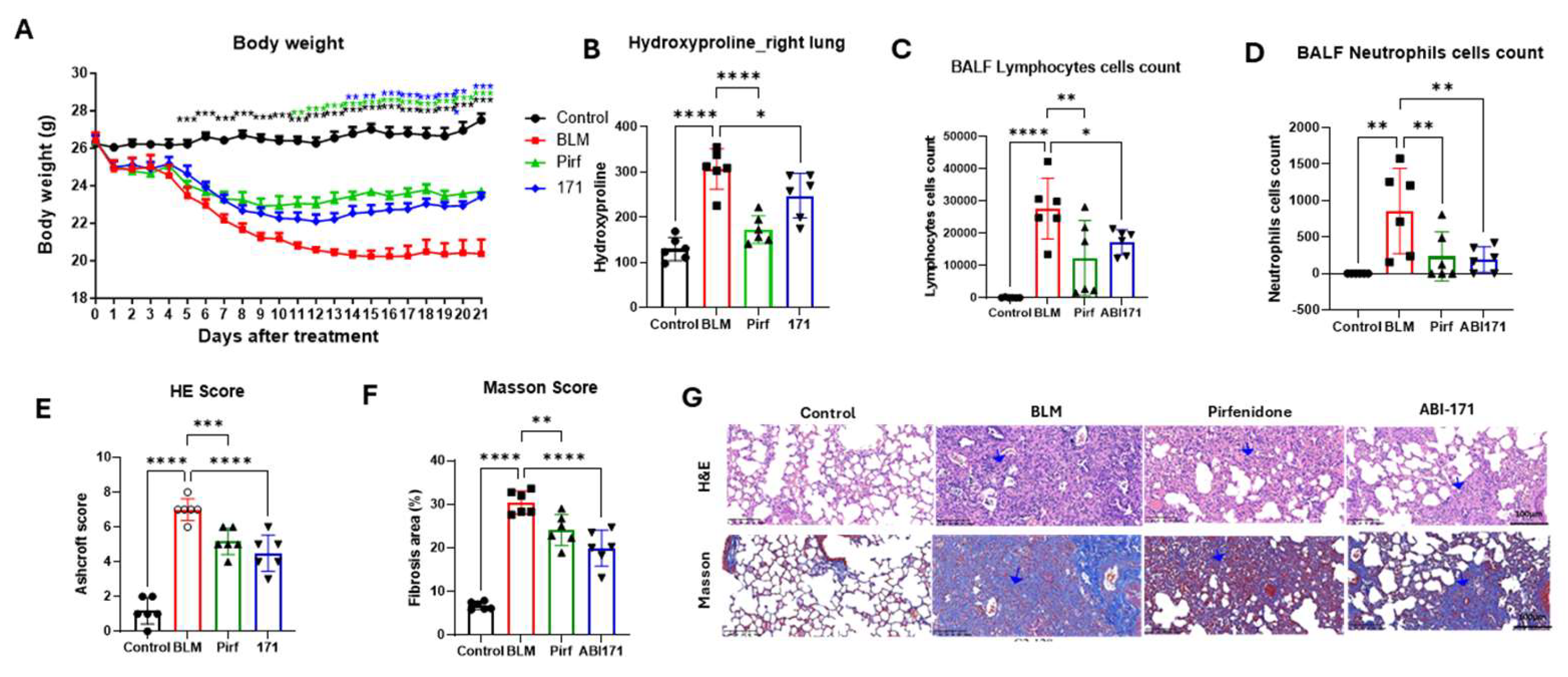
Compared with the BLM group, the hydroxyproline content significantly decreased in the BLM + ABI-154 and ABI-171 groups (Figure 1 B). Lymphocytes and neutrophils increased significantly in the BLM group but were significantly reduced in the ABI-171 group (P<0.05), indicating a potent anti-inflammatory effect (Figure 1 C, D). To explore the impact of our fluoro-catechins on bleomycin-mediated PF, pathological alterations of lung tissue were observed by H&E and Masson staining in each group. HE staining was used to determine fibrosis, while collagen deposition within different sections was analyzed by Masson’s trichrome staining (Figure 1 E,F). After bleomycin administration, a remarkable increase in alveolar septa thickening, as well as collagen deposition within lung tissue, was observed (BLM group, Figure 1 G). However, drug treatment had a reverse effect, with a decrease in the alveolitis and PF degrees relative to the bleomycin group (154 and 171 cohorts, Figure 1 G). ABI-171 exhibited superior and positive effects on body weight, inflammation, hydroxyproline levels, and lung pathology in the context of IPF when compared to ABI-154. As a result, ABI-171 was selected for further investigation in follow-up studies (see below).
3.2.2. Therapeutic IPF Animal Model (Treatment Start @ Day 7 after BLM Instillation)
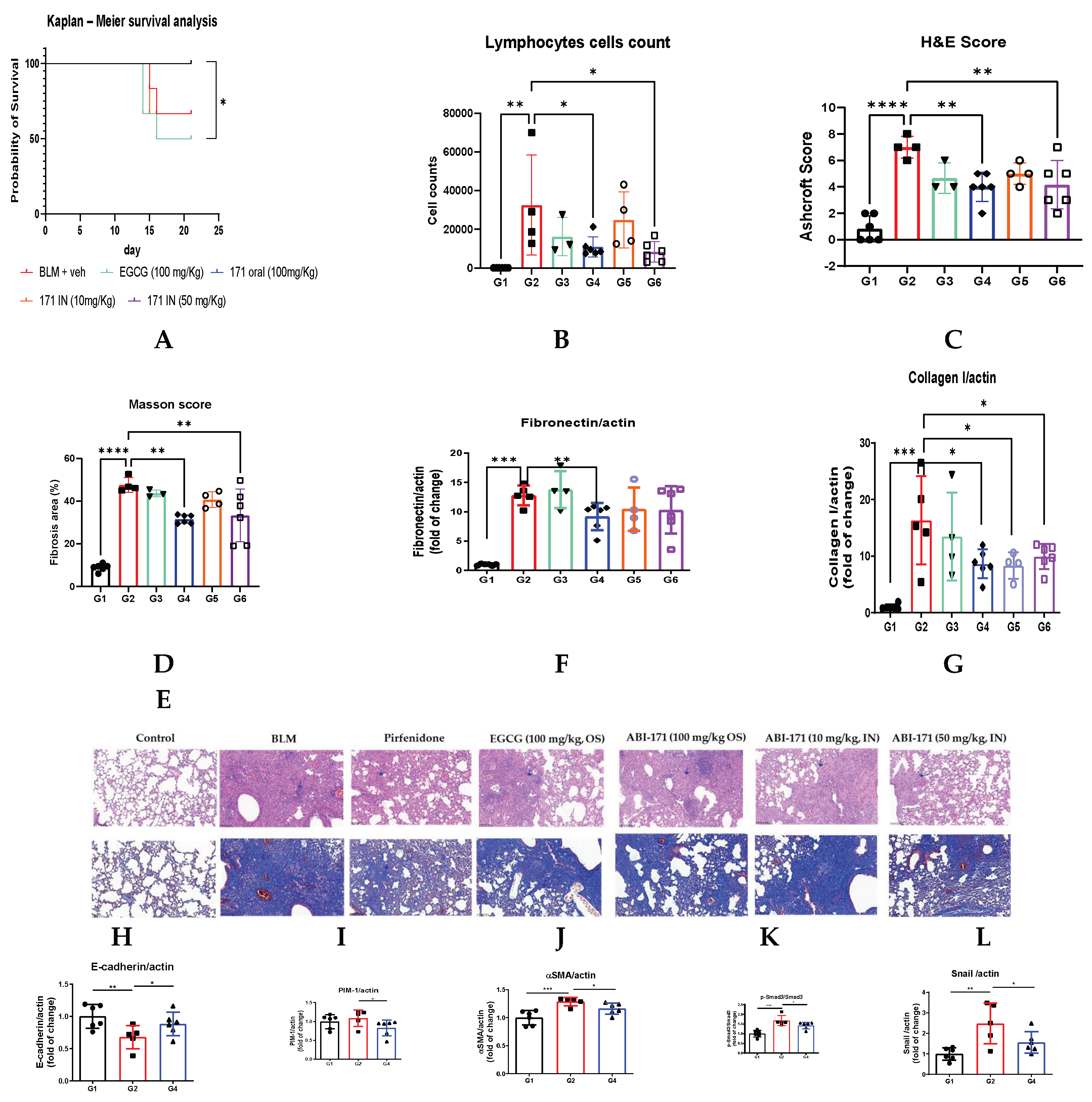
Based on the positive outcomes of our preliminary study, we designed a subsequent study to evaluate the therapeutic potential of ABI-171. In this study, drug treatment commenced 7 days post-induction of pulmonary fibrosis using BLM, employing a therapeutic model. We assessed the efficacy of ABI-171 administered intranasally at two dosages (10 mg/kg and 50 mg/kg, QD) and orally (100 mg/kg, QD), comparing it to oral EGCG (100 mg/kg, once daily), natural catechin currently undergoing Phase 1 clinical trials for IPF. Additionally, Pirfenidone (100 mg/kg, TID, orally) was included as a control, with treatment initiated on day zero. Unexpectedly, EGCG demonstrated limited to moderate activity in various assessments, notably a survival rate with 50% mortality. Conversely, both intranasal (50 mg/kg) and oral administration of ABI-171 resulted in complete survival and good recovery in body weight.
ABI-171 effectively mitigated BLM-induced lung injuries, as indicated by improvements in Masson’s and H&E scores (Figure 2 C-E). Notably, oral administration of ABI-171 (100 mg/kg) had similar efficacy compared to intranasal delivery (10 and 50 mg/kg doses), suggesting that both routes are viable, although the intranasal route seems to be more dose-effective. By day 21, mice treated with ABI-171 exhibited significant reductions in BLM-induced lung collagen and fibronectin levels (Figure 2 F,G), underscoring the importance of targeting fibronectin in IPF management. Considering the similar results obtained between group 4, 5, and 6 we have further investigated the mechanism of action using only tissues from Group 4. PIM1 is upregulated consistently in the fibrotic lung tissues of IPF patients and in mice’s lungs induced by intratracheal BLM administration; treatment with ABI-171 significantly reduced the level of this enzyme (Figure 2 I). E-cadherin, crucial for maintaining epithelial integrity, is often downregulated in pulmonary fibrosis, leading to EMT and subsequent fibrotic tissue remodeling. Our analysis revealed that oral treatment with ABI-171 significantly elevated E-cadherin levels (Figure 2 H), suggesting its potential to halt pulmonary fibrosis progression by preserving epithelial integrity.
Figure 2.
K-L showed a significant reduction in p-Smad3, αSMA, and Snail protein levels, indicating a significant decrease in fibrotic activity. p-Smad3 is a key signaling molecule in the TGF-β pathway, which promotes fibrosis; thus, its reduction suggests less active TGF-β signaling. αSMA, a marker of myofibroblasts—the primary cells responsible for fibrosis—being reduced implies decreased myofibroblast presence or activity. Snail, a transcription factor involved in EMT, also shows lower levels, indicating inhibition of EMT and fewer epithelial cells converting into fibrotic mesenchymal cells. Collectively, these reductions demonstrate that the therapeutic intervention effectively mitigates the processes driving fibrosis, potentially ameliorating IPF progression.
Figure 2.
K-L showed a significant reduction in p-Smad3, αSMA, and Snail protein levels, indicating a significant decrease in fibrotic activity. p-Smad3 is a key signaling molecule in the TGF-β pathway, which promotes fibrosis; thus, its reduction suggests less active TGF-β signaling. αSMA, a marker of myofibroblasts—the primary cells responsible for fibrosis—being reduced implies decreased myofibroblast presence or activity. Snail, a transcription factor involved in EMT, also shows lower levels, indicating inhibition of EMT and fewer epithelial cells converting into fibrotic mesenchymal cells. Collectively, these reductions demonstrate that the therapeutic intervention effectively mitigates the processes driving fibrosis, potentially ameliorating IPF progression.
4. Discussion
The results of this study highlight the significant therapeutic potential of ABI-171 in treating idiopathic pulmonary fibrosis (IPF), a disease for which there are currently no curative treatments. Although our results are based on a bleomycin-induced pulmonary fibrosis model in mice, it is important to acknowledge that both nintedanib and pirfenidone—the two FDA-approved antifibrotic drugs for IPF—were developed using similar models.[21],[22] Ongoing clinical trials for IPF continue to rely on these models to target the pathways involved in disease pathogenesis.[23],[24] In this context, the dual inhibition of DYRK1A and PIM1 kinases by ABI-171 provides a comprehensive approach to addressing the critical pathological processes of IPF, including inflammation, fibroblast activation, epithelial-mesenchymal transition (EMT), and extracellular matrix (ECM) remodeling.
ABI-171 significantly improved survival rates and reduced collagen accumulation in both preventive and therapeutic models of bleomycin-induced IPF. In the preventive model, where treatment began immediately after bleomycin administration, ABI-171 effectively mitigated early fibrotic changes and inflammation. This was evidenced by the significant reduction in lymphocyte and neutrophil counts in the BALF, suggesting a robust anti-inflammatory effect mediated by DYRK1A inhibition. DYRK1A plays a pivotal role in regulating inflammatory responses and fibroblast activation, both essential factors in fibrosis progression (ref).
In the therapeutic model, where treatment began after fibrosis had been established, ABI-171 continued to demonstrate strong efficacy. The compound reduced lung collagen and fibronectin levels, both key markers of fibrosis, and improved histopathological scores. Notably, ABI-171 outperformed EGCG, a naturally occurring catechin derived from green tea, which is currently undergoing Phase 1 clinical trials for IPF.[25] While EGCG has shown promise due to its anti-inflammatory and antifibrotic properties, its clinical application is limited by poor bioavailability and potential safety concerns with long-term use. Furthermore, EGCG is widely available as a dietary supplement and through green tea consumption, which complicates its positioning as a pharmaceutical-grade therapeutic. ABI-171, a fluoro-catechin derivative, addresses these limitations by offering enhanced bioavailability, selectivity, and potency, making it a more viable candidate for clinical use in IPF.
Mechanistically, ABI-171 exerts its anti-fibrotic effects through the inhibition of both DYRK1A and PIM1 kinases and potentially including LOXL2 and TGF-β signaling as well. By inhibiting DYRK1A, ABI-171 modulates critical signaling pathways, including STAT3, reducing the transcription of pro-fibrotic genes and inhibiting fibroblast activation. PIM1 inhibition by ABI-171 disrupts the EMT process, a crucial event in fibrosis progression, by downregulating transcription factors like Snail and αSMA. The observed increase in E-cadherin levels, which are essential for maintaining epithelial integrity, suggests that ABI-171 helps preserve epithelial characteristics and prevents their transition into a fibrogenic state.
Although direct inhibition of LOXL2 by ABI-171 as well as inhibition of TGF-β signaling by the metabolite of ABI-171 resulted from interacting with LOXL2 has not yet been demonstrated, the structural characteristics of the compound raise the hypothesis that ABI-171 could affect LOXL2 and TGF-β signaling similarly as described for polyphenolic compounds.7 LOXL2 is a key enzyme involved in collagen cross-linking and tissue stiffening, both hallmarks of fibrosis. Inhibition of LOXL2 has been associated with decreased fibrotic tissue formation in various models, and its regulation is tied to the TGF-β signaling pathway. Given that ABI-171 significantly reduces levels of Smad3—a critical downstream effector of TGF-β signaling—and other transcription factors involved in EMT, it is plausible that ABI-171 may also reduce LOXL2-mediated collagen deposition and ECM remodeling. This hypothesis is further supported by ABI-171’s ability to reduce Snail expression, which is known to be regulated by LOXL2 as well as by TGF-β during EMT (ref). However, further studies are required to confirm any direct or indirect inhibition of LOXL2 and TGF-β signaling by ABI-171.
An intriguing observation in this study was the differential impact of intranasal administration on body weight in the preventive versus therapeutic models. In the preventive model, intranasal ABI-171 significantly improved body weight, likely due to its potent anti-inflammatory effects during the early stages of fibrosis. However, in the therapeutic model, where fibrosis had already been established, the primary benefit of ABI-171 was a reduction in fibrosis rather than body weight restoration. This variance highlights the importance of early intervention in IPF and underscores the distinct pathological processes at play in the early versus late stages of the disease.
ABI-171’s superior pharmacokinetic profile, characterized by improved bioavailability and lung tissue penetration, also plays a crucial role in its efficacy. Intranasal administration ensures high local concentrations of the drug in the lungs, the primary site of action, while minimizing systemic exposure and potential side effects. This route of administration bypasses the challenges associated with first-pass metabolism and food-related interactions, common limitations of oral delivery systems, particularly for polyphenols like EGCG.
In addition to ABI-171, we also evaluated ABI-154, another catechin derivative. While ABI-154 demonstrated some efficacy in initial preclinical tests by modulating inflammatory and fibrotic pathways, its poor pharmacokinetic properties and limited bioavailability restricted its therapeutic impact. These limitations highlight the enhanced potential of ABI-171, which was developed to overcome the issues seen with ABI-154.
In conclusion, ABI-171 represents a promising disease-modifying therapy for IPF by addressing multiple pathological processes, including inflammation, fibroblast activation, EMT, and ECM remodeling through the dual inhibition of DYRK1A and PIM1 kinases, as well as LOXL2 and TGF-β signaling inhibition. Its superior pharmacokinetic properties and multi-targeted mechanism of action make ABI-171 a transformative therapeutic agent. Further clinical trials are warranted to validate these preclinical findings and fully explore the therapeutic potential of ABI-171 in patients with IPF. The success of ABI-171 in these preclinical models provides a strong foundation for its advancement into clinical development, with the potential to significantly improve outcomes for patients suffering from this devastating disease.
Author Contributions
Conceptualization:,A.G. and Y.H. resources, A.G..; writing—original draft preparation, A.G.; writing—review and editing, YH, and G.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
Author Gian Luca Araldi was employed by the company Avanti Biosciences. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Mei Q, Liu Z, Zuo H, Yang Z, Qu J. Idiopathic Pulmonary Fibrosis: An Update on Pathogenesis. Front Pharmacol. 2022 Jan 19;12:797292. [CrossRef]
- American Thoracic Society. Idiopathic Pulmonary Fibrosis: Diagnosis and Treatment Guidelines.
- Saha P, Talwar P. Idiopathic pulmonary fibrosis (IPF): disease pathophysiology, targets, and potential therapeutic interventions. Mol Cell Biochem. 2023 Sep 14. [CrossRef]
- King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011 Dec 3;378(9807):1949-61. [CrossRef] [PubMed]
- Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, Cottin V, Flaherty KR, Hansell DM, Inoue Y, Kim DS, Kolb M, Nicholson AG, Noble PW, Selman M, Taniguchi H, Brun M, Le Maulf F, Girard M, Stowasser S, Schlenker-Herceg R, Disse B, Collard HR; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014 May 29;370(22):2071-82. doi: 10.1056/NEJMoa1402584. Epub 2014 May 18. Erratum in: N Engl J Med. 2015 Aug 20;373(8):782. doi: 10.1056/NEJMx150012. PMID: 24836310.
- Sriram N., Kalayarasan S., Sudhandiran G. Epigallocatechin-3-gallate exhibits anti-fibrotic effect by attenuating bleomycin-induced glycoconjugates, lysosomal hydrolases and ultrastructural changes in rat model pulmonary fibrosis. Chemico-Biol. Interact. 2009;180:271–280. [CrossRef]
- Wei Y, Kim TJ, Peng DH, Duan D, Gibbons DL, Yamauchi M, Jackson JR, Le Saux CJ, Calhoun C, Peters J, Derynck R, Backes BJ, Chapman HA. Fibroblast-specific inhibition of TGF-β1 signaling attenuates lung and tumor fibrosis. J Clin Invest. 2017 Oct 2;127(10):3675-3688. [CrossRef]
- Wei Y, Dong W, Jackson J, Ho TC, Le Saux CJ, Brumwell A, Li X, Klesney-Tait J, Cohen ML, Wolters PJ, Chapman HA. Blockng LOXL2 and TGFβ1 signalling induces collagen I turnover in precision-cut lung slices derived from patients with idiopathic pulmonary fibrosis. Thorax. 2021 Jul;76(7):729-732. [CrossRef]
- Estornut C, Milara J, Bayarri MA, Belhadj N, Cortijo J. Targeting Oxidative Stress as a Therapeutic Approach for Idiopathic Pulmonary Fibrosis. Front Pharmacol. 2022 Jan 21;12:794997. [CrossRef]
- Chapman HA, Wei Y, Montas G, Leong D, Golden JA, Trinh BN, Wolters PJ, Le Saux CJ, Jones KD, Hills NK, Foster E, Oldham JM, Linderholm AL, Kotak P, Decaris M, Turner S, Song JW. Reversal of TGFβ1-Driven Profibrotic State in Patients with Pulmonary Fibrosis. N Engl J Med. 2020 Mar 12;382(11):1068-1070. [CrossRef]
- Cai ZY, Li XM, Liang JP, Xiang LP, Wang KR, Shi YL, Yang R, Shi M, Ye JH, Lu JL, Zheng XQ, Liang YR. Bioavailability of Tea Catechins and Its Improvement. Molecules. 2018 Sep 13;23(9):2346. [CrossRef]
- Ramachandran B, Jayavelu S, Murhekar K, Rajkumar T. Repeated dose studies with pure Epigallocatechin-3-gallate demonstrated dose and route dependant hepatotoxicity with associated dyslipidemia. Toxicol Rep. 2016 Mar 5;3:336-345. [CrossRef]
- Araldi GL, Hwang YW. Development of Novel Fluorinated Polyphenols as Selective Inhibitors of DYRK1A/B Kinase for Treatment of Neuroinflammatory Diseases including Parkinson’s Disease. Pharmaceuticals (Basel). 2023 Mar 15;16(3):443. [CrossRef]
- Deboever E, Fistrovich A, Hulme C, Dunckley T. The Omnipresence of DYRK1A in Human Diseases. Int J Mol Sci. 2022 Aug 19;23(16):9355. [CrossRef]
- Li YL, Zhang MM, Wu LW, Liu YH, Zhang ZY, Zeng LH, Lin NM, Zhang C. DYRK1A reinforces epithelial-mesenchymal transition and metastasis of hepatocellular carcinoma via cooperatively activating STAT3 and SMAD. J Biomed Sci. 2022 Jun 2;29(1):34. [CrossRef]
- Warfel NA, Kraft AS. PIM kinase (and Akt) biology and signaling in tumors. Pharmacol Ther 151: 41–49, 2015. [CrossRef]
- Zhang XY, Zou Y, Liu YQ, Cao YM, Zhu JL, Zhang JH, Chen X, Zhang R, Li JB. Inhibition of PIM1 kinase attenuates bleomycin induced pulmonary fibrosis in mice by modulating the ZEB1/E-cadherin pathway in alveolar epithelial cells. Mol Immunol 125: 15–22, 2020. [CrossRef]
- Pham TX, Lee J, Guan J, Caporarello N, Meridew JA, Jones DL, Tan Q, Huang SK, Tschumperlin DJ, Ligresti G. Transcriptional analysis of lung fibroblasts identifies PIM1 signaling as a driver of aging-associated persistent fibrosis. JCI Insight. 2022 Mar 22;7(6):e153672. [CrossRef]
- Eerola SK, Santio NM, Rinne S, Kouvonen P, Corthals GL, Scaravilli M, Scala G, Serra A, Greco D, Ruusuvuori P, Latonen L, Rainio EM, Visakorpi T, Koskinen PJ. Phosphorylation of NFATC1 at PIM1 target sites is essential for its ability to promote prostate cancer cell migration and invasion. Cell Commun Signal. 2019 Nov 15;17(1):148. [CrossRef]
- Wei Y, Kim TJ, Peng DH, Duan D, Gibbons DL, Yamauchi M, Jackson JR, Le Saux CJ, Calhoun C, Peters J, Derynck R, Backes BJ, Chapman HA. Fibroblast-specific inhibition of TGF-β1 signaling attenuates lung and tumor fibrosis. J Clin Invest. 2017 Oct 2;127(10):3675-3688. [CrossRef]
- Liu Y, Lu F, Kang L, Wang Z, Wang Y. Pirfenidone attenuates bleomycin-induced pulmonary fibrosis in mice by regulating Nrf2/Bach1 equilibrium. BMC Pulm Med. 2017 Apr 18;17(1):63. [CrossRef]
- Pan L, Cheng Y, Yang W, Wu X, Zhu H, Hu M, Zhang Y, Zhang M. Nintedanib Ameliorates Bleomycin-Induced Pulmonary Fibrosis, Inflammation, Apoptosis, and Oxidative Stress by Modulating PI3K/Akt/mTOR Pathway in Mice. Inflammation. 2023 Aug;46(4):1531-1542. [CrossRef]
- Liu T, De Los Santos FG, Phan SH. The Bleomycin Model of Pulmonary Fibrosis. Methods Mol Biol. 2017;1627:27-42. [CrossRef]
- Kolb P, Upagupta C, Vierhout M, Ayaub E, Bellaye PS, Gauldie J, Shimbori C, Inman M, Ask K, Kolb MRJ. The importance of interventional timing in the bleomycin model of pulmonary fibrosis. Eur Respir J. 2020 Jun 11;55(6):1901105. [CrossRef]
- https://clinicaltrials.gov/study/NCT05195918?cond=Idiopathic%20Pulmonary%20Fibrosis&intr=EGCG&rank=3.
Table 2.
Best fluoro catechins pharmacokinetic parameters.
 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



