Submitted:
02 October 2024
Posted:
04 October 2024
You are already at the latest version
Abstract
BACKGROUND. Invasive aspergillosis (IA) is a deadly fungal lung infection. Antifungal resistance and treatment side effects are major concerns. Iron chelators are vital for IA management, but systemic use can cause side effects. We developed nanoparticles (NPs) to selectively deliver the iron chelator deferasirox (DFX) for IA treatment. METHODS DFX was encapsulated in poly(lacticco‐ glycolic acid) (PLGA) NPs using a single emulsion solvent evaporation method. NPs were characterized by light scattering and electron microscopy. DFX loading efficiency and release were assessed spectrophotometrically. Toxicity was evaluated using SRB, luciferase, and XTT assays. Therapeutic efficacy was tested in an IA mouse model, assessing fungal burden by qPCR and biodistribution via imaging.RESULTS DFX‐NPs had a size of ~50nm and a charge of ~‐30mV, with a loading efficiency of ~90%. Release kinetics showed DFX release via diffusion and bioerosion. The EC50 of DFX‐NPs was significantly lower (p<0.001) than the free drug, and they were significantly less toxic (p<0.0001) in mammalian cell cultures. In vivo, NPs treatment significantly reduced Af burden (p<0.05). CONCLUSION The designed DFX‐NPs effectively target and kill Af with minimal toxicity to mammalian cells. The significant in vivo therapeutic efficacy suggests these NPs could be a safe and effective treatment for IA.
Keywords:
Invasive aspergillosis
; Antifungal resistance
; Nanoparticles
; Deferasirox
; Poly(lactic-co-glycolic acid)
; Iron chelators
; Safe and effective treatment
1. Introduction
Aspergillus spp. can cause a range of respiratory diseases called “aspergillosis” that extend from hypersensitivity to invasive disease [1]. The immune system plays a crucial role in the risk of aspergillosis. Individuals with weakened immune systems are more susceptible to the disease [2]. However, the factors that determine severity and type of aspergillosis are poorly understood. Recent epidemiological studies estimate that aspergillosis affects about 15 million people annually. Over 2113000 people develop invasive aspergillosis (IA), with a crude annual mortality of 85.2% [3]. Despite the growing concern, fungal infections receive very little attention and resources, leading to research and development unmet need [4]. Recognizing the increasing fungal infection burden the World Health Organization (WHO) recently published the first ever list of priority fungal pathogens and ranked infectious fungi on critical, high, and medium priority groups based on severity of disease and treatment challenges. Aspergillus fumigatus (Af), a ubiquitous opportunistic airborne mold was ranked as one of the four critical priority fungi [4]. Aspergillosis treatment and management is challenged by limited access to quality diagnostics and therapeutics as well as emergence of antifungal resistance. Antifungal resistance has major implications for human health. It generally leads to prolonged therapy and hospital stays, and an increased need for expensive and often highly toxic second line antifungal medicines [5]. Agricultural use is the main cause for the rising rates of azole-resistant Aspergillus fumigatus infections. Azole-resistant strains of Aspergillus have been reported around the globe, but the true prevalence and rate of triazole resistance remain underestimated and partly unknown [6,7]. Furthermore, environmental changes contribute to Aspergillus global burden. There is an increase in (i) the bioavailability of Aspergillus conidia in the air due to increased growth of fungi as temperatures rise and (ii) the conidia dispersal due to storms. Additionally, several pollutants are also known to cause direct damage to the lung epithelia and the local microbiome that provides an innate barrier to infection, this damage creates a favorable micro-environment for Aspergillus infection [8,9]. Finally, pulmonary viral infections seem to be associated with aspergillosis. Viral invasive aspergillosis (VIA) has emerged in intensive critical care (ICU) settings following the recent influenza H1N1 and COVID-19 pandemics. VIA seems to differ from IA in terms of pathogenicity and clinical presentation [10,11]. Considering current challenges in IA treatment and the incidence increase of this fungal infection, there is an urgent need to develop novel therapeutic approaches to effectively treat IA.
Iron chelators are being explored as a potential therapeutic strategy for treating Af infections, including invasive aspergillosis. Iron is essential for the growth and virulence of Aspergillus fumigatus [12]. Iron chelators work by binding to iron, making it unavailable to the fungus, thereby inhibiting its growth and biofilm formation [13]. Studies have shown that common iron chelators like deferiprone (DFP), deferasirox (DFX), can inhibit biofilm formation in a dose-dependent manner [13].
In clinical settings the use of iron chelators requires careful consideration of the specific chelator (Deferoxamine is bioavailable to Aspergillus species since the fungus can use it as siderophore) and the susceptibility of the Aspergillus strain. Individual assessments are often necessary to determine the most effective treatment [13]. The ongoing research in the therapeutic potential of iron chelators is focused on understanding the complex interactions between iron metabolism and fungal virulence, as well as developing new iron chelators that are more effective and have fewer side effects [14]. In addition, some studies have explored the use of iron chelators in combination with traditional antifungal drugs (azoles and polyene antibiotics). For example, combining DFP with antifungal agents has shown synergistic effects, improving treatment outcomes [15]. Preliminary results suggest that iron chelation therapy could be a viable adjunct to conventional antifungal treatments. Clinical trials aim to determine the optimal dosing, safety, and efficacy of iron chelators in patients with invasive aspergillosis. While there is a Phase2 clinical trial using radiolabeled deferoxamine for the visualization of pulmonary Aspergillus infection [16].
DFX is an FDA approved iron chelator for the treatment of chronic iron overload due to blood transfusions in patients aged 2 years and older, and for chronic iron overload in non-transfusion-dependent thalassemia syndromes in patients aged 10 years and older1. Systemic administration of DFX can lead to several adverse events (AEs), some of which can be serious. Common AEs include skin reactions and gastrointestinal issues (nauseas, vomiting, diarrhea, stomach pain) while severe AEs include renal failure, liver failure, often fatal gastrointestinal hemorrhage, severe allergic reactions and vision and hearing problems. IA usually impacts immunocompromised individuals [17]. While there is no FDA guidance on the use of DFX in immunocompromised patients, given their status, it is likely that the AEs of DFX will be pronounced in these individuals. Thus, targeted delivery of DFX to the site of Af infection would be a better therapeutic strategy. DFX is a good candidate to treat IA since it has DFX has a high affinity for ferric iron (Fe³⁺), which is essential for the growth and metabolism of Af, the primary pathogen in IA [18,19]. Furthermore, its safety profile is well-documented, thus it is a viable option for repurposing in the treatment of IA. By binding to ferric iron, DFX deprives the fungus of this critical nutrient, inhibiting its growth and proliferation.
It is well known that drug nanodelivery offers several advantages over traditional systemic administration. There is a plethora of FDA approved NPs (doxil, Abraxane, onivyde, ambisome, vyxeos, marqibo) that are now preferred treatments over the conventional drugs. NPs can improve the solubility and stability of drugs enhancing their absorption and effectiveness [20]. Additionally, NPs can be designed to release their payload in a controlled manner over time. This sustained release can maintain therapeutic drug levels for extended periods, reducing the frequency of dosing and improving patient compliance [21]. By concentrating the drug at the target site and reducing systemic exposure, nanodelivery systems can lower the risk of adverse effects and toxicity associated with high systemic drug levels [20,22]. Encapsulation within nanoparticles can protect drugs from degradation by enzymes or other biological processes, extending their shelf life and effectiveness23. Furthermore, the behavior of NPs in the respiratory system is influenced by their size, shape, and surface properties. NPs with size of 50nm, can penetrate deep into the lungs and may accumulate there24; thus, targeted delivery in the lungs can be achieved by NPs size manipulation.
The objective of this study is to develop NPs for the targeted delivery of DFX in the lungs to treat IA. DFX loaded into PLGA NPs represents a novel therapeutic approach for IA due to several key advantages (i) PLGA nanoparticles can be engineered to target the lungs, enhancing the accumulation of DFX at the infection site. This targeted delivery helps in maximizing the therapeutic effect while minimizing systemic side effects; (ii) PLGA NPs provide a controlled and sustained release of DFX, ensuring a steady therapeutic concentration over an extended period. This reduces the need for frequent dosing and helps maintain effective drug levels, (iii) Encapsulating DFX in PLGA nanoparticles protects the drug from degradation and enhances its stability, improving its shelf life and efficacy; (iv) PLGA safely degrades into lactic acid and glycolic acid, which are naturally metabolized by the body (iv) PLGA NPs can improve the pharmacokinetic profile of DFX, enhancing its absorption, distribution, and retention in the body. These attributes make DFX PLGA NPs a promising strategy for treating IA, potentially improving patient outcomes and reducing the burden of this serious fungal infection.
2. Materials and Methods
2.1. Nanoparticle Formulation
For formulation of PLGA NPs, we employed a single emulsion-solvent evaporation technique. Briefly, DFX (10 mg/mL) was dissolved in DMSO. 100mg of PLGA and were dissolved in oil phase 10ml ethyl acetate. Then DFX was added to the oil phase. 200mg of Pluronic F68 was dissolved in 20ml of double distilled water. The oil phase was added dropwise to the water phase while stirring. The solution was sonicated on ice at 60% power for 1 minute with 10 second break (4 cycles) using a probe sonicator (VCX 130; Sonics and Materials, Inc, Newtown, CT, USA). Then, the oil phase was eliminated by evaporation under reduced pressure (150 mbar) using a rotary evaporator (30 rpm) (Hei-VAP Advantage ML/G5B; Heidolph North America, Grove Village, IL, USA). Drug-free PLGA NPs were formulated using the same procedure.
2.2. Particle Size and Zeta Potential Analyses
To determine the size and zeta potential of NPs, 20 μL of each sample was diluted to 1 mL of distilled water and the size and zeta potential of the NPs were analyzed using a Zetasizer Nano-ZS (Malvern Instruments, Malvern, UK).
2.3. Transmission Electron Microscopy (TEM) & Scanning Electron Microscopy (SEM)
For morphological characterization, the formulated NPs were evaluated by TEM. A 10 μL of NPs sample was suspended 1 mL distilled water. One drop of this suspension was placed over a carbon 400 mesh TEM grid and allowed to dry. Images were visualized at 120 KeV at indicated magnifications on a Tecnai 12 microscope (FEI, Hillsboro, OR, USA) equipped with a Gatan, Inc (Pleasanton, CA, USA) 896 2.2.1 US1000 camera.
2.4. Drug Entrapment (DE%) and Drug Loading (DL%) Efficiencies
NPs content of DFX was estimated by ultraviolet (UV) spectrophotometric method. Briefly, SHK-loaded NPs were dissolved in acetonitrile (1 mg/mL) and kept on a shaker at 37°C for 72 hours until the complete release of the entrapped drug. Then, samples were removed and centrifuged at 10,000× g for 5 minutes at 4°C to extract the drug present in the solution. The supernatant (700 μL) was collected and 125µl were added to 175µl imaging solution (Gibb’s reagent and Borate Buffer pH 9.5). The absorbance of the supernatant was measured by a plate reader (BioTek Cytation 5, Agilent, CA, USA) at a wavelength of 658nm. Drug concentration was calculated based on DFX standard curve (range from 0mg to 10mg). We used the following equations to calculate drug entrapment and loading efficiencies:
DL% = ((Dt mass − Df mass)/NP mass)×100
DE% = ((Dt mass−Df mass)/Dt mass)×100
Df: free drug; Dt: total drug.
2.5. Release Kinetics Analysis
In vitro release kinetics of DFX from PLGA NPs was determined using FBS (fetal bovine serum) at various pHs (pH 4.4, 5.4, 6.4, and 7.4) at 37°C. The NPs (25 mg) were dispersed in 5 mL of the FBS buffer and divided into equal aliquots (1 mL each). These tubes were kept on a shaker at 37°C and 150 rpm. At designated time intervals (1, 2, 4, 8, 12, 24, 48, and 72 hours), these tubes were taken from the shaker and centrifuged at 10,000× g at 4°C for 5 minutes. Then, the supernatant was removed to estimate the amount of released drug was estimated as above using an imaging reagent and absorbance at 658nm. The release data were analyzed using the Korsmeyer Peppas model (Mt/M∞=ktn, Mt=drug release at time t, M∞= total amount of drug, k= release rate constant, n=release exponent).
2.6. Mammalian Cell Culture
Cell lines H292 and Raw264.7 were cultured onto well-plates using RPMI 1640 and DMEM media supplemented with 10% fetal bovine serum, 100 units/mL penicillin G, and 100 μg/mL streptomycin. Cells were kept in a humidified incubator with 5% CO2 at 37°C during cultivation and experiments.
2.7. Fungal Culture
Aspergillus fumigatus Af293 strain was cultured in potato dextrose agar petri dishes for 7 days prior to harvesting conidia for experimental use. The Af293 strain was purchased by ATCC.
2.8. Cytotoxicity Evaluation
2.8.1. Mammalian CELLS
The cytotoxicity of free DFX, empty NPs and DFX NPs was evaluated in mammalian cell lines by the well-established SRB assay using a commercially available kit (CytoScanTM SRB Cell Cytotoxicity Assay, G Biosciences, USA). Briefly, cells were seeded in 96-well plates (50.000 cells/well). After 24 hours, the cells were treated with designated concentrations (0mM-control, 2mM, 4mM, 8mM and 16mM) of DFX or equivalent of empty and DFX NPs for 24 and 48 hours. At the end of incubation, the treated cells were fixed using 10% trichloroacetic acid for 60 minutes at 4°C. After washing four times with distilled water, the plates were left to air-dry and then stained with SRB staining solution (0.057% w/v in 1% v/v acetic acid solution) for 30 minutes at room temperature. The cells were washed four times with 1% acetic acid solution. After drying, the cells were solubilized with 200 μL of 10 mM Tris base solution (pH 10.5) on a shaker for 30 minutes. The absorbance was measured using the BioTek Cytation 5 plate reader at 570 nm. The cell cytotoxicity was calculated using the following equation: % Cytotoxicity= (100 x (Cell Control – Experimental)) ÷ (Cell Control). The software GraphPad Prism 10 was used to determine the EC50.
2.8.2. Fungal Cultures
The inhibition of fungal proliferation was evaluated by XTT assay. We used a commercially available XTT assay kit (CyQUANTTM XTT Cell Viability Assay, Invitrogen, USA). Briefly, conidia were cultured at a seeding density of 106 conidia/well onto 96-well plates and treatments were added as described above. After 24h and 48h, 50 µl of XXT labeling mixture was added and cells were incubated for additional 4h at 5% CO2 and 37°C. The absorbance was measured using an BioTek Cytation 5 plate reader at 450nm and 660nm. Specific absorbance was calculated using the formula: Specific Absorbance = [Abs450nm(Sample) – Abs450nm(Blank)] – Abs660nm(Sample). Growth inhibition was calculated via the following equation: %growth inhibition = [1 – (AbsSample/AbsControl)] *100. The software GraphPad Prism 10 was used to determine the EC50.
2.9. In vivo Biodistribution Model
PLGA NPs loaded with indocyanine green dye (IGC) (Cardiogreen powder, Sigma-Aldrich, USA) were injected (100μl/injection) intravenously in female athymic nu/nu mice (strain NU/J #002019, 6-weeks old, The Jackson Laboratory) (n=5/group). Infrared detection images were taken before injection and immediately after injection, 20min, 1h, 2h, 4h, 6h, 12h, 24h, 48h and 72h after injection. 72h after injection no signal was detected, mice were sacrificed, organs were harvested (Brain, Heart, Lungs, Thyroid, Kidneys, Spleen, Liver, Fallopian tubes, Ovaries) and infrared detection images of the organs were taken taken using the Li-COR Pearl Impulse instrumentation. ImageJ was used to quantify the signal of each organ in the images taken.
2.10. Neutropenic Mouse Model of IA
Both male and female 6 weeks old C57BL/6 mice were used (strain C57BL/6J #000664, The Jackson Laboratory) (n=5/group). Neutropenia in mice was established via cyclosphosphamide solution IP injections (150mg/kg) two days before injections and three days after infection. On day 0 mice were infected via inhalation of Af293 conidia. Briefly the mice were placed in an inhalation chamber and 108/ml conidia were nebulized for 30 minutes to ensure adequate exposure. Immediately after infection on day 0 and daily until day 7, mice were treated with PBS (control), DFX 20mg/kg (free drug), empty NPs (NP control) and DFX NPs 20mg/kg intravenously. On day 8 mice were sacrificed, and lungs were collected for further analyses. Mice were monitored daily for changes in weight and signs of distress. Supportive care including hydration and nutritional support was provided as needed.
2.11. qPCR to determine fungal burden
Infected mouse lungs collected were cut in small pieces using surgical scissors. PBS (1ml per 100mg tissue) was added to lung pieces and tissue was homogenized using a dounce homogenizer followed by a rotor stator homogenizer (Benchmark Scientific D1000). Lung homogenates were centrifuged to remove debris. Supernatant was collected. A DNA extraction kit (Qiagen DNeasy Blood & Tissue Kit, Qiagen, USA) and followed the manufacturer’s instructions to extract DNA from the sample. Alpco Ltd GeneProof Aspergillus PCR kit (Alpco, Ltd) was used according to manufacturer’s instructions for Aspergillus detection. The fungal burden was normalized to the amount of host DNA using mouse GAPDH as a housekeeping gene.
2.12. Study Approval
All animal studies were approved by the IACUC and Albany Medical College.
2.13. Statistical Analysis
All statistical analyses were performed using GraphPad Prism 10 software. P value was set at p > 0.5 was for statistically significant differences.
3. Results
3.1. Nanoparticles Characterization
Based upon formulation methods and components, the size of the engineered PLGA NPs varied from 20 to 70 nm with average diameter of 50nm. The zeta potential of DFX NPs was −32.9 mV (Figure 1).
The TEM and SEM micrographs displayed that the DFX NPs had a spherical shape and smooth surface (Figure 2).
The DFX NPs loading, and entrapment (DL% and DE%) efficiency were 77.9% and 79.2% (Table 1).
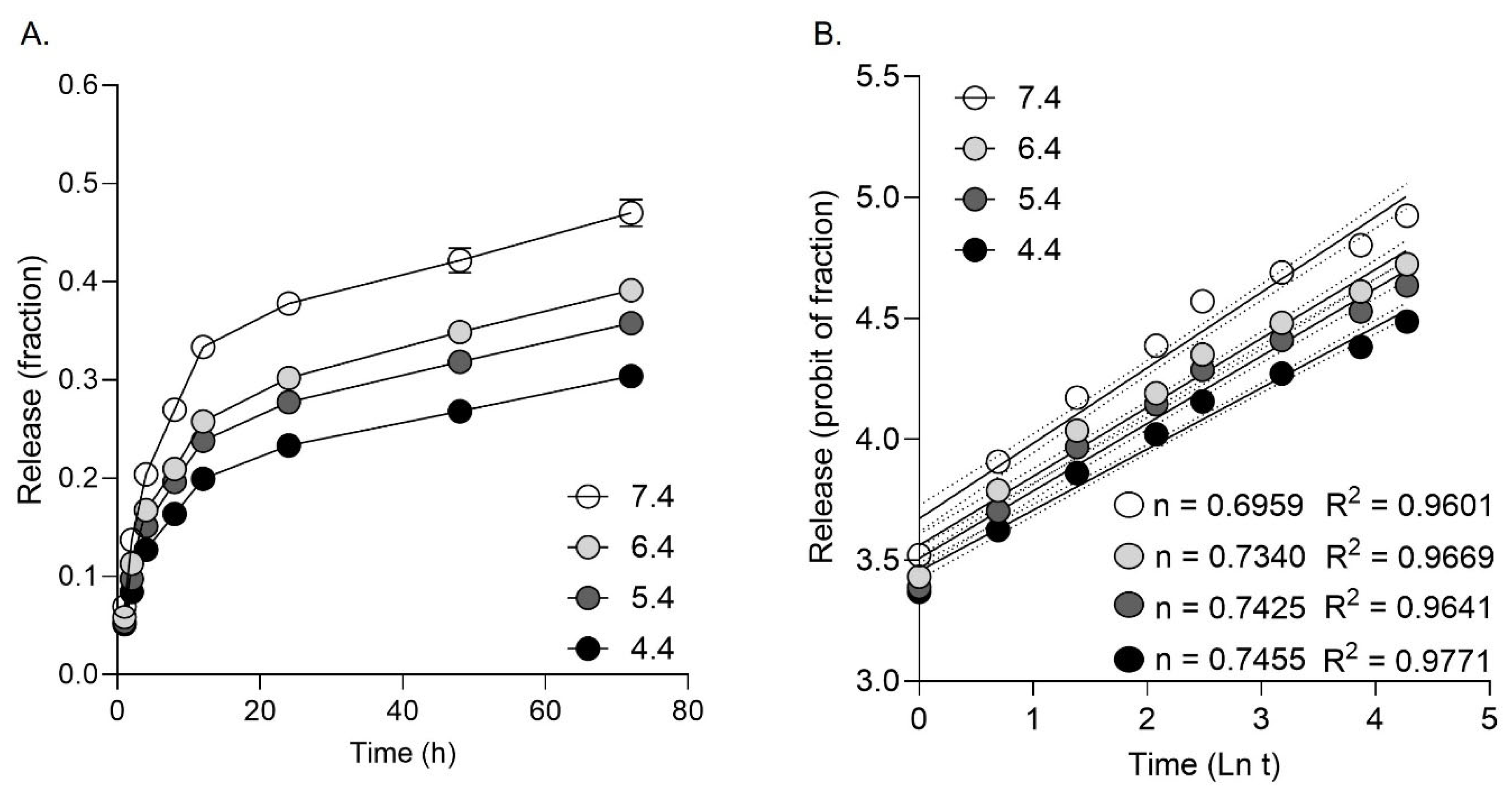
The pattern of drug release revealed that 20% of drug was released within the first 2 hours, while 80% of drug was released after 48 hours. The release of DFX from NPs appeared to be somewhat pH-dependent, with highest release at pH 7.4 (Figure 1A). The DFX release profile at pH range 4.4-7.4 fitted the Korsmeyer-Peppas model, with drug release at pH 4.4 best fitting the model (R2 = 0.9771). The release exponent (n) ranged from 0.6959 (pH 7.4) to 0.7455 (pH 4.4) suggesting drug release due a combination of diffusion and bioerosion (Figure 1B).
Figure 3.
The DFX release profile from PLGA NPs in FBS. A. DFX release over 72h in pH 4.4, 5.4, 6.4 and 7.4. B. Release data fit with the Korsmeyer-Peppas drug release kinetic model.
Figure 3.
The DFX release profile from PLGA NPs in FBS. A. DFX release over 72h in pH 4.4, 5.4, 6.4 and 7.4. B. Release data fit with the Korsmeyer-Peppas drug release kinetic model.
We tested the effect of the DFX NPs treatment in cell viability in lung epithelial cell line H292 and the macrophage cell line Raw264.7 cells, since the bronchi are primary consistent from epithelial cells and macrophages. In the Raw264.7 cells free DFX significant reduced cell viability (p <0.0001) compared to DFX NPs. Suggesting that DFX nanodelivery has a prophylactic effect in this cell population (Figure 4A). In the H292 cells free DFX did not reduce cell viability. However, DFX NPs were significantly less toxic (p = 0.02) compared to free DFX suggesting that the NPs had a prophylactic effect (Figure 4B). Finally, both DFX and DFX NPs treatments were toxic to the Af293 cultures compared to control and control NPs. However, DFX NPs were significantly (p<0.0001) more toxic to the Af293 culture compared to free DFX, suggesting effective drug delivery to the fungus (Figure 4C). Finally, empty PLGA NPs had no effect in the cell or the Af293 cultures.
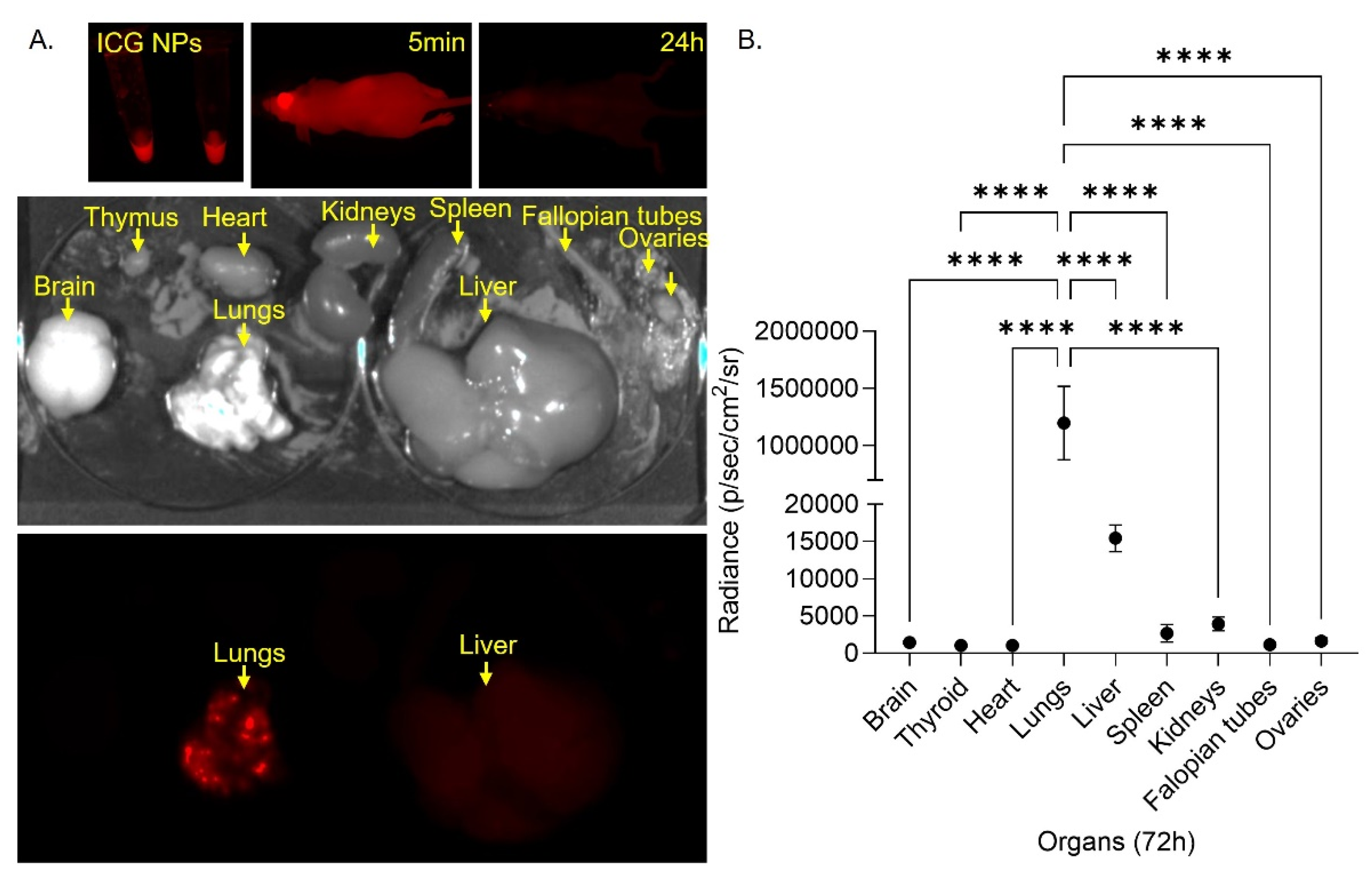
We evaluated the biodistribution of these NPs in vivo by formulating indocyanine green loaded PLGA NPs and administering these NPs intravenously (IV) to female nude mice. We monitored biodistribution by infrared imaging over 72h. At 24h no signal was detected in the mouse. Mice were sacrificed at 72h, and organs were harvested and imaged for infrared signal (Figure 6A). We identified very strong and significantly (p <0.0001) higher ICG signal in the lungs compared to other organs. Some ICG signal was also detected in the liver (Figure 6B). These results imply that NPs are delivered and sustained in the lungs, suggesting targeted delivery of the NPs in the lungs, where IA manifests.
Figure 5.
Biodistribution study in nude mice. A. Infrared images of the NPs formulation, the mice after 5min and 24h and the harvested organs (brain, thymus, heart, lung, kidneys, spleen, liver, cervix and ovaries) 72h after treatment PLGA NPs loaded with Indocyanine green (ICG) 0.5nmol. B. Quantification of infrared signal from each organ after 72h (**** p <0.0001).
Figure 5.
Biodistribution study in nude mice. A. Infrared images of the NPs formulation, the mice after 5min and 24h and the harvested organs (brain, thymus, heart, lung, kidneys, spleen, liver, cervix and ovaries) 72h after treatment PLGA NPs loaded with Indocyanine green (ICG) 0.5nmol. B. Quantification of infrared signal from each organ after 72h (**** p <0.0001).
Finally, we tested the efficacy of the DFX NPs in a mouse model of IA (Figure 7A). We used PBS and empty PLGA NPs as controls and compared the therapeutic efficacy of the free DFX compared to DFX NPs after 7 days treatment post Af293 infection. We found that the DFX NPs significantly (p = 0.0001) reduced the Af293 burden in the lungs compared to free DFX. Furthermore, there was no significance in the burden between the uninfected mice and the infected DFX NPs treated mice, indicating that fungus was almost cleared from the lungs after the DFX NPs treatment (Figure 7B). Additionally, we monitored the animals’ weight throughout the treatments as an indirect method to evaluate potential toxicity. The weight monitoring data demonstrate that the DFX NPs had no impact in the weight of the mice. However, the mice treated with PBS, control NPs and DFX showed weight loss (Figure 7C). These results imply that the DFX NPs were not toxic.
4. Discussion
We formulated PLGA NPs loaded with DFX with size ~50nm and surface charge of ~ -30mV. Aspergillus cell wall consists of negatively charged components such as glucans, chitin, and glycoproteins [25,26]. These components contribute to the overall negative charge of the cell wall, which can influence interactions. Mammalian cell membrane is also negatively charged. If the NPs were positively charged, they would likely have electrostatic interactions with both mammalian cells and fungal hyphae, thus negative charge was preferred. The DFX loading efficiency was high ~80%. High loading efficiency means that more DFX can be delivered to the target site, potentially increasing its therapeutic effect. Furthermore, with more drug loaded into each nanoparticle, the frequency of dosing can be reduced, increasing the efficiency of use. Additionally, by delivering a higher concentration of the drug directly to the target site, systemic side effects can be minimized. PLGA degrades through hydrolysis of its ester bonds, which is influenced by the pH of the surrounding environment. In acidic conditions, the hydrolysis rate increases, leading to faster degradation of the polymer and, consequently, quicker drug release. In neutral or basic conditions, the degradation is slower, resulting in a more sustained release. However, DFX has poor water solubility and is highly sensitive to pH changes. In acidic conditions, such as in gastric fluid, DFX is practically insoluble. This explains why most of the DFX was released at pH 7.4. The release exponent (n) at the Korsmeyer-Peppas model of drug release can provide information about how the DFX is being released from the NPs. n =0.5 indicates that the drug is being released due to passive diffusion. 0.5 < n < 1 indicates that the drug is being released due to a combination of diffusion and bioerosion, while n = 1 indicates controlled release due to bioerosion. Since bioerosion happens in acidic environments and DFX is insoluble in acidic conditions we aimed for a drug release that would combine drug diffusion and bioerosion. Furthermore, 20% of the DFX was released in serum within the first 2 hours. The time it takes for NPs to reach the lungs after intravenous (IV) administration can vary based on several factors, including the size, surface properties, and composition of the nanoparticles. Generally, nanoparticles can reach the lungs within minutes after IV administration. Thus, we anticipate this initial passive DFX diffusion to have an impact in the lungs as well as a systemic effect. The bronchi, where aspergillosis is established, are comprised of epithelial cells and macrophages. Thus, we decided to evaluate the toxicity of the DFX NPs in epithelial cells and macrophages. NPs with size of 50nm are expected to be phagocytosed more easily compared to NPs of bigger size. However, NPs with a negative surface charge, such as -30mV, tend to be less readily phagocytosed. This explains the DFX NPs prophylactic effect observed in mammalian cells compared to free DFX. While it was not within the scope of this work to investigate internalization of the NPs by Aspergillus hyphae, we observed that DFX nanodelivery had higher toxicity to the Af293 culture compared to free DFX, implying that the DFX NPs can interact with the fungal hyphae and deliver DFX. However, additional research is needed to define these interactions and confirm the NPs internalization. Furthermore, DFX NPs at concentration 8mM were completely toxic to the fungus while had no toxicity to mammalian cells, indicating that (i) nanodelivery of DFX is more effective compared to free DFX (ii) via nanodelivery we can use lower concentrations of DFX and still achieve high fungal toxicity. Our biodistribution study demonstrated that PLGA NPs with size ~50nm can accumulate and be retained in the lungs after 72h. This targeted delivery is essential since it can enhance the efficacy of the drug in the site of infection while reduce toxicity to other tissues. On the other hand, some ICG signal was also observed in the liver, the NPs retained in the liver might induce liver dysfunction and toxicity. While our data indicate targeted delivery additional studies are needed to confirm drug clearance and toxicity. Finally, we observed significant therapeutic efficacy of the DFX NPs in the mouse model of IA. The DFX NPs seem to clear the fungal infection from the lungs as a monotherapy. We anticipate that if these NPs are used in combination with known antifungals to increase their therapeutic efficacy. Furthermore, for the scope of this work we have not tested these NPs in Af azole resistant strains. It is important to investigate if these NPs have toxic effect to azole resistant strains and if the DFX-NPs can re-sensitize these stains to azoles.
In conclusion, our study demonstrates that DFX-loaded nanoparticles (NPs) offer a promising approach for treating invasive aspergillosis (IA) through a combination of drug diffusion and bioerosion. The initial rapid release of DFX in serum, coupled with the targeted delivery and retention of NPs in the lungs, suggests a dual impact on both local and systemic levels. The observed prophylactic effect of DFX NPs in mammalian cells, along with their higher toxicity to Aspergillus hyphae compared to free DFX, highlights the potential of nanodelivery to enhance therapeutic efficacy while minimizing toxicity. Our findings indicate that DFX NPs can effectively clear fungal infections in a mouse model of IA, and further research is warranted to explore their impact on azole-resistant strains and potential synergistic effects with existing antifungals. Additionally, while some liver accumulation was noted, further studies are needed to confirm drug clearance and assess long-term toxicity. Overall, DFX NPs represent a significant advancement in targeted antifungal therapy, offering a more effective and safer alternative to conventional treatments.
Acknowledgments
We would like to thank Dr. Nathaniel Cady, Dr. Natalya Tokranova for their assistance with the electron microscopy images.
References
- Takazono, T. & Sheppard, D. C. Aspergillus in chronic lung disease: Modeling what goes on in the airways. in Medical Mycology vol. 55 39–47 (Oxford University Press, 2017).
- Cadena, J., Thompson, G. R. & Patterson, T. F. Aspergillosis: Epidemiology, Diagnosis, and Treatment. Infectious Disease Clinics of North America vol. 35 415–434 Preprint at (2021). [CrossRef]
- Denning, D. W. Global incidence and mortality of severe fungal disease. The Lancet Infectious Diseases vol. 24 e428–e438 Preprint at (2024). [CrossRef]
- 9789240060241-eng.
- Fisher, M. C. et al. Tackling the emerging threat of antifungal resistance to human health. Nature Reviews Microbiology vol. 20 557–571 Preprint at (2022). [CrossRef]
- Chowdhary, A., Sharma, C. & Meis, J. F. Azole-resistant aspergillosis: Epidemiology, molecular mechanisms, and treatment. Journal of Infectious Diseases 216, S436–S444 (2017).
- Burks, C., Darby, A., Londoño, L. G., Momany, M. & Brewer, M. T. Azole-resistant Aspergillus fumigatus in the environment: Identifying key reservoirs and hotspots of antifungal resistance. PLoS Pathogens vol. 17 Preprint at (2021). [CrossRef]
- Hay, J. et al. Assessing Thermal Adaptation of a Global Sample of Aspergillus Fumigatus: Implications for Climate Change Effects.
- Van Rhijn, N. & Bromley, M. The consequences of our changing environment on life threatening and debilitating fungal diseases in humans. Journal of Fungi 7, (2021).
- Albrich, W. C. & Lamoth, F. Viral-associated Pulmonary Aspergillosis: Have We Finally Overcome the Debate of Colonization versus Infection? American Journal of Respiratory and Critical Care Medicine vol. 208 230–231 Preprint at (2023). [CrossRef]
- Marr, K. A. et al. Aspergillosis complicating severe coronavirus disease. Emerg Infect Dis 27, 18–25 (2021). [CrossRef]
- Matthaiou, E. I., Sass, G., Stevens, D. A. & Hsu, J. L. Iron: An essential nutrient for Aspergillus fumigatus and a fulcrum for pathogenesis. Current Opinion in Infectious Diseases vol. 31 506–511 Preprint at (2018). [CrossRef]
- Nazik, H. et al. Effects of iron chelators on the formation and development of Aspergillus fumigatus biofilm. Antimicrob Agents Chemother 59, 6514–6520 (2015). [CrossRef]
- Haas, H. Iron - a key nexus in the virulence of Aspergillus fumigatus. Front Microbiol 3, (2012).
- Zarember, K. A., Cruz, A. R., Huang, C. Y. & Gallin, J. I. Antifungal activities of natural and synthetic iron chelators alone and in combination with azole and polyene antibiotics against Aspergillus fumigatus. Antimicrob Agents Chemother 53, 2654–2656 (2009). [CrossRef]
- iMagIng pulmonaRy Aspergillosis Using Gallium-68-dEferoxamine (MIRAGE).
- Fang, W. et al. Diagnosis of invasive fungal infections: challenges and recent developments. Journal of Biomedical Science vol. 30 Preprint at (2023). [CrossRef]
- Sun, J., Xiao, S. & Xue, C. The tug-of-war on iron between plant and pathogen. Phytopathology Research vol. 5 Preprint at (2023). [CrossRef]
- Miethke, M. Molecular strategies of microbial iron assimilation: From high-affinity complexes to cofactor assembly systems. Metallomics vol. 5 15–28 Preprint at (2013). [CrossRef]
- Kumar, A. et al. Recent Trends in Nanocarrier-Based Drug Delivery System for Prostate Cancer. AAPS PharmSciTech vol. 25 Preprint at (2024). [CrossRef]
- Li, S., Chen, L. & Fu, Y. Nanotechnology-based ocular drug delivery systems: recent advances and future prospects. Journal of Nanobiotechnology vol. 21 Preprint at (2023). [CrossRef]
- Maghsoudnia, N., Eftekhari, R. B., Sohi, A. N., Zamzami, A. & Dorkoosh, F. A. Application of nano-based systems for drug delivery and targeting: a review. Journal of Nanoparticle Research vol. 22 Preprint at (2020). [CrossRef]
- Patra, J. K. et al. Nano based drug delivery systems: Recent developments and future prospects. Journal of Nanobiotechnology vol. 16 Preprint at (2018). [CrossRef]
- Forest, V. & Pourchez, J. Human biological monitoring of nanoparticles, a new way to investigate potential causal links between exposure to nanoparticles and lung diseases? Pulmonology vol. 29 4–5 Preprint at (2023). [CrossRef]
- Gow, N. A. R., Latge, J.-P. & Munro, C. A. The Fungal Cell Wall: Structure, Biosynthesis, and Function. Microbiol Spectr 5, (2017).
- Beauvais, A., Fontaine, T., Aimanianda, V. & Latgé, J. P. Aspergillus cell wall and biofilm. Mycopathologia 178, 371–377 (2014). [CrossRef]
Figure 1.
Charge and size distribution of DFX NPs. A. Zeta potential of DFX loaded PLGA NPs. B. Size distribution of DFX loaded PLGA NPs.
Figure 1.
Charge and size distribution of DFX NPs. A. Zeta potential of DFX loaded PLGA NPs. B. Size distribution of DFX loaded PLGA NPs.
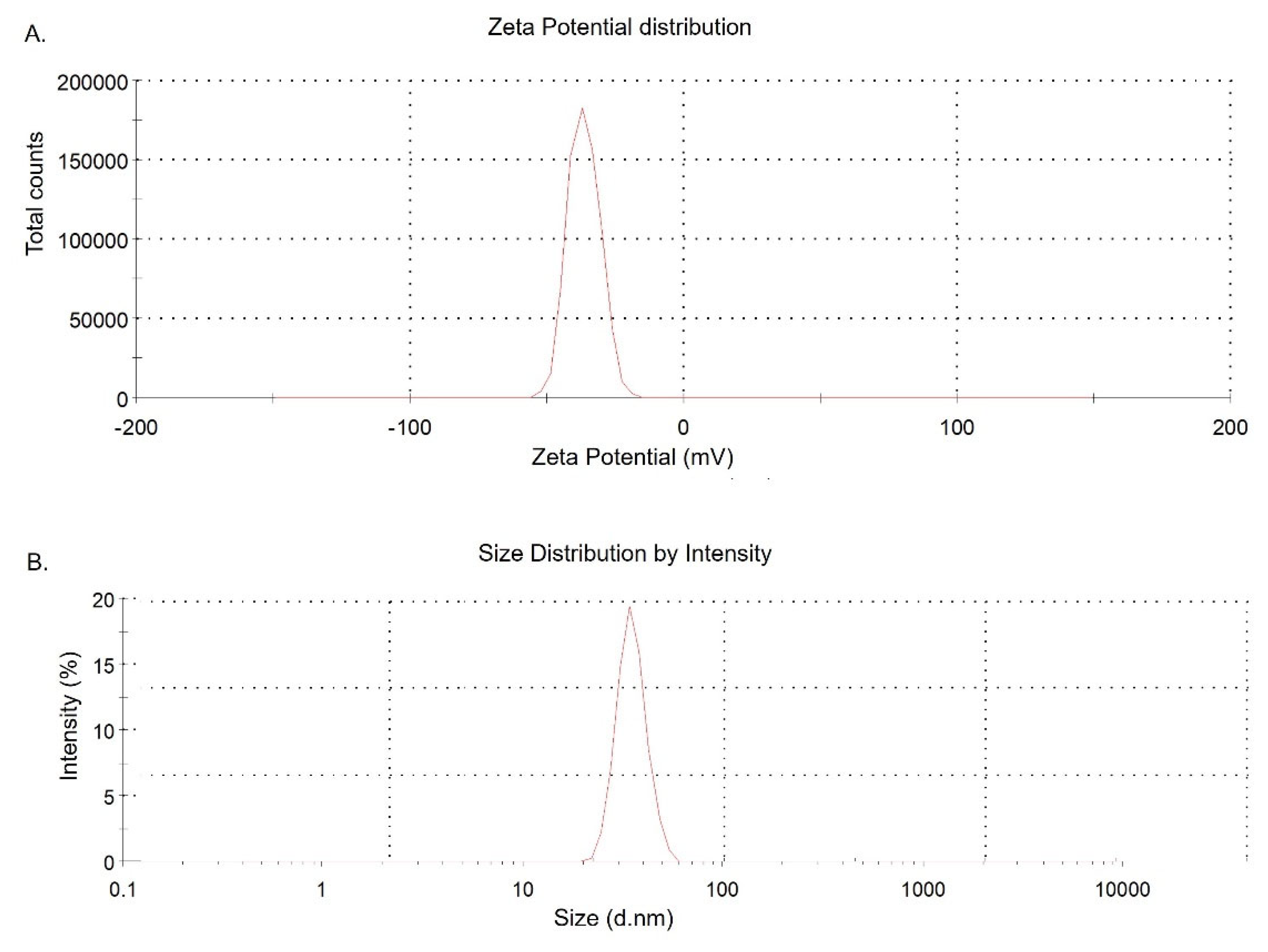
Figure 2.
TEM and SEM micrographs of the DFX-loaded PLGA NPs. A. TEM micrograph showing shape and size distribution. B. TEM micrograph showing absence of cracks in the NPs surface. C. SEM micrograph showing smooth spherical surface and size distribution.
Figure 2.
TEM and SEM micrographs of the DFX-loaded PLGA NPs. A. TEM micrograph showing shape and size distribution. B. TEM micrograph showing absence of cracks in the NPs surface. C. SEM micrograph showing smooth spherical surface and size distribution.

Figure 4.
Viability assays in mammalian cells and Af cultures. A. SRB assay data in Raw264.7 cells (**** p < 0.0001). B. SRB assay data in H292 cells (**** p <0.0001). B. XTT assay data in Af239 culture ( ** p = 0.0016, *** p = 0.0003, **** p <0.0001).
Figure 4.
Viability assays in mammalian cells and Af cultures. A. SRB assay data in Raw264.7 cells (**** p < 0.0001). B. SRB assay data in H292 cells (**** p <0.0001). B. XTT assay data in Af239 culture ( ** p = 0.0016, *** p = 0.0003, **** p <0.0001).
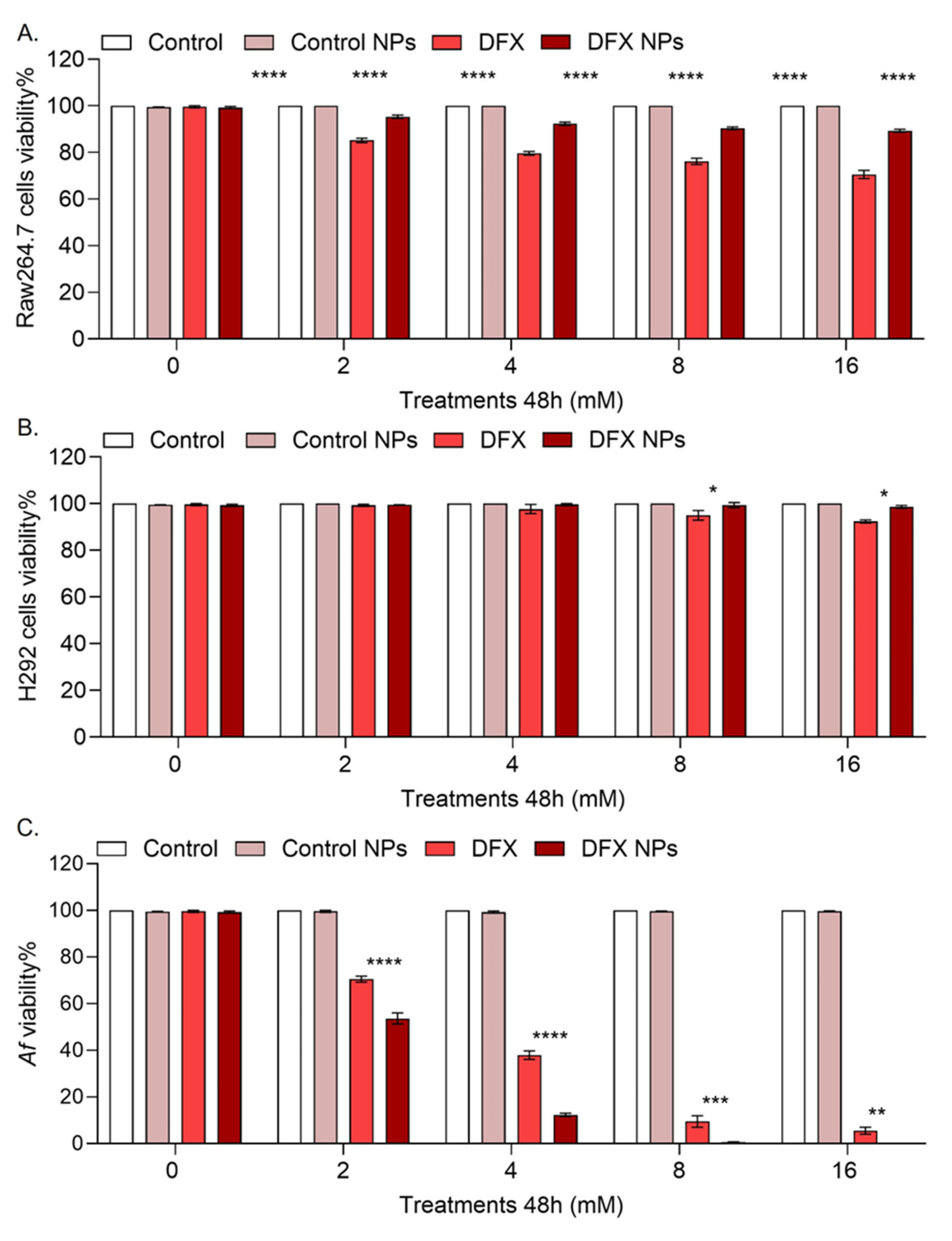
Figure 7.
In vivo efficacy of treatments in the neutropenic mouse model of IA. A. Schematic mouse model and treatments. B. Weight monitoring. C. Fungal CFUs estimated via 18S RNA qPCR (*** p = 0.0001, **** p < 0.0001).
Figure 7.
In vivo efficacy of treatments in the neutropenic mouse model of IA. A. Schematic mouse model and treatments. B. Weight monitoring. C. Fungal CFUs estimated via 18S RNA qPCR (*** p = 0.0001, **** p < 0.0001).
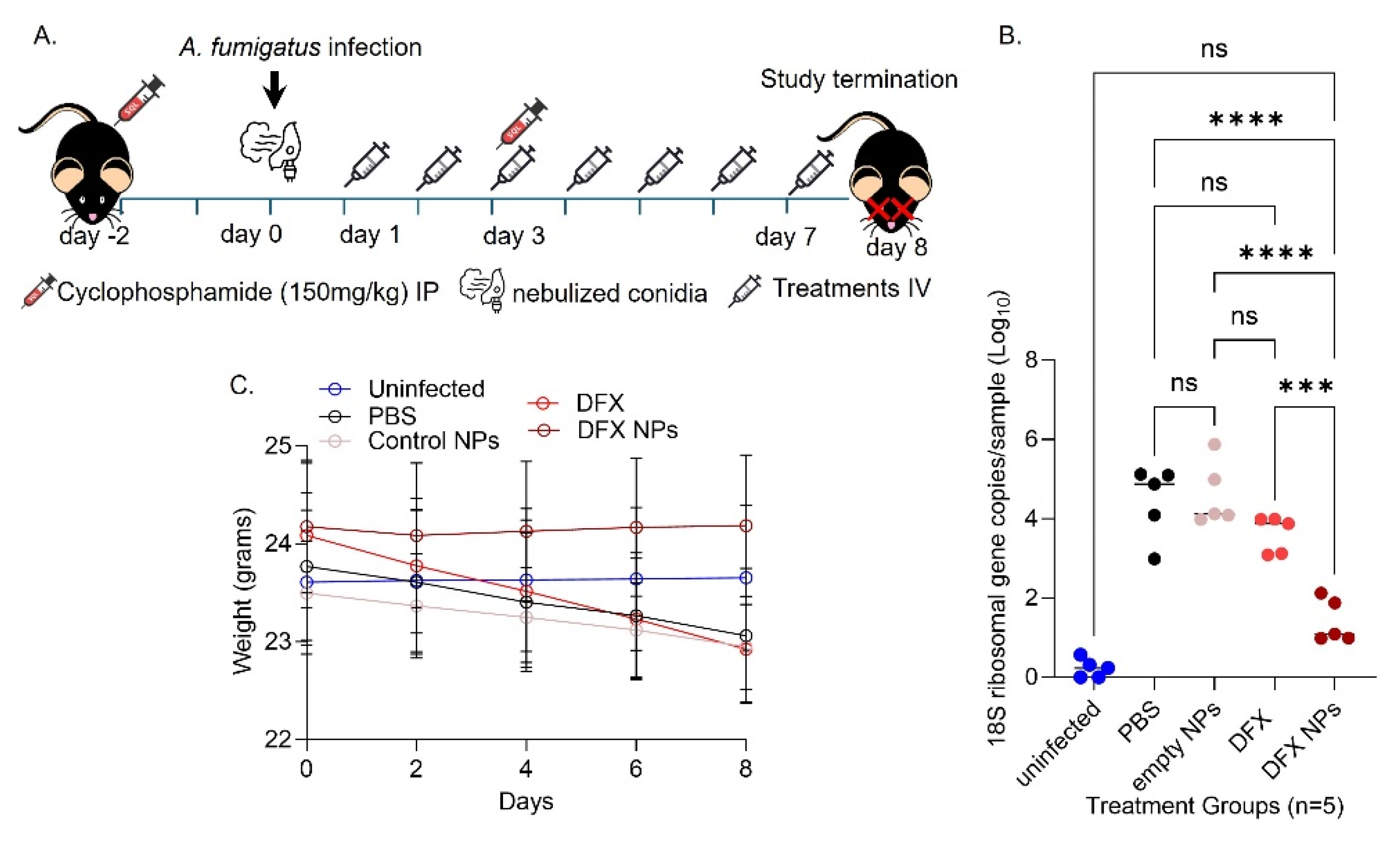

Table 1.
DL% and DE% of the DFX-loaded NPs.
 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



