
Submitted:
02 October 2024
Posted:
04 October 2024
Read the latest preprint version here
Abstract
Background/Objectives: Community-acquired methicillin-resistant Staphylococcus aureus (CA-MRSA) greatly complicates treatment of skin and soft tissue infections (SSTI). It was previ-ously found that subcutaneous (SQ) treatment with the mononuclear phagocyte (MP)-selective activator, complement peptide-derived immunostimulant-02 (CPDI-02; formerly EP67), increases prophylaxis of outbred CD-1 mice against SQ infection with CA-MRSA. Here, we determined if treatment with CPDI-02 also increases curative protection. Methods: Female CD-1 mice were challenged SQ with CA-MRSA USA300 LAC then CPDI-02 or inactive scCPDI-02 was administered by a topical, SQ, IM, or IV route at 6- or 24-hrs post-challenge. Dermal abscess sizes were compared over 10 days and CA-MRSA burden, neutrophils, MP, and pro-inflammatory cytokines were compared in dermal abscesses. CPDI-02 PK and distribution in female CD-1 mice were compared after IM or IV dosing and CPDI-02 toxicity was determined by IM dose escalation and repeat IM dosing in male and female CD-1 mice. Results: Repeat IM treatment with CPDI-02 starting at 6-hrs post-challenge decreased maximum abscess surface area, CA-MRSA burden, and time to resolu-tion, whereas repeat treatment by the topical, SQ, or IV route had no effect. Starting CPDI-02 treatment at 24-hrs post-challenge was ineffective regardless of route. Single IM treatment with CPDI-02 starting at 6-hrs post-challenge was as effective as repeat IM treatment, increased systemic exposure, and decreased initial levels of IL-1β and increased MP in dermal abscesses. CPDI-02 was tolerated to between 130-170 mg/kg for single IM dosing and 65-130 mg/kg for repeat IM dosing with males being more tolerant. Conclusions: Single early-stage IM treatment with CPDI-02 may increase curative protection against SSTI caused by CA-MRSA or other pathogens controlled by activated MP.
Keywords:
anti-microbial resistance
; AMR
; antibiotic resistance
; ABR
; drug-resistant bacteria
; antibi-otic-resistant bacteria
; multidrug resistance
; MDR
; host-directed therapy
; EP67
; complement peptide-derived immunostimulant
; administration route
; CPDI-02 toxicity
; CPDI-02 dosing
; IM dosing
1. Introduction
Skin and soft tissue infections (SSTI) of healthy skin (primary infection), damaged skin (secondary infection), and/or underlying fat, fascia, and muscle are the most common infections encountered in community or healthcare settings [1,2,3]. In the United States alone, 9.1 million cases of SSTI were diagnosed in 5.4 million patients from 2010 to 2020 with an average biennial recurrence of 26.3% and average annual treatment costs of $19.6 billion [4]. Over the same period, SSTI were diagnosed in 1,065,444 emergency room visits and required 264,856 hospitalizations [4]. Worldwide, the age-standardized incidence of SSTI was estimated to be ~294 million in 2019 [5]. Thus, SSTI are a tremendous global burden on healthcare infrastructure, resources, and costs.
Staphylococcus aureus (S. aureus) is a gram-positive bacterium commonly found on the skin and mucous membranes of healthy individuals [6,7,8] that is one of the most frequently isolated pathogens from monomicrobial or polymicrobial SSTI in the United States [1,6,9,10] and globally [10,11,12,13]. SSTI that involve S. aureus can be acute, chronic, recurrent, or persistent [14] and potentially lead to a wide range of minor to life-threatening diseases such as cellulitis, primary and secondary abscesses, infected ulcers and wounds, toxic shock syndrome, catheter-associated infections, necrotizing fasciitis, and bacteremia [6,15].
Although S. aureus is sensitive to most antibiotics [16], the ability to quickly develop antibiotic resistance (ABR) through genomic mutations or horizontal transfer of ABR genes from other bacteria and the widespread use of antibiotics [17] has led to the emergence of hospital-associated (HA) and community-associated (CA) strains of S. aureus with multidrug resistance (MDR) [6,11]. ABR strains of S. aureus were first reported in hospitalized patients shortly after the introduction of penicillin in 1942 [18] with >95% of isolates being penicillin-resistant today [17,19,20]. MDR strains of methicillin-resistant S. aureus (MRSA) were later reported in hospitalized patients (HA-MRSA) shortly after the introduction of methicillin in 1961 [20]. HA-MRSA strains then began to spread and evolve within community settings (CA-MRSA) in the 1990s and started correlating with a global increase in the incidence of SSTI in 2000 [6,19]. MRSA strains have since become resistant to other classes of antibiotics including lincosamides, aminoglycosides, glycopeptides, macrolides, quinolones, oxazolidinones, and streptogramins [19,20] with frequencies and patterns of MDR varying by region over time [10]. As such, given its global impact on public health, the US Centers for Disease Control (CDC) and the World Health Organization (WHO) have designated MRSA as a “serious threat” that is a “high priority” for increased development of novel antibiotics and therapeutic approaches to overcome MDR [13,21].
Complement Peptide-Derived Immunostimulant-02 (CPDI-02) (formerly EP67) is a novel, second-generation host-derived decapeptide agonist of C5a Receptor 1 (C5aR1 / C5a1R / CD88) that is based on the C-terminal pharmacophore of human complement peptide C5a (hC5a) [22,23,24]. Unlike hC5a (EC50 ~7 nM in primary human mononuclear phagocytes (MP) and neutrophils (NP) [25]), CPDI-02 minimizes potential NP-mediated toxicity by selectively activating MP (monocytes, macrophages, dendritic cells) with 103-fold greater potency than NP (EC50 167 nM in primary human MP vs. 160 μM in primary human NP) [25]. CPDI-02 is also immunostimulatory in mice [22,26,27,28,29,30,31,32], rabbits [33], and domestic pigs (unpublished). Given that activated MP (especially monocytes and macrophages) broadly protect against most pathogens that cause monomicrobial and polymicrobial SSTI including gram-positive bacteria, gram-negative bacteria, and fungi [34,35,36,37], treatment with CPDI-02 alone or in combination with antimicrobials (“adjunct immunotherapy”) may improve the treatment of SSTI caused by high priority ESKAPE bacterial pathogens like MRSA [38,39,40] and/or other pathogens regardless of MDR status.
It was previously found that repeat subcutaneous (SQ) treatment with CPDI-02 to outbred mice before and after SQ challenge with CA-MRSA USA300 (3 injections total) significantly decreased maximum dermal abscess surface area, CA-MRSA load per gram of tissue, and time to abscess resolution vs. inactive, scrambled scCPDI-02 [23]. Consistent with protection, SQ treatment with CPDI-02 increased local levels of IL-1β, IL-6, and CXCL1 and NP recruitment to the abscess [23]. In contrast, SQ treatment of C5aR1 knockout mice with CDPI-02 had no effect against SQ infection with CA-MRSA up to 48-hours post-challenge vs. inactive scCPDI-02 [23]. Thus, IM treatment with CPDI-02 increases protection against SQ infection with CA-MRSA in a prophylactic setting. It remains unclear, however, if treatment with CPDI-02 increases protection against SQ infection with CA-MRSA in a curative setting.
In this study, we determined (i.) if CPDI-02 increases curative protection against SQ infection with CA-MRSA by comparing the effect of administration route, treatment time post-challenge, and dose frequency of CPDI-02 vs. inactive scrambled scCPDI-02 on maximum dermal abscess surface area and time to resolution in female outbred CD-1 mice; (ii.) if single IM treatment with CPDI-02 or scCDPI-02 at 6-hours post-challenge affects levels of pro-inflammatory cytokines, neutrophils, and mononuclear phagocytes in dermal abscesses in female outbred CD-1 mice; (iii.) if IM administration affects systemic exposure and distribution of CPDI-02 by comparing PK profiles and distributions of CPDI-02 after IM or IV administration in female outbred CD-1 mice; and (iv.) preliminary toxicity of CPDI-02 after IM administration by IM dose escalation and repeat IM dosing in male and female CD-1 mice.
2. Materials and Methods
2.1. Large-Scale Synthesis of CPDI-02 and inactive, scrambled scCPDI-02
Machine-assisted solid phase peptide synthesis of CPDI-02 (Table 1) was performed on an AAPPTEC Apex 396 instrument employing an eight well reactor block. Assembly of CPDI-02 occurred on a 1 mmol scale on two aliquots of Fmoc-Arg(Pbf)-Wang resin (2 x 1.85 g, 0.27 mmol/g) in two separate wells in parallel. Prior to assembly, each aliquot of resin was vortexed in DMF (14 mL) to swell the resin. After 30 minutes, the resin was filtered, and vortexed with a 20% solution of piperidine in DMF, twice (14 mL, 1 x 3 mins, 1 x 9 mins) to deprotect the Fmoc-group. After washing with DMF (14 mL, 8 x 1 min), the resin was vortexed with a solution of Fmoc DAla-OH (0.62g, 2 mmol) and HATU (0.76 g, 2 mmol) in DMF (12 mL) for 30 seconds. 1 M DIEA in DMF (4 mL, 4 mmol) was added and the reaction mixture was vortexed for two hours. The resin was filtered and washed with DMF (14 mL, 5 x 1 min). This protocol was repeated using Fmoc-NMeLeu-OH, Fmoc-Pro-OH, Fmoc-Met-OH, Fmoc-Asp(tBu)-OH, Fmoc-Lys(tBoc)-OH, Fmoc-Phe-OH, Fmoc-Ser(tBu)-OH and Fmoc-Tyr(tBu)-OH to assemble CPDI-02. All coupling times were two hours except for the addition of Fmoc-Pro-OH to the resin-bound N-methyl-leucyl-residue, which required 16 hours for a negative chloranil test [41]. After deprotection of the final Fmoc- group, the resulting peptide-resin was washed with DMF (14 mL, 5 x 1 min), dichloromethane (14 mL, 5 x 1 min) and dried in vacuo. scCPDI-02 (Table 1) was assembled on a 0.5 mmol scale on Fmoc-Ser(tBu)-Wang resin using the same protocols with the exception that all couplings were complete within two hours.
For the cleavage of each peptide, a chilled cocktail of trifluoroacetic acid, phenol, water, and triisopropylsilane (88/5/5/2 v/v/v/v, 10 mL/g of peptide resin) was added with stirring to the peptide-resin at room temperature. After two and a half hours, the resin was filtered, and crude peptide in the filtrate was precipitated in chilled diethyl ether, filtered, and lyophilized.
Crude peptides were purified by preparative reversed-phase high performance liquid chromatography (RP-HPLC) on a Waters XSelect® Prep CSH130 C18 column (50 × 250 mm, 5 μm, 130 Å pore size, catalog number 186007029) eluted at a flow rate of 100 mL/min using a solvent gradient of 0.25 N triethylammonium phosphate (TEAP, pH 2.25, Solvent A) and a mixture of the TEAP buffer and acetonitrile (2/3, v/v. Solvent B). Crude CPDI-02 (1.05 g) was dissolved in a mixture of Solvent A and Solvent B (77/23, v/v) and loaded on to the column, which had been previously equilibrated with the same solvent mixture. The product was eluted from the column by raising the content of Solvent B in the eluent to 43% over 75 minutes, and column effluent was continuously monitored at 214 nm and collected in fractions. Fractions containing the major peak were pooled, diluted two-fold with water and loaded onto the same column now equilibrated with a mixture of 10 mM HCl (Solvent C) and acetonitrile (Solvent D) (95/5, v/v) at 100 mL/min. Raising the content of Solvent D in the eluent to 60% over 30 minutes eluted the product, which was collected in fractions. Lyophilization of pooled fractions containing ≥ 95% of the major product by RP-HPLC afforded the hydrochloride salt of CPDI-02 as a fluffy white powder (565 mg, yield 46%, [M+H]+ found 1241.652, theoretical 1241.635, Bruker autoflex maX). Crude scCPDI-02 was fractionated on the same column using a solvent gradient of 15% - 85% Solvent B over 75 minutes followed by desalting to the hydrochloride salt as described previously to yield the desired peptide as a fluffy white powder (232 mg, yield 37%, [M+H]+ found 1241.6315, theoretical 1241.6352, Thermo Q Exactive).
Peptide purity was determined by RP-UPLC using three different columns with the same gradient elution conditions; an ACQUITY BEH C18 column (150 × 2.1 mm, 1.7 μm, 130 Å pore size, catalog number 186003556), an ACQUITY BEH Phenyl column (150 × 2.1 mm, 1.7 μm, 130 Å pore size, catalog number 186003378), and an ACQUITY BEH C4 column (150 × 2.1 mm, 1.7 μm, 130 Å pore size, catalog number 186004497) from Waters (Milford, MA) [25]. Solvents used were water containing 0.1 % TFA (Solvent E) and acetonitrile containing 0.1 % TFA (Solvent F). Peptides were eluted by increasing the percentage of Solvent F from 5 to 60 % over 30 min at 0.2 mL/min and column effluent was continuously monitored at 214 nm. Both peptides were ≥95% pure on all three reversed-phase columns.
2.2. Topical Ointment Formulation of CPDI-02
For topical administration, CPDI-02 was formulated in an ointment of 3% (w/v) hydroxypropyl methylcellulose (HPMC) in 30% (v/v) propylene glycol [42] within 24 hours of the first topical treatment. HPMC [270 mg] was added to propylene glycol [2.70 mL] at 100 mg HPMC/mL and mixed at 4°C for 1 hour and CPDI-02 [270 mg] was dissolved in deionized water [6.5 mL] at 41.5 mg CPDI-02/mL. The CPDI-02 solution [6.3 mL] was added to the HPMC solution [2.4 mL] and mixed overnight to produce an ointment of 3% CPDI-02 (w/v) [3 mg/mL], aliquoted using a positive displacement pipette, and stored at 4°C until use.
2.3. Bacterial Strain & Culturing
CA-MRSA USA300 LAC (Staphylococcus aureus Rosenbach, ATCC BAA-1756) was stored in 50 % glycerol at -80°C until use. To start a culture, a Tryptic Soy Agar (TSA; Remel) plate was streaked and incubated overnight at 37°C. A single colony from the streaked agar plate was transferred to a 50-mL conical tube containing 30 mL of Tryptic Soy Broth (TSB; Remel) and incubated [37°C] overnight while shaking [200 rpm]. The overnight culture was diluted [1:10] in TSB, incubated at 37°C for ~2 hours until A600 = 0.5 to 0.55, and pelleted [3,000 g, 10 min] at 4°C. The pellet was resuspended in PBS and pelleted as before, then resuspended in 0.05% (w/v) Cytodex-1 in sterile PBS (typical yield ~5 x 108 CFU/mL). MRSA concentration was confirmed by serial dilution in sterile PBS, dot plating on TSA plates, and incubation at 37°C for 24 hours before counting.
2.4. Curative CPDI-02 treatment of healthy female outbred mice subcutaneously infected with CA-MRSA
All procedures were approved by the University of Nebraska Medical Center Institutional Animal Care and Use Committee. Female outbred CD-1 mice (Charles Rivers, Strain Code 022, 4-6 weeks old) were acclimated in the animal facility at least one week and rear flanks were shaved 24 hours before the study. Mice were anesthetized with isoflurane and 0.100 mL of CA-MRSA stock solution (Section 2.3) was injected SQ (5 x 107 CFU total) from the left rear flank into the dorsal side. Repeat treatment: Vehicle alone or vehicle containing the indicated dose of CPDI-02 or inactive scCPDI-02 was administered (n=10 mice) at 6-hours post-challenge, then daily over 5 days starting at 24 -hours post-challenge (6-, 24-, 48-, 72-, 96-, and 120-hours post-challenge) by the indicated administration route (6 treatments total). For IM and SQ administration, CPDI-02 was administered in 0.050 mL of sterile PBS [20 mg/mL] (volume adjusted to 50 mg/kg dose). For topical administration, CPDI-02 was applied to abscesses in 0.100 mL of ointment (3% w/v) (Section 2.2) using a positive displacement pipette [150 mg/kg dose]. For IV administration, CPDI-02 was administered in 0.050 mL sterile PBS [5 mg/mL] (volume adjusted to 12.5 mg/kg dose). Single IM treatment: At 6-hours post-challenge, vehicle alone (PBS) or vehicle containing the indicated dose of CPDI-02 or inactive scrambled CPDI-02 (scCPDI-02) was administered (n=10 mice) by the IM route as above (1 treatment total). Mouse abscesses were imaged with reference scales daily over 10 days starting at 24 -hours post-challenge. Abscess surface areas were determined by image analysis (ImageJ®) and compared between vehicle and treatment group for each administration route over ten days post-challenge by repeated measurement Two-Way ANOVA with Geisser-Greenhouse correction.
2.5. CA-MRSA burden of dermal abscesses after single IM treatment with CPDI-02
At 24 hours post-challenge (Section 2.4: Single IM treatment), mice were euthanized (n=5 per cohort) and rear flanks were disinfected with 70 % EtOH. Abscess & skin biopsies (Tru-Punch Sterile Disposable Biopsy Punch Razors (8 mm2, Sklar instruments 961130) were suspended in TSB [2 mL] and homogenized in C tubes (Milteni Biotec 130-093-237) using a gentleMACS Dissociator (Milteni Biotec 130-093-235, program B). Homogenized abscess tissues were serially diluted in PBS and plated on TSA plates, incubated at 37°C for 24 hours, and bacterial colonies were counted. Bacterial colony counts per abscess were normalized to starting tissue weights and compared using a Mann-Whitney test (nonparametric t-test).
2.6. Proportions of neutrophils and mononuclear phagocytes in dermal abscesses after single IM treatment with CPDI-02
At 24 hours post-challenge (Section 2.4: Single IM treatment), mice were euthanized (n=5 per cohort) and abscess and skin biopsies were collected (Section 2.5) and fixed in 10 % formalin for 48 hours. After tissue fixation, tissues were prepared into paraffin tissue sections by the Tissue Science Facility at the University of Nebraska - Medical Center. Paraffin tissue sections were deparaffinated and immunostained for neutrophils (Ly6G) and mononuclear phagocytes (F4/80). Primary antibodies were Rat / IgG2a anti-F4/80 (14-4801-82 Invitrogen (BM8), 1:50) and Rat / IgG2b anti-Ly6 G/Ly6C (MA1-40038 Invitrogen (NIMP-R14), 1:50) diluted in TBS + 1 % BSA, overnight incubation. Secondary antibodies were Anti-Rat IgG2a – FITC (11-4817-82 Invitrogen, 1:200) and Anti-Rat IgG2b - eFluor 660 (50-4815-82 Invitrogen, 1:200), incubated for 2 hours. Sections were mounted on cover slides using 4’6’-diamidino-2-phenylindole (DAPI) and imaged on a Zeiss 710 Confocal Laser Scanning Microscope under 20x magnification. Image files (.czi) were analyzed with ImageJ [43] using the following parameters: Import Options: Hyperstack / ROI manager / Color mode: Colorized / Autoscale and Split channels options checked; Adjust / Threshold/ Otsu / Red / Dark Background; Process/Binary / Make Binary; Process / Binary / Fill Holes; Process / Binary / Watershed; Analyze / Analyze Particles / Size (micro2): 7 to 315 μm2 / Circularity: 0.00 to 1.00 / Show: Outlines / Display Results / Clear Results / Summarize / Exclude on Edges / Overlay.
2.7. Pro-inflammatory cytokines and chemokines in dermal abscesses
At 3 hours & 24 hours post-challenge (Section 2.4: Single IM treatment), mice were euthanized (n = 5 per cohort per time point) then abscess and skin biopsies were collected (Section 2.5), suspended in PBS [2 mL], and homogenized in C tubes (Milteni Biotec 130-093-237) using a gentleMACS Dissociator (Milteni Biotec 130-093-235, program B). Tissue homogenates were stored at -20 °C until analysis. Homogenized abscess tissues were then analyzed using a V-PLEX Proinflammatory Panel 1 Mouse Kit (Meso Scale Diagnostics K15048D-1) on a MESO QuickPlex SQ 120MM to measure the inflammatory markers of the abscesses.
2.7. Dose escalation of CPDI-02 by the IM route in healthy outbred CD-1 mice
All procedures were approved by the University of Nebraska Medical Center Institutional Animal Care and Use Committee. Twenty-four CD-1 mice, approximately 4 weeks old were randomized by weight and sex into four cohorts and then intramuscularly administered either vehicle alone (filter-sterilized 0.2 μm endotoxin-free PBS) or vehicle containing CPDI-02 twice a week over 4 weeks (8 doses total). CPDI-02 doses began at 15 mg/kg and subsequent doses were increased by a conventional modified Fibonacci scheme (Increase over previous dose: 100 %, 67 %, 50 %, 40 %, 30 %, 33 %, 33 %) to a final dose of 225 mg/kg. Mice were weighed prior to the study and prior to each dose administration. On each dose administration date, mice were assigned a Pain and Distress Assessment Score (Table S1). Four days after the last treatment, mice were euthanized, whole blood was collected for Complete Blood Count (CBC) with differential (VetScan® HM5 Hematology Analyzer) and the thymus, heart, lungs, brain, liver, spleen, kidney, femur bone marrow, popliteal lymph nodes, Peyer’s patches, and injection site muscle (caudal thigh muscle) were collected. Collected tissues were fixed in 10 % formalin for 48 hours and then embedded in paraffin. Before embedment in paraffin, femur bone marrow samples underwent decalcification by shaking in 10 % formic acid for 8 hours. Paraffin samples were cut into sections and underwent a Hematoxylin and Eosin (H&E) stain, then analyzed for signs of inflammation and other tissue abnormalities utilizing a Nikon Eclipse E600 at 20x magnification and imaged using a Lumenera Infinity 2 Microscope Camera. Mouse weights between cohorts were compared by repeated measurement 2-Way ANOVA. CBC and differential results were analyzed by Mann-Whitney Test (nonparametric T-tests). For all statistical analyses, cohorts were grouped by treatment (PBS vs. CPDI-02, mixed genders) or within genders (Male PBS vs. CPDI-02 separate from female PBS vs. CPDI-02).
287. Repeat dosing of CPDI-02 by the IM route in healthy outbred CD-1 mice
All procedures were approved by the University of Nebraska Medical Center Institutional Animal Care and Use Committee. Sixty CD-1 mice (thirty male and thirty female) approximately six weeks old were randomized into six cohorts (three cohorts for each gender) and then intramuscularly administered either vehicle alone (filter-sterilized 0.2 μm endotoxin-free PBS), or vehicle containing CPDI-02 at 50 % the observed MTD [65 mg/kg], or CPDI-02 suspended in PBS at 100 % the observed MTD [130 mg/kg] twice a week over 32 days (9 doses total). Mice were weighed prior to each dose administration. On each dose administration date, mice were assigned a Pain and Distress assessment score (Table S1). Four days after the last treatment, through a terminal cardiac bleed, whole blood was collected for Complete Blood Count (CBC) with differential, which was analyzed on an Abaxis VetScan® HM5 Hematology Analyzer. In addition, injection site muscle tissues were collected, fixed in 10 % formalin for 48 hours, then embedded in paraffin. Paraffin samples were cut into sections and underwent an H&E stain, then analyzed for signs of inflammation and other tissue abnormalities utilizing a Nikon Eclipse E600 at 20x magnification and imaged using a Lumenera Infinity 2 Microscope Camera. Mouse weights between cohorts were compared by repeated measurement 2-Way ANOVA. CBC and differential results were analyzed by Kruskal-Wallis Test (nonparametric One-Way ANOVA). For all statistical analyses, cohorts were grouped by treatment (PBS vs. 50 % OMTD vs. 100 % OMTD, mixed genders) or within genders (Male PBS vs. 50 % OMTD vs. 100 % OMTD separate from female PBS vs. 50 % OMTD vs. 100 % OMTD).
3. Results
3.1. Repeat treatment with CPDI-02 starting at an early stage of subcutaneous infection with CA-MRSA increases curative protection of healthy female outbred mice depending on the route of administration
To establish that CPDI-02 increases curative protection against subcutaneous infection with CA-MRSA, we (i.) subcutaneously injected CA-MRSA USA300 LAC into the rear dorsal side of healthy female outbred CD-1 mice, (ii.) administered vehicle alone or vehicle containing CPDI-02 (Table 1) at the indicated dose by a topical (TOP), subcutaneous (SQ), intramuscular (IM), or intravenous (IV) route at 6 -hours post-challenge, then daily over five days post-challenge (6 treatments total), and (iii.) compared surface areas of dermal abscesses daily over 10 days post-challenge by quantitative image analysis (Figure 1).
Compared to vehicle alone (Figure 1C, white circles), repeat treatment with 50 mg CPDI-02/kg by the IM route (Figure 1C, black circles) decreased maximum dermal abscess surface area (Day 1 post-challenge) by 58% [31 ± 8 (SEM) vs. 74 ± 11 mm2, P = 0.0052] and time to resolution by approximately 1 to 2 days. In contrast, repeat treatment with CPDI-02 by TOP (Figure 1A, black circles), SQ (Figure 1B, black circles), or IV (Figure 1D, black circles) routes had no effect on maximum abscess surface area or time to resolution vs. vehicle alone (Figure 1A,B,D, white circles). Repeat treatment with inactive scrambled CPDI-02 (scCPDI-02; Table 1) by the IM route at 50 mg/kg (Figure S2, black circles) also had no effect vs. vehicle alone (Figure S2, white circles), indicating that curative protection by IM treatment is due to the immunostimulatory activity of CPDI-02. Furthermore, repeat treatment with CPDI-02 starting at 24 -hours post-challenge had no effect regardless of administration route (not shown). Thus, repeat treatment with CPDI-02 by an IM but not a TOP, SQ, or IV route increases curative protection against SQ infection with CA-MRSA when initiated early enough during infection under these experimental conditions.
3.2. Single early-stage IM treatment with CPDI-02 is sufficient to increase curative protection of healthy female outbred mice against subcutaneous infection with CA-MRSA in a dose-dependent manner
The first IM treatment with CPDI-02 at 6 -hours post-challenge decreased maximum dermal abscess surface area by 58% vs. vehicle alone (Figure 1C, Day 1 post-challenge, black vs. white circle) before the second IM treatment. This suggested that subsequent IM treatments with CPDI-02 are not required to increase curative protection against SQ infection with CA-MRSA.
To determine if single early-stage IM treatment with CPDI-02 is sufficient to increase curative protection against subcutaneous infection with CA-MRSA, we (i.) subcutaneously injected CA-MRSA USA300 LAC into the rear dorsal side of healthy female outbred CD-1 mice, (ii.) administered vehicle alone or vehicle containing between 3.125 mg to 50 mg CPDI-02/kg by the IM route at 6 -hours post-challenge, (iii.) compared dermal abscess surface areas daily over 10 days post-challenge by quantitative image analysis (Figure 2A), and (iv.) on Day 1 post-challenge, compared colony forming units (CFU) of CA-MRSA/g of dermal abscess biopsies by dilution plating (Figure 2B) and the presence of CA-MRSA in the epidermis and dermis of dermal abscesses by histochemistry (HC) with gram staining (Figure 2C).
Single early-stage IM treatment with CPDI-02 (Figure 2A, black symbols) decreased maximum dermal abscess surface area (Day 1 post-challenge) and time to resolution (Day 10 post-challenge) vs. vehicle alone (Figure 2A, white circles) as dose of CPDI-02 increased with an ED50 of ~4 mg CDPI-02/kg. Single early-stage IM treatment with CPDI-02 at 50 mg/kg (Figure 2A, black circles) also decreased maximum dermal abscess surface area (60% vs. 59% decrease on Day 1 post-challenge) and time to resolution to a similar extent as repeated early-stage IM treatment at the same dose (Figure 1C, black circles) despite 50% larger maximum abscess surface areas in vehicle-treated mice vs. repeated IM treatment (Figure 2A vs. Figure 1C, black circles, Day 1 post-challenge) [111 ± 37 (SD) vs. 74 ± 36 mm2, P = 0.0433]. Single early-stage treatment with 50 mg CPDI-02/kg by the IM route also decreased CA-MRSA CFU/g of abscess tissue on Day 1 post-challenge (Figure 2B, CPDI-02) by 1.9-fold vs. vehicle alone (Figure 2B, Vehicle) [1.8 x 106 ± 0.8 x 106 (SD) vs. 3 x 106 ± 2 x 106 CFU/g of abscess tissue] and greatly decreased detectable CA-MRSA in the epidermis and dermis of dermal abscess (Figure 2C, CPDI-02 vs. Vehicle). Thus, single early-stage treatment with CPDI-02 by the IM route is sufficient to increase curative protection against subcutaneous infection with CA-MRSA in a dose-dependent manner under these experimental conditions.
3.3. Single early-stage IM treatment with CPDI-02 increases mononuclear phagocytes in the epidermis and dermis of dermal abscesses of female outbred mice during subcutaneous infection with CA-MRSA
To determine if single early-stage IM treatment with CPDI-02 affects the number of neutrophils and/or mononuclear phagocytes in dermal abscesses after subcutaneous infection with CA-MRSA, we (i.) subcutaneously injected CA-MRSA USA300 LAC into the rear dorsal side of healthy female outbred CD-1 mice, (ii.) administered vehicle alone or vehicle containing 50 mg CPDI-02/kg by the IM route at 6 -hours post-challenge, then (iii.) compared ratios of neutrophils (Ly6G/Ly6C+) and mononuclear phagocytes (F4/80+) to total cells (DAPI+) in the epidermis and dermis of dermal abscesses by quantitative immunohistochemistry (IHC) image analysis on Day 1 post-challenge (Figure 3).
Single early-stage IM treatment with CPDI-02 (Figure 3B,C) or vehicle alone (Figure 3G,H) had similar ratios of neutrophils (Ly6G/Ly6C+) to total cells (DAPI+) in the epidermis and dermis of dermal abscesses (Figure 3K). In contrast, CPDI-02 (Figure 3D,E) increased ratios of mononuclear phagocytes (F4/80+) to total cells (DAPI+) by 4-fold (Figure 3L) and mononuclear phagocytes to neutrophils (Ly6G/Ly6C+) by 7-fold (Figure 3M) vs. vehicle alone (Figure 3I,J). Thus, single early-stage IM treatment with CPDI-02 increases mononuclear phagocytes in the epidermis and dermis of dermal abscesses at an early stage of subcutaneous infection with CA-MRSA under these experimental conditions.
3.4. Single early-stage IM treatment with CPDI-02 decreases early levels of IL-1β in dermal abscesses during subcutaneous infection with CA-MRSA
To determine if single early-stage IM treatment with CPDI-02 affects cytokines and chemokines potentially involved in local inflammation of dermal abscesses after subcutaneous infection with CA-MRSA, we (i.) subcutaneously injected CA-MRSA USA300 LAC into the rear dorsal side of healthy female outbred CD-1 mice, (ii.) administered vehicle alone or vehicle containing 50 mg CPDI-02/kg by the IM route at 6 -hours post-challenge, then (iii.) compared major inflammatory cytokines and chemokines by multiplex ELISA (Figure 4 and Figure S3) at 3 -hours post-treatment (9 -hours post-challenge) or 18 -hours post-treatment (24 -hours post-challenge).
Single IM treatment with CPDI-02 at 6-hours post-challenge decreased levels of IL-1β in dermal abscesses at 3 -hours post-treatment (Figure 4A, black bar) but not 18-hours post-treatment (Figure 4B, black bar) vs. vehicle alone (Figure 4, white bars) and had no effect on levels of IFN-γ, IL-2, IL-4, IL-5, IL-6, IL-10, IL-12p70, CXCL-1 [KC/GRO], or TNF-α at either timepoint (Figure S3). Thus, single early-stage IM treatment with CPDI-02 decreases early levels of IL-1β in dermal abscesses after subcutaneous infection with CA-MRSA but not most other cytokines / chemokines potentially involved in the inflammation of dermal abscesses under these experimental conditions.
3.5. Comparison of IV and IM administration on CPDI-02 plasma levels and distribution to organs and dermal abscesses in female outbred mice
To determine if IM administration affects systemic exposure and/or distribution of CPDI-02 during subcutaneous infection with CA-MRSA, we (i.) subcutaneously injected CA-MRSA USA300 LAC into the rear dorsal side of healthy female outbred CD-1 mice, (ii.) administered vehicle alone or vehicle containing CPDI-02 by the IV [12.5 mg/kg] or IM [50 mg/kg] route at 6 -hours post-challenge, then (iii.) compared plasma concentrations of CPDI-01 at 2, 4, 8, and 24 -hours post-treatment (Figure 5A) and normalized mass of CPDI-02 in organs and dermal abscesses at 24 -hours post-challenge by LC-MS/MS (Figure 5B. We chose these IM and IV doses of CPDI-02, respectively, because repeat IM administration of 50 mg CPDI-02/kg to female outbred mice decreased maximum dermal abscess surface area and time to resolution (Figure 1C), whereas repeat IV administration of 12.5 CPDI-02/kg had no effect (Figure 1A) vs. vehicle alone.
IM administration of CPDI-02 increased plasma concentrations (Figure 5A, black circles) vs. IV administration (Figure 5A, white circles) over 24 -hours but had no effect on the relative distribution of CPDI-02 to the heart (HT), lungs (LG), liver (LV), kidneys (KD), spleen (SP), or brain (BR) (Figure 5B, black vs. white circles). IM administration also increased levels of CPDI-02 in some dermal abscesses (AB) (Figure 5B, AB –black circles) vs. IV administration (Figure 5B, AB – white circles) at 24 -hours post-challenge but the overall difference between administration routes was not statistically significant (P=0.0636). Thus, single IM administration of CPDI-02 may increase curative protection against subcutaneous infection with CA-MRA, in part, by increasing systemic exposure and subsequent localization of CPDI-02 to dermal abscesses.
3.6. IM dose escalation of CPDI-02 in healthy male and female outbred mice
To begin establishing a maximally tolerated dose (MTD) of CPDI-02 in outbred mice after IM administration, we (i.) injected healthy male and female outbred CD-1 mice with vehicle alone or vehicle containing increasing doses of CPDI-02 up to 225 mg/kg in the (left) caudal thigh muscle twice per week over 28 days (8 injections total) then (ii.) compared body weights by gravimetric analysis (Figure 6A) and pain and distress (Figure 6B) by single-blinded scoring assessment (Table S1) on the day of each injection and (iii.) compared complete blood count with differential by hematology analyzer (Figure 6C,D; Table S2) and signs of inflammation in tissues and organs by histochemistry with H&E staining (Figure S4) 4 days after the final injection. We started with ~4-week-old mice to ensure that later body weights would be low enough at escalated doses of CPDI-02 to accommodate the solubility of CPDI-02 in PBS (~100 mg/mL) at the maximum volume allowed for single IM injections in mice (0.05 mL).
IM dose escalation with PBS vehicle alone (Figure 6A, white circles [males] and squares [females]) or vehicle containing up to 225 mg CPDI-02/kg over 28 days (Figure 6A, black circles [males] and squares [females]) did not affect weight gain or survival of healthy outbred CD-1 mice. IM administration of 170 or 225 mg CPDI-02/kg, however, increased signs of mild (Score 2) to moderate distress (Score 3) in male and female mice within 15 minutes of injection that resolved after 1 to 2 hours (Figure 6B, black circles [males] and squares [females]).
IM dose escalation with CPDI-02 decreased platelets in male and female mice (Figure 6C, black bars) and decreased platecrit in male mice (Figure 6D, black bars) 4 days after the final injection (Day 28) vs. vehicle alone (Figure 6C,D, white bars). It also increased signs of mild inflammation in 83% of female mice and 50% of male mice (Figure S4B, black bars), whereas vehicle alone increased signs of signs of (mild?) inflammation around the injection site in 17% of female mice but not in male mice (Figure S4B, black bars). IM dose escalation with CPDI-02 or vehicle alone, however, did not increase signs of inflammation in the thymus, heart, lungs, brain, liver, spleen, kidney, femur bone marrow, popliteal lymph nodes, or Peyer’s patches in male or female mice (not shown). Thus, IM administration of CPDI-02 is tolerated by healthy male and female outbred mice to somewhere between 130 and 170 mg CPDI-02/kg with males likely tolerating a higher dose than females.
3.7. Repeat IM dosing of CPDI-02 in healthy male and female outbred mice
To begin determining the toxicity of CPDI-02 in healthy outbred mice after repeat IM dosing, we (i.) injected healthy male and female outbred CD-1 mice with vehicle alone (endotoxin-free PBS) or vehicle containing CPDI-02 at 50% [65 mg/kg] or 100% of the observed MTD above [130 mg/kg] in the left caudal thigh muscle twice per week over 32 days (9 injections total) then (ii.) compared body weights by gravimetric analysis (Figure 7A) and pain and distress by single-blinded scoring assessment (Table S1) on the day of each injection and (iii.) compared complete blood count with differential by hematology analyzer (Table S3; Figure 7C,D), blood chemistry by blood chemistry analyzer (Table S4), and inflammation in tissues and organs by histochemistry with H&E staining (Figure S5) 4 days after the final injection.
Repeat IM dosing of vehicle alone (Figure 7A, white circles [males] and squares [females]) or vehicle containing 65 mg CPDI-02/kg (Figure 7A, black circles [males] and squares [females]) or 130 mg CPDI-02/kg (Figure 7A, black circles [males] and squares [females]) over 32 days had no effect on weight gain by healthy male or female outbred mice. Unlike dose escalation to 225 mg CPDI-02/kg (Figure 6B), repeat dosing with CPDI-02 did not increase signs of distress up to 2 hours after injection or affect CBC (Table S3), including platelets (Figure 7C, grey and black bars) or platecrit (Figure 7D, grey and black bars), or affect blood chemistry (Table S4) in male or female outbred mice vs. vehicle alone (Figure 7C,D, white bars) up to 4 days after the final injection. Repeat IM dosing with 130 mg CPDI-02/kg, however, increased mortality in female mice (Figure 7, grey line) but not male mice (Figure 7, black line) after the 8th and 9th IM injection. Repeat IM dosing with CPDI-02 also increased signs of mild to severe inflammation around the injection site compared to vehicle alone (Figure S5B, grey and black bars) in a dose-dependent manner and in a greater proportion of female mice than male mice at both doses (Figure S5B, grey and black bars). There were, however, no signs of inflammation in the thymus, heart, lungs, brain, liver, spleen, kidney, femur bone marrow, popliteal lymph nodes, or Peyer’s patches (not shown). Thus, repeat IM treatment with CPDI-02 is tolerated by healthy male and female outbred mice to somewhere between 65 and 130 mg CPDI-02/kg with males likely tolerating a higher repeat dose than females.
4. Discussion
It was previously found that SQ treatment with the mononuclear phagocyte (MP)-selective activator, CPDI-02 (formerly EP67) [25], increases prophylaxis of outbred mice against SQ infection with CA-MRSA [23]. It remains unclear, however, if treatment with CPDI-02 also increase curative protection.
Our study provides evidence that indicates (i.) repeat treatment with CPDI-02 increases curative protection of outbred against SQ infection with CA-MRSA by the IM route but not TOP, SQ, or IV routes when initiated at an early stage of infection; (ii.) single early-stage IM treatment with CPDI-02 increases curative protection of outbred against SQ infection with CA-MRSA to the same extent as repeat IM treatment and is dose-dependent; and (iii.) single and repeat IM dosing of CPDI-02 is tolerated by healthy male and female outbred mice at supratherapeutic doses with males tolerating higher doses than females. We found that (i.) unlike repeat topical, SQ, or IV treatment, repeat IM treatment with 50 mg CPDI-02/kg starting at 6-hours post-SQ challenge with CA-MRSA USA300 LAC decreased maximum dermal abscess surface area and time to resolution over 10 days in female outbred CD-1 mice (Figure 1), whereas repeat IM treatment with 50 mg inactive scCPDI-02/kg starting at 6-hours post-challenge (Figure S2) or repeat CPDI-02 treatment starting at 24 hours post-challenge by the same four administration routes (not shown) had no effect; (ii.) single IM treatment with CPDI-02 up to 50 mg/kg starting at 6-hours post-challenge decreased maximum dermal abscess surface areas and time to resolution in female outbred CD-1 mice as dose increased (ED50 ~4 mg CPDI-02/kg) (Figure 2) and to a similar extent as repeat IM treatment using the same dose [50 mg CPDI-02/kg] (Figure 1); and (iii.) single IM dosing was tolerated by healthy male and female outbred CD-1 mice to somewhere between 130-170 mg CPDI-02/kg (Figure 6, Table S2, Figure S4), whereas repeat IM dosing with CPDI-02 was tolerated to somewhere between 65-130 mg CPDI-02/kg (Figure 7, Table S3, Figure S5) with males being significantly less affected by higher doses than females.
Our study also provides evidence that single early-stage IM treatment with CPDI-02 increases protection against SQ infection with CA-MRSA, in part, by increasing systemic exposure to CPDI-02 and subsequent numbers of mononuclear phagocytes (MP) at the site of infection. We found that single IM treatment with 50 mg CPDI-02/kg at 6-hours post-SQ challenge with CA-MRSA USA300 LAC increased the AUC of CPDI-02 vs. IV administration of 15 mg CPDI-02/kg over 24 hours (Figure 5) and increased ratios of MP to total cells and neutrophils in the epidermis and dermis of dermal abscess biopsies at 24-hours post-challenge (Figure 3).
5. Conclusions
Our study indicates that single early-stage IM treatment with CPDI-02 increases curative protection against SQ infection of outbred mice with CA-MRSA. Our study also suggests that the increase in curative protection occurs, in part, by increasing systemic exposure to CPDI-02 and subsequent number of mononuclear phagocytes (MP) at the site of infection. Given that activated MP (especially monocytes and macrophages) broadly protect against most pathogens that cause monomicrobial and polymicrobial SSTI, early-stage treatment with CPDI-02 alone or in combination with antimicrobials may improve the treatment of SSTI caused by high priority ESKAPE bacterial pathogens like MRSA and/or other pathogens regardless of MDR status.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Representative images of dermal abscesses on the rear dorsal region of CPDI-02-treated and untreated healthy female outbred mice 24 hours after subcutaneous challenge with CA-MRSA; Figure S2: Repeat IM treatment with inactive, scrambled CPDI-02 does not increase curative protection of healthy female outbred mice against subcutaneous challenge with CA-MRSA; Figure S3: Single curative IM treatment with CPDI-02 does not affect early levels of many cytokines and chemokines potentially involved in the inflammation of dermal abscesses after subcutaneous infection of healthy outbred female mice with CA-MRSA; Figure S4: H Histological comparison of inflammation around the injection site in caudal thigh muscles from healthy male and female outbred mice after IM dose escalation of CPDI-02; Figure S5: Histological comparison of inflammation around the injection site in caudal thigh muscles from healthy male and female outbred mice after repeat IM dosing of CPDI-02; Table S1: Pain and Distress Scoring in Mice; Table S2: Complete Blood Count with Differential 4 Days after IM Dose Escalation of CPDI-02 in Healthy Male and Female Outbred CD-1 Mice; Table S3: Complete Blood Count with Differential 4 Days after Repeat Dosing of CPDI-02 in Healthy Male and Female Outbred CD-1 Mice; Table S4: Blood Chemistry 4 Days after Repeat IM Dosing of CPDI-02 in Healthy Male and Female Outbred CD-1 Mice.
Author Contributions
Conceptualization, J.P.S., J.A.P., R.J.M., S.M.Cohen, and J.A.V.; methodology, J.P.S, S.M.C., and J.A.V.; software, J.P.S., and J.A.V.; validation, J.P.S., S.M.C., and J.A.V.; formal analysis, J.P.S., S.M.C., S.M.Cohen, Y.A., and J.A.V.; investigation, J.P.S., C.M.S, J.E.P, R.S., C.N., S.D.; resources, J.A.V.; data curation, J.P.S., S.M.C., and J.A.V.; writing—original draft preparation, J.P.S. and J.A.V.; writing—review and editing, J.P.S., S.M.C., J.A.P., R.J.M., S.M.Cohen, D.D.S., and J.A.V.; visualization, J.P.S. and J.A.V.; supervision, S.M.C., Y.A., K.F.L., and J.A.V.; project administration, J.A.V.; funding acquisition, J.A.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by NIH/NIAID 5R01AI125137 (J.A.V.), NIH/NIAID 1R01AI121050 (J.A.V.), the Weitz Family Foundation (J.A.V.), and NIH Office of the Director UG1OD024953 (R.J.M.). This research was partially conducted at the Auditory and Vestibular Technology Core (AVT) at Creighton University, Omaha, NE. This facility is supported by the Creighton University School of Medicine and grants GM103427 and GM139762 from the National Institute of General Medical Science (NIGMS), a component of the National Institutes of Health (NIH). This investigation is solely the responsibility of the authors and does not necessarily represent the official views of NIGMS or NIH.
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Animal Care and Use Committee of the University of Nebraska Medical Center (19-011-03-FC, 01 Jan 2019).
Data Availability Statement
We encourage all authors of articles published in MDPI journals to share their research data. In this section, please provide details regarding where data supporting reported results can be found, including links to publicly archived datasets analyzed or generated during the study. Where no new data were created, or where data is unavailable due to privacy or ethical restrictions, a statement is still required. Suggested Data Availability Statements are available in section “MDPI Research Data Policies” at https://www.mdpi.com/ethics.
Acknowledgments
We thank Dr. Tammy Kielian for CA-MRSA USA300 LAC strain.
Conflicts of Interest
Declare conflicts of interest or state “The authors declare no conflicts of interest.” Authors must identify and declare any personal circumstances or interest that may be perceived as inappropriately influencing the representation or interpretation of reported research results. Any role of the funders in the design of the study; in the collection, analyses or interpretation of data; in the writing of the manuscript; or in the decision to publish the results must be declared in this section. If there is no role, please state “The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results”.
References
- Kaye, K.S.; Petty, L.A.; Shorr, A.F.; Zilberberg, M.D. Current Epidemiology, Etiology, and Burden of Acute Skin Infections in the United States. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America 2019, 68, S193–S199. [Google Scholar] [CrossRef] [PubMed]
- Esposito, S.; Noviello, S.; Leone, S. Epidemiology and microbiology of skin and soft tissue infections. Curr Opin Infect Dis 2016, 29, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Fish, D.N. Skin and Soft Tissue Infections. In DiPiro’s Pharmacotherapy: A Pathophysiologic Approach, 12th Edition, DiPiro, J.T., Yee, G.C., Haines, S.T., Nolin, T.D., Ellingrod, V.L., Posey, L.M., Eds.; McGraw Hill: New York, NY, 2023.
- Vella, V.; Derreumaux, D.; Aris, E.; Pellegrini, M.; Contorni, M.; Scherbakov, M.; Bagnoli, F. The Incidence of Skin and Soft Tissue Infections in the United States and Associated Healthcare Utilization Between 2010 and 2020. Open Forum Infect Dis 2024, 11, ofae267. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Zhou, J.; Xu, B.N.; Li, Y.; Bao, W.; Cheng, X.L.; He, Y.; Xu, C.P.; Ren, J.; Zheng, Y.R.; et al. Global Burden of Bacterial Skin Diseases: A Systematic Analysis Combined With Sociodemographic Index, 1990-2019. Front Med (Lausanne) 2022, 9, 861115. [Google Scholar] [CrossRef]
- Linz, M.S.; Mattappallil, A.; Finkel, D.; Parker, D. Clinical Impact of Staphylococcus aureus Skin and Soft Tissue Infections. Antibiotics (Basel) 2023, 12. [Google Scholar] [CrossRef]
- Gehrke, A.E.; Giai, C.; Gomez, M.I. Staphylococcus aureus Adaptation to the Skin in Health and Persistent/Recurrent Infections. Antibiotics (Basel) 2023, 12. [Google Scholar] [CrossRef]
- Al Kindi, A.; Alkahtani, A.M.; Nalubega, M.; El-Chami, C.; O'Neill, C.; Arkwright, P.D.; Pennock, J.L. Staphylococcus aureus Internalized by Skin Keratinocytes Evade Antibiotic Killing. Front Microbiol 2019, 10, 2242. [Google Scholar] [CrossRef]
- Ray, G.T.; Suaya, J.A.; Baxter, R. Incidence, microbiology, and patient characteristics of skin and soft-tissue infections in a U.S. population: a retrospective population-based study. BMC Infect Dis 2013, 13, 252. [Google Scholar] [CrossRef]
- Mariani, F.; Galvan, E.M. Staphylococcus aureus in Polymicrobial Skinand Soft Tissue Infections: Impact of Inter-Species Interactionsin Disease Outcome. Antibiotics (Basel) 2023, 12. [Google Scholar] [CrossRef]
- Grundmann, H.; Aires-de-Sousa, M.; Boyce, J.; Tiemersma, E. Emergence and resurgence of meticillin-resistant Staphylococcus aureus as a public-health threat. Lancet 2006, 368, 874–885. [Google Scholar] [CrossRef]
- Antimicrobial Resistance, C. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- WHO Bacterial Priority Pathogens List, 2024: bacterial pathogens of public health importance to guide research, development and strategies to prevent and control antimicrobial resistance. 2024.
- Howden, B.P.; Giulieri, S.G.; Wong Fok Lung, T.; Baines, S.L.; Sharkey, L.K.; Lee, J.Y.H.; Hachani, A.; Monk, I.R.; Stinear, T.P. Staphylococcus aureus host interactions and adaptation. Nat Rev Microbiol 2023, 21, 380–395. [Google Scholar] [CrossRef] [PubMed]
- Del Giudice, P. Skin Infections Caused by Staphylococcus aureus. Acta Derm Venereol 2020, 100, adv00110. [Google Scholar] [CrossRef] [PubMed]
- Chambers, H.F.; Deleo, F.R. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat Rev Microbiol 2009, 7, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Lobanovska, M.; Pilla, G. Penicillin's Discovery and Antibiotic Resistance: Lessons for the Future? Yale J Biol Med 2017, 90, 135–145. [Google Scholar]
- Rammelkamp, C.H.; Maxon, T. Resistance of Staphylococcus aureus to the Action of Penicillin. Proceedings of the Society for Experimental Biology and Medicine 1942, 51, 386–389. [Google Scholar] [CrossRef]
- Lowy, F.D. Antimicrobial resistance: the example of Staphylococcus aureus. J Clin Invest 2003, 111, 1265–1273. [Google Scholar] [CrossRef]
- Gajdacs, M. The Continuing Threat of Methicillin-Resistant Staphylococcus aureus. Antibiotics (Basel) 2019, 8. [Google Scholar] [CrossRef]
- Antibiotic resistance threats in the United States, 2019. 2019.
- Morgan, E.L.; Morgan, B.N.; Stein, E.A.; Vitrs, E.L.; Thoman, M.L.; Sanderson, S.D.; Phillips, J.A. Enhancement of in vivo and in vitro immune functions by a conformationally biased, response-selective agonist of human C5a: implications for a novel adjuvant in vaccine design. Vaccine 2009, 28, 463–469. [Google Scholar] [CrossRef]
- Sheen, T.R.; Cavaco, C.K.; Ebrahimi, C.M.; Thoman, M.L.; Sanderson, S.D.; Morgan, E.L.; Doran, K.S. Control of methicillin resistant Staphylococcus aureus infection utilizing a novel immunostimulatory peptide. Vaccine 2011, 30, 9–13. [Google Scholar] [CrossRef]
- Klos, A.; Wende, E.; Wareham, K.J.; Monk, P.N. International Union of Basic and Clinical Pharmacology. [corrected]. LXXXVII. Complement peptide C5a, C4a, and C3a receptors. Pharmacological reviews 2013, 65, 500–543. [Google Scholar] [CrossRef] [PubMed]
- Alshammari, A.M.; Smith, D.D.; Parriott, J.; Stewart, J.P.; Curran, S.M.; McCulloh, R.J.; Barry, P.A.; Iyer, S.S.; Palermo, N.; Phillips, J.A.; et al. Targeted Amino Acid Substitution Overcomes Scale-Up Challenges with the Human C5a-Derived Decapeptide Immunostimulant EP67. ACS Infectious Diseases 2020. [Google Scholar] [CrossRef] [PubMed]
- Tallapaka, S.B.; Karuturi, B.V.K.; Yeapuri, P.; Curran, S.M.; Sonawane, Y.A.; Phillips, J.A.; David Smith, D.; Sanderson, S.D.; Vetro, J.A. Surface conjugation of EP67 to biodegradable nanoparticles increases the generation of long-lived mucosal and systemic memory T-cells by encapsulated protein vaccine after respiratory immunization and subsequent T-cell-mediated protection against respiratory infection. Int J Pharm 2019, 565, 242–257. [Google Scholar] [CrossRef]
- Karuturi, B.V.K.; Tallapaka, S.B.; Yeapuri, P.; Curran, S.M.; Sanderson, S.D.; Vetro, J.A. Encapsulation of an EP67-Conjugated CTL Peptide Vaccine in Nanoscale Biodegradable Particles Increases the Efficacy of Respiratory Immunization and Affects the Magnitude and Memory Subsets of Vaccine-Generated Mucosal and Systemic CD8+ T Cells in a Diameter-Dependent Manner. Molecular pharmaceutics 2017, 14, 1469–1481. [Google Scholar] [CrossRef]
- Karuturi, B.V.; Tallapaka, S.B.; Phillips, J.A.; Sanderson, S.D.; Vetro, J.A. Preliminary evidence that the novel host-derived immunostimulant EP67 can act as a mucosal adjuvant. Clinical immunology 2015, 161, 251–259. [Google Scholar] [CrossRef]
- Phillips, J.A.; Morgan, E.L.; Dong, Y.; Cole, G.T.; McMahan, C.; Hung, C.Y.; Sanderson, S.D. Single-step conjugation of bioactive peptides to proteins via a self-contained succinimidyl bis-arylhydrazone. Bioconjug Chem 2009, 20, 1950–1957. [Google Scholar] [CrossRef]
- Morgan, E.L.; Thoman, M.L.; Sanderson, S.D.; Phillips, J.A. A novel adjuvant for vaccine development in the aged. Vaccine 2010, 28, 8275–8279. [Google Scholar] [CrossRef]
- Kollessery, G.; Nordgren, T.M.; Mittal, A.K.; Joshi, S.S.; Sanderson, S.D. Tumor-specific peptide-based vaccines containing the conformationally biased, response-selective C5a agonists EP54 and EP67 protect against aggressive large B cell lymphoma in a syngeneic murine model. Vaccine 2011, 29, 5904–5910. [Google Scholar] [CrossRef]
- Hung, C.Y.; Hurtgen, B.J.; Bellecourt, M.; Sanderson, S.D.; Morgan, E.L.; Cole, G.T. An agonist of human complement fragment C5a enhances vaccine immunity against Coccidioides infection. Vaccine 2012, 30, 4681–4690. [Google Scholar] [CrossRef]
- Tempero, R.M.; Hollingsworth, M.A.; Burdick, M.D.; Finch, A.M.; Taylor, S.M.; Vogen, S.M.; Morgan, E.L.; Sanderson, S.D. Molecular adjuvant effects of a conformationally biased agonist of human C5a anaphylatoxin. Journal of immunology 1997, 158, 1377–1382. [Google Scholar] [CrossRef]
- Lauvau, G.; Loke, P.; Hohl, T.M. Monocyte-mediated defense against bacteria, fungi, and parasites. Semin Immunol 2015, 27, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Park, M.D.; Silvin, A.; Ginhoux, F.; Merad, M. Macrophages in health and disease. Cell 2022, 185, 4259–4279. [Google Scholar] [CrossRef] [PubMed]
- Lionakis, M.S.; Drummond, R.A.; Hohl, T.M. Immune responses to human fungal pathogens and therapeutic prospects. Nature reviews. Immunology 2023, 23, 433–452. [Google Scholar] [CrossRef]
- Yakupu, A.; Aimaier, R.; Yuan, B.; Chen, B.; Cheng, J.; Zhao, Y.; Peng, Y.; Dong, J.; Lu, S. The burden of skin and subcutaneous diseases: findings from the global burden of disease study 2019. Front Public Health 2023, 11, 1145513. [Google Scholar] [CrossRef]
- Rice, L.B. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J Infect Dis 2008, 197, 1079–1081. [Google Scholar] [CrossRef]
- Vale de Macedo, G.H.R.; Costa, G.D.E.; Oliveira, E.R.; Damasceno, G.V.; Mendonca, J.S.P.; Silva, L.D.S.; Chagas, V.L.; Bazan, J.M.N.; Alianca, A.; Miranda, R.C.M.; et al. Interplay between ESKAPE Pathogens and Immunity in Skin Infections: An Overview of the Major Determinants of Virulence and Antibiotic Resistance. Pathogens 2021, 10. [Google Scholar] [CrossRef]
- Pidwill, G.R.; Gibson, J.F.; Cole, J.; Renshaw, S.A.; Foster, S.J. The Role of Macrophages in Staphylococcus aureus Infection. Frontiers in immunology 2020, 11, 620339. [Google Scholar] [CrossRef]
- Vojkovsky, T. Detection of secondary amines on solid phase. Pept Res 1995, 8, 236–237. [Google Scholar]
- Nibbering, P.H.; Goblyos, A.; Adriaans, A.E.; Cordfunke, R.A.; Ravensbergen, B.; Rietveld, M.H.; Zwart, S.; Commandeur, S.; van Leeuwen, R.; Haisma, E.M.; et al. Eradication of meticillin-resistant Staphylococcus aureus from human skin by the novel LL-37-derived peptide P10 in four pharmaceutical ointments. Int J Antimicrob Agents 2019, 54, 610–618. [Google Scholar] [CrossRef]
- Shihan, M.H.; Novo, S.G.; Le Marchand, S.J.; Wang, Y.; Duncan, M.K. A simple method for quantitating confocal fluorescent images. Biochem Biophys Rep 2021, 25, 100916. [Google Scholar] [CrossRef]
Figure 1.
Repeat treatment with CPDI-02 increases curative protection of healthy female outbred mice against subcutaneous challenge with CA-MRSA depending on route of administration. On Day 0, CA-MRSA (USA300 LAC, 5×107 CFU) was injected SQ from the left rear flank into the dorsal side of 6-week-old female outbred CD-1 mice. Vehicle alone (white circles) or vehicle containing the indicated dose of CPDI-02 (black circles) was administered (↑) at 6 -hours post-challenge, then daily over 5 days starting 24 -hours post-challenge by the (A) topical (directly onto abscess, volume vehicle), (B) subcutaneous (left rear flank, PBS, volume), (C) intramuscular (left caudal thigh muscle, sterile PBS, 50 μL), or (D) intravenous (tail vein, sterile PBS, volume) route. Average dermal abscess surface areas ± SEM (n=10 mice) from vehicle alone or vehicle containing CPDI-02 were determined daily by quantitative image analysis (Figure S1) and compared for each administration route by repeated measurement Two-Way ANOVA with Geisser-Greenhouse correction.
Figure 1.
Repeat treatment with CPDI-02 increases curative protection of healthy female outbred mice against subcutaneous challenge with CA-MRSA depending on route of administration. On Day 0, CA-MRSA (USA300 LAC, 5×107 CFU) was injected SQ from the left rear flank into the dorsal side of 6-week-old female outbred CD-1 mice. Vehicle alone (white circles) or vehicle containing the indicated dose of CPDI-02 (black circles) was administered (↑) at 6 -hours post-challenge, then daily over 5 days starting 24 -hours post-challenge by the (A) topical (directly onto abscess, volume vehicle), (B) subcutaneous (left rear flank, PBS, volume), (C) intramuscular (left caudal thigh muscle, sterile PBS, 50 μL), or (D) intravenous (tail vein, sterile PBS, volume) route. Average dermal abscess surface areas ± SEM (n=10 mice) from vehicle alone or vehicle containing CPDI-02 were determined daily by quantitative image analysis (Figure S1) and compared for each administration route by repeated measurement Two-Way ANOVA with Geisser-Greenhouse correction.
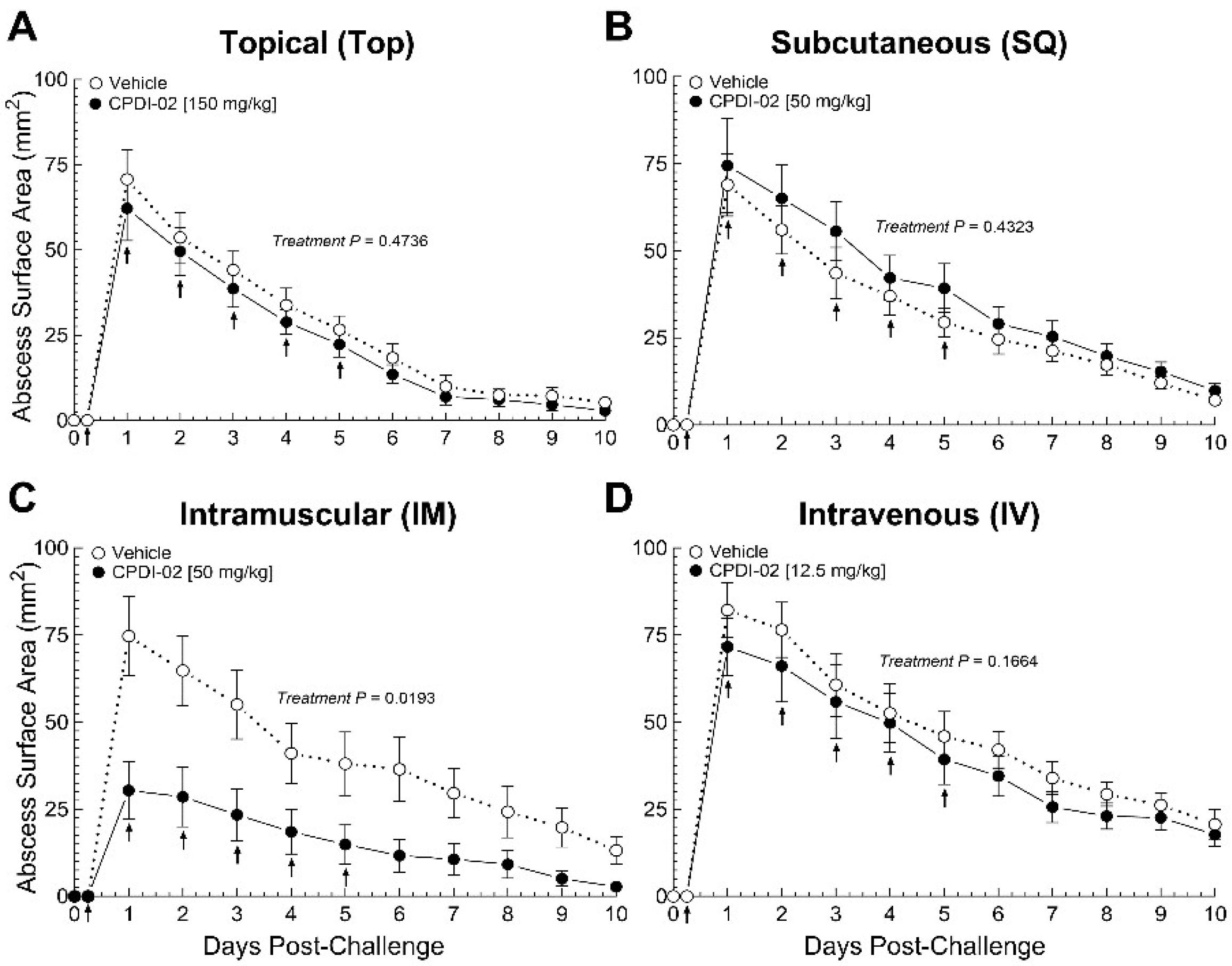
Figure 2.
Single early-stage IM treatment with CPDI-02 is sufficient to increase curative protection of healthy female outbred mice against subcutaneous infection with CA-MRSA in a dose-dependent manner. On Day 0, community-acquired methicillin-resistant Staphylococcus aureus (CA-MRSA, USA300 strain, 5×107 CFU) was administered SQ from the left rear flank into the dorsal side of 6-week-old female outbred CD-1 mice. (A) At 6 -hours post-challenge, vehicle alone (PBS, white circles) or vehicle containing the indicated dose of CPDI-02 (black symbols) was administered IM (↑) to the left caudal thigh muscle. Average dermal abscess surface areas ± SEM (n=10 mice) were then determined daily starting 24 -hours post-challenge by quantitative image analysis (Figure S1) and compared between doses by repeated measurement 2-Way ANOVA with Geisser-Greenhouse correction and Tukey post-test. On Day 1 post-challenge, (B) average colony forming units (CFU) of CA-MRSA/g of abscess biopsy ± SD (n=10 mice) after treatment with vehicle alone (white circles) or vehicle containing CPDI-02 at 50 mg/kg (black circles) were determined by dilution plating and compared by two-tailed Mann-Whitney test and (C) the presence of CA-MRSA in the epidermis and dermis of abscess cross-sections was determined by histochemistry (HC) with gram staining and imaging (40X magnification). HC images are representative of three mice from each treatment group.
Figure 2.
Single early-stage IM treatment with CPDI-02 is sufficient to increase curative protection of healthy female outbred mice against subcutaneous infection with CA-MRSA in a dose-dependent manner. On Day 0, community-acquired methicillin-resistant Staphylococcus aureus (CA-MRSA, USA300 strain, 5×107 CFU) was administered SQ from the left rear flank into the dorsal side of 6-week-old female outbred CD-1 mice. (A) At 6 -hours post-challenge, vehicle alone (PBS, white circles) or vehicle containing the indicated dose of CPDI-02 (black symbols) was administered IM (↑) to the left caudal thigh muscle. Average dermal abscess surface areas ± SEM (n=10 mice) were then determined daily starting 24 -hours post-challenge by quantitative image analysis (Figure S1) and compared between doses by repeated measurement 2-Way ANOVA with Geisser-Greenhouse correction and Tukey post-test. On Day 1 post-challenge, (B) average colony forming units (CFU) of CA-MRSA/g of abscess biopsy ± SD (n=10 mice) after treatment with vehicle alone (white circles) or vehicle containing CPDI-02 at 50 mg/kg (black circles) were determined by dilution plating and compared by two-tailed Mann-Whitney test and (C) the presence of CA-MRSA in the epidermis and dermis of abscess cross-sections was determined by histochemistry (HC) with gram staining and imaging (40X magnification). HC images are representative of three mice from each treatment group.
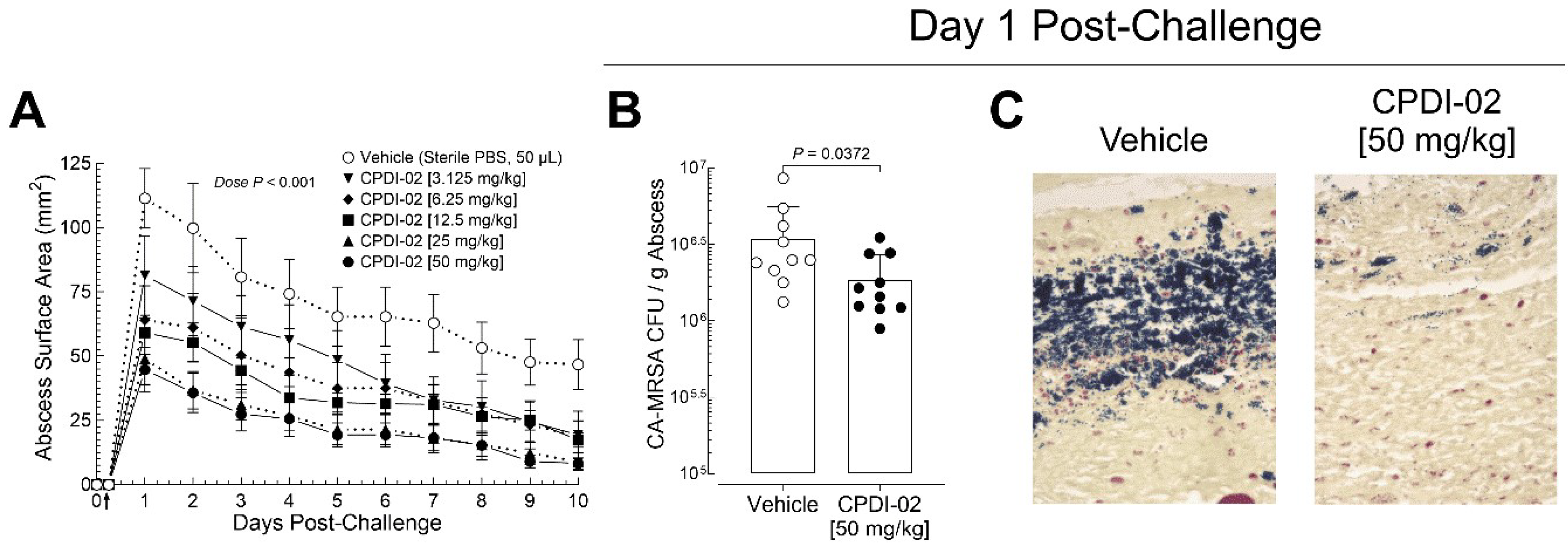
Figure 3.
Single early-stage IM treatment with CPDI-02 increases the number of mononuclear phagocytes in the epidermis and dermis of dermal abscesses from healthy female outbred mice 24 hours after subcutaneous infection with CA-MRSA. On Day 0, community-acquired methicillin-resistant Staphylococcus aureus (CA-MRSA, USA300 strain, 5×107 CFU) was administered SQ from the left rear flank into the dorsal side of 6-week-old healthy female outbred CD-1 mice. At 6 -hours post-challenge, vehicle alone (PBS) or vehicle containing CPDI-02 at 50 mg/kg was administered IM to the [left] caudal thigh muscle. On Day 1 post-challenge, the presence of (A,F) cell nuclei (DAPI+ cells, blue), (B,G) neutrophils (Ly6G/Ly6C+ cells, red), and (D,I) mononuclear phagocytes (F4/80+ cells, green) in the epidermis and dermis of dermal abscesses was determined by IHC at 20X magnification. Average ratios ±SD (n=3 mice) of (K) neutrophils (Ly6G+/Ly6C+) to nuclei (DAPI+), (L) mononuclear phagocytes (F4/80+) to nuclei (DAPI+), and (K) mononuclear phagocytes (F4/80+) to neutrophils (Ly6G/Ly6C+) in the epidermis and dermis were then determined by quantitative IHC image analysis and compared by unpaired t test with Welch’s correction. IHC images are representative of 3 mice from each treatment group.
Figure 3.
Single early-stage IM treatment with CPDI-02 increases the number of mononuclear phagocytes in the epidermis and dermis of dermal abscesses from healthy female outbred mice 24 hours after subcutaneous infection with CA-MRSA. On Day 0, community-acquired methicillin-resistant Staphylococcus aureus (CA-MRSA, USA300 strain, 5×107 CFU) was administered SQ from the left rear flank into the dorsal side of 6-week-old healthy female outbred CD-1 mice. At 6 -hours post-challenge, vehicle alone (PBS) or vehicle containing CPDI-02 at 50 mg/kg was administered IM to the [left] caudal thigh muscle. On Day 1 post-challenge, the presence of (A,F) cell nuclei (DAPI+ cells, blue), (B,G) neutrophils (Ly6G/Ly6C+ cells, red), and (D,I) mononuclear phagocytes (F4/80+ cells, green) in the epidermis and dermis of dermal abscesses was determined by IHC at 20X magnification. Average ratios ±SD (n=3 mice) of (K) neutrophils (Ly6G+/Ly6C+) to nuclei (DAPI+), (L) mononuclear phagocytes (F4/80+) to nuclei (DAPI+), and (K) mononuclear phagocytes (F4/80+) to neutrophils (Ly6G/Ly6C+) in the epidermis and dermis were then determined by quantitative IHC image analysis and compared by unpaired t test with Welch’s correction. IHC images are representative of 3 mice from each treatment group.
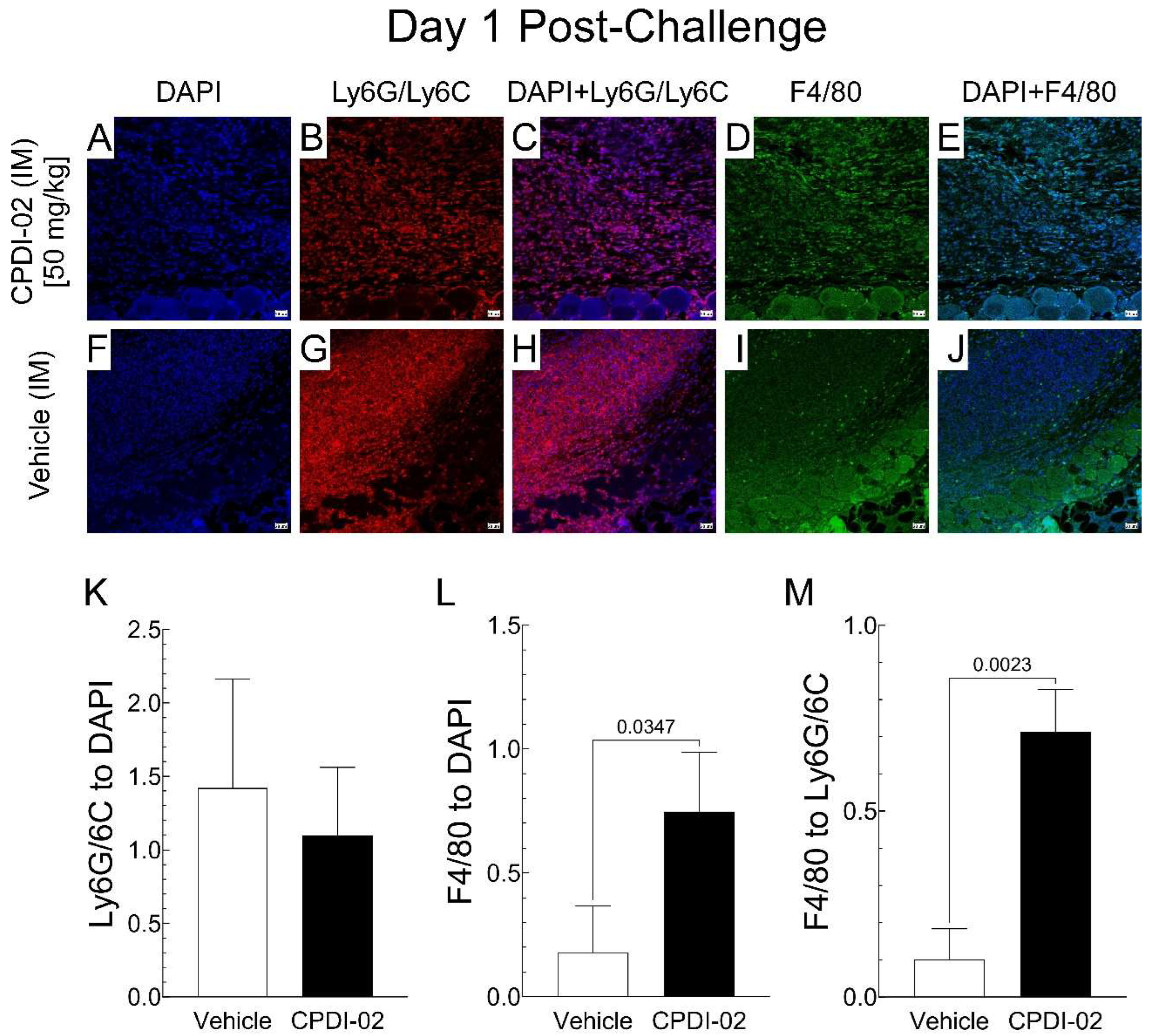
Figure 4.
Single curative treatment with CPDI-02 by the IM route decreases early levels of IL-1β in dermal abscesses of healthy female outbred mice after subcutaneous infection with CA-MRSA. On Day 0, community-acquired methicillin-resistant Staphylococcus aureus (CA-MRSA, USA300 strain, 5×107 CFU) was administered SQ from the left rear flank into the dorsal side of 6-week-old healthy female outbred CD-1 mice. At 6-hours post-challenge, vehicle alone (sterile PBS, white bars) or vehicle containing CPDI-02 at 50 mg/kg (black bars) was administered IM to the left caudal thigh muscle. Average concentrations of pro-inflammatory markers in dermal abscess biopsies ±SD (n=5 mice per time point) at (A) 3 -hours post-treatment (9 -hours post-challenge) or (B) 18 -hours post-treatment (24 -hours post-challenge) were determined by multiplex ELISA and compared by two-tailed t test with Mann-Whitney post-test (P value shown). No differences in other major murine cytokines / chemokines involved in inflammation (IFN-γ, IL-2, IL-4, IL-5, IL-6, IL-10, IL-12p70, CXCL-1 [KC/GRO], TNF-α) were observed at these timepoints (Figure S3).
Figure 4.
Single curative treatment with CPDI-02 by the IM route decreases early levels of IL-1β in dermal abscesses of healthy female outbred mice after subcutaneous infection with CA-MRSA. On Day 0, community-acquired methicillin-resistant Staphylococcus aureus (CA-MRSA, USA300 strain, 5×107 CFU) was administered SQ from the left rear flank into the dorsal side of 6-week-old healthy female outbred CD-1 mice. At 6-hours post-challenge, vehicle alone (sterile PBS, white bars) or vehicle containing CPDI-02 at 50 mg/kg (black bars) was administered IM to the left caudal thigh muscle. Average concentrations of pro-inflammatory markers in dermal abscess biopsies ±SD (n=5 mice per time point) at (A) 3 -hours post-treatment (9 -hours post-challenge) or (B) 18 -hours post-treatment (24 -hours post-challenge) were determined by multiplex ELISA and compared by two-tailed t test with Mann-Whitney post-test (P value shown). No differences in other major murine cytokines / chemokines involved in inflammation (IFN-γ, IL-2, IL-4, IL-5, IL-6, IL-10, IL-12p70, CXCL-1 [KC/GRO], TNF-α) were observed at these timepoints (Figure S3).
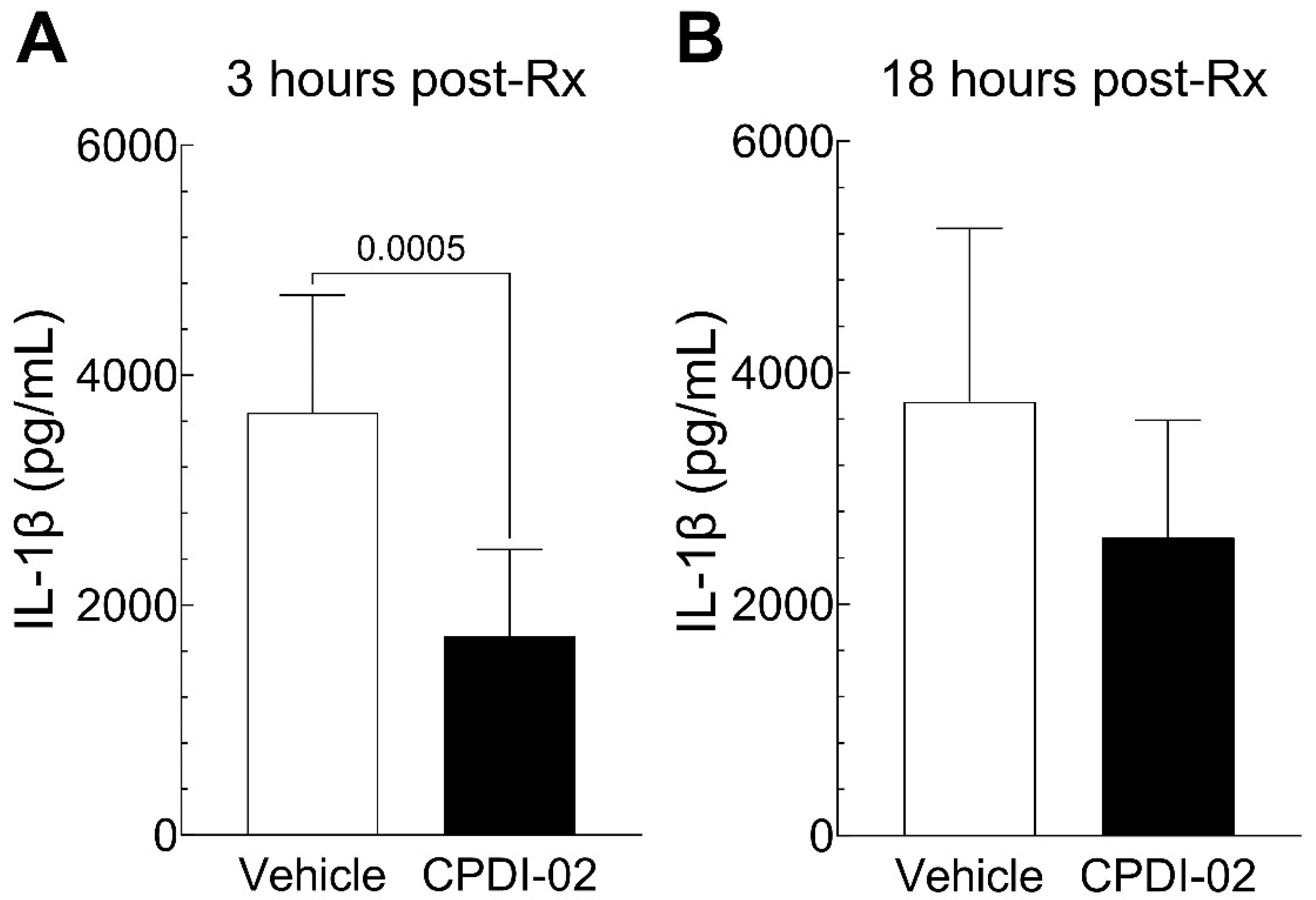
Figure 5.
Preliminary PK and distribution of CPDI-02 in dermal abscess-bearing female outbred mice after intravenous or intramuscular administration. On Day 0, community-acquired methicillin-resistant Staphylococcus aureus (CA-MRSA, USA300 strain, 5×107 CFU) was administered SQ from the left rear flank into the dorsal side of 6-week-old healthy female outbred CD-1 mice. At 6 -hours post-challenge, vehicle (PBS, white circles) or vehicle containing CPDI-02 (black circles) was injected into the tail vein [12.5 mg/kg] (IV) or left caudal thigh muscle [50 mg/kg] (IM) of healthy female outbred CD-1 mice (X weeks old) then (A) average plasma concentrations of CPDI-02 ±SD (n=5 mice) over 24 -hours post-treatment and (B) average mass of CPDI-02/g of abscess or indicated organ ±SD (n=5) at 24 -hours post-challenge were determined by LC-MS/MS and compared by 2-way ANOVA. AB – abscess; HT – heart; LG – lung; LV – liver; KD – kidneys; SP – spleen; BR – brain.
Figure 5.
Preliminary PK and distribution of CPDI-02 in dermal abscess-bearing female outbred mice after intravenous or intramuscular administration. On Day 0, community-acquired methicillin-resistant Staphylococcus aureus (CA-MRSA, USA300 strain, 5×107 CFU) was administered SQ from the left rear flank into the dorsal side of 6-week-old healthy female outbred CD-1 mice. At 6 -hours post-challenge, vehicle (PBS, white circles) or vehicle containing CPDI-02 (black circles) was injected into the tail vein [12.5 mg/kg] (IV) or left caudal thigh muscle [50 mg/kg] (IM) of healthy female outbred CD-1 mice (X weeks old) then (A) average plasma concentrations of CPDI-02 ±SD (n=5 mice) over 24 -hours post-treatment and (B) average mass of CPDI-02/g of abscess or indicated organ ±SD (n=5) at 24 -hours post-challenge were determined by LC-MS/MS and compared by 2-way ANOVA. AB – abscess; HT – heart; LG – lung; LV – liver; KD – kidneys; SP – spleen; BR – brain.
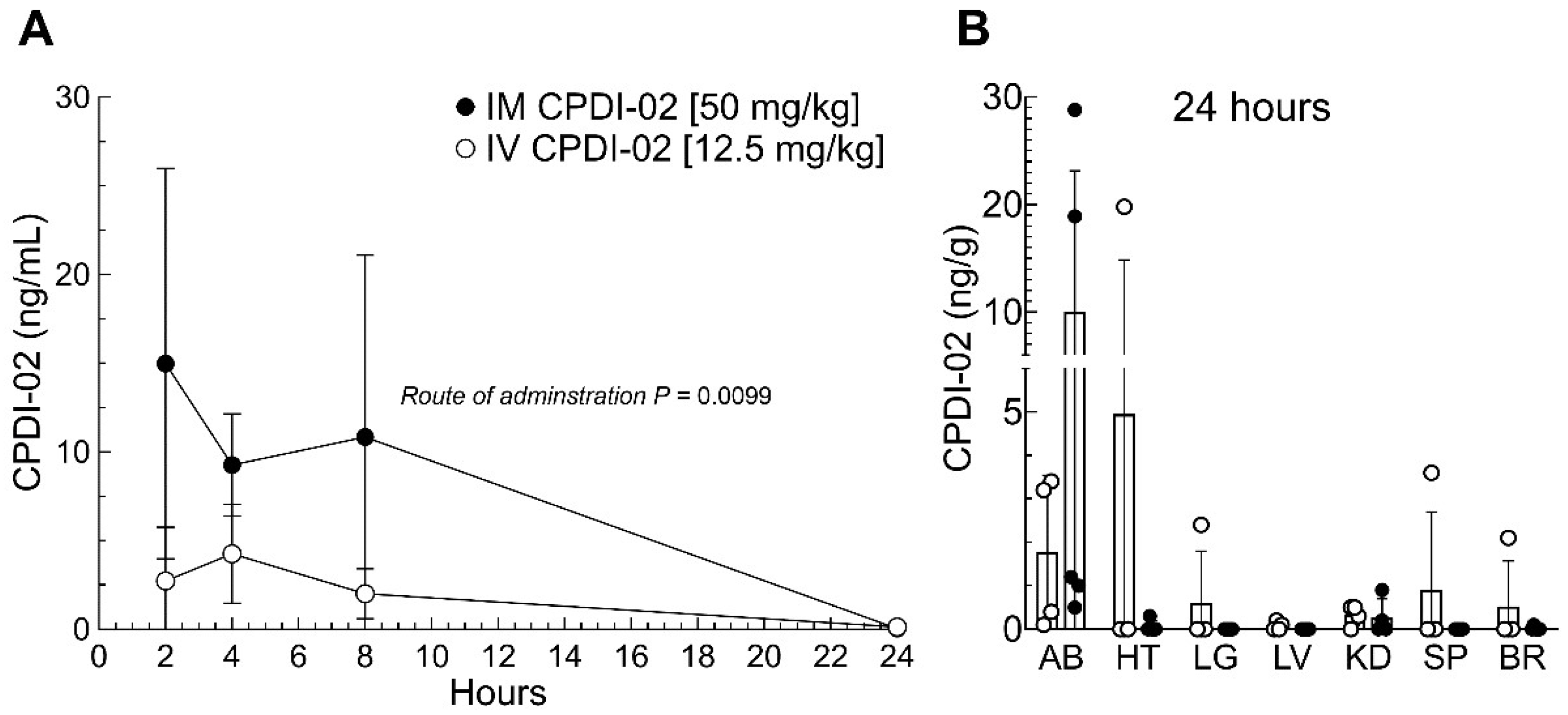
Figure 6.
IM dose escalation of CPDI-02 in healthy male and female outbred CD-1 mice. (A) Vehicle alone (PBS, white symbols) or vehicle containing CPDI-02 (black symbols) was injected biweekly (↓) into the left caudal thigh muscle of healthy male and female outbred CD-1 mice (4 weeks old) at the indicated dose (black arrows) and average daily body weights ±SEM (n=6 mice) were compared within each sex to vehicle alone by 2-way ANOVA. (B) Pain and distress were scored on the day of each injection based on standardized signs in mice (Table S1). Signs of mild (score 2) to moderate (score 3) distress were observed starting 15 minutes after injection with CPDI-02 and resolved after 1 to 2 hours. Four days after the final injection (Day 28), average values for complete blood count (CBC) with differential ±SD (n=X mice) were determined by hematology analyzer (Table S2) and compared by ordinary two-way ANOVA with uncorrected Fisher’s LSD and single pooled variance. Differences in CBC were only observed between (C) platelet concentrations and (D) platelet volumes.
Figure 6.
IM dose escalation of CPDI-02 in healthy male and female outbred CD-1 mice. (A) Vehicle alone (PBS, white symbols) or vehicle containing CPDI-02 (black symbols) was injected biweekly (↓) into the left caudal thigh muscle of healthy male and female outbred CD-1 mice (4 weeks old) at the indicated dose (black arrows) and average daily body weights ±SEM (n=6 mice) were compared within each sex to vehicle alone by 2-way ANOVA. (B) Pain and distress were scored on the day of each injection based on standardized signs in mice (Table S1). Signs of mild (score 2) to moderate (score 3) distress were observed starting 15 minutes after injection with CPDI-02 and resolved after 1 to 2 hours. Four days after the final injection (Day 28), average values for complete blood count (CBC) with differential ±SD (n=X mice) were determined by hematology analyzer (Table S2) and compared by ordinary two-way ANOVA with uncorrected Fisher’s LSD and single pooled variance. Differences in CBC were only observed between (C) platelet concentrations and (D) platelet volumes.
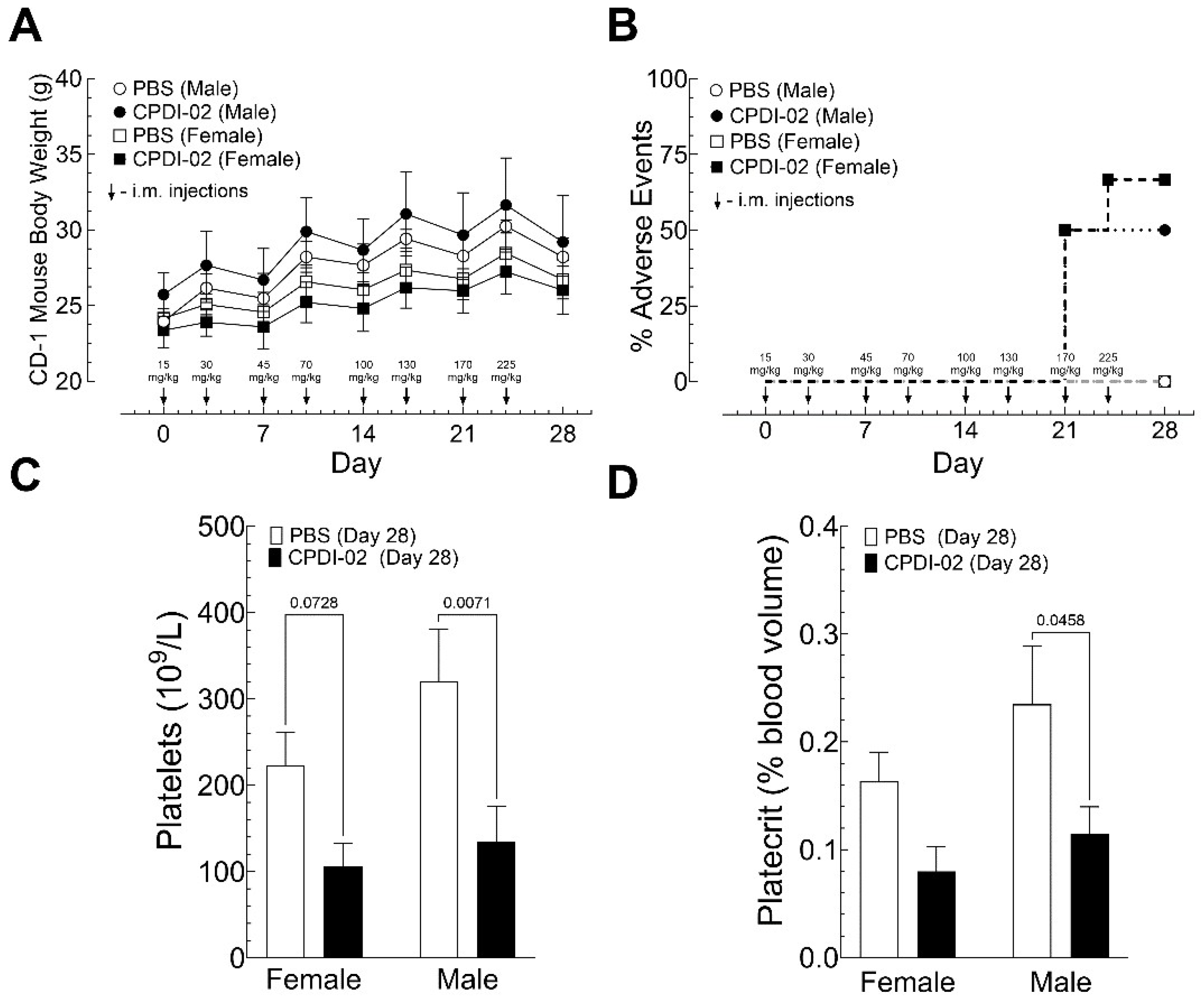
Figure 7.
Repeat IM dosing of CPDI-02 in healthy male and female outbred mice. Vehicle alone (PBS) or vehicle containing CPDI-02 was injected biweekly(↓) into the [left?] caudal muscle of healthy male and female outbred CD-1 mice (6 weeks old) at the indicated dose (black arrows) and(A) average daily body weights ±SEM (n=10 mice) were compared within each sex to vehicle alone by 2-way ANOVA and (B) survival for each treatment group was compared by simple Kaplan-Meier analysis with Mantel-Cox Log-rank test (Log-rank P values shown where relevant). No signs of distress were observed in any treatment group up to 2 hours after injection. Four days after the final injection (Day 32), average values for complete blood count (CBC) with differential ±SD (n=X mice) were determined by hematology analyzer (Table S3) and compared by compared by ordinary two-way ANOVA with uncorrected Fisher’s LSD and single pooled variance.
Figure 7.
Repeat IM dosing of CPDI-02 in healthy male and female outbred mice. Vehicle alone (PBS) or vehicle containing CPDI-02 was injected biweekly(↓) into the [left?] caudal muscle of healthy male and female outbred CD-1 mice (6 weeks old) at the indicated dose (black arrows) and(A) average daily body weights ±SEM (n=10 mice) were compared within each sex to vehicle alone by 2-way ANOVA and (B) survival for each treatment group was compared by simple Kaplan-Meier analysis with Mantel-Cox Log-rank test (Log-rank P values shown where relevant). No signs of distress were observed in any treatment group up to 2 hours after injection. Four days after the final injection (Day 32), average values for complete blood count (CBC) with differential ±SD (n=X mice) were determined by hematology analyzer (Table S3) and compared by compared by ordinary two-way ANOVA with uncorrected Fisher’s LSD and single pooled variance.
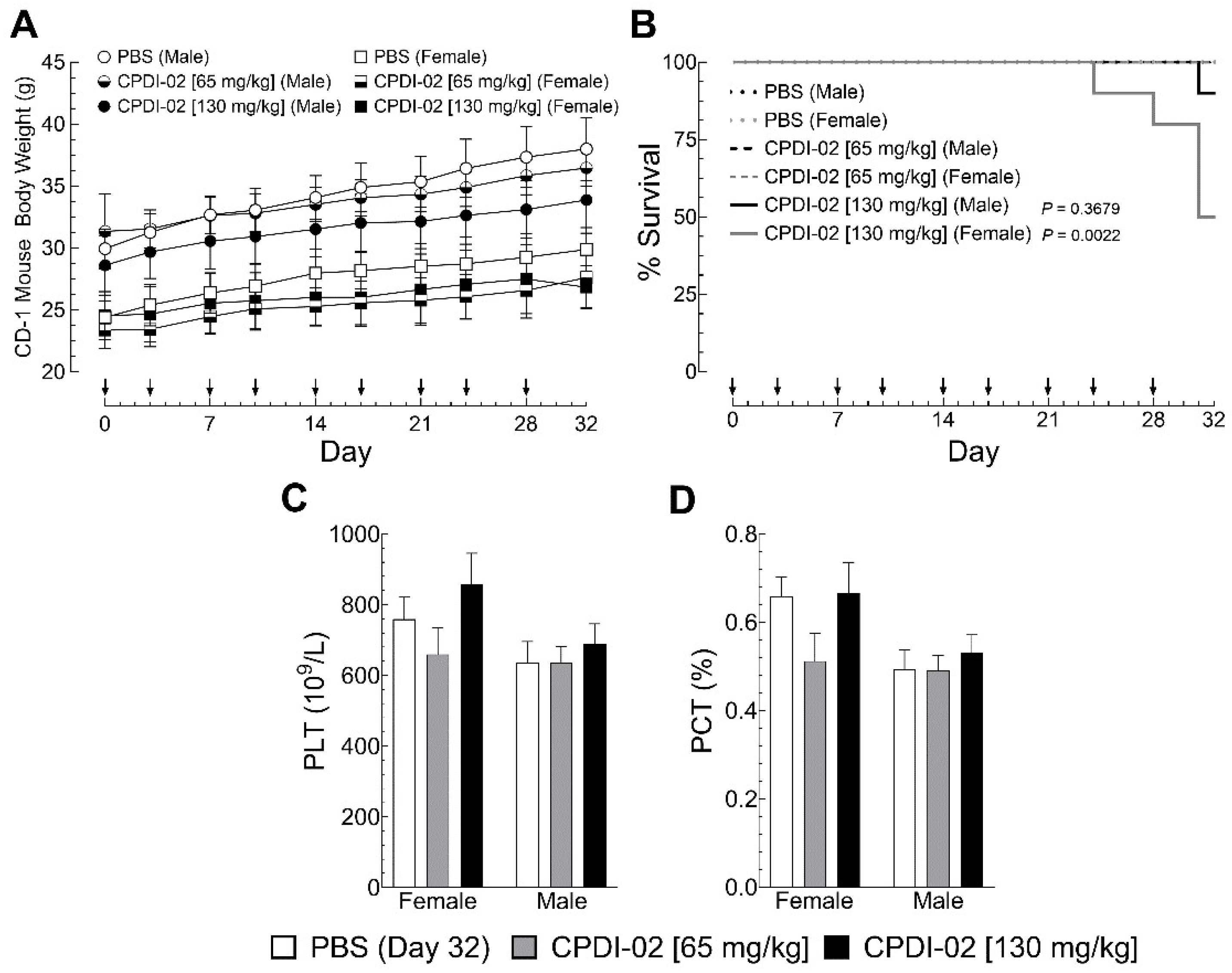

Table 1.
Molecular masses and sequences of CPDI-02 and inactive, scrambled scCPDI-02.
| Peptide | Molecular mass (g/mol) | Amino Acid Sequence |
|---|---|---|
| CPDI-02 | 1241.6 | Tyr01Ser02Phe03Lys04Asp05Met06Pro07(nme-Leu)08(D-Ala)09Arg10 |
| scCPDI-02 | 1241.6 | (nme-Leu)01Arg02Met03Tyr04Lys05Pro06(D-Ala)07Phe08Asp09Ser10 |
1 nme-Leu = N-methyl-leucine
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



