Submitted:
03 October 2024
Posted:
05 October 2024
You are already at the latest version
Abstract
The S1 subunit of SARS-CoV-2 Spike has been found in the blood of covid patients and vaccinated individuals. From BioGRID, we selected 146 significant human proteins experimentally interacting with S1. Then, we derived an interactome model that facilitated the study of functional activities. Through a reverse engineering approach, we identified 27 specific one-to-one interac-tions of S1 with the human proteome. S1 interacts in this manner independently from the biological context in which it operates, be it infection or vaccination. Instead, when it works together with viral proteins, carry multiple attacks on single human proteins out, showing a different functional engagement. Through Cytoscape we showed functional implications and its tropism to human organs/tissues, such as nervous system, liver, blood, and lungs. As a single protein, S1 operates in a complex metabolic landscape which includes 2557 GO biological processes, much more than the 1430 terms controlled when operating in a group. A Data-Merging approach shows that the total proteins involved by S1 in the cell are over 60,000 with an average involvement per single bio-logical process of 26.19. However, many human proteins get entangled in over 100 biological different activities each. Clustering analysis showed statistically significant activations of many molecular mechanisms, like those related to hepatitis-B infections. This suggests potential in-volvement in carcinogenesis, based on a viral strategy that uses the ubiquitin system to impair the tumor suppressor and antiviral functions of TP53, as well as the role of RPS27A in protein turnover and cellular stress responses.
Keywords:
1. Introduction
2. Materials and Methods
2.1. BioGRID
2.2. STRING
2.3. Protein Enrichment
2.4. Cytoscape and Network Topology Analysis.
2.5. CentiScaPe.
2.6. GO and KEGG Pathway Analyses
2.7. SARS2-Human Proteome Interaction Database (SHPID)
2.8. Highlighting the Nodes of a STRING Network Involved in the Same Biological Process (GO)
2.9. Evaluation of the HUB-and-Spoke Model
2.10. Cluster Analysis
2.11. Protein Intrinsic Disorder and Secondary Structure Prediction
2.12. Data Merging
2.13. CIDER
3. Results
3.1. A Brief Analysis of the Behavior that we Expect for S1 Free in Solution
3.2. Data Source
3.3. The Interactome-1060
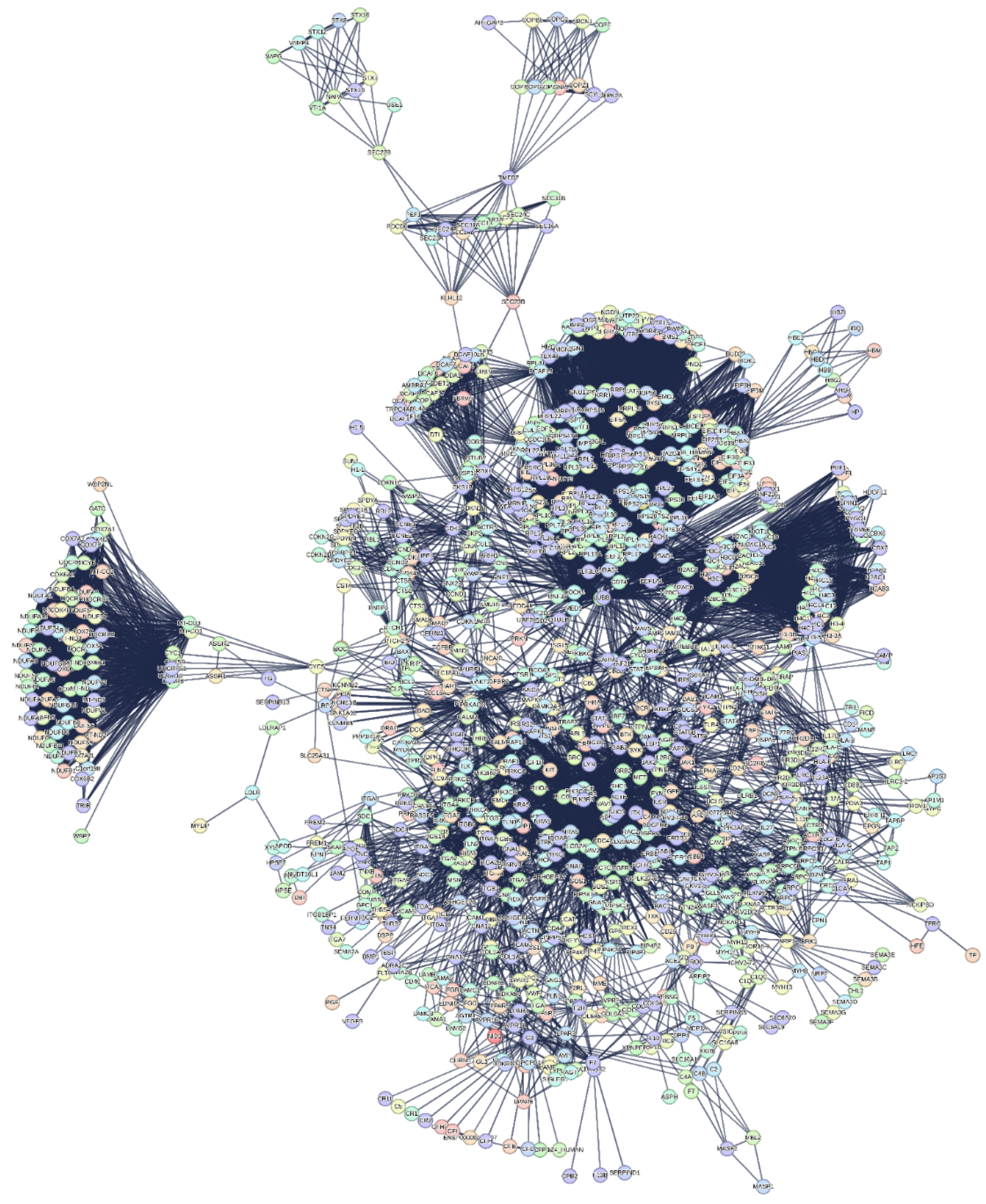
2.3.1. Quantitative Aspects of Interactome-1060 Functional Processes

| Biological Process | Terms significantly enriched | |
|---|---|---|
| Biological Process (Gene Ontology) | 1430 terms | |
| Molecular Function (Gene Ontology) | 165 terms | |
| Cellular Component (Gene Ontology) | 283 terms | |
| Reference publications (PubMed) | >10,000 publications | |
| Local network cluster (STRING) | 251 clusters | |
| KEGG Pathways | 202 pathways | |
| Reactome Pathways | 693 pathways | |
| WikiPathways | 302 pathways | |
| Disease-gene associations (DISEASES) | 114 diseases | |
| Tissue expression (TISSUES) | 167 tissues | |
| Subcellular localization (COMPARTMENTS) | 287 compartments | |
| Human Phenotype (Monarch) | 787 phenotypes | |
| Annotated Keywords (UniProt) | 103 keywords | |
| Protein Domains (Pfam) | 17 domains | |
| Protein Domains and Features (InterPro) | 144 domains | |
| Protein Domains (SMART) | 44 domains | |
| All enriched terms (without PubMed) | 4989 enriched terms | |
2.3.2. Significant Topological Parameters of the Interactome-1060
| Number of nodes | 1060 |
| Number of edges | 17493 ** |
| Average node degree | 33 |
| Avg. local clustering coefficient | 0.679 |
| Expected number of edges | 8382 |
| PPI enrichment p-value | <1.0✕10‒16 |
| Confidence score | 0.900 |
| Source channels | 6 |
| Network diameter | 10 |
| Network radius | 5 |
| Characteristic path-length | 3.717 |
| Network heterogeneity | 1.187 |
| Network density | 0.33 |
| Network centralization | 0.189 |
| Connected components | 1*** |
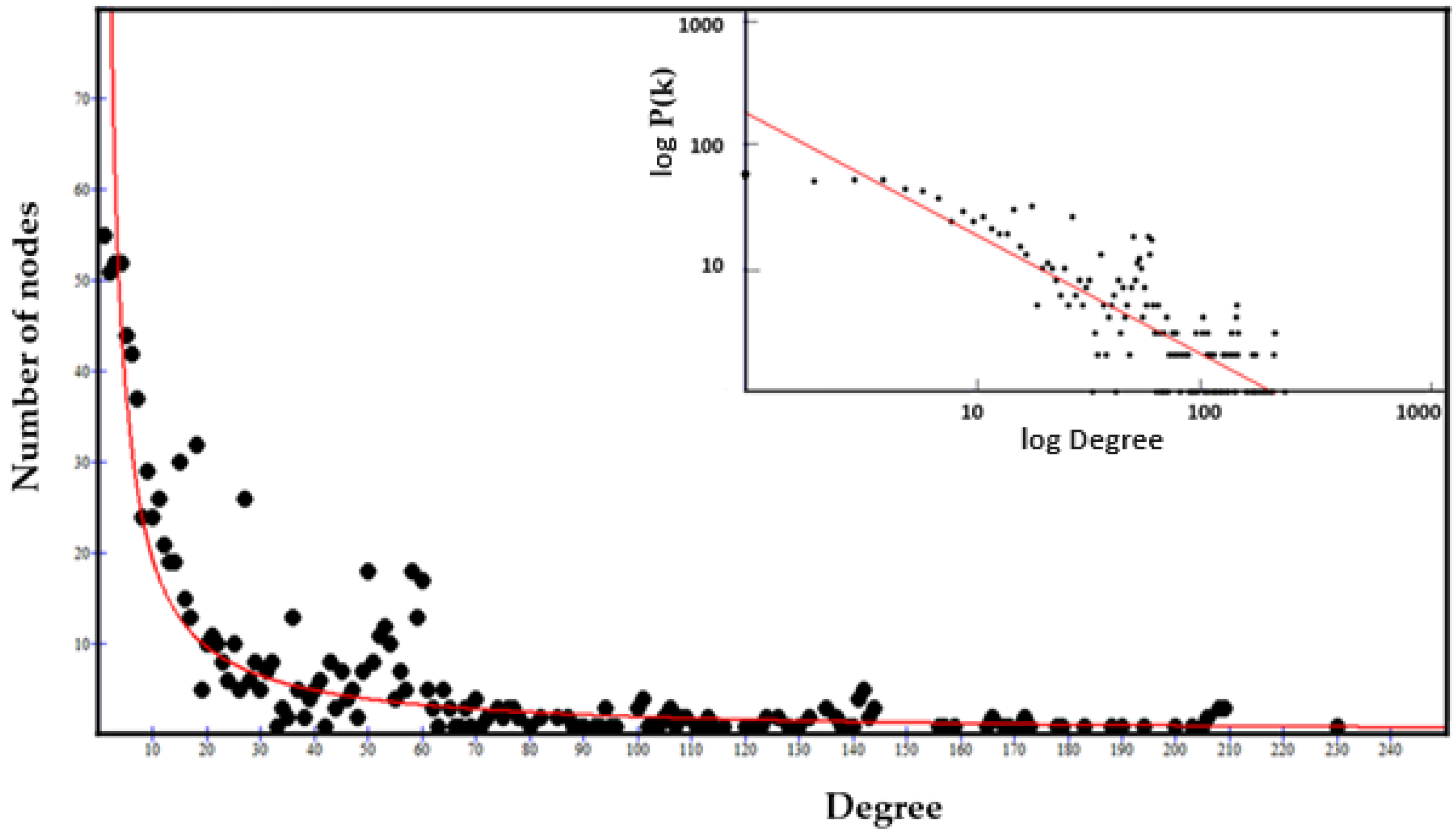
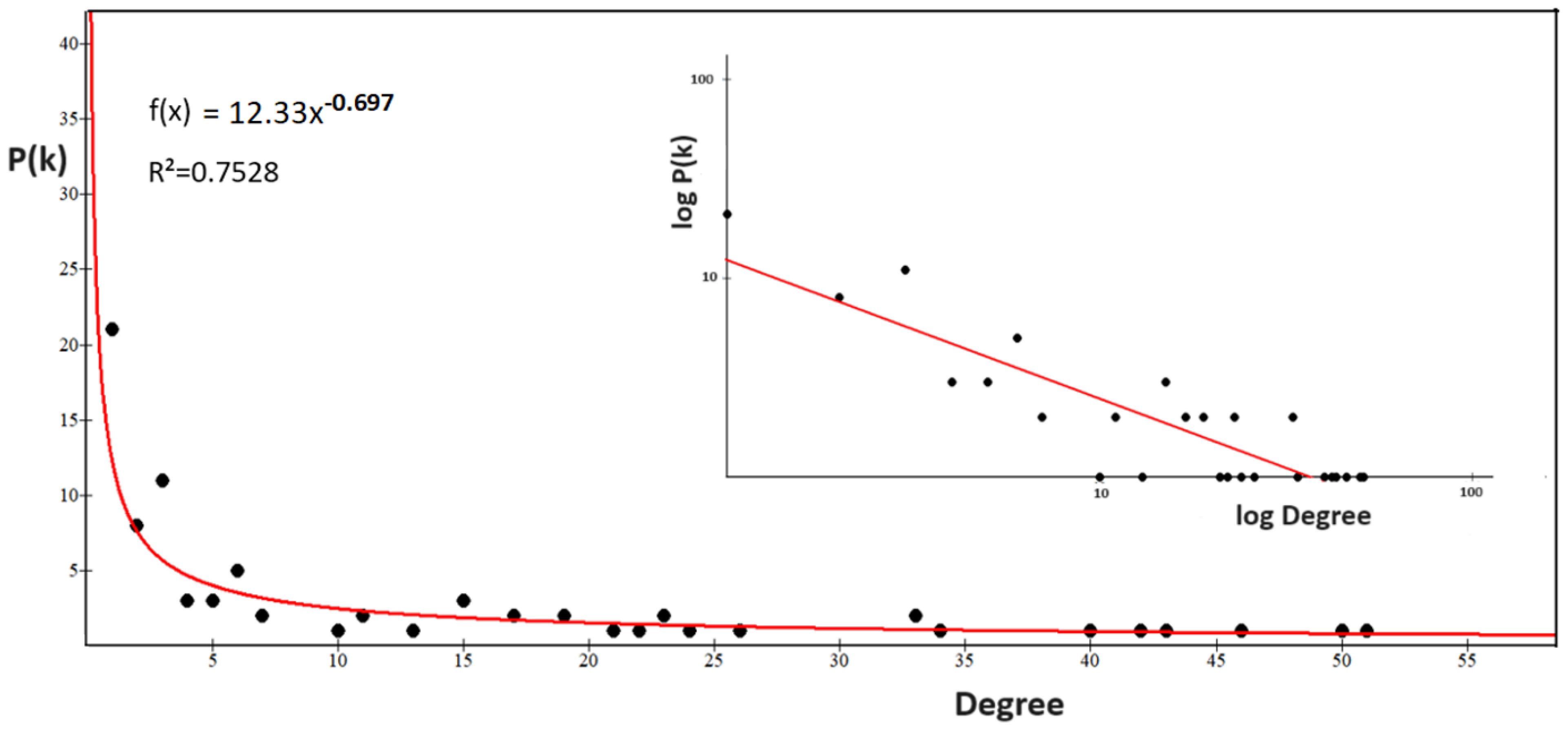
2.3.3. The Power Law of the Interactome-1060
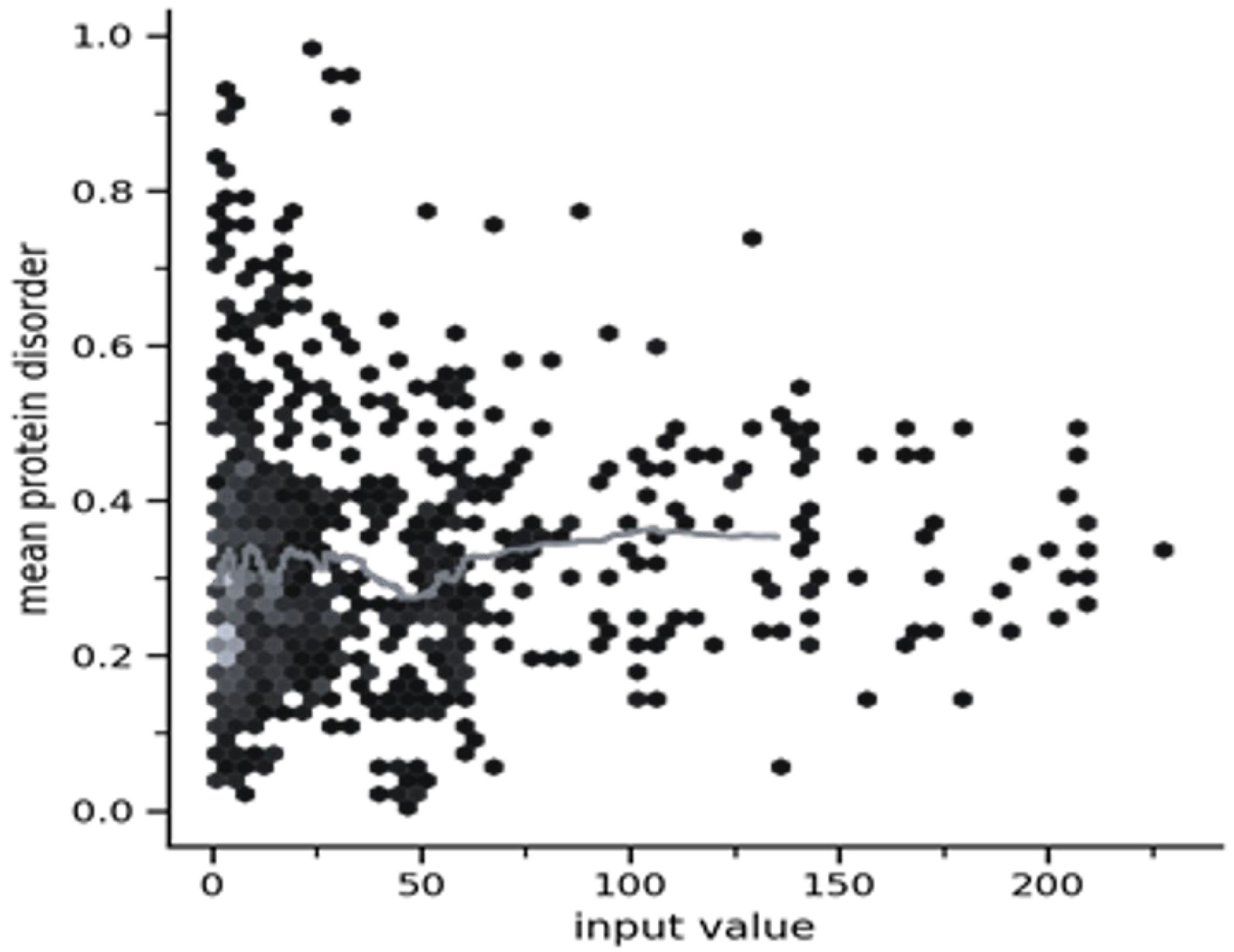
2.3.4. Origin of the Node Fitness in the Interactome-1060
2.3.5. Centralities Based Analysis of the Interactome-1060
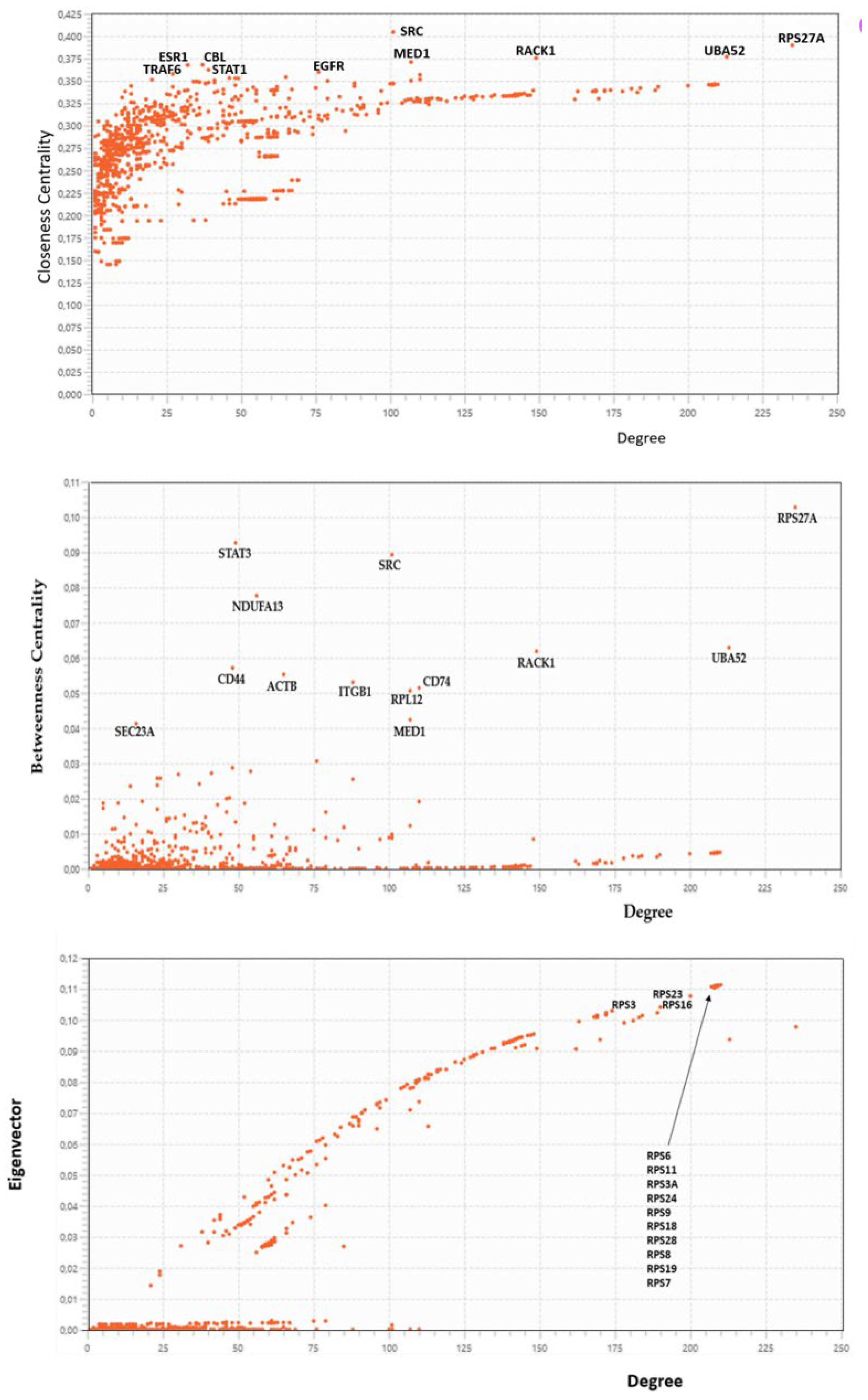
| HUB nodes | Degree | Bottleneck | Degree |
| RPS6 | 210 | RPS27A | 235 |
| RPS11 | 209 | UBA52 | 213 |
| RPS3A | 209 | RACK1 | 149 |
| RPS24 | 209 | CD74 | 110 |
| RPS9 | 209 | MED1 | 107 |
| RPS18 | 208 | SRC | 101 |
| RPS28 | 209 | EEF1A1 | 88 |
| RPS8 | 208 | EGFR | 76 |
| RPS19 | 207 | ACTB | 65 |
| RPS7 | 208 | CD44 | 48 |
| RPS23 | 200 | STAT3 | 49 |
| RPS16 | 190 | CBL | 37 |
| RPS3 | 174 | ||
| RPS15 | 172 | ||
| RPS5 | 189 | ||
| FAU | 172 | ||
| RPS13 | 184 | ||
| RPS21 | 169 | ||
| RPS17 | 169 | ||
| RPS14 | 183 | ||
| RPS27 | 181 |
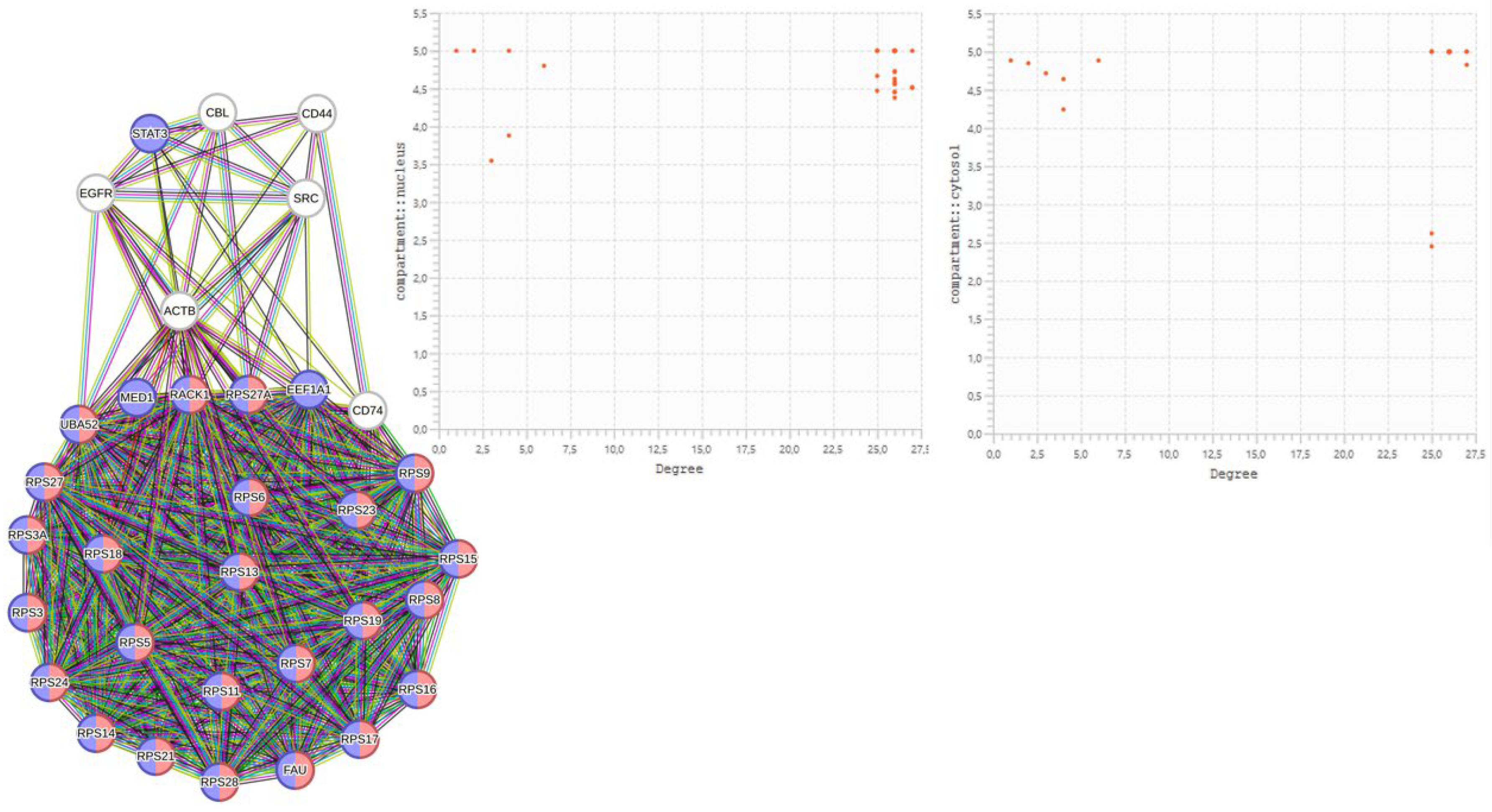
2.4.1. Justifications for a One-to-One Study
2.5.1. Reverse Engineering
2.6.1. The Interactome-814
| Number of nodes | 814 |
| Number of edges | 7409 |
| Average node degree | 15.9 |
| Avg. local clustering coefficient | 0.547 |
| Expected number of edges | 2285 |
| PPI enrichment p-value | <1.0✕10‒16 |
| Confidence score | 0.900 |
| Source channels | 6 |
| Network diameter | 7 |
| Network radius | 4 |
| Characteristic path length | 3.189 |
| Network heterogeneity | 1.042 |
| Network density | 0.22 |
| Network centralization | 0.138 |
| Connected components | 1** |
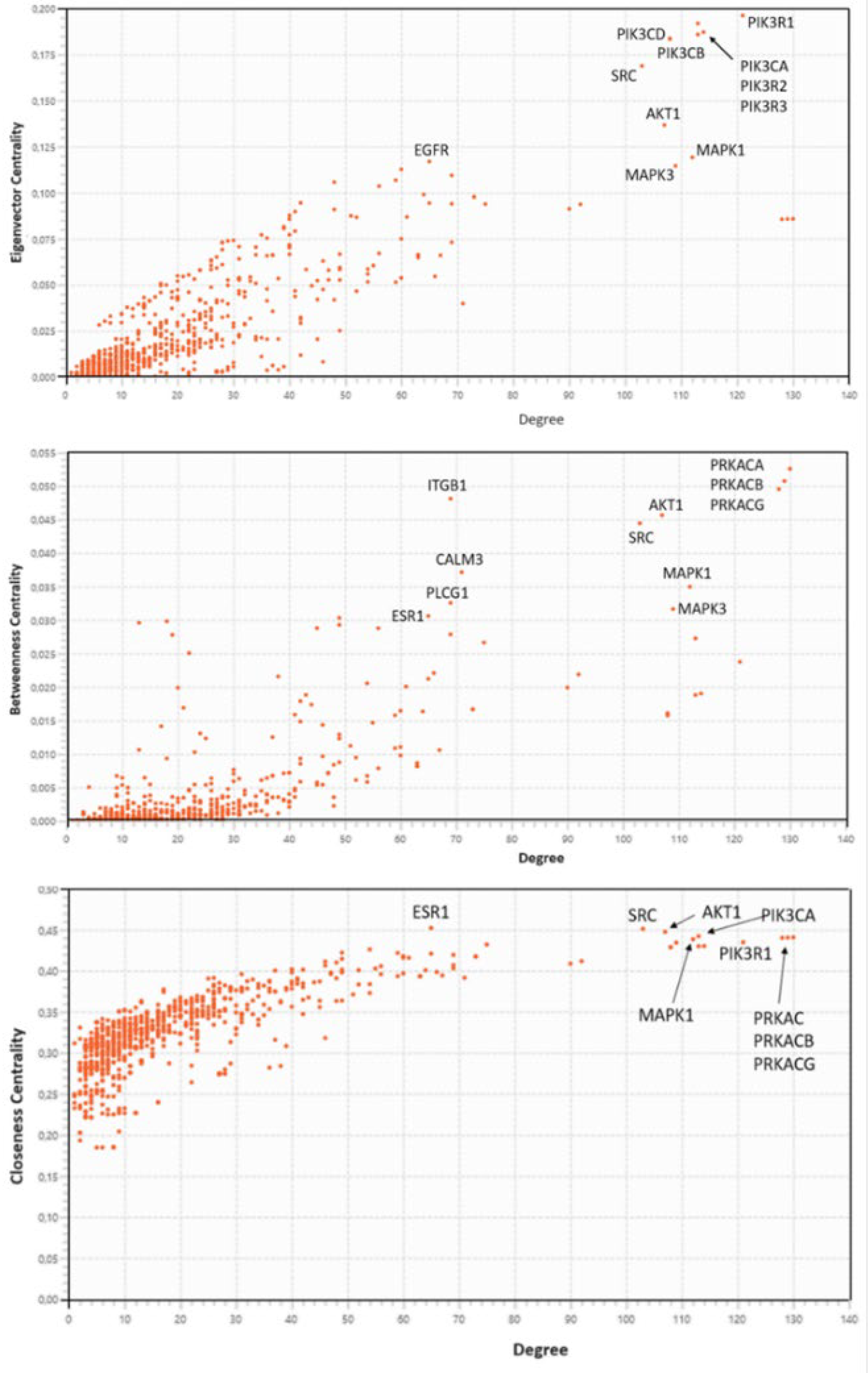
| HUB nodes | Degree | Bottleneck nodes | Degree |
|---|---|---|---|
| PIK3R1 | 121 | AKT1 | 65 |
| PIK3CA | 113 | EGFR | 103 |
| PIK3R2 | 114 | ESR1 | 107 |
| PIK3R3 | 113 | MAPK1 | 113 |
| PIK3CD | 108 | MAPK3 | 130 |
| PIK3CB | 108 | PIK3CA | 129 |
| SRC | 103 | PIK3R1 | 128 |
| AKT1 | 107 | PRKACA | 112 |
| MAPK1 | 112 | PRKACB | 121 |
| EGFR | 65 | PRKACG | 109 |
| MAPK3 | 109 | PTK2 | 75 |
| AKT3 | 73 | RHOA | 49 |
| AKT2 | 73 | SRC | 65 |
| ESR1 | 65 | TP53 | 69 |
| PLCG1 | 69 | ||
| TP53 | 75 | ||
| MAPK8 | 92 | ||
| MAPK9 | 90 |
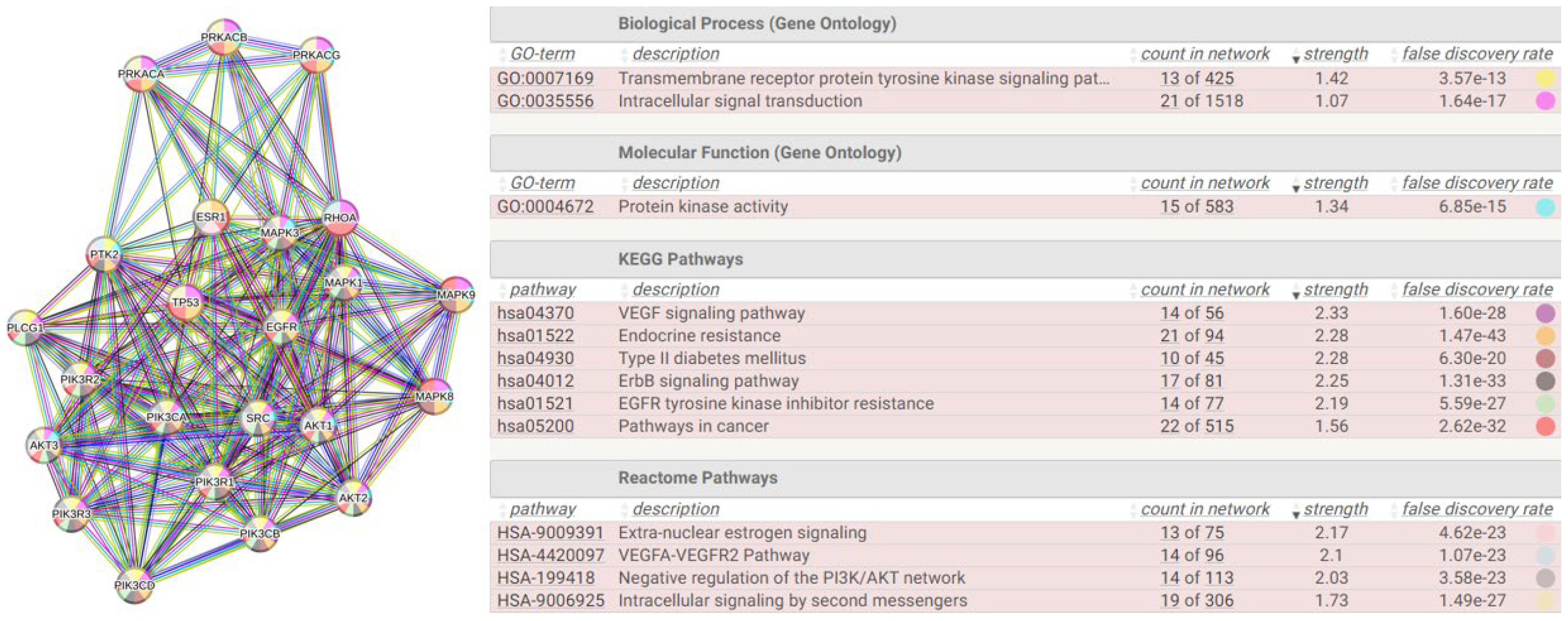
| Biological Process | Terms significantly enriched | |
|---|---|---|
| Biological Process (Gene Ontology) | 2557 terms | |
| Molecular Function (Gene Ontology) | 321 terms | |
| Cellular Component (Gene Ontology) | 231 terms | |
| Reference publications (PubMed) | >10,000 publications | |
| Local network cluster (STRING) | 246 clusters | |
| KEGG Pathways | 213 pathways | |
| Reactome Pathways | 828 pathways | |
| WikiPathways | 453 pathways | |
| Disease-gene associations (DISEASES) | 222 diseases | |
| Tissue expression (TISSUES) | 223 tissues | |
| Subcellular localization (COMPARTMENTS) | 218 compartments | |
| Human Phenotype (Monarch) | 1196 phenotypes | |
| Annotated Keywords (UniProt) | 124 keywords | |
| Protein Domains (Pfam) | 14 domains | |
| Protein Domains and Features (InterPro) | 222 domains | |
| Protein Domains (SMART) | 52 domains | |
| All enriched terms (without PubMed) | 7120 enriched terms | |
2.7.1. Data Merging
| Number of Biological Processes (GO) (%) |
Redundant genes (%) * |
Coding genes |
Average genes per single process. |
Genes found >100 times |
|
| Merging of 1060+814 (after pruning) ** | 2837 (total) |
68,003 (total) |
--- | 23.97 |
----- |
| Coupled processes in the merging of 1060+814 | 554 (39) *** | 24,301 (35.8) |
944 | 21.9 | ABL1, AGT, AKT1, APOE, BCL2, BTK, CD28, EGFR, FYN, HLA, HRAS, IL12A, IL12B, IL12RB1, IL23A, JAK2, KDR, KIT, LYN, MAPK1, RHOA, SRC, SYK, THBS1, TICAM1, TLR4, TNF, TYK2, ZAP70. |
| Uncoupled processes in 814 | 1515 (53) | 39,691 (58.3) |
771 | 26.19 | ADA, ADCY8, ADRA1A, ADRA2A, AGT, AGTR2, AKT1, AKT2, APOE, APP, AR, ASPH, ATF2, ATF4, ATP2B4, AVP, AVPR, BAD, BAK1, BAX, BCL2, CALM1, CTNNB1, CYBA, DLG1, EDNRA, EGFR, EP300, FOS, FOXO1, FOXO3, FYN, GNAI2, GSK3A, GSK3B, HIF1A, HSP90AA1, HSP90AB1, HSPA5, IGF1R, IL12B, IL2, INSR, IRAK1, ITGB1, JAK2, JUN, KCNE1, KCNQ1, KDR, KIT, LYN, MALT1, MAP2K1, MAPK1, MAPK14, MAPK3, MAPK8, MED1, MMP9, MTOR, MYD88, NFKB1, NKX3-1, NOS1, PODGFRA, PIK3CA, PIK3CG, PLCG2, PPARA, PPARG, PPP3CA, PRKCD, PTEN, PTK2B, PTPN2, RELA, RHOA, RIPK1, RIPK2, RACK1, RPTOR, SLC8A1, SMAD3, SNCA, SRC, STAT3, SYK, TGFB1, THBS1, TIRAP, TLR2, TLR4, TNF, TP53, |
| Uncoupled processes in 1060 | 214 (8) | 4,011 (5.9) |
701 | 18.74 | Family EIF, Eukaryotic initiation factors gene family, (230), histones (295), family NDUF (352), family RPL (516), family RPS (411). **** |
2.8.1. Clustering Analysis
| 1-CLUSTERS OF UNCOUPLED FUNCTIONS OF INTERACTOME-1060 | ||||
|---|---|---|---|---|
| Cluster No. | Primary description | GO-term | p-value | Gene count * |
| 1 | Cytoplasmic translation | GO:0002181 | 4.83 × 10−83 | 266 |
| 2 | Focal adhesion | GO:0005925 | 7.61 × 10−48 | 189 |
| 3 | Aerobic electron transport chain | GO:0019646 | 1.49 × 10−47 | 75 |
| 4 | DNA replication-dependent chromatin assembly | GO:0006335 | 6.67 × 10−19 | 44 |
| 5 | Antigen processing and presentation | GO:0019882 | 6.67 × 10−16 | 33 |
| 6 | Complement activation, classical pathway | GO:0006958 | 1.67 × 10−11 | 23 |
| 7 | COPII vesicle coat | GO:0030127 | 2.46 × 10−12 | 20 |
| 8 | Activation of phospholipase C activity | GO:0007202 | 3.30 × 10−06 | 18 |
| 9 | COPI vesicle coat | GO:0030126 | 1.90 × 10−09 | 11 |
| 10 | Cholesterol metabolism | hsa04979 | 2.70 × 10−04 | 10 |
| Cluster No. | Secondary description | GO-term | p-value | Gene count |
| 1 | Formation of a pool of free 40S subunits | HAS-72689 | 7.09 × 10−91 | - |
| 3 | Respiratory chain complex | GO:0098803 | 7.29 × 10−52 | - |
| 4 | CENP-A containing nucleosome | GO:0043505 | 5.51 × 10−15 | - |
| 6 | Complement and coagulation cascades | hsa04610 | 4.06 × 10−09 | - |
| 8 | G alpha (q) signaling events | HAS-418597 | 1.11 × 10−03 | - |
| 10 | Plasma lipoprotein particle clearance | GO:0034381 | 5.60 × 10−03 | - |
| Cluster No. | Tertiary description | GO-term | p-value | Gene count |
| 1 | Ribosome | GO:0005848 | 2.08 × 10−79 | - |
| ________________________________________________________________________________________________________ | ||||
| 2—CLUSTERS OF COUPLED FUNCTIONS OF INTERACTOMES-1060+814 | ||||
| Cluster No. | Primary description | GO-term | p-value | Gene count |
| 1 | Positive regulation of transferase activity | GO:0051347 | 2.76 × 10−63 | 409 |
| 2 | Focal adhesion | GO:0005925 | 5.66 × 10−44 | 232 |
| 3 | ECM-receptor interaction | hsa04512 | 9.88 × 10−36 | 79 |
| 4 | Long-term potentiation | HAS-9620244 | 7.01 × 10−06 | 54 |
| 5 | Rho protein signal transduction | GO:00072666 | 9.12 × 10−08 | 43 |
| 6 | Formation of Fibrin Clot (Clotting Cascade) | CL:18784 | 1.09 × 10−06 | 37 |
| 7 | Antigen processing and presentation | GO:0019882 | 7.05 × 10−13 | 35 |
| 8 | Complement activation | GO:006956 | 1.33 × 10−18 | 33 |
| 9 | Cholesterol metabolism | hsa04979 | 1.80 × 10−03 | 13 |
| 10 | Renin-angiotensin system | hsa4614 | 2.09 × 10−03 | 9 |
| Cluster No. | Secondary description | GO-term | p-value | Gene count |
| 1 | Cellular responses to stress | 7.56 × 10−11 | - | |
| 2 | Mixed, incl. Constitutive Signaling by Aberrant PI3K in Cancer, and FCERI mediated Ca+2 mobilization | CL:17328 | 2.28 × 10−34 | - |
| 3 | Protein complex involved in cell adhesion | GOCC:0098636 | 1.09 × 10−27 | - |
| 4 | Calmodulin-binding | KW.0112 | 5.05 × 10−15 | - |
| 5 | G alpha (12/13) signaling events | HAS-416482 | 1.69× 10−09 | - |
| 6 | Blood coagulation | GO:0007596 | 7.29 × 10−24 | - |
| 9 | Regulation of plasma lipoprotein particle levels | GO:0097006 | 2.16 × 10−05 | - |
| Cluster No. | Tertiary description | GO-term | p-value | Gene count |
| 1 | Protein kinase binding | GO:0019901 | 6.94 × 10−74 | - |
| 5 | Mixed, incl. Sema4D in semaphorin signaling, and ARHGEF1-like, PH domain. | CL:17973 | 1.765× 10−06 | - |
| 9 | Protein-lipid complex | GO:0032994 | 6946 × 10−74 | - |
| 3-CLUSTERS OF UNCOUPLED FUNCTIONS OF INTERACTOME-814 | ||||
| Cluster No. | Primary description | GO-term | p-value | Gene count |
| 1 | Hepatitis B | hsa055161 | 4.98 × 10−73 | 259 |
| 2 | mTOR signaling pathway | hsa04150 | 2.05 × 10−36 | 139 |
| 3 | Fc gamma R-mediated phagocytosis | hsa04555 | 6.62 × 10−32 | 113 |
| 4 | Long-term depression | hsa04730 | 1.72 × 10−29 | 72 |
| 5 | Blood vessels diameter maintenance | GO:0097746 | 3.61 × 10−13 | 61 |
| 6 | ECM-receptor interaction | hsa04512 | 9.96 × 10−24 | 56 |
| 7 | Complement activation | GO:0006956 | 3.73 × 10−18 | 32 |
| 8 | Renin-angiotensin system | hsa04614 | 1.10 × 10−04 | 14 |
| 9 | Glycerophospholipid metabolism | Hsa00564 | 2.71 × 10−08 | 13 |
| 10 | Plasma lipoprotein particle remodeling | GO:0034369 | 9.94 × 10−05 | 12 |
| Cluster No. | Secondary description | GO-term | p-value | Gene count |
| 3 | Constitutive Signaling by Aberrant PI3K in Cancer | 1.12 × 10−33 | - | |
| 4 | Calmodulin binding | GO:0005516 | 1.11 × 10−21 | - |
| 5 | Mixed, incl. Heterotrimeric G-protein complex, and Signaling transduction inhibitor | CL24307 | 6.90 × 10−13 | - |
| 6 | Cell adhesion mediated by integrin | GO:0033627 | 2.32 × 10−14 | - |
| 7 | Initial triggering of complement | HAS-166663 | 5.26 × 10−12 | - |
| 8 | Dipeptidyl-peptidase activity, and Meprin A complex | CL31769 | 6.08 × 10−03 | - |
| 10 | Cholesterol metabolism | hsa04979 | 1.98 × 10−49 | |
| Cluster No. | Tertiary description | GO-term | p-value | Gene count |
| 3 | GPVI-mediated activation cascade, and SH2 domain superfamily | CL:17470 | 1.53 × 10−27 | - |
| 5 | Vascular smooth muscle contraction | hsa04270 | 8.57 × 10−39 | - |
| 6 | Integrin | KW-0401 | 1.88 × 10−16 | - |
| 10 | Protein-lipid complex | GO:0032994 | 5.40 × 10−03 | - |
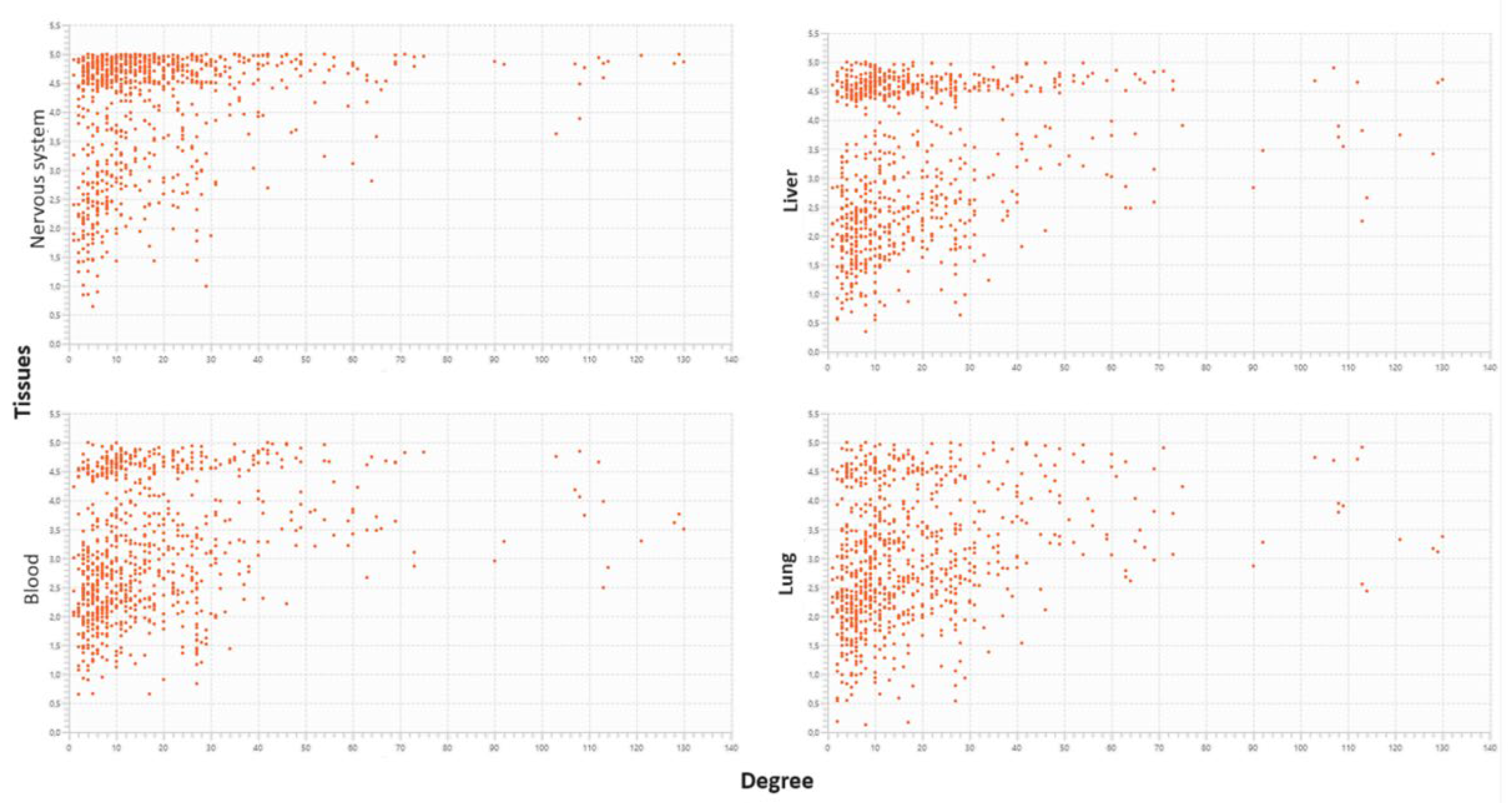
2.8.1.1. The liver aspects
2.8.1.2. Vascular aspects
2.8.1.3. Cumulative effects may originate cancerous involvement
2.8.1.4. Neural effects
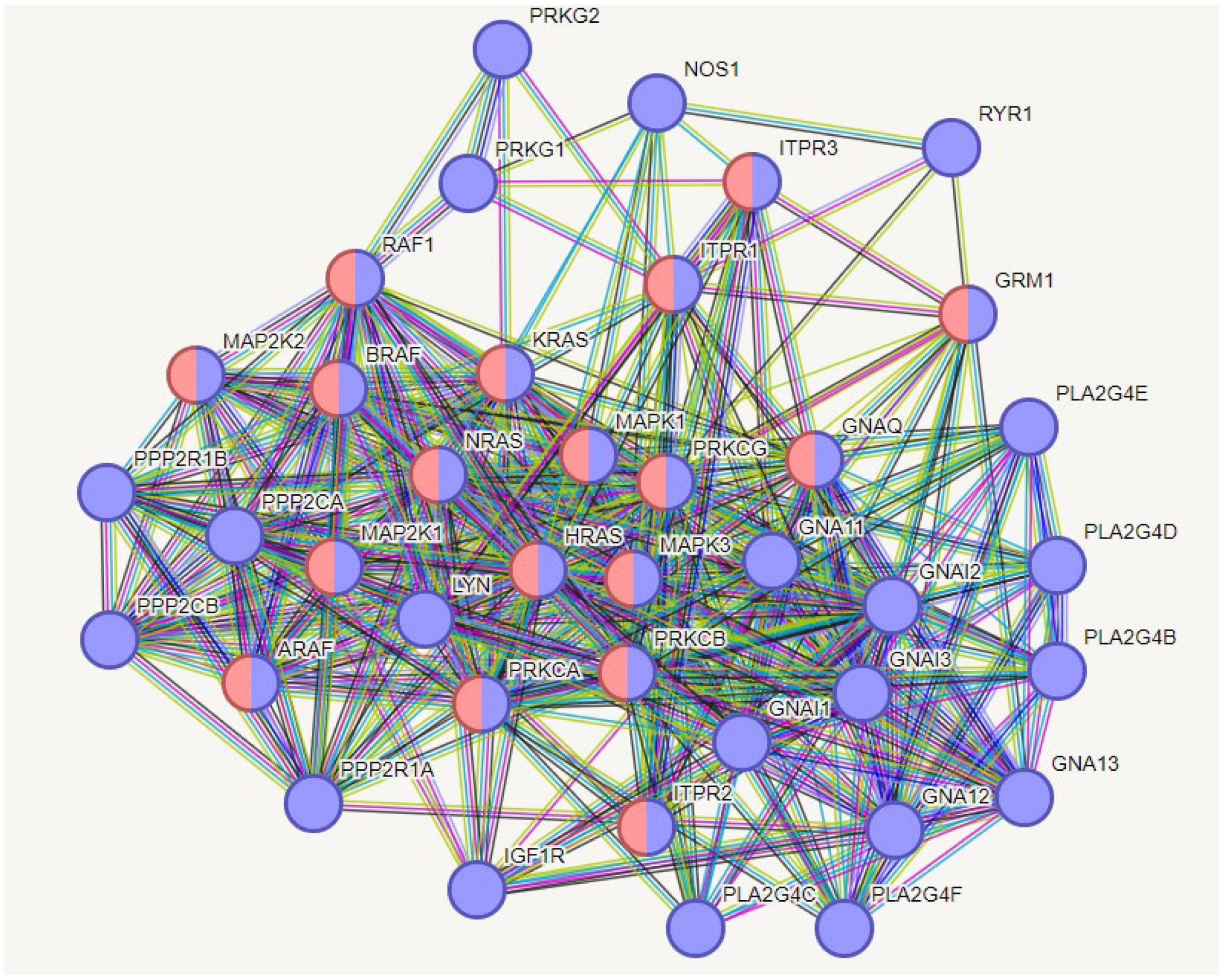
3. Discussion
3.1. Considerations on Cancer Development
3.2. Other Observations that Support Cancer Development
3.3. TP53 Interactions
3.3.1. RPS27A Interactions
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Perimeter Limits of Interactomic Analysis
| Interaction type | Abundance | Confidence score | Incidence% |
|---|---|---|---|
| Total interactions | 34,986* | - | - |
| No experimental characterization | 3,016 | - | 8.62 |
| Highest score experimentally proven interactions | 19,556 | Score ≥ 0.9 | 55.89 |
| High score experimentally proven interactions | 5,233 | 0.7 ≤ score < 0.9 | 14.94 |
| Medium score experimentally proven interactions | 2,722 | 0.4 ≤ score < 0.7 | 7.78 |
| Low score interactions | 4,462 | Score ≤ 0.4 | 12.75 |
| Combined experimental interactions used in this study | 24,789 | high and highest | 70.85 |
References
- Pietzner, M., Denaxas, S., Yasmeen, S., Ulmer, M. A., Nakanishi, T., Arnold, M., Kastenmüller, G,. Hemingway, H., & Langenberg, C. (2024). Complex patterns of multimorbidity associated with severe COVID-19 and long COVVID. Communications Medicine, 4(1), 94.
- Ewing, A. G., Salamon, S., Pretorius, E., Joffe, D., Fox, G., Bilodeau, S., & Bar-Yam, Y. (2024). Review of organ damage from COVID and Long COVID: a disease with a spectrum of pathology. Medical Review.
- Yu, G., & Huang, H. (2024). The logic of coronavirus infection: revealing the heterogeneity of disease progression and treatment outcomes in COVID patients. Res. Square, preprint. [CrossRef]
- Fernández-de-Las-Peñas, C., Torres-Macho, J., Catahay, J. A., Macasaet, R., Velasco, J. V., Macapagal, S., et al. & Notarte, K. I. (2024). Is antiviral treatment at the acute phase of COVID-19 effective for decreasing the risk of long-COVID? A systematic review. Infection, 52(1), 43-58.
- Greene, C., Connolly, R., Brennan, D., Laffan, A., O’Keeffe, E., Zaporojan, L., O’Callaghan, O., Thomson, B., Connolly, E., Argue, R., et al., (2024). Blood–brain barrier disruption and sustained systemic inflammation in individuals with long COVID-associated cognitive impairment. Nature neuroscience, 27(3), 421-432.
- Strongin, S. R., Stelson, E., Soares, L., Sukhatme, V., Dasher, P., Schito, M., Challa, AP., Geng, LN., & Walker, T. A. (2024). Using real-world data to accelerate the search for long COVID therapies. Life Sciences, 122940.
- Nahalka, J. (2024). 1-L Transcription of SARS-CoV-2 Spike Protein S1 Subunit. International Journal of Molecular Sciences, 25(8), 4440.
- Parry, PI; Lefringhausen, A.; Turni, C.; Neil, CJ; Cosford, R.; Hudson, NJ; Gillespie, J. 'Spikeopathy': la proteina spike del COVID-19 è patogena, sia dal virus che dall'mRNA del vaccino. Biomedicines 2023, 11, 2287. [ Google Scholar] [ CrossRef ].
- Mulroney, T. E., Pöyry, T., Yam-Puc, J. C., Rust, M., Harvey, R. F., Kalmar, L., Horne, E., Booth, L., Ferreira, AP., Stoneley, M., et al., (2024). N 1-methylpseudouridylation of mRNA causes+ 1 ribosomal frameshifting. Nature, 625(7993), 189-194.
- Colonna, G. (2024). Understanding the SARS-CoV-2–Human Liver Interactome Using a Comprehensive Analysis of the Individual Virus–Host Interactions. Livers, 4(2), 209-239.
- Mansueto, G., Fusco, G., & Colonna, G. (2024). A Tiny Viral Protein, SARS-CoV-2-ORF7b: Functional Molecular Mechanisms. Biomolecules, 14(5), 541.
- Sun, Z., Ren, K., Zhang, X., Chen, J., Jiang, Z., Jiang, J., ... & Li, L. (2021). Mass spectrometry analysis of newly emerging coronavirus HCoV-19 spike protein and human ACE2 reveals camouflaging glycans and unique post-translational modifications. Engineering, 7(10), 1441-1451.
- Mouliou, D. S., & Dardiotis, E. (2022). Current evidence in SARS-CoV-2 mRNA vaccines and post-vaccination adverse reports: knowns and unknowns. Diagnostics, 12(7), 1555.
- Cosentino, M., & Marino, F. (2022). The spike hypothesis in vaccine-induced adverse effects: questions and answers. Trends in molecular medicine, 28(10), 797-799.
- Tan, X., Lin, C., Zhang, J., Khaing Oo, M. K., & Fan, X. (2020). Rapid and quantitative detection of COVID-19 markers in micro-liter sized samples. BioRxiv, 2020-04.
- Bošnjak, B., Stein, S. C., Willenzon, S., Cordes, A. K., Puppe, W., Bernhardt, G., Ravens, I., Ritter, C., Schultze-Florey, C., Godecke, N., et al (2021). Low serum neutralizing anti-SARS-CoV-2 S antibody levels in mildly affected COVID-19 convalescent patients revealed by two different detection methods. Cellular & molecular immunology, 18(4), 936-944.
- Yonker, L. M., Swank, Z., Bartsch, Y. C., Burns, M. D., Kane, A., Boribong, B. P., Davis, JP., Loiselle, M., Novak, T., Senussi, Y., et al., (2023). Circulating spike protein detected in post–COVID-19 mRNA vaccine myocarditis. Circulation, 147(11), 867-876.
- Yang, Y.; Fang, Q.; Shen, H.-B. Predicting gene regulatory interactions based on spatial gene expression data and deep learning. PLoS Comput. Biol. 2019, 15, e1007324. [CrossRef]
- Chikofsky, E.; Cross, J. Reverse engineering and design recovery: A taxonomy. IEEE Softw. 1990, 7, 13–17. [CrossRef]
- Green, S. Can biological complexity be reverse engineered? Stud. Hist. Philos. Sci. Part C Stud. Hist. Philos. Biol. Biomed. Sci. 2015, 53, 73–83.
- Natale, J.L.; Hofmann, D.; Hernández, D.G.; Nemenman, I. Reverse-engineering biological networks from large data sets. arXiv 2017, arXiv:1705.06370.
- Oughtred R, Rust J, Chang C, Breitkreutz BJ, Stark C, Willems A, Boucher L, Leung G, Kolas N, Zhang F, Dolma S, Coulombe-Huntington J, Chatr-Aryamontri A, Dolinski K, Tyers M. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021 Jan;30(1):187-200. Epub 2020 Nov 23. PMID: 33070389; PMCID: PMC7737760. [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2020, 49, D605–D612; Erratum in: Nucleic Acids Res. 2021, 49, 10800. [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2022, 51, D638–D646. [CrossRef]
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J. Proteome Res. 2018, 18, 623–632. [CrossRef]
- Chung, F.; Lu, L.; Dewey, T.G.; Galas, D.J. Duplication Models for Biological Networks. J. Comput. Biol. 2003, 10, 677–687. [CrossRef]
- Scardoni, G.; Tosadori, G.; Faizan, M.; Spoto, F.; Fabbri, F.; Laudanna, C. Biological network analysis with CentiScaPe:Centralities and experimental dataset integration. F1000Research 2015, 3, 139. [CrossRef]
- Perera, S.; Perera, H.N.; Kasthurirathna, D. Structural characteristics of complex supply chain networks. In Proceedings of the 2017 Moratuwa Engineering Research Conference (MERCon), Moratuwa, Sri Lanka, 29–31 May 2017; pp. 135–140. hps://doi.org/10.1109/MERCon.2017.7980470.
- Barabási, A.-L. Network Science, 1st ed.; Cambridge University Press: Cambridge, UK, 2016.
- Syakur, M.A.; Khotimah, BK.; Rochman, EMS.; Satoto, BD. Integration K-Means Clustering Method and Elbow Method for Identification of The Best Customer Profile Cluster — IOP Conference Series. Mater. Sci. Eng. 2018, 336, 012017. [CrossRef]
- Erdős, G.; Dosztányi, Z. Analyzing Protein Disorder with IUPred2A. Curr. Protoc. Bioinform. 2020, 70, e99. [CrossRef]
- Drozdetskiy, A.; Cole, C.; Procter, J.; Barton, G.J. JPred4: A protein secondary structure prediction server. Nucleic Acids Res. 2015, 43, W389–W394. hps://doi.org/10.1093/nar/gkv332.
- Arakawa, K., Tomita, M. (2013). Merging Multiple Omics Datasets In Silico: Statistical Analyses and Data Interpretation. In: Alper, H. (eds) Systems Metabolic Engineering. Methods in Molecular Biology, vol 985. Humana Press, Totowa, NJ. [CrossRef]
- Das, R. K., & Pappu, R. V. (2013). Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proceedings of the National Academy of Sciences, 110(33), 13392-13397.
- A.S. Holehouse, R.K. Das, J.N. Ahad, M.O.G. Richardson, R.V. Pappu (2017) CIDER: Resources to analyze sequence-ensemble relationships of intrinsically disordered proteins. Biophysical Journal, 112: 16-21.
- Das, Rahul K.; Ruff, Kiersten M.; Pappu, Rohit V. Relating sequence encoded information to form and function of intrinsically disordered proteins. Current Opinion in Structural Biology (2015), 32 (), 102-112CODEN: COSBEF; ISSN:0959-440X. (Elsevier Ltd.).
- Theillet, F. X., Kalmar, L., Tompa, P., Han, K. H., Selenko, P., Dunker, A. K., ... & Uversky, V. N. (2013). The alphabet of intrinsic disorder: I. Act like a Pro: On the abundance and roles of proline residues in intrinsically disordered proteins. Intrinsically Disordered Proteins, 1(1), e24360.
- Ali SA, Ayalew H, Gautam B, Selvaraj B, She JW, Janardhanan JA, Yu HH. Detection of SARS-CoV-2 Spike Protein Using Micropatterned 3D Poly(3,4-Ethylenedioxythiophene) Nanorods Decorated with Gold Nanoparticles. ACS Appl Mater Interfaces. 2024 Jan 9. Epub ahead of print. PMID: 38193284. [CrossRef]
- Letarov AV, Babenko VV, Kulikov EE. Free SARS-CoV-2 Spike Protein S1 Particles May Play a Role in the Pathogenesis of COVID-19 Infection. Biochemistry (Mosc). 2021 Mar;86(3):257-261. PMID: 33838638; PMCID: PMC7772528. [CrossRef]
- V’kovski, Philip; Kratzel, Annika; Steiner, Silvio; Stalder, Hanspeter; Thiel, Volker (March 2021). "Coronavirus biology and replication: implications for SARS-CoV-2". Nature Reviews Microbiology. 19 (3): 155–170. [CrossRef]
- Cortés-Sarabia K, Luna-Pineda VM, Rodríguez-Ruiz HA, Leyva-Vázquez MA, Hernández-Sotelo D, Beltrán-Anaya FO, Vences-Velázquez A, Del Moral-Hernández O, Illades-Aguiar B. Utility of in silico-identified-peptides in spike-S1 domain and nucleocapsid of SARS-CoV-2 for antibody detection in COVID-19 patients and antibody production. Sci Rep. 2022 Sep 5;12(1):15057. PMID: 36064951; PMCID: PMC9442563. [CrossRef]
- Feng Y, Yi K, Gong F, Zhang Y, Shan X, Ji X, Zhou F, He Z. Ultra-sensitive detection of SARS-CoV-2 S1 protein by coupling rolling circle amplification with poly(N-isopropylacrylamide)-based sandwich-type assay. Talanta. 2024 Jul 14;279:126572. Epub ahead of print. PMID: 39024855. [CrossRef]
- Dunker, A. K., Lawson, J. D., Brown, C. J., Williams, R. M., Romero, P., Oh, J. S., Oldfield, C., Campen, AM., Ratlif, C., Hipps, K., Ausio, J., et al., (2001). Intrinsically disordered protein. Journal of molecular graphics and modelling, 19(1), 26-59. [CrossRef]
- Ragone, R., Facchiano, F., Facchiano, A., Facchiano, A. M., & Colonna, G. (1989). Flexibility plot of proteins. Protein Engineering, Design and Selection, 2(7), 497-504. [CrossRef]
- Mao, A. H., Lyle, N., & Pappu, R. V. (2013). Describing sequence–ensemble relationships for intrinsically disordered proteins. Biochemical Journal, 449(2), 307-318.
- Campen A, Williams RM, Brown CJ, Meng J, Uversky VN, Dunker AK. TOP-IDP-scale: a new amino acid scale measuring propensity for intrinsic disorder. Protein Pept Lett. 2008;15(9):956-63. PMID: 18991772; PMCID: PMC2676888. [CrossRef]
- Zhang XW, Yap YL. The 3D structure analysis of SARS-CoV S1 protein reveals a link to influenza virus neuraminidase and implications for drug and antibody discovery. Theochem. 2004 Jul 26;681(1):137-141. Epub 2004 Jul 9. PMID: 32287547; PMCID: PMC7126208. [CrossRef]
- Bozhilova, L. V., Whitmore, A. V., Wray, J., Reinert, G., & Deane, C. M. (2019). Measuring rank robustness in scored protein interaction networks. BMC bioinformatics, 20, 1-14.
- Guidotti, R.; Gardoni, P.; Chen, Y. Network reliability analysis with link and nodal weights and auxiliary nodes. Struct. Saf. 2017, 65, 12–26. [CrossRef]
- De Vico Fallani, F.; Richiardi, J.; Chavez, M.; Achard, S. Graph analysis of functional brain networks: Practical issues in translational neuroscience. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130521. [CrossRef]
- Li, V.; Silvester, J. Performance Analysis of Networks with Unreliable Components. IEEE Trans. Commun. 1984, 32, 1105–1110. [CrossRef]
- 52] Knight, S.; Nguyen, H.X.; Falkner, N.; Bowden, R.; Roughan, M. The Internet Topology Zoo. IEEE J. Sel. Areas Commun. 2011, 29, 1765–1775. [CrossRef]
- Swarthout JT, Lobo S, Farh L, Croke MR, Greentree WK, Deschenes RJ, Linder ME. DHHC9 and GCP16 constitute a human protein fatty acyltransferase with specificity for H- and N-Ras. J Biol Chem. 2005 Sep 2;280(35):31141-8. Epub 2005 Jul 6. PMID: 16000296. [CrossRef]
- Marom R, Burrage LC, Venditti R, Clément A, Blanco-Sánchez B, Jain M, Scott DA, Rosenfeld JA, Sutton VR, Shinawi M, et al., Undiagnosed Diseases Network; Westerfield M, De Matteis MA, Lee B. COPB2 loss of function causes a coatopathy with osteoporosis and developmental delay. Am J Hum Genet. 2021 Sep 2;108(9):1710-1724. Epub 2021 Aug 26. PMID: 34450031; PMCID: PMC8456174. [CrossRef]
- Sheikhahmadi, A., Nematbakhsh, M. A., & Shokrollahi, A. (2015). Improving detection of influential nodes in complex networks. Physica A: Statistical Mechanics and its Applications, 436, 833-845.
- Kazemzadeh, Farzaneh & Safaei, Ali & Mirzarezaee, Mitra. (2022). Optimal selection of seed nodes by reducing the influence of common nodes in the influence maximization problem. 1-7. 10.1109/IKT57960.2022.10039040. Conference article: 13th International Conference on Information and Knowledge Technology (IKT).
- Tandel, S. S., Jamadar, A., & Dudugu, S. (2019, March). A survey on text mining techniques. In 2019 5th International Conference on Advanced Computing & Communication Systems (ICACCS) (pp. 1022-1026). IEEE.
- Salloum, S.A., Al-Emran, M., Monem, A.A., Shaalan, K. (2018). Using Text Mining Techniques for Extracting Information from Research Articles. In: Shaalan, K., Hassanien, A., Tolba, F. (eds) Intelligent Natural Language Processing: Trends and Applications. Studies in Computational Intelligence, vol 740. Springer, Cham. [CrossRef]
- Y. Hasin, M. Seldin and A. Lusis, Multi-omics approaches to disease, Genome Biol., 2017, 18(1), 83.
- Graw, S., Chappell, K., Charity L. Gies, A., Bird, J., Robeson, M., Stephanie D., Multi-omics data integration considerations and study design for biological systems and disease, Mol. Omics, 2021, 17, 2, 170-185, The Royal Society of Chemistry, doi="10.1039/D0MO00041H". [CrossRef]
- Spirin, V., Minry, LA., Protein complexes and functional modules in molecular networks. Proceedings of the National Academy of Sciences, 2003, 100.21: 12123-12128.
- Morea, F., & De Stefano, D. (2024). Enhancing Stability and Assessing Uncertainty in Community Detection through a Consensus-based Approach. arXiv preprint arXiv:2408.02959.
- Barabási, A. L. (2013). Network science. Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences, 371(1987), Chapter 9, 20120375.
- Wimsatt, W. C. (2007). Re-engineering philosophy for limited beings: Piecewise approximations to reality. Harvard University Press.
- Chen, D., Lü, L., Shang, M. S., Zhang, Y. C., & Zhou, T. (2012). Identifying influential nodes in complex networks. Physica a: Statistical mechanics and its applications, 391(4), 1777-1787.
- Barabási, A. L. (2013). Network science. Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences, 371(1987), 20120375.
- Barabási, A.-L. Network Science, 1st ed.; Cambridge University Press: Cambridge, UK, 2016.
- Mao J, O'Gorman C, Sutovsky M, Zigo M, Wells KD, Sutovsky P. Ubiquitin A-52 residue ribosomal protein fusion product 1 (Uba52) is essential for preimplantation embryo development. Biol Open. 2018 Oct 2;7(10):bio035717. PMID: 30135083; PMCID: PMC6215406. [CrossRef]
- Eastham MJ, Pelava A, Wells GR, Watkins NJ, Schneider C. RPS27a and RPL40, Which Are Produced as Ubiquitin Fusion Proteins, Are Not Essential for p53 Signalling. Biomolecules. 2023 May 28;13(6):898. PMID: 37371478; PMCID: PMC10296562. [CrossRef]
- van den Heuvel J, Ashiono C, Gillet LC, Dörner K, Wyler E, Zemp I, Kutay U. Processing of the ribosomal ubiquitin-like fusion protein FUBI-eS30/FAU is required for 40S maturation and depends on USP36. Elife. 2021 Jul 28;10:e70560. PMID: 34318747; PMCID: PMC8354635. [CrossRef]
- Park C, Walsh D. RACK1 Regulates Poxvirus Protein Synthesis Independently of Its Role in Ribosome-Based Stress Signaling. J Virol. 2022 Sep 28;96(18):e0109322. Epub 2022 Sep 13. PMID: 36098514; PMCID: PMC9517738. [CrossRef]
- Jha S, Rollins MG, Fuchs G, Procter DJ, Hall EA, Cozzolino K, Sarnow P, Savas JN, Walsh D. Trans-kingdom mimicry underlies ribosome customization by a poxvirus kinase. Nature. 2017 Jun 29;546(7660):651-655. Epub 2017 Jun 21. PMID: 28636603; PMCID: PMC5526112. [CrossRef]
- Mauro V.P., Edelman G.M. The ribosome filter hypothesis. Proc. Natl Acad. Sci. USA. 2002; 99:12031–12036.
- Elhamamsy, A.R., Metge, B.J., Alsheikh, H.A., Shevde, L.A., Samant, R.S. Ribosome biogenesis: a central player in cancer metastasis and therapeutic resistance. Cancer Res. 2022; 82:2344–2353.
- Lee A.S., Burdeinick-Kerr, R., Whelan, S.P. A ribosome-specialized translation initiation pathway is required for cap-dependent translation of vesicular stomatitis virus mRNAs. Proc. Natl Acad. Sci. USA. 2013; 110:324–329.
- Shi Z., Fujii K., Kovary K.M., Genuth N.R., Rost H.L., Teruel M.N., Barna M. Heterogeneous ribosomes preferentially translate distinct subpools of mRNAs genome-wide. Mol. Cell. 2017; 67:71–83.
- Tu C., Meng L., Nie H., Yuan S., Wang W., Du J., Lu G., Lin G., Tan Y.Q. A homozygous RPL10L missense mutation associated with male factor infertility and severe oligozoospermia. Fertil. Steril. 2020; 113:561–568.
- Dong, J. & Horvath, S. Understanding network concepts in modules. BMC Syst. Biol. 1, 24 (2007).
- Stelzl, U. et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell 122, 957–968 (2005).
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. hps://doi.org/10.1101/gr.1239303.
- Assenov, Y.; Ramírez, F.; Schelhorn, S.-E.; Lengauer, T.; Albrecht, M. Computing topological parameters of biological networks. Bioinformatics 2008, 24, 282–284. hps://doi.org/10.1093/bioinformatics/btm554.
- Li, V.; Silvester, J. Performance Analysis of Networks with Unreliable Components. IEEE Trans. Commun. 1984, 32, 1105–1110. [CrossRef]
- A.-L. Barabási, R.Albert, and H. Jeong. Mean-field theory of scale-free random networks. Physica A 272:173-187, 1999.
- H. Jeong, B. Tombor, R. Albert, Z. N. Oltvai, and A.-L. Barabási. The large-scale organization of metabolic networks. Nature 407: 651-654, 2000.
- Wagner, A. and D.A. Fell. The small world inside large metabolic networks. Proc. R. Soc. Lond. B 268: 1803–1810, 2001.
- L.A.N. Amaral, A. Scala, M. Barthelemy and H.E. Stanley. Classes of small-world networks. Proceeding National Academy of Sciences U. S. A. 97:11149-11152, 2000.
- K.-I. Goh, B. Kahng, and D. Kim. Universal behavior of load distribution in scale-free networks. Phys. Rev. Lett. 87: 278701, 2001.
- Strogatz, S. H. Exploring complex networks. Nature 410, 268–276 (2001).
- Chen, B., Fan, W., Liu, J., & Wu, F. X. (2014). Identifying protein complexes and functional modules—from static PPI networks to dynamic PPI networks. Briefings in bioinformatics, 15(2), 177-194.
- V. Havel. A remark on the existence of finite graphs. Casopis Pest. Mat., 80:477-480, 1955.
- Charo Del Genio, G. Thilo, and K.E. Bassler. All scale-free networks are sparse. Phys. Rev. Lett. 107:178701, 10 2011.
- G. Bianconi and A.-L. Barabási. Competition and multiscaling in evolving networks. Europhysics Letters, 54: 436-442, 2001.
- A.-L. Barabási, R. Albert, H. Jeong, and G. Bianconi. Power-law distribution of the world wide web. Science, 287: 2115, 2000.
- M. Medo, G. Cimini, and S. Gualdi. Temporal effects in the growth of networks. Phys. Rev. Lett., 107:238701, 2011.
- S. N. Dorogovtsev, J.F.F. Mendes, and A.N. Samukhin. Structure of growing networks with preferential linking. Phys. Rev. Lett., 85: 4633, 2000.
- G. Bianconi and A.-L. Barabási. Bose-Einstein condensation in complex networks. Phys. Rev. Lett., 86: 5632–5635, 2001.
- Yukalov, V I (2005). "Number-of-particle fluctuations in systems with Bose-Einstein condensate". Laser Physics Letters. 2 (3): 156–161.
- Pizzuti, C. (2017). Evolutionary computation for community detection in networks: A review. IEEE Transactions on Evolutionary Computation, 22(3), 464-483.
- Mardikoraem M, Woldring D. Protein Fitness Prediction Is Impacted by the Interplay of Language Models, Ensemble Learning, and Sampling Methods. Pharmaceutics. 2023 Apr 25;15(5):1337. PMID: 37242577; PMCID: PMC10224321. [CrossRef]
- Golinski A.W., Mischler K.M., Laxminarayan S., Neurock N.L., Fossing M., Pichman H., Martiniani S., Hackel B.J. High-Throughput Developability Assays Enable Library-Scale Identification of Producible Protein Scaffold Variants. Proc. Natl. Acad. Sci. USA. 2021;118:e2026658118. [CrossRef]
- Wang S., Liu D., Ding M., Du Z., Zhong Y., Song T., Zhu J., Zhao R. SE-OnionNet: A Convolution Neural Network for Protein–Ligand Binding Affinity Prediction. Front. Genet. 2021;11:607824. [CrossRef]
- Kuzmin K., Adeniyi A.E., DaSouza A.K., Lim D., Nguyen H., Molina N.R., Xiong L., Weber I.T., Harrison R.W. Machine Learning Methods Accurately Predict Host Specificity of Coronaviruses Based on Spike Sequences Alone. Biochem. Biophys. Res. Commun. 2020;533:553–558. [CrossRef]
- Das S., Chakrabarti S. Classification and Prediction of Protein–Protein Interaction Interface Using Machine Learning Algorithm. Sci. Rep. 2021;11:1761. [CrossRef]
- Stiuso, P., Ragone, R., & Colonna, G. (1990). Molecular organization and structural stability of. beta. s-crystallin from calf lens. Biochemistry, 29(16), 3929-3936.
- Vazquez, R. Pastor-Satorras, and A. Vespignani. Large-scale topological and dynamical properties of Internet. Phys. Rev., E 65: 066130, 2002.
- R. Xulvi-Brunet and I. M. Sokolov. Changing correlations in networks: assortativity and dissortativity. Acta Phys. Pol. B, 36: 1431, 2005.
- M. Posfai, Y Y. Liu, J-J Slotine, and A.-L. Barabási. Effect of correlations on network controllability. Scientific Reports, 3: 1067, 2013.
- Li, J., & Convertino, M. (2021). Inferring ecosystem networks as information flows. Scientific reports, 11(1), 7094.
- Iakoucheva, L.M., Brown, C.J., Lawson, J.D., Obradovic, Z., and Dunker, A.K. (2002). Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 323, 573–584.
- Higurashi, M., Ishida, T., and Kinoshita, K. (2008). Identification of transient hub proteins and the possible structural basis for their multiple interactions. Protein Sci. 17, 72–78.
- Han, J.D., Bertin, N., Hao, T., Goldberg, D.S., Berriz, G.F., Zhang, L.V., Dupuy, D., Walhout, A.J., Cusick, M.E., Roth, F.P., and Vidal, M. (2004). Evidence for dynamically organized modularity in the yeast protein-protein interaction network. Nature 430, 88–93.
- Hu G, Wu Z, Uversky VN, Kurgan L. Functional Analysis of Human Hub Proteins and Their Interactors Involved in the Intrinsic Disorder-Enriched Interactions. Int J Mol Sci. 2017 Dec 19;18(12):2761. PMID: 29257115; PMCID: PMC5751360. [CrossRef]
- Lun XK, Zanotelli VR, Wade JD, Schapiro D, Tognetti M, Dobberstein N, Bodenmiller B. Influence of node abundance on signaling network state and dynamics analyzed by mass cytometry. Nat Biotechnol. 2017 Feb;35(2):164-172. Epub 2017 Jan 16. PMID: 28092656; PMCID: PMC5617104. [CrossRef]
- Perovic, V., Sumonja, N., Marsh, L. A., Radovanovic, S., Vukicevic, M., Roberts, S. G., & Veljkovic, N. (2018). IDPpi: Protein-protein interaction analyses of human intrinsically disordered proteins. Scientific reports, 8(1), 10563.
- Hwang, W., Cho, Y. R., Zhang, A., & Ramanathan, M. (2006, March). Bridging centrality: identifying bridging nodes in scale-free networks. In Proceedings of the 12th ACM SIGKDD international conference on knowledge discovery and data mining (pp. 20-23).
- Tripathi LP, et al. (2013) Understanding the Biological Context of NS5A-Host Interactions in HCV Infection: A Network-Based Approach. J Proteome Res. [PubMed:23682656].
- Soofi A, Taghizadeh M, Tabatabaei SM, Rezaei Tavirani M, Shakib H, Namaki S, Safari Alighiarloo N. Centrality Analysis of Protein-Protein Interaction Networks and Molecular Docking Prioritize Potential Drug-Targets in Type 1 Diabetes. Iran J Pharm Res. 2020 Fall;19(4):121-134. PMID: 33841528; PMCID: PMC8019861. [CrossRef]
- Zhang A. Protein interaction networks: computational Analysis. Cambridge University Press; 2009.
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003; 13:2498–504.
- Bu D, Zhao Y, Cai L, Xue H, Zhu X, Lu H, Zhang J, Sun S, Ling L, Zhang N, Li G, Chen R. Topological structure analysis of the protein-protein interaction network in budding yeast. Nucleic Acids Res. 2003 May 1;31(9):2443-50. PMID: 12711690; PMCID: PMC154226. [CrossRef]
- Negre CFA, Morzan UN, Hendrickson HP, Pal R, Lisi GP, Loria JP, Rivalta I, Ho J, Batista VS. Eigenvector centrality for characterization of protein allosteric pathways. Proc Natl Acad Sci U S A. 2018 Dec 26;115(52):E12201-E12208. Epub 2018 Dec 10. PMID: 30530700; PMCID: PMC6310864. [CrossRef]
- Fletcher, J. M., & Wennekers, T. (2018). From structure to activity: Using centrality measures to predict neuronal activity. International journal of neural systems, 28(02), 1750013.
- Chen, SJ., Liao, DL., Chen, CH. et al. Construction and Analysis of Protein-Protein Interaction Network of Heroin Use Disorder. Sci Rep 9, 4980 (2019). [CrossRef]
- Vallabhajosyula RR, Chakravarti D, Lutfeali S, Ray A, Raval A. Identifying hubs in protein interaction networks. PLoS One. 2009;4(4):e5344. Epub 2009 Apr 28. PMID: 19399170; PMCID: PMC2670494. [CrossRef]
- Kadoki M, Patil A, Thaiss CC, Brooks DJ, Pandey S, Deep D, Alvarez D, von Andrian UH, Wagers AJ, Nakai K, Mikkelsen TS, Soumillon M, Chevrier N. Organism-Level Analysis of Vaccination Reveals Networks of Protection across Tissues. Cell. 2017 Oct 5;171(2):398-413.e21. Epub 2017 Sep 21. PMID: 28942919; PMCID: PMC7895295. [CrossRef]
- López CB, and Hermesh T (2011). Systemic responses during local viral infections: type I IFNs sound the alarm. Current Opinion in Immunology 23, 495–499.
- Manz MG, and Boettcher S (2014). Emergency granulopoiesis. Nature Reviews Immunology 14, 302–314.
- Schenkel JM, and Masopust D (2014). Tissue-resident memory T cells. Immunity 41, 886–897.
- Kadoki M, Patil A, Thaiss CC, Brooks DJ, Pandey S, Deep D, Alvarez D, von Andrian UH, Wagers AJ, Nakai K, Mikkelsen TS, Soumillon M, Chevrier N. Organism-Level Analysis of Vaccination Reveals Networks of Protection across Tissues. Cell. 2017 Oct 5;171(2):398-413.e21. Epub 2017 Sep 21. PMID: 28942919; PMCID: PMC7895295. [CrossRef]
- Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, and Kupper TS (2012). Skin infection generates non-migratory memory CD8+ TRM cells providing global skin immunity. Nature 483, 227–231.
- Stary G, Olive A, Radovic-Moreno AF, Gondek D, Alvarez D, Basto PA, Perro M, Vrbanac VD, Tager AM, Shi J, et al. (2015). VACCINES. A mucosal vaccine against Chlamydia trachomatis generates two waves of protective memory T cells. Science 348, aaa8205.
- Scardoni, G. and Laudanna, C. (2012) ‘Centralities based analysis of complex networks’, New Frontiers in Graph Theory, InTech Open.
- Melé M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M, Young TR, Goldmann JM, Pervouchine DD, Sullivan TJ, et al. (2015). The human transcriptome across tissues and individuals. Science 348, 660–665.
- Dobrin R, Zhu J, Molony C, Argman C, Parrish ML, Carlson S, Allan MF, Pomp D, and Schadt EE (2009). Multi-tissue coexpression networks reveal unexpected subnetworks associated with disease. Genome Biol 10, R55.
- Ariotti S, Hogenbirk MA, Dijkgraaf FE, Visser LL, Hoekstra ME, Song J-Y, Jacobs H, Haanen JB, and Schumacher TN (2014). T cell memory. Skin-resident memory CD8⁺ T cells trigger a state of tissue-wide pathogen alert. Science 346, 101–105.
- Braun, E., & Marom, S. (2015). Universality, complexity and the praxis of biology: Two case studies. Studies in History and Philosophy of Science Part C: Studies in History and Philosophy of Biological and Biomedical Sciences, 53, 68-72.
- Green, S. (2015). Can biological complexity be reverse engineered? Studies in History and Philosophy of Science Part C: Studies in History and Philosophy of Biological and Biomedical Sciences, 53, 73-83.
- Krohs, U. (2012). Convenience experimentation. Studies in History and Philosophy of Science Part C: Studies in History and Philosophy of Biological and Biomedical Sciences, 43(1), 52-57.
- Nithya, C., Kiran, M., & Nagarajaram, H. A. (2021). Comparative analysis of Pure Hubs and Pure Bottlenecks in Human Protein-protein Interaction Networks. bioRxiv, 2021-04.
- Pang E, Hao Y, Sun Y, Lin K. Differential variation patterns between hubs and bottlenecks in human protein-protein interaction networks. BMC Evol Biol. 2016 Dec 1;16(1):260. PMID: 27903259; PMCID: PMC5131443. [CrossRef]
- Yu H, Kim PM, Sprecher E, Trifonov V, Gerstein M. The importance of bottlenecks in protein networks: correlation with gene essentiality and expression dynamics. PLoS Comput Biol. 2007 Apr 20;3(4):e59. Epub 2007 Feb 14. PMID: 17447836; PMCID: PMC1853125. [CrossRef]
- Zimmermann M.G., Eguíluz V.M., San Miguel M., Spadaro A. Cooperation in an adaptive network Adv. Complex Syst., 03 (01n04) (2011), pp. 283-297, 10.1142/S0219525900000212.
- Skyrms B., Pemantle R. A dynamic model of social network formation Proc. Natl. Acad. Sci., 97 (16) (2000), pp. 9340-9346, 10.1073/pnas.97.16.9340.
- Paul, C. P., Good, P. D., Winer, I., & Engelke, D. R. (2002). Effective expression of small interfering RNA in human cells. Nature biotechnology, 20(5), 505-508.
- Wang, E. T., Cody, N. A., Jog, S., Biancolella, M., Wang, T. T., Treacy, D. J., ... & Burge, C. B. (2012). Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscle-blind proteins. Cell, 150(4), 710-724.
- Bauer, N. C., Doetsch, P. W., & Corbett, A. H. (2015). Mechanisms regulating protein localization. Traffic, 16(10), 1039-1061.
- Huang, H. Y., & Hopper, A. K. (2015). In vivo biochemical analyses reveal distinct roles of β-importins and eEF1A in tRNA subcellular traffic. Genes & development, 29(7), 772-783.
- Gasparski, A. N., Moissoglu, K., Pallikkuth, S., Meydan, S., Guydosh, N. R., & Mili, S. (2023). mRNA location and translation rate determine protein targeting to dual destinations. Molecular Cell, 83(15), 2726-2738.
- Komar, A. A., Samatova, E., & Rodnina, M. V. (2023). Translation Rates and Protein Folding. Journal of Molecular Biology, 168384.
- Barabási, A. L. (2007). Network medicine—from obesity to the “diseasome”. New England Journal of Medicine, 357(4), 404-407.
- Gene Ontology C. The gene ontology resource: enriching a gold mine. Nucleic Acids Res. 2021;49(D1):D325–D334.
- Gillis J, Pavlidis P. Assessing identity, redundancy and confounds in Gene Ontology annotations over time. Bioinformatics. 2013 Feb 15;29(4):476-82. Epub 2013 Jan 6. PMID: 23297035; PMCID: PMC3570208. [CrossRef]
- Thomas PD. The Gene Ontology and the Meaning of Biological Function. Methods Mol Biol. 2017; 1446:15-24. PMID: 27812932. [CrossRef]
- Martucci, D; Masseroli, M; Pinciroli, F. Gene ontology application to genomic functional annotation, statistical analysis and knowledge mining. In: Ontologies in Medicine. IOS Press, 2004. p. 108-131.
- Paci, P., Fiscon, G., Conte, F. et al. Gene co-expression in the interactome: moving from correlation toward causation via an integrated approach to disease module discovery. npj Syst Biol Appl 7, 3 (2021). [CrossRef]
- Przytycka, T. M., Singh, M., & Slonim, D. K. (2010). Toward the dynamic interactome: it's about time. Briefings in bioinformatics, 11(1), 15-29.
- Nagaraj N, Wisniewski JR, Geiger T, Cox J, Kircher M, Kelso J, Pääbo S, Mann M. Deep proteome and transcriptome mapping of a human cancer cell line. Mol Syst Biol. 2011 Nov 8;7:548. PMID: 22068331; PMCID: PMC3261714. [CrossRef]
- Wiggins, P., Choi, J., Huang, D., & Lo, T. (2024). Noise robustness and metabolic load determine the principles of central dogma regulation. Bulletin of the American Physical Society.
- Lo, T., Choi, J., Huang, D., & Wiggins, P., Noise robustness and metabolic load determine the principles of central dogma regulation. (2024), Science Advances, Vol. 10, No. 34. [CrossRef]
- Hausser J, Mayo A, Keren L, Alon U. Central dogma rates and the trade-off between precision and economy in gene expression. Nat Commun. 2019 Jan 8;10(1):68. PMID: 30622246; PMCID: PMC6325141. [CrossRef]
- Dekel, E., & Alon, U. (2005). Optimality and evolutionary tuning of the expression level of a protein. Nature, 436 (7050), 588-592.
- Gallagher, L. A., Bailey, J., & Manoil, C. (2020). Ranking essential bacterial processes by speed of mutant death. Proceedings of the National Academy of Sciences, 117(30), 18010-18017.
- Lengeler, Joseph W.; Drews, Gerhart; Schlegel, Hans G. (ed.). Biology of the Prokaryotes. John Wiley & Sons, 2009.
- Vidal M, Cusick ME, Barabási AL. Interactome networks and human disease. Cell. 2011 Mar 18;144(6):986-98. PMID: 21414488; PMCID: PMC3102045. [CrossRef]
- Satam H, Joshi K, Mangrolia U, Waghoo S, Zaidi G, Rawool S, Thakare RP, Banday S, Mishra AK, Das G, Malonia SK. Next-Generation Sequencing Technology: Current Trends and Advancements. Biology (Basel). 2023 Jul 13;12(7):997. Erratum in: Biology (Basel). 2024 Apr 24;13(5):286. https://doi.org/10.3390/biology13050286. PMID: 37508427; PMCID: PMC10376292. [CrossRef]
- Caudai, C., Galizia, A., Geraci, F., Le Pera, L., Morea, V., Salerno, E., Via, A., & Colombo, T. (2021). AI applications in functional genomics. Computational and Structural Biotechnology Journal, 19, 5762-5790.
- Koumakis, L. (2020). Deep learning models in genomics; are we there yet? Computational and Structural Biotechnology Journal, 18, 1466-1473.
- Asp M, Bergenstråhle J, Lundeberg J (October 2020). "Spatially Resolved Transcriptomes-Next Generation Tools for Tissue Exploration". BioEssays. 42 (10): e1900221. PMID 32363691. S2CID 218492475. [CrossRef]
- Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, et al. (July 2016). "Visualization and analysis of gene expression in tissue sections by spatial transcriptomics". Science. 353 (6294): 78–82. PMID 27365449. [CrossRef]
- Keskin, O., Tuncbag, N., & Gursoy, A. (2016). Predicting protein–protein interactions from the molecular to the proteome level. Chemical reviews, 116(8), 4884-4909.
- Koh, G. C., Porras, P., Aranda, B., Hermjakob, H., & Orchard, S. E. (2012). Analyzing protein–protein interaction networks. Journal of proteome research, 11(4), 2014-2031.
- Su, Y. J., Chang, C. W., Chen, M. J., & Lai, Y. C. (2021). Impact of COVID-19 on liver. World journal of clinical cases, 9(27), 7998.
- Diao, Y., Tang, J., Wang, X., Deng, W., Tang, J., & You, C. (2023). Metabolic syndrome, nonalcoholic fatty liver disease, and chronic hepatitis B: A narrative review. Infectious Diseases and Therapy, 12(1), 53-66.
- Ali, F. E., Mohammedsaleh, Z. M., Ali, M. M., & Ghogar, O. M. (2021). Impact of cytokine storm and systemic inflammation on liver impairment patients infected by SARS-CoV-2: Prospective therapeutic challenges. World journal of gastroenterology, 27(15), 1531.
- Frank SA. Immunology and Evolution of Infectious Disease. Princeton (NJ): Princeton University Press; 2002. Chapter 4, Specificity and Cross-Reactivity.
- You, H., Qin, S., Zhang, F., Hu, W., Li, X., Liu, D., ... & Tang, R. (2022). Regulation of pattern-recognition.
- Xia, Z., & Storm, D. R. (2005). The role of calmodulin as a signal integrator for synaptic plasticity. Nature Reviews Neuroscience, 6(4), 267-276.
- Harrison-Bernard, L. M. (2009). The renal renin-angiotensin system. Advances in physiology education, 33(4), 270-274.
- Iwamoto DV, Calderwood DA. Regulation of integrin-mediated adhesions. Curr Opin Cell Biol. 2015 Oct;36:41-7. Epub 2015 Jul 17. PMID: 26189062; PMCID: PMC4639423. [CrossRef]
- Nunes-Hasler, P., Kaba, M., & Demaurex, N. (2020). Molecular mechanisms of calcium signaling during phagocytosis. Molecular and Cellular Biology of Phagocytosis, 103-128.
- Mylvaganam S, Freeman SA, Grinstein S. The cytoskeleton in phagocytosis and macropinocytosis. Curr Biol. 2021 May 24;31(10):R619-R632. PMID: 34033794. [CrossRef]
- Jaumouillé V, Waterman CM. Physical Constraints and Forces Involved in Phagocytosis. Front Immunol. 2020 Jun 12;11:1097. PMID: 32595635; PMCID: PMC7304309. [CrossRef]
- Guertin, D. A., & Sabatini, D. M. (2007). Defining the role of mTOR in cancer. Cancer cell, 12(1), 9-22.
- Huang, J., Wang, C., Hou, Y., Tian, Y., Li, Y., Zhang, H., ... & Li, W. (2023). Molecular mechanisms of Thrombospondin-2 modulates tumor vasculogenic mimicry by PI3K/AKT/mTOR signaling pathway. Biomedi-cine & Pharmacotherapy, 167, 115455.
- Lichner, Z., Ding, Q., Samaan, S., Saleh, C., Nasser, A., Al-Haddad, S., ... & Yousef, G. M. (2015). miRNAs dysregulated in association with Gleason grade regulate extracellular matrix, cytoskeleton and androgen receptor pathways. The Journal of pathology, 237(2), 226-237.
- Jiao, L., Liu, Y., Yu, X. Y., Pan, X., Zhang, Y., Tu, J., ... & Li, Y. (2023). Ribosome biogenesis in disease: new players and therapeutic targets. Signal Transduction and Targeted Therapy, 8(1), 15.
- Piazzi, M., Bavelloni, A., Gallo, A., Faenza, I., & Blalock, W. L. (2019). Signal transduction in ribosome biogenesis: a recipe to avoid disaster. International journal of molecular sciences, 20(11), 2718.
- Solà, C., Barrón, S., Tusell, J. M., & Serratosa, J. (1999). The Ca2+/calmodulin signaling system in the neural response to excitability. Involvement of neuronal and glial cells. Progress in neurobiology, 58(3), 207-232.
- Wu, H.-Y., Tomizawa, K., and Matsui, H. (2007). Calpain-calcineurin signaling in the pathogenesis of calci-um-dependent disorder. Acta Med. Okayama 61, 123–137. [CrossRef]
- Gonçalves, C. A., Sesterheim, P., Wartchow, K. M., Bobermin, L. D., Leipnitz, G., & Quincozes-Santos, A. (2022). Why antidiabetic drugs are potentially neuroprotective during the Sars-CoV-2 pandemic: The focus on astroglial UPR and calcium-binding proteins. Frontiers in Cellular Neuroscience, 16, 905218.
- Yapici-Eser, H., Koroglu, Y. E., Oztop-Cakmak, O., Keskin, O., Gursoy, A., & Gur-soy-Ozdemir, Y. (2021). Neuropsychiatric symptoms of COVID-19 explained by SARS-CoV-2 proteins’ mimicry of human protein interactions. Frontiers in Human Neuroscience, 15, 656313.
- Li, Y., Pehrson, A. L., Waller, J. A., Dale, E., Sanchez, C., & Gulinello, M. (2015). A critical evaluation of the activity-regulated cytoskeleton-associated protein (Arc/Arg3. 1)'s putative role in regulating dendritic plasticity, cogni-tive processes, and mood in animal models of depression. Frontiers in neuroscience, 9, 279.
- Bekhbat M, Treadway MT, Goldsmith DR, Woolwine BJ, Haroon E, Miller AH, Felger JC. Gene signatures in peripheral blood immune cells related to insulin resistance and low tyrosine metabolism define a sub-type of depression with high CRP and anhedonia. Brain Behav Immun. 2020 Aug;88:161-165. Epub 2020 Mar 18. PMID: 32198016; PMCID: PMC7415632. [CrossRef]
- Cusato, J., Manca, A., Palermiti, A., Mula, J., Costanzo, M., Antonucci, M., ... & Cal-cagno, A. (2023). COVID-19: a possible contribution of the MAPK pathway. Biomedi-cines, 11(5), 1459.
- Ghasemnejad-Berenji, M., & Pashapour, S. (2021). SARS-CoV-2 and the possible role of Raf/MEK/ERK pathway in viral survival: is this a potential therapeutic strategy for COVID-19?. Pharmacology, 106(1-2), 119-122.
- Almutairi, M. M., Sivandzade, F., Albekairi, T. H., Alqahtani, F., & Cucullo, L. (2021). Neuroinflammation and Its Impact on the Pathogenesis of COVID-19. Frontiers in medicine, 8, 745789.
- Shiravand, Y., Walter, U., & Jurk, K. (2021). Fine-Tuning of Platelet Responses by Serine/Threonine Protein Kinases and Phosphatases—Just the Beginning. Hämostaseologie, 41(03), 206-216.
- Guergnon, J., Godet, A. N., Galioot, A., Falanga, P. B., Colle, J. H., Cayla, X., & Garcia, A. (2011). PP2A targeting by viral proteins: a widespread biological strategy from DNA/RNA tumor viruses to HIV-1. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease, 1812(11), 1498-1507.
- Dahlman, I., Belarbi, Y., Laurencikiene, J., Pettersson, A. M., Arner, P., & Kulyté, A. (2017). Comprehensive functional screening of miRNAs involved in fat cell insulin sensitivity among women. American Journal of Physiology-Endocrinology and Metabolism, 312(6), E482-E494.
- Todorovic, S., Simeunovic, V., Prvulovic, M., Dakic, T., Jevdjovic, T., Sokanovic, S., ... & Mladenovic, A. (2024). Dietary restriction alters insulin signaling pathway in the brain. BioFactors, 50(3), 450-466.
- Verger, A., Kas, A., Dudouet, P. et al. Visual interpretation of brain hypometabolism related to neurological long COVID: a French multicentric experience. Eur J Nucl Med Mol Imaging 49, 3197–3202 (2022). [CrossRef]
- Guedj, E., Campion, J.Y., Dudouet, P. et al. 18F-FDG brain PET hypometabolism in patients with long COVID. Eur J Nucl Med Mol Imaging 48, 2823–2833 (2021). [CrossRef]
- Bockaert, J., Perroy, J., Bécamel, C., Marin, P., & Fagni, L. (2010). GPCR interacting proteins (GIPs) in the nervous system: Roles in physiology and pathologies. Annual review of pharmacology and toxicology, 50(1), 89-109.
- Theobald, S. J., Simonis, A., Georgomanolis, T., Kreer, C., Zehner, M., Eisfeld, H. S., Albert, MC., Chen, J., Motameny, S., Erger, F., Fischer, J., et al., (2021). Long-lived macrophage reprogramming drives spike protein-mediated inflammasome activation in COVID-19. EMBO molecular medicine, 13(8), e14150.
- Li, X., Wu, K., Zeng, S., Zhao, F., Fan, J., Li, Z., Yi, L., Ding, H., Zhao, M., Fan, S., et al., (2021). Viral infection modulates mitochondrial function. International Journal of Molecular Sciences, 22(8), 4260.
- 206] Theofilis, P., Sagris, M., Oikonomou, E., Antonopoulos, A. S., Siasos, G., Tsioufis, C., & Tousoulis, D. (2021). Inflammatory mechanisms contributing to endothelial dysfunction. Biomedicines, 9(7), 781.
- Batabyal, R., Freishtat, N., Hill, E., Rehman, M., Freishtat, R., & Koutroulis, I. (2021). Metabolic dysfunction and immunometabolism in COVID-19 pathophysiology and therapeutics. International Journal of Obesity, 45(6), 1163-1169.
- Wheeler, S. E., Shurin, G. V., Yost, M., Anderson, A., Pinto, L., Wells, A., & Shurin, M. R. (2021). Differential antibody response to mRNA COVID-19 vaccines in healthy subjects. Microbiology spectrum, 9(1), 10-1128.
- Huang, S., & Houghton, P. J. (2003). Targeting mTOR signaling for cancer therapy. Current opinion in pharmacology, 3(4), 371-377.
- Yuan, T. L., & Cantley, L. (2008). PI3K pathway alterations in cancer: variations on a theme. Oncogene, 27(41), 5497-5510.
- Zhao, J., & Guan, J. L. (2009). Signal transduction by focal adhesion kinase in cancer. Cancer and Metastasis Reviews, 28, 35-49.
- Ding, X., Zhang, W., Li, S., & Yang, H. (2019). The role of cholesterol metabolism in cancer. American journal of cancer research, 9(2), 219.
- Chauhan, A. J., Wiffen, L. J., & Brown, T. P. (2020). COVID-19: a collision of complement, coagulation and inflammatory pathways. Journal of Thrombosis and Haemostasis, 18(9), 2110-2117.
- Milani, D., Caruso, L., Zauli, E., Al Owaifeer, A. M., Secchiero, P., Zauli, G., ... & Tisato, V. (2022). p53/NF-kB balance in SARS-CoV-2 infection: From OMICs, genomics and pharmacogenomics insights to tailored therapeutic perspectives (COVIDomics). Frontiers in Pharmacology, 13, 871583.
- Gioia, U., Tavella, S., Martínez-Orellana, P., Cicio, G., Colliva, A., Ceccon, M., ... & d’Adda di Fagagna, F. (2023). SARS-CoV-2 infection induces DNA damage, through CHK1 degradation and impaired 53BP1 recruitment, and cellular senescence. Nature Cell Biology, 25(4), 550-564.
- Cao, M., Wang, L., Xu, D., Bi, X., Guo, S., Xu, Z., ... & Li, K. (2022). The synergistic interaction landscape of chromatin regulators reveals their epigenetic regulation mechanisms across five cancer cell lines. Computational and Structural Biotechnology Journal, 20, 5028-5039.
- Icard, P., Lincet, H., Wu, Z., Coquerel, A., Forgez, P., Alifano, M., & Fournel, L. (2021). The key role of Warburg effect in SARS-CoV-2 replication and associated inflammatory response. Biochimie, 180, 169-177.
- Shi, D., & Gu, W. (2012). Dual roles of MDM2 in the regulation of p53: ubiquitination dependent and ubiquitination independent mechanisms of MDM2 repression of p53 activity. Genes & cancer, 3(3-4), 240-248.
- Zhang S, El-Deiry WS. Transfected SARS-CoV-2 spike DNA for mammalian cell expression inhibits p53 activation of p21(WAF1), TRAIL Death Receptor DR5 and MDM2 proteins in cancer cells and increases cancer cell viability after chemotherapy exposure. Oncotarget. 2024 May 3;15:275-284. PMID: 38709242; PMCID: PMC11073320. [CrossRef]
- Wang X, Liu Y, Li K, Hao Z. Roles of p53-Mediated Host-Virus Interaction in Coronavirus Infection. Int J Mol Sci. 2023 Mar 28;24(7):6371. PMID: 37047343; PMCID: PMC10094438. [CrossRef]
- Pal, A., Tripathi, S. K., Rani, P., Rastogi, M., & Das, S. (2024). p53 and RNA viruses: The tug of war. Wiley Interdisciplinary Reviews: RNA, 15(1), e1826.
- Chen, L., & Wang, H. (2019). Nicotine promotes human papillomavirus (HPV)-immortalized cervical epithelial cells (H8) proliferation by activating RPS27a-Mdm2-P53 pathway in vitro. Toxicological Sciences, 167(2), 408-418.
- Nanduri, B., Suvarnapunya, A. E., Venkatesan, M., & Edelmann, M. J. (2013). Deubiquitinating enzymes as promising drug targets for infectious diseases. Current pharmaceutical design, 19(18), 3234-3247.
- Valerdi, K. M., Hage, A., van Tol, S., Rajsbaum, R., & Giraldo, M. I. (2021). The role of the host ubiquitin system in promoting replication of emergent viruses. Viruses, 13(3), 369.
- Liu, X. M., Yang, F. F., Yuan, Y. F., Zhai, R., & Huo, L. J. (2013). SUMOylation of mouse p53b by SUMO-1 promotes its pro-apoptotic function in ovarian granulosa cells. PloS one, 8(5), e63680.
- Matteo D. Parco, Jessica Le Berichel, Paolina Hamon, C. Matthias Wilk, Meriem Belabed , Nader Yatim, Alexis Zafferano, Jesse Boumediene, Chiara Falcomatà, Miriam Merad et al., (2024). Hematopoietic aging promotes cancer by fueling IL-1⍺–driven emergency myelopoiesis. Science, eadn0327.
- Shannon C.E., Weaver W. The Mathematical Theory of Communication. University of Illinois Press; Champaign, IL, USA: 1949.
- Prigogine I. What is Entropy? Naturwissenschaften. 1989; 76:1–8. [CrossRef]
- Skene K.R. Life’s a Gas: A Thermodynamic Theory of Biological Evolution. Entropy. 2015; 17:5522–5548. [CrossRef]
- Dewar R.C. Maximum Entropy Production as an Inference Algorithm that Translates Physical Assumptions into Macroscopic Predictions: Don’t Shoot the Messenger. Entropy. 2009; 11:931–944. [CrossRef]
- Feistel R., Ebeling W. Entropy and the self-organization of information and value. Entropy. 2016; 18:193. [CrossRef]
- Ebeling W., Frömmel C. Entropy and predictability of information carriers. BioSystems. 1998; 46:47–55. [CrossRef]
- Calmet, J., & Daemi, A. (2004). From entropy to ontology. na.
- Daemi, A., & Calmet, J. (2004, November). From Ontologies to Trust through Entropy. In Proc. of the Int. Conf. on Advances in Intelligent Systems-Theory and Applications.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



