
Submitted:
04 October 2024
Posted:
04 October 2024
You are already at the latest version
Abstract
Background/Objectives: Congenital Myasthenic Syndromes (CMS) are caused by variants in >30 genes with increasing numbers of variants of unknown significance (VUS) discovered by next generation sequencing. Establishing VUS pathogenicity requires in vitro studies that slow diagno-sis and treatment initiation. Recent protein structure prediction softwares, AlphaFold2/ColabFold, have revolutionised structural biology; such predictions have also been leveraged in AlphaM-issense which predicts ClinVar variant pathogenicity with 90% accuracy. Few reports, however, have tested these tools on rigorously-characterised clinical data. We therefore assessed ColabFold and AlphaMissense as diagnostic aids for CMS, using variants of the CHRN genes that encode the nicotinic acetylcholine receptor (nAChR).
Methods: Utilising a dataset of 61 clinically-validated CHRN variants, (1) we evaluated the pos-sibility of a ColabFold metric (either predicted structural disruption, prediction confidence, and prediction quality), that distinguishes variant pathogenicity. (2) We assessed AlphaMissenses’ ability to differentiate variant pathogenicity. (3) We compared AlphaMissense to existing patho-genicity prediction programmes, AlamutVP and EVE.
Results: Analysing variant effects on ColabFold CHRN structure prediction, prediction confi-dence, and prediction quality, doesn’t yield any pathogenicity-indicative metric. However, Al-phaMissense predicts variant pathogenicity with 63.93% accuracy on our dataset – a much greater proportion than AlamutVP (27.87%) and EVE (28.33%).
Conclusions: Emerging in silico tools can revolutionise genetic disease diagnosis – however, im-provement, refinement, and clinical validation is imperative prior to practical acquisition.
Keywords:
CMS
; AlphaFold2
; ColabFold
; AlphaMissense
; Pathogenicity
; Prediction
1. Introduction
Congenital myasthenic syndromes (CMS) are a group of rare mendelian monogenic disorders, characterised by abnormal signal transmission from nerve to muscle, across the neuromuscular junction (NMJ) [1]. Fatigable weakness of skeletal muscles is the main symptom of CMS patients, although severity ranges from mild fatigability to severe disability, respiratory crises, and premature death [2]. Pathogenic variants in >30 causative genes have been identified [3], reflecting the number of proteins essential for the development, maintenance and function of the NMJ.
Amongst the causative CMS genes identified, variants in the CHRN genes that encode the subunits of the muscle nicotinic acetylcholine receptor (nAChR) are responsible for ~50% of all CMS cases [4]. Muscle nAChR is a pentameric ligand-gated ion-channel, comprised of α1 (x2), β1, δ, and ε subunits [5]. Following binding of acetylcholine (released from the pre-synaptic nerve terminal), nAChR open, permeating an influx of Na+ and Ca2+ ions into myocytes, initiating an endplate potential that induces muscular contraction [6]. CMS-causing variants of the nAChR do not cause homogenous pathophysiology - rather, different variants disrupt nAChR function and neuromuscular signal transmission in different ways, precipitating three distinct CMS clinical subgroups, classified according to their underlying pathophysiology: receptor deficiency syndromes (RDS, OMIM numbers: CHRNA1 = 253290, CHRNB1 = 616314, CHRND = 616323, CHRNE = 608931), slow-channel syndromes (SCS, OMIM numbers: CHRNA1 = 601462, CHRNB1 = 616313, CHRND = 616321, CHRNE = 605809), and fast-channel syndromes (FCS, OMIM numbers: CHRNA1 = 608930, CHRND = 616322, CHRNE = 616324) [7].
A generic myasthenia diagnosis can be made based upon clinical presentation. However, treatments for certain subtypes have detrimental effects on other subtypes [8]. It’s therefore imperative that a genetic diagnosis is made, in which the causative gene, variant, and its pathophysiological effect is elucidated to optimise treatment [9]. Rapid advancements in our understanding of human genetics over the last 20 years has made non-sense, frameshift and splice boundary variants easily identified as pathogenic. However, it is still challenging to determine whether missense variants are pathogenic. Whilst many CMS-causing variants are already characterised, novel candidate variants require in vitro expression studies and NMJ functional studies to determine pathogenicity (and if pathogenic, their specific pathophysiological mechanism). This manual process is lengthy, and can only be performed at a handful of clinics worldwide, delaying treatment optimisation for patients.
Since 1994, biennial Critical Assessment of Structure Prediction (CASP) competitions have tracked the progress of attempts to predict the 3D-shape of proteins from their amino acid sequence [10]. In 2021, DeepMinds’ AlphaFold2 recorded unprecedented accuracy in this challenge of structure prediction, demonstrating “accuracy competitive with experimental structures” [11]. This ability to accurately predict the 3D structure of a protein from its primary sequence has sparked interest into possible applications of AlphaFold2 for predicting variant effects on protein structure, stability, and function. Despite DeepMind claiming that “AlphaFold has not been validated for predicting the effect of variants” [12], with certain studies backing this claim [13,14], others have provided evidence for such a use case [15], (McBride JM, et al. AlphaFold2 can predict single-mutation effects on structure and phenotype. arXiv [Preprint] 2022. [Accessed 15 August 2024] https://arxiv.org/abs/2204.06860).
ColabFold is an open-source, accelerated version of AlphaFold2, available as a Jupyter notebook within Google Colaboratory that reportedly doesn’t compromise the quality of structure prediction [16]. It doesn’t require the large local computing power, nor extensive bioinformatics skills that Alphafold2 needs to set up, making it a more accessible tool for CMS clinicians worldwide.
In September 2023, DeepMind advanced upon AlphaFold2 to release AlphaMissense, a missense variant pathogenicity prediction model that leverages AlphaFold2 structural predictions at its core. Upon its’ release, AlphaMissense claimed to predict variant pathogenicity with 90% accuracy on variants within the ClinVar database [17].
The aim of this study was to test whether ColabFold and/or Alphamissense could accurately predict the pathogenicity of missense variants in CHRN genes that produce the different subunits of the muscle nAChR receptor. To do so, we used a clinically and experimentally validated database of genetic variants observed in our CMS referral service, containing 42 pathogenic variants (including RDS, SCS, and FCS variants), as well as 19 non-pathogenic variants of the nAChR (spanning all adult subunits). For each variant, we ran the wild-type, and pathogenic/non-pathogenic nAChR subunit sequences through ColabFold, before assessing how the variants’ ColabFold ‘signature’ (i.e. the variants’ effect on ColabFolds’ prediction) differed from the wild-type. This was based on the output data from each ColabFold prediction, which are the same as Alphafold2 – position of amino acid residues, confidence in the predicted position of each amino acid, and the overall predicted quality of template modelling.
We wanted to investigate whether any of the ColabFold data outputs distinguished CHRN CMS variant pathogenicity. Therefore, for each metric, we first tested for differences between pathogenic and non-pathogenic variants, and secondly, split the pathogenic variants into their respective syndrome groups (RDS/SCS/FCS), and compared once more.
Subsequently, in order to compare the ability of AlphaMissense to predict CHRN variant pathogenicity with established non-structural in silico methods, we finally assessed the capability of AlphaMissense, AlamutVP and EVE to differentiate pathogenic from non-pathogenic variants within our dataset. AlamutVP is currently widely used by clinical geneticists, and suggests an ACMG classification [18] for variants based upon a multiplicity of different sources, including computational (REVEL, MaxEntScan, NNSPLICE, SIFT, and PolyPhen-2 predictors) and population data [19]. For further comparison, we also tested EVE, an independent, evolutionary-based pathogenicity prediction programme, not trained directly on ClinVar, that has recently been demonstrated as a leading programme in the field [20].
2. Materials and Methods
2.1. Variant Dataset
We obtained an anonymised dataset from our CMS referral service, containing a list of genetic variants that had been tested for CMS pathogenicity, as well as their functional effects and ensuing clinical syndrome (if pathogenic). This dataset was then curated to contain only CHRN variants, of which existed 42 pathogenic (23 RDS, 14 SCS, and 5 FCS) variants, and 19 non-pathogenic variants spanning each nAChR subunit.
The National Congenital Myasthenia Service defines variant pathogenicity using functional diagnostic experiments. RDS variants result in <25% wild-type nAChR expression, SCS variants induce significantly longer opening times than wild-type receptors in single channel recordings, whilst FCS variants induce significantly shorter opening times than wild-type receptors in single channel recordings. Non-pathogenic variants were tested by similar expression tests and single channel-recordings, but didn’t significantly affect receptor expression or kinetics.
2.2. ColabFold
Wild-type protein sequences of the nAChR genes were obtained from UniProt, using the canonical sequences of the nAChR subunits (UniProt ID: CHRNA1: P02708-2, CHRNB1: P11230-1, CHRND: Q07001-1, CHRNE: Q04844). Variant protein sequences were then produced manually by altering wild-type protein sequences appropriately.
We then ran the individual wild-type subunit sequences, and each individual mutated sequence through ColabFold Batch (available at https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/batch/AlphaFold2_batch.ipynb#Instructions), with parameters outlined in Table 1.
Predicted models with the highest global Predicted Local Distance Difference Test (pLDDT) scores were taken from ColabFold as output for that specific sequence.
2.3. Measuring Variant-Induced Predicted Structural Disruption
To calculate Root Mean Square Deviation (RMSD), both wild-type and mutated ColabFold-predicted subunits were loaded into PyMol, before the ‘align’ feature was used, with PyMol providing an RMSD measurement in the process.
Following this, we used the ‘measurement’ tool on PyMol to individually calculate the Euclidean distance between corresponding residues on the wild-type and mutated predicted nAChR subunit structure. To determine 10aa Local Distance Difference (LDD) scores, we calculated the average distance between the α-carbon of corresponding amino acids both 5 residues upstream and downstream of the mutated residue, along with the mutated residue itself. Similarly for 5Å LDD scores, we measured the average distance between all corresponding, neighbouring residues that fell within a 5Å 3D radius of the mutated α-carbon atom (as determined by PyMol using the command: ‘select all within 5 of Residue/CA’).
2.4. Measuring Variant-Induced Structure Prediction Confidence Changes
Global pLDDT scores were taken from the ColabFold output file and % change was calculated from wild-type to mutated nAChR subunit structure predictions.
Per-residue pLDDT scores were obtained from the B-factor column within the PDB file for each predicted structure. We calculated the average pLDDT score of residues both 5 amino acids upstream/downstream of the mutated residue (as well as the mutated residue itself), as well as those residues that had previously been deemed as falling within a 5Å radius of the mutated residue during the structural disruption analysis. This process was performed on the wild-type and mutated ColabFold-predicted subunits, before the % change in average pLDDT scores for each local proximity metric from wild-type to mutated nAChR subunit, was calculated.
2.5. Measuring Variant-Induced Structure Prediction Quality Changes
pTM scores for individual wild-type and mutated structures were taken from the ColabFold output file, before the % change in pTM was calculated in each case.
2.6. AlphaMissense
AlphaMissense variant pathogenicity predictions were obtained manually from https://console.cloud.google.com/storage/browser/dm_alphamissense;tab=objects?prefix=&forceOnObjectsSortingFiltering=false, for each variant, using the canonical CHRN subunit sequences.
2.7. AlamutVP
We used the Open Gene tab in Alamut Visual Plus v1.7.1 to load the MANE Select gene transcript within the GRCh37 genome for each CHRN gene (RefSeq; CHRNA1: NM_000079.4 CHRNB1: NM_000747.3, CHRND: NM_000751.3, CHRNE: NM_000080.4). For each variant, we queried the variant using corresponding cDNA coordinates. Suggested ACMG classifications were subsequently generated for each variant - ACMG standards were grouped such that ‘Pathogenic’ (Score >=10) and ‘Likely Pathogenic’ (Score between [6,9]) were both considered as ‘Pathogenic’ for our analysis. Variants with a predicted pathogenicity class rated as ‘Uncertain Significance’ (Score between [0,5]) were considered ‘Ambiguous’. Variants predicted as either ‘Likely Benign’ (Scores between [-6,-1]), and ‘Benign’ (Score <=-7) were considered ‘Non-pathogenic’ for our analysis.
2.8. EVE
EVE variant pathogenicity predictions were obtained manually from https://evemodel.org. Canonical CHRN subunit sequences (described in 2.2) were used for all subunits other than CHRNA1, for which the non-canonical P02708 sequence had to be used – variant nomenclature was adjusted accordingly.
2.9. Statistical Analyses
All statistical analyses were performed in GraphPad prism. All ColabFold data was checked for normality using the Shapiro-Wilk test; for the majority of datasets that were non-normally distributed, a Mann-Whitney U test was used to compare pathogenic and non-pathogenic variants, whilst the Kruskal-Wallis test was used to compare between individual CHRN CMS subgroups, and non-pathogenic variants. In each case, we report the median and interquartile range of this data. The % change in global pLDDT data was normally-distributed; therefore, Welch’s unpaired T-test was performed to compare pathogenic and non-pathogenic variant sets, whilst Welch’s ANOVA was used for multiple group comparisons for this metric. We report the mean and standard deviation for this data. Where significant results were found in Kruskal-Wallis tests, a post-hoc Dunn’s test was used to perform pairwise multiple comparisons and reveal group-specific significant differences. Statistical significance was set at 0.05. Receiver Operating Characteristic (ROC) analysis was also performed in GraphPad prism using the Wilson-Brown method.
3. Results
3.1. Variant-Induced ColabFold-Predicted Structural Disruption
3.1.1. Global Structural Disruption
Firstly, we analysed the ColabFold-predicted global structural disruption caused by a variant of the nAChR, recording the root-mean-square deviation (RMSD) between ColabFold-predicted wild-type and mutated nAChR subunits. RMSD is the predominant quantitative measure of structural similarity between distinct molecular structures, giving the global structural disruption elicited by a variant (Figure 1A).
3.1.2. Local Structural Disruption
For a more nuanced measure of predicted structural disruption, we measured the variant-induced structural disruption to the immediate area surrounding the variant site. AlphaFold2 has been demonstrated as sensitive enough to predict the local structural effects of variants, with local-distance-difference (LDD) scores shown to correlate between experimental and AlphaFold2-predicted pairs of wild-type and mutated proteins (McBride JM, et al. AlphaFold2 can predict single-mutation effects on structure and phenotype. arXiv [Preprint] 2022. [Accessed 15 August 2024] https://arxiv.org/abs/2204.06860). Similarly to RMSD, LDD scores provide a measure of average distance between respective residues in two different structures following alignment, but for atoms/residues within pre-defined localities. We recorded LDD scores between ColabFold-predicted wild-type and mutated nAChR subunit structures by sequence as well as structural proximity – five amino acids upstream and downstream of the mutated residue as well as the mutated residue itself (10aa LDD), and all amino acids within 5Å of the mutated residue (5Å LDD) (Figure 1D).
We performed a Mann-Whitney U test to compare both LDD scores between pathogenic and non-pathogenic variants, finding no statistically significant difference for either metric (10aa p-value = 0.8926, 5Å p-value = 0.8804) (Figure 1E-F). We then performed a Kruskal-Wallis test to compare both LDD scores between the different CHRN CMS subgroups and non-pathogenic variants, again finding no statistically significant difference for either LDD score (10aa p-value = 0.7738, 5Å p-value = 0.8407) (Figure 1G-H).
3.2. Variant-Induced ColabFold Prediction Confidence Change
3.2.1. Global Prediction Confidence Change
As well as providing a protein structure prediction, ColabFold also produces a per-residue confidence metric, predicted local distance differece test, pLDDT, which reflects its own confidence in the predicted position of individual amino acids. Previously proposed as a potential proxy measure for positional stability [13], (Zhang et al. Applications of AlphaFold beyond Protein Structure Prediction. bioRxiv [Preprint] 2021. [Accessed 15 August 2024] https://www.biorxiv.org/content/10.1101/2021.11.03.467194v1.), we also wanted to measure the impact that variants of different CMS classifications had on ColabFolds’ global average prediction confidence, thus measuring the % change in average global pLDDT, from the wild-type, to mutated nAChR subunit predictions.
We compared this metric between pathogenic and non-pathogenic variants using Welch’s unpaired T-test, finding no statistically significant difference between the two groups (p-value = 0.7832) (Figure 2A). Subsequent comparison of this metric between CHRN CMS subgroups and non-pathogenic variants using Welch’s ANOVA also revealed no statistically significant differences (p-value = 0.3593) (Figure 2B).
3.2.2. Local Prediction Confidence Change
Like with our structural change metrics, we also deemed it sensible to measure not just how a variant affects the confidence of ColabFold in its prediction of the entire protein structure, but also how it alters local structure prediction confidence. Therefore, we also measured the % change in average pLDDT from wild-type to mutated nAChR subunit predictions at two local proximity definitions – for the 5 amino acids directly upstream/downstream of the mutated residue as well as the mutated residue itself (% change in average 10aa pLDDT), and all amino acids within a 5Å proximity of the mutated residue (% change in average 5Å pLDDT) (Figure 2C).
We compared both of these metrics between the combined pathogenic variant group with non-pathogenic variants using a Mann-Whitney U test, finding no statistically significant difference between the groups in either of the two local prediction confidence change measures (10aa p-value = 0.7634, 5Å p-value = 0.9569) (Figure 2D-E).
However, when comparing these metrics between each of the RDS, SCS, FCS and non-pathogenic variant groups using a Kruskal-Wallis test, a statistically significant result was found for the % change in average 10aa and 5Å pLDDT (10aa: p = 0.0083**, Figure 2F; 5Å: p = 0.0185*, Figure 2G). A post-hoc Dunn’s test subsequently revealed that for both metrics there was a statistically significant difference between the RDS and SCS variant groups (adjusted 10aa p = 0.0066**, adjusted 5Å p = 0.0126*). In both cases, no other pairwise comparisons were statistically significant.
Following this finding, and considering the relatively low number of FCS variants in our dataset (thus limiting statistical power), we combined the SCS and FCS variants into one pathogenic ‘kinetic’ variant group (i.e. variants that alter the kinetic properties of the receptor as opposed to RDS variants that primarily affect the receptors’ stability). We then performed an additional Kruskal-Wallis test to compare the % change in average 10aa pLDDT, and % change in average 5Å pLDDT, across pathogenic deficiency variants, pathogenic ‘kinetic’ variants, and non-pathogenic variants. In both tests, a greater statistically significant difference was revealed (10aa: p = 0.0031**, Figure 2H; 5Å p = 0.0079**, Figure 2I). A post-hoc Dunn’s test thereafter revealed that in both cases, the statistically significant difference existed between the RDS, and the new ‘kinetic’ variant groups (adjusted 10aa p = 0.0021**, adjusted 5Å p = 0.0056**). No other statistically significant differences were identified.
3.3. Variant-Induced ColabFold Prediction Quality Change
A further output metric that ColabFold provides with each structure prediction is the pTM, which represents the quality of protein structure prediction relative to an existing solved target-structure [21] (with this step forming an indispensable step of the ColabFold algorithm). Whilst predominantly a measure of topological accuracy, pTM scores have also been associated with dynamic protein information [22], and therefore we deemed it relevant to assess the effect that variants of the nAChR will have on this ColabFold output value.
We used a Mann-Whitney U test to compare this metric between pathogenic and non-pathogenic groups, with no statistically significant difference between the two groups (p = 0.6513) (Figure 3A).
We then compared the pTM data between individual CHRN CMS subgroups, and non-pathogenic variants using a Kruskal-Wallis test. No statistically significant difference was found (p = 0.3689) (Figure 3B).
3.4. AlphaMissense Variant Pathogenicity Prediction
Following the release of AlphaMissense, we wanted to analyse its acclaimed ability to accurately predict missense variant pathogenicity. In our rigorous dataset of 61 CHRN variants, AlphaMissense predicted the pathogenicity of 39 variants correctly (63.93%), rated 8 variants as ambiguous (13.11%), and predicted 14 variants incorrectly (22.95%, Figure 4A).
Specifically for the 42 pathogenic variants of the nAChR, AlphaMissense correctly predicted 31 as pathogenic (73.81%), 6 as ambiguous (14.29%), and classified 5 variants incorrectly as benign (11.90%, Figure 4B). Whereas for the 19 non-pathogenic variants of the nAChR, AlphaMissense correctly predicted 8 of these variants as benign (42.11%), 2 as ambiguous (10.53%), and 9 incorrectly as pathogenic (47.37%, Figure 4C).
3.5. AlphaMissense Comparison with AlamutVP and EVE
Finally, in order to provide a benchmark with which to compare our AlphaMissense results, we assessed the ability of AlamutVP and EVE to predict the pathogenicity of our nAChR variants. In total, AlamutVP predicted 17 of the variants correctly (27.87%), predicted 43 as ambiguous (70.49%), and 1 variant incorrectly (1.64%, Figure 4A). EVE did not provide a prediction for the CHRNA1 S289I pathogenic variant - however, for the 60 pathogenicity predictions provided, EVE predicted 17 correctly (28.33%), 29 as ambiguous (48.33%), and 14 incorrectly (23.33%, Figure 4A). Regarding pathogenic variants, AlamutVP predicted 17 correctly (40.48%), 25 ambiguously (59.52%), and 0 incorrectly (0.00%), whilst EVE predicted 12 correctly (29.27%), 18 ambiguously (43.90%), and 11 incorrectly (26.83%) (Figure 4B). For non-pathogenic variants, AlamutVP predicted 0 correctly (0.00%), 18 ambiguously (94.74%) and 1 incorrectly (5.26%), and EVE predicted 5 correctly (26.32%), 11 ambiguously (57.89%), and 3 incorrectly (15.79%) (Figure 4C). ROC analysis can be used to directly evaluate and compare the accuracy of different diagnostic tests; thus, we finally performed ROC analysis to compare AlphaMissense, AlamutVP, and EVE. Despite AlphaMissense predicting a greater proportion of our variant dataset correctly, AlamutVP had the highest discriminatory power for our dataset based on ROC analysis, with an area under the curve (AUC) of 0.7688, compared to AlphaMissense (0.7231), and EVE (0.5944) (Figure 4D-F).
Analysing variant effects on ColabFold CHRN structure prediction, prediction confidence, and prediction quality, doesn’t yield any pathogenicity-indicative metric. However, AlphaMissense predicts variant pathogenicity with 63.93% accuracy on our dataset – a much greater proportion than AlamutVP (27.87%) and EVE (28.33%).
4. Discussion
Currently, determination of novel CMS genetic variant pathogenicity relies on manual diagnostic techniques that can only be performed by a handful of diagnostic centres worldwide. Identification of new, reliable, and accessible CMS diagnostic tools would permit faster treatment optimisation for patients globally. Following AlphaFold2s’ release in 2021, and the subsequent development of derivative programmes such as ColabFold, ambiguity has surrounded the application of these protein-structure prediction softwares to functional variant studies. More recently, AlphaMissense was reported to demonstrate great accuracy in this challenge of protein variant pathogenicity classification on a broad ClinVar dataset. However, no studies to our knowledge have used clinically and experimentally validated data to test whether ColabFold and AlphaMissense are useful tools for predicting missense variant pathogenicity. In this study, we compared the accuracy of AlphaMissense with established pathogenicity prediction programmes AlamutVP and EVE.
As AlphaFold2/ColabFold predicts structures based on peptide sequence homology to published experimental structures, it was unlikely that a single missense variant would significantly disrupt the predicted overall structure. Therefore, we investigated whether these algorithms could predict significant structural disruption in the vicinity of the variant. Indeed, there were no statistical significances between any of the data outputs for the overall structure (RMSD, pLDDT, pTM), but we did observe some statistically significant differences between the local (10aa and 5Å) pLDDT scores, with RDS pathogenic variants producing significantly higher values than SCS or the combined “kinetic variants” group. This is intriguing, because RDS variants would be expected to disrupt structural stability, and pLDDT has been thought to serve as a proxy for residue stability [13,23], (Zhang et al. Applications of AlphaFold beyond Protein Structure Prediction. bioRxiv [Preprint] 2021. [Accessed 15 August 2024] https://www.biorxiv.org/content/10.1101/2021.11.03.467194v1.). Conversely kinetic variants would be expected to stabilise a particular conformation of the protein – either an active state, or an inactive state. However, there was no significant difference between the RDS variants and non-pathogenic variants, meaning that this metric was not useful for determining RDS variant pathogenicity. Investigations into local structural changes (10aa and 5Å LDD scores) did not show any significant differences between groups.
We next showed that the more recently released AlphaMissense model is able to predict CHRN variant pathogenicity accurately 63.93% of the time. Our results replicate similar findings to other validatory studies of AlphaMissense (Ljungdahl et al. AlphaMissense is better correlated with functional assays of missense impact than earlier prediction algorithms. bioRxiv [Preprint] 2023. https://doi.org/10.1101/2023.10.24.562294), in that we note AlphaMissenses’ propensity to overestimate variant pathogenicity. In our dataset, AlphaMissense correctly predicted 73.81% of pathogenic variants as pathogenic, and only 11.90% as non-pathogenic. Conversely, AlphaMissense more commonly predicted non-pathogenic variants of the nAChR as pathogenic (47.37%), than non-pathogenic (42.11%).
Crucially, we compared our AlphaMissense results with the ability of AlamutVP and EVE to predict variant pathogenicity using our dataset. AlphaMissense does predict a significantly greater proportion of variants correctly (63.93%) compared to Alamut (27.87%), and EVE (28.33%). However, ROC analysis demonstrated a greater AUC for AlamutVP (0.7688), compared to AlphaMissense (0.7231) and EVE (0.5944); this discrepancy could perhaps reflect overly conservative classification parameters within AlamutVP and the ACMG classification used, implicating that a lower threshold for pathogenic classification prediction within AlamutVP could be employed to improve prediction accuracy. Considering the % of correct pathogenicity predictions was our primary measure of accuracy, we deem AlphaMissense in its current state a more useful tool than both AlamutVP and EVE.
AlphaMissense thus constitutes a welcome extension upon prior structural-prediction softwares, targeted to this specific problem of variant pathogenicity prediction. However, whilst initial genome-wide results are encouraging, our results provide reason for caution regarding the clinical utility of AlphaMissense. How to improve such programmes remains a somewhat empirical question – certainly, the relative scarcity of training data on which to train such programmes remains a challenge upon which attention should be focused. Indeed, the very recent use of a widely more expansive training dataset in the development of BaseFold has been shown to dramatically improve protein structure prediction accuracy from AlphaFold2 levels (Geraldene Munsamy, et al. IMPROVING ALPHAFOLD2 PERFORMANCE WITH A GLOBAL METAGENOMIC & BIOLOGICAL DATA SUPPLY CHAIN. bioRxiv [Preprint] 2024. doi:https://doi.org/10.1101/2024.03.06.583325), and the integration of improved ligand docking, as well as protein-protein interaction information in Alphafold3 could yet improve pathogenicity prediction [24]. Incorporation of such increasingly accurate protein structural information within variant pathogenicity predictions, as with AlphaFold2 structures and AlphaMissense, is likely to improve the accuracy of such programmes, potentially to clinically useful levels.
We believe our study to be one of the first investigations to use a clinically validated dataset into the ability of new-age protein-structure prediction softwares, as well as the recently released AlphaMissense to predict functional variant effects. By comparing the ColabFold signature of variants across different CHRN CMS subgroups and non-pathogenic variants, we demonstrate that for all metrics tested, substantial variance within, and overlap between variant groups means that we fail to provide evidence for the incorporation of ColabFold into CMS diagnostic procedures. We also show that whilst the recently-released AlphaMissense does predict a greater proportion of variants accurately than AlamutVP and EVE, it fails to reach the degree of accuracy reported in seminal studies using large datasets of ClinVar variants [17]. The muscle nAChR may be a particularly challenging protein for prediction algorithms, as it is a heteromultimeric membrane protein with numerous conformational states. However, structural prediction algorithms are developing rapidly, and with the recent release of AlphaFold3 [24], the ability to look at predicted differences in protein complex formation, conformational stability, and ligand docking efficiencies are all on the horizon. Therefore, although we show here that ColabFold and Alphamissense were unable to predict CMS missense variant pathogenicity with a clinically useful level of accuracy, structure-based in silico approaches hold promise for helping diagnosis of CMS, and other genetic diseases more broadly.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. excel file: Raw Data, excel file: Results, excel file: Variants, and excel file: Variant Validator.
Author Contributions
Conceptualization, F.R-P. and Y.D.; methodology, F.R-P. and Y.D.; formal analysis, F.R-P.; investigation, F.R-P.; resources, Y.D.; data curation, F.R-P.; writing—original draft preparation, F.R-P.; writing—review and editing, F.R-P. and Y.D.; visualization, F.R-P. and Y.D.; supervision, Y.D.; project administration, Y.D.; funding acquisition, Y.D. All authors have read and agreed to the published version of the manuscript.
Funding
FRP was funded by the University of Oxford Medical Sciences Final Honour School (FHS) program. YYD was funded by the MRC, grant reference MR/S007180/1.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Human variant data included in this study will be deposited in ClinVar prior to publication. Data collected and analysed during this study can be obtained from the corresponding author upon request.
Acknowledgments
We acknowledge Prof David Beeson, Dr Judith Cossins, Dr Anjali Lloyd and Dr Michael Oldridge for identification of genetic variants. We acknowledge Dr Weiwei Liu, and Ms Susan Maxwell for expression testing of nAChR variants, Dr Richard Webster for carrying out single channel studies of variant channels. We acknowledge Dr Anjali Lloyd for help using AlamutVP.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Vanhaesebrouck An E, Beeson D. The congenital myasthenic syndromes: expanding genetic and phenotypic spectrums and refining treatment strategies. Curr Opin Neurol. 2019; 32(5): 696-703. [CrossRef]
- Finsterer, J. Congenital myasthenic syndromes. Orphanet J Rare Dis. 2019; 14(1): 57. [CrossRef]
- Rodríguez Cruz P, Palace J, Beeson D. The Neuromuscular Junction and Wide Heterogeneity of Congenital Myasthenic Syndromes. Int. J. Mol. Sci. 2018; 19(6): 1677. [CrossRef]
- Abicht A, Müller JS, Lochmüller, H. Congenital Myasthenic Syndromes Overview. GeneRev. 2003. [Accessed 07 January 2024]. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1168/.
- Rahman Md M, Teng J, Worrell B T, et al. Structure of the Native Muscle-type Nicotinic Receptor and Inhibition by Snake Venom Toxins. Neuron. 2020; 106(6): 952-962.e5. [CrossRef]
- Omar A, Marwaha K, Bollu PC. Physiology, Neuromuscular Junction. Nih.gov. 2021. [Accessed 07 January 2024]. Available online: https://www.ncbi.nlm.nih.gov/books/NBK470413/.
- Engel AG, Shen XM, Selcen D, et al. What Have We Learned from the Congenital Myasthenic Syndromes. J Mol Neurosci. 2010; 40: 143-153. [CrossRef]
- Thompson R, Bonne G, Missier P, et al. Targeted therapies for congenital myasthenic syndromes: systematic review and steps towards a treatabolome. Emerg Top Life Sci. 2019; 3(1): 19-37. [CrossRef]
- Engel, G. Congenital Myasthenic Syndromes in 2018. Curr Neurol Neurosci Rep. 2018; 18(8): 46. [CrossRef]
- Moult J, Fidelis K, Kryshtafovych A, et al. Critical assessment of methods of protein structure prediction (CASP) – round x. Proteins. 2013; 82(S2): 1-6. [CrossRef]
- Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021; 596: 583-589. [CrossRef]
- AlphaFold. AlphaFold Protein Structure Database: Frequently Asked Questions. [Accessed 07 January 2024]. Available online: https://alphafold.ebi.ac.uk/faq.
- Pak MA, Markhieva KA, Novikova MS, et al. Using AlphaFold to predict the impact of single mutations on protein stability and function. PLoS ONE. 2021; 18(3): e0282689. [CrossRef]
- Buel GR, Walters KJ. Can AlphaFold2 predict the impact of missense mutations on structure? Nat Struct Mol Biol. 2022; 29(1): 1-2. [CrossRef]
- Xiao Q, Xu M, Wang W, et al. Utilization of AlphaFold2 to Predict MFS Protein Conformations after Selective Mutation. Int J Mol Sci. 2022; 23(13): 7235. [CrossRef]
- Mirdita M, Schütze K, Moriwaki Y, et al. ColabFold: making protein folding accessible to all. Nat Methods. 2022; 19(6): 679-682. [CrossRef]
- Cheng. J, Guido. Novati, Pan. J, et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Sci. 2023; 381(6664). [CrossRef]
- Masson. E, Zou. W-B, Génin. E, et al. Expanding ACMG variant classification guidelines into a general framework. Hum Genomics. 2022; 16(1). [CrossRef]
- SOPHiA GENETICS. Alamut Visual Plus. [Accessed 03 April 2024]. Available online: https://www.sophiagenetics.com/platform/alamut-visual-plus/.
- Frazer. J, Notin. P, Dias. M, et al. Disease variant prediction with deep generative models of evolutionary data. Nature. 2021; 599(7883): 91–95. [CrossRef]
- Zhang, Y. & Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins. 2004; 57(4): 702-710. [CrossRef]
- Guo H-B, Perminov A, Bekele S, et al. AlphaFold2 models indicate that protein sequence determines both structure and dynamics. Sci. Rep. 2022; 12: 10696. [CrossRef]
- Ruff KM, Pappu RV. AlphaFold and Implications for Intrinsically Disordered Proteins. J Mol Biol. 2021; 433(20): 167208. [CrossRef]
- Abramson. J, Adler. J, Dunger. J, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature. 2024; 1–3. [CrossRef]
Figure 1.
Structural-based metrics of nAChR subunit variant predicted effect. (A) Alignment of predicted wild-type and mutated nAChR subunits, from which RMSD is calculated. (B) Box plots comparing the RMSD of pathogenic and non-pathogenic variants from WT (p=0.3885: Mann-Whitney U test, n=42/19), and (C) between functional CHRN CMS subgroups and non-pathogenic variants (p=0.8345: Kruskal-Wallis test, n=23/14/5/19). (D) Example measurements of a variants’ local structural effect on corresponding residues in wild-type and P265L mutated CHRNE (from which LDD scores are derived). Box plots showing 10aa/5Å LDD scores of ColabFold-predicted wild-type and mutated nAChR subunits, compared between (E-F) pathogenic and non-pathogenic variants (10aa p=0.8926, 5Å p=0.8804: Mann Whitney U test, n=42/19), and (G-H) between functional CHRN CMS subgroups and non-pathogenic variants (10aa p=0.7738, 5Å p=0.8407: Kruskal-Wallis test, n=23/14/5/19).
Figure 1.
Structural-based metrics of nAChR subunit variant predicted effect. (A) Alignment of predicted wild-type and mutated nAChR subunits, from which RMSD is calculated. (B) Box plots comparing the RMSD of pathogenic and non-pathogenic variants from WT (p=0.3885: Mann-Whitney U test, n=42/19), and (C) between functional CHRN CMS subgroups and non-pathogenic variants (p=0.8345: Kruskal-Wallis test, n=23/14/5/19). (D) Example measurements of a variants’ local structural effect on corresponding residues in wild-type and P265L mutated CHRNE (from which LDD scores are derived). Box plots showing 10aa/5Å LDD scores of ColabFold-predicted wild-type and mutated nAChR subunits, compared between (E-F) pathogenic and non-pathogenic variants (10aa p=0.8926, 5Å p=0.8804: Mann Whitney U test, n=42/19), and (G-H) between functional CHRN CMS subgroups and non-pathogenic variants (10aa p=0.7738, 5Å p=0.8407: Kruskal-Wallis test, n=23/14/5/19).
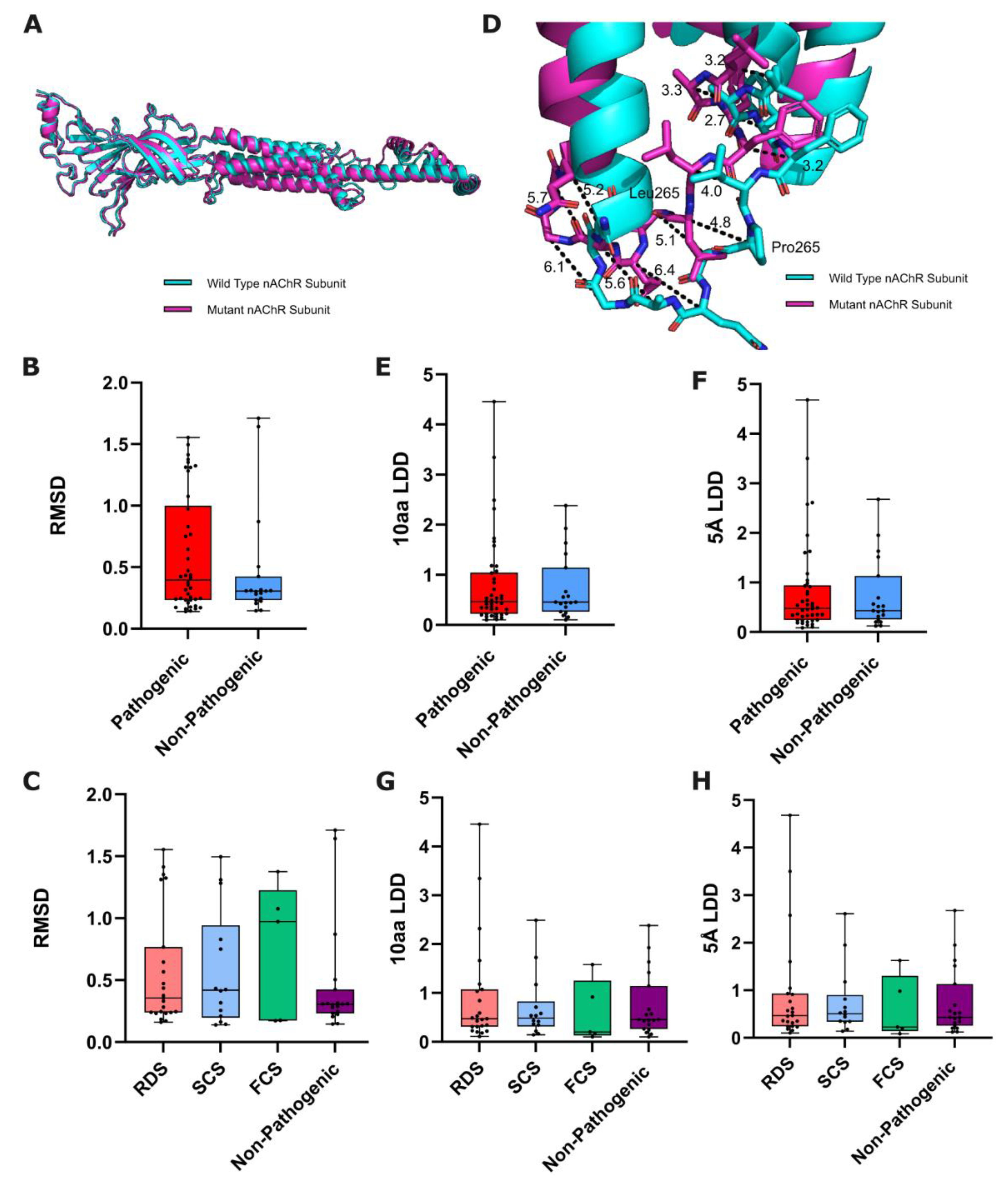
Figure 2.
Prediction confidence-based metrics of nAChR subunit variant effect. % change in global pLDDT was calculated from ColabFold-predicted wild-type to mutated nAChR subunit structures, and compared between (A) pathogenic and non-pathogenic variants (p=0.7832: Welch’s unpaired T-test, n=42/19), and (B) functional CHRN CMS subgroups and non-pathogenic variants (p=0.3593: Welch’s ANOVA, n=23/14/5/19). Error bars represent mean ± SD. (C) Representation of differences in pLDDT between WT and P265L mutant CHRNE, rainbow coloured based on B-factor (confidence) values from ColabFold .pdb file output, red = more confident to green = less confident. Box plots showing % change in 10aa and 5Å pLDDT from ColabFold-predicted wild-type to mutated nAChR subunit structures, compared between pathogenic and non-pathogenic variants (10aa p=0.7634, 5Å p=0.9569: Mann-Whitney U test, n=42/19) (D-E), and between functional CHRN CMS subgroups and non-pathogenic variants (10aa p=0.0083**, 5Å p=0.0185*, Kruskal-Wallis test, n=23/14/5/19) (F-G). Post-Hoc Dunn’s Test for the % change in average 10aa/5Å pLDDT revealed a statistically significant difference between RDS and SCS variants for both metrics (10aa adjusted p-value = 0.0066**, 5Å adjusted p-value = 0.0126*). To increase statistical power of comparisons, SCS and FCS variants were subsequently combined into a ‘kinetic’ variant group. Box plots showing % change in average (H) 10aa pLDDT, and (I) 5Å pLDDT, compared between RDS, kinetic (SCS+FCS), and non-pathogenic variants (10aa p=0.0031**, 5Å p=0.0079**: Kruskal-Wallis test, n=23/19/19). Post-Hoc Dunn’s Test for the % change in both 10aa/5Å average pLDDT revealed a statistically significant difference between RDS and kinetic (SCS+FCS) variants (10aa adjusted p-value =0.0021**, 5Å adjusted p-value = 0.0056**).
Figure 2.
Prediction confidence-based metrics of nAChR subunit variant effect. % change in global pLDDT was calculated from ColabFold-predicted wild-type to mutated nAChR subunit structures, and compared between (A) pathogenic and non-pathogenic variants (p=0.7832: Welch’s unpaired T-test, n=42/19), and (B) functional CHRN CMS subgroups and non-pathogenic variants (p=0.3593: Welch’s ANOVA, n=23/14/5/19). Error bars represent mean ± SD. (C) Representation of differences in pLDDT between WT and P265L mutant CHRNE, rainbow coloured based on B-factor (confidence) values from ColabFold .pdb file output, red = more confident to green = less confident. Box plots showing % change in 10aa and 5Å pLDDT from ColabFold-predicted wild-type to mutated nAChR subunit structures, compared between pathogenic and non-pathogenic variants (10aa p=0.7634, 5Å p=0.9569: Mann-Whitney U test, n=42/19) (D-E), and between functional CHRN CMS subgroups and non-pathogenic variants (10aa p=0.0083**, 5Å p=0.0185*, Kruskal-Wallis test, n=23/14/5/19) (F-G). Post-Hoc Dunn’s Test for the % change in average 10aa/5Å pLDDT revealed a statistically significant difference between RDS and SCS variants for both metrics (10aa adjusted p-value = 0.0066**, 5Å adjusted p-value = 0.0126*). To increase statistical power of comparisons, SCS and FCS variants were subsequently combined into a ‘kinetic’ variant group. Box plots showing % change in average (H) 10aa pLDDT, and (I) 5Å pLDDT, compared between RDS, kinetic (SCS+FCS), and non-pathogenic variants (10aa p=0.0031**, 5Å p=0.0079**: Kruskal-Wallis test, n=23/19/19). Post-Hoc Dunn’s Test for the % change in both 10aa/5Å average pLDDT revealed a statistically significant difference between RDS and kinetic (SCS+FCS) variants (10aa adjusted p-value =0.0021**, 5Å adjusted p-value = 0.0056**).
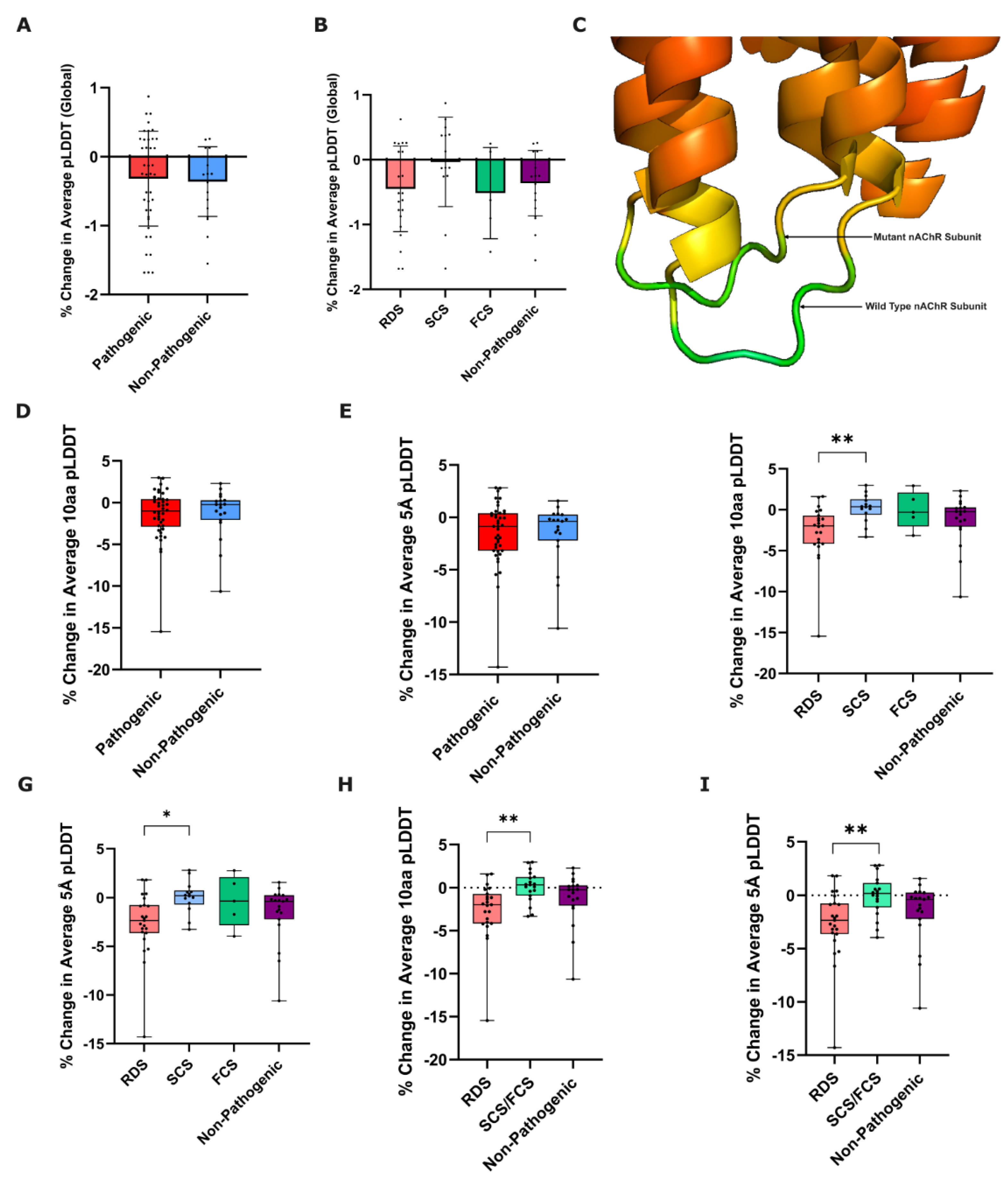
Figure 3.
% Change in pTM as a metric of nAChR subunit variant effect. Box plots showing % change in pTM from wild-type to mutated ColabFold-predicted nAChR subunit, compared between (A) pathogenic and non-pathogenic variants (p=0.6513: Mann-Whitney U test, n=42/19), and (B) functional CHRN CMS subgroups and non-pathogenic variants (p=0.3689: Kruskal-Wallis test, n=23/14/5/19).
Figure 3.
% Change in pTM as a metric of nAChR subunit variant effect. Box plots showing % change in pTM from wild-type to mutated ColabFold-predicted nAChR subunit, compared between (A) pathogenic and non-pathogenic variants (p=0.6513: Mann-Whitney U test, n=42/19), and (B) functional CHRN CMS subgroups and non-pathogenic variants (p=0.3689: Kruskal-Wallis test, n=23/14/5/19).
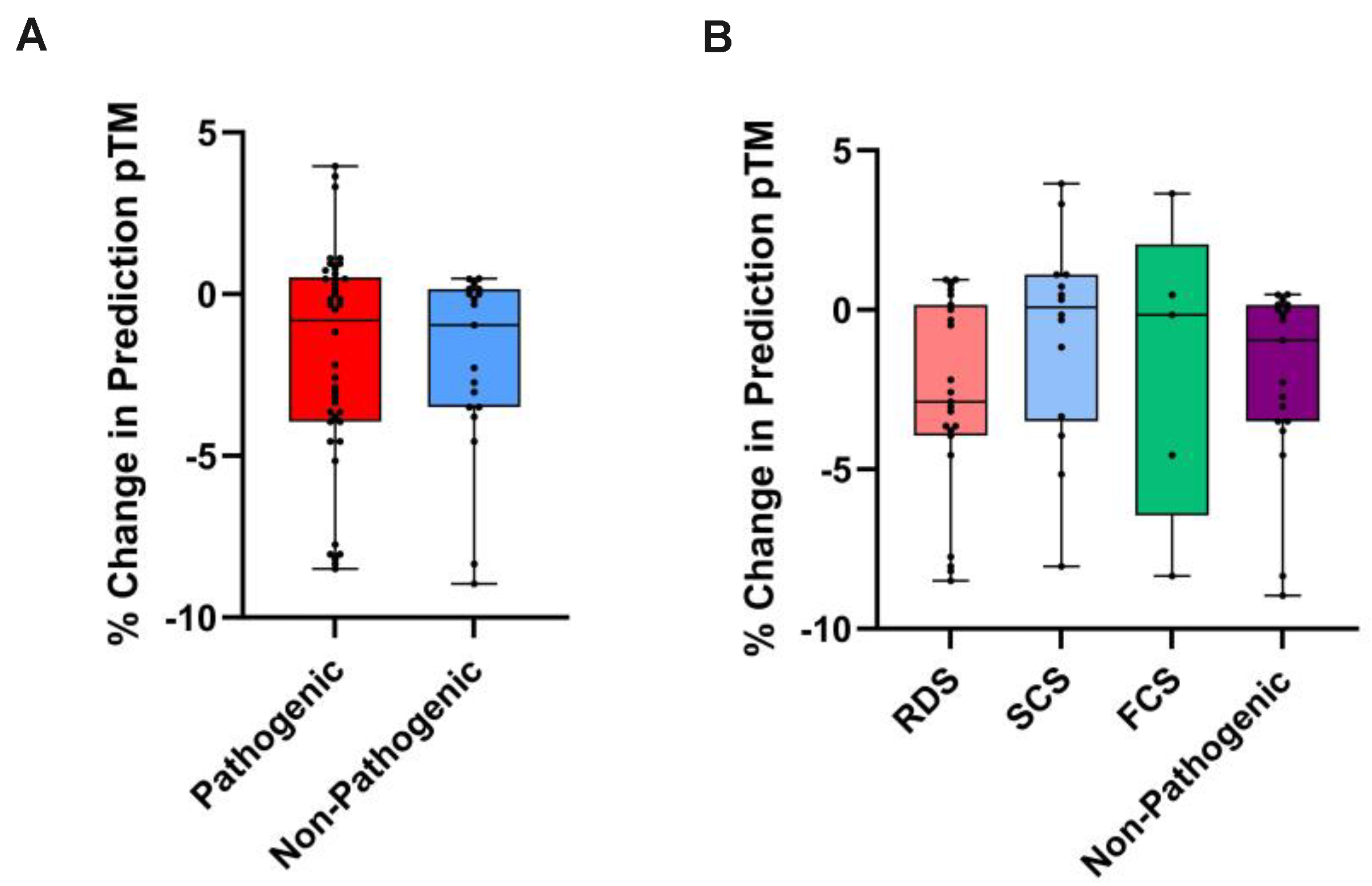
Figure 4.
AlphaMissense comparison with AlamutVP and EVE. Column charts comparing the number of variants that AlphaMissense, AlamutVP, and EVE predicted as correct/ambiguous/incorrect, for (A) all variants combined, (B) for pathogenic variants only, and (C) for non-pathogenic variants only. ROC analyses demonstrated that AlamutVP had the greatest area under the curve (AUC); 0.7688 (D), compared to AlphaMissense (0.7231) (E), and EVE (0.5944) (F).
Figure 4.
AlphaMissense comparison with AlamutVP and EVE. Column charts comparing the number of variants that AlphaMissense, AlamutVP, and EVE predicted as correct/ambiguous/incorrect, for (A) all variants combined, (B) for pathogenic variants only, and (C) for non-pathogenic variants only. ROC analyses demonstrated that AlamutVP had the greatest area under the curve (AUC); 0.7688 (D), compared to AlphaMissense (0.7231) (E), and EVE (0.5944) (F).

Table 1.
ColabFold parameter values utilised in our study.
| Parameter | Value |
|---|---|
| MSA Mode | MMseqs2 (UniRef+Environmental) |
| Number of Models | 5 |
| Number of Recycles | 3 |
| Stop at Score | 100 |
| Use Amber | No |
| Use Templates | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



