Submitted:
04 October 2024
Posted:
07 October 2024
You are already at the latest version
Abstract
In this article we focus on kynurenic acid metabolism, a biochemical part of L-tryptophan catabolism, in processes related to memory and cognitive impairment and its subsequent relevance to neuropsychiatric disorders and the ageing process. Kynurenic acid is synthesised from L-kynurenine by kynurenine aminotransferases.Experimental pharmacological studies have shown that elevated levels of kynurenic acid in the brain are associated with impaired learning and that lowering kynurenic acid levels can improve these symptoms. The discovery of new compounds with the ability to block kynurenine aminotransferase opens up new therapeutic avenues against memory impairment and dementia. The newly developed Helix pomatia snail model of memory is being used for this pharmacological approach. In conclusion, diet, exercise and physical activity have a significant impact on the endogenous pharmacology of kynurenic acid and significantly modulate steady-state conditions and delay the events of the ageing process.
Keywords:
Kynurenic Acid
; Dementia
; Cerebrolysin
; D-Cycloserine
; Glial Depressing Factor
; Jerusalem Balsam
; Herbs
; Snail Helix pomatia
; Memory
- Tryptophan metabolism via the kynurenine pathway
- NMDARs and α-7nAChRs, and neuroprotection
- Kynurenic acid and significant abnormalities
- Kynurenic acid metabolism throughout life
- Glutamatergic and acetyl cholinergic activity and dementia
- Kynurenic acid and neuropsychiatric disorders and dementia
- Different types of pathology after HIV-1
- Glia depressing Factor
-
Anti-dementia approaches
- 9.1
- Stochastic Resonance Therapy (SRT)
- 9.2
- Antidementia drugs
- 9.3
- Herbal medicines - prophylaxis - protection
- Future perspectives
1. Tryptophan Metabolism via the Kynurenine Pathway
In tryptophan catabolism, one of the pathways of particular biological interest is that leading to the formation of niacin, the kynurenine pathway [1], (see Figure 1). Tryptophan catabolism along this pathway has a remarkable function, namely the synthesis of neuroactive metabolites, 3-hydroxykynurenine with neurotoxic [2], quinolinic acid with excitatory [3] and kynurenic acid (KYNA) with inhibitory properties [4]. L-kynurenine, the first major metabolite of tryptophan catabolism along the kynurenine pathway, crosses the blood-brain barrier well [5], is actively accumulated in various tissues (brain, heart, liver) [5], and is taken up by various cells (astrocytes, neurons, macrophages) including their organelles (mitochondria) [6,7].
In the kynurenine pathway, L-kynurenine is catabolised by kynurenine-3-hydroxylase, followed by kynureninase and 3-hydroxyanthranilic acid oxygenase to quinolinic acid [4]. And quinolinic acid is catabolised by quinolinic acid phosphoribosyltransferase to nicotinic acid mononucleotide and finally to the active coenzyme nicotinamide adenine dinucleotide (NAD) [1,4].
The other possibility of L-kynurenine degradation is the formation of KYNA by irreversible transamination of L-kynurenine [8], or the formation of anthranilic acid by kynureninase [1], followed by hydroxylation of anthranilic acid to 3-hydroxyanthranilic acid [9]. KYNA synthesis from tryptophan using indole-3-pyruvic acid has also been described [10,11,12], but its significance remains to be elucidated.
The capacity for KYNA formation from L-kynurenine by kynurenine aminotransferase (KAT) is very high and several KATs have been discovered in different mammalian organs [13,14].
In rat and human peripheral tissues, four types of proteins are capable of catalysing the kynurenine-2-oxoacid transamination reaction to form KYNA [13,14,15,16,17]. Very low activity of KYNA formation has been reported in peripheral tissues from pigs [17] or the snail Helix pomatia [18], suggesting species differences. The central nervous system (CNS) is also significantly involved in KYNA formation and three proteins, KAT I, KAT II and KAT III, have been found in rat, mouse, piglet, snail and human brain tissue [19,20,21,22,23,24,25].
They are characterised by specific enzymatic properties and different biochemical activities, and their different physiological roles have been suggested. KAT I, characterised by a high pH optimum of 9.6, may be particularly important in pathological conditions [19,20,21], whereas KAT II, with a neutral pH of 7.4, may operate essentially under physiological conditions [21,22], and KAT III, with a pH around 8.0-9.0 [14,23,24,25], may share its action between physiological and pathological conditions. There are data suggesting that human KAT I is a protein with multifunctional activities and may also be an important protein in KYNA synthesis under physiological conditions [20]. Interestingly, KATs appear to have the ability to change their chemical properties and presumably their actions under physiological and pathological conditions [26,27]. KYNA formation has been shown to occur preferentially in glia, astrocytes and to a lesser extent in neurons [7,16,26,27,28].
2. NMDARs and α-7nAChRs, and Neuroprotection
In the last decades of the last century, important information about the action of KYNA was revealed. KYNA acts as an endogenous antagonist of the glutamate ionotropic excitatory amino acid receptors N-methyl-D-aspartate (NMDAR), alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid, kainate [4,29,30], and the nicotinic acetylcholinergic subtype alpha-7 receptor (α-7nAChR) [31], but is also an agonist at the orphan G-protein coupled receptor GPR-35 [30] and has anticonvulsant and neuroprotective activities [32,33]. In the brain, KYNA levels range from low nanomolar to low micromolar concentrations [4,10] and there is evidence that these physiologically relevant concentrations of KYNA are likely to block α-7nACh more effectively than NMDA receptors [31,34,35]. With regard to KYNA inhibition of glutamatergic neurotransmission, accumulated data suggest that the glycine likely co-agonist site of the NMDA receptor is not saturated [4,36,37,38], so physiological concentrations of KYNA may be sufficient to inhibit NMDA receptor activity.
With regard to KYNA inhibition of cholinergic neurotransmission, KYNA inhibits α-7nAChRs non-competitively (IC 50 approximately 7 µM) [31,34,35], the inhibition of α-7nAChRs by KYNA is likely to be voltage independent [34,35]. Speculatively, it has been suggested that KYNA may act via intracellular second messengers that affect α-7nAChR function [34,35]. Interestingly, KYNA also increases the expression of non-α-7nAChRs, α-4-ß-2nAChRs [30,34,35], suggesting a notable interaction between KYNA and the receptor functions of cholinergic neurons. Importantly, α-7nAChRs play multiple roles in modulating the glutamatergic system in both normal and diseased nervous systems [38].
On the other hand, a biphasic change in KYNA formation has been reported after prolonged nicotine application [39], suggesting a down- and/or up-regulation by nicotine.
The neuroprotective and anticonvulsant activities of KYNA have been documented [32], and this role is most likely related to the modulation of both glutamatergic and acetylcholinergic neurotransmission [3], since the overall effects of NMDA receptor antagonists and the effects of α-7nAChR antagonists on neuronal plasticity and also on viability are similar and close to those of KYNA, as demonstrated by different pharmacological approaches. For example, KYNA and α-7nAChR antagonists can block neurite outgrowth [40,41] or both can reduce apoptotic neuronal death [40,42] or both have anticonvulsant activity [30,43,44]. Accumulating data suggest that both NMDA and nACh receptors are involved in the regulation of neuronal plasticity and survival in the brain. An antioxidant activity of KYNA may also play a role [33].
3. Kynurenic Acid and Significant Abnormalities
Dysfunction of KYNA synthesis in the brain has been suggested as an important factor contributing to neuronal degeneration [4,30,32]. Indeed, in vivo experimental studies have shown that reducing KYNA synthesis in the rat brain using non-specific inhibitors can lead to neurotoxic effects [45,46,47]. Importantly, a preferential loss of layer III of the entorhinal cortex after local injection of a non-specific inhibitor of KYNA synthesis has been shown to be an important factor in the pathophysiology of human temporal lobe epilepsy [46,48]. On the other hand, increases in brain and serum KYNA levels have been observed in rats exposed to kainic acid-induced stereotypic behaviour and seizures [49], or during oxygen deprivation [50], or in a genetic model of dystonia [51], or in chronic kainic acid rats with spontaneous seizures [52], or after encephalomyocarditis infection in piglets [53], or after HIV-1 infection in humans [54]. Respiratory distress, bronchopneumonia, lobar pneumonia, pulmonary oedema or even tuberculosis occur after various infections such as EMCV, HIV-1, influenza A or Corona virus [53,54]. In particular, high mortality has been reported in infected mammals, including humans [53,54,55,56,57,58,59]. It would therefore be reasonable to consider the importance of activation of kynurenine metabolism during events associated with impaired oxygen consumption capacity. Indeed, a marked increase in brain KYNA has been found during asphyxia due to oxygen deprivation [50]. Of particular interest in this work was the observation that the longer the period of oxygen deprivation, the higher the observed peak in brain KYNA and the higher the lethality [50]. The decrease in ATP synthesis also correlates significantly with the duration of asphyxia [50,60]. Of particular note is the dramatic increase in brain KYNA levels during the 15-20 min period of asphyxia, characterised by almost 100% lethality [50], which may be relevant to ischaemic events in stroke or even sudden death [53,61]. Experimental work in vivo has shown that administration of KYNA (icv) to rats resulted in ataxia and stereotypy in a dose-dependent manner (0.025-1.6 µmol) [62,63]. Importantly, the authors demonstrated that administration of 0.8 µmol KYNA resulted in drowsiness and approximately 25% animal mortality, and at a dose of 1.6 µmol, all animals died of cardiorespiratory failure within 2-5 minutes [62,63].
These data suggest that spontaneous synthesis of KYNA in the brain due to infection and/or unknown cause could lead to cardiorespiratory dysfunction followed by acute death. Our data indicate that kynurenine metabolism is also activated in the brain after EMCV infection [64,65], and it is reasonable to assume that this increase may be responsible for the occurrence of cardiorespiratory dysfunction and sudden death. However, this proposed mechanism requires further investigation. We have previously demonstrated differences in the effect of tryptophan metabolites on cardiac and brain mitochondrial respiratory parameters. KYNA increases the oxygen consumption of rat heart mitochondria [66,67] and this observation suggests an essential role for KYNA in cellular mitochondrial function, at least in cardiac myocytes. While cardiac mitochondria are sensitive to KYNA, brain mitochondria are only slightly affected by this kynurenine metabolite [67].
3-hydroxykynurenine and 3-hydroxyanthranilic acid have been shown to significantly affect mitochondrial function in the brain and heart. No effect was reported in the presence of the potent neurotoxin quinolinic acid [67]. Furthermore, the sensitivity of brain and heart mitochondria to these metabolites was insignificantly affected by ageing, at least in healthy rats [68], indicating a high availability of mitochondrial energy supply throughout life.
4. Kynurenic Acid Metabolism throughout Life
KYNA metabolism in the CNS shows a characteristic pattern of changes throughout the lifespan in different animal species and in humans [69,70,71,72,73,74]. A marked increase in KYNA levels in the CNS occurs before birth, accompanied by a dramatic decrease on the day of birth [69,73]. Low KAT activity was found during the first week after birth [70] and a slow and progressive increase was observed during maturation [70,71] and ageing [71,72]. The data published at that time showed the changes in KAT II activity [70], as the changes in KAT I or KAT III during ontogeny and ageing were not elucidated at that time.
The remarkable change in KYNA metabolism during ontogeny and maturation in the mammalian brain has been suggested to be a consequence of the development of neuronal connectivity organisation, synaptic plasticity and receptor recognition [4,30,34,35,40,48]. It is likely that KYNA has different functions throughout life, depending on the neuronal network currently available. Furthermore, the dramatic decrease in KYNA on the day of birth [69,73] suggests that KYNA is involved in biochemical processes/events that regulate lung function.
Consistent with an increase in KYNA throughout life, an increase in L-kynurenine, the bioprecursor for KYNA synthesis, has been reported in aged rats [72] and also in the cerebrospinal fluid of elderly humans [74]. Tryptophan has also been found to increase with age [75]. The high levels of tryptophan in the elderly may be due to uncontrolled consumption, e.g. of chocolate, but also to a reduced capacity for biochemical degradation during ageing.
5. Glutamatergic and Acetylcholinergic Activity and Dementia
In the early 1970s, Olney, Rothman, Choi and others showed that excessive activation of NMDA receptors, a cellular phenomenon involved in neuronal signalling in both neurotrophic and neuronal plasticity, can trigger a series of intracellular and/or extracellular events involved in neuronal death [4,30]. At the same time, this phenomenon was thought to provide a mechanism for memory formation in the brain. According to the hypothesis proposed by Greenmayer et al in 1988 [76] and supported by others, glutamatergic transmission plays an important role in the neuropath mechanism and symptomatology of dementia [4,30]. Whitehouse et al, 1981 showed that the nucleus basalis of Meynert provides a diffuse cholinergic input to the neocortex [77].
Compared with an age- and sex-matched control, the nucleus basalis of a patient with Alzheimer's disease showed a significant reduction in neurons. The loss of this neuronal population was an anatomical correlate of the well-documented cholinergic dysfunction in AD [77]. Neurochemically, a reduction in choline acetyltransferase has emerged as an important marker of dementia [78]. Loss or impairment of cholinergic neurons has been described in Alzheimer's disease, vascular dementia, Down syndrome and during ageing [78,79,80,81,82,83].
6. Kynurenic Acid and Neuropsychiatric Disorders and Dementia
The suggestion that elevated levels of KYNA in the CNS may be involved in cognitive decline is supported by the fact that elevated levels of KYNA have been found in patients with Alzheimer's disease [84,85,86], patients with DOWN syndrome [87] patients with hydrocephalus [75], patients with subcortical sclerotic encephalopathy [88], patients infected with the HIV-1 virus [54,89], patients with early stage Huntington's disease [90], patients with schizophrenia [91,92], and also in normal elderly people [74].
Interestingly, KYNA levels are significantly increased in demented PD patients but not in non-demented PD patients [93], and these observations raise the question of whether an increase in KYNA levels in the brain of demented PD patients is an important neurochemical marker of dementia. Nevertheless, the development of pathological events depends not only on the degree of dopamine depletion [94,95] but also on the degree of KYNA enhancement, processes that may be interdependent or separate.
It is known from pharmacological studies that KYNA can improve cognition and memory [96], but it has also been shown to impair working memory [97], and increasing endogenous KYNA levels induces spatial memory deficits [98].
In Alzheimer's disease, increased KYNA levels have been observed in several regions, the frontal cortex, hippocampus, putamen, caudate nucleus and cerebellum, ranging from 123 to 192% of CO [86], suggesting a general increase in KYNA levels in the brain. However, a study of the enzyme activity of KATs showed a significant increase in KAT I in the putamen and caudate nucleus, but not in KAT II, which was only moderately affected, Figure 2, adapted from [86].
One might ask whether increased KYNA metabolism in the basal ganglia compensates for hyperactivity in the striato-frontal lobe, as we have previously suggested in Alzheimer's disease [86]. The interaction between high KYNA levels and dopamine reduction in the striatum has been demonstrated [99]. We therefore suggest that the decrease in dopaminergic neurotransmission of the nigrostriatal loop, which is characteristic not only of PD but also of ageing [94,95], is associated with an increase in KYNA metabolism in basal ganglia regions, as has been observed not only in PD patients [85,93] but also in people with Alzheimer's disease, vascular encephalopathy, cerebral infarction [85] and ageing [74,85].
It is therefore reasonable to assume that dopaminergic neurotransmission in the caudate nucleus is upregulated by astrocyte-derived KYNA, especially with advancing age [85,94]. Some interesting findings and correlations can also be drawn from experimental data: the reduction of dopamine levels in various rat brain regions, such as the limbic and basal ganglia regions of rats in the kainic acid epileptic model [100], in the acute phase is also associated with a significant increase in KYNA [49].
Levels of the neurotransmitter serotonin were not altered during the acute phase, although there was a marked increase in the turnover of both catecholamine and serotoninergic neurons. The lack of serotonin lowering in the kainic acid model was one of the miracles of my study and work," commented Prof Oleh Hornykiewicz, my PhD supervisor at the time.
A pronounced increase in KYNA levels (Figure 3, adapted from [87]), decreased KAT I activity and normal KAT II (Figure 4, adapted from [87]) in the frontal and temporal cortex were measured in Down's syndrome subjects, indicating a remarkable discrepancy between high KYNA levels and low KAT activities in the frontal and temporal cortex of Down's syndrome subjects [87]. It is likely that substrate availability, i.e. L-kynurenine or cellular KYNA, or reduced KYNA clearance may be responsible for the remarkably high KYNA levels. The question arises as to whether the genetic background of Down's syndrome plays a mitigated role in Alzheimer's disease, as they share similar neurochemical events related to the deficit in nACh receptor expression/function and the increase in KYNA in the brain (frontal cortex) [82,83,86,87].
It is also important to note the pathological state of the patients used in the Down's syndrome study. According to clinical findings, the control patients and especially the Down's syndrome patients had pathology related to infarction and bronchopneumonia, respectively [87], processes characterised by increased KYNA levels [86,110]. This could be the reason for such a strong KYNA enhancement, indicating a very good correlation between oxygen impairment and early lethality [53,60,62,63].
Microglial activation is an important pathological feature of a variety of neurological disorders, including ageing, and correlates with increased KYNA metabolism [101,102]. There is a predominant astrocytic localisation of KAT in the rat brain [16,28] and the major intermediate filament component of astrocytes, glial fibrillary acidic protein (GFAP), increases with age [102,103,104].
In addition, the interaction between glial and immune cells and the secretion of various cytokines such as interferon or interleukin-1 and 6, tumour necrosis factor [105] in response to injury or infection or during the ageing process play a prominent role in the initiation and propagation of CNS damage [104].
Activation of tryptophan degradation by tryptophan 2,3-dioxygenase by interferon has also been observed in human monocytes/macrophages and a variety of human cells and cell lines in vitro [107,108]. The positive correlation between an increase in CSF ß2-microglobulin and KYNA levels during the ageing process [74] suggests an activation of immune cells. Increased CSF ß2-microglobulin levels have been reported in Alzheimer's disease [108], cerebral infarction and meningitis [53], and HIV-1 infection [89,109].
7. Different Types of Pathology after HIV-1
The assessment of kynurenine metabolism, e.g. L-kynurenine and KYNA levels and the activity of KYNA, KAT I and KAT II synthesising enzymes in the frontal cortex and cerebellum of HIV-1 infected patients in relation to different types of pathology classified as HIV in the brain (HIV), opportunistic infection (OPP), cerebral infarction (INF), malignant lymphoma of the brain (LY) and glial dystrophy (GD) and in controls (CO) showed significant alterations [110]. Importantly, of all the pathologies studied, OPP was the most common (65%), followed by HIV (26%), LY, INF and GD (22% each). In addition, 68% of HIV-1 patients had bronchopneumonia, the highest incidence of which was 60% in the OPP and LY groups.
KYNA was significantly increased in the frontal cortex in LY (392% of CO), HIV (231% of CO) and GD (193% of CO) and in the cerebellum in GD (261% of CO). Concerning the enzyme KAT I, the activity was significantly increased in the frontal cortex of all pathological subgroups, i.e. OPP = 420% > INF > LY > HIV > GD = 192% of CO [110]. Similar changes were found in the cerebellum, where KAT I activity was significantly increased in all pathological subgroups (OPP = 320% > LY, HIV > GD > INF = 176% of CO). In contrast, KAT II activity was only moderately but significantly higher in the frontal cortex of INF and OPP; in the cerebellum of HIV, OPP and LY it was comparable to that of controls, while in INF and GD it was even slightly reduced [110].
On the other hand, L-kynurenine, a bioprecursor of KYNA, was increased in the frontal cortex of LY (385% of CO) and INF (206% of CO) and in the cerebellum of GD, LY, OPP and HIV (between 177 and 147% of CO). Correlation analyses between kynurenine parameters showed an association between a high KAT I/KAT II ratio and increased KYNA levels and lower L-kynurenine in the frontal cortex and cerebellum of the HIV and LY subgroups. The data revealed dramatic biochemical variability in the brain between the pathological groups [110].
Interestingly, among the normal subjects we used as controls, we found some who had been diagnosed with bronchopneumonia [110], and they were indeed characterised by high KYNA metabolism in the brain as well. KAT I activity in the frontal cortex and cerebellum was markedly increased by about 877% and 479% of CO, respectively. This finding suggests a remarkable correlation between impaired conditions of oxygen availability and increased KYNA formation in the human brain. These observations may also have implications for understanding the pathological processes in the brain following HIV-1 infection, as well as other infections associated with the development of neuropsychiatric and neurological symptoms, including memory and cognitive impairment, as well as lethality [110].
8. Glia Depressing Factor
Our previous data showed that human cerebrospinal fluid and even human serum significantly reduced KAT activities [111] and we have suggested the presence of a "glia depressing factor" (GDF), but its exact mechanism of action/function needs to be further elucidated. It is known that dietary restriction can reduce the incidence of memory loss and/or cognitive impairment, as dietary restriction reduces GFAP transcription and microglial activation during ageing [112], which increases significantly with age [104]. In this context, a reduction in KYNA formation has also been reported [112]. There are many different factors that control the wide variety of biochemical machinery in the mammalian body and their properties/function are characterised by common events.
It is questionable whether GDF may lose its effectiveness in controlling KYNA metabolism during life, and this abnormality is partly reflected in Down syndrome, Alzheimer's disease, various forms of dementia, ageing and even schizophrenia. Interestingly, CSF from multiple sclerosis patients showed significantly weaker inhibition of KAT I activity [111], an enzyme whose role has been linked to pathological conditions.
9. Anti-Dementia Approaches
Several approaches have been approved to treat dementia or improve memory and cognition, including drugs that interact with cholinergic activities, i.e. acetylcholine esterase inhibitors such as galantamine, donepezil, rivastigmine, or drugs that block glutamate neurotransmission such as NMDA receptor blockers - memantine or nootropics: Piracetam; or Ginkgo biloba or cerebrolysine and many others [30,108,113,114]. Interestingly, a significant interaction between KYNA action and/or metabolism and galantamine treatment has been demonstrated; the mechanism of action of galantamine in the treatment of dementia has been suggested to be due to its action as a nicotinic allosteric potentiating ligand and also as a competitive antagonist of KYNA-induced inhibition of α-7nACh receptors [113]. The development of this task is described in the review by Stone, et al, 2014 [30]. In addition, experimental animal studies have provided important evidence that exercise also has a significant impact on the learning process.
9.1. Stochastic Resonance Therapy (SRT)
The first therapeutic indication for exercise was introduced as early as 1880 by Jean-Martin Charcot, who described that the use of a rocking chair in Parkinson's disease patients led to an improvement in symptoms, particularly tremor and stability [115]. A marked deficit in dopaminergic neurotransmission and its importance in PD symptoms was introduced by Hornykiewicz [94,95]. The therapeutic potential of vibration in PD patients has also been confirmed by many studies [116,117,118]. Dopamine is significantly affected by exercise [119] and endogenous KYNA has been shown to control extracellular dopamine levels in rat striatum in vivo [120].
A phenomenon called stochastic resonance, in which oscillations enhance the response of a non-linear system to a weak signal, can affect molecular biological machinery and physiological responses. The first report on the mechanism of stochastic resonance was made by Benzi et al [121], and its significance was applied to a theoretical explanation of the periodic recurrence of the Earth's ice ages.
Interestingly, in biology, stochastic resonance has been experimentally demonstrated in several sensory neural systems, including crayfish, shark, cricket [122], and also in humans [123,124]. Collins et al. showed that the tactile sensation of the human fingertip can be enhanced by mechanical vibration [123], and Collins' finding suggests that mechanoreceptors can be affected by stochastic resonance [124].
Stochastic resonance therapy (SRT) is now being used to rehabilitate patients with several neuropsychiatric disorders, including Parkinson's disease [125], multiple sclerosis [126], Alzheimer's disease [127,128], stroke [129], depression and schizophrenia [130]. Exercise has significant effects on learning and long-term potentiation [131], axonal regeneration of sensory neurons [132], restoration of synaptic plasticity [133], and also on tryptophan metabolism [134,135].
We have shown that SRT affects tryptophan metabolism by significantly reducing serum L-tryptophan, L-kynurenine and KYNA levels in healthy human subjects [135]. Importantly, following SRT, subjects reported increased stability and ease of walking over the following hours. The effect on tryptophan metabolites was time-dependent and the reduction was measured 60 minutes after SRT, indicating a longer lasting effect. The changes in L-tryptophan metabolism suggest an increased incorporation of the amino acid into ongoing biochemical processes. As L-tryptophan crosses the blood-brain barrier significantly, changes in serum may also affect NAD and/or serotonin synthesis not only in the periphery but also in the CNS [4,5]. The well-being and good feeling reported by subjects after SRT would suggest an antidepressant effect.
The decrease in KYNA levels after SRT may also involve the activation of the glial-decreasing factor GDF, as proposed [111]. GDF has the ability to block KAT, the kynurenine aminotransferase, and probably simultaneously block glial activities, thereby directly or indirectly exerting its neurotrophic function. On the other hand, reducing KYNA could be beneficial for glutamatergic and acetyl cholinergic neurotransmission.
A transient decrease in blood KYNA levels was observed in rats subjected to treadmill exercise [134]. This effect was dependent on the duration of exercise and the transient decrease was observed on the 1st and 14th day of the experiment, whereas on the 21st day of the experiment, KYNA levels were comparable to the control.
9.2. Antidementia Drugs
The study of cerebrolysin and D-cycloserine in competition with kynurenine metabolism was a great scientific experience.
9.2.1. Cerebrolysin
The therapeutic effects of cerebrolysin in dementia and brain injury have been proposed due to the neurotrophic and neuroprotective activities of this compound [114]. In our previous research, we found that piglet brain has very low levels of KATs and has the ability to block rat KATs activity when present in the reaction mixture for the KAT assay [136]. As piglet brain is used to synthesise cerebrolysin [137,138], we also investigated cerebrolysin and found that indeed cerebrolysin significantly blocked brain KAT I, II and III activities [139]. We proposed that inhibition of KAT activity could modulate KYNA levels at the receptor site and lead to an increase in acetyl cholinergic and glutamatergic neurotransmission.
Another mechanism of therapeutic action of cerebrolysin may be related to a reduction in microglial proliferation, and thus the therapeutic efficacy of cerebrolysin may involve a blockade of KAT-associated microgliosis [138]. Regarding the cellular localisation of KATs, in situ hybridisation studies have shown that KAT-I mRNA activity is expressed in the mitochondria of glial and neuronal cells, as well as in the cytosol of choroid plexus epithelial cells [28] and in human astrocytes [26]. A significant correlation between microglial proliferation and a concomitant increase in KAT activity/expression and GFAP expression in astrocytes and some neurons has been demonstrated [16,28,48,102,103,104]. Interestingly, therapeutic efficacy of cerebrolysin has also been observed in schizophrenia patients.
9.2.2. D-Cycloserine
D-cycloserine is a partial agonist at the NMDA receptor site [140], has a developmental effect on learning [141], and showed anticonvulsant activity in the kainic acid rat model of temporal lobe epilepsy [142]. We demonstrated significant formation of brain and plasma KYNA in rats with developed seizures [49]. Surprisingly, we also observed increased KYNA in kainic acid seizure-resistant rats. D-cycloserine significantly blocked kainic acid-induced seizures [142], comparable to MK-801 and diazepam. Given these observations, it was important to further elucidate the mechanism of D-cycloserine's anticonvulsant activity, as several other enzymes are affected by D-cycloserine, such as GABA transaminase [143]. The most surprising finding was that D-cycloserine induces inhibition of KYNA formation by blocking KAT I, KAT II and KAT III in the frontal cortex [144], and this biochemical event may be of potential benefit for CNS treatment of cognition and/or memory in various neurological and neuropsychiatric disorders [145]. Interestingly, the remarkable efficacy of D-cycloserine treatment in schizophrenia has been noted in the presence of neuroleptics [141].
With this in mind, it is compelling that the reduction of dopamine neurotransmission by neuroleptics and the reduction of KYNA levels by D-cycloserine treatment represent a promising therapeutic paradigm in patients with schizophrenia. The most important factor is the dose of D-cycloserine. Treatment with low doses gives a better therapeutic outcome [145], I suggested to Prof Goff.
The blocking effect of cerebrolysin or D-cycloserine on KAT activity to reduce KYNA formation was recently investigated in the snail Helix pomatia, and these data were consistent with results observed in rats and humans, suggesting a high degree of similarity between these different species and stages of brain development (Baran, H.; Kronsteiner, C. MS in sub.). One striking finding is that all species show increased KYNA synthesis in the CNS with age, but not in the Helix pomatia snail [18]. One speculation could be that the endogenous compound, such as GDF, that blocks KYNA synthesis is not affected along the lifespan in the snail.
High doses of D-cycloserine had no effect on KAT I activity, but actually tended to increase KYNA. This finding may be crucial for therapeutic efficacy (Baran observation).
9.3. Herbal Medicines - Prophylaxis – Protection
Old European reference books on medicinal plants document a number of other plants, such as Salvia officinalis (Sage) and Melissa officinalis (Balm), with memory enhancing properties. In our preliminary study, we investigated the effect of Salvia on KYNA synthesis. Interestingly, we found that Salvia significantly reduced KYNA formation (Baran observation). These data suggest a link between Salvia's ability to inhibit KYNA synthesis and its memory enhancing properties. Hawthorn berries 'haws' are used to make wine, jelly and flavour brandy, and this plant has been used as a remedy for heart problems and also to treat Alzheimer's disease. We have found that hawthorn berries also block KYNA synthesis [146]. We have evaluated the effect of a remedy, Jerusalem Balsam, a herbal mixture, for its ability to block the biosynthetic machinery of KYNA synthesis, e.g. the activity of the enzyme that synthesises KYNA, KAT II, in an in vitro study [147]. Jerusalem Balsam is recommended for the improvement of lung function. We found that Jerusalem Balsam was a dose-dependent and significant inhibitor of KAT II activity in an in vitro assay.
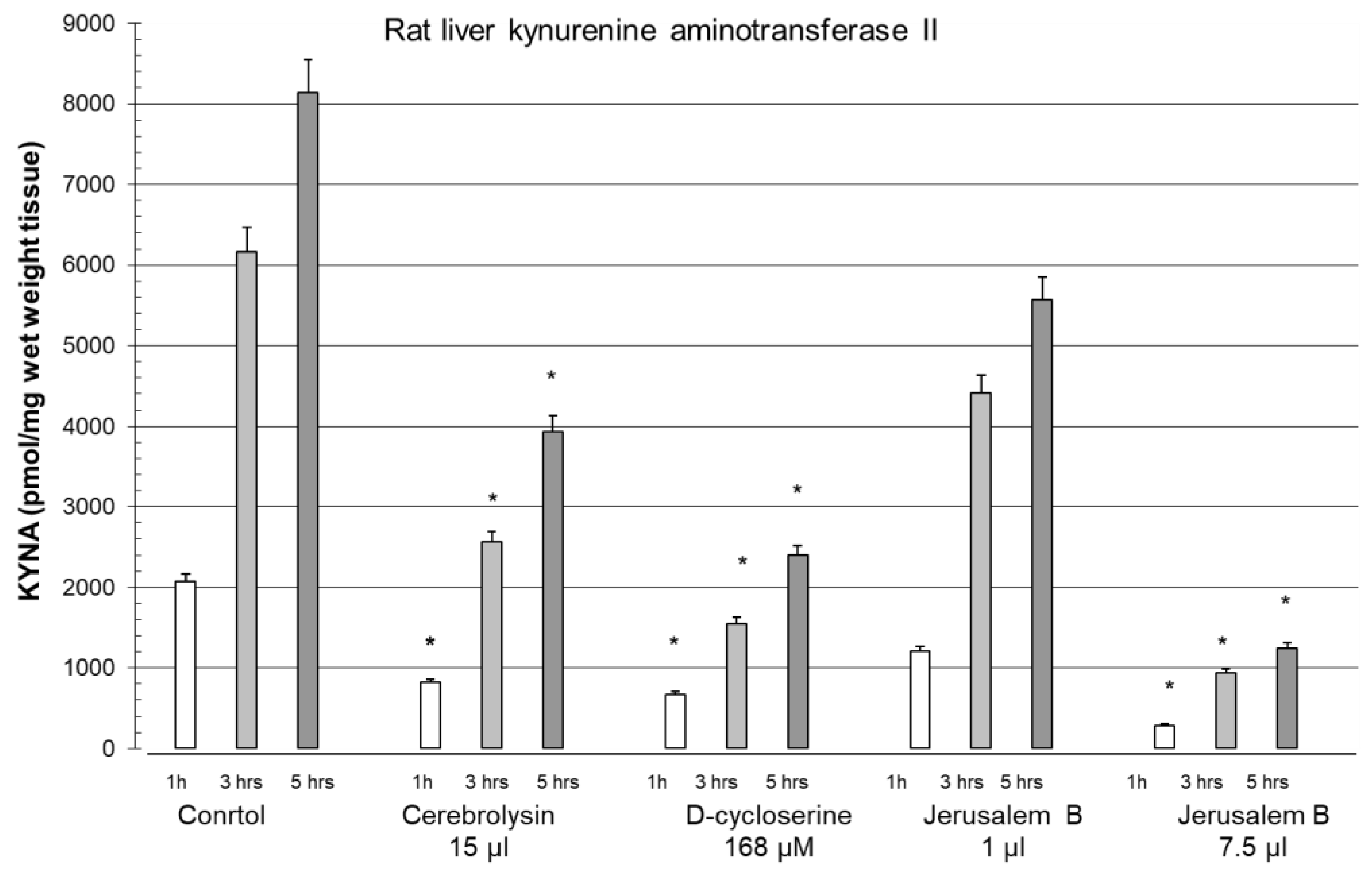
We compared the effect of Jerusalem Balsam with that of cerebrolysine and D-cycloserine and found that the inhibitory effect of Jerusalem Balsam on KAT activity was significant and the inhibition was seen for up to 5 hours under the experimental conditions (Figure 7, adapted from [147]). Jerusalem Balsam was the most effective, followed by D-cycloserine and cerebrolysine. The reduction in KYNA synthesis by Jerusalem Balsam was a noteworthy biochemical effect, as it may act in part as an anti-dementia agent.
Finally, we believe that the frequent use of plants and/or berry extracts can significantly prevent the progression of ageing and the development of pathological conditions associated with the ageing process, with the main advantage being the absence of side effects.
I once asked Prof Kido what was more important to know about a molecule, the effect of the molecule or the mechanism of action of the molecule. Prof Kido's answer was - the effect.
Regarding the mechanism of action of KYNA on cholinergic neurons, Prof Stone TW concludes that there is no confirmed, reliable evidence of antagonistic activity of KYNA at the nicotinic receptor and therefore the results should only be interpreted in terms of the confirmed site of action, the blocking effect on glutamatergic neurons [148].
10. Future Perspectives
In the light of the accumulated data, we conclude that modulation of KYNA-induced inhibition of glutamatergic (and cholinergic) activities may be causally related to the efficacy of both drugs and herbs in the ageing process and in pathological conditions such as Alzheimer's disease, dementia, Down's syndrome or schizophrenia. A study by Vohra et al. showed that depletion of KYNA by dietary restriction can lead to activation of the NMDAR of a specific pair of interneurons with a critical role in learning [112]. In the future, using the Helix pomatia snail model of memory, we may be able to select more herbs and/or drugs that support attention, alertness and learning, and find new meaning in the depletion of the metabolism of KYNA formation.
Authors' contributions
The review was structured by HB, and all authors reviewed and approved the final manuscript.
Acknowledgments
I would like to thank everyone who contributed to my scientific work, especially my supervisors, Univ.-Prof. MD Hornykiewicz, O. Institute of Biochemical Pharmacology, Medical University of Vienna, Austria, and Univ.-Prof. Dr. Schwarcz, R. University of Maryland, USA, and Univ.-Prof. MD Kido, R. Wakayama Medical College, Japan, and especially Prim. MD Kepplinger, B. MSc, Clinical Director of the Neuropsychiatric Clinic Mauer, Austria, for his research interest. I would like to thank Mag. Carina Kronsteiner for her technical assistance.
This work was supported in part by the Austrian FWF (project P15371 to HB); the Austrian National Bank (Jubilee Fund, grant 12316 to HB); the Austrian NFB Life Science Project Nr. LS10-032 (to BK and HB); and by NÖ Forschung und Bildung, Austria (to HB).
Disclosure statement
This manuscript has been read and approved by the authors. The authors report no competing interests.
Abbreviations
α-7 nAChR: α-7 nicotinic acetylcholine receptor; AD: Alzheimer's disease; CNS: central nervous system; CO: control; DS: Down syndrome; GDF: glia depressing factor; GFAP: glial fibrillary acidic protein; GPR-35: G-protein-coupled receptor 35; KAT: kynurenine aminotransferase; KYNA: kynurenic acid; L-KYN: L-kynurenine; NAD: nicotinamide adenine dinucleotide; NMDAR: N-methyl-D-aspartate receptor; SRT: stochastic resonance therapy; TRP: tryptophan.
References
- Brown, R.R. Biochemistry and pathology of tryptophan metabolism and its regulation by amino acids, vitamin B6 and steroid hormone. Am J Clin Nutr. 1971, 24, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Eastmann, C.L.; Guilarte, T.R. Cytotoxicity of 3-hydroxykynurenine in aneuronal hybrid cell line. Brain Res. 1989, 495, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Perkins, M.N.; Stone, T.W. An iontophoretic investigation of the actions of convulsant kynurenines and their interaction with the endogenous excitant quinolonic acid. Brain Res. 1982, 247, 184–187. [Google Scholar] [CrossRef]
- Stone, T.W. Neuropharmacology of quinolinic and kynurenic acids. Pharmacol Rev. 1993, 45, 309–379. [Google Scholar]
- Fukui, S.; Schwarcz, R.; Rapoport, S.I.; Takada, Y.; Smith, Q.R. Blood-brain barrier transport of kynurenines: implications for brain synthesis and metabolism. J Neurochem. 1991, 56, 2007–2015. [Google Scholar] [CrossRef]
- Speciale, C.; Hares, K.; Schwarcz, R.; Brookes, N. High-affinity uptake of L-kynurenine by a Na+-independent transport of neutral amino acids in astrocytes. J Neurosci. 1989, 9, 2066–2072. [Google Scholar] [CrossRef] [PubMed]
- Turski, W.A.; Gramsbergen, J.B.P.; Traitler, H.; Schwarcz, R. Rat brain slices produce and liberate kynurenic acid upon expose to L-kynurenine. J Neurochem. 1989, 52, 1629–1636. [Google Scholar] [CrossRef]
- Gal, E.M.; Sherman, A.D. L-Kynurenine: its synthesis and possible regulatory function in brain. Neurochem Res. 1980, 5, 223–239. [Google Scholar] [CrossRef]
- Baran, H.; Schwarcz, R. Presence of 3-hydroxyanthranilic acid in rat tissue and evidence for its production from anthranilic acid in the brain. J Neurochem. 1990, 55, 738–744. [Google Scholar] [CrossRef]
- Moroni, F.; Russi, P.; Lombardi, G.; Beni, M.; Carla, V. Presence of kynurenic acid in the mammalian brain. J Neurochem. 1988, 51, 177–180. [Google Scholar] [CrossRef]
- Moroni, F.; Russi, P.; Carla, V.; De Luca, G.; Politi, V. The regulation of brain kynurenic acid content: Focus on indole-3-pyruvic acid. In: Schwarcz, R.; Young, S.N.; Brown, R.R. (eds). Kynurenine and serotonin pathway progress in tryptophan research. Adv Exp Med Biol. Plenum Press, New York, London. 1991, 294, 299–308. [Google Scholar] [PubMed]
- Politi, V.; Lavaggi, M.V.; DiStazio, G.; Margonelli, A. Indole-3-pyruvic acid as a direct precursor of kynurenic acid. In: Schwarcz, R.; Young, S.N.; Brown, R.R. (eds). Kynurenine and serotonin pathway progress in tryptophan research. Adv Exp Med Biol. 1991, 294, 515–518. [Google Scholar] [PubMed]
- Kido, R. Kynurenate forming enzymes in liver, kidney and brain. In Schwarcz, R.; Young, S.N.; Brown, R.R. (eds). Kynurenine and serotonin pathways: Progress in tryptophan Research. Adv Exp Med Biol, Plenum Publishing Corporation, New York, ISBN 0-306-43929-8. 1989, Vol 294, 201-205. ISBN 0-306-43929-8.
- Baran, H.; Amann, G.; Lubec, B.; Lubec, G. Kynurenic acid and kynurenine aminotransferase in heart. Pediatr Res. 1997, 41, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Okuno, E.; Tsujimoto, M.; Nakamura, M.; Kido, R. Kynurenine-pyruvate aminotransferase in rat kidney and brain. In Schwarcz, R.; Young, S.N.; Brown, R.R. (eds). Kynurenine and serotonin pathways: Progress in tryptophan Research. Adv Exp Med Biol, Plenum Publishing Corporation, New York, ISBN 0-306-43929-8. 1989, Vol 294, 567-572.
- Okuno, E.; Du, F.; Ishikawa, T.; Tsujimoto, M.; Nakamura, M.; Schwarcz, R.; Kido, R. Purification and characterisation of kynurenine-pyruvate aminotransferase from rat kidney and brain. Brain Res. 1990, 534, 37–44. [Google Scholar] [CrossRef]
- Baran, H.; Kepplinger, B.; Draxler, M.; Ferraz-Leite, H. Kynurenic acid metabolism in rat, piglet and human tissues. 7th Congress of the European Society for Clinical Neuropharmacology Battistin, L. (ed) Medimond Srl, International Proceedings. 2004, 227-231.
- Kronsteiner, C.; Baran, H.; Kepplinger, B. Kynurenic acid levels and kynurenine aminotransferase I, II and III activities in ganglia, heart and liver of snail Helix Pomatia. Cell Physiol Biochem. 2023, 57, 279–297. [Google Scholar] [CrossRef] [PubMed]
- Baran, H.; Okuno, E.; Kido, R.; Schwarcz, R. Purification and characterisation of kynurenine aminotransferase I from human brain. J Neurochem. 1994, 62, 730–738. [Google Scholar] [CrossRef]
- Han, Q.; Li, J.; Li, J. pH dependence, substrate specificity and inhibition of human kynurenine aminotransferase I. Eur J Biochem. 2004, 271, 4804–4814. [Google Scholar] [CrossRef]
- Okuno, E.; Nakamura, M.; Schwarcz, R. Two kynurenine aminotransferases in human brain. Brain Res. 1991, 542, 307–312. [Google Scholar] [CrossRef]
- Schmidt, W.; Guidetti, P.; Okuno, E.; Schwarcz, R. Characterization of human brain kynurenine aminotransferases using [3H]kynurenine as a substrate. Neuroscience. 1993, 55, 177–184. [Google Scholar] [CrossRef]
- Guidetti, P.; Amori, L.; Sapko, M.T.; Okuno, E.; Schwarcz, R. Mitochondrial aspartate aminotransferase: a third kynurenate-producing enzyme in the mammalian brain. J Neurochem. 2007, 102, 103–111. [Google Scholar] [CrossRef]
- Yu, P.; Li, Z.; Zhang, L.; Tagle, D.A.; Cai, T. Characterization of kynurenine aminotransferase III, a novel member of a phylogenetically conserved KAT family. Gene. 2006, 365, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Robinson, H.; Cai, T.; Tagle, D.A.; Li, J.Y. Biochemical and Structural properties of mouse kynurenine aminotransferase III. Mol Cell Biol. 2009, 29, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Kiss, C.; Ceresoli-Borroni, G.; Guidetti, P.; Zielke, C.L.; Zielke, H.R.; Schwarcz, R. Kynurenate production by cultured human astrocytes. J Neural Transm. 2003, 110, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rossi, F.; Schwarcz, R.; Rizzi, M. Curiosity to kill the KAT (kynurenine aminotransferase): structural insights into brain kynurenic acid synthesis. Curr Opin Struct Biol. 2008, 18, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.C.; Du, F.; McCarthy, K.E.; Okuno, E.; Schwarcz, R. Immunocytochemical localization of kynurenine aminotransferase in the rat striatum: A light and electron microscopic study. J Comp Neurol. 1992, 326, 82–90. [Google Scholar] [CrossRef]
- Birch, P.J.; Grossman, C.J.; Hayes, A.G. Kynurenic acid antagonizes responses to NMDA via an action at the strychnine-insensitive glycine receptor. Eur J Pharmacology. 1988, 154, 85–87. [Google Scholar] [CrossRef]
- Stone, T.W.; Stoy, N.; Darlington, L.G. An Expanding range of targets for kynurenine metabolism of tryptophan. Trends in Paharmacological Science. 2013, 34, 34,136–143. [Google Scholar]
- Hilmas, C.; Pereira, E.F.R.; Alkondon, M.; Rassoulpour, A.; Schwarcz, R.; Albuquerque, E.X. The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: physiopathological implications. J Neurosci. 2001, 21, 7463–7473. [Google Scholar] [CrossRef]
- Foster, A.C.; Vezzani, A.; French, E.D.; Schwarcz, R. Kynurenic acid blocks neurotoxicity and seizures induced in rats by the related brain metabolite quinolinic acid. Neurosci Lett. 1984, 48, 273–278. [Google Scholar] [CrossRef]
- Bratek-Gerej, E.; Ziembowicz, A.; Godlewski, J.; Salinska, E. The Mechanism of the Neuroprotective Effect of Kynurenic Acid in the Experimental Model of Neonatal Hypoxia–Ischemia: The Link to Oxidative Stress. Antioxidants. 2021, 10, 1775. [Google Scholar] [CrossRef]
- Albuquerque, E.X.; Alkondon, M.; Pereira, E.F.R.; Castro, N.G.; Schrattenholz, A.; Barbosa, C.T.F.; Bonfante-Carbarcas, R.; Aracava, Y.; Eisenberg, H.M.; Maelike, A. Properties of neuronal nicotinic acetylcholine receptors: pharmacological characterization and modulation of synaptic function. J Pharmacol Exp Ther. 1997, 280, 1117–1136. [Google Scholar] [PubMed]
- Albuquerque, E.X.; Pereira, E.F.; Alkondon, M.; Rogers, S.W. Mammalian nicotinic receptors: from structure to function. Physiol Rev. 2009, 89, 73–120. [Google Scholar] [CrossRef]
- Bergeron, R.; Meyer, T.M.; Coyle, J.T.; Greene, R.W. Modulation of N-methyl-D-aspartate receptor function by glycine transport. Proc Natl Acad Sci USA. 1998, 22, 15730–15734. [Google Scholar] [CrossRef] [PubMed]
- D´Angelo, E.; Rossi, P.; Garthwaite, J. Dual component NMDA receptor currents at a single central synapse. Nature. 1990, 346, 467–470. [Google Scholar] [CrossRef]
- Koukouli, F.; Maskos, U. The multiple roles of the α7 nicotinic acetylcholine receptor in modulating glutamatergic systems in the normal and diseased nervous system. Biochem Pharmacol. 2015, 4, 378–387. [Google Scholar] [CrossRef]
- Rassoulpour, A.; Wu, H.Q.; Albuquerque, E.X.; Schwarcz, R. Prolonged nicotine administration results in biphasic, brain-specific changes in kynurenate levels in the rat. Neuropsychopharmacol. 2005, 30, 697–704. [Google Scholar] [CrossRef]
- Pearce, I.A.; Cambray-Deakin, M.A.; Burgoyne, R.D. Glutamate acting on NMDA receptors stimulates neurite outgrowth from cerebellar granule cells. FEBS Lett. 1987, 223, 143–147. [Google Scholar] [CrossRef]
- Pugh, P.C.; Berg, D.K. Neuronal acetylcholine receptors that bind alpha-bungarotoxin mediate neurite retraction in a calcium-dependent manner. J Neurosci. 1994, 14, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Damaj, M.E.; Glassco, W.; Dukat, M.; Martin, B.R. Pharmacological characterization of nicotine-induced seizures in mice. J Pharmacol Exp Ther. 1999, 291, 1284–1291. [Google Scholar]
- Leeson, P.D.; Iversen, L.L. The glycine site on the NMDA receptor: structure-activity relationships and therapeutic potential. J Med Chem. 1994, 37, 4053–4067. [Google Scholar] [CrossRef]
- Sun, A.; Cheng, J. Novel targets for therapeutic intervention against ischemic brain injury. Clin Neuropharmacol. 1999, 22, 164–171. [Google Scholar] [PubMed]
- Du, F.; Eid, T.; Schwarcz, R. Neuronal damage after the injection of aminooxy acetic acid into the entorhinal cortex: a silver impregnation. Neuroscience. 1988, 82, 1165–1178. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Whetsell, W.O., Jr; Abou-Khalil, B.; Lothman, E.W.; Schwarcz, R. Preferential neuronal loss in layer III of the entorhinal cortex in patients with temporal lobe epilepsy. Epilepsy Res. 1993, 16, 223–233. [Google Scholar] [CrossRef] [PubMed]
- McMaster, O.G.; Baran, H.; Wu, H.Q.; Du, F.; French, E.; Schwarcz, R. Gamma-Acetylenic GABA produces axon-sparing neurodegeneration after focal injection into the rat hippocampus. Exp Neurol. 1993, 124, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.-Q. Kynurenines in the mammalian brain: when physiology meets pathology. Nat Rev Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef]
- Baran, H.; Gramer, M.; Hönack, D.; Löscher, W. Systemic administration of kainate induces marked increase of endogenous kynurenic acid in various brain regions and plasma of rats. Eur J Pharmacol. 1995, 286, 167–175. [Google Scholar] [CrossRef]
- Baran, H.; Kepplinger, B.; Herrera-Marschitz, M.; Stolze, K.; Lubec, G.; Nohl, H. Increased kynurenic acid in the brain after neonatal asphyxia. Life Sci. 2001, 69, 1249–1256. [Google Scholar] [CrossRef]
- Richter, A.; Löscher, W.; Baran, H.; Gramer, M. Increased levels of kynurenic acid in brains of genetically dystonic hamsters. Dev Brain Res. 1996, 92, 111–116. [Google Scholar] [CrossRef]
- Baran, H.; Du, F.; Schwarcz, R. Transient increase in striatal kynurenate synthesis following systemic kainate administration in rat. Soc for Neurosci Abs 1991, 17, 356.10. [Google Scholar]
- Baran, H.; Draxler, M.; Kronsteiner, C.; Kepplinger, B. Increase of kynurenic acid after Encephalomyocarditis virus infection and ist significances. Neurosignals. 2021, 29, 24–34. [Google Scholar]
- Baran, H.; Hainfellner, J.A.; Kepplinger, B.; Mazal, P.R.; Schmid, H.; Budka, H. Kynurenic acid metabolism in the brain of HIV-1 infected patients. J Neural Transm. 2000, 107, 1127–1138. [Google Scholar] [CrossRef] [PubMed]
- Kerr-Pontes, L.R.; Oliveira, F.A.; Freire, C.A. Tuberculosis associated with AIDS: the situation in a Northeastern region of Brazil. Rev Saude Publica 1997, 31, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Gordin, F.M.; Roediger, M.P.; Girard, P.M.; Lundgren, JD.; Miro, J.M.; Palfreeman, A.; Rodriguez-Barradas, M.C.; Wolff, M.J.; Easterbrook, P.J.; Clezy, K.; Slater, L.N. Pneumonia in HIV-infected persons: increased risk with cigarette smoking and treatment interruption. Am J Respir Crit Care Med. 2008, 178, 630–636. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.Y.; Wang, X.T.; Zhang, L.N. Chinese Critical Care Ultrasound Study Group (CCUSG): Findings of lung ultrasonography of novel corona virus pneumonia during the 2019–2020 epidemic. Intensive Care Med Letter. 2020, 12, 1–2. [Google Scholar]
- Peters, C.E.; Carette, J.A. Return of the neurotropic enteroviruses: Co-opting cellular pathways for infection. Viruses. 2021, 13, 1–23. [Google Scholar] [CrossRef]
- Lucas, S.B.; Hounnou, A.; Peacock, C.; Beaumel, A.; Djomand, G.; N'Gbichi, J.M.; Yeboue, K.; Hondé, M.; Diomande, M.; C Giordano, C.; et al. The mortality and pathology of HIV infection in a West African city. AIDS. 1993, 7, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Lubec, B.; Marx, M.; Herrera-Marschitz, M.; Labudowa, O.; Hoeger, H.; Gille, L.; Nohl, H.; Mosgoeller, W.; Lubec, G. Decrease of heart protein kinase C and cyclin-dependent kinase precedes death in perinatal asphyxia of the rat. The FASEB Journal. 1997, 11, 482–492. [Google Scholar] [CrossRef]
- Urbańska, E.M.; Luchowski, P.; Luchowska, E.; Pniewski, J.; Woźniak, R.; Chodakowska-Zebrowska, M.; Lazarewicz, J. Serum kynurenic acid positively correlates with cardiovascular disease risk factor, homocysteine: a study in stroke patients. Pharmacol Rep. 2006, 58, 507–511. [Google Scholar]
- Vécsei, L.; Beal, M.F. Intracerebroventricular injection of kynurenic acid, but not kynurenine induces ataxia and stereotyped behaviour in rats. Brain Res Bull. 1990, 25, 623–627. [Google Scholar] [CrossRef]
- Vécsei, L.; Beal, M.F. Comparative behavioral and pharmacological studies with centrally administered kynurenine and kynurenic acid in rats. Eur J Pharmacol. 1991, 196, 239–246. [Google Scholar] [CrossRef]
- Baran, H.; Draxler, M.; Kalbacher, H.; Nowotny, N.; Schmidt, P.; Kepplinger, B.; Url, A.; Schuh, M.; Hofecker, G. Kynurenine aminotransferase I and II (KAT I and KAT II) in piglet’s brain after Encephalomyocarditis (EMCV) infection. Soc Neurosci Abstr. 200, 33, 215.8. [Google Scholar]
- Baran, H.; Draxler, M.; Kepplinger, B.; Kahlbacher, H.; Nowotny, N.; Schmidt, P.; Url, A.; Schuh, M.; Hofecker, G. Kynurenine metabolism in piglet brain due to EMCV infection. Pharmacology. 2003, 69, 212–217. [Google Scholar]
- Baran, H.; Staniek, K.; Kepplinger, B.; Stur, I.; Draxler, M.; Nohl, H. Kynurenines and the respiratory parameters in rat heart mitochondria. Life Sci. 2003, 72, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Baran, H.; Staniek, K.; Bertignol-Spörr, M.; Attam, M.; Kronsteiner, C.; Kepplinger, B. Effects of various kynurenine metabolites on respiratory parameters of rat brain, liver and heart mitochondria. Int J Tryptophan Res. 2016, 9, 17–29. [Google Scholar] [CrossRef]
- Baran, H.; Staniek, K.; Bertignol, Attam, M.; Kepplinger, B. Influence of tryptophan metabolites on the respiratory parameters of rat brain mitochondria during aging. Soc for Neurosci Abs 2010, 406.9. [Google Scholar]
- Beal, M.F.; Swartz, K.J.; Isacson, O. Developmental changes in brain kynurenic acid concentrations. Dev Brain Res. 1992, 68, 136–139. [Google Scholar] [CrossRef]
- Baran, H.; Schwarcz, R. Regional differences in the ontogenetic pattern of kynurenine aminotransferase in the rat brain. Dev Brain Res. 1993, 74, 283–286. [Google Scholar] [CrossRef]
- Gramsbergen, J.B.P.; Schmidt, W.; Turski, W.A.; Schwarcz, R. Age-related changes in kynurenic acid production in rat brain. Brain Res. 1992, 588, 1–5. [Google Scholar] [CrossRef]
- Wada, H.; Ito, H.; Orimo, H.; Sato, A. Kynurenine specifically increases in the cerebrospinal fluid of the aged rats. Biogenic Amines. 1994, 19, 221–225. [Google Scholar]
- Walker, D.W.; Curtis, B.; Lacey, B.; Nitsos, I. Kynurenic acid in brain and cerebrospinal fluid of fetal, newborn, and adult sheep and effects of placental embolization. Pediatr Res. 1999, 45, 820–826. [Google Scholar] [CrossRef]
- Kepplinger, B.; Baran, H.; Kainz, A.; Ferraz-Leite, H.; Newcombe, J.; Kalina, P. Age-related increase of kynurenic acid in human cerebrospinal fluid: Positive correlation with IgG and beta2-microglobulin changes. Neurosignals. 2005, 14, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Kepplinger, B.; Baran, H.; Kronsteiner, C.; Reuss, J. Increased levels of kynurenic acid in the cerebrospinal fluid in patients with hydrocephalus. Neurosignals. 2019, 27, 1–11. [Google Scholar]
- Greenamyre, J.T.; Maragos, W.F.; Albin, R.L.; Penney, J.B.; Young, A.B. Glutamate transmission and toxicity in Alzheimer’s disease. Review Prog Neuropsychopharmacol Biol Psychiatry. 1988, 12, 421–430. [Google Scholar] [CrossRef]
- Whitehouse, P.J.; Price, D.L.; Clark, A.W.; Coyle, J.T.; DeLong, M.R. Alzheimer disease: Evidence for selective loss of cholinergic neurons in the nucleus basalis. Annals of Neurology. 1981, 10, 122–126. [Google Scholar] [CrossRef] [PubMed]
- DeKosky, S.T.; Ikonomovic, M.D.; Styren, S.D.; Beckett, L.; Wisniewski, S.; Bennett, D.A.; Cochran, E.J.; Kodower, J.H.; Mufson, E.J. Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann Neurol. 2002, 51, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Frölich, L.; Dirr, A.; Götz, M.E.; Gsell, W.; Reichmann, H.; Riederer, P.; Maurer, K. Acetylcholine in human CSF: methodological considerations and levels in dementia of Alzheimer type. J Neural Transm. 1988, 105, 961–973. [Google Scholar] [CrossRef]
- Jia, J.P.; Jia, J.M.; Zhou, W.D.; Xu, M.; Chu, C.B.; Yan, C.; Sun, Y.X. Differential acetylcholine and choline concentrations in the cerebrospinal fluid of patients with Alzheimer’s disease and vascular dementia. Chinese Med J. 2004, 117, 1161–1164. [Google Scholar]
- Ikeda, Y.; Okuyama, S.; Fujiki, Y.; Tomoda, K.; Ohshiro, K.; Itoh, T.; Yamauchi, T. Changes of acetylcholine and choline concentrations in cerebrospinal fluids of normal subjects and patients with dementia of Alzheimer-type. J Neural Transm Suppl. 1990, 30, 25–32. [Google Scholar]
- Enquidawork, E.; Gulesserian, T.; Balic, N.; Cairns, N.; Lubec, G. Changes in nicotinic acetylcholine receptor subunits expression in brain of patients with Down syndrome and Alzheimer’s disease. J Neural Transm Suppl. 2001, 61, 211–222. [Google Scholar]
- Brugge, K.L.; Nichols, S.L.; Salmon, D.P.; Hill, L.R.; Delis, D.C.; Aaron, L.; Trauner, D.A. Cognitive impairment in adults with Down's syndrome: similarities to early cognitive changes in Alzheimer's disease. Neurology. 1994, 44, 232–238. [Google Scholar] [CrossRef]
- Baran, H.; Jellinger, K. Does increased kynurenic acid levels in the brain contribute to the impairment of memory in Alzheimer’s disease? Second International Oxidative Stress and Brain Damage Symposium, Abs., Chicago, Illinois, USA. 1997, Lecture.
- Baran, H.; Jellinger, K. Kynurenine metabolism in Alzheimer disease brain. Eur. J. of Neuroscience. 1998, 10 Suppl. 10, 32.09. [Google Scholar]
- Baran, H.; Jellinger, K.; Deecke, L. Kynurenine metabolism in Alzheimer’s disease. J Neural Transm. 1999, 106, 165–181. [Google Scholar] [CrossRef] [PubMed]
- Baran, H.; Cairns, N.; Lubec, B.; Lubec, G. Increased kynurenic acid levels and decreased brain kynurenine aminotransferase I in patients with DOWN syndrome. Life Sci. 1996, 58, 1891–1899. [Google Scholar] [CrossRef]
- Kepplinger, B.; Baran, H.; Kainz, A.; Newcombe, J.; Nohl, H. Altered kynurenic acid levels in CSF and serum of patients with multiple sclerosis. Multiple Sclerosis. 2001, 7 Supl. 1, S118, P400. [Google Scholar]
- Heyes, M.P.; Brew, B.J.; Saito, K.; Quearry, B.J.; Price, R.W.; Lee, K.; Bhalla, R.B.; Der, M.; Markey, S.P. Inter-relationships between quinolinic acid, neuroactive kynurenines, neopterin and beta2-microglobulin in cerebrospinal fluid and serum of HIV-1-infected patients. J Neuroimmunol. 1992, 40, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Connick, J.H.; Carla, V.; Moroni, F.; Stone, T.W. Increase in kynurenic acid in Huntington's disease motor cortex. J Neurochem. 1989, 52, 958–987. [Google Scholar] [CrossRef]
- Erhardt, S.; Blennow, K.; Nordin, C.; Skogh, E.; Lindström, L.H.; Engberg, G. Kynurenic acid levels are elevated in the cerebrospinal fluid of patients with schizophrenia. Neurosci Lett. 2001, 313, 96–98. [Google Scholar] [CrossRef]
- Schwarcz, R.; Rassoulpour, A.; Wu, H.Q.; Medoff, D.; Tamminga, C.A.; Robert, R.C. Increased cortical kynurenate content in schizophrenia. Biol Psychiatry. 2001, 50, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Matson, W.R.; Beal, M.F.; Myers, R.H.; Bird, E.D.; Milbury, P.; Saso, S. Kynurenine pathway abnormalities in Parkinson’s disease. Neurology. 1992, 42, 1702–1706. [Google Scholar] [CrossRef]
- Hornykiewicz, O. Topography and behaviour of noradrenaline and dopamine(3-hydroxytryptamine) in the substantia nigra of normal and Parkinsonian patients (In German). Wien Klin Wochenschr. 1963, 75, 309–312. [Google Scholar]
- Hornykiewicz, O.; Kish, S.J. Neurochemical basis of dementia in Parkinson’s disease. Can J Neurol Sci. 1984, 11 (1 Suppl), 185–190. [Google Scholar] [CrossRef] [PubMed]
- Hlinak, Z.; Krejci, I. Kynurenic acid and 5,7-dichlorokynurenic acids improve social and object recognition in male rats. Psychopharmacol. 1995, 120, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Steele, R.J.; Stewart, M.G. 7-Chlorokynurenate, an antagonist of the glycine binding site on the NMDA receptor, inhibits memory formation in day-old chicks (Gallus domesticus). Behav Neural Biol. 1993, 60, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Chess, A.C.; Simoni, M.K.; Alling, T.E.; Bucci, D.J. Elevations of kynurenic acid produce working memory deficits. Schizophr Bull. 2007, 33, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Rassoulpour, A.; Wu, H.Q.; Ferré, S.; Schwarcz, R. Nanomolar concentrations of kynurenic acid reduce extracellular dopamine levels in the striatum. J Neurochem. 2005, 93, 762–765. [Google Scholar] [CrossRef]
- Sperk, G.; Lassmann, H.; Baran, H.; Kish, S.J.; Seitelberger, F.; Hornykiewicz, O. Kainic acid induced seizures: Neurochemical and histopathological changes. Neuroscience. 1983, 4, 1301–1315. [Google Scholar] [CrossRef]
- Ting, K.K.; Brew, B.; Guillemin, G. The involvement of astrocytes and kynurenine pathway in Alzheimer’s disease. Neurotox Res. 2007, 12, 247–262. [Google Scholar] [CrossRef]
- Nichols, N.R.; Day, J.R.; Laping, N.J.; Johnson, S.A.; Finch, C.E. GFAP mRNA increases with age in rat and human brain. Neurobiol of Aging. 1993, 421–429. [Google Scholar] [CrossRef]
- Ajenikoko, M.K.; Ajagbe, A.O.; Onigbinde, O.A.; Okesina, A.A.; Tijani, A.A. Review of Alzheimer’s disease drugs and their relationship with neuron-glia interaction. IBRO Neurosci Rep. 2023, 14, 64–76. [Google Scholar] [CrossRef]
- Monzón-Mayor, M.; Álvarez, M.I.; Arbelo-Galván, J.F.; Romero-Alemán, M.M.; Yanes, C.; Plaza, M.L.; Rodriguez, J.R.; Rodriguez, J.J.; Toledano, A. Long–term evolution of local, proximal and remote astrocyte responses after diverse nucleus basalis lesioning (an experimental Alzheimer model): GFAP immunocytochemical study. Brain Res. 2000, 865, 235–258. [Google Scholar] [CrossRef]
- Du Laping, N.J.; Teter, B.; Nichols, N.R.; Rozovsky, I.; Finch, C.E. Glial fibrillary acidic protein: regulation by hormones, cytokines, and growth factors. Brain Pathol. 1994, 4, 259–275. [Google Scholar] [CrossRef] [PubMed]
- Byrne, G.; Lehmann, L.K.; Kischbaum, J.G.; Borden, E.C.; Lee, C.M.; Brown, R.R. Induction of tryptophan degradation in vitro and in vivo: A gamma-interferon stimulated activity. J Interferon Res. 1986, 6, 389–398. [Google Scholar] [CrossRef]
- Werner, E.R.; Bitterlich, G.; Fuchs, D.; Hausen, A.; Reibneger, G.; Szabo, G.; Dierich, M.; Wachter, H. Human macrophages degrade tryptophan upon induction by interferon gamma. Life Sci. 1987, 42, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.; Frank, A.; Hernanz, A. Relationship of interleukin-1 beta and beta2-microglobulin with neuropeptides in cerebrospinal fluid of patients with dementia of the Alzheimer type. J Neuroimmunol. 1993, 48, 235–240. [Google Scholar] [CrossRef]
- Brew, B.J.; Bhalla, R.B.; Fleisher, M.; Paul, M.; Khan, A.; Schwartz, M.K.; Price, R.W. Cerebrospinal fluid ß2-microglobulin in patients infected with human immunodeficiency virus. Neurology. 1989, 39, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Baran., H.; Hainfellner, J.A.; Kepplinger, B. Baran. H.; Hainfellner, J.A.; Kepplinger, B. Kynurenic Acid Metabolism in Various Types of Brain Pathology in HIV-1 Infected Patients. Int J Tryptophan Res 2012, 5, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Baran, H.; Kepplinger, B.; Draxler, M. Endogenous kynurenine aminotransferases inhibitor is proposed to act as "Glia Depressing Factor" (GDF). Int J Tryptophan Res. 2010, 3, 13–22. [Google Scholar] [CrossRef]
- Vohra, M.; Lemieux, G.A.; Lin, L.; Ashrafi, K. The beneficial effects of dietary restriction on learning are distinct from its effects on longevity and mediated by depletion of a neuroinhibitory metabolite. PLoS Biol 2017, 15, 1–24. [Google Scholar] [CrossRef]
- Lopes, C.; Pereira, E.F.; Wu, H.Q.; Purushottamachar, P.; Njar, V.C.; Schwarcz, R.; Albuquerque, E.X. Competitive antagonism between the nicotinic allosteric potentiating ligand galantamine and kynurenic acid at alpha 7 nicotinic receptors. J Pharmacol Exp Ther. 2007, 322, 48–58. [Google Scholar] [CrossRef]
- Rockenstein, E.; Adame, A.; Mante, M.; Larrea, G.; Crews, L.; Windisch, M.; Moessler, H.; Masliah, E. Amelioration of the cerebrovascular amyloidosis in a transgenic model of Alzheimer’s disease with the neurotrophic compound Cerebrolysin. J Neural Transm. 2005, 112, 269–282. [Google Scholar] [CrossRef]
- Goetz, C.G. Jean-Martin Charcot and his vibratory chair for Parkinson disease. Neurology. 2009, 73, 475–478. [Google Scholar] [CrossRef] [PubMed]
- Haas, C.T.; Buhlmann, A.; Turbanski, S.; Schmidtbleicher, D. Proprioceptive and sensorimotor performance in Parkinson's disease. Res Sports Med. 2006, 14, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, M.A.; Farley, B.G. Exercise and neuroplasticity in persons living with Parkinson's disease. Eur J Phys Rehabil Med. 2009, 45, 215–229. [Google Scholar]
- Turbanski, S.; Haas, C.T.; Schmidtbleicher, D.; Friedrich, A.; Duisberg, P. Effects of random whole-body vibration on postural control in Parkinson's disease. Res Sports Med. 2005, 13, 243–256. [Google Scholar] [CrossRef]
- Hattori, S.; Naoi, M.; Nishino, H. Striatal dopamine turnover during treadmill running in the rat: relation to the speed of running. Brain Res Bull. 1994, 35, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.Q.; Rassoulpour, A.; Schwarcz, R. Kynurenic acid leads, dopamine follows: a new case of volume transmission in the brain? J Neural Transm. 2007, 114, 33–41. [Google Scholar] [CrossRef]
- Benzi, R.; Sutera, A.; Vulpiani, A. The mechanism of stochastic resonance. J. Phys. 1981, A14, L453.457. [Google Scholar] [CrossRef]
- Levin, J.E.; Miller, J.P. Broadband neural encoding in the cricket cercal sensory system enhanced by stochastic resonance. Nature. 1996, 380, 165–168. [Google Scholar] [CrossRef]
- Collins, J.J.; Imhoff, T.T.; Grigg, P. Noise-enhanced tactile sensation. Nature. 1996, 383, 770. [Google Scholar] [CrossRef]
- Collins, J.J.; Priplata, A.A.; Gravelle, D.C.; Niemi, J.; Harry, J.; Lipsitz, L.A. Noise-enhanced human sensorimotor function. IEEE Eng Med Biol Mag. 2003, 22, 76–83. [Google Scholar] [CrossRef]
- Yousefi, B.; Tadibim, V.; Khoei, A.F.; Montazeri, A. Exercise therapy, quality of life, and activities of daily living in patients with Parkinson disease: a small scale quasi-randomised trial. Trials. 2009, 10, 67. [Google Scholar] [CrossRef] [PubMed]
- Wunderer, K.; Schabrun, S.M.; Chipchase, L.S. Effects of whole body vibration on strength and functional mobility in multiple sclerosis. Physiother Theory Pract. 2010, 26, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Andel, R.; Crowe, M.; Pedersen, N.L.; Fratiglioni, L.; Johansson, B.; Gatz, M. Physical exercise at midlife and risk of dementia three decades later: a population-based study of Swedish twins. J Gerontol A Biol Sci Med Sci. 2008, 63, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Pitkala, K.H.; Raivio, M.M.; Laakkonen, M.L.; Tilvis, R.S.; Kautiainen, H.; Strandberg, T.E. Exercise rehabilitation on home-dwelling patients with Alzheimer's disease-a randomized, controlled trial. Study protocol. Trials. 2010, 11, 92. [Google Scholar] [CrossRef]
- Kluding, P.M.; Tseng, B.Y.; Billinger, S.A. Exercise and executive function in individuals with chronic stroke: a pilot study. J Neurol Phys Ther. 2011, 35, 11–17. [Google Scholar] [CrossRef]
- Kucyi, A.; Alsuwaidan, M.T.; Liauw, S.S.; McIntyre, R.S. Aerobic physical exercise as a possible treatment for neurocognitive dysfunction in bipolar disorder. Postgrad Med. 2010, 122, 122,107–116. [Google Scholar] [CrossRef]
- van Praag, H.; Christie, B.R.; Sejnowski, T.J.; Gage, F.H. Running enhances neurogenesis, learning, and long-term potentiation in mice. Proc Natl Acad Sci USA. 1999, 96, 13427–13431. [Google Scholar] [CrossRef]
- Molteni, R.; Zheng, J.Q.; Ying, Z.; Gómez-Pinilla, F.; Twiss, J.L. Voluntary exercise increases axonal regeneration from sensory neurons. Proc Natl Acad Sci USA. 2004, 101, 8473–8478. [Google Scholar] [CrossRef]
- Ying, Z.; Roy, R.R.; Edgerton, V.R.; Gomez-Pinilla, F. Exercise restores levels of neurotrophins and synaptic plasticity following spinal cord injury. Exp Neurol. 2005, 19, 283–295. [Google Scholar]
- Kepplinger, B.; Kalina, P.; Zeiner, D.; Eigner, S.; Baran, H. Influence of exercise on kynurenic acid levels in the serum. Amino acids. 2007, 33, LVII. [Google Scholar]
- Kepplinger, B.; Baran, H.; Sedlnitzky-Semler, B.; Badawi, N.R.; Erhart, H. Stochastic Resonance Activity Influences Serum Tryptophan Metabolism in Healthy Human Subjects. Int J Tryptophan Res. 2011, 4, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Baran, H.; Kepplinger, B. Porcine tissues influence kynurenic acid formation. Parkinsonism Relat Disord. 2006, Suppl.1, Suppl. [Google Scholar]
- Crook, T.H.; Ferris, S.H.; Alvarez, X.A.; Laredo, M.; Moessler, H. Effect of N-PEP-12 on memory among older adults. Int Clin Psychopharmacol. 2005, 20, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Riley, C.; Hutter Paier, B.; Windisch, M.; Doppler, E.; Moessler, H.; Wrowski, R. A peptide preparation protects cells in organotypic brain slices against cell death after glutamate intoxication. J Neural Transm. 2006, 113, 103–110. [Google Scholar] [CrossRef]
- Baran, H.; Kepplinger, B. Cerebrolysin lowers kynurenic acid formation - An in vitro study. Eur Neuropsychopharmacol. 2009, 19, 161–168. [Google Scholar] [CrossRef]
- Kemp, J.A.; Lesson, P.D. The glycine site of the NMDA receptor – five years on. Trends Pharmacol Sci. 1993, 14, 20–25. [Google Scholar] [CrossRef]
- Goff, D.C. D-Cycloserine: An evolving role in learning and neuroplasicity in Schizophrenia. Schizophrenia Bulletin. 2012, 38, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Baran, H.; Löscher, W.; Mevissen, M. The glycine/NMDA receptor partial agonist D-cycloserine blocks kainate-induced seizures in rats. Comparison with MK-801 and diazepam. Brain Res. 1994, 652, 195–200. [Google Scholar] [CrossRef]
- Baran, H.; Gramer, M.; Löscher, W. Alterations in plasma and brain amino acids after administration of the glycine/NMDA receptor partial agonist, D-cycloserine, to mice and rats. Eur J Pharmacol. 1995, 373, 197–201. [Google Scholar] [CrossRef]
- Baran, H.; Kepplinger, B. D-cycloserine lowers kynurenic acid formation – New mechanism of action. Eur Neuropsychopharmacol. 2014, 4, 639–644. [Google Scholar] [CrossRef]
- Goff, D.C. D-cycloserine in Schizophrenia: New Strategies for Improving Clinical Outcomes by Enhancing Plasticity. Curr Neuropharmacol. 2017, 15, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Kepplinger, B.; Kronsteiner, C.; Więcek, M.; Baran, H. Hawthorn berry extract lowers kynurenic acid and anthranilic acid formation. 15th ANA Meeting at the IST Austria, 2017 September 24th-26th, Abstract/Lecture.
- Baran, H.; Pietryja, M.J.; Kronsteiner, C.; Kepplinger, B. Jerusalem balsam lowers kynurenic acid formation: an in vitro study. J Tradit Med Clin Natur. 2017, 6, 3. [Google Scholar] [CrossRef]
- Stone, T.W. Does kynurenic acid act on nicotinic receptors? An assessment o the evidence. J Neurochem. 2020, 152, 627–649. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Tryptophan catabolism along the kynurenine pathway.
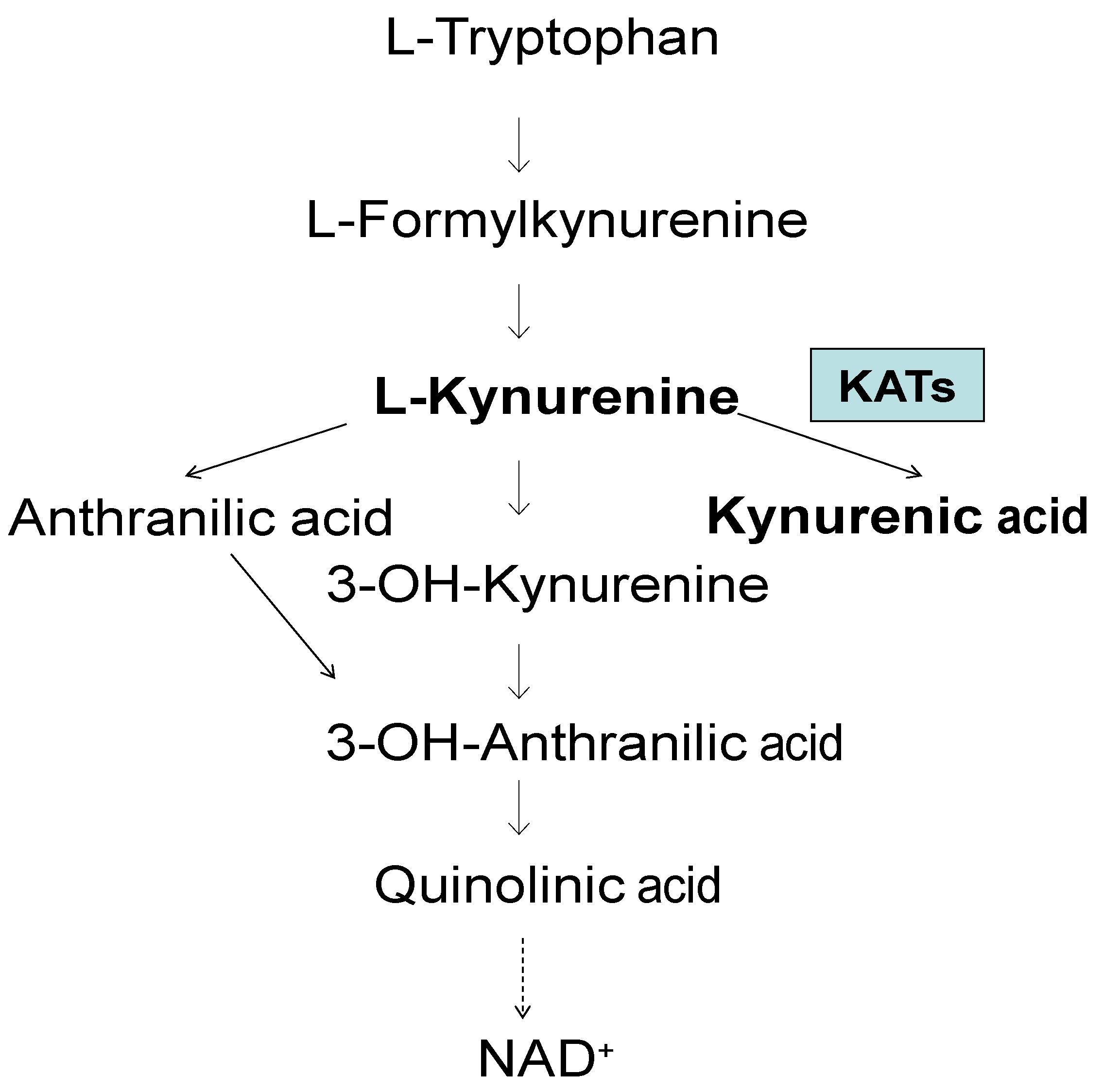
Figure 2.
Kynurenine aminotransferase I and II activities in the brain of patients with Alzheimer disease (AD) and controls (CO). Data represent a mean ± SEM. Significances: *p < 0.05, vs CO.
Figure 2.
Kynurenine aminotransferase I and II activities in the brain of patients with Alzheimer disease (AD) and controls (CO). Data represent a mean ± SEM. Significances: *p < 0.05, vs CO.
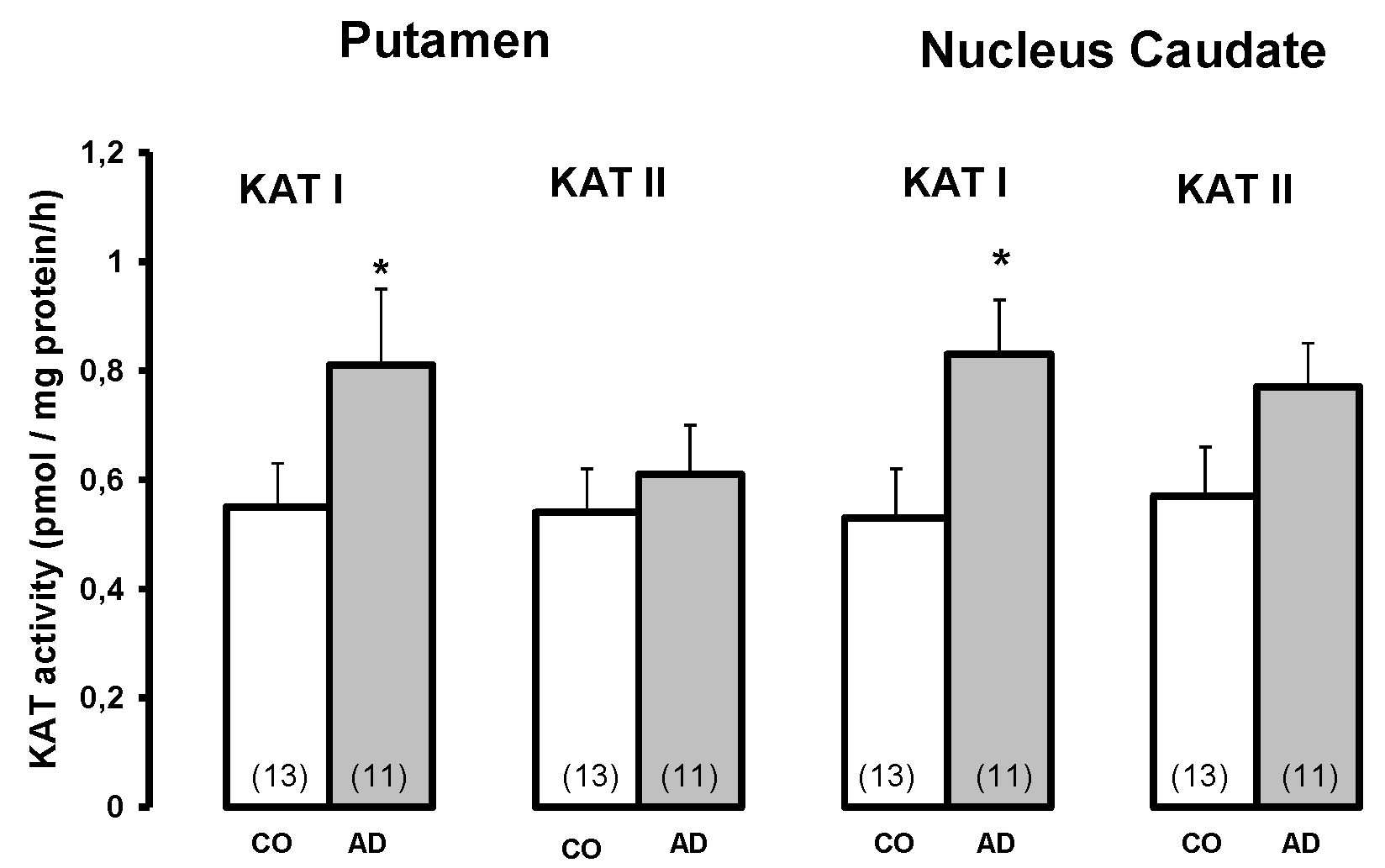
Figure 3.
Kynurenic acid levels in the brain of Down syndrome (DS) patients and controls (CO). Data represent a mean ± SEM. Significances: *p < 0.05; **p < 0.01 vs CO.
Figure 3.
Kynurenic acid levels in the brain of Down syndrome (DS) patients and controls (CO). Data represent a mean ± SEM. Significances: *p < 0.05; **p < 0.01 vs CO.
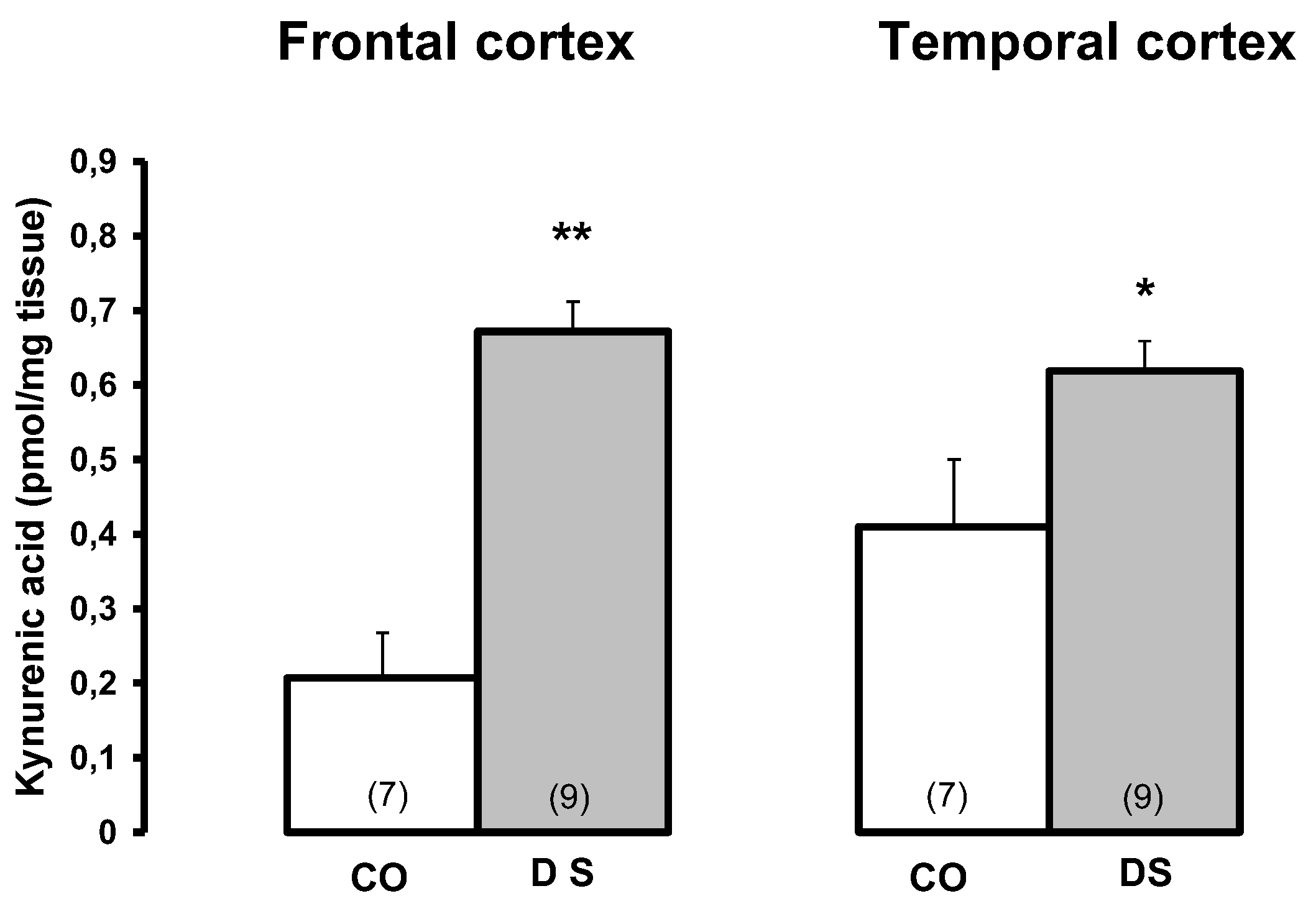
Figure 4.
Kynurenine aminotransferase I and II activities in the brain of Down syndrome (DS) patients and controls (CO). Data represent a mean ± SEM. Significances: **p < 0.01 vs CO.
Figure 4.
Kynurenine aminotransferase I and II activities in the brain of Down syndrome (DS) patients and controls (CO). Data represent a mean ± SEM. Significances: **p < 0.01 vs CO.
Figure 5.
Brain kynurenic acid (KYNA) levels in patients infected with HIV-1 virus (HIV-1) and controls (CO). Data are mean ± SEM. Significances: *p < 0.05 vs. CO.
Figure 5.
Brain kynurenic acid (KYNA) levels in patients infected with HIV-1 virus (HIV-1) and controls (CO). Data are mean ± SEM. Significances: *p < 0.05 vs. CO.
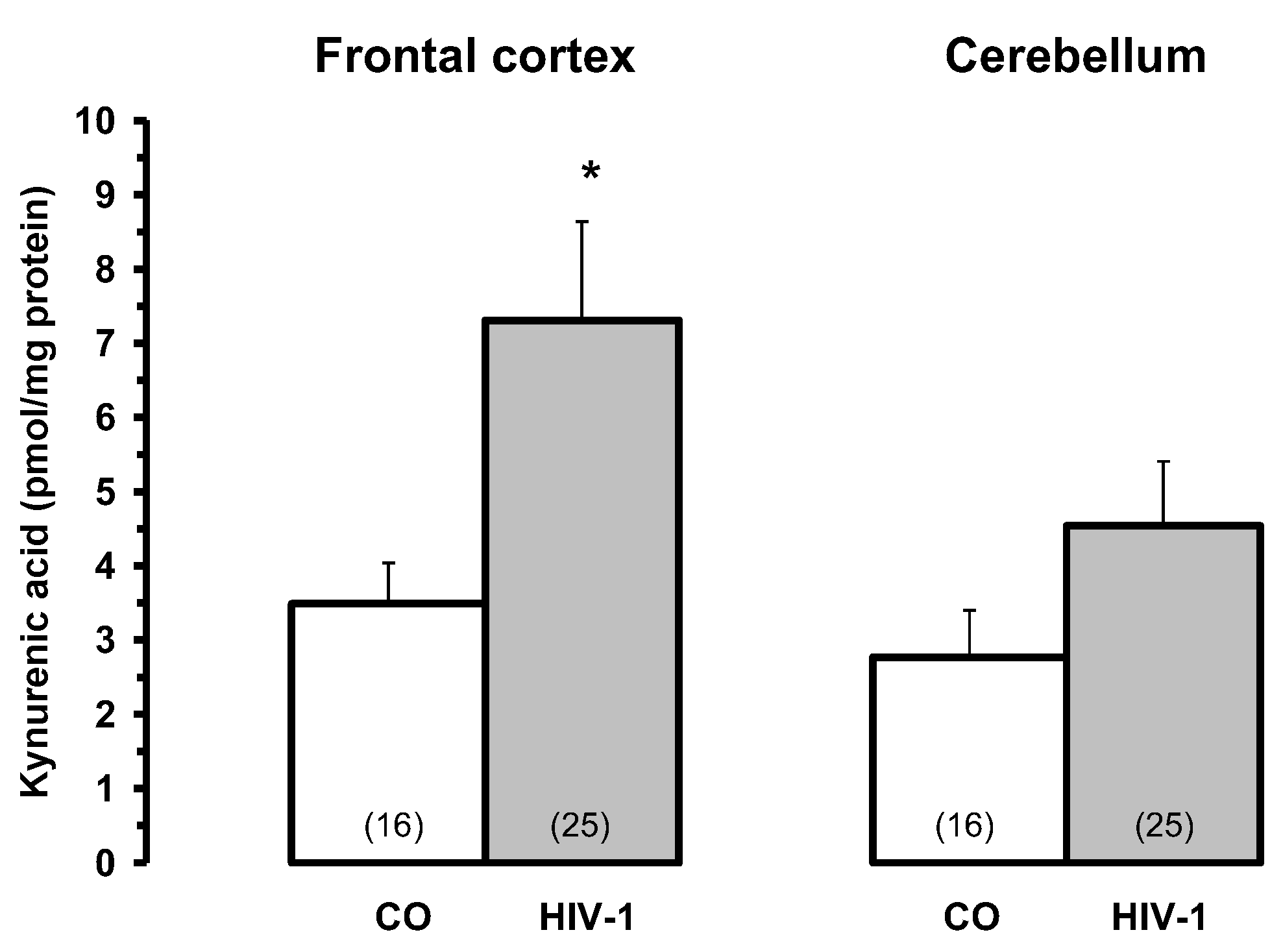
Figure 6.
Kynurenine aminotransferase I and II activities in HIV-1 infected brain and controls (CO). Data are mean ± SEM. Significances: *p < 0.05 vs. CO.
Figure 6.
Kynurenine aminotransferase I and II activities in HIV-1 infected brain and controls (CO). Data are mean ± SEM. Significances: *p < 0.05 vs. CO.
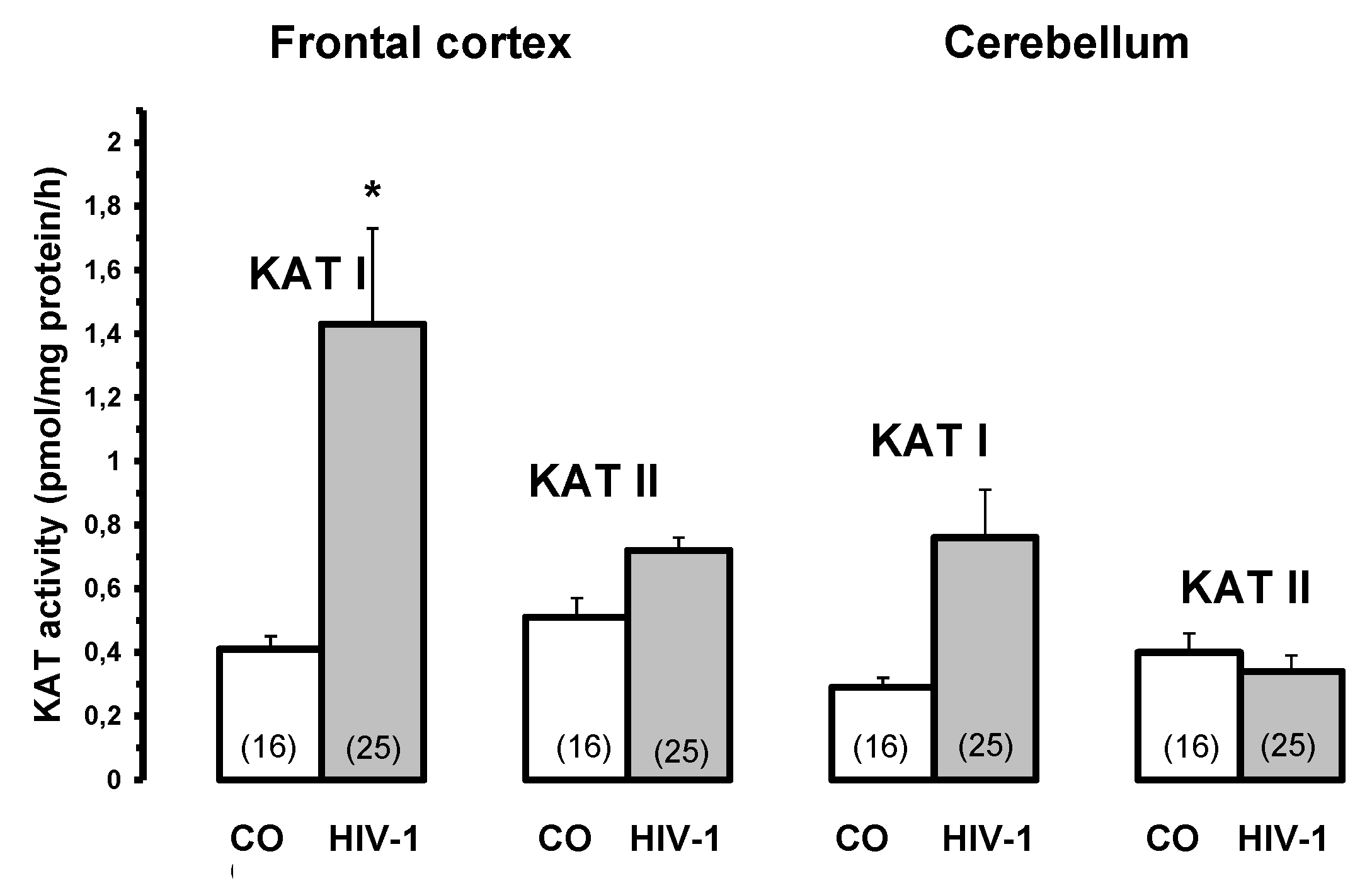
Figure 7.
Inhibition of kynurenine aminotransferase II activity in rat liver in the presence of cerebrolysin (Cer) 15 µl, D-cycloserine (D-C) 168 µM and Jerusalem Balsam 1 and 7.5 µl. Different times of incubation, 1, 3 and 5 hrs. Abbreviation: Jerusalem Balsam (Jerusalem B). Data are mean ± SEM. Significances: *p < 0.05 vs. CO.
Figure 7.
Inhibition of kynurenine aminotransferase II activity in rat liver in the presence of cerebrolysin (Cer) 15 µl, D-cycloserine (D-C) 168 µM and Jerusalem Balsam 1 and 7.5 µl. Different times of incubation, 1, 3 and 5 hrs. Abbreviation: Jerusalem Balsam (Jerusalem B). Data are mean ± SEM. Significances: *p < 0.05 vs. CO.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



