
Submitted:
05 October 2024
Posted:
07 October 2024
You are already at the latest version
Abstract
Since the discovery that Histone deacetylase inhibitors (HDCAi) could enhance radiation response, a number of HDACi, mainly pan-HDAC inhibitors, have been studied either as monotherapy or in combination with photon irradiation or chemotherapeutic drugs in the management of breast cancer. Studies on combination of HDACi and particle type of radiation remain limited. CUDC-101 is a multitarget inhibitor of Histone deacetylases (HDACs), epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2(HER-2). In this paper, the effectiveness of CUDC-101 in enhancing radiation response to protons irradiation was studied in MCF-7, MDA-MB-231 and MCF-10A cell lines, using X-rays as reference radiation. γ-H2AX foci assays showed increased sensitivity to CUDC-101 in MDA-MB-231 cell line compared to the MCF-7 cell line. In both cell lines, induction of apoptosis was enhanced in CUDC-101 pre-treated cells compared to radiation (protons or X-rays) alone. Increased apoptosis was also noted in CUDC-101 pre-treated cells in the MCF-10A cell line. Cell cycle analysis showed increased G2/M arrest by CUDC-101 mono-treatment as well as combination of CUDC-101 and X-irradiation in the MDA-MB-231 cell line. Collectively, the findings indicate that CUDC-101 effectively enhances response to both X-radiation and proton irradiation, in the triple negative MDA-MB-231 cell line. The study highlights that CUDC-101 holds potential in the management of triple negative breast cancer as monotherapy or in combination with X-irradiation.
Keywords:
histone deacetylase inhibitors
; CUDC‐101
; proton therapy
; proton irradiation
1. Introduction
Radiation therapy is commonly indicated as adjuvant therapy to reduce the risk of loco-regional recurrence of breast cancer and to improve disease-free survival [1]. As a result, megavoltage (MV) photon-based radiation therapy remains to be an important treatment modality in breast cancer and the recent advances in treatment planning and delivery techniques such as intensity modulated radiotherapy (IMRT) and volumetric modulated arc therapy (V-MAT) have improved dose distribution and sparing of healthy tissues [2]. However, associated late side-effects such as secondary malignancies and cardiopulmonary toxicities are still observed in breast cancer survivors after photon-based therapy, particularly in women treated for left-sided breast cancer [3,4,5]. Photon-based therapy is widely available, however rapid expansion in the number of proton therapy centers across the globe have improved access for breast cancer patients. In general, the number of cancer patients treated with proton therapy is increasing rapidly with an estimated 190 000 in 2018, and an expected increase to over 300,000 in 2030 [6]. Further, the superior dose distribution of protons, has been reported to be a suitable modality for re-irradiation in a number of tumour types [7]. The physical aspects of proton therapy are well understood, but the biological aspects remain under-explored for protons alone as well as in combination therapies with drugs and concomitant therapies [5,8,9]. In recent years, combination therapies of Histone deacetylase inhibitors (HDACi) and photon irradiation have been a focus of many studies [10,11,12,13,14,15,16]. However, combination therapies with particle type of radiation and HDCAi remain limited.
Histone deacetylase inhibitors (HDACi) are epigenetic drugs that can sensitise cancer cells to ionising radiation with little effect on healthy cells [17,18]. To date, five HDACi SAHA (generic name vorinostat), belinostat, panabinostat, chidamide, romidepsin have been approved by the Food and Drug Administration (FDA) for cancer therapy [19,20]. More than 20 different HDACi are in different phases of clinical trials as monotherapy or in combination with other DNA damaging agents [21]. HDACi are classified into several classes according to their chemical structure namely benzamides (e.g., chidamide, entinostat), hydroxamic acids (e.g., SAHA, belinostat, panabinostat, CUDC-101), cyclic tetrapeptides (e.g., romidepsin) and aliphatic acids (e.g., butyrate, valproic acid)[16,22]. Of these classes, hydroxamic acids is the main class that has been used and is continuously being in most studies [23]. Hydroxamic acids are preferred as they inhibit a broad range of HDACs (HDACs1-11), and they can cause cellular effects at low (nM) concentrations[21].
Evidence from pre-clinical studies has revealed that the combination of photon radiation and HDACi results in increased cell kill in a number of cell lines including lung, melanoma, prostate, glioma, colon, non-small cell lung cancer (NSCLC), osteosarcoma and breast to name a few [10,11,12,13,14,15,16,24,25,26,27]. Reports on combination of HDACi with proton irradiation remain limited [28,29,30,31]. The need for further studies on HDACi and particle therapy has been expressed [32]. For treatment of breast cancer, several studies have explored using pan-HDACi SAHA or Panobinostat in combination with chemotherapeutic drugs, but studies on CUDC-101 are lacking [33,34,35,36]. CUDC-101 is a hydroxamic acid that inhibits HDACs, epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER-2)[37]. EGFR and HER-2 have been recognized and are regarded as a biomarkers for resistance in tumours. EGFR is reported to be expressed in all molecular sub-types of breast cancer, and over-expressed in triple-negative breast cancers [38,39]. Therefore, the dual targeting of EGFR and HER-2 may be beneficial for EGFR over-expressing triple-negative breast cancer [40], for which CUDC-1010 would be an appealing HDACi candidate . To the best of our knowledge, only one in vitro study has been conducted on the effect of CUDC-101 and photon-irradiation in the triple-negative breast cancer cell line MDA-MB-231 [14]. Considering the growing interest to use proton therapy for breast cancer patients, we aimed to quantify and compare the efficacy of CUDC-101 in combination proton and X-radiation, in MCF-7, MCF-10A and MDA-MB-231 cell lines.
2. Results
2.1. Determination of IC50 and Timepoint of Irradiation in Relation to the Drug
To determine the half maximal inhibitory concentration (IC50) of HDACi, MCF-7, MDA-MB-231 and MCF-10A cells were pre-treated with CUDC-101 at concentrations that ranged from 0.16 µM to 20µM. Cell proliferation was assesed 72 hours post-treatment using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) cell viability assays. The determined half-maximal inhibitory concentration (IC50) values are presented in Table 1.
To determine the optimal sequence of administration of HDACi and radiation, cells were irradiated with X-rays (2Gy) at 8 hours, 16 hours, and 24 hours before treatment with CUDC-101 as well as immediately, at 8 hours, 16 hours and 24 hours post-treatment with CUDC-101. Cell proliferation was determined at 72 hours post-irradiation using MTT assays. Pre-treatment with HDACi 24-hours before irradiation resulted in the least cell survival for all three cell lines and was subsequently used for all experiments (Figure 1).
2.2. Effect of CUDC-101 in Enhancing Radiation Response in Breast Cell Lines
Colony survival assays were performed to determine the effect of CUDC-101 in enhancing radiation-mediated cell killing (or inhibition of cell proliferation). For all three cell lines, comparison of survival curves showed an increased cell killing after proton irradiation compared to X-irradiated cells (Figure 2a–f). This resulted in relative biologic effectiveness at 10% survival (RBE10) values of 1.51,1.31 and 1.20, in MCF-7, MDA-MB-231 and MCF-10A cell lines, respectively. All observed RBE values are close to the RBE value of 1.1, which is used in clinical practice. The proton RBE was further enhanced by pre-treatment with CUDC-101, from 1.15 to 1.34 in the MCF-7 cell line and from 1.2 to 1.34 in MCF-10A cell line (Table 2). In the case of the MDA-MB-231 cell line, combination treatment of 2Gy protons and CUDC-101 and 2Gy X-rays and CUDC-101 yielded similar values, Figure 2d, which inferred that the differential effect of two radiation types might be obscured by the effect of CUDC-101. Accordingly, the sensitization enhancement ratio (SER) after X-irradiation was higher (2.09) than the proton SER (1.77), which suggested that in the MDA-MB-231 cell line, an increased biologic effect can be anticipated after CUDC-101 and X-rays compared to combination therapy of protons and CUDC-101. The MDA-MB-231 cell line also exhibited a higher RBE compared to the other two cell lines, suggesting increased sensitivity to proton irradiation. The SER values observed for the MCF-7 and MCF-10A cell lines were higher for protons compared to X-rays, which indicates increased sensitisation after proton irradiation compared to X-irradiation. The proton SER for MCF-7 was higher (1.5) than that determined in MCF-10A cell line (1.23) and the X-ray SER values of the cell lines were comparable (1.16 and 1.10 in MCF-7 and MCF-10A cell lines, respectively.
2.2. Effect of CUDC-101 on Radiation-Induced DNA DSB Formation and Repair
To assess DNA damage induction and repair after combination treatment with CUDC-101 and irradiation, γ-H2AX foci assays were performed as molecular markers of DNA double strand break (DSB) and repair. Cells were pre-treated with IC50 concentrations of HDACi for 24 hours and irradiated with protons or X-rays. γ-H2AX foci assays were performed at 1 hour and 24 hours post-irradiation with 2Gy 200MeV mid-spread-out Bragg peak (SOBP) protons or 250 kVp x-rays. Overall, an increased number of γ-H2AX foci were noted post-irradiation with protons compared to X-irradiation in all three cell lines (Figure 3a–f). Further, in comparison to X-ray irradiated cells, an increased number of persisting γ-H2AX foci at 24 hours post-proton irradiated cells was observed, which suggested that the type of damage induced by protons is complex in nature and more difficult to repair (Figure 3a–f).
In the MCF-7 cell line, at 24 hours post irradiation, a significant reduction in the number of γ-H2AX foci was noted after irradiation protons (p<0.0047) or x-rays(p<0.0009) as well as in CUDC-101 pre-treated cells (p<0.0001), which suggests that addition of the CUDC-101 did not impair the repair of the DNA DSB (Figure 3a,b). Comparison of the combination treatment (CUDC-101 and 2Gy) and irradiation alone (2Gy protons), resulted in a non-significant result with p values of 0.1580 and 0.1319 at 1 hour and 24 hours post-irradiation, respectively. Comparison of combination treatment of 2Gy and X-irradiation and X-rays also yielded a non-significant result with p values of 0.0718 and 0.3018 at 1 hour and 24 hours post-irradiation, respectively. Although not statistically significant, it was noted that proton irradiation alone yielded a higher number of γ-H2AX foci as compared to combination treatment of proton and CUDC-101 at 1-hour post-irradiation in this cell line (Figure 3a). This was not observed after irradiation with X-rays (Figure 3b).
In the MDA-MB-231 cell line, the number of γ-H2AX foci induced by radiation alone (protons or X-rays) and those induced by combination of radiation and CUDC-101 were not statistically significant (p =0.7672) at 1-hour post-irradiation (Figure 3c,d). A significantly reduced (p=0.0003), but notable number of retained γ-H2AX foci was observed after combination therapy with proton and CUDC-101 as well as proton irradiation alone (Figure 3c). For X-ray irradiation, the number of retained γ-H2AX foci at 24 hours after combined treatment remained high and the decrease in the number of γ-H2AX foci was not statistically significant(p=0.4262) compared to the 1 hour time point (Figure 3d).Taken together, the findings suggest repair impairment in the MDA-MB-231 cell line and a high sensitivity to CUDC-101, which is also observable in the unirradiated CUDC-101 control samples. This is reflected as an increase in the number of γ-H2AX foci induced by CUDC-101 monotherapy at 24 hours as compared to 1 hour (Figure 3c,d).
In MCF-10A cell line, the only non-malignant cell line included in the study, an overall reduced number of γ-H2AX foci were noticed compared to the other two cell lines. A statistically significant (p=0.008) higher number of remaining γ-H2AX foci at 24 hours was noted with 2 Gy protons and CUDC-101 compared to proton irradiation alone (Figure 3e). Almost complete repair was noted after proton and X-ray irradiation alone and in the combination treatment of X-rays and CUDC-101 at 24 hours (Figure 3f). Representative immunohistochemistry images of MCF-7 and MDA-MB-231 cell lines are shown in Figure 3g and Figure 3h, respectively.
2.3. Impact of CUDC-101 and Radiation on Apoptosis in Breast Cell Lines
To investigate the induction of apoptosis after treatment with CUDC-101, radiation (protons or x-rays) or combination therapy of CUDC-101 and radiation, the Annexin V/PI apoptosis assay was performed. Apoptosis and necrosis were assessed at 48 hours post-irradiation with 2 Gy and 6 Gy protons or X-rays, as well as after combination of CUDC-101 and radiation (proton or X-rays). In all three cell lines, increased apoptosis levels were observed post proton-irradiations as compared to X-ray irradiations (Figure 4a–c). Pre-treatment with CUDC-101 significantly increased the level of proton-induced apoptosis after 2Gy (p = 0.0020) and after 6Gy (p=0.0011) in MCF-7 cell line (Figure 4a). Similarly, in the MDA-MB-231 cell line, a significantly increase was observed in CUDC-101 pre-treated samples after 2Gy (p=0.0219) and 6Gy (p= 0.0216) (Figure 4b). In the spontaneously immortalised MCF-10A cell samples, increased apoptotic fractions were observed in the CUDC-101 treated MCF-10A cells (Figure 4c). Further, treatment with 1µM of apoptosis inducer staurosporine induced apoptosis in MCF-7 and MCF-10A cell lines, whereas necrosis was induced in the MDA-MB-231 cell line at 24 hours after treatment (Figure 4d).
In addition, in the MCF-7 cell line, increased necrosis was noted after proton irradiations compared to X-irradiations (p=0.0063). Pre-treatment with CUDC-101 further increased levels of necrosis after X-irradiation in this cell line (p=0.0051) (Figure 5a,b). A different observation was made in the MDA-MB-231 cell line, pre-treatment with CUDC-101 reduced levels of necrosis after proton irradiation(p=0.0136) and X-rays (p = 0.3647). Comparison of levels of necrosis between the two cell lines showed increased amounts of necrotic cell populations in the MCF-7 cell line compared to the MDA-MB-231 cell line after protons (p= 0.0174) and protons and CUDC-101 (p=0.0095) (Figure 5g). Representative images of apoptosis profiles are included in Supplementary Material (Figure S1).
Cell cycle progression after treatment with CUDC-101 and radiation was assessed using propidium iodide with RNase staining. For all cell lines, an increased fraction of cells was observed in the G2/M phase of the cell cycle at 24 hours post-irradiation with 6 Gy protons or X-rays, indicating a G2/M cell cycle arrest (Figure 6a–f). Also, in all three cell lines, mono-treatment with CUDC-101 induced G2/M cell cycle arrest at 24 hours, which persisted at 48 hours (Figure 6a–f). In the MCF-7 and MDA-MB-231 cell lines, an increase in fraction of G2/M cells was also observed at 48 hours in X-ray irradiated cells compared to proton-irradiated cells at doses of 2Gy (p=0.0012) and at 6Gy(p=0.0007). This increase was more evident in the MDA-MB-231 cell line compared to the MCF-7 cell line (Figure 5g,h). Furthermore, compared to radiation treatment alone, pre-treatment with CUDC-101 had a minimal effect on the cell cycle progression in MCF-7 cells at neither 24 hours (p=0.4929 for 2Gy and p=0.0532 for 6Gy) nor 48 hours (p=0.6985 for 2Gy and p=0.3118 for 6Gy) post proton-irradiation as evidenced by comparable G2/M fractions at these timepoints (Figure 5g). However, in the MDA-MB-231 cell line, pre-treatment with CUDC-101 increased the G2/M fraction after exposure to both 2 Gy p=0.0030) and 6 Gy(p=0.0027) X-rays which was maintained at 48 hours post irradiation (Figure 5h). It seems sensible to associate the increased fraction of G2/M cells after X-ray irradiations to the reduced levels of apoptosis and necrosis which were seen in the MCF-7 and MDA-MB-231 cell lines at 48 hours post-irradiation. In this instance, the increased G2/M could be an indicator of mitotic catastrophe as a mode of cell death after x-irradiations. Similarly, in the MCF-10A cells, increasedG2/M fractions were noted in CUDC-101 pre-treated compared to radiation alone. Cell cycle profiles are included in the Supplementary Material (Figure S2).
3. Discussion
3.1. CUDC-101 Increases Sensitivity of MCF-7, MDA-MB-231 and MCF-10A Cell Lines to Proton and X-ray Irradiation
Our data shows that the multi-target inhibitor CUDC-101 enhances the response to both protons and X-rays in MCF-7 and MCF-10A breast cell lines. The enhancement was most notable after proton irradiation, with proton SER values of 1.50 and 1.23; and X-ray SER values of 1.16 and 1.10 in MCF-7 and MCF-10A cell lines, respectively. Similarly, in hepatocellular carcinoma (HCC) cell lines, Choi et al. reported higher proton SER values of 1.25 and 1.21 compared to X-ray SER values of 1.15 and 1.11, using Panobinostat in Huh7 and Hep3B cells, respectively. Yu et al. also reported Valproic acid (VPA)-mediated RBE10 value of 1.17 compared to RBE10 value of 1.08 without VPA in Hep3B cells after treatment with 6MV photons [31]. In another study, Gerelchuluun et al. reported an RBE10 value of 1.24 and proton SER values of 1.31 and 1.16 compared to γ-ray SER values of 1.43 and 1.08 in lung carcinoma (A549) and normal fibroblast (AG1522) cell lines, respectively, following treatment with 2µM of SAHA and respectively [29]. Although different HDACi and different cell lines were used in the mentioned studies, the findings are consistent with those of the current study, where higher RBE and SER values were reported for proton compared to X-rays. The enhanced radiation response in MCF-7 cell line indicate a potential benefit of using CUDC-101 and protons in treatment of breast tumours with estrogen, progesterone and her-2 positive molecular sub-types. Previous studies have asserted that HDACs are not overexpressed in normal tissues which lead to minimal effect of HDACi on normal tissues [17,41,42]. While our study indicates SER of 1.10 and 1.23 in the normal breast cell line (MCF-10A) following pre-treatment with CUDC-101, these values were lower than the ones observed in the malignant cell lines. It should be noted that the former studies used SAHA which inhibits HDACs only, whereas in the current study CUDC-101 which inhibits HDACs, EGFR and HER-2, was used. Therefore, the slight effect of CUDC-101 observed in the normal cell line can be attributed to inhibition of EGFR, which is important for growth and maintenance of MCF-10A cells [43].
A different effect was seen in the MDA-MB-231 cell line in the current study, with a higher X-ray SER value of 2.09 compared to proton SER value of 1.77, which implies that in this cell line, CUDC-101 is best combined with X-rays for maximal therapeutic benefit of the combination treatment. It is yet to be determined if a similar result will be seen using other triple negative breast cell lines. In addition, the RBE10 value of 1.31 for the MDA-MB-231 cell line indicates that protons are more effective than X-rays to inhibit the proliferation capacity, at a higher rate than the RBE10 values observed in the MCF-7 and MCF-10Acell lines, in the. Previous studies have associated higher RBE values to DNA damage repair capacity of the cell line. In other words, cell lines with higher RBE values, were reported to be deficient in DNA repair capacity [44,45].
3.2. Proton Irradiation Induces Increased DNA Damage That Is Not Easily Repaired in Breast Cell Lines
The complexity of DNA damage induction and repair after proton therapy remains a subject of discussion [46]. Contrary to X-rays that have no mass and no charge, protons are charged particles with a larger mass which can create more direct and complex DNA damage. Previous studies reported an increased number of γ-H2AX foci that are larger in size after proton irradiation as compared to X-rays in different cell lines [47,48,49,50,51,52,53]. Other studies reported that SOBP protons induced increased γ-H2AX foci, which were larger in size and reached maximum point at 1-hour post irradiation, whereas maximal γ-H2AX foci count was reached at 30 minutes post irradiation with X-rays and plateau protons, which were also smaller in size and were resolved at 6-hours post irradiation [47,50,54]. Gerelchuulun et al. reported a 1.2-1.6-fold increase in γ-H2AX foci in ONS76 medulloblastoma and MOLT4 leukemia cells after proton irradiation compared to 10MV X-rays [51]. In another study, irradiation with SOBP protons induced more clustered DNA damage, whereas entrance plateau protons induced mixed-type damage that consisted of clustered and non-clustered DNA damage [50]. Consistent with these studies, a significantly increased number (1.4-1.5-fold) of γ-H2AX foci was observed at 1-hour post-irradiation with 2 Gy SOBP protons compared to 2 Gy X-rays in all three cell lines (Figure 3a–h). The fact that persisting γ-H2AX foci at 24 hours post-irradiation were observed mainly after proton irradiation (Figure 3b,c), suggests that the type of DNA damage induced by protons is difficult to repair, supporting the assertions of complex DNA damage [47,50,51,53,55,56,57].
Of relevance is also the ongoing discussions about differential requirement of DNA DSB repair pathways following protons and x-rays [47,55]. Previous studies reported that post-proton irradiation, the error free homologous repair (HR) is preferred, and non-homologous end joining (NHEJ) is preferred after irradiation with x-rays [53,55]. Latter reports indicated that HR is required post-proton irradiation due to the complex nature of the DNA damage, but NHEJ is also indispensable for repair of proton-induced DSB [45,47,57]. Further evidence pointed out that irrespective of the type of radiation, the initial fast repair is conducted by NHEJ, and HR occurs at a later stage [53,58,59].In a recent report by Lohberger et al., mismatch repair (MMR) and nucleotide excision repair (NER) repair pathways together with HR and NHEJ pathways were found to be activated mainly post-proton irradiation in chondrosarcoma cells [52]. These studies bear relevance to the observed prolonged appearance of γ-H2AX foci after proton irradiation as compared to x-irradiated cells, particularly in the MDA-MB-231 cell line in the current study. Consistent with the notion that cell lines with higher RBE values have defective repair pathways, Lee et al. reported that MDA-MB-231 cells are deficient in HR, base excision and nucleotide excision repair (NER)[60]. This would explain the increased retention of γ-H2AX foci in MDA-MB-231 cell line and increased sensitivity to proton irradiation (RBE10 of 1.31) compared to the MCF-7 cell line (RBE10 of 1.15). As previously mentioned, the HDACi-mediated RBE was however lower than the proton RBE due compounding effect of DNA damage that is induced CUDC-101 in the MDA-MB-231 cell line, which was not seen in the other cell lines. Several studies also reported having observed increased sensitivity to proton irradiation in cells that are deficient in HR machinery [47,53,55,56,61].
Limited studies have been conducted on combination therapy of HDACi and proton irradiation with respect to DNA DSB induction and repair. A 3 hour pre-treatment with 1mM HDACi Valproic acid (VPA) and mid-SOBP protons prolonged appearance of γ-H2AX foci in Hep3B and Huh7 hepatocellular carcinoma cell lines [31]. Pre-treatment with 5nM Panobinostat increased the γ-H2AX foci yield at 24 hours post irradiation with 6 Gy mid-SOBP protons in Huh7 and Hep3B hepatocellular carcinoma cell lines [28]. In NFF28 normal fibroblasts cells, Johnson et al. reported resolution of γ-H2AX foci to near background levels at 24 hours post-treatment with 10µM SAHA and irradiation with 200 MeV protons [30]. The results of these earlier studies are consistent with the observation in the current study, since the retention of γ-H2AX foci was in general higher after proton irradiation in malignant cell lines compared the MCF-10A cell line. It is also worth noting that in the study by Johnson et al., 200MeV entrance plateau protons were used, whereas 200 MeV mid-SOBP protons were used in the current study. Persisting γ-H2AX foci were detected at 24 hours post treatment with 2 Gy protons and CUDC-101 in the MCF-10A cell line, which could be an indication of increased normal tissue effect.
Treatment with CUDC-101 alone resulted in an induction of γ-H2AX foci at 1 hour, which increased at 24 hours post treatment in MDA-MB-231 cell line (Figure 3c–e), but not in the MCF-7 and MCF-10A cell lines. Similarly, in the MDA-MB-231 cell line, an increased G2/M phase fraction in comparison to the untreated control was seen at 24 hours (p=0.003) and at 48 hours (p=0.0002). The increase in the number of γ-H2AX foci at 24 hours suggests that additional γ-H2AX foci might have been induced by cell death mechanisms. Induction of DNA damage by sole treatment with HDACi has previously been reported mainly in leukemia cells [62,63], which would explain the success of HDACi monotherapies in treating haematological malignancies with poor performance in solid tumours. In a study by Choi et al., Panobinostat alone did not induce γ-H2AX foci in hepatocellular carcinoma cell lines [28]. Further investigation is required to confirm the observations of CUDC-101 induced DNA DSB formation in the current study.
3.3. CUDC-101 Enhances Protons-Induced Apoptosis
The type of cell death after irradiation is mainly determined by the cell type and type of radiation. Apoptosis was formerly reported to be the main mode of cell death in haematological cancer cells whereas mitotic catastrophe was reported to be the main mode of cell kill in solid tumours after irradiation [64]. Further, several studies asserted that apoptosis would be the main mode of cell death in solid tumours, through either the intrinsic or extrinsic apoptotic pathways, whereas the main mode of cell death post X-ray irradiation would be mitotic catastrophe [51,64,65,66,67,68,69,70]. Consistent with these assertions made in these reports, increased apoptosis was observed post proton irradiations, whereas minimal levels of apoptosis were noted after X-irradiation in all three cell lines (Figure 4a–c). Apoptosis was most notable in the MDA-MB-231 cell line at 2 Gy and 6 Gy proton irradiations, as well as after combination therapy of proton irradiation (2 Gy and 6 Gy) and CUDC-101. Although a marked number of unresolved DSB was noted at 24 hours after treatment with CUDC-101 monotherapy in this cell line, minimal levels of apoptosis were noted, suggesting a different type of cell death. Indeed, cell cycle analysis in both MCF-7 and MDA-MB-231 cell lines showed increased G2/M arrest at 48 hours after CUDC-101 monotherapies, as well as after combination treatments of CUDC-101 and X-ray irradiation (Figure 5a–d), which suggested induction of mitotic catastrophe. Similar observations were made by Schlaff et al. who reported induction of mitotic catastrophe in glioblastoma cell line after CUDC-101 treatment [14]. Keeping with the argument that CUDC-101 and X-ray monotherapies induces mitotic catastrophe, the combination therapy of X-rays and CUDC-101 was expected to result in higher levels of mitotic catastrophe (G2/M) compared to proton irradiations and CUDC-101. Certainly, Figure 5d–e show an even higher proportion of G2/M cells after combination therapy of 2 Gy X-rays and CUDC-101 compared to monotherapies with either x-rays or CUDC-101 in the MDA-MB-231 cell line.
In the MCF-7 cell line, the levels of apoptosis and necrosis were comparable at the lower dose of 2 Gy (Figure 4a–b). The levels of necrosis exceeded that of apoptosis at doses of 6 Gy, Figure 4c. Several reports have asserted that MCF-7 cells are deficient in caspase 3 and therefore lacks the morphological features associated with apoptosis [71,72]. Natarajan et al. reported that MCF-7 cells can switch to necroptosis pathway after treatment with HDACi SAHA[73]. Necroptosis is a caspase-independent mechanism of programmed cell death that is activated when apoptosis is blocked and it bears mechanistic similarity to apoptosis and morphological similarity to necrosis [74]. Treatment with 1µM of apoptosis inducer staurosporine, induced more apoptosis in MCF-7 cell line compared to the MDA-MB-231 cell line at 24 hours post-treatment (Figure 4j). In earlier studies, staurosporine, at a concentration of 1µM, was reported to induce apoptosis in MCF-7 cells through partial activation of caspase-6 [71]. The authors also noted that apoptosis occurred earlier (16 hours), with the absence of typical apoptotic morphology and absence of apoptotic bodies in the MCF-7 cell line compared to T47D cells [71]. To the contrary, Poliseno et al. reported that at 5 hours post-treatment with staurisporine at 1µM induced necrosis in MCF-7 cell lines expressing low anti-apoptotic Bcl-2 protein [75]. Taken together, these studies imply that necrosis is induced earlier, and apoptosis is only detectable at a later stage in MCF-7 cells. In view of the mentioned overlapping similarities between necroptosis, necrosis and partial apoptosis in this cell line, it seems reasonable to assume that what was reported in previous literature, as well as in the current study, might have been necroptosis.
The increased apoptosis seen in CUDC-101 treated cells in the MCF-10A cell line is thought to be due to inhibition of EGFR (Figure 4g–i). Increased apoptosis was seen mainly after treatment with protons and CUDC-101 compared to X-ray irradiated cells (Figure 4g). Further, lower levels of apoptosis was seen after treatment 2 Gy X-rays and CUDC-101 compared to 6 Gy X-rays and CUDC-101. These findings imply that, if CUDC-101 is considered for use in triple negative breast cancer, it might be better to combine it with X-rays to reduce normal tissue reactions. Notwithstanding, in a Phase 1 study of 275mg/m2 CUDC-101 in combination with cisplatin and X-ray radiation in squamous cell head and neck cancers, out of the 12 patients that enrolled for the study, 5 patients discontinued CUDC-101 due to adverse side effects. CUDC-101 was administered 3 times a week for one week before starting with radiation and cisplatin and was concurrently administered with cisplatin and radiation in a fractionated regime up to a total dose of 70 Gy. The authors suggested alternate scheduling of CUDC-101 and using different routes of administration to minimize adverse effects [76]. Subsequently, in another Phase I trial in advanced solid tumours, Schlaff et al. reported that intravenous administration of CUDC-101 for 1 hour for 5 consecutive days, every 2 weeks were well tolerated. The authors recommended a dose of 275mg/m2 to be used [77]. A Phase I study (NCT01702285) to assess safety and tolerability of orally administered CUDC-101 was terminated for unknown reasons.
3.3. CUDC-101 Induces G2/M Cell Cycle Arrest and Enhances x-Irradiation-Induced G2/M Cell Cycle Arrest
Previous studies highlighted the important role that HDACs play in cell cycle progression. In particular, HDAC 3 and HDAC 10 have been implicated in mediating progression through the G2/M phase of the cell cycle [78,79,80]. HDACs 2,3,5 and SIRT2 have also been implicated in fascilitating exit from mitosis stage[78,81,82,83]. HDACi, therefore, induces CDK-inhibitor p21, to induce cell cycle arrest [78]. Monotherapy with 5nM of HDACi panobinostat induced G2/M arrest from 24.3% to 51.4% at 24 hours post-treatment in Huh7 HCC cell lines. Pre-treatment with 5nM panobinostat also increased G2/M proportions of cells after 6Gy X-ray or proton irradiation [28].The amounts of G2/M cells seen after single panobinostat treatment and after combination therapy of panobinostat and proton- or X-ray irradiation were not statistically signinficant [28]. In another study, 6 Gy photons or 6 Gy protons increased proportions of G2/M cell to 71% and 70%, respectively in Hep3 HCC cell line [31]. Pre-treatment with 1mM of HDACi VPA before radiation further increased G2/M fraction from 73.4% to 80.1% at 24 hours and from 59.9% to 58.6% at 72 hours[31]. In the current study, similar to the mentioned studies, 6 Gy protons and 6 Gy x-rays increased G2/M cells at 24 hours compared to the untreated control. Also, similar to Choi et al., treatment with CUDC-101 monotherapy increased G2/M cell cycle arrest in all three breast cell lines at 24 hours. The increase was most notable in the MDA-MB-231 and MCF-10A cell lines, indicating increased toxicity of CUDC-101 in these cell lines. Contrary to the mentioned studies, increased G2/M was seen in X-ray irradiated cells compared to the proton-irradiated cells, with or without HDACi pre-treatment in the current study. As previously mentioned, this differential increase in G2/M fraction after X-rays, suggested mitotic catastrophe as a mode of cell death.
4. Conclusion
The marked response seen in the MDA-MB-231 and MCF-10A cell lines in this study can be attributed to inhibition of EGFR by CUDC-101. Positive EGFR status has long been recognised as negative prognostic factors in breast cancer, and evidence has pointed out that EGFR is overexpressed in triple negative cancer [40]. The current results draw attention to the potential benefit of CUDC-101 in the management of triple negetive breast cancers as monotherapy or when combined with X-ray irradiation or proton irradiation. Due to the increased toxicity of CUDC-101 on normal cells particularly when combined with protons, it is advisable that CUDC-101 in combination with X-rays rather than with protons, be considered. CUDC-101 shows potential to enhance treatment efficacy in combination treatment with radiation, but future preclinical in vivo research and clinical trials are warranted to confirm these in vitro findings.
5. Materials and Methods
5.1. Cell Cultures
MCF-7 and MCF-10A (gifted by the Physiology Department, University of Pretoria) cells were cultured in Dulbecco’s Modified Eagle’s Medium F-12 (DMEM-F12; GibcoTM, Thermo Fisher Scientific, Sandton, SA) and Ham’s F-12 (GibcoTM, Thermo Fisher Scientific, Sandton, SA) supplemented with 10% fetal bovine serum (FBS) (GibcoTM, Thermo Fisher Scientific, Sandton, SA), 100 μg/mL penicillin (GibcoTM, Thermo Fisher Scientific, Sandton, SA) and 100 μg/mL streptomycin for bacterial contamination. MCF-10A medium was further supplemented with epidermal growth factor (EGF) (20 ng/mL final concentration) (GibcoTM, Thermo Fisher Scientific, Sandton, SA) and hydrocortisone (0.5 mg/mL final concentration) (Sigma-Aldrich, Missouri, USA).
MDA-MB-231 cells (gifted by the Department of Natural Sciences, University of Western Cape) were cultured in Roswell Park Memorial Institute (RPMI) 1640 (GibcoTM, Thermo Fisher Scientific, Sandton, SA) supplemented with 10% FBS, 100 μg/L penicillin and 100 μg/mL streptomycin (Sigma-Aldrich, Missouri, USA).
All cell lines were cultured in T275 or T75 cell culture flasks (Corning® T-75 flasks (ATCC catalogue #430641, USA) under standard conditions in a humidified incubator at 37ºC, 5% CO2 (Forma series 3 water jacketed incubator, Thermo Fisher Scientific, Waltham, Massa-chusetts, USA). Cell growth was assessed over 24 hour intervals and sub-cultured once 80% confluence was reached.
5.2. Histone Deacetylase Inhibitor
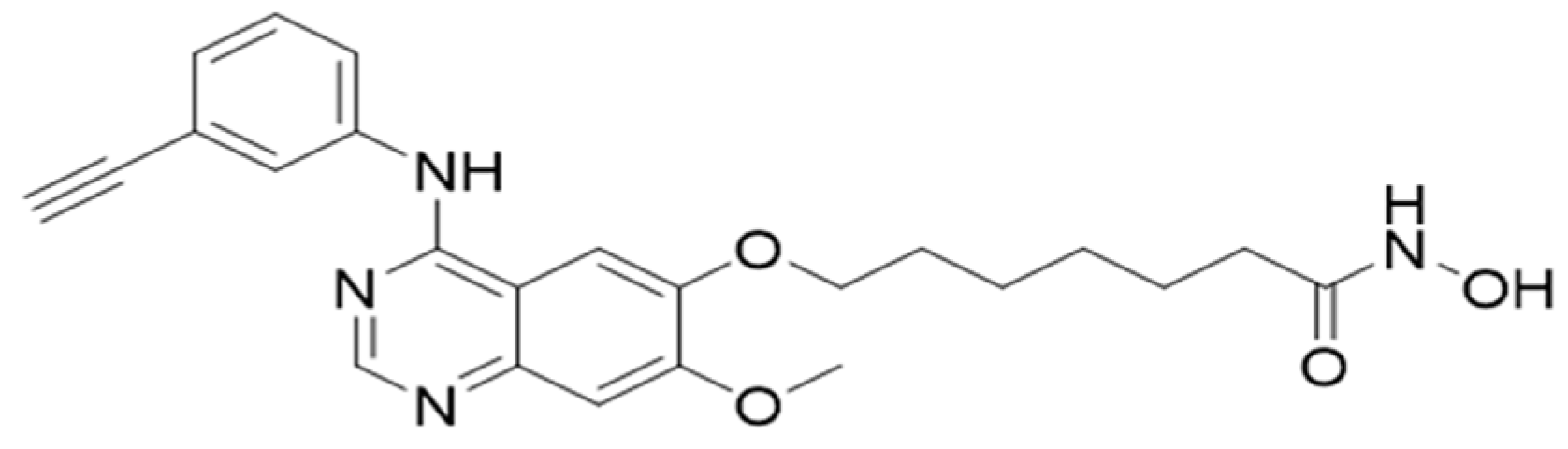
CUDC-101 (molecular weight of 434.49) was purchased from Sigma Aldrich (Sigma-Aldrich, Missouri, USA) and resolved in dimethylsulfide (DMSO) and stored at stock concentration of 1mM (Biotechnology Hub, Johannesburg, S.A) at -80° for long term storage.
Figure 7.
Molecular structure of CUDC-101.
5.3. Irradiations
Photon irradiations were performed using the 250 kVp X-Rad 320 unit (Precision X-ray, Madison, US) at a mean dose rate of 0.69 Gy/min at a Source Surface Distance (SSD) of 50 cm. Calibrations of the unit were performed according to the Technical report series-398 (TRS-398) protocol, with a Farmer 117 chamber for which a chamber calibration factor has been obtained from the National Metrology Institute of South Africa (NMISA). Proton irradiations were performed using 200MeV mid-SOBP beam extracted from 106 MHz cyclotron, at a dose rate of 2 Gy/minute, located at the Trento Institute for fundamental Physics and Application (TIFPA).
5.4. Cell Proliferation Assays
Cell proliferation assays were conducted using the thiazolyl blue tetrazoliumbromide (MTT) cell viability assay kit. Cells were seeded at a pre-determined density of 3000 cells/well in 96-well plates and allowed to attach. Cells were treated with different concentrations of the HDACi (SAHA or CUDC-101) ranging from 0 µM to 20µM and incubated for a further 72 hours. A 20 μl of a 5 mg/mL stock solution of MTT (Sigma-Aldrich, Missouri, USA) was added to each well and incubated for a further 4 hours to allow formazan formation. MTT containing media was carefully removed and 100 μL of DMSO was added to each well. The formation of formazan in the viable cells was monitored by measuring absorbance at wavelengths of 595 nm on a spectrophotometer to determine the half-maximal inhibitory concentrations (IC50) values for HDACi SAHA and CUDC-101 for the different cell lines.
5.5. Colony Survival Assays (CSA) and RBE Analysis
Cells (250-1500) were seeded in 6-well plates (Whitehead Scientific, Cape Town, SA) and allowed to attach overnight. Cells were treated with CUDC-101 at a concentration of 0.6µM, 2.7 µM and 0.3 µM for MCF-7, MCF-10A and MDA-MB-231 respectively for 24 hours and irradiated with 0, 2, 4, 6 and 8 Gy of 250KeV X-rays or 200 MeV SOBP protons. The cells were returned for incubation for a further 8-14 days to allow for colony development. Once colonies of approximately 50 cells were formed, they were fixed with methanol and stained with 2% crystal violet dissolved in methanol and left to dry overnight. Colonies were manually counted and the size was validated by microscopic inspection. Plating efficiency was calculated under untreated conditions using the equation:
Plating efficiency was used to normalise the surviving fractions for HDACi and radiation induced cell death. Surviving fraction of cells was calculated using the equation:
Survival curves were plotted and analysed using Graphpad Prism Software Version 10.00 for Windows (GraphPad Software, San Diego, CA, USA). The RBE of the different treatment conditions was calculated at a survival fraction of 10%:
4.5. Annexin V-FITC/Propidium Iodide Apoptosis and Cell Cycle Analysis Assays
Apoptosis and cell cycle progression were analysed using flow cytometry. For the apoptosis assays, at 48 hours post-irradiation, the media in which the cells were incubated was retained and combined with harvested cells before centrifugation. The cell pellet was resuspended in 100µl 1X annexin-binding buffer and stained with with 2.5µlAnnexin V FITC and 2.5µl propidium iodide (PI) (catalogue number 13242, Invitrogen, Thermo Fisher Scientific, Sandton, S.A) according to the manufacturer’s instructions. Cells were incubated at room temperature (25⁰C) for 15 minutes in the dark. An additional annexin-binding buffer was added after incubation and the samples analysed using the BD AccuriTM C6 Plus (BD Biosciences, SA) with 15 000-20 000 cells per measurement.
For analysis of the cell cycle, cells were harvested at 24 hours and 48 hours post treatments and the cell pellet was resuspended in a solution of 100µl propidium iodide and RNase (FxCycleTM PI/RNase, Invitrogen, Massachusetts, USA). Propidium iodide stains for both DNA and RNA, therefore the ribonuclease (RNase) digests and removes RNA to ensure that only DNA content is analysed [84]. The samples were analysed using FACSort (Beckton Dickinson, San Jose, CA, USA), with 15 000-20 000 events per measurement. Fluorescence measurements were done at 495 nm and 519 nm (peak emission) at fluorescein isothiocyanate (FITC) channel for Annexin V FITC; 536 nm and 616 nm (peak emmision) at FL2 or FL3 channels for propidium iodide for all flow cytometry assays.
4.6. Gamma-H2AX foci Assay
Treated cells were harvested 1 hour and at 24 hours post-irradiation and a suspension of approximately 120000 cells/0.25 mL were centrifuged onto coated slides (X-tra adhesive slides, Leica Biosystems, Buffalo Grove, IL, USA). Three slides were prepared for each treatment condition. The slides were fixed in freshly prepared 4% paraformaldehyde (PFA) for 20 min and washed in PBS for 5 minutes. Cells were then permeabilised with PBS-triton X-100 solution (GibcoTM, Thermo Fisher Scientific, Sandton, SA) for 10 minutes and blocking of no-specific antibody binding by washing 3 times in 1% bovine serum albumin (BSA) solution (Roche, Sigma-Aldrich/Merck, St. Louis, Missouri, USA) for 10 minutes per wash. Cells were incubated with phospho-histone H2A.X (Ser139) Monoclonal Antibody (3F2) antibody (Invitrogen, Biocom Africa (Pty) Ltd., Centurion, South Africa) for 1 hour at room temperature, followed by washing 3 times in 1% bovine serum albumin (BSA) (Roche, Sigma-Aldrich/Merck, St. Louis, Missouri, USA) to remove any unbound primary antibody. Cells were then incubated for a further 1-hour in the dark with Rabbit anti-Mouse IgG (H+L) FITC Secondary Antibody, secondary antibody (Invitrogen, Thermo Fisher Scientific, Sandton, SA) in a humidified chamber. Nuclear counterstaining was done with Prolong diamond anti-fade with DAPI (Thermo Fisher Scientific, Sandton, SA). Slides were stored at room temperature for a minimum of 24 hours and scanned automatically using the MetaCyte software module of the Metafer 4 scanning system with a 40X objective. For each slide, a minimum of 1000 cells were captured, and the average number of γ-H2AX foci per scanned slide was derived from the MetaCyte software.
4.7. Statistical Analysis
Statistical analysis was performed using Graphpad Prism version 10.2. All data was expressed as the mean the mean ± SD of three independent experiments (n=3). Statistical significance was determined using two-tailed Student’s t-test, and p<0.05 was considered statistically significant.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Representative images for apoptosis profiles; Figure S2: Representative images of the cell cycle profile after different treatment conditions.
Author Contributions
Conceptualization, E.N.S, S.N., C.V. and A.J.; methodology, E.N.S, S.N., C.V. and A.J.; formal analysis, E.N.S, S.N., C.V. and A.J.; investigation, E.N.,S.N., C.V.,A.B.,A.J.,S.; resources, X.X.; data curation, X.X.; writing—original draft preparation, E.N.S; writing—review and editing, E.N.S, S.N., C.V. and A.J.; supervision, S.N., C.V. and A.J.; funding acquisition, E.N.S, S.N., C.V., A.B., A.J. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by NRF iThemba Laboratories and Department of Higher Education and Training, South Africa. The APC was funded by the University of Pretoria, Cape Peninsula University of Technology, NRF iThemba Laboratories and GSI Helmholtzzentrum für Schwerionenforschung.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Ethics Committee of University of Pretoria (Ethics number: 689/2021).
Informed Consent Statement
Not applicable
Data Availability Statement
Not applicable
Acknowledgments
We acknowledge the Radiation biology team in the Biophysics Division at iThemba NRF Laboratories, South Africa for assistance with proton data collection. We also acknowledge the Trento Institute of Fundamental Physics (TIFPA) for provision of the proton beam, and the Cellular, Computational and Integrative Biology (CIBIO) Laboratories where the proton experiments were conducted.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhao, X.; Tang, Y.; Wang, S.; Yang, Y.; Fang, H.; Wang, J.; Jing, H.; Zhang, J.; Sun, G.; Chen, S.; et al. Locoregional recurrence patterns in women with breast cancer who have not undergone post-mastectomy radiotherapy. Radiat Oncol 2020, 15, 212. [Google Scholar] [CrossRef] [PubMed]
- Kowalchuk, R.O.; Corbin, K.S.; Jimenez, R.B. Particle Therapy for Breast Cancer. Cancers (Basel) 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Corbin, K.S.; Mutter, R.W. Proton therapy for breast cancer: progress & pitfalls. Breast Cancer Management 2018, 7, BMT06. [Google Scholar] [CrossRef]
- Chowdhary, M.; Lee, A.; Gao, S.; Wang, D.; Barry, P.N.; Diaz, R.; Bagadiya, N.R.; Park, H.S.; Yu, J.B.; Wilson, L.D.; et al. Is Proton Therapy a “Pro” for Breast Cancer? A Comparison of Proton vs. Non-proton Radiotherapy Using the National Cancer Database. Front Oncol 2018, 8, 678. [Google Scholar] [CrossRef]
- Mutter, R.W.; Choi, J.I.; Jimenez, R.B.; Kirova, Y.M.; Fagundes, M.; Haffty, B.G.; Amos, R.A.; Bradley, J.A.; Chen, P.Y.; Ding, X.; et al. Proton Therapy for Breast Cancer: A Consensus Statement From the Particle Therapy Cooperative Group Breast Cancer Subcommittee. Int J Radiat Oncol Biol Phys 2021, 111, 337–359. [Google Scholar] [CrossRef]
- Luo, W.; Ali, Y.F.; Liu, C.; Wang, Y.; Liu, C.; Jin, X.; Zhou, G.; Liu, N.-A. Particle Therapy for Breast Cancer: Benefits and Challenges. Frontiers in Oncology 2021, 11. [Google Scholar] [CrossRef]
- Verma, V.; Rwigema, J.M.; Malyapa, R.S.; Regine, W.F.; Simone, C.B. , 2nd. Systematic assessment of clinical outcomes and toxicities of proton radiotherapy for reirradiation. Radiother Oncol 2017, 125, 21–30. [Google Scholar] [CrossRef]
- Lühr, A.; von Neubeck, C.; Pawelke, J.; Seidlitz, A.; Peitzsch, C.; Bentzen, S.M.; Bortfeld, T.; Debus, J.; Deutsch, E.; Langendijk, J.A.; et al. “Radiobiology of Proton Therapy”: Results of an international expert workshop. Radiotherapy and Oncology 2018, 128, 56–67. [Google Scholar] [CrossRef]
- Ahmad, R.; Barcellini, A.; Baumann, K.; Benje, M.; Bender, T.; Bragado, P.; Charalampopoulou, A.; Chowdhury, R.; Davis, A.J.; Ebner, D.K.; et al. Particle Beam Radiobiology Status and Challenges: A PTCOG Radiobiology Subcommittee Report. International Journal of Particle Therapy 2024, 13, 100626. [Google Scholar] [CrossRef]
- Baschnagel, A.; Russo, A.; Burgan, W.E.; Carter, D.; Beam, K.; Palmieri, D.; Steeg, P.S.; Tofilon, P.; Camphausen, K. Vorinostat enhances the radiosensitivity of a breast cancer brain metastatic cell line grown in vitro and as intracranial xenografts. Mol Cancer Ther 2009, 8, 1589–1595. [Google Scholar] [CrossRef]
- Camphausen, K.; Scott, T.; Sproull, M.; Tofilon, P.J. Enhancement of xenograft tumor radiosensitivity by the histone deacetylase inhibitor MS-275 and correlation with histone hyperacetylation. Clin Cancer Res 2004, 10, 6066–6071. [Google Scholar] [CrossRef] [PubMed]
- Camphausen, K.; Tofilon, P.J. Inhibition of Histone Deacetylation: A Strategy for Tumor Radiosensitization. Journal of Clinical Oncology 2007, 25, 4051–4056. [Google Scholar] [CrossRef] [PubMed]
- Chinnaiyan, P.; Cerna, D.; Burgan, W.E.; Beam, K.; Williams, E.S.; Camphausen, K.; Tofilon, P.J. Postradiation sensitization of the histone deacetylase inhibitor valproic acid. Clin Cancer Res 2008, 14, 5410–5415. [Google Scholar] [CrossRef] [PubMed]
- Schlaff, C.D.; Arscott, W.T.; Gordon, I.; Tandle, A.; Tofilon, P.; Camphausen, K. Radiosensitization Effects of Novel Triple-Inhibitor CUDC-101 in Glioblastoma Multiforme and Breast Cancer Cells In Vitro. International Journal of Radiation Oncology, Biology, Physics 2013, 87, S650. [Google Scholar] [CrossRef]
- Groselj, B.; Sharma, N.L.; Hamdy, F.C.; Kerr, M.; Kiltie, A.E. Histone deacetylase inhibitors as radiosensitisers: effects on DNA damage signalling and repair. Br J Cancer 2013, 108, 748–754. [Google Scholar] [CrossRef]
- Damaskos, C.; Garmpis, N.; Valsami, S.; Kontos, M.; Spartalis, E.; Kalampokas, T.; Kalampokas, E.; Athanasiou, A.; Moris, D.; Daskalopoulou, A.; et al. Histone Deacetylase Inhibitors: An Attractive Therapeutic Strategy Against Breast Cancer. Anticancer Res 2017, 37, 35–46. [Google Scholar] [CrossRef]
- Lee, J.H.; Choy, M.L.; Ngo, L.; Foster, S.S.; Marks, P.A. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc Natl Acad Sci U S A 2010, 107, 14639–14644. [Google Scholar] [CrossRef]
- Armeanu, S.; Pathil, A.; Venturelli, S.; Mascagni, P.; Weiss, T.S.; Göttlicher, M.; Gregor, M.; Lauer, U.M.; Bitzer, M. Apoptosis on hepatoma cells but not on primary hepatocytes by histone deacetylase inhibitors valproate and ITF2357. J Hepatol 2005, 42, 210–217. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb Perspect Med 2016, 6. [Google Scholar] [CrossRef]
- Jenke, R.; Reßing, N.; Hansen, F.K.; Aigner, A.; Büch, T. Anticancer Therapy with HDAC Inhibitors: Mechanism-Based Combination Strategies and Future Perspectives. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef]
- Smalley, J.P.; Cowley, S.M.; Hodgkinson, J.T. Bifunctional HDAC Therapeutics: One Drug to Rule Them All? Molecules 2020, 25. [Google Scholar] [CrossRef] [PubMed]
- Dokmanovic, M.; Clarke, C.; Marks, P.A. Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res 2007, 5, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Rajak, H.; Singh, A.; Raghuwanshi, K.; Kumar, R.; Dewangan, P.K.; Veerasamy, R.; Sharma, P.C.; Dixit, A.; Mishra, P. A structural insight into hydroxamic acid based histone deacetylase inhibitors for the presence of anticancer activity. Curr Med Chem 2014, 21, 2642–2664. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.W.; Yeh, Y.L.; Wang, Y.C.; Huang, W.J.; Chen, Y.A.; Chiou, Y.S.; Ho, S.Y.; Lin, P.; Wang, Y.J. Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, enhances radiosensitivity and suppresses lung metastasis in breast cancer in vitro and in vivo. PLoS One 2013, 8, e76340. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wong, P.; Radany, E.; Wong, J.Y. HDAC inhibitor, valproic acid, induces p53-dependent radiosensitization of colon cancer cells. Cancer Biother Radiopharm 2009, 24, 689–699. [Google Scholar] [CrossRef]
- Munshi, A.; Kurland, J.F.; Nishikawa, T.; Tanaka, T.; Hobbs, M.L.; Tucker, S.L.; Ismail, S.; Stevens, C.; Meyn, R.E. Histone deacetylase inhibitors radiosensitize human melanoma cells by suppressing DNA repair activity. Clin Cancer Res 2005, 11, 4912–4922. [Google Scholar] [CrossRef]
- Munshi, A.; Tanaka, T.; Hobbs, M.L.; Tucker, S.L.; Richon, V.M.; Meyn, R.E. Vorinostat, a histone deacetylase inhibitor, enhances the response of human tumor cells to ionizing radiation through prolongation of gamma-H2AX foci. Mol Cancer Ther 2006, 5, 1967–1974. [Google Scholar] [CrossRef]
- Choi, C.; Lee, G.H.; Son, A.; Yoo, G.S.; Yu, J.I.; Park, H.C. Downregulation of Mcl-1 by Panobinostat Potentiates Proton Beam Therapy in Hepatocellular Carcinoma Cells. Cells 2021, 10. [Google Scholar] [CrossRef]
- Gerelchuluun, A.; Maeda, J.; Manabe, E.; Brents, C.A.; Sakae, T.; Fujimori, A.; Chen, D.J.; Tsuboi, K.; Kato, T.A. Histone Deacetylase Inhibitor Induced Radiation Sensitization Effects on Human Cancer Cells after Photon and Hadron Radiation Exposure. International journal of molecular sciences 2018, 19, 496. [Google Scholar] [CrossRef]
- Johnson, A.M.; Bennett, P.V.; Sanidad, K.Z.; Hoang, A.; Jardine, J.H.; Keszenman, D.J.; Wilson, P.F. Evaluation of Histone Deacetylase Inhibitors as Radiosensitizers for Proton and Light Ion Radiotherapy. Front Oncol 2021, 11, 735940. [Google Scholar] [CrossRef]
- Yu, J.I.; Choi, C.; Shin, S.-W.; Son, A.; Lee, G.-H.; Kim, S.-Y.; Park, H.C. Valproic Acid Sensitizes Hepatocellular Carcinoma Cells to Proton Therapy by Suppressing NRF2 Activation. Scientific Reports 2017, 7, 14986. [Google Scholar] [CrossRef] [PubMed]
- Antrobus, J.; Parsons, J.L. Histone Deacetylases and Their Potential as Targets to Enhance Tumour Radiosensitisation. Radiation 2022, 2, 149–167. [Google Scholar] [CrossRef]
- Abdel-Ghany, S.; Raslan, S.; Tombuloglu, H.; Shamseddin, A.; Cevik, E.; Said, O.A.; Madyan, E.F.; Senel, M.; Bozkurt, A.; Rehman, S.; et al. Vorinostat-loaded titanium oxide nanoparticles (anatase) induce G2/M cell cycle arrest in breast cancer cells via PALB2 upregulation. 3 Biotech 2020, 10, 407. [Google Scholar] [CrossRef] [PubMed]
- Lyu, H.; Hou, D.; Liu, H.; Ruan, S.; Tan, C.; Wu, J.; Hicks, C.; Liu, B. HER3 targeting augments the efficacy of panobinostat in claudin-low triple-negative breast cancer cells. npj Precision Oncology 2023, 7, 72. [Google Scholar] [CrossRef] [PubMed]
- NOURIEMAMZADEN, F.; WORD, B.; COTTON, E.; HAWKINS, A.; LITTLEJOHN, K.; MOORE, R.; MIRANDA-CARBON, G.; ORISH, C.N.; LYN-COOK, B. Modulation of Estrogen α and Progesterone Receptors in Triple Negative Breast Cancer Cell Lines: The Effects of Vorinostat and Indole-3-Carbinol In Vitro. Anticancer Research 2020, 40, 3669–3683. [Google Scholar] [CrossRef]
- Wawruszak, A.; Luszczki, J.J.; Grabarska, A.; Gumbarewicz, E.; Dmoszynska-Graniczka, M.; Polberg, K.; Stepulak, A. Assessment of Interactions between Cisplatin and Two Histone Deacetylase Inhibitors in MCF7, T47D and MDA-MB-231 Human Breast Cancer Cell Lines - An Isobolographic Analysis. PLoS One 2015, 10, e0143013. [Google Scholar] [CrossRef]
- Lai, C.-J.; Bao, R.; Tao, X.; Wang, J.; Atoyan, R.; Qu, H.; Wang, D.-G.; Yin, L.; Samson, M.; Forrester, J.; et al. CUDC-101, a Multitargeted Inhibitor of Histone Deacetylase, Epidermal Growth Factor Receptor, and Human Epidermal Growth Factor Receptor 2, Exerts Potent Anticancer Activity. Cancer Research 2010, 70, 3647–3656. [Google Scholar] [CrossRef]
- Masuda, H.; Zhang, D.; Bartholomeusz, C.; Doihara, H.; Hortobagyi, G.N.; Ueno, N.T. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res Treat 2012, 136, 331–345. [Google Scholar] [CrossRef]
- Hossein-Nejad-Ariani, H.; Althagafi, E.; Kaur, K. Small Peptide Ligands for Targeting EGFR in Triple Negative Breast Cancer Cells. Scientific Reports 2019, 9, 2723. [Google Scholar] [CrossRef]
- O’Donovan, N.; Crown, J. EGFR and HER-2 antagonists in breast cancer. Anticancer Res 2007, 27, 1285–1294. [Google Scholar]
- Qiu, L.; Burgess, A.; Fairlie, D.P.; Leonard, H.; Parsons, P.G.; Gabrielli, B.G. Histone deacetylase inhibitors trigger a G2 checkpoint in normal cells that is defective in tumor cells. Mol Biol Cell 2000, 11, 2069–2083. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Bae, S.C. Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. Am J Transl Res 2011, 3, 166–179. [Google Scholar] [PubMed]
- Bessette, D.C.; Tilch, E.; Seidens, T.; Quinn, M.C.; Wiegmans, A.P.; Shi, W.; Cocciardi, S.; McCart-Reed, A.; Saunus, J.M.; Simpson, P.T.; et al. Using the MCF10A/MCF10CA1a Breast Cancer Progression Cell Line Model to Investigate the Effect of Active, Mutant Forms of EGFR in Breast Cancer Development and Treatment Using Gefitinib. PLoS One 2015, 10, e0125232. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.; Son, A.; Lee, G.H.; Shin, S.W.; Park, S.; Ahn, S.H.; Chung, Y.; Yu, J.I.; Park, H.C. Targeting DNA-dependent protein kinase sensitizes hepatocellular carcinoma cells to proton beam irradiation through apoptosis induction. PLoS One 2019, 14, e0218049. [Google Scholar] [CrossRef]
- Bright, S.J.; Flint, D.B.; Chakraborty, S.; McFadden, C.H.; Yoon, D.S.; Bronk, L.; Titt, U.; Mohan, R.; Grosshans, D.R.; Sumazin, P.; et al. Nonhomologous End Joining Is More Important Than Proton Linear Energy Transfer in Dictating Cell Death. Int J Radiat Oncol Biol Phys 2019, 105, 1119–1125. [Google Scholar] [CrossRef]
- Vitti, E.T.; Parsons, J.L. The Radiobiological Effects of Proton Beam Therapy: Impact on DNA Damage and Repair. Cancers (Basel) 2019, 11. [Google Scholar] [CrossRef]
- Szymonowicz, K.; Krysztofiak, A.; Linden, J.V.; Kern, A.; Deycmar, S.; Oeck, S.; Squire, A.; Koska, B.; Hlouschek, J.; Vüllings, M.; et al. Proton Irradiation Increases the Necessity for Homologous Recombination Repair Along with the Indispensability of Non-Homologous End Joining. Cells 2020, 9. [Google Scholar] [CrossRef]
- Costes, S.V.; Boissière, A.; Ravani, S.; Romano, R.; Parvin, B.; Barcellos-Hoff, M.H. Imaging features that discriminate between foci induced by high- and low-LET radiation in human fibroblasts. Radiat Res 2006, 165, 505–515. [Google Scholar] [CrossRef]
- Leatherbarrow, E.L.; Harper, J.V.; Cucinotta, F.A.; O’Neill, P. Induction and quantification of gamma-H2AX foci following low and high LET-irradiation. Int J Radiat Biol 2006, 82, 111–118. [Google Scholar] [CrossRef]
- Oeck, S.; Szymonowicz, K.; Wiel, G.; Krysztofiak, A.; Lambert, J.; Koska, B.; Iliakis, G.; Timmermann, B.; Jendrossek, V. Relating Linear Energy Transfer to the Formation and Resolution of DNA Repair Foci After Irradiation with Equal Doses of X-ray Photons, Plateau, or Bragg-Peak Protons. Int J Mol Sci 2018, 19. [Google Scholar] [CrossRef]
- Gerelchuluun, A.; Hong, Z.; Sun, L.; Suzuki, K.; Terunuma, T.; Yasuoka, K.; Sakae, T.; Moritake, T.; Tsuboi, K. Induction of in situ DNA double-strand breaks and apoptosis by 200 MeV protons and 10 MV X-rays in human tumour cell lines. Int J Radiat Biol 2011, 87, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Bracalente, C.; Ibañez, I.L.; Molinari, B.; Palmieri, M.; Kreiner, A.; Valda, A.; Davidson, J.; Durán, H. Induction and persistence of large γH2AX foci by high linear energy transfer radiation in DNA-dependent protein kinase-deficient cells. Int J Radiat Oncol Biol Phys 2013, 87, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Grosse, N.; Fontana, A.O.; Hug, E.B.; Lomax, A.; Coray, A.; Augsburger, M.; Paganetti, H.; Sartori, A.A.; Pruschy, M. Deficiency in homologous recombination renders Mammalian cells more sensitive to proton versus photon irradiation. Int J Radiat Oncol Biol Phys 2014, 88, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Hojo, H.; Dohmae, T.; Hotta, K.; Kohno, R.; Motegi, A.; Yagishita, A.; Makinoshima, H.; Tsuchihara, K.; Akimoto, T. Difference in the relative biological effectiveness and DNA damage repair processes in response to proton beam therapy according to the positions of the spread out Bragg peak. Radiat Oncol 2017, 12, 111. [Google Scholar] [CrossRef]
- Fontana, A.O.; Augsburger, M.A.; Grosse, N.; Guckenberger, M.; Lomax, A.J.; Sartori, A.A.; Pruschy, M.N. Differential DNA repair pathway choice in cancer cells after proton- and photon-irradiation. Radiother Oncol 2015, 116, 374–380. [Google Scholar] [CrossRef]
- Liu, Q.; Ghosh, P.; Magpayo, N.; Testa, M.; Tang, S.; Gheorghiu, L.; Biggs, P.; Paganetti, H.; Efstathiou, J.A.; Lu, H.M.; et al. Lung cancer cell line screen links fanconi anemia/BRCA pathway defects to increased relative biological effectiveness of proton radiation. Int J Radiat Oncol Biol Phys 2015, 91, 1081–1089. [Google Scholar] [CrossRef]
- Gerelchuluun, A.; Manabe, E.; Ishikawa, T.; Sun, L.; Itoh, K.; Sakae, T.; Suzuki, K.; Hirayama, R.; Asaithamby, A.; Chen, D.J.; et al. The major DNA repair pathway after both proton and carbon-ion radiation is NHEJ, but the HR pathway is more relevant in carbon ions. Radiat Res 2015, 183, 345–356. [Google Scholar] [CrossRef]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 2008, 7, 2902–2906. [Google Scholar] [CrossRef]
- Mladenov, E.; Kalev, P.; Anachkova, B. The Complexity of Double-Strand Break Ends is a Factor in the Repair Pathway Choice. Radiation Research 2009, 171, 397–404. [Google Scholar] [CrossRef]
- Lee, K.J.; Mann, E.; Wright, G.; Piett, C.G.; Nagel, Z.D.; Gassman, N.R. Exploiting DNA repair defects in triple negative breast cancer to improve cell killing. Therapeutic Advances in Medical Oncology 2020, 12, 1758835920958354. [Google Scholar] [CrossRef]
- Rostek, C.; Turner, E.L.; Robbins, M.; Rightnar, S.; Xiao, W.; Obenaus, A.; Harkness, T.A. Involvement of homologous recombination repair after proton-induced DNA damage. Mutagenesis 2008, 23, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Gaymes, T.J.; Padua, R.A.; Pla, M.; Orr, S.; Omidvar, N.; Chomienne, C.; Mufti, G.J.; Rassool, F.V. Histone deacetylase inhibitors (HDI) cause DNA damage in leukemia cells: a mechanism for leukemia-specific HDI-dependent apoptosis? Mol Cancer Res 2006, 4, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Petruccelli, L.A.; Dupéré-Richer, D.; Pettersson, F.; Retrouvey, H.; Skoulikas, S.; Miller, W.H., Jr. Vorinostat induces reactive oxygen species and DNA damage in acute myeloid leukemia cells. PLoS One 2011, 6, e20987. [Google Scholar] [CrossRef] [PubMed]
- Vanderwaeren, L.; Dok, R.; Verstrepen, K.; Nuyts, S. Clinical Progress in Proton Radiotherapy: Biological Unknowns. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, C.; Piro, S.; Tabbì, G.; Ragusa, M.; Di Pietro, V.; Zimmitti, V.; Cuda, F.; Anello, M.; Consoli, U.; Salinaro, E.T.; et al. Cellular and molecular effects of protons: Apoptosis induction and potential implications for cancer therapy. Apoptosis 2006, 11, 57–66. [Google Scholar] [CrossRef]
- Lee, K.B.; Lee, J.S.; Park, J.W.; Huh, T.L.; Lee, Y.M. Low energy proton beam induces tumor cell apoptosis through reactive oxygen species and activation of caspases. Exp Mol Med 2008, 40, 118–129. [Google Scholar] [CrossRef]
- Alan Mitteer, R.; Wang, Y.; Shah, J.; Gordon, S.; Fager, M.; Butter, P.P.; Jun Kim, H.; Guardiola-Salmeron, C.; Carabe-Fernandez, A.; Fan, Y. Proton beam radiation induces DNA damage and cell apoptosis in glioma stem cells through reactive oxygen species. Sci Rep 2015, 5, 13961. [Google Scholar] [CrossRef]
- Zhang, X.; Lin, S.H.; Fang, B.; Gillin, M.; Mohan, R.; Chang, J.Y. Therapy-resistant cancer stem cells have differing sensitivity to photon versus proton beam radiation. J Thorac Oncol 2013, 8, 1484–1491. [Google Scholar] [CrossRef]
- Finnberg, N.; Wambi, C.; Ware, J.H.; Kennedy, A.R.; El-Deiry, W.S. Gamma-radiation (GR) triggers a unique gene expression profile associated with cell death compared to proton radiation (PR) in mice in vivo. Cancer Biol Ther 2008, 7, 2023–2033. [Google Scholar] [CrossRef]
- Ristic-Fira, A.M.; Todorovic, D.V.; Koricanac, L.B.; Petrovic, I.M.; Valastro, L.M.; Cirrone, P.G.; Raffaele, L.; Cuttone, G. Response of a human melanoma cell line to low and high ionizing radiation. Ann N Y Acad Sci 2007, 1095, 165–174. [Google Scholar] [CrossRef]
- Mooney, L.M.; Al-Sakkaf, K.A.; Brown, B.L.; Dobson, P.R. Apoptotic mechanisms in T47D and MCF-7 human breast cancer cells. Br J Cancer 2002, 87, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Jänicke, R.U. MCF-7 breast carcinoma cells do not express caspase-3. Breast Cancer Res Treat 2009, 117, 219–221. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, U.; Venkatesan, T.; Radhakrishnan, V.; Samuel, S.; Rathinavelu, A. Differential Mechanisms of Cell Death Induced by HDAC Inhibitor SAHA and MDM2 Inhibitor RG7388 in MCF-7 Cells. Cells 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Ma. Luisa, E.; Olga, M.E.; Gerardo, H.V.-N. Necrosis as Programmed Cell Death. In Cell Death, Tobias, M.N., Ed.; IntechOpen: Rijeka, 2015; p. Ch. 19.
- Poliseno, L.; Bianchi, L.; Citti, L.; Liberatori, S.; Mariani, L.; Salvetti, A.; Evangelista, M.; Bini, L.; Pallini, V.; Rainaldi, G. Bcl2-low-expressing MCF7 cells undergo necrosis rather than apoptosis upon staurosporine treatment. Biochem J 2004, 379, 823–832. [Google Scholar] [CrossRef]
- Galloway, T.J.; Wirth, L.J.; Colevas, A.D.; Gilbert, J.; Bauman, J.E.; Saba, N.F.; Raben, D.; Mehra, R.; Ma, A.W.; Atoyan, R.; et al. A Phase I Study of CUDC-101, a Multitarget Inhibitor of HDACs, EGFR, and HER2, in Combination with Chemoradiation in Patients with Head and Neck Squamous Cell Carcinoma. Clin Cancer Res 2015, 21, 1566–1573. [Google Scholar] [CrossRef]
- Schlaff, C.; Arscott, W.; Gordon, I.; Camphausen, K.; Tandle, A. Human EGFR-2, EGFR and HDAC triple-inhibitor CUDC-101 enhances radiosensitivity of GBM cells. Biomedical Research Journal 2015, 2, 105. [Google Scholar] [CrossRef]
- Li, G.; Tian, Y.; Zhu, W.-G. The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Frontiers in Cell and Developmental Biology 2020, 8. [Google Scholar] [CrossRef]
- Bhaskara, S.; Chyla, B.J.; Amann, J.M.; Knutson, S.K.; Cortez, D.; Sun, Z.W.; Hiebert, S.W. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol Cell 2008, 30, 61–72. [Google Scholar] [CrossRef]
- Bhaskara, S.; Knutson, S.K.; Jiang, G.; Chandrasekharan, M.B.; Wilson, A.J.; Zheng, S.; Yenamandra, A.; Locke, K.; Yuan, J.L.; Bonine-Summers, A.R.; et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell 2010, 18, 436–447. [Google Scholar] [CrossRef]
- Park, S.H.; Ozden, O.; Liu, G.; Song, H.Y.; Zhu, Y.; Yan, Y.; Zou, X.; Kang, H.J.; Jiang, H.; Principe, D.R.; et al. SIRT2-Mediated Deacetylation and Tetramerization of Pyruvate Kinase Directs Glycolysis and Tumor Growth. Cancer Res 2016, 76, 3802–3812. [Google Scholar] [CrossRef]
- Dryden, S.C.; Nahhas, F.A.; Nowak, J.E.; Goustin, A.S.; Tainsky, M.A. Role for human SIRT2 NAD-dependent deacetylase activity in control of mitotic exit in the cell cycle. Mol Cell Biol 2003, 23, 3173–3185. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liu, P.Y.; Scarlett, C.J.; Malyukova, A.; Liu, B.; Marshall, G.M.; MacKenzie, K.L.; Biankin, A.V.; Liu, T. Histone deacetylase 5 blocks neuroblastoma cell differentiation by interacting with N-Myc. Oncogene 2014, 33, 2987–2994. [Google Scholar] [CrossRef] [PubMed]
- Comşa, Ş.; Cîmpean, A.M.; Raica, M. The Story of MCF-7 Breast Cancer Cell Line: 40 years of Experience in Research. Anticancer Res 2015, 35, 3147–3154. [Google Scholar] [PubMed]
Figure 1.
Pre-treatment withCUDC-101 at 24 hours before 250kV X-irradiation offered maximal sensitization. Cells were pre-treated with 0.3 µM, 0.6 µM and 2.7 µM CUDC-101 in MCF-7, MDA-MB-231and MCF-10Acell lines. Cell proliferation was evaluated at 24, 16, 8 hours before radiation; immediately and at 8 and 24 hours after irradiation, as depicted on the X-axis. Cell proliferation was assessed with MTT assay at 72 hours post-irradiation.
Figure 1.
Pre-treatment withCUDC-101 at 24 hours before 250kV X-irradiation offered maximal sensitization. Cells were pre-treated with 0.3 µM, 0.6 µM and 2.7 µM CUDC-101 in MCF-7, MDA-MB-231and MCF-10Acell lines. Cell proliferation was evaluated at 24, 16, 8 hours before radiation; immediately and at 8 and 24 hours after irradiation, as depicted on the X-axis. Cell proliferation was assessed with MTT assay at 72 hours post-irradiation.
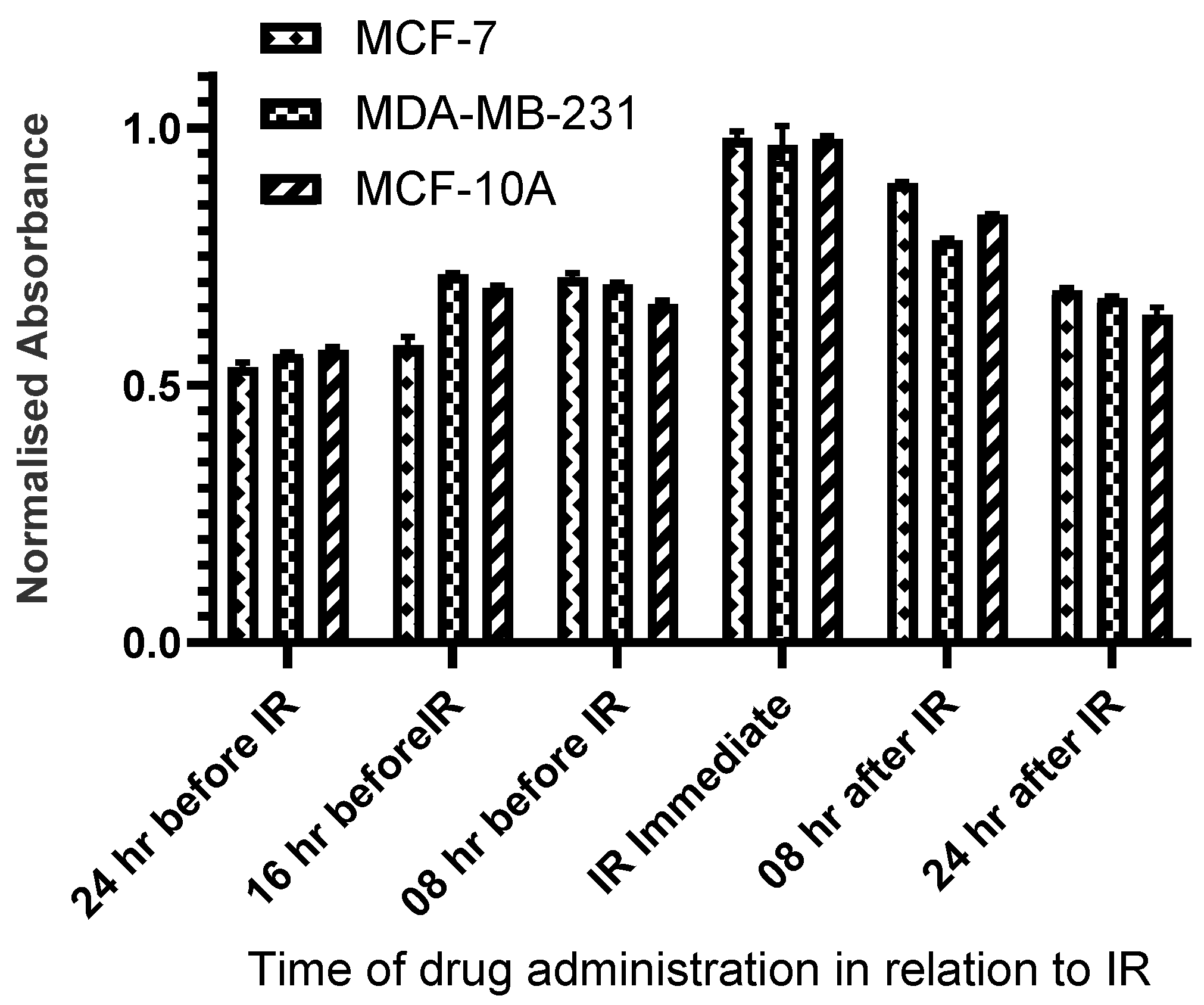
Figure 2.
CUDC-101 sensitises MCF-7 (a) and (b), MDA-MB-231 (c) and (d) and MCF-10A (e) and (f) cells to proton irradiation. Data is expressed as the mean ± SD of three independent experiments.
Figure 2.
CUDC-101 sensitises MCF-7 (a) and (b), MDA-MB-231 (c) and (d) and MCF-10A (e) and (f) cells to proton irradiation. Data is expressed as the mean ± SD of three independent experiments.
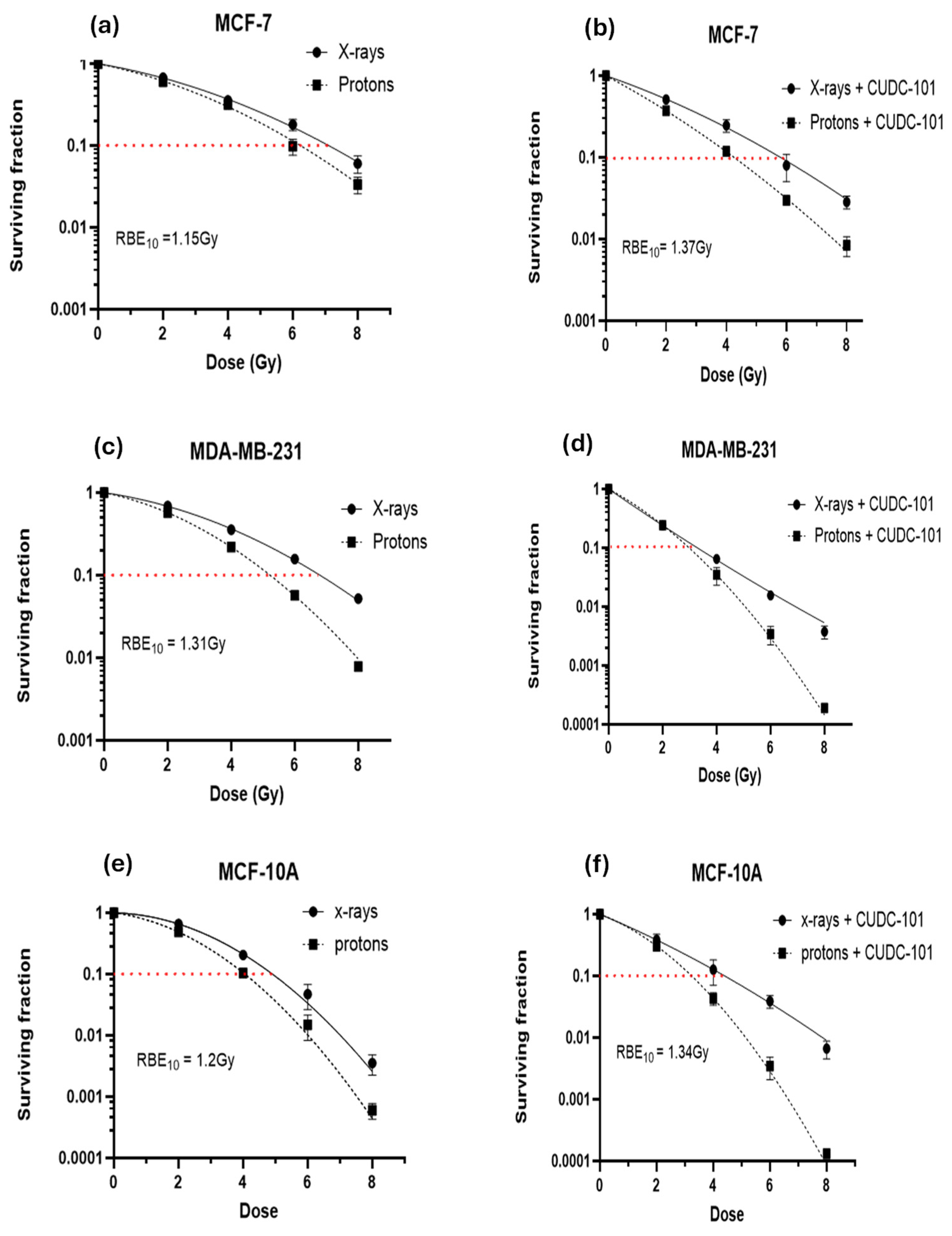
Figure 3.
Effect of CUDC-101 combined with protons or X-rays in MCF-7 (a-b), MDA-MB-231 (c-d) and MCF-10A (e-f) cell lines. Histograms show the mean ± SD of three independent experiments (n=3) **p = 0.0037 in MDA-MB-231 and **p = 0.0088 in MCF-10A cell lines. Images are obtained at 40X magnification using a Metafer 4 scanning system. Analysis was done using unpaired two-tailed Student’s t test, **p (<0.002).
Figure 3.
Effect of CUDC-101 combined with protons or X-rays in MCF-7 (a-b), MDA-MB-231 (c-d) and MCF-10A (e-f) cell lines. Histograms show the mean ± SD of three independent experiments (n=3) **p = 0.0037 in MDA-MB-231 and **p = 0.0088 in MCF-10A cell lines. Images are obtained at 40X magnification using a Metafer 4 scanning system. Analysis was done using unpaired two-tailed Student’s t test, **p (<0.002).
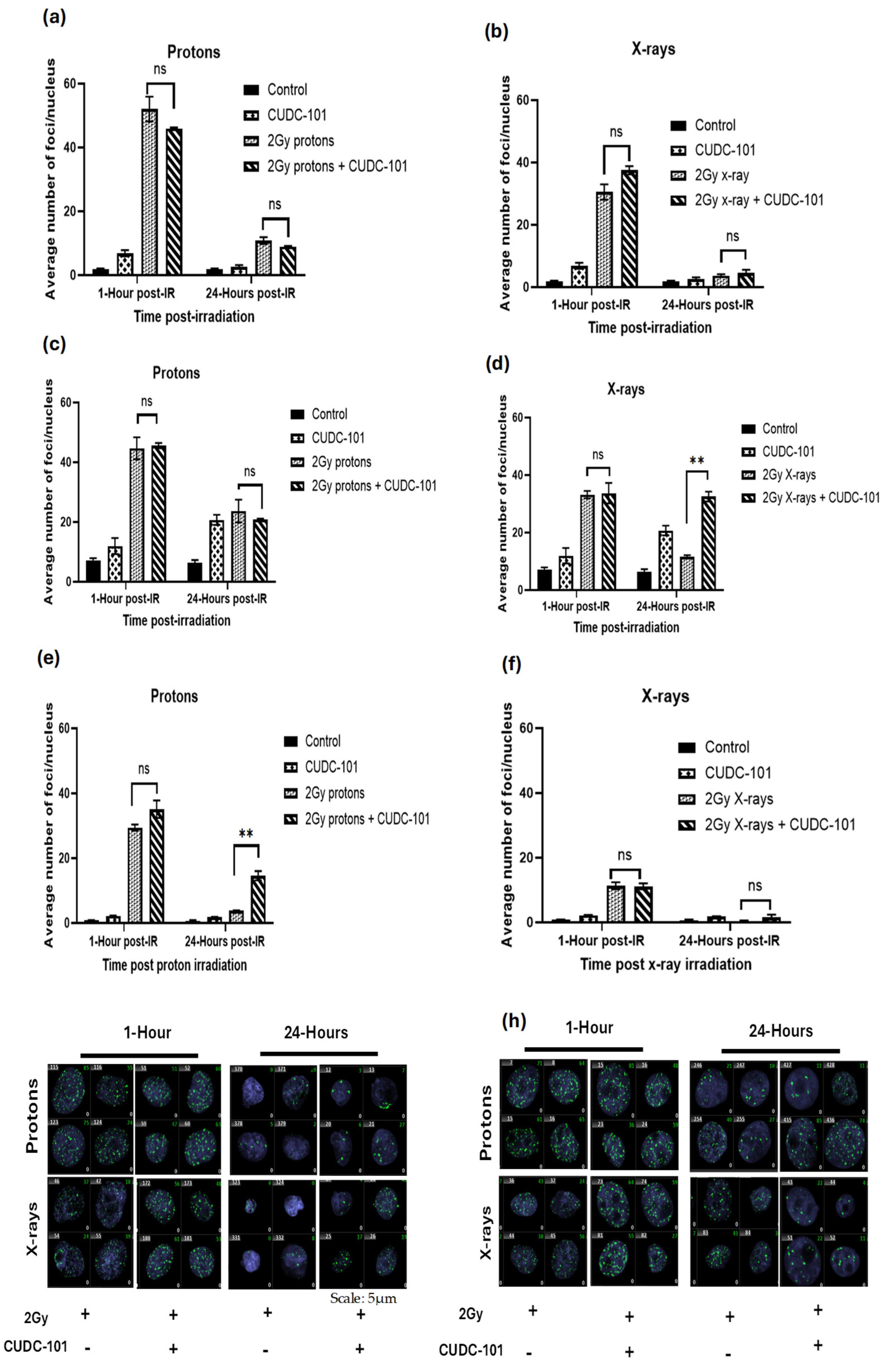
Figure 4.
Induction of apoptosis and necrosis at 48 hours post treatment with CUDC-101 combined with protons or X-rays in MCF-7 (a-b), MDA-MB-231 (c-d) and MCF-10A (e-f) cell lines. Induction of apoptosis and necrosis at 24 hours after treatment with 1µM staurosporine in the three cell lines (f). Histograms show the mean ± SD of three independent experiments (n=3). Comparisons were conducted using unpaired two-tailed Student t test, ***p (<0.0003) **p (<0.002), *p (<0.05).
Figure 4.
Induction of apoptosis and necrosis at 48 hours post treatment with CUDC-101 combined with protons or X-rays in MCF-7 (a-b), MDA-MB-231 (c-d) and MCF-10A (e-f) cell lines. Induction of apoptosis and necrosis at 24 hours after treatment with 1µM staurosporine in the three cell lines (f). Histograms show the mean ± SD of three independent experiments (n=3). Comparisons were conducted using unpaired two-tailed Student t test, ***p (<0.0003) **p (<0.002), *p (<0.05).
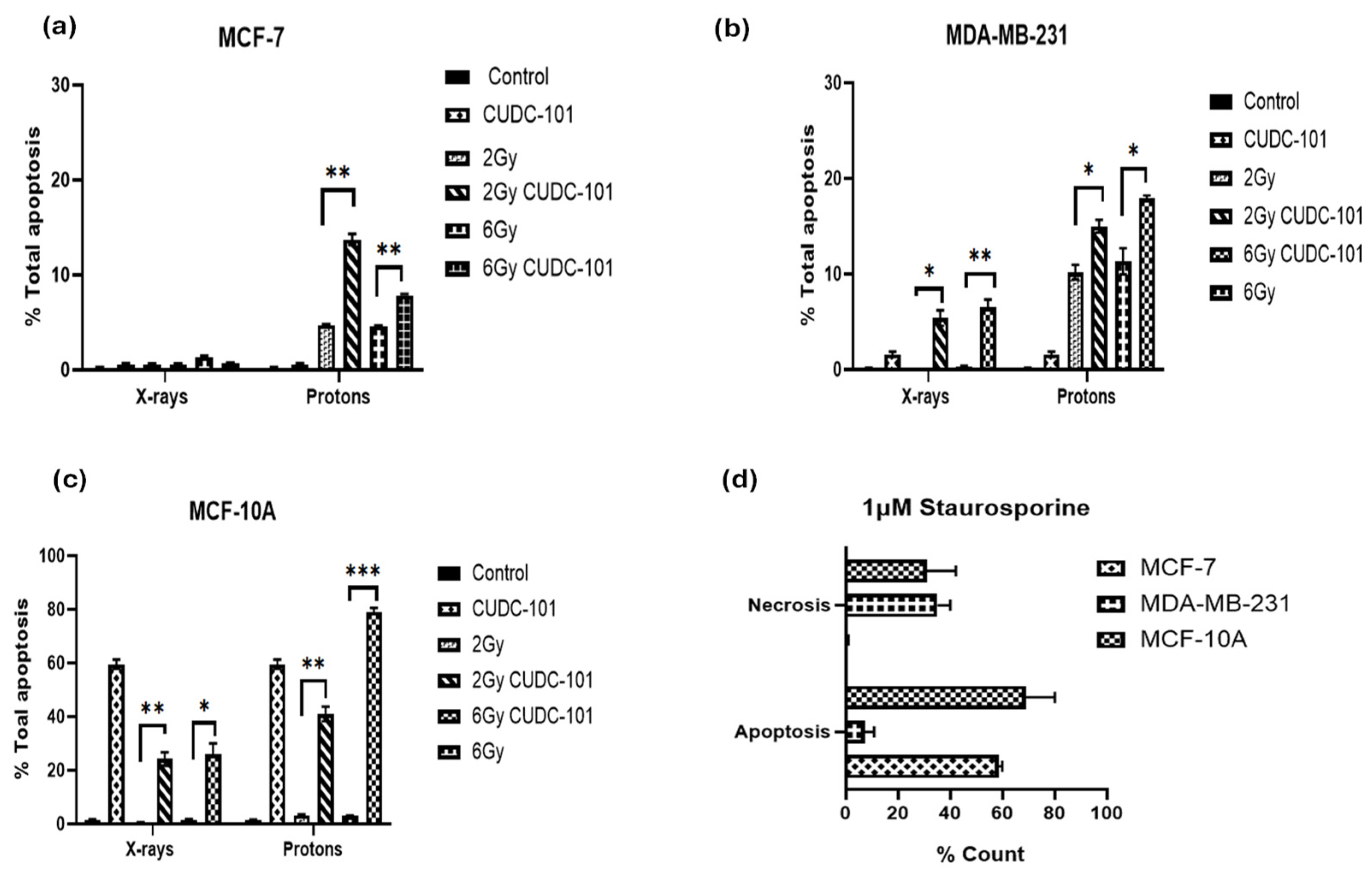
Figure 5.
Induction of necrosis at 48 hours post treatment with CUDC-101 combined with protons or X-rays in MCF-7 (a-b), MDA-MB-231 (c-d) and MCF-10A (e-f) cell lines. Increased amounts of necrosis after treatment with 2Gy protons and 2Gy protons and CUDC-101 in MCF-7 cell line compared to MDA-MB-231 cell line(g). Histograms show the mean ± SD of three independent experiments (n=3). Comparisons were conducted using unpaired two-tailed Student t test, ***p (<0.0003) **p (<0.002), *p (<0.05).2.4. Effect of CUDC-101 and radiation on cell cycle progression.
Figure 5.
Induction of necrosis at 48 hours post treatment with CUDC-101 combined with protons or X-rays in MCF-7 (a-b), MDA-MB-231 (c-d) and MCF-10A (e-f) cell lines. Increased amounts of necrosis after treatment with 2Gy protons and 2Gy protons and CUDC-101 in MCF-7 cell line compared to MDA-MB-231 cell line(g). Histograms show the mean ± SD of three independent experiments (n=3). Comparisons were conducted using unpaired two-tailed Student t test, ***p (<0.0003) **p (<0.002), *p (<0.05).2.4. Effect of CUDC-101 and radiation on cell cycle progression.
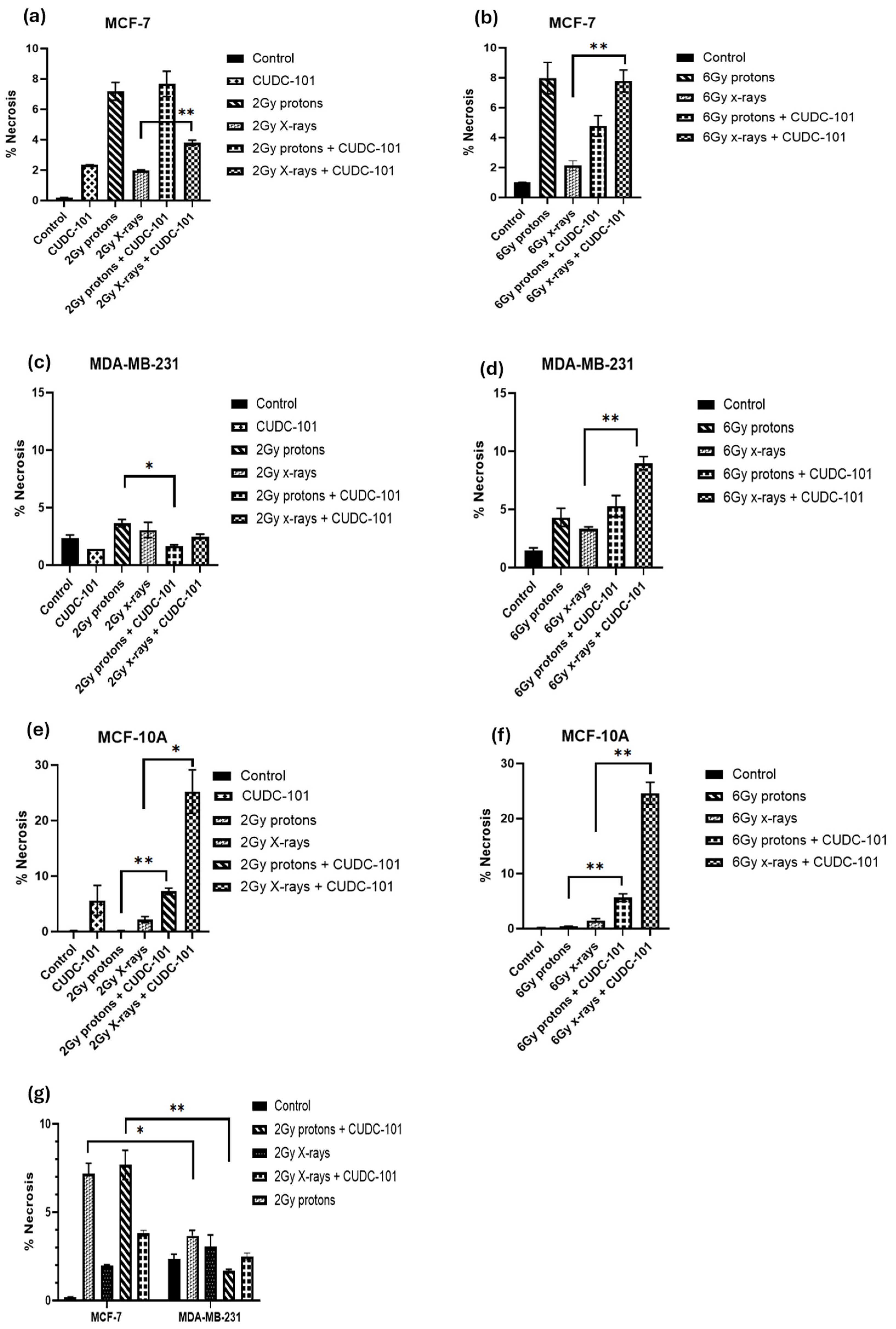
Figure 6.
Quantification of the effect of CUDC-101 alone and in combination with X-rays and protons on cell cycle progression in MCF-7 (a-b) MDA-MB-231 (c-d); and MCF-10A (e-f) cell lines. Data represents the mean ± SD of three independent experiments (n=3). Comparisons were conducted using two-tailed Student t test, **p < 0.008.
Figure 6.
Quantification of the effect of CUDC-101 alone and in combination with X-rays and protons on cell cycle progression in MCF-7 (a-b) MDA-MB-231 (c-d); and MCF-10A (e-f) cell lines. Data represents the mean ± SD of three independent experiments (n=3). Comparisons were conducted using two-tailed Student t test, **p < 0.008.
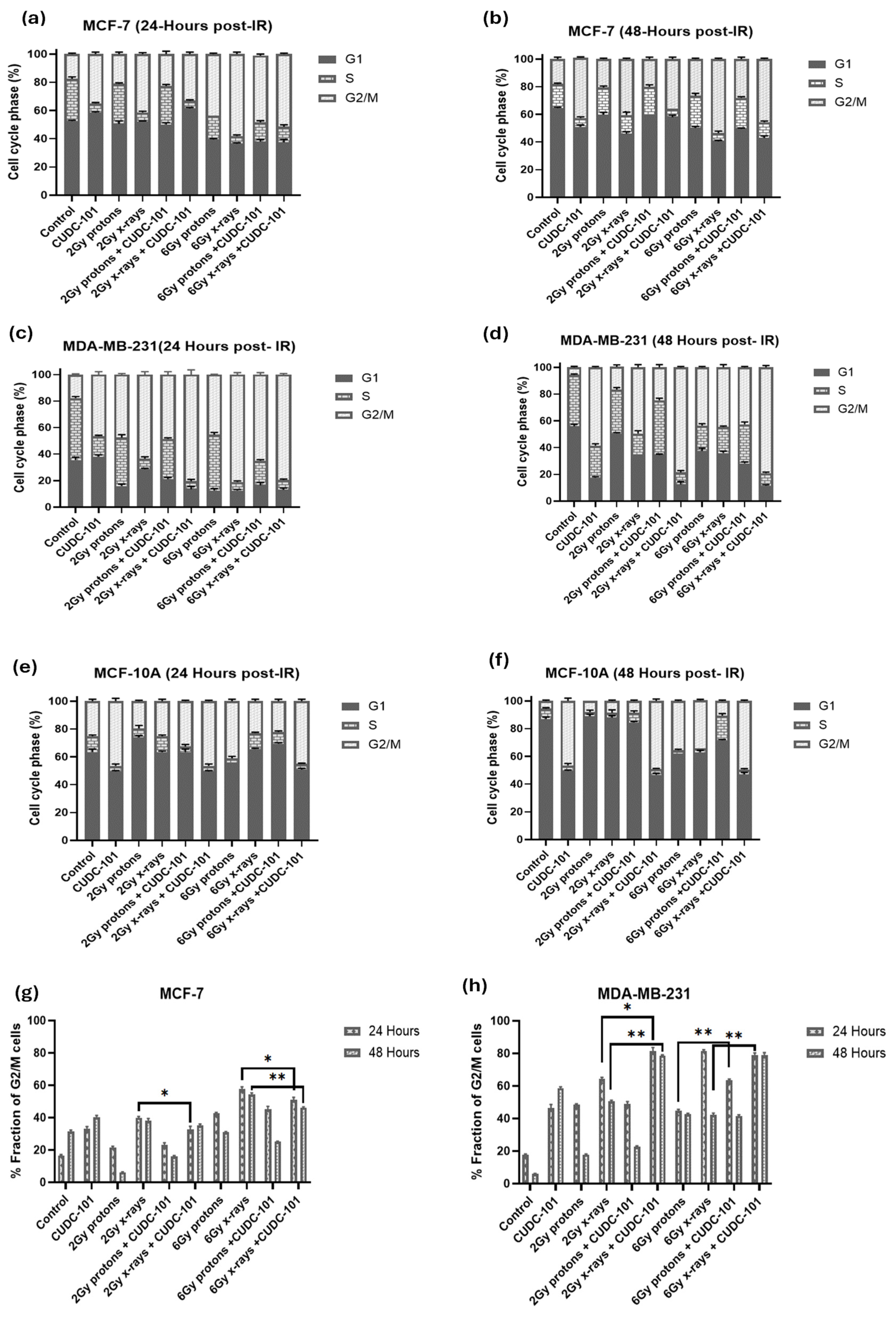

Table 1.
Half maximal Inhibitory Concentration (IC50) values for the different cell lines.
| Cell line | CUDC-101 (µM) |
|---|---|
| MCF-7 | 0.31 |
| MDA-MB-231 | 0.60 |
| MCF-10A | 2.7 |
Table 2.
Radiation response parameters of MCF-7, MDA-MB-231 and MCF-10A cell lines.
| Cell line | RBE10 | HDACi-mediated RBE | SER10 (Protons) |
SER10 (X-rays) |
|---|---|---|---|---|
| MCF-7 | 1.15 ± 0.03 | 1.37 ± 0.004 | 1.50± 0.01 | 1.16± 0.03 |
| MDA-MB-231 | 1.31± 0.01 | - | 1.77± 0.03 | 2.09± 0.02 |
| MCF-10A | 1.20 ±0.01 | 1.34 ± 0.02 | 1.23± 0.04 | 1.10± 0.01 |
Data represents the mean ± SD. RBE: Relative biologic effectiveness; SER: the sensitisation enhancement ratio.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



