
Submitted:
07 October 2024
Posted:
08 October 2024
You are already at the latest version
Abstract
Tear fluid is emerging as a valuable resource for biomarker discovery, but its low sample volume and dynamic composition pose significant challenges for analytical testing. Most tear proteomics studies have focused on samples collected with Schirmer strips, while capillary tube collection has received less attention. To address these challenges, we developed a novel in-capillary trypsin digestion workflow that requires as little as 0.5 μL of tear fluid for bottom-up shotgun proteomics. This method uses liquid chromatography-tandem mass spectrometry (LC-MS/MS) to identify a higher number of proteins with enhanced efficiency compared to previously reported methods, which typically required 5-10 μL of pooled tear fluid. Using the same microcentrifuge tube for both tear collection and sample processing, this streamlined workflow simplifies sample handling and minimizes both sample loss and experimental errors associated with sample transfer. This method also efficiently reduced sample processing time to under 2 hours prior to overnight trypsin digestion, compared to the 5-8 hours required by other methods. With this workflow, we identified 500-800 proteins per 0.5 μL sample without the need for fractionation, allowing for at least three technical replicates. This optimized workflow greatly enhances the efficiency of tear proteomics for biomarker discovery.

Keywords:
tear proteomics
; tear biomarker
; biomarker discovery
; point-of-care testing
; POCT
; LC-MS/MS
1. Introduction
Tear fluid holds great promise as a non-invasive source of biomarkers for both ophthalmic and systemic diseases [1,2,3,4,5,6,7,8,9,10]. Unlike other biofluids, such as blood and cerebrospinal fluid, tear fluid collection is non-invasive and causes little discomfort, making it ideal for real-time, point-of-care testing (POCT) [11]. However, the small volume of tear fluid available and the dynamic compositional changes, often trigged by stimulation of reflex tearing from irritation during collection, pose challenges for tear proteomics and biomarker discovery. Current collection methods, such as Schirmer strips and sponges, usually require anesthetic drops and lengthy collection times, which can alter tear composition, trigger reflex tearing, and introduce eyelid cellular debris contamination, complicating the interpretation of proteomic results [3,11,12,13,14,15,16]. On the other hand, capillary tubes offer a superior method for collecting “cleaner and purer” tear samples by significantly reducing reflex tearing, minimizing cellular contamination, and enhancing patient comfort [11,13,17,18]. Notably, 0.5 μL of tear fluid can usually be collected using a single capillary tube in under 1 minute, thereby avoiding the eye irritation and reflex tearing that occur with large volumes collected over longer periods or pooled tear samples from consecutive collections. To date, methods for processing and analyzing 0.5 μL tear samples collected using capillary tubes have not been reported. Even when pooling larger tear volumes, previous studies using capillary tubes reported lower protein numbers compared to other methods [19,20,21]. However, these results may reflect limitations in processing techniques rather than actual protein content. In addition, obtaining sufficient tear volumes from individuals with dry eye disease or reduce tear production remains a challenge, limiting the broader application of tear proteomics in clinical settings [2,14,15,16,22,23,24,25,26] .
To address these limitations, we have developed a rapid and efficient in-capillary digestion workflow tailored specifically for tear fluid proteomics. This novel method requires only 0.5 μL of tear fluid and eliminates sample transfer between collection, processing, and analysis, thereby significantly reducing the risk of sample loss, contamination, and associated errors. Using LC-MS/MS, this new method simplifies experimental procedures, shortens the sample preparation and handling time and consistently identifies a higher number of proteins in tear fluid samples compared to previously reported approaches. By streamlining this tear proteomics workflow, our method enhances the feasibility of using tear fluid for proteomics research and biomarker discovery, opening new avenues for real-time POCT, disease detection and management, and personalized medicine.
2. Results
2.1. Development of a Novel In-Capillary Digestion Workflow for Enhanced Tear Proteome in Biomarker Discovery
We developed a novel proteomic strategy specifically tailored for tear film fluids in small volume capillaries to enhance LC-MS/MS protein identification (Figure 1). Tear samples were collected using 0.5 μL capillary tubes and transported to the laboratory on
By optimizing these sample handling and processing procedures, we were able to streamline the entire workflow, reducing the total sample preparation time (excluding the overnight trypsin digestion) to under 2 hours, a significant improvement over conventional protocols, which typically require 5-8 hours [10,14,16,21,27]. Notably, this streamlined workflow eliminated the need for peptide fractionation and desalting, steps that are usually time-consuming and prone to sample loss. These optimizations translated into more rapid and reproducible analyses, ultimately improving the sensitivity and efficiency of tear proteomics for biomarker discovery in research and clinical applications.
2.2. Optimization of Protein Quantities and LC Gradients for LC-MS/MS Analysis
To determine the optimal amount of tear peptides for loading onto an Evosep tip for LC-MS/MS analysis, we prepared peptide solutions from pooled tear samples and tested three different peptide amounts—200 ng, 500 ng and 1,000 ng—using LC gradients of 21-, 44-, and 88-minitues (Figure 2). Each experiment was repeated in triplicate. The average number of proteins identified from each experiment ranged from 171 to 615, with the highest number (615 proteins) observed using a 500 ng loading mass and an 88-min gradient (Figure 2 & Table S1). Based on these results, we selected 500 ng peptides with the 88-minute gradient as the optimal condition for inclusion in our tear proteomics method.
2.3. Urea Volume Optimization
In our initial workflow, 50 µL of 8 M urea was initially added to tear samples to denature proteins prior to digestion. However, urea must be removed during later stages of sample processing before LC-MS/MS analysis. To streamline the tear proteomics workflow, we opted to remove urea by using an Evosep C18 tip washing step, avoiding additional desalting steps. An insufficient urea volume may prevent thorough mixing with the tear fluid, while an excessive amount could lead to incomplete desalting during the Evosep C18 tip washing step, which can negatively impact LC-MS/MS performance and reduce the number of identified proteins. To address this, we set out to determine the optimal volume of 8 M urea for our workflow. We used four capillary tubes, each containing 0.5 µL of pooled tear sample, and tested four different volumes of 8 M urea: 5, 10, 25, and 50 µL, following the procedures outlined in Figure 1. The average number of tear proteins identified for each condition was 547, 816, 766, and 564, respectively (Figure 3 & Table S3). The results showed that 10 μL of 8 M urea yielded the highest number of protein identifications, making it the optimal volume for this workflow.
2.4. Protein Concentration Measurement before DTT Reduction and IAA Alkylation
Accurate protein quantification is essential for translational and clinical quantitative proteomics, as well as biomarker discovery. Due to the small volume of tear samples, direct protein concentration measurements on the original tear samples are not feasible. Instead, these measurements should be performed during sample processing, once sufficient volume is achieved after buffer addition. However, certain reagents used in biomedical sample processing can interfere with protein measurements, especially when measurement times vary due to differing sample quantities in each experiment [28,29]. There are several steps in the workflow where the sample volume is sufficiently large for protein concentration measurement. These steps include: (i) after denaturation when the urea concentration is diluted from 8 M to 1.6 M, (ii) after addition of DTT for reduction, and (iii) after addition of IAA for alkylation (Figure 1b). To determine the optimal step for protein measurements, we prepared solutions corresponding to these three steps and evaluated tear protein concentration measurements under different reagent conditions. Measurements were taken at 0, 5, 15, 30, 60, 90, and 120 minutes after mixing 0.5 μL of pooled and aliquoted tear samples mixed with the respective buffer solutions (Figure S1a & 1b). We used A280 absorbance measurements on a NanoDrop spectrophotometer, which requires minimal sample volumes for protein quantification. Our results showed that tear protein concentration measurement remained stable even after 2 hours at room temperature in the presence of 1.6 M urea. However, the addition of DTT and IAA significantly altered the A280 absorbance of the buffer solutions, which impacted the tear sample measurements. In addition, A280 absorbance changed with the longer incubation times in the presence of DTT and IAA (Figure S1a & 1b). These findings suggest that the most suitable step for A280 protein measurement is after the denaturation step, when the urea concentration has been diluted from 8 M to 1.6 M, but before the addition of DTT and IAA (Figure 1a & S2). This step provides the most reliable and stable measurements with minimal reagent interference, regardless of the timing of sample preparation. Using this method, we measured the protein concentrations of 22 tear samples, yielding an average protein concentration of 10.3 ± 2.9 µg/µL (Table S2).
Considering that the protein concentration in some tear samples may be less than 10 µg/µL—as indicated by the average concentration of 10.3 ± 2.9 µg/µL obtained above—this could result in A280 absorbance readings below 0.1 on a NanoDrop spectrophotometer. To address this, we further optimized the workflow. Initially, 20 µL of 50 mM ammonium bicarbonate buffer was added to the tear sample containing 10 µL of urea to allow for more accurate protein measurement. The protein concentration measurement was then performed in the presence of 2.67 M urea (Figure S1c&1d). After the protein concentration was measured, an additional 20 µL of 50 mM ammonium bicarbonate was added to reduce the urea concentration to 1.6 M before proceeding with DTT reduction and IAA alkylation (Figure S2).
2.5. Tear Proteome and Tear Protein Functions
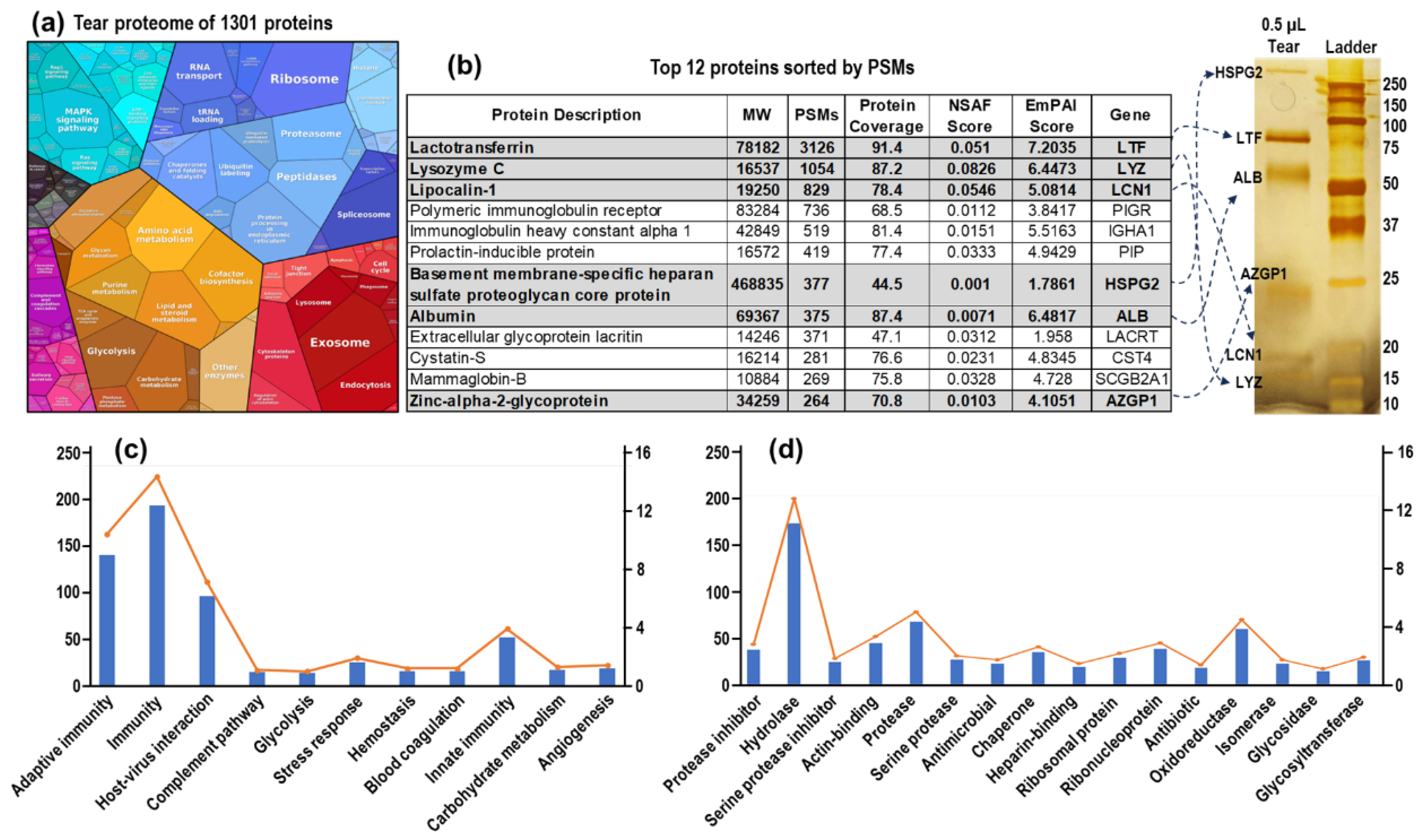
In this study, we collected 39 LC-MS/MS datasets from tear samples: 27 from amount and gradient optimization experiments and 12 from tests involving different volume of urea. By integrating these datasets, we identified a total of 1,301 proteins in tear fluid (Figure 4a & Table S4). To gain deeper insight into the tear proteome and its dynamic range, we performed SDS-PAGE analysis using 0.5 µL of tear sample, followed by silver staining. The visible bands were excised, subjected to in-gel digestion, and analyzed by LC-MS/MS (Figure 4b). We compared these results with the top 12 proteins from a representative dataset obtained using our in-capillary tear proteomics workflow. All visible bands from the silver-stained SDS-PAGE gel corresponded to the top 12 proteins, which were among the 825 total identified proteins identified that dataset, ranked by PSMs. Additionally, we conducted Gene Ontology (GO) analysis to explore the biological processes and molecular functions of the 1,301 identified tear proteins (Figure 4c, 4d & & Table S5). GO biological process analysis indicated that tear proteins are involved in immunity, host-virus interaction, complementary pathway, and angiogenesis (Figure 4c). GO molecular function analysis revealed that the tear proteome includes protease inhibitors, hydrolases, proteases, antimicrobial and antibiotic proteins, chaperones, and glycosidases (Figure 4d). Further proteomics and bioinformatics analysis of tear fluids will not only enhance our understanding of tear fluid pathophysiology but also aid in identifying biomarkers for both eye and system diseases.
3. Discussion and Conclusions
The study developed a rapid and efficient in-capillary digestion workflow for tear proteomics that overcomes the challenges associated with small sample volumes and complex tear fluid composition. By utilizing only 0.5 µL of tear fluid, our method streamlines sample handling, minimizes sample loss and data errors, and enhances protein identification without the need for extensive desalting or fractionation. Coupled with LC-MS/MS analysis, this streamlined workflow enables high-throughput tear proteomics investigations and biomarker discovery with improved sensitivity and efficiency. This innovative approach unlocks the potential for tear fluid as a non-invasive source of biomarker source, paving the way for advancements in POCT diagnostics and personalized medicine in both ophthalmic and systemic diseases. Moving forward, our focus is on further improving this approach by incorporating Parallel Reaction Monitoring (PRM)-based targeted mass spectrometry technique to achieve a more precise quantitation of a number of potential biomarkers in tear fluid samples.
4. Materials and Methods
4.1. Tear Fluid Sample Collection and Processing
The study was approved by Advarra IRB (number Pro00072069), and written informed consent was obtained from all individuals who participated. Tear samples were collected from volunteers using capillary method [2,17,30,31]. Drummond™ Short-Length Microcaps™ Micropipets (Fisher Scientific, Catalog No.22-249512), were used to collect 0.5 µL of tear film fluid from each volunteer. The capillaries containing tear samples were put into 1.5 mL Protein LoBind microcentrifuge tubes and frozen in a -20 °C freezer for short-time storage (several hours to overnight) in the eye clinics. The tear samples were transported to the research laboratory on dry ice and then stored in an -80 °C freezer before processing.
4.2. Initial Workflow for In-Capillary Digestion of Tear Fluid Samples
For pooled sample preparation, multiple capillaries containing 0.5 μL of tear film samples were combined in a single Protein LoBind microcentrifuge tube and centrifuged at 3,000 x g for 1 minute. The tear samples were then vortexed thoroughly to ensure a uniform mixture before aliquoting using capillaries with 0.5 μL of tear film sample. Each capillary was placed in a 1.5 mL Protein LoBind microcentrifuge tube, and 49.5 μL of 8M urea in 50 mM ammonium bicarbonate (Millipore Sigma, Catalog No. 09830-500G) was added to each tube. The microcentrifuge tubes were vortexed vigorously and then centrifuge in the benchtop microcentrifuge at ~20,000 x g for 5 minutes to break the capillary tube into small pieces and thoroughly mix the tear fluid samples with the urea solution. This procedure was repeated three times. The samples were then incubated at room temperature for 30 minutes to promote protein denaturation. Following this, the samples were diluted fivefold to reduce the urea concentration to 1.6 M prior to protein quantification. Protein concentrations of tear samples were measured by A280 measurements collected on a NanoDrop One Microvolume UV-Vis Spectrophotometer using 1.6 M urea in 50 mM ammonium bicarbonate as blank.1 μL of 500 μM DL-Dithiothreitol (DTT) (Millipore Sigma, Catalog No. 3860-5GM) was then added to the above solution to make a final DTT concentration of 10 mM. The samples were incubated at 56 °C for 30 minutes for reduction. After reduction, iodoacetamide (IAA) (Millipore Sigma, Catalog No. I6125-5G) was added to the sample to a final concentration of 25 mM and incubated for 30 min at room temperature in the dark. Trypsin/Lys-C Mix, Mass Spec Grade (Promega, Catalog No. V5071) was added to the tear samples at a 1:20 protease-to-protein ratio. The samples were then digested for 16 hours at 37°C.
4.3. Optimization of Protein Quantities and LC Gradients for LC-MS/MS Analysis
After digestion, the enzyme reaction was quenched by adding 3 µL of 50% formic acid, lowering the pH to 3–4. The tryptic peptide samples were then prepared in amounts of 200 ng, 500 ng, and 1,000 ng, based on the protein concentrations measured in the earlier steps. The peptide samples were loaded onto Evosep tips (EV2011) following the manufacturer’s instructions prior to LC-MS/MS analysis. LC gradients of 21, 44, and 88 minutes were used for peptide separation.
4.4. Optimization of 8M Urea Volume for Complete Trypsin Digestion of Tear Proteins and Maximizing Protein Identifications
Pooled tear samples were aliquoted into capillary tubes, with 0.5 μL of tear film sample in each, and placed into 1.5 mL Protein LoBind microcentrifuge tubes. To each microcentrifuge tube, 10 μL of 8M urea in 50 mM ammonium bicarbonate was added. To ensure thorough mixing of the small volume of tear samples with the urea solution, the microcentrifuge tubes were vortexed vigorously and then centrifuged in a benchtop microcentrifuge at ~20,000 x g for 5 minutes to break the capillary tubes into small pieces. This procedure was repeated three times. The samples were then incubated at room temperature for 30 minutes to promote protein denaturation. Following this, 20 μL of 50 mM ammonium bicarbonate was added to dilute the urea concentration to 2.67 M and bring the total volume to 30 μL. Protein concentrations of the tear samples were measured using A280 on a NanoDrop One Microvolume UV-Vis Spectrophotometer, with the 2.67 M urea solution as the blank. An additional 20 μL of 50 mM ammonium bicarbonate was added, followed by 1 μL of 500 μM DTT (Millipore Sigma, Catalog No. 3860-5GM) to achieve a final DTT concentration of 10 mM. The samples were incubated at 56 °C for 30 minutes for reduction. After reduction, IAA (Millipore Sigma, Catalog No. I6125-5G) was added to the samples to a final concentration of 25 mM, and they were incubated for 30 minutes at room temperature in the dark. Finally, Trypsin/Lys-C Mix, Mass Spec Grade (Promega, Catalog No. V5071), was added to the tear samples at a 1:20 protease-to-protein ratio, and the samples were digested for 16 hours at 37 °C.
4.5. Test the Effect of DTT and IAA on Tear Protein Concentration Measurements
Pooled tear samples were aliquoted into capillary tubes, each containing 0.5 µL of tear fluid, and placed in individual Protein LoBind microcentrifuge tubes. To each tube, 10 µL of 8 M urea solution was added, and the samples were incubated at room temperature for 30 minutes to denature the proteins. Following this, either 40 µL or 20 µL of 50 mM ammonium bicarbonate solution was added to dilute the urea concentration. DTT and IAA were then added to the tubes as follows: S1 (no DTT or IAA), S2 (10 mM DTT), and S3 (10 mM DTT followed by 25 mM IAA). The volumes in all tubes were adjusted to be equal using 50 mM ammonium bicarbonate solution. Corresponding buffer controls (B1, B2, and B3) were prepared without tear proteins to match the experimental conditions of the tear samples. The tear protein and buffer solutions were incubated at room temperature, and A280 absorbance was measured at 0, 5, 15, 30, 45, 60, 90, and 120 minutes using a NanoDrop spectrophotometer.
4.6. LC-MS/MS Analysis
The MS data were collected with a timsTOF Pro 2 mass spectrometer coupled with an Evosep One LC system connected to an 8 cm Evosep performance column (EV-1109) using the 60 samples-per-day extended gradient. The tear peptide samples were analyzed with a standard data-dependent acquisition method with parallel accumulation-serial fragmentation (DDA-PASEF) with the instrument operating in the positive mode. A full scan was first collected with the mass range of 100-1700 m/z and the TIMS 1/k0 range of 0.60-1.60 V·s/cm2. During a full 1.17s cycle, 10 PASEF ramps were performed with the ramp and accumulation time both set to 100 ms. The following parameters were applied for all DDA-PASEF HCD data collection: precursor intensity threshold: 2.5E3; charge state: +1 to +5; dynamic exclusion: 0.4 min; target intensity for fragmentation: 2E4; isolation window (linear): 2 m/z at 700 m/z and 3 m/z at 800 m/z; collision energy (linear): 20 eV at1/k0 of 0.60 V·s/cm2 and 59 eV at 1/k0 of 1.60 V·s/cm2.
4.7. LC-MS/MS Database Search
All DDA data were searched against the Uniprot reviewed human protein database (downloaded 3/30/2023) with BPS (Bruker ProteoScape) (ver. 2024). The trypsin activity was set to be fully specific with one missed cleavage site allowed. A maximum of two of the following variable modifications were allowed on each peptide: C [+57.021464], STY [+79.966331], and M [+15.994915]. The precursor and the fragment mass tolerances were set to ± 10 ppm and ± 20 ppm respectively. For protein identification, at least one associated peptide was identified within ± 5 ppm of its theoretical m/z value. The Xcorr score cutoff at 1 was applied to +1 peptides, whereas for peptides with a higher charge state, the Xcorr score cutoff was set at 0.8, with deltaCN cutoff set at 0.1 for all peptides. Additionally, only peptides with spectral evidence on at least 40% of the fragments (to aim for ≥ 80% sequence coverage) were considered for protein statistical calculations.
4.8. Silver Staining Analysis of Tear Samples
0.5 μL of tear film sample was mixed with 10 μL of 2× SDS-PAGE sample buffer and separated by SDS-PAGE electrophoresis. The gel was fixed in 10% methanol and 10% acetic acid for 30 minutes or overnight. The gel was then washed four times in water, with each wash lasting at least 5 minutes, taking care not to over wash. Next, the gels were incubated in sodium thiosulfate (1 or 2 pellets, approximately 400 mg, per 500 mL of water) for exactly 90 seconds. Reserve 20 mL of this sodium thiosulfate solution for the developing solution. After incubation, the gels were washed quickly three times with water (fill the dish with water and then dump it out at the sink). The gels were then immersed in silver nitrate solution (0.9 g of silver nitrate in 500 mL of water) and allowed to stain for 10 minutes, until they turned slightly yellow. The silver nitrate solution can be recovered and reused. After staining, the gels were washed quickly three times with water (again, fill the dish with water and dump it out). Finally, the developer solution was prepared by mixing 10 g of potassium carbonate, 20 mL of the sodium thiosulfate solution from the previous step, and 250 µL of 40% formaldehyde in 500 mL of water. The reaction was stopped by adding destaining solution (10% methanol and 5% acetic acid). The gels were washed in water 2–3 times and left in water until further analysis.
4.9. In-Gel Trypsin Digest from Silver-Stained Gels
Pooled tear samples were prepared by excising bands from silver-stained gels, which were first washed twice in ddH2O for 15 minutes. Bands of interest were cut out using a clean razor blade, chopped into 5–6 small pieces, and transferred to 1.5 mL Protein LoBind microcentrifuge tubes containing 100 µL of HPLC-grade water. After a 10-minute incubation at room temperature with gentle vortexing, the gel pieces were destained using destaining solution (0.4g potassium ferricyanide (K3Fe(CN)6) in 200 ml sodium thiosulphate (0.2g/L Na2S2O3•5H2O) until no bands were visible, then washed 4–5 times with Milli-Q H2O until transparent. The gel pieces were equilibrated in 100 µL of 50 mM ammonium bicarbonate buffer for 20 minutes, followed by two washes with 100 µL of 25 mM ammonium bicarbonate in 50% acetonitrile, each for 10 minutes, and a final wash in 100% acetonitrile for 10 minutes. The dehydrated gel slices were dried in a speed-vac for 5 minutes. For reduction and alkylation, the slices were covered with 10 mM DTT and incubated at room temperature for 30 minutes, then treated with an equal volume of 55 mM iodoacetamide for 45 minutes in the dark. The gel slices were washed with 25 mM ammonium bicarbonate for 10 minutes, dehydrated again with acetonitrile, and dried. Trypsin/Lys-C (20 ng/µL in 25 mM ammonium bicarbonate) was added to cover the gel slices (~30 µL), and the samples were incubated at 4 °C for 10–15 minutes before being covered with 25–30 µL of 25 mM ammonium bicarbonate for overnight digestion at 37 °C. To extract the tryptic peptides, 50 µL of acetonitrile was added, followed by 10 minutes of shaking at room temperature. The solution was transferred to a clean tube, and the gel slices were rehydrated with 30 µL HPLC-grade water for 10 minutes, followed by another extraction with 50 µL acetonitrile. The extracted solutions were combined, frozen at –80 °C, and lyophilized. The samples were cleaned using StageTips, reconstituted in 20 µL of Solution A (0.1% formic acid in HPLC water), and loaded onto Evosep tips for subsequent LC-MS/MS analysis.
4.10. Gene Ontology (GO) Analysis
Gene Ontology (GO) analysis of the 1,301 tear proteins was performed using The Database for Annotation, Visualization and Integrated Discovery (DAVID) Bioinformatics software (https://david.ncifcrf.gov/) [32]. Only biological process and molecular function entries with P-Value < 0.05 and % > 1% were listed and plotted. The Proteomap was generated using Proteomaps software (https://www.proteomaps.net/).
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1-S2; Table S1-S5.
Author Contributions
J.X. designed and conducted the experiments, and analyzed the data. J.X., J.K., and L.T.L. wrote the manuscript. K.F. performed the silver staining, and K.C.G. assisted with data analysis. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was approved by Advarra IRB (number Pro00072069).
Informed Consent Statement
Written informed consent has been obtained from all individuals who participated to publish this paper.
Data Availability Statement
The datasets generated during and/or analyzed during the current
study are available from the corresponding author upon reasonable request.
Acknowledgments
We thank Jackie Sikora of Sewickley Eye Group for assistance with tear sample collection.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Koduri, M.A.; Prasad, D.; Pingali, T.; Singh, V.K.; Shanbhag, S.S.; Basu, S.; Singh, V. Optimization and evaluation of tear protein elution from Schirmer’s strips in dry eye disease. Indian J Ophthalmol 2023, 71, 1413–1419. [Google Scholar] [CrossRef] [PubMed]
- Nattinen, J.; Aapola, U.; Jylha, A.; Vaajanen, A.; Uusitalo, H. Comparison of Capillary and Schirmer Strip Tear Fluid Sampling Methods Using SWATH-MS Proteomics Approach. Transl Vis Sci Technol 2020, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Vergouwen, D.P.C.; Schotting, A.J.; Endermann, T.; van de Werken, H.J.G.; Grashof, D.G.B.; Arumugam, S.; Nuijts, R.; Ten Berge, J.C.; Rothova, A.; Schreurs, M.W.J.; et al. Evaluation of pre-processing methods for tear fluid proteomics using proximity extension assays. Sci Rep 2023, 13, 4433. [Google Scholar] [CrossRef]
- Gijs, M.; Arumugam, S.; van de Sande, N.; Webers, C.A.B.; Sethu, S.; Ghosh, A.; Shetty, R.; Vehof, J.; Nuijts, R. Pre-analytical sample handling effects on tear fluid protein levels. Sci Rep 2023, 13, 1317. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Du, C.X.; Pan, X.D. The use of in-strip digestion for fast proteomic analysis on tear fluid from dry eye patients. PLoS One 2018, 13, e0200702. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Rao, J.; Zheng, Z.; Yu, Y.; Lou, S.; Liu, L.; He, Q.; Wu, L.; Sun, X. Integrated Tear Proteome and Metabolome Reveal Panels of Inflammatory-Related Molecules via Key Regulatory Pathways in Dry Eye Syndrome. J Proteome Res 2019, 18, 2321–2330. [Google Scholar] [CrossRef]
- Ananthi, S.; Venkatesh Prajna, N.; Lalitha, P.; Valarnila, M.; Dharmalingam, K. Pathogen induced changes in the protein profile of human tears from Fusarium keratitis patients. PLoS One 2013, 8, e53018. [Google Scholar] [CrossRef]
- Li, N.; Wang, N.; Zheng, J.; Liu, X.M.; Lever, O.W.; Erickson, P.M.; Li, L. Characterization of human tear proteome using multiple proteomic analysis techniques. J Proteome Res 2005, 4, 2052–2061. [Google Scholar] [CrossRef]
- Hagan, S.; Martin, E.; Enriquez-de-Salamanca, A. Tear fluid biomarkers in ocular and systemic disease: potential use for predictive, preventive and personalised medicine. EPMA J 2016, 7, 15. [Google Scholar] [CrossRef]
- Boerger, M.; Funke, S.; Leha, A.; Roser, A.E.; Wuestemann, A.K.; Maass, F.; Bahr, M.; Grus, F.; Lingor, P. Proteomic analysis of tear fluid reveals disease-specific patterns in patients with Parkinson’s disease - A pilot study. Parkinsonism Relat Disord 2019, 63, 3–9. [Google Scholar] [CrossRef]
- Rentka, A.; Koroskenyi, K.; Harsfalvi, J.; Szekanecz, Z.; Szucs, G.; Szodoray, P.; Kemeny-Beke, A. Evaluation of commonly used tear sampling methods and their relevance in subsequent biochemical analysis. Ann Clin Biochem 2017, 54, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Fullard, R.J.; Tucker, D.L. Changes in human tear protein levels with progressively increasing stimulus. Invest Ophthalmol Vis Sci 1991, 32, 2290–2301. [Google Scholar]
- Murube, J. Basal, reflex, and psycho-emotional tears. Ocul Surf 2009, 7, 60–66. [Google Scholar] [CrossRef]
- Jones, G.; Lee, T.J.; Glass, J.; Rountree, G.; Ulrich, L.; Estes, A.; Sezer, M.; Zhi, W.; Sharma, S.; Sharma, A. Comparison of Different Mass Spectrometry Workflows for the Proteomic Analysis of Tear Fluid. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Tse, J.S.; Sze, Y.H.; Ka-Wai Cheung, J.; Li, K.K.; Lam, T.C. A Protein Suspension-Trapping Sample Preparation for Tear Proteomics by Liquid Chromatography-Tandem Mass Spectrometry. J Vis Exp 2023. [Google Scholar] [CrossRef]
- Harkness, B.M.; Hegarty, D.M.; Saugstad, J.A.; Behrens, H.; Betz, J.; David, L.L.; Lapidus, J.A.; Chen, S.; Stutzman, R.; Chamberlain, W.; et al. Experimental design considerations for studies of human tear proteins. Ocul Surf 2023, 28, 58–78. [Google Scholar] [CrossRef] [PubMed]
- Bachhuber, F.; Huss, A.; Senel, M.; Tumani, H. Diagnostic biomarkers in tear fluid: from sampling to preanalytical processing. Sci Rep 2021, 11, 10064. [Google Scholar] [CrossRef]
- Xiao, J.; Fu, Y.; Frenia, K.; Sikora, J.; Garwood, K.; Mental, R.; Labriola, L.T. Tear fluid proteomic analysis with improved LC-MS/MS protocol. Invest. Ophthalmol. Vis. Sci. 2024, 65, 6557. [Google Scholar]
- Ponzini, E.; Santambrogio, C.; De Palma, A.; Mauri, P.; Tavazzi, S.; Grandori, R. Mass spectrometry-based tear proteomics for noninvasive biomarker discovery. Mass Spectrom Rev 2022, 41, 842–860. [Google Scholar] [CrossRef]
- Zhan, X.; Li, J.; Guo, Y.; Golubnitschaja, O. Mass spectrometry analysis of human tear fluid biomarkers specific for ocular and systemic diseases in the context of 3P medicine. EPMA J 2021, 12, 449–475. [Google Scholar] [CrossRef]
- Green-Church, K.B.; Nichols, K.K.; Kleinholz, N.M.; Zhang, L.; Nichols, J.J. Investigation of the human tear film proteome using multiple proteomic approaches. Mol Vis 2008, 14, 456–470. [Google Scholar]
- Lepine, M.; Zambito, O.; Sleno, L. Targeted Workflow Investigating Variations in the Tear Proteome by Liquid Chromatography Tandem Mass Spectrometry. ACS Omega 2023, 8, 31168–31177. [Google Scholar] [CrossRef]
- Li, B.; Sheng, M.; Li, J.; Yan, G.; Lin, A.; Li, M.; Wang, W.; Chen, Y. Tear proteomic analysis of Sjogren syndrome patients with dry eye syndrome by two-dimensional-nano-liquid chromatography coupled with tandem mass spectrometry. Sci Rep 2014, 4, 5772. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Beuerman, R.W.; Chan, C.M.; Zhao, S.Z.; Li, X.R.; Yang, H.; Tong, L.; Liu, S.; Stern, M.E.; Tan, D. Identification of tear fluid biomarkers in dry eye syndrome using iTRAQ quantitative proteomics. J Proteome Res 2009, 8, 4889–4905. [Google Scholar] [CrossRef]
- Aluru, S.V.; Shweta, A.; Bhaskar, S.; Geetha, K.; Sivakumar, R.M.; Utpal, T.; Padmanabhan, P.; Angayarkanni, N. Tear Fluid Protein Changes in Dry Eye Syndrome Associated with Rheumatoid Arthritis: A Proteomic Approach. Ocul Surf 2017, 15, 112–129. [Google Scholar] [CrossRef]
- Jungert, K.; Paulsen, F.; Jacobi, C.; Horwath-Winter, J.; Garreis, F. Prolactin Inducible Protein, but Not Prolactin, Is Present in Human Tears, Is Involved in Tear Film Quality, and Influences Evaporative Dry Eye Disease. Front Med (Lausanne) 2022, 9, 892831. [Google Scholar] [CrossRef]
- Aydin, E.; Dhar, P.; Gokhale, M.; Chong, L.; Azizoglu, S.; Suphioglu, C. A Review of Emerging Tear Proteomics Research on the Ocular Surface in Ocular Allergy. Biology (Basel) 2022, 11. [Google Scholar] [CrossRef]
- Olson, B.J.; Markwell, J. Assays for determination of protein concentration. Curr Protoc Protein Sci Chapter 3, Unit 3 4. 2007. [Google Scholar] [CrossRef]
- Kielkopf, C.L.; Bauer, W.; Urbatsch, I.L. Methods for Measuring the Concentrations of Proteins. Cold Spring Harb Protoc 2020, 2020, 102277. [Google Scholar] [CrossRef] [PubMed]
- Pieczynski, J.; Szulc, U.; Harazna, J.; Szulc, A.; Kiewisz, J. Tear fluid collection methods: Review of current techniques. Eur J Ophthalmol 2021, 31, 2245–2251. [Google Scholar] [CrossRef] [PubMed]
- Krajcikova, K.; Glinska, G.; Tomeckova, V. Effect of tear fluid sampling and processing on total protein quantity and electrophoretic pattern. Taiwan J Ophthalmol 2022, 12, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res 2022, 50, W216–W221. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A novel in-capillary digestion workflow for enhanced tear proteomics in biomarker discovery. The LC-MS/MS workflow was optimized for tear samples as small as 0.5 µL, collected using glass capillary tubes. (a) Tear fluid sample collection using 0.5 µL glass capillary tubes, with an average protein concentration of 10.3 ± 2.9 µg/µL. (b) Experimental procedures for protein denaturation, reduction, alkylation, and trypsin digestion. To minimize potential sample loss and errors associated with transferring small-volume samples, 8 M urea solution was directly added to the microcentrifuge tube containing the capillary tube with 0.5 μL tear fluid, without flushing out the tear fluid sample. The tube was then rigorously vortexed and centrifuged at the maximum speed (~20,000 x g) in a benchtop microcentrifuge to break the capillary tube into small pieces and thoroughly mix the tear samples with the urea solution. The sample was incubated at room temperature for 30 minutes to promote protein denaturation and then diluted fivefold to reduce the urea concentration to 1.6 M prior to protein quantification via A280 measurement on a NanoDrop spectrophotometer. Subsequently, proteins were reduced with dithiothreitol (DTT) at 56 °C for 30 minutes and alkylated with iodoacetamide (IAA) at room temperature in the dark before overnight trypsin digestion. After digestion and pH adjustment to 2-3, 500 ng of digested peptides were directly loaded onto an Evosep tip for LC-MS/MS analysis (c), followed by data analysis (d). ice or dry-ice (Figure 1a). The direct addition of 8 M urea into the microcentrifuge tubes, without the need to flush the sample out of the capillaries, allowed for seamless protein solubilization and denaturation. Rigorous vortexing and high-speed centrifugation ensured thorough mixing of the tear fluid with the urea solution, efficiently fragmenting the capillary and maximizing protein recovery (Figure 1b). This method not only preserved the integrity of the tear sample but also minimize the processing steps that usually contribute to inefficiencies in traditional workflows. The peptide samples were then directly loaded onto an Evosep tip using a modified protocol, including extensive washing steps to remove any remaining urea in the samples, before undergoing LC-MS/MS and data analysis (Figure 1c & 1d).
Figure 1.
A novel in-capillary digestion workflow for enhanced tear proteomics in biomarker discovery. The LC-MS/MS workflow was optimized for tear samples as small as 0.5 µL, collected using glass capillary tubes. (a) Tear fluid sample collection using 0.5 µL glass capillary tubes, with an average protein concentration of 10.3 ± 2.9 µg/µL. (b) Experimental procedures for protein denaturation, reduction, alkylation, and trypsin digestion. To minimize potential sample loss and errors associated with transferring small-volume samples, 8 M urea solution was directly added to the microcentrifuge tube containing the capillary tube with 0.5 μL tear fluid, without flushing out the tear fluid sample. The tube was then rigorously vortexed and centrifuged at the maximum speed (~20,000 x g) in a benchtop microcentrifuge to break the capillary tube into small pieces and thoroughly mix the tear samples with the urea solution. The sample was incubated at room temperature for 30 minutes to promote protein denaturation and then diluted fivefold to reduce the urea concentration to 1.6 M prior to protein quantification via A280 measurement on a NanoDrop spectrophotometer. Subsequently, proteins were reduced with dithiothreitol (DTT) at 56 °C for 30 minutes and alkylated with iodoacetamide (IAA) at room temperature in the dark before overnight trypsin digestion. After digestion and pH adjustment to 2-3, 500 ng of digested peptides were directly loaded onto an Evosep tip for LC-MS/MS analysis (c), followed by data analysis (d). ice or dry-ice (Figure 1a). The direct addition of 8 M urea into the microcentrifuge tubes, without the need to flush the sample out of the capillaries, allowed for seamless protein solubilization and denaturation. Rigorous vortexing and high-speed centrifugation ensured thorough mixing of the tear fluid with the urea solution, efficiently fragmenting the capillary and maximizing protein recovery (Figure 1b). This method not only preserved the integrity of the tear sample but also minimize the processing steps that usually contribute to inefficiencies in traditional workflows. The peptide samples were then directly loaded onto an Evosep tip using a modified protocol, including extensive washing steps to remove any remaining urea in the samples, before undergoing LC-MS/MS and data analysis (Figure 1c & 1d).
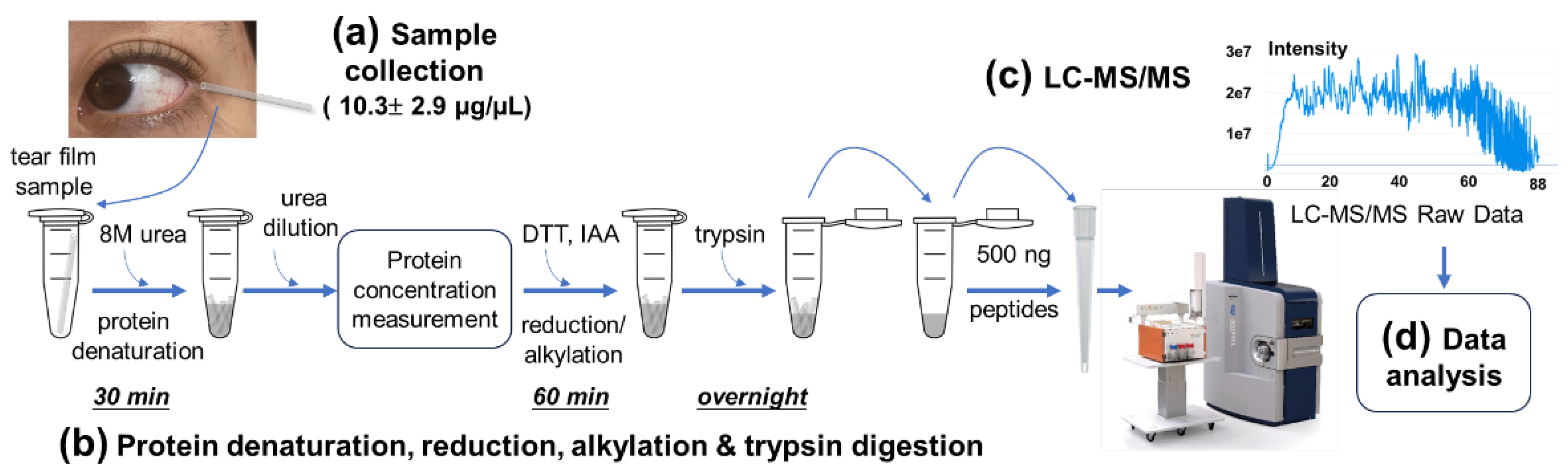
Figure 2.
Determining optimal protein quantities and LC gradients for tear fluid samples. Three different amounts of tear protein peptides (200, 500, and 1000ng) were tested using three different LC gradients (21, 44 and 88 mins). The experiments were repeated for three times.
Figure 2.
Determining optimal protein quantities and LC gradients for tear fluid samples. Three different amounts of tear protein peptides (200, 500, and 1000ng) were tested using three different LC gradients (21, 44 and 88 mins). The experiments were repeated for three times.
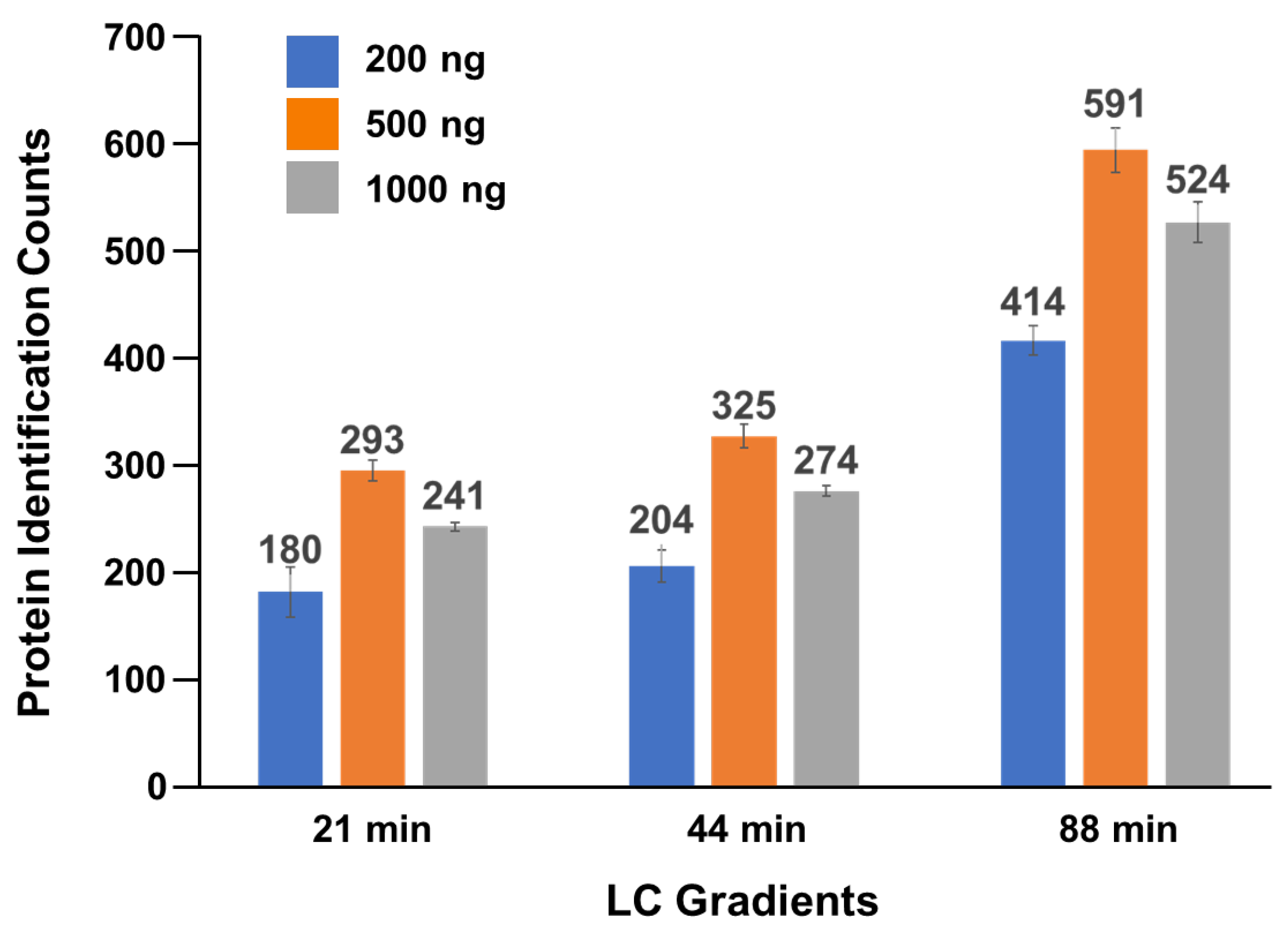
Figure 3.
Optimization of 8M urea solution volume. Pooled tear samples were collected in 0.5 µL glass capillary tubes and transferred to Protein LoBind microcentrifuge tubes. Different volumes of 8M urea (5, 10, 25, and 50 µL) were added to each tube, followed by the workflow described in Figure 1. The tube with 10 µL of 8M urea resulted in the highest number of identified tear proteins (816 ± 9 proteins). The experiments were repeated three times.
Figure 3.
Optimization of 8M urea solution volume. Pooled tear samples were collected in 0.5 µL glass capillary tubes and transferred to Protein LoBind microcentrifuge tubes. Different volumes of 8M urea (5, 10, 25, and 50 µL) were added to each tube, followed by the workflow described in Figure 1. The tube with 10 µL of 8M urea resulted in the highest number of identified tear proteins (816 ± 9 proteins). The experiments were repeated three times.
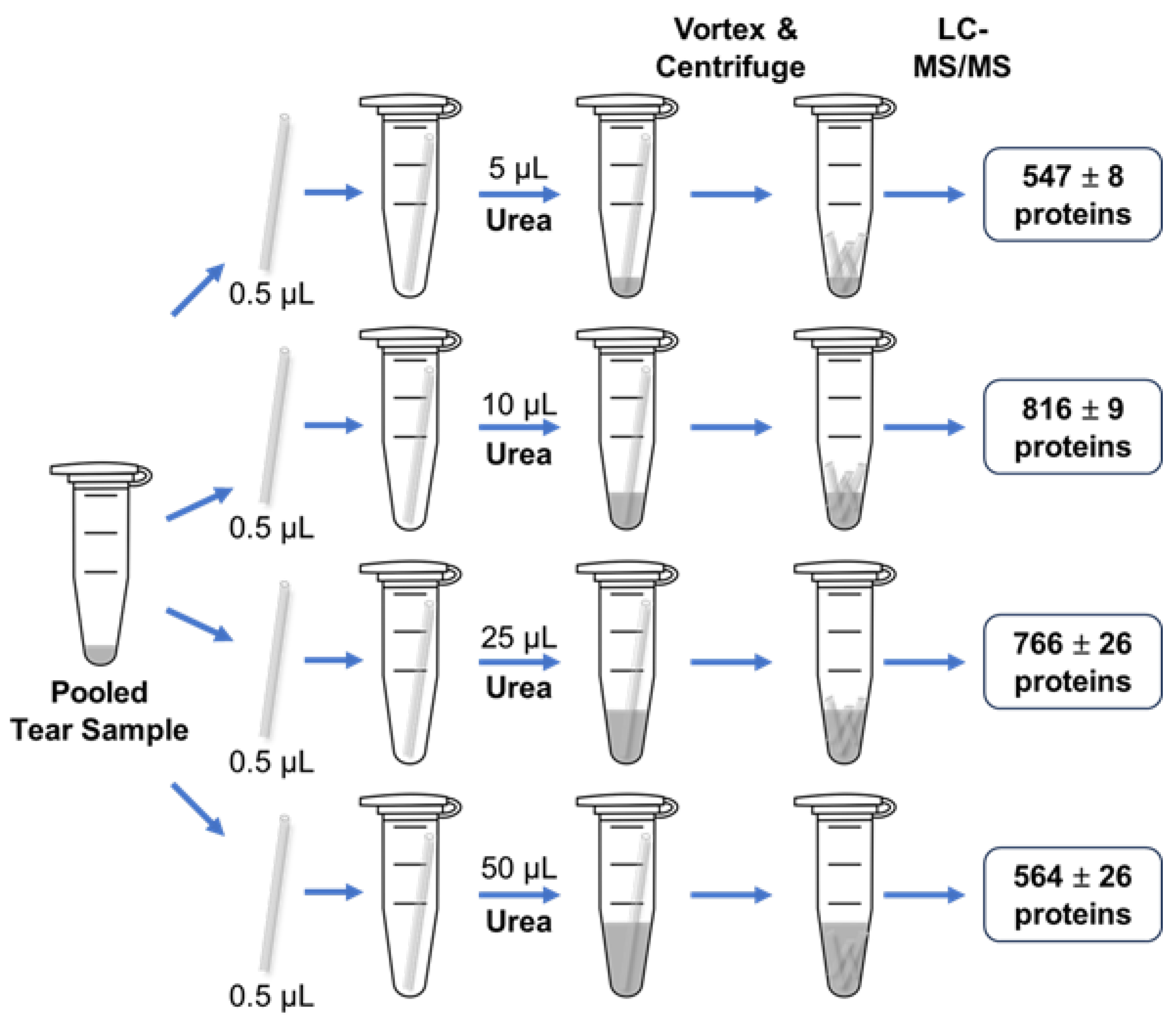
Figure 4.
Overview of Tear Proteome--Protein Functions and Classifications. (a) Proteomap depicting the 1,301 proteins identified in this study, collected using the glass capillary tube method. (b) Top 12 Tear Proteins ranked by PSMs (peptide-spectrum matches) in a representative tear proteomics dataset. Six of these proteins correspond to the prominent bands visible on the silver-stained SDS-PAGE gel from 0.5 µL of tear samples. (c & d) Gene Ontology (GO) analysis of the 1,301 tear proteins, revealing their involvement in biological processes (c) and molecular functions (d), was performed using DAVID Bioinformatics software.
Figure 4.
Overview of Tear Proteome--Protein Functions and Classifications. (a) Proteomap depicting the 1,301 proteins identified in this study, collected using the glass capillary tube method. (b) Top 12 Tear Proteins ranked by PSMs (peptide-spectrum matches) in a representative tear proteomics dataset. Six of these proteins correspond to the prominent bands visible on the silver-stained SDS-PAGE gel from 0.5 µL of tear samples. (c & d) Gene Ontology (GO) analysis of the 1,301 tear proteins, revealing their involvement in biological processes (c) and molecular functions (d), was performed using DAVID Bioinformatics software.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



