
Submitted:
08 October 2024
Posted:
10 October 2024
You are already at the latest version
Abstract
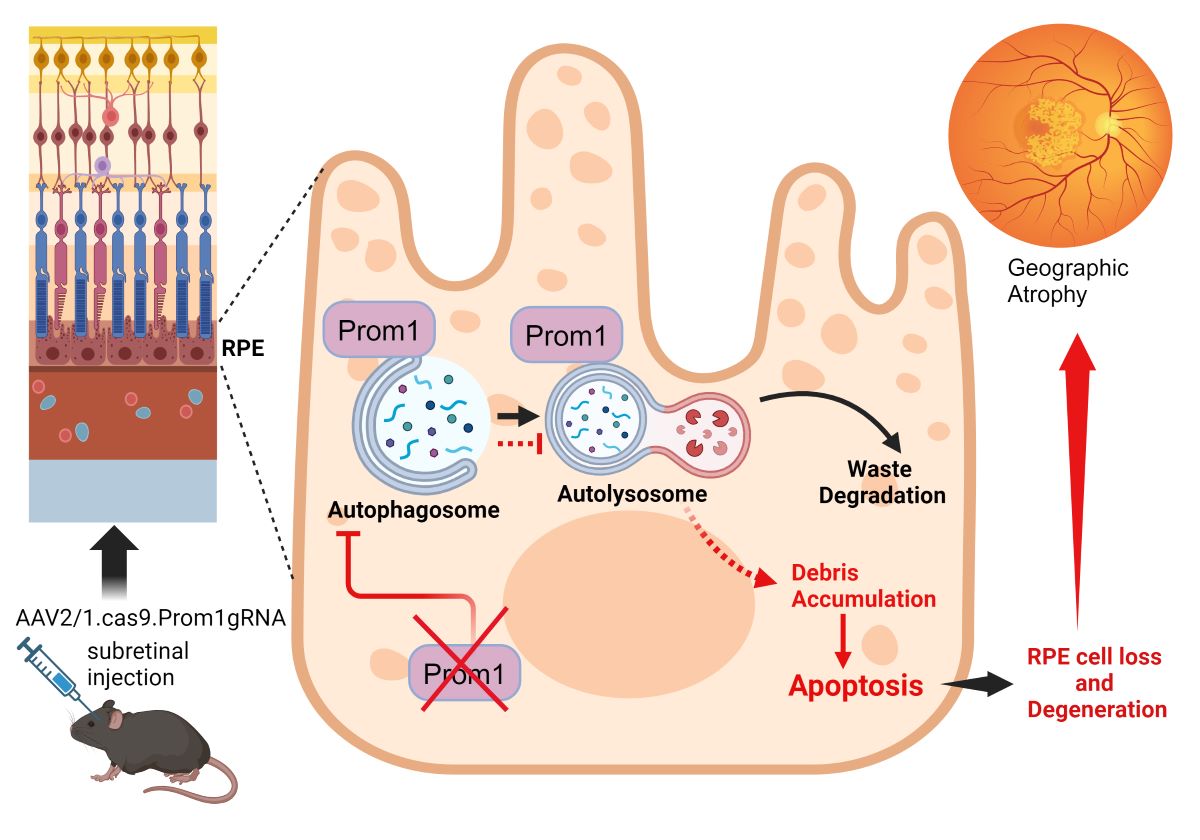
There are currently no effective treatments for retinal pigment epithelial (RPE) cell loss in atrophic AMD (aAMD). However, our research on Prom1, a known structural protein in photoreceptors (PRs), has revealed its distinct role in RPE and offers promising insights. While pathogenic Prom1 mutations have been linked to macular diseases with RPE atrophy, the broader physiological impact of dysfunctional Prom1 in RPE loss is unclear. We have shown that Prom1 plays a crucial role in regulating autophagy and cellular homeostasis in human and mouse RPE (mRPE) cells in vitro. Nevertheless, a comprehensive understanding of its in vivo expression and function in mRPE remains to be elucidated. To characterize Prom1 expression in RPE in situ, we used RNAscope assays and immunogold electron microscopy. Our use of chromogenic and fluorescent RNAscope assays in albino and C57BL/6J mice retinal sections has revealed Prom1 mRNA expression in perinuclear regions in mRPE in situ. Immunogold imaging showed Prom1 expression in RPE mitochondria. To confirm Prom1 expression in RPE, we interrogated human RPE single-cell RNA-sequencing datasets using an online resource, Spectacle. Our analysis showed Prom1 expression in human RPE. To investigate Prom1's function in RPE homeostasis, we performed RPE-specific Prom1-knockdown (KD) using subretinal injections of AAV2/1.CMV.saCas9.U6.Prom1gRNA in male and female mice. Our data show that RPE-specific Prom1-KD in vivo resulted in abnormal RPE morphology, sub-retinal fluid accumulation, and secondary PR loss. These changes were associated with patchy RPE cell death and reduced a-wave amplitude, indicating retinal degeneration. Our findings underscore the central role of Prom1 in cell-autonomous mRPE homeostasis. The implications of Prom1-KD in causing aAMD-like RPE defects and retinal degeneration in a mouse model are significant and could lead to novel treatments for aAMD.
Keywords:
atrophic age-related macular degeneration
; geographic atrophy
; mitochondria
; microglia
; subretinal injections
; adeno-associated virus
Introduction
The Prominin-1 (Prom1) gene encodes a transmembrane glycoprotein [1,2], which is widely recognized as an antigenic marker for stem cells and cancer stem cells [3,4]. Prom1 is expressed in differentiated epithelial and non-epithelial cells [5], glial cells [5], and the adult retina [6], suggesting that Prom1 plays a general role beyond stemness and differentiation status and is not limited to specific cell types. In the retina, Prom1 is present in the photoreceptor outer segments [7] and plays an essential role in photoreceptor disk morphogenesis [8]. The loss of function Prom1 mutations result in inherited retinal dystrophies, including autosomal dominant and autosomal recessive retinitis pigmentosa [7,9], cone-rod dystrophies [10,11,12], and macular dystrophies [8,13]. Prom1-associated macular dystrophy, also known as Stargardt disease 4 (STGD4), has clinical and pathophysiological features similar to ABCA4-related Stargardt disease 1 (STGD1) and the atrophic (dry) form of age-related macular degeneration (AMD), where abnormal cytotoxic lipofuscin bisretinoid accumulation triggers degeneration of macular rods, cones, and retinal pigment epithelial (RPE) cells [14,15,16]. This indicates that Prom1 dysfunction causes photoreceptor and RPE degeneration, primarily in the macula, but that disease progression may or may not involve increased bisretinoid lipofuscin. Understanding how Prom1 regulates RPE health and homeostasis is crucial for developing effective therapies.
We have previously shown that Prom1 is expressed in human and mouse RPE cell cultures in vitro. Somewhat surprisingly, Prom1 is predominantly a cytoplasmic protein in human RPE cells, and Prom1-knockout (K.O.) in human RPE cells activates mTORC1/2 activities and impairs the trafficking of autophagosomes to lysosomes [17]. Our studies also showed that cytosolic Prom1 interacts with p62 and HDAC6 in the developing autophagosome in human RPE cells, confirming its role in autophagy regulation [17]. We have recently shown that Prom1-KO activates mTORC1, reduces TFEB activity, and induces epithelial-mesenchymal transition (EMT) in mouse RPE (mRPE) cells, demonstrating that Prom1-mTORC1-TFEB signaling is a central driver of cell-autonomous mRPE homeostasis and suggesting a possible role in the development of geographic atrophy (G.A.) [18].
Prom1-related retinopathies are associated with various pathogenic Prom1 variants and heterogeneous phenotypical characteristics. The main phenotypic distinction lies between recessive and dominant forms of the disease [19]. While the recessive disease is associated with early-onset retinal degeneration, the dominant disease is associated with late-onset dystrophy, predominantly involving the macula, demonstrating that Prom1 mutations and inheritance patterns differentially impact multiple cell types in the outer retina. It is unclear whether Prom1 dysfunction primarily affects the photoreceptor cells with secondary involvement of the RPE or whether the RPE is a primary origin of the disease. Increasing evidence suggests that loss of function Prom1 mutations, including c.400C>T, p.R373C, and c.869delG mutants, cause RPE granular mottling, thinning of the outer retina, and parafoveal RPE atrophy in the macula [20,21,22]. In some younger patients with a mean age of 42 years, SD-OCT showed thinning of the RPE/Bruch’s membrane, indicative of RPE cell loss and early progression to GA [23]. Consistent with these observations, ophthalmic findings in younger patients with Prom1 R373C mutation exhibit distinct macular phenotypes, including central G.A., multifocal G.A., and bull’s eye maculopathy [24]. In subsequent follow-up studies of these young patients, the G.A. area was significantly enlarged in a time-dependent manner. Profound degeneration of the outer retinal layer, accompanied by extensive loss of RPE cells, was consistent with the longitudinal progression of Prom1-associated retinal degeneration. While these findings suggest that Prom1 dysfunction primarily impacts the RPE, additional investigations are necessary to understand how Prom1 function differentially regulates photoreceptor versus RPE homeostasis.
To address this knowledge gap, this study focused on understanding Prom1’s expression and its significance in RPE biology. To validate Prom1 expression in the pigmented RPE in situ, we used chromogenic and fluorescent RNAscope assays and immunogold electron microscopy. To demonstrate that loss of Prom1 in the mouse leads to RPE degeneration in vivo and recapitulates clinical features of atrophic AMD, we used AAV2/1-mediated Prom1 gene knockdown (K.D.). Our studies demonstrate that Prom1 plays distinct roles in the photoreceptors vs. RPE and begin to demonstrate how Prom1 preserves the physiologic functions of the RPE.
Materials and Methods
Materials
The backbone replication-deficient all-in-one purified experimental viral particles AAV2/1.CMV.saCas9.U6.Prom1gRNA (titer of 6.76x1022 Genome Copies/ml) and control viral particles AAV2/1.CMV.saCas9.U6.scrambledgRNA (titer of 7.96x1012 Genome Copies/ml) gRNA were commercially obtained from GeneCopoeia (Rockville, MD). The AAV particles were generated following a standardized protocol using highly purified plasmids and Endofectin-AAV reagents. The mouse Prom1-transcript variant 4 mRNA-RNAscope probe (catalog # 412221) and RNAscope assay kit were purchased from ACDBio (Newark, CA). The cleaved caspase-3 (Asp175) (D3E9) rabbit mAb- Alexa Fluor 647 conjugate (catalog# 9602), goat serum (catalog # 5425), and Prolong Gold AntiFade Reagent with DAPI (catalog #8961), anti-p62 antibody (catalog# 23214), and anti-LC3-I/II antibody (catalog# 12741) were obtained from Cell Signaling Technology (Danvers, MA).
Mice and Colony Management
C57BL/6J mice were obtained from the Jackson Laboratory (JAX, stock #000664), and the albino sentinel mice were from Charles River (CD-1 strain code 022). Mice were housed, maintained on a 12-h light-dark cycle, and provided food and water ad libitum. The Institutional Animal Care and Use Committee of Vanderbilt University Medical Center (VUMC) approved all experiments. All animal procedures followed the guidelines of the Association for Research in Vision and Ophthalmology Statement on the Use of Animals in Ophthalmic and Vision Research. Both male and female mice (4-6 weeks old) were used for this project.
Subretinal Injections
Both male and female C57BL/6J mice (4-6 weeks of age) were anesthetized by intraperitoneal injection with a mixture of 12.5 mg/Kg xylazine and 62.5 mg/kg ketamine. Topical ocular anesthesia was performed using 0.5% proparacaine, and the pupils were dilated with 0.5% tropicamide. Following dilation, the animals were placed under a dissecting microscope (Nikon), and the fundus was visualized with a drop of 2.5% methylcellulose. The sclera was punctured posterior to the limbus with a 33-gauge hypodermic Hamilton needle, avoiding trauma to the iris and the lens. The needle was placed at the inferior site of the ora serrata and advanced transsclerally into the subretinal space, as described earlier [25]. The contents of the syringe, 1ml of the viral vector solution (AAV2/1-saCas9-Prom1-gRNA or AAV2/1-saCas9-scrambled-gRNA) were slowly released into the subretinal space.
Electroretinogram (ERG) in Mice
Mice were dark-adapted overnight, dilated with 1% tropicamide, and anesthetized with 20/8/0.8 mg/kg ketamine/xylazine/urethane according to previously published methods [26]. To prevent hypothermia in anesthetized mice, they were placed on the heated surface of the ERG system. Corneal electrodes with integrated stimulators (Celeris System, Diagnosys LLC, Lowell, MA) were placed on eyes lubricated with GenTeal drops. The subdermal platinum needle electrodes were placed in the snout and back of the head at the location of the visual cortex. A ground electrode was placed in the back of the mouse. For ERGs, mice were exposed to 50 flashes of 1 Hz, 0.05 cd s/m2 white light with a pulse frequency of 1, as published earlier [26]. Each experimental group had 13-15 eyes.
Chromogenic and Fluorescent Prom1 RNAscope Assays in Mouse Retina Sections
The freshly isolated eyeballs from the euthanized C57BL6J and sentinel mice were fixed in 10% neutral-buffered formalin (NBF) overnight, embedded in paraffin, cut into 3.5-4mm sections, and placed on glass slides. In some cases, the ocular posterior cup was dissected from the mouse eye for in-situ localization of Prom1 in mouse RPE. The slides were then placed on the Leica Bond-RX IHC stainer, and all steps besides coverslipping were performed on the Bond IHC stainer. Slides were baked and deparaffinized, and heat-induced antigen retrieval was performed using the Lecia Epitope Retrieval 2 solution at 95oC and the ACD 2.5 LSx protease enzyme using the ACD RNAscope® protocol. Slides were hybridized with the Prom1 probe (Cat#412228, 23214, ACD-Bio-Techne (Newark, CA) for 2 hours. Both negative (Dapb) and positive (PPIB) control slides were used alongside the target probe. The ACD RNAscope® 2.5 LSx Reagent Kit- chromogenic RED detection system was used to visualize RPE and P.R.s of the sentinel albino mouse retina sections. The slides were counterstained with hematoxylin, a Leica Bond detection system component. Slides were dehydrated, coverslipped, and used for 40X brightfield microscopy.
To visualize Prom1 expression in pigmented RPE in the C57/BL6J mice, we used the ACD RNAscope® Multiplex Fluorescent Reagents Kit system containing OPAL570 and Cy5 fluorescent markers. DAPI was used as a counterstain to visualize nuclei, an ACD/Leica RNAscope kit component. Slides were coverslipped, and images were taken using the Nikon widefield microscope (100X) and the LSM 880 confocal microscope (60X) at the Cell Imaging Shared Resource at Vanderbilt. Images were also captured using a 40X Leica fluorescent whole slide imager at Vanderbilt’s Digital Histology Shared Resource.
H&E Staining and Histology
Mice subretinally injected with scrambled or Prom1-gRNA were housed and fed ad libitum at the Vanderbilt University Department of Animal Care. After 10-11 weeks post-injection, mice were euthanized by CO2 asphyxiation followed by thoracotomy, and eyes were collected and fixed in 4% PFA. Eyes were subsequently embedded in paraffin, cut into 3.5-4mm sections, and stained with hematoxylin-eosin (H&E). The stained sections were analyzed using the Translational Pathology Shared Resource (TPSR), and images were captured using a high throughput Leica SCN400 40X Brightfield slide scanner automated digital image system from Leica Microsystems at Vanderbilt’s Digital Histology Shared Resource (DHSR). All whole slides were imaged at 40X magnification to a 0.25 mm/pixel resolution. The slides were viewed and annotated, and images were analyzed using the Aperio ImageScope (v.12.4.6.5003) available through the Digital Slide Archive (DSA) at VUMC.
Immunohistochemistry
The formalin-fixed-paraffin-embedded mouse retinal sections were placed on slides, and all steps involving immunohistochemistry (IHC) were performed on the Bond IHC stainer at Vanderbilt’s TPSR. Slides were deparaffinized, and heat-induced antigen retrieval was performed using the Epitope Retrieval 1 solution for 20 minutes. Slides were then incubated with anti LC3A/B (Cat#23214, Cell Signaling, Danvers, MA) and anti-p62 (Cat#23214, Cell Signaling, Danvers, MA) for 1 hour at a dilution of 1:2500 and 1:500, respectively. The Bond Polymer Refine Detection system was used for chromogenic visualization. The slides were dehydrated, coverslipped with hematoxylin as a counterstain, and scanned using the Leica 40X SCN400 Brightfield scanner at DHSR. Images were analyzed using the Aperio ImageScope available through DSA at VUMC.
To detect active caspase-3 levels in mouse retinal sections, antigen retrieval was performed, and the slides were blocked with goat serum. Subsequently, the slides were incubated overnight with an anti-cleaved-caspase3-Alexa Fluor 647 conjugate antibody at 40C. The slides were washed three times with 1X PBS and coverslipped with an antifade mounting medium containing DAPI. The fluorescent immunostained tissue slides were imaged on an Aperio Versa 200 automated slide scanner (Leica Biosystems) at 40X magnification to a 0.162 mm/pixel resolution. The images were analyzed using the Aperio ImageScope as Vanderbilt’s DHSR.
Transmission Electron Microscopy (TEM)
TEM was performed using methods described earlier with modifications [27]. Briefly, eyecups were fixed in 4% PFA and 0.5% glutaraldehyde. After fixation, the samples were cryoprotected by gradual equilibrium with 30% glycerol followed by plunge freezing in liquid ethane. Samples were freeze-substituted in 1.5% uranyl acetate in methanol for 48 hours at -800C, followed by infiltration with HM20 at -300C. The HM20 was polymerized with UV light for 48 hours at -300C. Samples were sectioned at 100 nm nominal thickness of a UC7 ultramicrotome and collected onto 300 mesh Ni grids. For immunogold labeling, the grids were fixed, and antigens were retrieved using 0.1% sodium borohydride with 50 mM glycine and then blocked in 10% goat serum. Samples were incubated with rabbit polyclonal primary antibodies against Prom1 from Origene (Rockville, MD) (catalog number TA354470) and Abcam (Waltham, MA) (catalog number ab19898) at 1:50 dilution for 2 hours, followed by the secondary antibody at 1:20 for 1 hour. The grids were poststained with 2% uranyl acetate. TEM imaging was performed on a JEOL 2100+ equipped with an AMT nanosprint15 MKII CMOS camera using AMT acquisition software.
Analysis of Single-Cell RNA-Sequencing RPE and Retina Datasets
All published single-cell RNA-sequencing and ATAC mouse and human RPE/retina datasets were analyzed using Spectacle, an interactive online resource for single-cell RNA data analysis (https://singlecell-eye.org/app/spectacle/). Spectacle uses retina, RPE, and choroid datasets from human and mouse samples to identify which cell types express a gene of interest and characterize gene expression changes across regions or disease states [28].
Statistical Analysis
All data were analyzed using the GraphPad Prism 9 program (GraphPad Software Inc., San Diego, CA). Data are expressed as mean ± S.E. An unpaired 2-tailed Student’s t-test and Bonferroni post-hoc testing were used to assess statistical significance. Unless otherwise stated, *P<0.05, **P<0.01, ****P<0.0001 values were considered significant.
Results
Expression of Prom1 in Mouse RPE in Situ
We have shown that Prom1 is expressed in human and mouse RPE cells in vitro, but its expression in mouse RPE in situ has not been fully characterized beyond its immunoreactivity in the RPE microvilli [7,17,18]. Consistent with these observations, Prom1 expression was detected in hTERT-RPE-1 cells and found to interact physically with rhodopsin, implying a role of Prom1 in regulating RPE homeostasis [29]. To examine Prom1 expression in mouse RPE in situ, we used a chromogenic Prom1 RNAscope assay in albino mouse eye cups after removing the cornea, lens, and neuroretina. The RNAscope (RNA in situ hybridization) assay is used to visualize gene expression within tissues and detect specific RNA (mRNA) molecules and their spatial distribution [30]. Our results show Prom1 mRNA expression in the mouse RPE in situ, as evidenced by the red chromogenic puncta in RPE (arrowheads) (Figure 1A). The mouse eye cup with the area used for imaging (red box) is shown in Figure 1B. However, Prom1 labeling was not observed in the negative control, confirming the specificity of Prom1 RNAscope (Figure 1C). To confirm Prom1 mRNA expression, we repeated chromogenic RNAscope in another mouse eyecup. We found Prom1 expression in RPE in situ (black arrowheads) (Figure 1D). The mouse eye cup used for this image is shown in Figure 1E (red box). To confirm its expression in non-pigmented mouse RPE, we used a fluorescent RNAscope assay (using Cy5 fluorophore) and found the localized and discrete puncta of Prom1 mRNA in situ (white arrowheads, in the mouse eyecup (Figure 1F). Widefield high-resolution confocal microscopy showed Prom1 magenta puncta in RPE (Figure 1G). The chromogenic RNAscope assay shows a strong Prom1 signal (red signal) in the photoreceptor inner segments (IS) (black arrowheads), outer nuclear layer (ONL), and, interestingly, a small cohort of cells in the inner retina (white arrowheads) (Figure 2A), but Prom1 labeling was not observed in the negative control (Figure 2B). However, it is challenging to see Prom1 in RPE using the chromogenic assay due to the pigmentation of RPE (Figure 2A). We used bleached retina sections to visualize Prom1 in RPE but bleaching interfered with the chromogenic RNAscope signal. To address this, we used a fluorescent RNAscope with a Cys5 fluorophore and widefield microscopy to detect the Prom1 signal in the pigmented RPE. Consistent with data in retinal sections from albino mice, we observed fluorescent Prom1 magenta puncta in pigmented RPE from C57BL6J mice, confirming its expression (white arrows) (Figure 2C). Furthermore, a strong Prom1 signal was observed in photoreceptor IS, ONL, as well as a small group of cells in the inner retina (white arrowheads), but not in the negative control (Figure 2C-D). This confirms that the Prom1 gene is highly expressed in the photoreceptors but is also detectable in RPE.
To visualize Prom1 gene expression in single RPE cells in the C57BL6J retinal sections, we used 100X objective high magnification widefield confocal microscopy. Our results show that the Prom1 mRNA magenta puncta is detectable in single RPE cells in the mouse retinal sections (white arrowheads) (Figure 2E). We observed the Prom1 signal near the DAPI-stained nuclei of RPE cells, predominantly localized in the basolateral region (Figure 2E). To further confirm Prom1’s expression in RPE, we used a different fluorophore, OPAL-570, which detects low-expressing mRNA, optimizes signal amplification, and reduces background noise [31]. The punctate Prom1 mRNA expression (red) was detected in the pigmented RPE, predominantly in the perinuclear region of RPE cells (Figure 2F). Consistent with data presented in Figure 2E, the Prom1 signal was mainly confined to the basolateral region of RPE cells and restricted from the apical processes (Figure 2F). These observations are consistent with our previous findings, showing that Prom1 is a cytoplasmic protein that does not localize to the apical membranes in cultured human RPE in vitro [17].
To confirm Prom1 expression in mouse RPE in situ, we used immunogold electron microscopy in C57BL/6J eyecups after removing the cornea, lens, and retina. Our data show sparse Prom1 labeling in RPE cells using a rabbit polyclonal antibody from Origene. Analysis of Prom1 labeling in RPE shows its presence in mitochondria (white arrows, Figure 3A-B). Additional immunogold TEM with an antibody from Aviva showed Prom1’s cytoplasmic localization in RPE (Figure 3C, white arrow, top left) and RPE mitochondria (Figure 3C, white arrow in middle left) and near mitochondria (Figure 3C, white arrow, bottom right). Prom1’s presence in mitochondria suggests it could influence mitochondrial dynamics, bioenergetics, and overall RPE health.
Since Prom1 regulates autophagy in mRPE cells [18], we also examined the in situ localization of two key autophagy mediators, p62 and LC3I/II proteins, in mouse retinal sections. Chromogenic immunohistochemical (IHC) analysis of mouse retinal sections using p62 and LC3-I/II antibodies shows positive immunolabeling of p62 (Figure 4A) and LC3-I/II (Figure 4C) in RPE. However, no labeling was detected in their respective negative controls. (Figure 4B and D). We found p62 immunoreactivity in the outer plexiform layer (OPL) and POS in the outer retina. Several other cell types in the inner retina showed p62 immunoreactivity, including the ganglion cell layer (GCL). We observed strong LC3-I/II immunoreactivity in the outer retina’s POS, IS, ONL, OPL, and GCL in the inner retina. These results demonstrate that the critical autophagy regulators are detectable in the mRPE in situ.
AAV2/1-Mediated Prom1-Knockdown (KD) in Vivo Using CRISPR/Cas9-gRNA
Our data show that mouse eyes injected with AAV2/1-scambled gRNA did not alter Prom1 expression in the RPE (Figure 5A-F, gray arrows, left panel). However, eyes injected with AAV2/1.CRISPR.Prom1.gRNA did not cause Prom1 gene deletion throughout the entire RPE layer (Figure 5G-M, gray arrows, right panel), probably because the AAV transduction is limited to the injection site. We used low- and high-magnification images (with different scale bars) to focus on areas of RPE degeneration after subretinal injections of AAV2/1.CRISPR.Prom1.gRNA in the mouse eye. While low-magnification images provide a more comprehensive view of the entire retinal section, allowing for the identification of general patterns and overall Prom1 expression levels in control vs. Prom1.gRNA-injected eyes, magnified areas of the retinal sections enable precise localization and characterization of Prom1 gene expression in mouse RPE in situ. Using images of different magnifications, we observed a Prom1 (Prom1-KD) loss in localized areas through the RPE layer (Figure 5J-M, right panel, white arrowheads). Some areas showed intact Prom1 expression, indicating that subretinal AAV2/1 cannot transduce the entire RPE layer (Figure 5J-M, gray arrows). Importantly, injection of AAV2/1-Prom1-gRNA did not cause downregulation of Prom1 expression in the mouse PRs, demonstrating that AAV2/1 specifically targets Prom1 in the non-dividing RPE without transducing other retinal cell types. There was no evidence of inflammatory response or subretinal neovascularization associated with subretinal delivery of AAV2/1-mediated gRNA. Some sections show artifactual detachment of the overlying retina with few areas of photoreceptor outer segments attached to the RPE.
AAV2 has been used effectively through subretinal injection to transduce the RPE in several gene therapy trials and animal models [32,33]. The subretinal injection of AAV2/1 into C57BL/6J mice can specifically target RPE genes without affecting other cell types in the murine retina [34,35]. To investigate if RPE-specific targeting of Prom1 in a mouse model leads to RPE degeneration and secondary photoreceptor loss, we performed subretinal injections of control-AAV2/1 carrying the scrambled gRNA and the experimental-AAV2/1 carrying the Prom1-gRNA. The control vector did not alter normal fundus (Figure 6A), whereas eyes injected with the Prom1-gRNA caused cloudy, patchy, and diffuse yellowish lesions (Figure 6B-C) consistent with RPE cell pathology and/or dysfunction. Non-invasive in-vivo scotopic full-field ERGs revealed waveform changes in mouse eyes injected with AAV2/1.CRISPR.Prom1.gRNA vs. control scrambled gRNA (Figure 6D). We observed a reduction in both the a- and b-wave amplitudes but focused on the a-wave. If the a-wave is compromised, the ON-bipolar cells will not receive the right input from the PRs’ initial signal to produce the b-wave. Therefore, the initial loss of a-wave disrupts the signal transmission pathway, which leads to a subsequent reduction in the b-wave. Analysis of ERGs showed a significant reduction in a-wave (derived from the PR layer) amplitude in mice 11 weeks after subretinal injection with Prom1-gRNA (Figure 6E) as compared with those of mice injected with control gRNA, revealing functional abnormalities in PRs with RPE-specific Prom1-KD. These observations suggest that localized RPE dysfunction can lead to PR loss, impacting ERG readings in mice.
To determine the corresponding anatomical changes that occur with RPE pathology and loss of PR function, we evaluated the morphology of the retina in mice injected with either control or Prom1-gRNA. Light microscopy-based H&E sections show significant differences in AAV2/1Prom1-gRNA-injected eyes compared to control AAV2/1.scr.gRNA at 11 weeks post-injection (Figure 7). We observed RPE vacuoles and degeneration with fluid accumulation in the subretinal space causing PRs to be detached from the RPE in AAV2/1.Prom1-gRNA injected (male and female) mice (Figure 7B, 7D), but not in age-matched mice injected with control-AAV2/1.scr.gRNA (Figure 7A, 7C). These effects were not observed throughout the entire retina. Instead, they appeared as patches of RPE degeneration/loss in well-demarcated areas with PR degeneration.
RPE-Specific Prom1-KD in a Mouse Model Causes RPE Cell Death
Activation (i.e., cleavage) of caspase-3 has been implicated in RPE cell degeneration in aAMD [36]. Cell death mechanisms, including apoptosis and pyroptosis, are activated in rodent RPE cells in vivo in response to amyloid beta, a drusen component [37]. Elevated proteolytic cleavage of caspase-3, a marker for apoptosis, was observed in the degenerating rodent RPE [37]. We tested whether patchy Prom1-KD in response to AAV2/1-Prom1 gRNA subretinal injection after 11 weeks is sufficient to trigger caspase-3 activation, leading to RPE death. Since RPE-specific Prom1-KD causes phenotypic changes in the RPE, resembling RPE degeneration, we tested if this is associated with RPE apoptosis. We used an antibody that detects active cleaved caspase-3 and found that subretinal injection of the control AAV1/2.CRISPR.scr.gRNA did not induce caspase-3 activation (Figure 8A, 8E, left panel). However, eyes injected with AAV2/1 carrying Prom1-gRNA showed active caspase-3 positive RPE cells (Figure 8B, 8F, right panel, micrographs showing white arrows), but the PRs were largely unaffected. A few images in the sections from Prom1-KD mouse retinas, where the active caspase-3 antibody shows a diffuse background in the POSs, indicate that degenerating RPE can concomitantly damage PRs, causing PR apoptosis. It is unclear whether the diffuse caspase-3 signal in the POSs is due to PR death or non-specific staining, and further investigation is warranted. To demonstrate if Prom1-KD in these regions causes RPE cell death, we used serial sections from these areas and labeled them for Prom1 using RNAscope. We found that the control AAV2/1.CRISPR.scr.gRNA did not affect Prom1 gene expression (Figure 8C, 8G, left panel, white arrowheads). However, AAV2/1.CRISPR.Prom1.gRNA injection in mouse eyes showed loss of the Prom1 gene in the areas where RPE cells labeled positive for active cleaved caspase-3 (Figure 8D, 8H, right panel). These results demonstrate that RPE-specific Prom1-KD causes RPE apoptosis, leading to cell loss and degeneration.
Prom1 Gene Expression in Human and Mouse Single-Cell RNA-Seq Datasets
To confirm Prom1 expression in human and mouse RPE single-cell datasets, we used Spectacle, an interactive web-based resource for exploring published single-cell RNA sequencing data [28]. Spectacle can identify which retinal cell type expresses a gene of interest, detect transcriptomic subpopulations within a cell type, and perform differential expression analyses. We used a human macular and peripheral RPE single-cell RNA and ATAC sequencing dataset to analyze Prom1 gene expression in human RPE by region and subpopulation [38]. The Spectacle interactive system was used to visualize graph-based clusters showing subpopulations of human RPE cells and their cluster identity in a dimensionality reduction plot where the dimensions are based upon the pre-computed values using uniform manifold approximation and projection (UMAP) [38]. Clusters showing single RPE cells (RPE_0 – RPE_4) are shaded in different colors in the dimensionality reduction plot [38]. To determine if the Prom1 gene is expressed in these various human RPE subpopulations, Spectacle was used to visualize gene expression as violin plots. Our analysis depicts the expression of the Prom1 gene present in each human RPE cluster (Figure 9A). Since Prom1 expression levels varied between these single-cell RPE clusters, it confirms heterogeneity within RPE tissue from human donors. We examined if the Prom1 gene was enriched in human RPE cells across macular and peripheral regions of the retina and found comparable levels of Prom1 expression in both areas (Figure 9B). These observations demonstrate that across anatomic regions, subpopulations of RPE have similar levels of Prom1 gene. To compare Prom1 expression in mouse RPE, we explored other mice RPE datasets (GSE263427 and GSE236220) and found Prom1 expression in them, confirming the presence of Prom1 gene in mouse RPE.
To confirm Prom1 gene expression in other published human RPE datasets, we used a single-cell RNA-sequencing dataset of human iPSC-derived RPE cells generated from patients with G.A. and age-matched healthy individuals [39]. In this study, the authors used iPSC-derived RPE cells for disease modeling because this approach has been widely used to demonstrate disease phenotypes, especially those observed in macular dystrophies and AMD [40,41]. The iPSC-RPE cells were characterized based on the expression levels of genes with known associations with G.A., including C3, CFH, CFHR1, APOE, and HTRA1, to confirm that GA patient-derived RPE cells are accurate disease models in vitro without aging these cells in culture [39]. Interrogating this dataset, we found robust Prom1 expression in all subpopulations of human RPE cells (Figure 9C). However, this plot does not directly indicate which clusters correspond to AMD or controls. The dimensionality reduction plot using the same dataset was used to distinguish AMD RPE from control RPE clusters (data not shown). Mapping the AMD RPE cells to their respective control conditions using Spectacle showed varied Prom1 expression in a few control cells vs. AMD RPE (i.e., RPE2, RPE3, and RPE4), highlighting Prom1’s important role in maintaining RPE function and health, and suggesting that its dysregulation can have significant implications for RPE homeostasis, particularly in the context of AMD (Figure 9D). To assess if the changes in Prom1 expression in control vs. AMD correlate with other well know AMD-risk genes, we interrogated the dataset for CFH gene expression, a major AMD-related risk locus [42]. CFH gene expression varied in a few control cells compared to AMD RPE cells (RPE2, RPE4, RPE5), confirming CFH’s involvement in RPE function.
The retina’s resident immune cells, including microglia, support RPE cell function by phagocytosing POSs and lipids, thereby protecting the RPE during outer retinal degenerative conditions [43]. Recently, a unique profile of microglia was uncovered at RPE atrophic sites in mouse and human models of retinal degeneration [44]. To examine whether Prom1 is expressed in retinal microglia, we interrogated published databases in Spectacle and found that the Prom1 gene is highly expressed in activated and proliferating activated microglia (heatmap, Figure 10A), which are known to phagocytose shed POSs at the site of damage and maintain retinal health [45]. Both light and dark conditions influence microglial behavior in the retina differently [45]. Darkness-induced microglial behavior has neuroprotective effects, especially during retinal degeneration. Our analysis showed that activated microglia in darkness in the degenerating retina have high levels of Prom1 (Figure 10B), suggesting that Prom1 can mediate microglial behavior in light and dark conditions. Interrogation of a separate mouse database [46] confirmed Prom1 gene expression in microglial cells from normal mouse retinas with high expression of initial undamaged microglial markers (MG0) and other clusters of microglia (i.e., lcMG1-3) from light-damaged retinas (Figure 10C). Other small clusters of microglia (srMG) also showed low Prom1 expression, suggesting that phenotypically heterogenous microglia with unique functional specializations at different locations of the mouse retina express Prom1.
Discussion
Despite emerging robust data demonstrating that Prom1 regulates RPE homeostasis [17,18], direct evidence of its presence in mouse RPE in situ has been lacking until now. This study convincingly shows that mouse RPE expresses the Prom1 gene in situ, at least to a sufficient level, to impact key RPE processes, including waste removal by autophagy and stabilizing lysosomal activity. While Prom1 mRNA expression is significantly higher in the mouse PR inner segments, the substantially greater number of PRs than RPE cells in the mouse retina [47] contributes partly to this observation. Prom1’s function in RPE is distinct from its role in PRs. In PRs, Prom1 is primarily involved in the structural organization of the OS and is localized to the disk membrane [6,48]. This localization supports its role in maintaining PR integrity and function. In contrast, in RPE cells, Prom1 is not a membrane-bound protein; its mRNA localizes to the perinuclear region, and we have shown that Prom1 functions as a signaling molecule within the cytoplasm to regulate autophagy and other intracellular processes integral to mouse RPE cell survival. These observations are consistent with our previous findings of cytoplasmic localization of Prom1 in human RPE cells [17]. Prom1 has been observed translocating from the membrane to the cytoplasm in response to high glucose levels, indicating that the cellular environment and metabolic state significantly influence Prom1’s localization [3]. Prom1 traffics to lysosomes occurs through its physical interactions with cytosolic histone deacetylase 6 (HDAC6) [49]. Our previous studies in human RPE cells revealed that cytosolic Prom1 interacts with autophagy-related proteins such as p62 and HDAC6, playing a crucial role in autophagosome biogenesis, followed by its trafficking to lysosomes [17]. Furthermore, our recent findings show that the loss of Prom1 in mouse RPE cultures disrupts autophagy and induces epithelial-mesenchymal transition, underscoring Prom1’s pivotal role as a regulator of cell-autonomous RPE homeostasis [18]. Collectively. these studies establish that Prom1 predominantly localizes in the cytoplasm under resting conditions in RPE cells and is essential for RPE waste removal, a function markedly distinct from its well-known role at the cell surface.
This study’s novel finding is the presence of Prom1 in RPE mitochondria. This suggests a role in mitochondrial dynamics, energy metabolism, and apoptosis. This finding could have implications for understanding metabolic disorders and degenerative diseases affecting the RPE. The localization studies expand our knowledge of Prom1’s functions and interactions within RPE cells, shedding light on RPE cellular processes and diseases.
Although Prom1 mutations are linked to macular diseases caused by RPE dysfunction, the precise role of Prom1 in RPE homeostasis remains unclear. We show that AAV2/1-Cas9-Prom1-gRNA-mediated targeting of Prom1 in mouse RPE in vivo leads to RPE abnormalities and secondary P.R. degeneration. Retinal section analysis revealed patches of caspase-3 positive RPE cells, confirming RPE degeneration and apoptosis due to loss of Prom1 function, mirroring the RPE degeneration observed in patients with G.A. These findings provide insights into the pathophysiology of RPE degeneration in macular diseases and suggest that an RPE-specific Prom1-KO mouse model will enhance our understanding of Prom1’s cell-autonomous role in maintaining RPE health and homeostasis. The global Prom1-KO mice of the pure C57BL/6J background showed complete P.R. degeneration by P20. However, the retinal degeneration in the C57BL/6xCBA/NSlc mice showed significantly slower retinal degeneration, demonstrating that retinal degeneration in mouse models depends on genetic background [50]. Although global K.O. mouse models are useful for studying human disease, tissue-specific gene targeting is required to understand the specific contribution of Prom1 in various retinal cell types and the molecular mechanisms associated with diverse disease phenotypes.
Prom1-related-retinal diseases, although relatively rare, significantly impact vision. The prevalence of these diseases varies based on specific phenotypes and genetic variants [19]. Most studies show early macular involvement, PR degeneration, and RPE atrophy are common features of Prom1-related inherited retinal disease phenotypes [21]. Prom1 is essential for maintaining the expression of ABCA4 and RDH12 in mouse RPE, which is crucial in regulating the visual cycle and preventing toxic bisretinoid accumulation [50]. We have shown that loss of Prom1 in mouse RPE in vitro impairs autophagy flux, activates mTORC1, and decreases transcription factor E.B. (TFEB) activity, leading to RPE defects similar to aAMD [18]. Furthermore, the loss of Prom1 function due to a pathogenic Prom1 mutation, combined with the heterozygous ABCA4 mutation, exacerbates RPE dysfunction, resulting in granular mottling and macula alterations, providing evidence for their shared pathophysiology [20]. Loss of Prom1 function may lead to lipofuscin accumulation and other toxic RPE metabolites through molecular crosstalk involving ABCA4 and mTORC1. The question remains—how does Prom1 participate in the expression of these genes? Since Prom1 is not a transmembrane protein in RPE, it cannot transduce the extracellular information intracellularly, but as a cytoplasmic protein, we have shown it can regulate numerous RPE cellular processes involving degradative pathways. Therefore, additional studies are needed to explore how their interplay impacts downstream target genes and upstream regulatory pathways, affecting RPE cell fate and disease outcomes.
It remains unclear whether Prom1-associated retinal diseases originate primarily in the RPE or the P.R.s with secondary RPE damage. Patients with the Prom1 R373C mutation (autosomal dominant Stargardt-like) exhibit macular dystrophy with three distinct phenotypes: central GA, multifocal GA, and bull’s eye maculopathy, suggesting an RPE cell-autonomous function of Prom1 in the human retina [24]. A recently identified loss-of-function Prom1 variant (c.1354dupT) has shown a multi-phenotypic effect linked to cone-rod dystrophy and retinitis pigmentosa (RP) [51]. This cohort suggests that Prom1-related pathology may also confer an RP phenotype. Interestingly, a cohort of young patients with this newly identified Prom1 variant displayed alterations in the RPE with a preserved PR layer. Older patients with more advanced conditions exhibited both PR and RPE degeneration, suggesting that in these patients, the disease initially affects the RPE [51]. Additionally, this study reported the early onset of an autosomal recessive form of STGD4, which differs from the late-onset autosomal dominant manifestation of STGD4 dystrophy. This confirms extensive genetic and phenotypic heterogeneity within Prom1-related inherited retinal diseases [19,52]. Some mutations lead to a complete loss of function, while others may result in a partially functional protein, leading to different degrees of retinal and RPE dystrophies. [53,54]. Modifier genes, epigenetic modifications, and other genetic factors can influence the heterogeneity of disease presentation, making diagnosis and management challenging.
We examined published human RPE single-cell RNA sequencing datasets using Spectacle and found the expression of the Prom1 gene in human RPE. Prom1 is expressed in the human retina’s macular and peripheral regions, indicating its fundamental role in maintaining RPE function. Importantly, we observed variations in Prom1 expression between distinct clusters of control RPE versus AMD RPE. These variations need further exploration to understand how Prom1 function influences RPE dysfunction and degeneration in aAMD and its potential significance in disease pathogenesis and RPE homeostasis. Analysis of a mouse retinal single-cell RNA sequencing dataset revealed the presence of Prom1 in RPE, amacrine cells, bipolar cells, early and late retinal progenitor cells, and Muller glia (data not shown), suggesting Prom1’s potential involvement in regulating retinal development, regeneration, and disease processes [55]. The presence of Prom1 in the synaptically active amacrine cells suggests its role in integrating and shaping the visual message presented to retinal ganglion cells. Prom1 in amacrine cells may contribute to retinal circuitry by enhancing the communication between P.R.s, bipolar cells, and ganglion cells, ensuring efficient visual processing and adaptation to changing environmental conditions. Additional studies are warranted to test Prom1’s emerging function in the amacrine cells, retinal progenitor cells, and Muller glia.
We also found Prom1 expression in advanced and proliferating retinal microglia, specialized immune cells critical for surveillance, phagocytosis, and retinal health. Interestingly, activated microglia in the degenerating retina exhibit high levels of the Prom1 gene, particularly during darkness, indicating that Prom1 may be involved in microglial responses to retinal stress. Furthermore, other small microglial populations also express Prom1, suggesting its unique functional roles in regulating phenotypically heterogeneous microglia at different retinal locations. The intricate relationship between Prom1 and microglia underscores the complexity and importance of understanding Prom1-related retinal pathologies.
In summary, our study provides comprehensive insights into the multifaceted role of Prom1 in RPE homeostasis and retinal health. The detailed characterization of Prom1’s localization, function, and implications in RPE/retinal health underscores its significance as a therapeutic target. Future research on elucidating the molecular mechanisms of Prom1’s interactions with other cellular pathways will be essential for developing effective therapies for aAMD and other Prom1-associated retinal diseases. Our findings highlight the importance of Prom1 as a central mediator in maintaining RPE health and offer promising directions for therapeutic advancements in retinal diseases.
Limitations
Our study has certain limitations associated with using subretinal injections of AAV2/1.saCas9.Prom1-gRNA to target Prom1 in the mouse RPE in situ. Subretinal injections typically result in the localized delivery of the vector, meaning the AAV2/1.saCas9.gRNA complex predominantly transduces RPE cells near the injection site. The restricted diffusion of the vector from the injection site can lead to uneven distribution, limiting Prom1 gene knockdown to specific areas. Additionally, the retinal structure and extracellular matrix act as barriers that hinder the widespread dissemination of the vector, resulting in more localized effects. Variability in the subretinal injection procedure, such as differences in injection volume and pressure, can also affect the vector’s spread and the pattern of Prom1 gene knockdown. Notably, the patchy nature of Prom1 knockdown and resulting RPE degeneration more closely mirrors the localized areas of RPE loss and dysfunction seen in human geographic atrophy (GA), a late-stage manifestation of dry AMD. This finding highlights the potential relevance of our model for studying GA progression and therapeutic interventions.
In our study, the eyes injected with Prom1-gRNA exhibited cloudy, patchy, and diffuse yellowish lesions by fundus imaging, consistent with RPE cell pathology and/or dysfunction—this was the only direct evidence of lesions observed. However, H&E staining did not reveal classic atrophic lesions. Nonetheless, the observed RPE degeneration, marked by active caspase-3 labeling, indicates significant RPE cell apoptosis, an early indicator of atrophy. Localized RPE dysfunction may also affect electroretinography (ERG) readings, suggesting photoreceptor (PR) degeneration. Further studies, including quantifying PR layer thickness, are necessary to confirm secondary PR loss. While these additional studies are crucial for fully characterizing the phenotypic changes following Prom1 knockdown in mouse RPE in situ, they were not performed in this study.
Data Availability Statement
The data supporting this study’s findings are available from the corresponding author upon reasonable request.
Acknowledgments
This work was supported by the International Retina Research Foundation, the Potocsnak family gift to the Vanderbilt Eye Institute, the Margy Ann and J Donald M Gass Chair endowment, and an unrestricted departmental research grant from Research to Prevent Blindness, Inc (New York, NY). The Translational Pathology Shared Resource (TPSR) is supported by NCI/NIH Cancer Center Support Grant P30CA068485, the Shared Instrumentation Grant S10OD023475-01A1 for the Leica Bond RX, the shared equipment grant S10 OD016355 for the Tissue Microarray (TMA) Grandmaster, and the shared equipment grant for the LCM: IS1BX003154. Whole slide imaging was performed in the Digital Histology Shared Resource at Vanderbilt University Medical Center (vumc.org/dhsr). Confocal microscopy and electron microscopy were performed in part through the use of the Vanderbilt Cell Imaging Shared Resource (supported by NIH grants CA68485, DK20593, DK58404, DK59637, EY08126, and 1S100D034315-01. We acknowledge Ms. Rachel Hart for her assistance with immunogold transmission electron microscopy (Vanderbilt, CISR).
Conflict of Interest
The authors declare no conflict of interest
References
- Miraglia, S.; Godfrey, W.; Yin, A.H.; Atkins, K.; Warnke, R.; Holden, J.T.; Bray, R.A.; Waller, E.K.; Buck, D.W. A novel five-transmembrane hematopoietic stem cell antigen: isolation, characterization, and molecular cloning. Blood 1997, 90, 5013-5021.
- Weigmann, A.; Corbeil, D.; Hellwig, A.; Huttner, W.B. Prominin, a novel microvilli-specific polytopic membrane protein of the apical surface of epithelial cells, is targeted to plasmalemmal protrusions of non-epithelial cells. Proc Natl Acad Sci U S A 1997, 94, 12425-12430. [CrossRef]
- Chen, H.; Luo, Z.; Dong, L.; Tan, Y.; Yang, J.; Feng, G.; Wu, M.; Li, Z.; Wang, H. CD133/prominin-1-mediated autophagy and glucose uptake beneficial for hepatoma cell survival. PLoS One 2013, 8, e56878. [CrossRef]
- Florek, M.; Haase, M.; Marzesco, A.M.; Freund, D.; Ehninger, G.; Huttner, W.B.; Corbeil, D. Prominin-1/CD133, a neural and hematopoietic stem cell marker, is expressed in adult human differentiated cells and certain types of kidney cancer. Cell Tissue Res 2005, 319, 15-26. [CrossRef]
- Pleskac, P.; Fargeas, C.A.; Veselska, R.; Corbeil, D.; Skoda, J. Emerging roles of prominin-1 (CD133) in the dynamics of plasma membrane architecture and cell signaling pathways in health and disease. Cell Mol Biol Lett 2024, 29, 41. [CrossRef]
- Ragi, S.D.; Lima de Carvalho, J.R., Jr.; Tanaka, A.J.; Park, K.S.; Mahajan, V.B.; Maumenee, I.H.; Tsang, S.H. Compound heterozygous novel frameshift variants in the PROM1 gene result in Leber congenital amaurosis. Cold Spring Harb Mol Case Stud 2019, 5. [CrossRef]
- Maw, M.A.; Corbeil, D.; Koch, J.; Hellwig, A.; Wilson-Wheeler, J.C.; Bridges, R.J.; Kumaramanickavel, G.; John, S.; Nancarrow, D.; Roper, K.; et al. A frameshift mutation in prominin (mouse)-like 1 causes human retinal degeneration. Hum Mol Genet 2000, 9, 27-34. [CrossRef]
- Yang, Z.; Chen, Y.; Lillo, C.; Chien, J.; Yu, Z.; Michaelides, M.; Klein, M.; Howes, K.A.; Li, Y.; Kaminoh, Y.; et al. Mutant prominin 1 found in patients with macular degeneration disrupts photoreceptor disk morphogenesis in mice. J Clin Invest 2008, 118, 2908-2916. [CrossRef]
- Zhang, Q.; Zulfiqar, F.; Xiao, X.; Riazuddin, S.A.; Ahmad, Z.; Caruso, R.; MacDonald, I.; Sieving, P.; Riazuddin, S.; Hejtmancik, J.F. Severe retinitis pigmentosa mapped to 4p15 and associated with a novel mutation in the PROM1 gene. Hum Genet 2007, 122, 293-299. [CrossRef]
- Pras, E.; Abu, A.; Rotenstreich, Y.; Avni, I.; Reish, O.; Morad, Y.; Reznik-Wolf, H.; Pras, E. Cone-rod dystrophy and a frameshift mutation in the PROM1 gene. Mol Vis 2009, 15, 1709-1716.
- Eidinger, O.; Leibu, R.; Newman, H.; Rizel, L.; Perlman, I.; Ben-Yosef, T. An intronic deletion in the PROM1 gene leads to autosomal recessive cone-rod dystrophy. Mol Vis 2015, 21, 1295-1306.
- Khan, A.O.; Bolz, H.J. Pediatric Cone-Rod Dystrophy with High Myopia and Nystagmus Suggests Recessive PROM1 Mutations. Ophthalmic Genet 2015, 36, 349-352. [CrossRef]
- Imani, S.; Cheng, J.; Shasaltaneh, M.D.; Wei, C.; Yang, L.; Fu, S.; Zou, H.; Khan, M.A.; Zhang, X.; Chen, H.; et al. Genetic identification and molecular modeling characterization reveal a novel PROM1 mutation in Stargardt4-like macular dystrophy. Oncotarget 2018, 9, 122-141. [CrossRef]
- Strauss, R.W.; Munoz, B.; Ahmed, M.I.; Bittencourt, M.; Schonbach, E.M.; Michaelides, M.; Birch, D.; Zrenner, E.; Ervin, A.M.; Charbel Issa, P.; et al. The Progression of the Stargardt Disease Type 4 (ProgStar-4) Study: Design and Baseline Characteristics (ProgStar-4 Report No. 1). Ophthalmic Res 2018, 60, 185-194. [CrossRef]
- Kniazeva, M.; Chiang, M.F.; Morgan, B.; Anduze, A.L.; Zack, D.J.; Han, M.; Zhang, K. A new locus for autosomal dominant stargardt-like disease maps to chromosome 4. Am J Hum Genet 1999, 64, 1394-1399. [CrossRef]
- Abalem, M.F.; Omari, A.A.; Schlegel, D.; Khan, N.W.; Jayasundera, T. Macular hyperpigmentary changes in ABCA4-Stargardt disease. Int J Retina Vitreous 2019, 5, 9. [CrossRef]
- Bhattacharya, S.; Yin, J.; Winborn, C.S.; Zhang, Q.; Yue, J.; Chaum, E. Prominin-1 Is a Novel Regulator of Autophagy in the Human Retinal Pigment Epithelium. Invest Ophthalmol Vis Sci 2017, 58, 2366-2387. [CrossRef]
- Bhattacharya, S.; Yin, J.; Huo, W.; Chaum, E. Loss of Prom1 impairs autophagy and promotes epithelial-mesenchymal transition in mouse retinal pigment epithelial cells. J Cell Physiol 2023. [CrossRef]
- Cehajic-Kapetanovic, J.; Birtel, J.; McClements, M.E.; Shanks, M.E.; Clouston, P.; Downes, S.M.; Charbel Issa, P.; MacLaren, R.E. Clinical and Molecular Characterization of PROM1-Related Retinal Degeneration. JAMA Netw Open 2019, 2, e195752. [CrossRef]
- Lee, W.; Paavo, M.; Zernant, J.; Stong, N.; Laurente, Z.; Bearelly, S.; Nagasaki, T.; Tsang, S.H.; Goldstein, D.B.; Allikmets, R. Modification of the PROM1 disease phenotype by a mutation in ABCA4. Ophthalmic Genet 2019, 40, 369-375. [CrossRef]
- Permanyer, J.; Navarro, R.; Friedman, J.; Pomares, E.; Castro-Navarro, J.; Marfany, G.; Swaroop, A.; Gonzalez-Duarte, R. Autosomal recessive retinitis pigmentosa with early macular affectation caused by premature truncation in PROM1. Invest Ophthalmol Vis Sci 2010, 51, 2656-2663. [CrossRef]
- Hwang, S.; Kang, S.W.; Jang, J.H.; Kim, S.J. Genetic and clinical characteristics of PROM1-related retinal degeneration in Korean. Sci Rep 2023, 13, 21877. [CrossRef]
- Paavo, M.; Lee, W.; Parmann, R.; Lima de Carvalho, J.R., Jr.; Zernant, J.; Tsang, S.H.; Allikmets, R.; Sparrow, J.R. Insights Into PROM1-Macular Disease Using Multimodal Imaging. Invest Ophthalmol Vis Sci 2023, 64, 27. [CrossRef]
- Ricca, A.M.; Han, I.C.; Hoffmann, J.; Stone, E.M.; Sohn, E.H. Macular Atrophy and Phenotypic Variability in Autosomal Dominant Stargardt-Like Macular Dystrophy Due to Prom1 Mutation. Retina 2023, 43, 1165-1173. [CrossRef]
- Muhlfriedel, R.; Michalakis, S.; Garcia Garrido, M.; Biel, M.; Seeliger, M.W. Optimized technique for subretinal injections in mice. Methods Mol Biol 2013, 935, 343-349. [CrossRef]
- Naguib, S.; Backstrom, J.R.; Gil, M.; Calkins, D.J.; Rex, T.S. Retinal oxidative stress activates the NRF2/ARE pathway: An early endogenous protective response to ocular hypertension. Redox Biol 2021, 42, 101883. [CrossRef]
- Petralia, R.S.; Wang, Y.X. Review of Post-embedding Immunogold Methods for the Study of Neuronal Structures. Front Neuroanat 2021, 15, 763427. [CrossRef]
- Voigt, A.P.; Whitmore, S.S.; Lessing, N.D.; DeLuca, A.P.; Tucker, B.A.; Stone, E.M.; Mullins, R.F.; Scheetz, T.E. Spectacle: An interactive resource for ocular single-cell RNA sequencing data analysis. Exp Eye Res 2020, 200, 108204. [CrossRef]
- Hori, A.; Nishide, K.; Yasukuni, Y.; Haga, K.; Kakuta, W.; Ishikawa, Y.; Hayes, M.J.; Ohnuma, S.I.; Kiyonari, H.; Kimura, K.; et al. Prominin-1 Modulates Rho/ROCK-Mediated Membrane Morphology and Calcium-Dependent Intracellular Chloride Flux. Sci Rep 2019, 9, 15911. [CrossRef]
- Chan, S.; Filezac de L’Etang, A.; Rangell, L.; Caplazi, P.; Lowe, J.B.; Romeo, V. A method for manual and automated multiplex RNAscope in situ hybridization and immunocytochemistry on cytospin samples. PLoS One 2018, 13, e0207619. [CrossRef]
- Nilsson, O.R.; Kari, L.; Rosenke, R.; Steele-Mortimer, O. Protocol for RNA fluorescence in situ hybridization in mouse meningeal whole mounts. STAR Protoc 2022, 3, 101256. [CrossRef]
- Bennett, J. Immune response following intraocular delivery of recombinant viral vectors. Gene Ther 2003, 10, 977-982. [CrossRef]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849-860. [CrossRef]
- Seo, S.J.; Krebs, M.P.; Mao, H.; Jones, K.; Conners, M.; Lewin, A.S. Pathological consequences of long-term mitochondrial oxidative stress in the mouse retinal pigment epithelium. Exp Eye Res 2012, 101, 60-71. [CrossRef]
- Auricchio, A.; Kobinger, G.; Anand, V.; Hildinger, M.; O’Connor, E.; Maguire, A.M.; Wilson, J.M.; Bennett, J. Exchange of surface proteins impacts on viral vector cellular specificity and transduction characteristics: the retina as a model. Hum Mol Genet 2001, 10, 3075-3081. [CrossRef]
- Kaneko, H.; Dridi, S.; Tarallo, V.; Gelfand, B.D.; Fowler, B.J.; Cho, W.G.; Kleinman, M.E.; Ponicsan, S.L.; Hauswirth, W.W.; Chiodo, V.A.; et al. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature 2011, 471, 325-330. [CrossRef]
- Gao, J.; Cui, J.Z.; To, E.; Cao, S.; Matsubara, J.A. Evidence for the activation of pyroptotic and apoptotic pathways in RPE cells associated with NLRP3 inflammasome in the rodent eye. J Neuroinflammation 2018, 15, 15. [CrossRef]
- Mullin, N.K.; Voigt, A.P.; Boese, E.A.; Liu, X.; Stone, E.M.; Tucker, B.A.; Mullins, R.F. Transcriptomic and Chromatin Accessibility Analysis of the Human Macular and Peripheral Retinal Pigment Epithelium at the Single-Cell Level. Am J Pathol 2023, 193, 1750-1761. [CrossRef]
- Senabouth, A.; Daniszewski, M.; Lidgerwood, G.E.; Liang, H.H.; Hernandez, D.; Mirzaei, M.; Keenan, S.N.; Zhang, R.; Han, X.; Neavin, D.; et al. Transcriptomic and proteomic retinal pigment epithelium signatures of age-related macular degeneration. Nat Commun 2022, 13, 4233. [CrossRef]
- Galloway, C.A.; Dalvi, S.; Hung, S.S.C.; MacDonald, L.A.; Latchney, L.R.; Wong, R.C.B.; Guymer, R.H.; Mackey, D.A.; Williams, D.S.; Chung, M.M.; et al. Drusen in patient-derived hiPSC-RPE models of macular dystrophies. Proc Natl Acad Sci U S A 2017, 114, E8214-E8223. [CrossRef]
- Hallam, D.; Collin, J.; Bojic, S.; Chichagova, V.; Buskin, A.; Xu, Y.; Lafage, L.; Otten, E.G.; Anyfantis, G.; Mellough, C.; et al. An Induced Pluripotent Stem Cell Patient Specific Model of Complement Factor H (Y402H) Polymorphism Displays Characteristic Features of Age-Related Macular Degeneration and Indicates a Beneficial Role for UV Light Exposure. Stem Cells 2017, 35, 2305-2320. [CrossRef]
- Schmitz-Valckenberg, S.; Fleckenstein, M.; Zouache, M.A.; Pfau, M.; Pappas, C.; Hageman, J.L.; Agron, E.; Malley, C.; Keenan, T.D.L.; Chew, E.Y.; et al. Progression of Age-Related Macular Degeneration Among Individuals Homozygous for Risk Alleles on Chromosome 1 (CFH-CFHR5) or Chromosome 10 (ARMS2/HTRA1) or Both. JAMA Ophthalmol 2022, 140, 252-260. [CrossRef]
- Karg, M.M.; Moorefield, M.; Hoffmann, E.; Philipose, H.; Krasniqi, D.; Hoppe, C.; Shu, D.Y.; Shirahama, S.; Ksander, B.R.; Saint-Geniez, M. Microglia preserve visual function loss in the aging retina by supporting retinal pigment epithelial health. Immun Ageing 2023, 20, 53. [CrossRef]
- Yu, C.; Lad, E.M.; Mathew, R.; Shiraki, N.; Littleton, S.; Chen, Y.; Hou, J.; Schlepckow, K.; Degan, S.; Chew, L.; et al. Microglia at sites of atrophy restrict the progression of retinal degeneration via galectin-3 and Trem2. J Exp Med 2024, 221. [CrossRef]
- Ronning, K.E.; Karlen, S.J.; Miller, E.B.; Burns, M.E. Molecular profiling of resident and infiltrating mononuclear phagocytes during rapid adult retinal degeneration using single-cell RNA sequencing. Sci Rep 2019, 9, 4858. [CrossRef]
- O’Koren, E.G.; Yu, C.; Klingeborn, M.; Wong, A.Y.W.; Prigge, C.L.; Mathew, R.; Kalnitsky, J.; Msallam, R.A.; Silvin, A.; Kay, J.N.; et al. Microglial Function Is Distinct in Different Anatomical Locations during Retinal Homeostasis and Degeneration. Immunity 2019, 50, 723-737 e727. [CrossRef]
- Jeon, C.J.; Strettoi, E.; Masland, R.H. The major cell populations of the mouse retina. J Neurosci 1998, 18, 8936-8946. [CrossRef]
- Corbeil, D.; Karbanova, J.; Fargeas, C.A.; Jaszai, J. Prominin-1 (CD133): Molecular and Cellular Features Across Species. Adv Exp Med Biol 2013, 777, 3-24. [CrossRef]
- Mak, A.B.; Nixon, A.M.; Kittanakom, S.; Stewart, J.M.; Chen, G.I.; Curak, J.; Gingras, A.C.; Mazitschek, R.; Neel, B.G.; Stagljar, I.; et al. Regulation of CD133 by HDAC6 promotes beta-catenin signaling to suppress cancer cell differentiation. Cell Rep 2012, 2, 951-963. [CrossRef]
- Dellett, M.; Sasai, N.; Nishide, K.; Becker, S.; Papadaki, V.; Limb, G.A.; Moore, A.T.; Kondo, T.; Ohnuma, S. Genetic background and light-dependent progression of photoreceptor cell degeneration in Prominin-1 knockout mice. Invest Ophthalmol Vis Sci 2014, 56, 164-176. [CrossRef]
- Puertas-Neyra, K.; Coco-Martin, R.M.; Hernandez-Rodriguez, L.A.; Gobelli, D.; Garcia-Ferrer, Y.; Palma-Vecino, R.; Telleria, J.J.; Simarro, M.; de la Fuente, M.A.; Fernandez-Bueno, I. Clinical exome analysis and targeted gene repair of the c.1354dupT variant in iPSC lines from patients with PROM1-related retinopathies exhibiting diverse phenotypes. Stem Cell Res Ther 2024, 15, 192. [CrossRef]
- Fujinami, K.; Oishi, A.; Yang, L.; Arno, G.; Pontikos, N.; Yoshitake, K.; Fujinami-Yokokawa, Y.; Liu, X.; Hayashi, T.; Katagiri, S.; et al. Clinical and genetic characteristics of 10 Japanese patients with PROM1-associated retinal disorder: A report of the phenotype spectrum and a literature review in the Japanese population. Am J Med Genet C Semin Med Genet 2020, 184, 656-674. [CrossRef]
- Liang, J.; She, X.; Chen, J.; Zhai, Y.; Liu, Y.; Zheng, K.; Gong, Y.; Zhu, H.; Luo, X.; Sun, X. Identification of novel PROM1 mutations responsible for autosomal recessive maculopathy with rod-cone dystrophy. Graefes Arch Clin Exp Ophthalmol 2019, 257, 619-628. [CrossRef]
- Wang, Y.; Wang, P.; Li, S.; Ouyang, J.; Jia, X.; Xiao, X.; Yang, J.; Li, X.; Sun, W.; Zhang, Q. Characterization of PROM1 p.Arg373Cys Variant in a Cohort of Chinese Patients: Macular Dystrophy Plus Peripheral Bone-Spicule Degeneration. Invest Ophthalmol Vis Sci 2021, 62, 19. [CrossRef]
- Clark, B.S.; Stein-O’Brien, G.L.; Shiau, F.; Cannon, G.H.; Davis-Marcisak, E.; Sherman, T.; Santiago, C.P.; Hoang, T.V.; Rajaii, F.; James-Esposito, R.E.; et al. Single-Cell RNA-Seq Analysis of Retinal Development Identifies NFI Factors as Regulating Mitotic Exit and Late-Born Cell Specification. Neuron 2019, 102, 1111-1126 e1115. [CrossRef]
Figure 1.
Prom1 mRNA expression in albino mouse RPE in situ. Representative brightfield images showing chromogenic Prom1 RNAscope (red) in albino mouse eyecup sections. The sections were counterstained with hematoxylin (blue). (A-B) The black arrowheads show Prom1 mRNA expression in mouse RPE in situ. Scale bar 60 mm. (B) The brightfield low-resolution image of the eyecup and the red box showing the area of the eyecup used for imaging. Scale bar 600 mm. (C) No Prom1 labeling in the negative control for Prom1 RNAscope Scale bar 60 mm. (D) Imaging from another mouse eyecup shows Prom1 mRNA expression in mouse RPE in situ (black arrowheads). (E) The brightfield low-resolution image of the eyecup and the red box showing the area of the eyecup used for imaging Scale bar 600 mm. (F) Representative fluorescent image showing Prom1 expression in an albino mouse eyecup (white arrows). The section was counterstained with DAPI (blue). Scale bar 1000 mm. (G) Confocal high-resolution micrograph of Prom1 expression in mouse RPE (white arrows; purple fluorescence). CC=choriocapillaris. Scale bar 100 mm.
Figure 2.
Prom1 RNAscope in pigmented C57BL/6J mouse retinal sections. Brightfield micrographs of (A) pigmented C57BL/6J retinal sections labeled by chromogenic RNAscope for Prom1 (red puncta). Labeling is present in the photoreceptor inner segments (IS) and outer nuclear layer (ONL) (black arrowheads) and a cohort of cells in the inner retina (white arrowheads). Scale bar 60 mm. (B) No labeling was detected in the negative control of chromogenic Prom1 RNAscope. Scale bar 60 mm. (C) Fluorescent confocal micrographs of Prom1 labeling with Cy5 fluorophore (magenta puncta) and DAPI counterstain in C57BL/6J retinal sections showing Prom1 mRNA expression (magenta puncta) in the mouse RPE (white arrows), photoreceptor inner segments (IS), ONL, and a cohort of cells in the inner retina (white arrowheads). Scale bar 200 mm. (D) Negative control of fluorescent Prom1 RNAscope in retinal sections shows no Prom1 labeling. Scale bar 200 mm. (E) Representative high-resolution widefield confocal microscopy using 100X objective showing Prom1 mRNA expression (magenta puncta) in single RPE cells in situ by a fluorescent RNAscope assay (white arrowheads). Sections were counterstained with DAPI (blue). Scale bar 200 mm. (F) Representative fluorescent images showing the presence of Prom1 mRNA in RPE (white arrows), photoreceptor IS, and ONL in mouse retinal sections. DAPI was used as a counterstain. Scale bar 50 mm. CC= choriocapillaris.
Figure 3.
Immunogold transmission electron microscopy in C57BL/6J mouse eyecups. Immunogold TEM of Prom1 in C57BL/6J mouse eyecups. (A-B) TEM micrographs showing positive Prom1 labeling in RPE mitochondria (white arrows). The scale bar is 500 nm. (C) Immunogold TEM of mouse RPE in situ showing cytoplasmic localization of Prom1 (white arrow, top left), mitochondria (white arrow, middle left), and in proximity to mitochondria (bottom right). The scale bar is 500 nm.
Figure 4.
Immunohistochemical detection of autophagy markers in mouse retinal sections. Representative brightfield micrographs showing (A) positive p62 immunolabeling in albino mouse retinal sections and (B) a negative control of p62 immunostaining. (C) LC3-I/II protein is expressed in mouse RPE and other cell types in the retina. (D) Hematoxylin was used to stain the nuclei for both p62 and LC3-I/II immunolabeling. Scale bar 20 mm.
Figure 5.
Subretinal injections of AAV2/1.Prom1.gRNA leads to Prom1-KD in mouse RPE in situ. Representative micrographs of retinal sections of C57BL/6J mice eyes labeled by fluorescent RNAscope for Prom1 (red puncta). (A-B) Low-magnification fluorescent micrographs with wider views of retinal sections injected with control AAV2/1.CRISPR.scr.gRNA. Scale bars of 60 mm and 100 mm. (C-F) Images of various magnifications showing Prom1 labeling (red puncta) in mouse RPE in control sections (gray arrows). Scale bars ranging from 50 mm to 100 mm. The sections were counterstained with DAPI. (G-I) Fluorescent micrographs with wider views of retinal sections injected with experimental AAV2/1.CRISPR.Prom1.gRNA. Scale bars 80 mm and 90 mm. (J-M) Low and high-magnification micrographs of retinal sections injected with AAV2/1.CRISPR.Prom1.gRNA shows patchy Prom1-KD (white arrowheads) in RPE, with areas showing unaltered Prom1 expression (grey arrows). Scale bars ranging from 50 mm to 100 mm.
Figure 6.
Fundus imaging and ERG of mouse eyes with subretinal injection of control or experimental viral vectors. Representative fundus images of mouse eyes injected with (A) control AAV2/1.CRISPR.scr.gRNA or (B-C) experimental AAV2/1.CRISPR.Prom1.gRNA. Images were obtained after 11 weeks of injection. Circles with dashed lines show areas of RPE degeneration in mouse eyes injected with Prom1-gRNA. (D) Representative ERG waveforms (a- and b-waves) in mouse eyes injected with scrambled (scr) or Prom1 gRNA. (E) Quantifying scotopic a-wave ERG responses from mice eyes injected with scr or Prom1-gRNA.
Figure 7.
Histology of mice eyes injected with control or experimental viral vectors. Brightfield micrographs of H&E-stained sections of C57BL/6J eyes injected with either (A) control AAV2/1.CRISPR.scr.gRNA or (B-D) experimental AAV2/1.CRISPR.Prom1.gRNA. Prom1-knockdown causes patchy RPE vacuolization and abnormalities with fluid accumulation between the RPE and PRs (back arrowheads). Scale bars for all micrographs, 60 mm.
Figure 8.
Prom1-knockdown in vivo causes RPE apoptosis and degeneration. Retinal sections of C57BL/6J mouse eyes injected with (A, E) control AAV2/1.CRISPR.scr.gRNA or (B, F) experimental AAV2/1.CRISPR.Prom1.gRNA were used to detect active cleaved caspase-3 by immunohistochemistry. White arrows show positive immunolabeling of RPE cells for active caspase-3 in retinal sections obtained from Prom1-gRNA-injected eyes but not in RPE from control eyes. Serial sections from the same mouse eyes were labeled by fluorescent RNAscope for Prom1. (C, G) Prom1 labeling (red puncta) was observed in eyes injected with control AAV2/1.CRISPR.scr.gRNA, but its expression was reduced in eyes injected with (D, H) experimental AAV2/1.CRISPR.Prom1.gRNA (white arrowheads). Scale bars for all micrographs, 50 mm.
Figure 9.
Interrogation of single-cell human RPE datasets using Spectacle shows Prom1 gene expression in human RPE. The single-nucleus ATAC human RPE dataset (PMID: 36775060) was used to analyze Prom1 gene expression. (A) Violin plot showing Prom1 gene expression in RPE single-cell clusters (RPE_0 to RPE_3). (B) Violin plot showing similar Prom1 gene expression levels across macular and peripheral regions. The patient iPSC-derived RPE dataset (PMID: 35882847) was used to analyze Prom1 gene expression. (C) Volcano plot showing Prom1 gene expression in multiple single-cell RPE clusters and progenitor/RPE cells. (D) Volcano plot showing Prom1 gene expression in control vs. AMD RPE. The differences between red (AMD) and blue (control) bars in some RPE subsets (RPE2, RPE3, and RPE4) suggest changes in Prom1 gene expression between control vs. disease conditions. (E) Plot showing AMD-risk HTRA1 gene expression levels in control vs. AMD conditions.
Figure 10.
Prom1 gene expression in mouse retinal microglia. Analysis of a mouse single-cell RNA dataset (PMID: 30890724) using Spectacle. (A) Heat map showing the Prom1 gene is highly expressed in proliferating and activated microglia; (B) Plot showing Prom1 gene expression in a subset of activated microglia in the degenerating mouse retina under dark conditions. A mouse retinal degeneration (induced by light damage) dataset (PMID: 30850344) was used to analyze Prom1 gene expression in adult retinal microglia. (C) The volcano plot shows the levels of Prom1 gene expression in undamaged microglia (MG0), other clusters of microglia (lcMG), and small clusters of microglia (sMG).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



