
Submitted:
08 October 2024
Posted:
09 October 2024
You are already at the latest version
Abstract
Recent studies have shown that Lipoprotein(a) (Lp(a)) is an important risk factor for a plethora of different cardiovascular diseases. It has been proven that Lp(a) levels are genetically determined, and correlate with risk of cardiovascular disease, independent of lifestyle factors. As of yet, treatment options to reduce Lp(a) levels are limited, but new research into Lp(a) reduction yields promising results. This review delves into Lp(a) biochemistry, mechanism of effect, association between Lp(a) and cardiovascular diseases, and possible therapies to minimize cardiovascular disease.
Keywords:
Lipoprotein(a)
; cardiovascular disease
; atherosclerosis
1. Introduction
Lipoprotein(a) has emerged as an independent and causal risk factor for atherosclerotic cardiovascular disease (CVD) and calcific aortic stenosis through mechanisms associated with atherogenesis, inflammation and thrombosis. Multiple, large-scale, prospective epidemiological studies demonstrate a robust association between elevated Lp(a) levels and increased incidence of ischaemic heart disease (IHD), myocardial infarction (MI), stroke and peripheral vascular disease. Further population-based data identified association of increased Lp(a) with incidence and rate of progression of calcific aortic stenosis. The value of these associations lies in their independence from traditional cardiovascular risk factors including diabetes, hypertension, and smoking. Lp(a) levels are largely determined by genetic factors with minimal influence from dietary or other factors. Therapeutic effects from established cardiovascular treatment strategies on Lp(a) levels are uncertain and do not result in optimal Lp(a) reduction. This review highlights the important role of Lp(a) in CVD.
2. Lipoprotein(a) Biochemistry
The structure of lipoprotein(a) [Lp(a)] (Figure 1) [13] consists of two parts: i) a large, hydrophilic apolipoprotein(a) [apo(a)] glycoprotein and ii) an LDL-like particle containing an ApoB100 glycoprotein on its surface [1]. The apo(a) glycoprotein is bound to the ApoB100 glycoprotein by a single disulfide bond and the presence of this apo(a) glycoprotein differentiates Lp(a) from LDL [1]. Apo(a) is genetically determined (encoded by the Lp(a) gene on chromosome 6) in an autosomal co-dominant pattern of inheritance [2].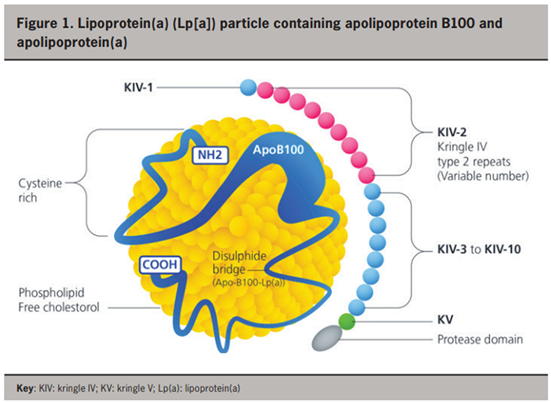
Within the apo(a) molecule are repeated domains called kringles. Polymorphisms within the hypervariable apo(a) gene arise due to varying numbers of kringle IV type 2 (IV2) repeats [3,4], resulting in over forty apo(a) isoforms. This finding is clinically relevant as smaller apo(a) isoforms (with less kringle IV2 repeats) are associated with higher plasma concentration of Lp(a), and increased incidence of coronary heart disease, ischaemic stroke, and calcific aortic stenosis [5,6]. Lp(a) plasma concentration exhibits high heritability and factors such as age, sex, diet, and exercise seemingly exert little influence on Lp(a) concentrations [6]. The variation in kringle IV2 repeats makes accurate measurement of plasma Lp(a) concentrations difficult [4].
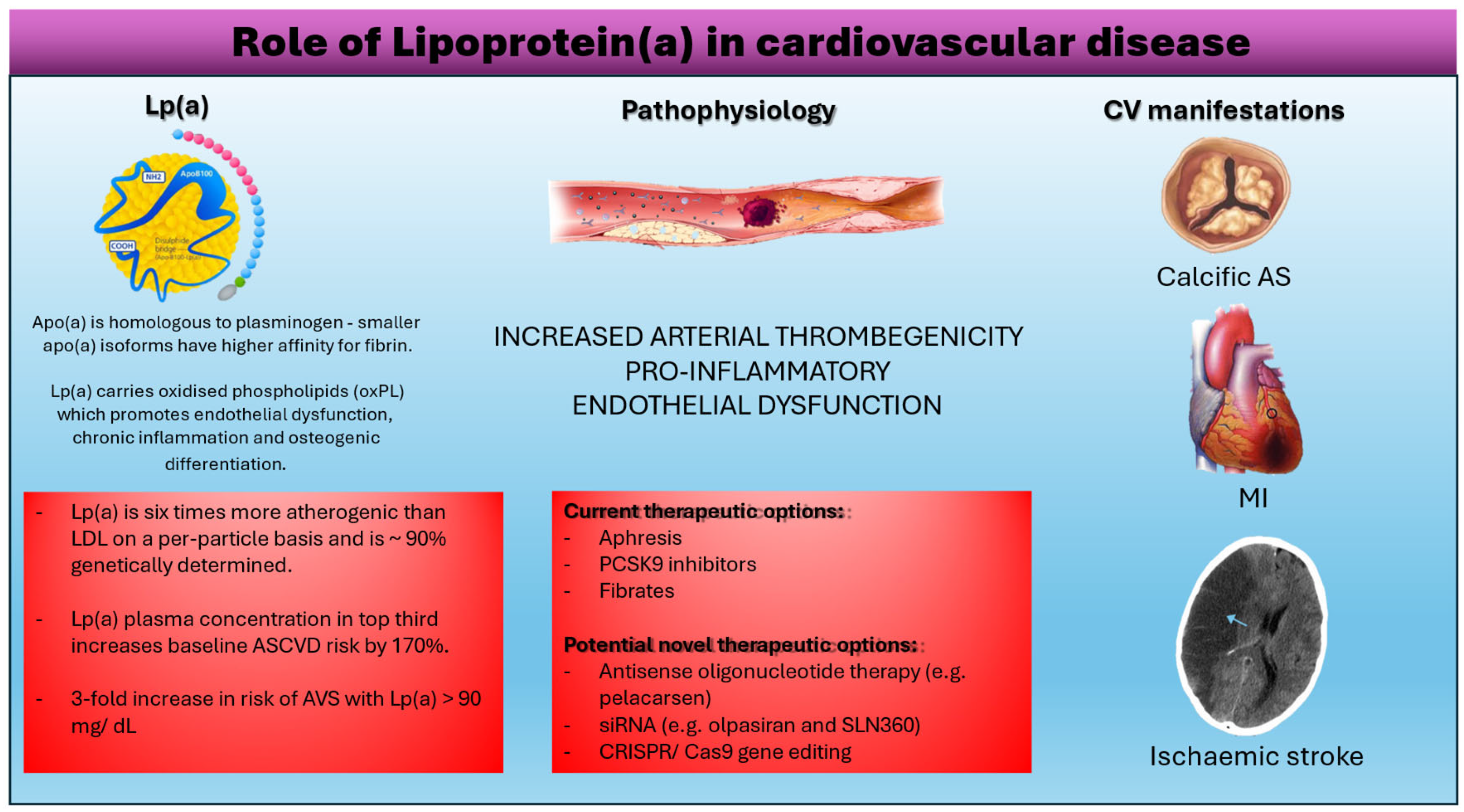
Figure 3.
The role of lipoprotein (a)in cardiovascular disease.
3. Lipoprotein(a) Biology and Genetic Variability
Amino acid sequencing of apo(a) shows remarkable structural similarity to plasminogen [7] with the kringle IV2 repeats of apo(a) being highly homologous to the kringle IV domains of the plasminogen gene. Phylogenetic analysis showed that the LP(a) gene evolved from the plasminogen gene, with Lp(a) only found in humans and hedgehogs [8]. An evolutionary explanation as to why humans evolved to produce high concentrations of Lp(a) is unclear, but research suggests Lp(a) provided a survival advantage through enabling thrombosis which is important in wound healing and increasing haemostasis during parturition as well as reducing major haemorrhage in the brain and airways [9]. In normal physiology, Lp(a) transports cholesterol esters and triglycerides in the bloodstream from liver to peripheral tissues to help maintain lipid homeostasis in the endogenous lipoprotein pathway. Lp(a) is synthesised and metabolised by the liver but whether Lp(a) is cleared by the LDL receptor is unknown [10].
There is extensive variability in Lp(a) levels between individuals and populations that cannot be fully explained by genetic factors alone. Randomized controlled clinical trials show that diets lower in saturated fats only modestly influence Lp(a) levels and often in the opposing direction to LDL cholesterol. The effect of physical activity/exercise is inconsistent, ranging from no change to moderate change in Lp(a) levels. However, this variation is likely modulated by age and type, intensity, and duration of exercise undertaken. Of interest, hormone replacement therapy (HRT) in postmenopausal women has been shown to lower Lp(a) levels with oral being more effective than transdermal estradiol; the type of HRT, dose of estrogen and addition of progestogen do not modify the Lp(a)-lowering effect of HRT. Kidney diseases also result in modulation of Lp(a) levels with increased levels seen in advancing disease stages, dialysis type and apolipoprotein(a) phenotypes. In contrast, Lp(a) levels are reduced in liver diseases in parallel with liver disease progression, although population studies have yielded conflicting results on the associations between Lp(a) levels and nonalcoholic fatty liver disease. Overall, current evidence supports the role of diet, hormones and chronic liver and kidney diseases in modifying Lp(a) levels [11].
Lp(a) is regulated mainly genetically by the LPA gene but involved genetic variants have not been fully elucidated. Improved understanding of the entanglements of genetic Lp(a) regulation may enhance genetic prediction of Lp(a) and CAD risk. Lp(a) concentrations are believed to be largely controlled by one single gene (LPA) that contains a complex interplay of several genetic elements with many effects. The effect of the apo(a) isoforms are, however, modified by many functional single nucleotide polymorphisms (SNPs) distributed over the complete range of allele frequencies (<0.1% to >20%) with pronounced effects on Lp(a) concentrations. A complex interaction is present between the apo (a) isoforms and LPA SNPs. Differences in the Lp(a) trait between ancestries may be caused by differences in the frequency and the isoform association of multiple LPA SNPs with large impact on Lp(a) concentrations and Lp(a) distribution. However, environmental exposures and inflammatory burden have also been proposed as causal factors. A comprehensive catalogue of the functional genetic variation in LPA across ancestries remains elusive but is needed to define the true remainder, which may then be attributable to polygenic influences or environment [12].
4. Mechanism of Lipoprotein(a) Effect
Whilst plasminogen can be broken down by tissue plasminogen activator (tPa), urokinase or streptokinase to form plasmin that degrades clots, apo(a) is resistant to proteolysis due to a single amino acid substitution in the apo(a) gene [1] resulting in an inactive protease domain. This gives apo(a) pro-thrombotic properties as it competes with plasminogen to bind to fibrin but apo(a) does not mediate fibrinolysis. Smaller apo(a) isoforms have a higher affinity for fibrin supporting their higher thrombogenic potential [10]. When this occurs at sites of plaque rupture, this may cause myocardial infarction (MI) and ischaemic stroke. Lp(a) also contains oxidised phospholipids (oxPL) [6] which co-localised with apo(a) in the arterial tunica intima and aortic valve annulus. This promotes endothelial dysfunction (leading to proliferation of vascular smooth muscle cells), increases macrophage apoptosis causing Lp(a) to accumulate in the arterial wall as well as increasing calcification [13]. These mechanisms contribute to the pathogenesis of atherosclerosis and calcific aortic stenosis. This could also explain why Lp(a) is about six times more atherogenic than LDL on a per particle basis [14].
5. Association of Elevated Lp(a) and Cardiovascular Disease
There is emerging consensus from population studies about the effect of Lp(a) levels on cardiovascular disease risk - there being a consistent correlation between Lp(a) levels and risk of atherosclerotic cardiovascular disease (ASCVD) (Table 1) [15]. However, the cardiovascular diseases studied have shared commonalities in their pathogenesis. Lp(a) has shown to have a baseline residual effect on the risk of atherosclerotic cardiovascular disease that currently cannot be fully compensated for purely through lifestyle changes [16]. A meta-analysis of twenty-seven international prospective studies identified “a clear association between Lp(a) and” [17]ASCVD, showing the baseline risk of ASCVD in the top third of Lp(a) concentrations to be 1.7 times higher than the lowest third [17]. Further epidemiological data showed a relationship between higher Lp(a) levels and the incidence of MI. The study involved over 12,000 patients stratified by ethnicity and adjusted for age and sex. The association of elevated Lp(a) concentration with new MI was independent of cardiovascular risk factors including diabetes, hypertension, and smoking. This observational study has also demonstrated an inverse association of isoform size and Lp(a) concentration, indicating a lower risk of MI with higher isoform size [15]. Observational data also suggests increased prevalence of recurrent ischaemic events and rates of repeat revascularisation in patients with previous percutaneous coronary intervention (PCI) [18]. Multiple studies confirmed the association of elevated Lp(a) levels as a predictor for major adverse cardiovascular events following PCI (Table 2) [18].
In 2022, the European Atherosclerosis Society (EAS) published a statement confirming the association between high Lp(a) levels and ASCVD and aortic valve stenosis- even with low LDL levels [19]. After reviewing the data, they concluded that Lp(a) elevation is associated with MI, strokes, and peripheral arterial disease in a primary prevention setting. The EAS also consolidated many theories about the pathogenic mechanisms of Lp(a) elevation with regards to ASCVD, stating that Lp(a)’s pro-inflammatory mechanisms can be attributed to: Lp(a)’s cell signaling properties, its affinity to oxidised phospholipids, and Lp(a)’s ability to promote pro-atherosclerotic chemical synthesis and secretion [19].
The AIM-HIGH sub study reported Lp(a) to be associated with carotid plaques and mural thrombi in 214 trial participants leading the authors to suggest that: “Lp(a), HDL-C or ApoA1 [are] independent factors associated with high-risk plaque features” [20]. Of note, Lp(a) levels do not appear to increase risk of venous thrombosis or affect fibrinolysis with data from the Copenhagen City Heart Study and the Copenhagen General Population Study showing no correlation between Lp(a) levels and risk of venous thrombosis [21].
6. Measurement of Plasma Lp(a) Concentration and Standardization
Circulating plasma Lp(a) levels rise after birth reaching a constant concentration in the first months of life [1,22]. Thereafter, individual Lp(a) concentrations remain relatively stable throughout life ranging widely from <1 to 200 mg/dL in the general population [23] . Many studies show women to be more prone to increased Lp(a) concentrations [24,25] and there are racial differences also reported with Lp(a) lowest in Caucasian patients and highest in patients of African ethnicity [26] The emergence of Lp(a) as a risk factor for cardiovascular disease has led to recommendations for measuring it at least once during lifetime, especially in high-risk populations [27,28]. However, there is controversy regarding standardization and validity of methods used to measure Lp(a) concentrations due to the highly variable size of Lp(a) which is largely determined by the apo(a) size. The resulting inter-individual and intra-individual variability in different populations is a consequence of the fact that most individuals are carriers of two different apo(a) alleles [29,30].This variability has resulted in different methodologies for Lp(a) measurement - either measurement of molarity of Lp(a) or quantification of the mass concentration of Lp(a). The latter are more prone to variation leading to diverging values of different mass-targeting kits, even within the same population with a standard Lp(a) molar concentration [31,32]. Despite this issue and the apo(a)-insensitive quantifying methods, [32,33] commercial kits measuring Lp(a) in mg/dL instead of estimating its molarity in nmol/L are still frequently referenced in practice and the literature [34,35] It is important to note that converting the mass concentration of Lp(a) to its molar equivalent (mg/dL to nmol/L) cannot produce accurate results, despite attempts suggesting an approximate 2–2.5× conversion factor [36].
7. Use of Pharmaceutical Agents to Reduce Lp(a) Levels
Lp(a) concentration is under primary genetic control with modest influence of environmental factors, pharmacologic and apheresis strategies to lower Lp(a) levels have po tential therapeutic importance. Lipid-modifying therapies including statins, ezetimibe, and fibrates reduce cardiovascular risk without substantially affecting Lp(a) levels. Conversely, lipid-modifying therapies such as niacin and CETP inhibitors lower Lp(a) levels without substantially influencing cardiovascular risk. PCSK9 monoclonal antibodies are the first class of pharmacologic agents to lower Lp(a) levels and substantially reduce cardiovascular risk. The clinical benefit of PCSK9 monoclonal antibodies is associated with baseline and on-treatment levels of Lp(a) but this effect is likely due to the dramatic reductions in LDL cholesterol.
Conflicting data exist regarding the effect of statins on Lp(a). Several studies show that the current “gold-standard” treatment to lower LDL cholesterol levels (statins) increase Lp(a) levels [22,34]. Although statins remain one of the most effective and safest drug category for atherosclerotic cardiovascular disease prevention, one study reported a mean 11% increase in Lp(a) levels with their use(37,38,39). The ILLUMINATE trial revealed that, in high-risk CVD patients, Lp(a) levels were positively and dose-dependently correlated with atorvastatin dose [40]. These data should be tempered by data from meta-analyses on the impact of different types and dose regimens of statins showing no clinically significant reduction in Lp(a) levels [41]. Despite this, at best, neutral effect of statins, European Atherosclerosis Society consensus statement suggests that statin therapy should not be discontinued as their cardioprotective actions overcome any risk associated with increased Lp(a) plasma concentrations [24].
As with statins, the data on the effect of ezetimibe on Lp(a) circulating levels is also inconclusive. A metanalysis of seven randomized controlled trials reported ezetimibe to reduce Lp(a) by 7%, an effect considered ineffective in reducing Lp(a)-related risk of CVD events [42]. Conflicting evidence was reported from a large meta-analysis from 10 randomized placebo-controlled clinical trials, demonstrating no effect of ezetimibe therapy on modulating plasma Lp(a) concentrations, either as a monotherapy or in combination with a statin [43].
Niacin (nicotinic acid) is an effective therapeutic agent for raising HDL levels and has been used for reduction in CVD events and mortality [44,45]. Niacin acts by silencing apo(a) gene expression in hepatocytes and is approved treatment for Lp(a) reduction, The effect of niacin is dose-dependent and leads to a 25% to 38% reduction in Lp(a) levels when niacin is administered at a 2 to 4 g daily dosage, respectively. Disappointingly, this reduction in Lp(a) levels has not been associated with any effect on CVD reduction [46]. Although a large meta-analysis of 14 randomized placebo-controlled clinical trials reported a significant reduction by 23% in plasma Lp(a) concentration, the prognostic relevance of this effect has yet to be clarified [47], while the Lp(a)-lowering effect of niacin has not been linked to any clinical benefit, in terms of ASCVD events, so far [48].
The failure of “first line” lipid lowering therapies to show reduction in Lp(a) levels or in the case of niacin, the reduction in Lp(a) not linked to CV event reduction has led to the development and study of agents that target reductions in serum Lp(a) concentrations.
6.1. Apheresis
Currently, the most effective treatment for high Lp(a) levels is apheresis - filtering patients’ blood to selectively remove lipoproteins containing ApoB100. This treatment is well tolerated by patients and reduces both serum LDL and Lp(a) concentrations by 60-70%, thus reducing cardiovascular disease risk by 54-90% [49]. HEART UK guidelines recommend apheresis in patients with Lp(a) levels higher than 60mg/dL, LDL-C levels above 125mg/dL on maximally tolerated cholesterol lowering therapy, and progressive coronary artery disease [50].
6.2. PCSK9 Inhibitors
PCSK9 inhibitors reduce serum LDL levels, but recent research shows that they are also effective in reducing serum Lp(a) concentrations [51]. With PCSK9 receptors inhibited, LDL is more readily absorbed into hepatocytes, thus decreasing serum LDL concentrations. In clinical trials, PCSK9 inhibitors reduce serum Lp(a) concentrations by up to 30% [51]. However, as these trials were not designed to observe changes in Lp(a) concentrations, we cannot assume that this Lp(a) lowering effect is durable or clinically significant.
6.3. Fibrates
Fibrates also lower both serum cholesterol and Lp(a). Fibrates are synthetic ligands of peroxisome proliferator- activated receptor alpha (PPAR alpha). PPAR alpha is a transcription factor predominantly found in fatty-acid metabolising tissues, e.g. liver and kidney tissue. PPAR alpha and fibrates enhance lipolysis in liver tissue by increasing lipoprotein lipase synthesis, whilst simultaneously inhibiting apoC-III gene expression. ApoB production is also decreased through fibrate action [52]. It has been shown in animal studies that free fatty acid uptake and catabolism is enhanced by fibrates, leading clinicians to the conclusion that fibrates promote the catabolism of cholesterol and Lp(a) in the liver, thus decreasing cardiovascular disease risk [52].
7. Trial Data of Targeted Lp(a) Reducing Agents
7.1. Pelacarsen
Pelacarsen is a promising antisense oligonucleotide therapy that reduces serum Lp(a) concentrations. This drug is an RNase H-competent antisense oligonucleotide (ASO). ASO’s bind to complementary mRNA strands using the gapmer approach; RNA-based sequences “frame” a DNA-based gap, allowing mRNA to bind. RNase H1 then cleaves the RNA at the site of RNA binding [53]. The effect of Pelacarsen is enhanced almost 30-fold by adding GalNAc [54]. Pelacarsen is currently in phase 3 clinical trials expected to finish May 2025. The results of early phase trials are promising: serum Lp(a) concentrations decreased between 35% and 80% (p value range 0.003 to <0.001) with multiple administrations, with results lasting for up to 16 weeks [54]. Others, who received weekly doses of 20mg Pelacarsen, measured a reduction in Lp(a) levels of at least 50mg/dL, with results persisting for at least 180 days post-administration [54].
7.2. siRNA
Short interfering RNA (siRNA) therapies, such as Olpasiran and SLN360, are also in development [55]. SiRNA is double stranded RNA, comprised of antisense and sense strands. The antisense strand is complementary to the target mRNA sequence. Once the siRNA enters the cell, the antisense strand is incorporated into the RNA-induced silencing complex (RISC). The activated antisense strand binds to the target mRNA (in this case LPA mRNA), signaling where argonaute proteins (e.g. AGO2) should cleave the target mRNA [56]. The target mRNA is then degraded and unable to synthesise proteins [27]. The antisense strand remains unchanged and can continue to degrade remaining copies of the target mRNA. Olpasiran activity is enhanced when conjugated by GalNAc. Olpasiran duration of action is prolonged, lasting on average between 3 and 6 months [31]. This is because the drug is renally excreted and is active in hepatocytes [57].
Olpasiran is currently undergoing clinical trials. Phase II trials reinforced from animal studies showing a decrease in Lp(a) concentrations of over 90% at doses more than 75mg, with effects lasting for several months [58]. Phase III clinical trials investigating the effectiveness of olpasiran in reducing risk of cardiovascular events in patients with pre-existing atherosclerotic cardiovascular disease and elevated Lp(a) is currently underway and expected to be complete by December 2026 [53].
SLN360 is a GalNAc-modified siRNA that targets Lp(a) mRNA [59]. Through various clinical trials, it has been shown that it also decreases both total cholesterol and Lp(a) concentrations in humans in a dose dependent manner [60]. In a phase I trial, adult patients with high Lp(a) and no known prior cardiovascular diseases, SLN360 reduced Lp(a) concentrations by more than 80% with doses of 600mg, with effects persisting for at least 150 days. It was well tolerated with adverse effects reported being low grade injection site events and headache. One patient also had elevated ALT and AST levels during the trial, which were attributed to the patient having the SARS-CoV-2 vaccine. SLN360 has been approved for phase II studies in patients with pre-existing ASCVD, with an estimated completion in 2024 [53].
8. Future Implications
Although existing pharmacology appears promising in reducing Lp(a), we await data to show whether this can effect cardiovascular events. If proven, then the possibility of a permanent reduction of Lp(a) through gene therapy would become clinically appealing.
8.1. CRISPR/Cas9
CRISPR/Cas9 involves a strand of antisense RNA and Cas9- an endonuclease. The antisense RNA binds to a section of complementary target DNA, which activates Cas9. Cas9 then makes double stranded breaks in the DNA, creating blunt ends in the strand. The target DNA is then excised and “melted,” with the remaining DNA strands repairing themselves by binding the two blunt ends together [61]. As current research points to PCSK9 inhibiting both cholesterol and Lp(a) clearance, research is being conducted in mice and non-human primates to determine whether CRISPR/Cas9 genome editing is effective in permanently lowering serum Lp(a) levels [62]. Current research into CRISPR/Cas9 efficacy is promising- Musunuru et al used nanoparticles with base editors and guide RNA in animals, showing a decrease in plasma PCSK9 levels of approximately 90%. Unlike previous studies, the side effects caused by the treatment-namely liver toxicity- were transient and the duration and efficacy of treatment outweighed these issues [63].
9. Conclusion
Lp(a) is considered a major risk factor for atherosclerotic CVD with several studies confirming an association between elevated Lp(a) levels and incident CAD in the general population. Additionally, observational data suggests Lp(a) is associated with increased adverse events in patients with established CAD and with aortic stenosis. Imaging data suggests the mechanism of Lp(a) and worse CV outcomes may be due to the adverse effect of high Lp(a) levels on plaque vulnerability. Promising pharmacotherapy intervention show reductions in serum Lp(a) levels but trial data of cardiovascular risk reduction is awaited.
Funding
The authors declare no funds were received for writing of this article.
Conflicts of interest
David Austin reports receiving speaker fees from , Astra Zeneca, Pfizer; research grants awarded to Newcastle University from TA Sciences, Kancera, and Astra Zeneca. AZ reports receiving speaker fees from: Amarin, Amgen, Sanofi, Astra Zeneca, Boehringer, Pfizer, Daiichi, Napp.
Abbreviations
Abbreviations in text
| CVD | cardiovascular disease |
| IHD | ischaemic heart disease |
| MI | myocardial infarction |
| LDL | low density lipoprotein |
| oxPL | oxidised lipoprotein |
| ASCVD | atherosclerotic cardiovascular disease |
| PCI | percutaneous coronary intervention |
| MACE | major adverse cardiovascular events |
| CV | cardiovascular |
| AVS | aortic valve stenosis |
| CHD | coronary heart disease |
| ISR | in-stent restenosis |
| CVA | cerebrovascular accident |
| EAS | european atherosclerosis society |
| PCSK9i | proprotein convertase subtilisin/kexin type 9 |
| PPAR alpha | peroxisome proliferator activated receptor alpha |
| ASO | antisense oligonucleotide |
| GalNAc | N-acetylgalactosamine |
| ALT | alanine transaminase |
| AST | aspartate transaminase |
| SARS-Cov-2 | Severe acute respiratory syndrome coronavirus 2 |
| CRISPR | clustered regularly interspaced short palindromic repeats |
References
- Utermann G. The mysteries of lipoprotein(a). Science. 1989 Nov 17;246(4932):904–10. [CrossRef]
- Berg K. A NEW SERUM TYPE SYSTEM IN MAN-THE Lp SYSTEM. Acta Pathologica Microbiologica Scandinavica. 2009 Aug 18;59(3):369–82. [CrossRef]
- Lackner C, Cohen JC, Hobbs HH. Molecular definition of the extreme size polymorphism in apolipoprotein(a). Human Molecular Genetics. 1993 Jan 1;2(7):933–40. [CrossRef]
- Kronenberg F. Lipoprotein(a) measurement issues: Are we making a mountain out of a molehill? Atherosclerosis. 2022 May;349:123–35. [CrossRef]
- Kraft HG, Arno Lingenhel, Silvano Köchl, Hoppichler F, Kronenberg F, Abe A, et al. Apolipoprotein(a) Kringle IV Repeat Number Predicts Risk for Coronary Heart Disease. Arteriosclerosis, Thrombosis, and Vascular Biology. 1996 Jun 1;16(6):713–9. [CrossRef]
- Kronenberg F. Human Genetics and the Causal Role of Lipoprotein(a) for Various Diseases. Cardiovascular Drugs and Therapy. 2016 Feb;30(1):87–100. [CrossRef]
- Eaton DL, Fless GM, Kohr WJ, McLean JW, Xu QT, Miller CG, et al. Partial amino acid sequence of apolipoprotein(a) shows that it is homologous to plasminogen. Proceedings of the National Academy of Sciences of the United States of America. 1987 May 1;84(10):3224–8. [CrossRef]
- Lawn RM, Schwartz K, Patthy L. Convergent evolution of apolipoprotein(a) in primates and hedgehog. Proceedings of the National Academy of Sciences. 1997 Oct 28;94(22):11992–7. [CrossRef]
- Langsted A, Kamstrup PR, Nordestgaard BG. High Lipoprotein(a) and Low Risk of Major Bleeding in Brain and Airways in the General Population: a Mendelian Randomization Study. Clinical Chemistry. 2017 Nov 1;63(11):1714–23. [CrossRef]
- McCormick SPA. Lipoprotein(a): Biology and Clinical Importance. The Clinical Biochemist Reviews. 2004 Feb 1;25(1):69–80.
- Enkhmaa B, Berglund L. [CrossRef]
- Coassin S, Kronenberg F. Atherosclerosis 2022:349;17-35. [CrossRef]
- Wilson DP, Jacobson TA, Jones PH, Koschinsky ML, McNeal CJ, Nordestgaard BG, et al. Use of Lipoprotein(a) in clinical practice: A biomarker whose time has come. A scientific statement from the National Lipid Association. Journal of Clinical Lipidology. 2022 Sep;16(5):e77–95. [CrossRef]
- Björnson E, Adiels M, Taskinen MR, Burgess S, Chapman MJ, Packard CJ, et al. Lipoprotein(a) Is Markedly More Atherogenic Than LDL: An Apolipoprotein B-Based Genetic Analysis. Journal of the American College of Cardiology. 2024 Jan 23;83(3):385–95. [CrossRef]
- Telyuk P;Austin D;Luvai A;Zaman.A “Lipoprotein(a): Insights for the Practicing Clinician.” Journal of clinical medicine. Accessed July 24, 2024. Available online: https://pubmed.ncbi.nlm.nih.gov/35806958/.
- Kronenberg F. Lipoprotein(a): from Causality to Treatment. Current Atherosclerosis Reports [Internet]. 2024 Jan 22 [cited 2024 Feb 7]. Available online: https://pubmed.ncbi.nlm.nih.gov/38252372/.
- Danesh J, Collins R, Peto R. Lipoprotein(a) and Coronary Heart Disease. Circulation. 2000 Sep 5;102(10):1082–5. [CrossRef]
- Telyuk, Pyotr. “Protected: Clinical Utility of Lipoprotein(a): An Interventionist’s Perspective.” The British Journal of Cardiology, July 1, 2024. Available online: https://bjcardio.co.uk/2024/07/clinical-utility-of-lipoproteina-an-interventionists-perspective/.
- Kronenberg F, Mora S, Stroes ESG, Ference BA, Arsenault BJ, Berglund L, et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and aortic stenosis: a European Atherosclerosis Society consensus statement. European Heart Journal. 2022 Aug 18;43(39):3925–46. [CrossRef]
- Zhao XQ, Hatsukami TS, Hippe DS, Sun J, Balu N, Isquith DA, et al. Clinical factors associated with high-risk carotid plaque features as assessed by magnetic resonance imaging in patients with established vascular disease (from the AIM-HIGH Study). The American Journal of Cardiology [Internet]. 2014 Nov 1 [cited 2024 Jun 18];114(9):1412–9. Available online: https://pubmed.ncbi.nlm.nih.gov/25245415.
- Kamstrup PR, Tybjærg -Hansen A, Nordestgaard BG. Genetic Evidence That Lipoprotein(a) Associates With Atherosclerotic Stenosis Rather Than Venous Thrombosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2012 Jul;32(7):1732–41. [CrossRef]
- Strandkjaer, N.; Hansen, M.K.; Nielsen, S.T.; Frikke-Schmidt, R.; Tybjaerg-Hansen, A.; Nordestgaard, B.G.; Tabor, A.; Bundgaard, H.; Iversen, K.; Kamstrup, P.R. Lipoprotein(a) Levels at Birth and in Early Childhood: The COMPARE Study. J. Clin. Endocrinol. Metab. 2022, 107, 324–335. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, M.L.; Fazio, S.; Giugliano, R.P.; Stroes, E.S.G.; Kanevsky, E.; Gouni-Berthold, I.; Im, K.; Lira Pineda, A.; Wasserman, S.M.; Ceska, R.; et al. Lipoprotein(a), PCSK9 Inhibition, and Cardiovascular Risk. Circulation 2019, 139, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, J.; Dobiasova, M.; Adler, L.; Francis, M. Gender differences in plasma levels of lipoprotein (a) in patients with angiographically proven coronary artery disease. Physiol. Res. 2004, 53, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Nago, N.; Kayaba, K.; Hiraoka, J.; Matsuo, H.; Goto, T.; Kario, K.; Tsutsumi, A.; Nakamura, Y.; Igarashi, M. Lipoprotein(a) levels in the Japanese population: Influence of age and sex, and relation to atherosclerotic risk factors. The Jichi Medical School Cohort Study. Am. J. Epidemiol. 1995, 141, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Enkhmaa, B.; Anuurad, E.; Berglund, L. Lipoprotein (a): Impact by ethnicity and environmental and medical conditions. J. Lipid Res. 2016, 57, 1111–1125. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Visseren, F.L.J.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Back, M.; Benetos, A.; Biffi, A.; Boavida, J.M.; Capodanno, D.; et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice. Eur. Heart J. 2021, 42, 3227–3337. [Google Scholar] [CrossRef]
- Ali, S.; Bunker, C.H.; Aston, C.E.; Ukoli, F.A.; Kamboh, M.I. Apolipoprotein A kringle 4 polymorphism and serum lipoprotein (a) concentrations in African blacks. Hum. Biol. 1998, 70, 477–490. [Google Scholar]
- Kraft, H.G.; Lingenhel, A.; Pang, R.W.; Delport, R.; Trommsdorff, M.; Vermaak, H.; Janus, E.D.; Utermann, G. Frequency distributions of apolipoprotein(a) kringle IV repeat alleles and their effects on lipoprotein(a) levels in Caucasian, Asian, and African populations: The distribution of null alleles is non-random. Eur. J. Hum. Genet. 1996, 4, 74–87. [Google Scholar] [CrossRef]
- Tsimikas, S.; Fazio, S.; Viney, N.J.; Xia, S.; Witztum, J.L.; Marcovina, S.M. Relationship of lipoprotein(a) molar concentrations and mass according to lipoprotein(a) thresholds and apolipoprotein(a) isoform size. J. Clin. Lipidol. 2018, 12, 1313–1323. [Google Scholar] [CrossRef]
- McConnell, J.P.; Guadagno, P.A.; Dayspring, T.D.; Hoefner, D.M.; Thiselton, D.L.; Warnick, G.R.; Harris, W.S. Lipoprotein(a) mass: A massively misunderstood metric. J. Clin. Lipidol. 2014, 8, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Marcovina, S.M.; Navabi, N.; Allen, S.; Gonen, A.; Witztum, J.L.; Tsimikas, S. Development and validation of an isoformindependent monoclonal antibody-based ELISA for measurement of lipoprotein(a). J. Lipid Res. 2022, 63, 100239. [Google Scholar] [CrossRef] [PubMed]
- De Boer, L.M.; Oorthuys, A.O.J.; Wiegman, A.; Langendam, M.W.; Kroon, J.; Spijker, R.; Zwinderman, A.H.; Hutten, B.A. Statin therapy and lipoprotein(a) levels: A systematic review and meta-analysis. Eur. J. Prev. Cardiol. 2022, 29, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Guddeti, R.R.; Patil, S.; Ahmed, A.; Sharma, A.; Aboeata, A.; Lavie, C.J.; Alla, V.M. Lipoprotein(a) and calcific aortic valve stenosis: A systematic review. Prog. Cardiovasc. Dis. 2020, 63, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Fazio, S.; Ferdinand, K.C.; Ginsberg, H.N.; Koschinsky, M.L.; Marcovina, S.M.; Moriarty, P.M.; Rader, D.J.; Remaley, A.T.; Reyes-Soffer, G.; et al. NHLBI Working Group Recommendations to Reduce Lipoprotein(a)-Mediated Risk of Cardiovascular Disease and Aortic Stenosis. J. Am. Coll. Cardiol. 2018, 71, 177–192. [Google Scholar] [CrossRef]
- Willeit, P.; Ridker, P.M.; Nestel, P.J.; Simes, J.; Tonkin, A.M.; Pedersen, T.R.; Schwartz, G.G.; Olsson, A.G.; Colhoun, H.M.; Kronenberg, F.; et al. Baseline and on-statin treatment lipoprotein(a) levels for prediction of cardiovascular events: Individual patient-data meta-analysis of statin outcome trials. Lancet 2018, 392, 1311–1320. [Google Scholar] [CrossRef]
- Tsimikas, S.; Gordts, P.; Nora, C.; Yeang, C.; Witztum, J.L. Statin therapy increases lipoprotein(a) levels. Eur. Heart J. 2020, 41, 2275–2284. [Google Scholar] [CrossRef]
- Arsenault, B.J.; Petrides, F.; Tabet, F.; Bao, W.; Hovingh, G.K.; Boekholdt, S.M.; Ramin-Mangata, S.; Meilhac, O.; DeMicco, D.; Rye, K.A.; et al. Effect of atorvastatin, cholesterol ester transfer protein inhibition, and diabetes mellitus on circulating proprotein subtilisin kexin type 9 and lipoprotein(a) levels in patients at high cardiovascular risk. J. Clin. Lipidol. 2018, 12, 130–136. [Google Scholar] [CrossRef]
- Wang, X.; Li, J.; Ju, J.; Fan, Y.; Xu, H. Effect of different types and dosages of statins on plasma lipoprotein(a) levels: A network meta-analysis. Pharmacol. Res. 2021, 163, 105275. [Google Scholar] [CrossRef]
- Awad, K.; Mikhailidis, D.P.; Katsiki, N.; Muntner, P.; Banach, M.; Lipid and Blood Pressure Meta-Analysis Collaboration Group. Effect of Ezetimibe Monotherapy on Plasma Lipoprotein(a) Concentrations in Patients with Primary Hypercholesterolemia: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Drugs 2018, 78, 453–462. [Google Scholar] [CrossRef]
- Sahebkar, A.; Simental-Mendia, L.E.; Pirro, M.; Banach, M.; Watts, G.F.; Sirtori, C.; Al-Rasadi, K.; Atkin, S.L. Impact of ezetimibe on plasma lipoprotein(a) concentrations as monotherapy or in combination with statins: A systematic review and meta-analysis of randomized controlled trials. Sci. Rep. 2018, 8, 17887. [Google Scholar] [CrossRef] [PubMed]
- Altschul, R.; Hoffer, A.; Stephen, J.D. Influence of nicotinic acid on serum cholesterol in man. Arch. Biochem. Biophys. 1955, 54, 558–559. [Google Scholar] [CrossRef] [PubMed]
- Guyton, J.R.; Blazing, M.A.; Hagar, J.; Kashyap, M.L.; Knopp, R.H.; McKenney, J.M.; Nash, D.T.; Nash, S.D. Extended-release niacin vs gemfibrozil for the treatment of low levels of high-density lipoprotein cholesterol. Niaspan-Gemfibrozil Study Group. Arch. Intern. Med. 2000, 160, 1177–1184. [Google Scholar] [CrossRef]
- Parish, S.; Hopewell, J.C.; Hill, M.R.; Marcovina, S.; Valdes-Marquez, E.; Haynes, R.; Offer, A.; Pedersen, T.R.; Baigent, C.; Collins, R.; et al. Impact of Apolipoprotein(a) Isoform Size on Lipoprotein(a) Lowering in the HPS2-THRIVE Study. Circ. Genom. Precis. Med. 2018, 11, e001696. [Google Scholar] [CrossRef]
- Sahebkar, A.; Reiner, Z.; Simental-Mendia, L.E.; Ferretti, G.; Cicero, A.F. Effect of extended-release niacin on plasma lipoprotein(a) levels: A systematic review and meta-analysis of randomized placebo-controlled trials. Metabolism 2016, 65, 1664–1678. [Google Scholar] [CrossRef]
- Julius, U.; Fischer, S. Nicotinic acid as a lipid-modifying drug—A review. Atheroscler. Suppl. 2013, 14, 7–13. [Google Scholar] [CrossRef]
- Enkhmaa B, Berglund L. Statins and Lp(a): The plot thickens. Atherosclerosis. 2019 Oct;289:173–5. [CrossRef]
- Vinci P, Di Girolamo FG, Panizon E, Tosoni LM, Cerrato C, Pellicori F, et al. Lipoprotein(a) as a Risk Factor for Cardiovascular Diseases: Pathophysiology and Treatment Perspectives. International Journal of Environmental Research and Public Health [Internet]. 2023 Sep 6 [cited 2023 Oct 3];20(18):6721. Available online: https://pubmed.ncbi.nlm.nih.gov/37754581/.
- Cegla J, Neely RDermotG, France M, Ferns G, Byrne CD, Halcox J, et al. HEART UK consensus statement on Lipoprotein(a): A call to action. Atherosclerosis [Internet]. 2019 Dec;291:62–70. Available online: https://www.atherosclerosis-journal.com/article/S0021-9150(19)31528-X/fulltext.
- Scheen AJ, Wallemacq C, Lancellotti P. [Inclisiran (Leqvio®), a potent cholesterol-lowering agent by inhibiting PCSK9 using small interfering RNA-based innovative therapy]. Revue Medicale De Liege [Internet]. 2022 Dec 1;77(12):745–51. Available online: https://pubmed.ncbi.nlm.nih.gov/36484754/#:~:text=Inclisiran%20is%20a%20novel%20small.
- Staels B, Dallongeville J, Auwerx J, Schoonjans K, Leitersdorf E, Fruchart JC. Mechanism of Action of Fibrates on Lipid and Lipoprotein Metabolism. Circulation. 1998 Nov 10;98(19):2088–93. [CrossRef]
- Thau H, Neuber S, Emmert MY, Nazari-Shafti TZ. Targeting Lipoprotein(a): Can RNA Therapeutics Provide the Next Step in the Prevention of Cardiovascular Disease? Cardiology and Therapy [Internet]. 2024 Feb 21 [cited 2024 Mar 6]. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10899152/.
- Hardy J, Niman S, Goldfaden RF, Majdi Ashchi, Bisharat M, Huston J, et al. A Review of the Clinical Pharmacology of Pelacarsen: A Lipoprotein(a)-Lowering Agent. American journal of cardiovascular drugs. 2021 Sep 7;22(1):47–54. [CrossRef]
- Tselepis AD. Treatment of Lp(a): Is It the Future or Are We Ready Today? Current Atherosclerosis Reports. 2023 Sep 5. [CrossRef]
- Roberts TC, Langer R, Wood MJA. Advances in oligonucleotide drug delivery. Nature Reviews Drug Discovery [Internet]. 2020 Aug 11;19(10):1–22. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7419031/.
- O’Donoghue ML, G. López JA, Knusel B, Gencer B, Wang H, Wu Y, et al. Study design and rationale for the Olpasiran trials of Cardiovascular Events And lipoproteiN(a) reduction-DOSE finding study (OCEAN(a)-DOSE). American Heart Journal. 2022 Sep;251:61–9. [CrossRef]
- Koren MJ, Moriarty PM, Baum SJ, Neutel J, Hernandez-Illas M, Weintraub HS, et al. Preclinical development and phase 1 trial of a novel siRNA targeting lipoprotein(a). Nature Medicine. 2022 Jan;28(1):96–103. [CrossRef]
- Rider DA, Eisermann M, Löffler K, Aleku M, Swerdlow DI, Dames S, et al. Pre-clinical assessment of SLN360, a novel siRNA targeting LPA, developed to address elevated lipoprotein (a) in cardiovascular disease. Atherosclerosis. 2022 May 1;349:240–7. [CrossRef]
- Nissen SE, Wolski K, Balog C, Swerdlow DI, Scrimgeour AC, Rambaran C, et al. Single Ascending Dose Study of a Short Interfering RNA Targeting Lipoprotein(a) Production in Individuals With Elevated Plasma Lipoprotein(a) Levels. JAMA. 2022 May 3;327(17):1679. [CrossRef]
- Asmamaw M, Zawdie B. Mechanism and Applications of CRISPR/Cas-9-Mediated Genome Editing. Biologics : Targets & Therapy [Internet]. 2021 Aug 21;15(1):353–61. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8388126/.
- Hoekstra M, Miranda Van Eck. Gene Editing for the Treatment of Hypercholesterolemia. Current Atherosclerosis Reports. 2024 Mar 18. [CrossRef]
- Musunuru K, Chadwick AC, Mizoguchi T, Garcia SP, DeNizio JE, Reiss CW, et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature [Internet]. 2021 May 1;593(7859):429–34. Available online: https://www.nature.com/articles/s41586-021-03534-y.

Table 1.
Epidemiological studies suggesting a causal role of Lp(a) in CVD.
| Epidemiological studies | Patient cohort | Study design | Outcome | Results |
|---|---|---|---|---|
|
The Copenhagen City Heart study https://doi.org/10.1161/CIRCULATIONAHA.107.715698 |
9330 randomly drawn patients from general population cohort study | 10 year follow up after blood sampling | Registry-based CV outcomes | stepwise increase in risk of MI with increasing levels of lipoprotein(a), with no evidence of threshold effect |
|
The Copenhagen General Population study Atherosclerosis 2022;349:166–174 |
12006 patients following CT 85884 patients to examine risk of heart disease |
Individuals who underwent cardiac computed tomography to measure mitral and aortic valve calcification and to examine risk of heart valve disease after blood sampling | Incidence of aortic and mitral valve disease | Elevated lipoprotein(a) was genetically and observationally associated with mitral and aortic valve calcification and aortic valve stenosis |
|
Danesh J et al: https://doi.org/10.1161/01.CIR.102.10.1082 |
5436 – non selected population |
Meta-analysis of 27 prospective studies | Mean follow up of 10 years | Confirmed association between Lp(a) and CHD |
|
Erqou et al:Apolipoprotein(a) isoforms and the risk of vascular disease JACC. 2010 May, 55 (19) 2160–2167 |
11,396 patients with vsacular disease and 46,938 controls | Meta-analysis of 40 prospective studies | Assess association of lipoprotein(a) isoforms with cardiovascular disease risk | People with smaller apo(a) isoforms have an approximately 2-fold higher risk of CHD or ischemic stroke than those with larger proteins |
|
Pare et al: Lipoprotein(a) levels and the risk of MI among 7 ethnic groups, INTERHEART study https://doi.org/10.1161/CIRCULATIONAHA.118.034311 |
6086 cases of first MI and 6857 controls from the INTERHEART study stratified by ethnicity and adjusted for age and sex | A total of 775 Africans, 4443 Chinese, 1352 Arabs, 1856 Europeans, 1469 Latin Americans, 1829 South Asians, and 1221 Southeast Asians were included | Incidence of MI | Lp(a) and isoform size varied markedly between ethnic groups. Higher Lp(a) associated with increased MI risk with especially high population burden in South Asians and Latin Americans. Isoform size inversely associated with Lp(a) but did not significantly contribute to risk |
Table 2.
Lp(a) as a predictor of MACE events following PCI.
| Study | Study population | Primary endpoint | Results |
|---|---|---|---|
| Zhang et al, 2023 https://doi.org/10.1016/j.jacl.2023.05.094. | Patients with PCI for ISR | MACE and repeat revascularisation | Increased risk of MACE and repeat revascularisation in high Lp(a) group (HR: 1.31, CI: 1.08 -1.58, P=0.007) |
| Yoon et al, 2021 J Am Coll Cardiol Intv. 2021 Sep, 14 (18) 2059–2068 | Patients with previous PCI | CV death, MI and ischaemic CVA, recurrent ischaemic events | Increased ischaemic events in elevated Lp(a) group (aHR: 1.17, CI: 1.05-1.30, P=0.004) |
| Qin et al, 2013 https://doi.org/10.1016/j.atherosclerosis.2013.01.014 | Meta-analysis, patients with previous PCI to ISR and de-novo lesions | Rate of in-stent restenosis | Increased ISR in patients with elevated Lp(a) (SMD=0.42, CI:0.14-0.71, P=0.003) |
| Liu et al, 2020 https://doi.org/10.1136/heartjnl-2020-316586 | Patients with low LDL following PCI | CV death, CVA, MI and repeat revascularisation | Increased incidence of repeat revascularisation in high Lp(a) group (13.3% vs 6.9%, P<0.05) |
| Park et al, 2015 https://doi.org/10.1111/1440-1681.12396 | Stable angina patients post PCI | MI and revascularisation | Increased rate of restenosis (19.8% vs 7.9%, P=0.001) |
| Kimura et al, 2022 https://doi.org/10.1016/j.jjcc.2022.03.004 | Patient with PCI for de novo lesions | CV death, MI, stent thrombosis, unplanned revascularisation | Higher incidence of MACE in Lp(a)>30mg/dl (33% vs 15.9%, P<0.001) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



