
Submitted:
08 October 2024
Posted:
09 October 2024
You are already at the latest version
Abstract
Microorganisms play a pivotal role in ecology, medicine, agriculture, and biotechnology. To comprehend their diversity, physiology, and functions, isolation and characterization are imperative. This review provides a concise overview of prevalent molecular biotechniques for microbial isolation. It describes the advantages, constraints, and practical applications of both culture-dependent and culture-independent approaches. Discussed methods encompass selective/differential media, 16S rRNA gene sequencing, metagenomics, fluorescence in situ hybridization (FISH), polymerase chain reaction (PCR)-based techniques, and single-cell genomics. Emerging high-throughput sequencing and single-cell isolation methods exemplify cutting-edge procedures explored herein. Integration of these molecular tools offers comprehensive insights into microbial-diversity, community structure, metabolic capabilities, and functional interplays.
Keywords:
molecular biotechniques
; FISH
; isolation
; microorganisms
; sequencing
1. Introduction
Microbes, including bacteria, archaea, fungi, viruses, and other microbes, play vital roles throughout the ecosystems of our world. Microorganisms play key roles in a multitude of activities, including but not limited to nutrient cycling, biodegradation, symbiotic relationships, and disease development (Prasad et al., 2021; Rowland et al., 2018). The isolation of microorganisms from their native habitats has significant relevance as it facilitates the investigation and comprehension of their biology, diversity, and prospective applications (Keller and Zengler, 2004). Nevertheless, the endeavor of microbial isolation encounters several obstacles owing to the intricate composition of microbial communities, the existence of unculturable or yet-to-be-cultivated microorganisms, and the constraints imposed by conventional culture-dependent methodologies (Bodor et al., 2020). To surmount these obstacles, the area of microbial isolation has been significantly transformed by the advent of molecular biotechniques. These approaches have facilitated the identification, characterization, and culture of microorganisms that were previously unattainable (Rashid and Stingl, 2015).
The isolation of microorganisms has significant relevance in several scientific areas and practical applications. Through the acquisition of pure cultures of microorganisms, scientists can explore the distinctive characteristics, metabolic capacities, and genetic attributes of these organisms (Navarrete-Bolaños, 2012). This information is crucial for comprehending the basic components of microbial life, including growth prerequisites, reproductive mechanisms, interactions with hosts, and methods for adaptability (Gould et al., 2018; Van De Veerdonk et al., 2017). Isolated microbes are also regarded as significant assets for biotechnological purposes, including the synthesis of enzymes, antibiotics, biofuels, and other beneficial substances (Yadav, 2021). Moreover, the isolation of pathogenic microbes plays a crucial role in the diagnosis of transmissible diseases, the development of efficient treatments, and the development of targeted medicinal products (Yang and Rothman, 2004). While the process of isolating microorganisms forms a foundational aspect for exploring their diversity, functions, and interactions, it is crucial to recognize the potential drawbacks of relying exclusively on genomic methods in microbiological and biotechnological research. Although genomic approaches provide valuable insights, there are notable disadvantages when used in isolation. For example, exclusive dependence on genomic methods may neglect the advantages of obtaining pure cultures, which play a vital role in achieving specific outcomes in research (Nagarajan and Loh, 2014). Furthermore, molecular methods in diagnosis may present alternatives that bypass the necessity for pathogen culture (Dong et al., 2008). Therefore, a balanced approach, considering both the strengths and potential limitations of isolation and genomic methods, is essential for comprehensive progress in microbiological and biotechnological knowledge. The microbial isolation process presents several problems, principally attributable to the intricate composition of microbial communities and the constraints imposed by conventional culture-based methodologies (Dror et al., 2020). Natural environments provide several obstacles, not the least of which is the high diversity of their microorganisms, many of which are either uncultivable or need growth conditions that are difficult to simulate in a laboratory setting (Pham and Kim, 2012). This causes certain microbial species to be underrepresented in culture collections, which hampers our ability to fully comprehend the diversity of microorganisms using culture techniques. It is also difficult to extract individual bacteria from microbial communities because of the intricate connections and interdependence that exist between various species (Stackebrandt and Embley, 2000). The difficulty of isolating microbes is compounded by factors such as their slow growth rate or the presence of viable but non-culturable (VBNC) cells (Jin et al., 2017). These challenges emphasize the need for novel methods that might circumvent these restrictions and improve microbial isolation (Streit and Schmitz, 2004).
Molecular biotechniques have revolutionized microbial isolation and identification, offering advanced alternatives that bypass the traditional culture-based methods, thereby significantly reducing the need for time-consuming and labor-intensive procedures (Rastogi and Sani, 2011). These methodologies use the molecular characteristics of microbes, including their DNA, RNA, and proteins, to discern, categorize, and investigate them in hitherto unknown ways. One such instance is the use of molecular methodologies, such as metagenomics and PCR amplification targeting certain genetic markers, such as the 16S rRNA gene. These approaches facilitate the identification and description of microorganisms that are either unable to be cultured or have not yet been cultivated (Amit Roy, 2014).
The aforementioned methodologies provide a culture-independent approach for investigating microbial communities, offering a valuable tool for overcoming challenges in the isolation of uncultured microbes. Unlike traditional culture-based methods, which may fail to capture the full diversity of microbial communities, these culture-independent techniques allow for the direct analysis of genetic material, enabling the identification of a wide range of microorganisms. This inclusiveness extends to previously unculturable microbes, as the methods do not rely on cultivation.
By circumventing the need for traditional culture techniques, these methodologies facilitate the process of studying microbial communities and enhance our ability to explore the functional capabilities of uncultured microbes. This not only accelerates the pace of microbial discovery but also contributes to a more comprehensive understanding of microbial ecology. Hence, these methodologies not only broaden our perspective on microbial communities but also enhance the accessibility and efficiency of culturing previously uncultured microbes (Gutleben et al., 2018; Liu et al., 2022; Ranjard et al., 2000). While it is true that selective media and enrichment methods have a historical foundation dating back to the early 20th century, notably pioneered by Martinus Beijerinck, the integration of molecular biotechniques in contemporary approaches has significantly enhanced the precision and efficiency of microbial isolation and growth. The foundational work of Beijerinck laid the groundwork for subsequent advancements, and acknowledging this historical perspective underscores the evolution of methodologies over time (Maukonen et al., 2003; Van Iterson et al., 2013). In addition, advanced methodologies such as whole-genome sequencing and transcriptomics provide valuable insights into the genetic composition, gene expression profiles, and metabolic capacities of microbes, hence aiding their separation and characterization (Maghembe et al., 2020; Maukonen et al., 2003; Sharma et al., 2022). Through the use of molecular biotechniques, scholars can enhance our comprehension of microorganisms, their interactions, and their impacts on several disciplines such as medicine, biotechnology, and environmental science. The primary objective of this narrative review is to provide a full comprehension of molecular biotechniques used in the identification of microorganisms. It highlights their applications, challenges, and prospects, demonstrating the crucial role of these techniques in advancing microbial research across various disciplines.
2. Culture-Dependent Techniques
Selective and differential media play a crucial role in the process of microbial isolation and identification. Selective media are formulated with certain components that possess the ability to impede the proliferation of specific microbes, while concurrently facilitating the development of desirable species. The constituents may include antibiotics, dyes, or compounds that selectively operate upon certain metabolic pathways or physiological attributes of undesired organisms (Kado and Heskett, 1970). Selective media are designed to inhibit the development of undesired microorganisms while facilitating the growth of certain target species, hence enhancing the efficiency of their isolation. For instance, Mannitol Salt Agar (MSA) is a selective medium used to isolate Staphylococcus aureus. The high salt concentration inhibits the growth of other bacteria, while S. aureus, a salt-tolerant pathogen, can thrive. The medium's inclusion of mannitol and a pH indicator aids in identifying S. aureus colonies by their characteristic yellow color resulting from mannitol fermentation.
In contrast, differential media are equipped with indicators or substrates that facilitate the distinction of microorganisms by evaluating their capacity to metabolize certain chemicals or generate distinctive biochemical processes. For instance, when isolating Escherichia coli from a complex sample, MacConkey Agar, a widely used differential medium, can be employed. The medium contains lactose as a substrate, and colonies of E. coli, capable of fermenting lactose, exhibit a distinctive pink coloration, allowing for easy differentiation from non-fermenting bacteria. (Hafeez and Aslanzadeh, 2018). The distinction is often detectable by changes in color or the development of distinct colonies. Differential media play a crucial role in elucidating the physiological and biochemical attributes of isolated microorganisms, hence facilitating their identification and characterization. Together, selective and differential media provide a robust methodology for the isolation and discrimination of microorganisms, hence permitting the examination of their varied characteristics, functionalities, and interrelationships (Na et al., 2015).
Enrichment cultures are a very effective technique used to isolate certain bacteria from intricate microbial populations (Dubey et al., 2006). The approach entails the provision of a tailored growth medium that exhibits selectivity in promoting the development of certain microorganisms while simultaneously impeding the growth of other microbial species. The primary objective of the enrichment culture is to provide optimal circumstances that promote the proliferation of the targeted microorganism by providing specialized nutrients or manipulating of environmental factors. Enrichment cultures facilitate the multiplication of target microorganisms by controlling variables such as temperature, pH, oxygen levels, or the introduction of certain substrates (Spieck and Lipski, 2011). For example, in the isolation of methane-producing methanogens, an enrichment culture may maintain anaerobic conditions, provide a carbon source like acetate, and optimize temperature and pH parameters to favor the growth of these specific microorganisms (Hengsbach et al., 2022). This method shines when used in a population of microorganisms when some are uncommon or grow slowly. Microorganisms of interest may be isolated and studied in detail to learn more about their features, metabolic pathways, and their uses via the use of enrichment cultures. Furthermore, enrichment cultures may be a useful tool for investigating the ecological processes and interactions within microbial populations (Pham and Kim, 2012).
Pure culture isolation is a basic microbiological method that includes obtaining a population of microorganisms composed of a single species or strain (Kaeberlein et al., 2002). The objective is to achieve the isolation and cultivation of a specific bacterium while ensuring its separation from other microbial contaminants or mixed populations. The use of pure cultures is essential in the investigation of the distinct traits, physiological processes, and genetic features of particular microorganisms. The isolation procedure often entails the use of a streaking technique to distribute a sample over a solid agar medium, therefore achieving the dispersion and segregation of individual cells. Single colonies may be created by using streaking or diluting procedures repeatedly, ensuring that each colony originates from a solitary microbial cell. Subsequently, these geographically distinct colonies may be subjected to comprehensive analysis and examination using diverse biochemical, molecular, and physiological methodologies (O’Neill et al., 2009). Pure culture isolation is a crucial technique that unveils the unique characteristics and behaviors of microorganisms, shedding light on their optimal conditions for development, metabolic capacities, pathogenicity, and potential applications in biotechnology and medicine (Overmann et al., 2017). Culturing microorganisms aids in understanding their nutritional needs, metabolic capacities, and antibiotic susceptibilities (Abraham et al., 2020). Furthermore, culture-dependent approach enables the isolation of viable microorganisms and supports various experiments, including genetic engineering and biochemical investigation (Solís-González and Loza-Tavera, 2019). Nevertheless, it is important to acknowledge that these strategies do possess significant limits. Certain bacteria might be difficult to cultivate because of their fastidious character or specialized growth needs, which can result in the underrepresentation of certain microbial taxa. Moreover, culture-dependent methodologies often fail to account for the presence of unculturable or yet-to-be-cultivated microorganisms, hence limiting our comprehension of the whole microbial diversity inside a given sample. Moreover, the process of isolating microorganisms by culture-dependent methods may be a time-intensive endeavor, necessitating several iterations of subculturing and selective procedures. Despite these constraints, culture-dependent methodologies continue to be a significant instrument in the separation of microorganisms and provide a robust basis for the examination and characterization of microorganisms in controlled laboratory environments.
3. 16S rRNA Gene Sequencing
The use of 16S rRNA gene sequencing is a common molecular methodology employed to identify and categorize bacteria. The 16S rRNA gene is ubiquitously found inside the ribosomes of bacteria and archaea. This gene includes areas that are conserved, facilitating primer binding, as well as regions that exhibit variability, offering valuable phylogenetic insights (Qurbani and Hamzah, 2020). The 16S rRNA gene is amplified and sequenced to generate a DNA sequence specific to each microbial species, which allows for precise taxonomic classification and phylogenetic analysis (Hussein et al., 2023). The 16S rRNA gene is amplified by polymerase chain reaction (PCR) utilizing universal primers that target the conserved regions. PCR amplification results in millions of copies of the target gene, facilitating its detection and sequencing. Various sequencing methods, including Sanger sequencing and next-generation sequencing (NGS) platforms, are used to sequence the purified PCR product after amplification (Zhu et al., 2020). Bioinformatics methods are used to check the derived 16S rRNA gene sequences against reference databases like the Ribosomal Database Project (RDP) and the Greengenes database. The Ribosomal Database Project (RDP) is widely recognized for its comprehensive coverage of microbial taxa and is frequently updated to incorporate the latest taxonomic information. On the other hand, the Greengenes database is known for its emphasis on phylogenetic diversity and is particularly valuable for studies focused on ecological relationships among microbial communities. RDP introduce taxonomic biases and computational challenges, while Greengenes, emphasizing phylogenetic diversity, offer limited taxonomic resolution and be more suited to ecological studies. Algorithms for sequence alignment can find the most closely related sequences, and then use that information to assign taxonomies. Analyzing the 16S rRNA gene sequences of various microorganisms allows for the construction of phylogenetic trees, which provide a graphical representation of the evolutionary connections between these species (Qurbani et al., 2022b). Despite its widespread use, 16S rRNA gene sequencing has limitations. It lacks strain-level resolution, may introduce bias due to universal primers, and is sensitive to contamination, especially in low-biomass samples. These factors should be considered for accurate interpretation in various scientific fields.
Data from the sequencing of the 16S rRNA gene may be analyzed using a number of different programs. Quantitative Insights into Microbial Ecology (QIIME), mothur, and DADA2 are some examples of such programs; they include extensive pipelines for quality control, sequence grouping, taxonomy assignment, and diversity analysis (Prodan et al., 2020). Reference databases like as SILVA, RDP, and Greengenes are widely recognized as invaluable tools for comparing and identifying microbial sequences (Glöckner et al., 2017). Numerous fields of microbiology may benefit from the sequencing of the 16S rRNA gene. It's used to identify and categorize bacterial and archaeal isolates, especially in situations when culture-based approaches have proven difficult or impossible to utilize (Church et al., 2020). It helps researchers find and describe new microbes, and it may provide information about the composition and distribution of microbial communities in various ecosystems (Fukuda et al., 2016). In addition, 16S rRNA gene sequencing has been used for a variety of purposes in the fields of medicine, microbiology, ecology, food science, and even environmental monitoring (Church et al., 2020; Ercolini, 2013). 16S rRNA gene sequencing has been used in case studies to examine the role of the gut microbiota in health and illness, to monitor the dissemination of infections, to evaluate the dynamics of microbial communities in soil and water systems, and to characterize the microbial composition of foods (Ju and Zhang, 2015).
4. Metagenomics
Metagenomics is a robust methodology used for the investigation of genetic material (DNA or RNA) that is directly obtained from environmental or clinical samples. In contrast to conventional culture-dependent approaches, metagenomics enables the comprehensive examination of whole microbial communities without necessitating the isolation and cultivation of individual microbes (Junca and Venegas, 2016; Liu et al., 2022). The workflow of metagenomics is summarized in (Figure 1.), which involves sample collection, DNA extraction, library preparation, sequencing, and bioinformatics analysis to generate high-quality metagenomically assembled genomes (MAGs).
In the past, conventional methods involving cloning were prevalent in illustrating metagenomics, However recent advancements have transformed the landscape, primarily through the adoption of direct next-generation sequencing (NGS) methods (Kulski, 2016).
Shotgun metagenomics is a technique that entails the sequencing of the whole genomic DNA inside a given sample, therefore offering a full perspective on the microbial population and its putative/potential functional capabilities. This technique facilitates the detection and classification of all microorganisms contained within a given sample, including both culturable and non-culturable species (Ju and Zhang, 2015). Short-read NGS technologies, such as Illumina sequencing, have been widely adopted due to their high-throughput and cost-effectiveness (Lee et al., 2013). However, the assembly contiguity of MAGs generated from short-read data is constrained by repeat elements, which has led to the exploration of long-read sequencing technologies, including single-tube long fragment read (stLFR), PacBio, and Nanopore (Liu et al., 2022). These long-read methods have proven instrumental in reconstructing complete genomes from diverse environmental samples, overcoming challenges associated with short-read technologies (van Dijk et al., 2023). While MAGs have become standard in metagenomic analysis, they are not without limitations, such as difficulties in resolving 16S rRNA genes and biosynthetic gene clusters. The use of long-read sequencing technologies and advanced binning algorithms has improved MAG quality, enhancing their value for functional interpretation and metabolic reconstruction (Singleton et al., 2021).
Additionally, metagenomics, with its evolving methodologies, is complemented by culturomics, a targeted strategy aimed at bringing previously uncultured organisms into laboratory cultivation (Li et al., 2023). Culturomics, an innovative approach, involves the use of diverse culture conditions, including variations in media composition, temperature, and atmospheric conditions, to simulate the complex and varied environments from which microbial samples are derived (Huang et al., 2023). This method has garnered notable success in isolating and characterizing previously elusive microbial species (Lagier et al., 2018). Advancements in culturomics, such as high-throughput culturing techniques, have expanded the scope of cultivation efforts. Culturomics, when integrated with metagenomic data analysis methodologies, provides a synergistic approach, allowing researchers to identify microbes and functions of interest more effectively (Huang et al., 2023).
Metagenomics provides insights into microbial communities and offers opportunities to bring uncultured organisms into cultivation. Strategies such as Culturomics, in situ culture, single-cell isolation, and iChip, coupled with advanced metagenomic data analysis methodologies, present new avenues for identifying target microbes and devising targeted approaches for isolation (Li et al., 2023; Yu et al., 2022). Despite these advancements, the interpretation of data to uncover nutritional or conditional requirements for cultivating uncultured organisms remains a significant challenge in the evolving field of metagenomics.
The capacity of metagenomics to evaluate the functional potential of microbial communities is one of its primary benefits. Researchers are able to discover additional information regarding the genes and metabolic pathways in a given microbial community by examining the collected DNA sequences. An understanding of the ecological functions, nutrient cycling capacities, and possible interactions of community microbes may be gained through this functional potential study (Oremland et al., 2005; Riesenfeld et al., 2004; Steele and Streit, 2005; Tringe et al., 2005). To conduct a comprehensive analysis of metagenomic data, various bioinformatics tools and pipelines are commonly employed. Taxonomic classification is often performed using tools such as Kraken2 (Wood et al., 2021) or MetaPhlAn2 (Truong et al., 2015), while functional annotation can be achieved using tools like HUMAnN3 (Beghini et al., 2021) or MG-RAST (Keegan et al., 2016). These tools aid in identifying the microbial composition and potential functional roles within a given community. Furthermore, it aids in comprehending the functional differences across various environments or pathological conditions (Martín et al., 2014). It facilitates the diagnosis of infectious illnesses, enables the detection of antibiotic resistance genes, and allows for the monitoring of alterations in microbial communities during disease development or therapy (Chen et al., 2019; Yasir et al., 2022). The ongoing progress in this area contributes to the expansion of our understanding of the microbial realm and has potential implications in other domains, including environmental surveillance, biotechnology, and medical diagnostics.
5. Fluorescence In Situ Hybridization
Fluorescence in situ Hybridization is a robust molecular methodology used for to identify, visualize, and quantify microorganisms inside their indigenous habitats (Amann et al., 2001). It is a useful technique for investigating microbial populations and has applications in many fields, including ecology, environmental science, and medical diagnostics (Caracciolo et al., 2010; Frickmann et al., 2017).FISH works by using fluorescently labeled DNA or RNA probes to bind to their respective target nucleic acids in the organisms of interest (Wagner et al., 2003). The probes have been precisely engineered to effectively complement and selectively attach to the specific or conserved sequences found within the nucleic acids of the target bacteria. The probes are affixed with fluorophores that produce fluorescence upon excitation with certain wavelengths of light. The hybridization of the probes with the nucleic acids of the target bacteria enables their precise identification and visualization (Azevedo et al., 2022).
FISH enables the direct observation of microorganisms in their native habitats, including soil, water, biofilms, and tissue specimens. Following the hybridization process, the samples are subjected to examination by fluorescence microscopy, wherein the fluorophore-labeled probes generate fluorescent signals that can be collected and then studied (Levsky and Singer, 2003). The visualization of individual cells or aggregates offers valuable insights into the spatial distribution, abundance, and interactions of microorganisms within the given sample. The quantification of microorganisms may be accomplished by two methods: counting cells that have been tagged with fluorescent markers, or by using image analysis software to evaluate the intensity of fluorescence and establish a correlation with the number of cells (García-Hernández et al., 2021). FISH proved excellent for identifying microorganisms that are difficult to grow or identify using standard techniques, delivering speedy and reliable diagnostic information at the genus to species level (Lewis et al., 2021). The design of FISH probes tailored for uncultured microbial groups involves a meticulous process wherein conserved regions, notably ribosomal RNA (rRNA) sequences, are strategically targeted. This design strategy hinges upon the comprehensive utilization of molecular databases housing sequences from studied unculturable microbial groups.
FISH may be used with other methods to improve its capabilities and deliver more in-depth insights. FISH, for example, may be used in conjunction with flow cytometry i.e. in FACS (fluorescence-activated cell sorting) to perform high-throughput analysis and sorting of microorganisms based on their fluorescence signals (Pereira et al., 2022). FISH may also be used in combination with other molecular approaches, such as metagenomics or transcriptomics, for connecting the identification and activity of microbes within complex communities (Dekas and Orphan, 2011; Nural Yaman et al., 2022). Such integrative techniques help us understand microbial activities, interactions, and community dynamics better. Researchers may acquire a more thorough knowledge of microbial populations and their roles by integrating FISH with other methods, opening the path for progress in research, diagnostics, and environmental monitoring. While FISH is a powerful tool for studying microbial populations, it comes with certain limitations. Accuracy can be influenced by probe specificity, leading to potential false-positive or false-negative results. Discrimination between closely related strains may pose challenges, and the technique provides a snapshot rather than capturing dynamic changes over time.
6. PCR-Based Techniques
PCR-based methods for microbe detection, identification, and quantification have revolutionized microbial isolation. PCR amplifies particular nucleic acid sequences, making these methods useful for investigating microbial diversity, identifying infectious diseases, and monitoring microbial populations (Steffan and Atlas, 1991; Valones et al., 2009). Traditional PCR is commonly used to identify microbes. It involves the amplification of a specified area of microbial DNA or RNA using specific primers. Researchers may selectively amplify and identify certain microbes in a sample by designing primers that target conserved microbial gene sequences. PCR uses unique genome sequences to identify and differentiate microorganisms (Rompré et al., 2002). Nested and multiplex PCR are variations of conventional PCR to improve microorganism identification sensitivity and specificity. Nested PCR uses internal primers to amplify a smaller portion of the first PCR amplicon in the second cycle (Šeligová et al., 2020). This method lowers false-positive rates and strengthens detection limits. Multiplex PCR uses several primer sets to amplify multiple target sequences. This method is excellent for identifying and differentiating several microorganisms in one reaction (Markoulatos et al., 2002). Advanced PCR methods like Quantitative PCR (qPCR) and digital PCR can quantify microorganisms in a sample. qPCR quantifies the original target sequence by measuring real-time amplification of target DNA or RNA. This method accurately quantifies microorganisms and tracks their population abundance (Smith and Osborn, 2009). Conversely, digital PCR allows absolute quantification of target sequences without the need for standard curves by dividing the PCR reaction into thousands of separate reactions. Even in diverse microbial populations, this method enables precise quantification (Ayhan et al., 2021).
PCR-based methodologies are extensively used in the identification and quantification of microorganisms. In the realm of diagnosing infectious diseases, these tools serve the purpose of identifying particular microbiological pathogens in patient samples (Yang and Rothman, 2004). These techniques are also utilized in environmental monitoring to detect and quantify microorganisms in various ecosystems, such as soil, water, or air (Peccia and Hernandez, 2006). The study of microbial ecology and biodiversity, as well as the safety of food supplies, rely heavily on these methods (Cai et al., 2014; Cocolin et al., 2011; Nadarajah and Kumar, 2019). PCR-based microbial identification has improved, yet still has drawbacks. Contamination by even a few target DNA copies may cause false-positive findings, hence, laboratory techniques and controls must be strict to reduce contamination. Another challenge is the limited information provided by PCR-based techniques alone. They can detect and measure microbes, although sequencing or culture may still be needed for full characterization. PCR-based methods may also struggle to discover new or unculturable microorganisms that lack specific target sequences. Advancements in PCR-based techniques continue to address these challenges. High-throughput sequencing methods like NGS can sequence and identify millions of DNA fragments, allowing microbial community study. PCR-based microbe identification is more sensitive, specific, and accurate due to advances in primer design, DNA extraction, and bioinformatics.
7. Single-Cell Genomics
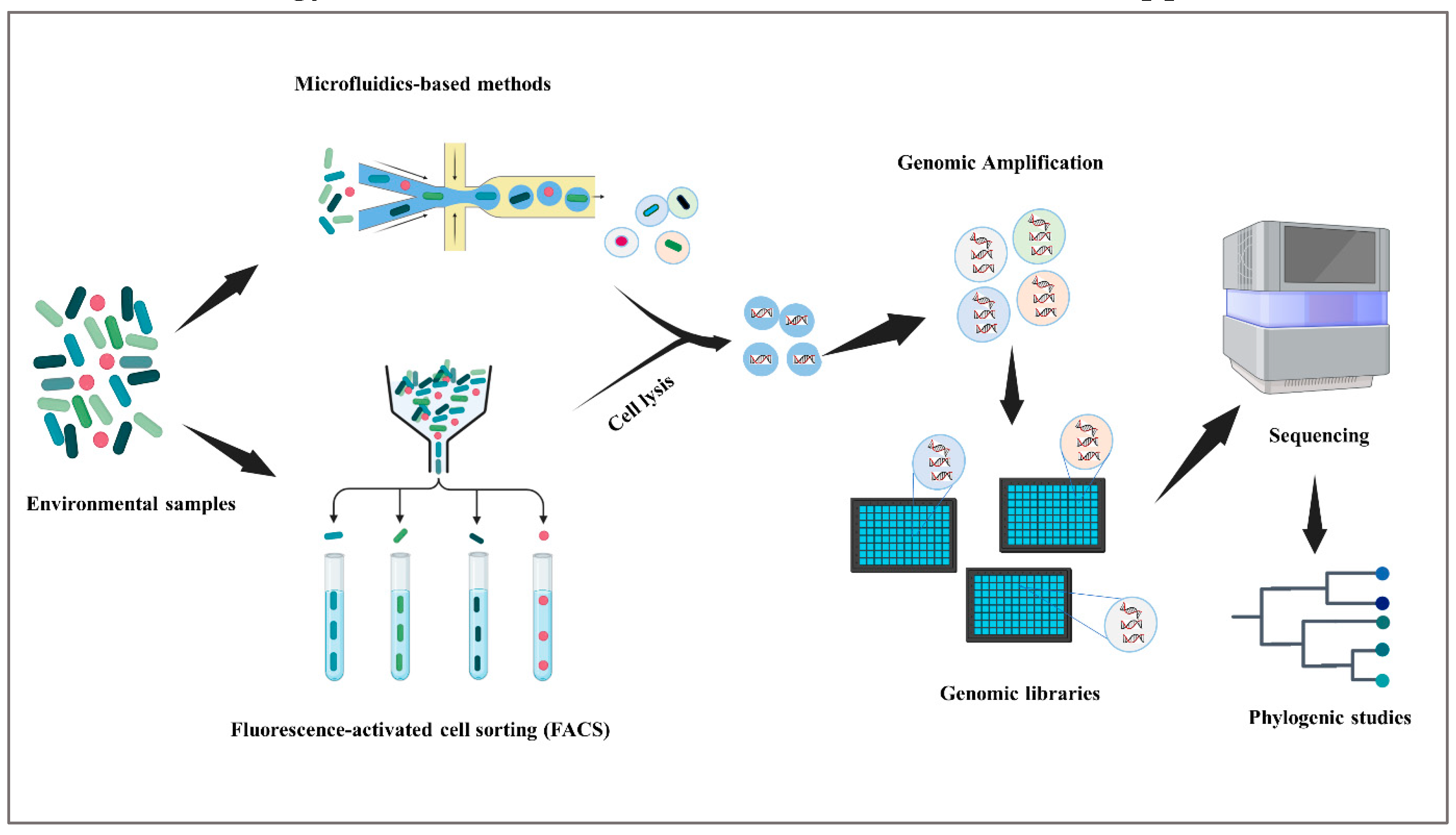
Single-cell genomics is a potent technology that has transformed the area of microbial identification by allowing the genetic study and characterization of individual microorganisms (Blainey, 2013). This particular approach (Figure 2) has played a pivotal role in the examination of the genomic variability among microorganisms, the comprehension of species that are difficult to cultivate, and the elucidation of intricate microbial communities (Gawad et al., 2016). Microfluidics-based technologies allow for high-throughput and very accurate single-cell separation. Cells may be encapsulated in small droplets or chambers on these platforms, making them amenable to controlled manipulation, lysis, and genetic study. The advantages of microfluidics-based single-cell isolation methods include enhanced cell viability, less contamination, and higher efficiency, all of which facilitate the simultaneous study of large numbers of isolated cells (Cao et al., 2023). Nonetheless, successful implementation of single-cell genomics often necessitates whole genome amplification (WGA) to obtain comprehensive genome sequences (Huang et al., 2015). This process is crucial for overcoming the limitations of low DNA content in individual cells. However, it is imperative to recognize potential challenges associated with WGA, including coverage bias, which may impact the accuracy and completeness of genomic data. Recent innovations, such as mini-metagenomics, have emerged to address these challenges by employing limited cell numbers, thereby improving coverage uniformity and reducing biases related to traditional WGA methods (Yu, 2016). Integrating this information seamlessly into the discussion enhances the overall understanding of single-cell genomics methodologies. The field of single-cell genomics offers significant contributions to understanding the genetic constituents, metabolic capabilities, and functional attributes of individual microorganisms. Through the examination of cellular genomes, scientists are able to discern and describe novel microbial species, ascertain their evolutionary origins, and get insights into their ecological functions within complex microbial communities (Müller and Nebe-Von-Caron, 2010). Furthermore, the utilization of single-cell genomics facilitates the recognition of genetic alterations, including single nucleotide polymorphisms (SNPs), copy number variations (CNVs), and genomic rearrangements. These genetic variations play a crucial role in the proliferation of microbial variety and the process of evolution (Grün and Van Oudenaarden, 2015). The use of this method also enables the identification of previously unknown genes and metabolic pathways, hence presenting prospects for the application of biotechnology and the advancement of innovative treatment approaches.
One of the major advantages associated with single-cell genomics is its capacity to investigate microorganisms that pose difficulties or are unfeasible to cultivate using conventional methodologies. Numerous microorganisms present in natural habitats resist culture, hence constraining our comprehension of their biological characteristics and functions (Yasen et al., 2020). The use of single-cell genomics enables researchers to get genomic data directly from microorganisms that are not amenable to cultivation. This approach offers valuable insights into the genetic composition, metabolic abilities, and possible interactions of these microbes with other species (Stepanauskas, 2012). This knowledge contributes to our comprehension of the microbial world and broadens our perspective on microbial diversity and evolutionary processes. The field of single-cell genomics is advancing rapidly, and the future holds tremendous promise. Single-cell isolation, amplification, and sequencing techniques continue to improve in precision and efficacy as a result of technological development. This permits a more in-depth investigation of microbial diversity and ecological interactions. In addition, the integration of single-cell genomics with other omics techniques, such as metagenomics and metatranscriptomics, will provide a deeper understanding of complex microbial communities (Cheng et al., 2019; Franzosa et al., 2015). The advancement of novel computational tools and analytical techniques will significantly augment the process of data interpretation and integration. This will empower researchers to untangle the complex associations between genotypes and phenotypes in individual microorganisms. The potential for expanding our knowledge of microbial diversity, evolution, and their ramifications in several fields such as environmental science, biotechnology, and human health is immense in the context of single-cell genomics.
8. Recent Advancements and Emerging Technologies
Due to technical advances and multidisciplinary methods, microorganism isolation has advanced significantly in recent years. This has enabled us to examine microbes in unprecedented depth. Advanced approaches include high-throughput sequencing systems, single-cell isolation and analysis, integration of several techniques, and artificial intelligence (AI)and data analysis tools (Franzosa et al., 2015; Gawad et al., 2016). High-throughput sequencing tools like NGS and third-generation sequencing have revolutionized microbial identification by sequencing huge numbers of genomes quickly and inexpensively. These systems have greatly improved DNA sequencing throughput, speed, and accuracy, enabling complete microbial community study and functional potential assessment (Mayo et al., 2014). They contributed to discovering new microbial species, characterizing complicated ecosystems, and studying microbial diversity and evolution (Bokulich et al., 2012). Modern single-cell isolation and analysis tools have improved our knowledge of microbes at the cell level (Gawad et al., 2016). Microfluidics-based platforms, such as droplet microfluidics and nanoliter-scale technologies, provide high-throughput and precise single-cell isolation, enabling the investigation of individual microorganisms (Cao et al., 2023). Single-cell genomics approaches like whole-genome amplification and sequencing reveal cell genetics, metabolic capacity, and function. These methods have transformed the study of unculturable microorganisms and microbial diversity (Cheng et al., 2019). Multi-method microbial identification and analysis has become effective. through combining metagenomics, metatranscriptomics, metaproteomics, and metabolomics improves a comprehensive understanding of microbial communities (Gutleben et al., 2018; Palazzotto and Weber, 2018). By integrating data from multiple omics levels, researchers can decipher the functional capabilities, interactions, and ecological roles of microorganisms within complex ecosystems (Franzosa et al., 2015). Integrative techniques provide a comprehensive perspective of microbial systems and a greater understanding of diversity and functions. Artificial intelligence and enhanced data analysis techniques have transformed large-scale microbiological data processing and interpretation. Machine learning algorithms and AI-based models are being used to analyze complicated microbial datasets, predict functions, and find community patterns (Li et al., 2021). These tools aid in the classification of microbial taxa, functional prediction, and the discovery of novel genes or metabolic pathways (Liu et al., 2023). In addition, the use of AI-driven techniques enables the investigation of microbial interactions, and ecological networks, and the discovery of possible microbial biomarkers for diverse applications, such as environmental monitoring and disease detection (He et al., 2022; Wani et al., 2022). These advances might help uncover the microbial world and boost environmental research, biotechnology, and human health.
9. The Role of Molecular Biotechniques in Addressing Antibiotic Resistance
Antibiotic resistance remains one of the most formidable challenges in modern healthcare, threatening to undermine decades of medical advances (Ahmed et al., 2024; Hussein et al., 2024; Kader et al., 2024; K. Qurbani et al., 2024). The rapid emergence of multidrug-resistant (MDR) pathogens necessitates advanced methods for their detection, isolation, and characterization. Molecular biotechniques have proven indispensable in this regard, offering precision and efficiency in identifying antibiotic-resistant microorganisms (Pauter et al., 2020). Techniques such as PCR, qPCR, and whole-genome sequencing (WGS) have become key tools in elucidating the genetic mechanisms underpinning resistance, such as mutations in target genes and the acquisition of resistance genes through horizontal gene transfer (Yamin et al., 2023). These molecular approaches allow for the identification of specific resistance genes, including those conferring resistance to critical antibiotic classes like β-lactams, aminoglycosides, and fluoroquinolones (Darby et al., 2023). Furthermore, the use of molecular techniques facilitates large-scale surveillance of antibiotic resistance in clinical and environmental contexts, providing a clearer understanding of how resistance genes spread within microbial communities (Struelens et al., 2024). This real-time detection and monitoring are essential for tracking the dissemination of MDR pathogens and for informing the development of novel therapeutic interventions, including next-generation antibiotics and innovative alternatives like bacteriophage therapy and nanoparticles (Aziz et al., 2023; Qurbani et al., 2022a; K. A. Qurbani et al., 2024).
Molecular biotechniques are thus not only vital for understanding resistance mechanisms but also play a pivotal role in combating the global threat of antibiotic resistance. Their ability to detect resistance at a genetic level offers unparalleled insights that inform both treatment strategies and public health interventions aimed at curbing the spread of resistant pathogens.
10. Molecular Biotechniques in Microbial Isolation and Bioremediation
In addition to their critical role in combating antibiotic resistance, molecular biotechniques have revolutionized the isolation and identification of microorganisms with significant biotechnological potential, particularly in the field of bioremediation. Microorganisms play a fundamental role in the biodegradation of pollutants and the detoxification of hazardous substances, including heavy metals, hydrocarbons, and industrial chemicals (Qurbani et al., 2022b; Qurbani and Hamzah, 2020). The application of molecular biotechniques, such as metagenomics, transcriptomics, and proteomics, has allowed for the identification and characterization of microbial communities responsible for these processes, even when traditional culture methods have proven insufficient (Mishra et al., 2021).
One of the most pressing environmental challenges is heavy metal contamination, which poses severe risks to ecosystems and human health. Toxic metals such as lead, mercury, cadmium, and arsenic accumulate in the environment due to industrial activities, threatening biodiversity and increasing the risk of cancers, neurotoxicity, and other severe health conditions in humans (Ali et al., 2024; Qurbani and Hamzah, 2021). The isolation and identification of microorganisms capable of tolerating and detoxifying heavy metals are critical to addressing this issue. Through the use of molecular techniques, researchers can unravel the genetic and metabolic pathways that allow certain microorganisms to thrive in metal-contaminated environments and transform or sequester these toxic substances (Barman et al., 2020).
Molecular biotechniques bypass the limitations of culture-based approaches by enabling the direct analysis of microbial communities within polluted environments. This provides a more comprehensive view of the microbial diversity and metabolic potential that underpins effective bioremediation processes (Sharma et al., 2021). Such insights are invaluable for optimizing microbial bioremediation strategies, which play a pivotal role in mitigating environmental pollution and reducing the risks associated with heavy metal toxicity, including their potential to cause cancer (Saravanan et al., 2023). By advancing our understanding of microbial ecology in polluted environments, molecular biotechniques enhance the development of sustainable and efficient bioremediation technologies.
11. Conclusions
Molecular biotechniques aid in the identification of microorganisms, helping researchers understand their diversity and complexity. This review encompasses various molecular approaches, including 16S rRNA gene sequencing, metagenomics, FISH, and PCR-based methods. These approaches have a number of benefits, including selective growth of target microorganisms, precise taxonomic identification, and the capacity to investigate unculturable species. They are used in a variety of fields, including ecology, medical diagnostics, environmental monitoring, and biotechnology. The importance of molecular biotechniques in microbial identification lies in their ability to provide insights into microbial diversity, functions, and interactions. Researchers may discover new species, and examine their genetics, metabolic capacity, and environmental functions using these methods. These methods help identify, diagnose, and monitor microbial infections, improving public health and disease control. Overall, molecular biotechniques have transformed microbial isolation, expanding our understanding of the microbial world and its importance in different sectors. These methods and future advances will help us understand microbes and uncover discoveries that might improve human health, the environment, and biotechnology.
Future Perspective
Molecular biotechniques for microorganism isolation and identification will undergo transformative advances. Innovations in high-throughput sequencing and single-cell analysis are set to significantly improve precision in characterizing microbial communities. Integration of multi-omics approaches will offer a comprehensive understanding of microbial functions. Artificial intelligence and machine learning will accelerate data analysis, aiding in the identification of novel microbial species. In the realm of personalized medicine, molecular biotechniques will contribute to tailoring therapeutic strategies by exploring the intricate relationships between the human microbiome and health. The future promises exciting prospects for gaining deeper insights into microbial diversity and functions in various ecosystems.
Author Contributions
K Qurbani, conceptualization and writing: original draft preparation, review and editing; SH Hussein: writing: original draft preparation, review and editing; SK Ahmed: conceptualization and writing: original draft preparation, review and editing.
Acknowledgements
I certify that no individuals other than the listed co-authors contributed to this publication.
Financial disclosure: I certify that no financial and/or material support was received for this research or the creation of this work.
Information pertaining to writing assistance: I certify that no funded writing assistance was utilized in the production of this manuscript.
Ethical disclosure: N/A
Data sharing statement: N/A
References
- Abraham, S. Abraham, S., O’Dea, M., Sahibzada, S., Hewson, K., Pavic, A., Veltman, T., Abraham, R., Harris, T., Trott, D.J., Jordan, D. Correction: Escherichia coli and Salmonella spp. Isolated from Australian meat chickens remain susceptible to critically important antimicrobial agents (PLoS ONE (2019) 14, 10 (e0224281). https://doi.org/10.1371/journal.pone.0224281. PLoS One, 2020; 15, e0224281. [Google Scholar] [CrossRef]
- Ahmed, S.K. , Hussein, S., Qurbani, K., Ibrahim, R.H., Fareeq, A., Mahmood, K.A., Mohamed, M.G. Antimicrobial resistance: Impacts, challenges, and future prospects. J. Med. Surgery Public Heal. 2024, 2, 100081. [Google Scholar] [CrossRef]
- Ali, A.S. , Nazar, M.E., Mustafa, R.M., Hussein, S., Qurbani, K., Ahmed, S.K. Impact of heavy metals on breast cancer. World Acad. Sci. J. 2024, 6, 1–12. [Google Scholar]
- Amann, R. , Fuchs, B.M., Behrens, S. The identification of microorganisms by fluorescence in situ hybridisation. Curr. Opin. Biotechnol. 2001, 12, 231–236. [Google Scholar] [CrossRef]
- Amit Roy, S.R. Molecular Markers in Phylogenetic Studies-A Review. J. Phylogenetics Evol. Biol. 2014, 02, 131. [Google Scholar] [CrossRef]
- Ayhan, K. , Coşansu, S., Orhan-Yanıkan, E., Gülseren, G. Advance methods for the qualitative and quantitative determination of microorganisms. Microchem. J. 2021, 166, 106188. [Google Scholar] [CrossRef]
- Azevedo, A.S. , Fernandes, R.M., Faria, A.R., Silvestre, O.F., Nieder, J.B., Lou, C., Wengel, J., Almeida, C., Azevedo, N.F. Spectral imaging and nucleic acid mimics fluorescence in situ hybridization (SI-NAM-FISH) for multiplex detection of clinical pathogens. Front. Microbiol. 2022, 13, 976639. [Google Scholar] [CrossRef]
- Aziz, D. , Hassan, S.A., Mamand, D.M., Qurbani, K. New azo-azomethine derivatives: Synthesis, characterization, computational, solvatochromic UV‒Vis absorption and antibacterial studies. J. Mol. Struct. 2023, 1284, 135451. [Google Scholar] [CrossRef]
- Barman, D. , Jha, D.K., Bhattacharjee, K., 2020. Metallotolerant bacteria: insights into bacteria thriving in metal-contaminated areas. Microb. Versatility Varied Environ. Microbes Sensitive Environ. 135–164.
- Beghini, F. , McIver, L.J., Blanco-Míguez, A., Dubois, L., Asnicar, F., Maharjan, S., Mailyan, A., Manghi, P., Scholz, M., Thomas, A.M., Valles-Colomer, M., Weingart, G., Zhang, Y., Zolfo, M., Huttenhower, C., Franzosa, E.A., Segata, N. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with biobakery 3. Elife 2021, 10, e65088. [Google Scholar] [CrossRef]
- Blainey, P.C. The future is now: Single-cell genomics of bacteria and archaea. FEMS Microbiol. Rev. 2013, 37, 407–427. [Google Scholar] [CrossRef]
- Bodor, A. , Bounedjoum, N., Vincze, G.E., Erdeiné Kis, Á., Laczi, K., Bende, G., Szilágyi, Á., Kovács, T., Perei, K., Rákhely, G. Challenges of unculturable bacteria: environmental perspectives. Rev. Environ. Sci. Biotechnol. 2020, 19, 1–22. [Google Scholar] [CrossRef]
- Bokulich, N.A. , Joseph, C.M.L., Allen, G., Benson, A.K., Mills, D.A. Next-generation sequencing reveals significant bacterial diversity of botrytized wine. PLoS One 2012, 7, e36357. [Google Scholar] [CrossRef]
- Cai, H.Y. , Caswell, J.L., Prescott, J.F. Nonculture Molecular Techniques for Diagnosis of Bacterial Disease in Animals: A Diagnostic Laboratory Perspective. Vet. Pathol. 2014, 51, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Cao, J. , Chen, X., Huang, S., Shi, W., Fan, Q., Gong, Y., Peng, Y., Wu, L., Yang, C. Microfluidics-based single cell analysis: from transcriptomics to spatiotemporal multi-omics. TrAC - Trends Anal. Chem. 2023, 158, 116868. [Google Scholar] [CrossRef]
- Caracciolo, A.B. , Bottoni, P., Grenni, P. Fluorescence in situ hybridization in soil and water ecosystems: A useful method for studying the effect of xenobiotics on bacterial community structure. Toxicol. Environ. Chem. 2010, 92, 567–579. [Google Scholar] [CrossRef]
- Chen, H. , Bai, X., Li, Y., Jing, L., Chen, R., Teng, Y. Source identification of antibiotic resistance genes in a peri-urban river using novel crAssphage marker genes and metagenomic signatures. Water Res. 2019, 167, 115098. [Google Scholar] [CrossRef]
- Cheng, M. , Cao, L., Ning, K. Microbiome Big-Data Mining and Applications Using Single-Cell Technologies and Metagenomics Approaches Toward Precision Medicine. Front. Genet. 2019, 10, 972. [Google Scholar] [CrossRef]
- Church, D.L. , Cerutti, L., Gürtler, A., Griener, T., Zelazny, A., Emler, S. Performance and application of 16S rRNA gene cycle sequencing for routine identification of bacteria in the clinical microbiology laboratory. Clin. Microbiol. Rev. 2020, 33, 1–74. [Google Scholar] [CrossRef]
- Cocolin, L. , Rajkovic, A., Rantsiou, K., Uyttendaele, M. The challenge of merging food safety diagnostic needs with quantitative PCR platforms. Trends Food Sci. Technol. 2011, 22, S30–S38. [Google Scholar] [CrossRef]
- Darby, E.M. , Trampari, E., Siasat, P., Gaya, M.S., Alav, I., Webber, M.A., Blair, J.M.A. Molecular mechanisms of antibiotic resistance revisited. Nat. Rev. Microbiol. 2023, 21, 280–295. [Google Scholar]
- Dekas, A.E. , Orphan, V.J., 2011. Identification of diazotrophic microorganisms in marine sediment via fluorescence in situ hybridization coupled to nanoscale secondary ion mass spectrometry (FISH-NanoSIMS), in: Methods in Enzymology. Elsevier, pp. 281–305. [CrossRef]
- Dong, J. , Olano, J.P., McBride, J.W., Walker, D.H. Emerging pathogens: Challenges and successes of molecular diagnostics. J. Mol. Diagnostics 2008, 10, 185–197. [Google Scholar] [CrossRef]
- Dror, B. , Jurkevitch, E., Cytryn, E. State-of-the-art methodologies to identify antimicrobial secondary metabolites in soil bacterial communities-A review. Soil Biol. Biochem. 2020, 147, 107838. [Google Scholar] [CrossRef]
- Dubey, S.K. , Tripathi, A.K., Upadhyay, S.N. Exploration of soil bacterial communities for their potential as bioresource. Bioresour. Technol. 2006, 97, 2217–2224. [Google Scholar] [CrossRef] [PubMed]
- Ercolini, D. High-throughput sequencing and metagenomics: Moving forward in the culture-independent analysis of food microbial ecology. Appl. Environ. Microbiol. 2013, 79, 3148–3155. [Google Scholar] [CrossRef]
- Franzosa, E.A. , Hsu, T., Sirota-Madi, A., Shafquat, A., Abu-Ali, G., Morgan, X.C., Huttenhower, C. Sequencing and beyond: Integrating molecular “omics” for microbial community profiling. Nat. Rev. Microbiol. 2015, 13, 360–372. [Google Scholar] [CrossRef]
- Frickmann, H. , Zautner, A.E., Moter, A., Kikhney, J., Hagen, R.M., Stender, H., Poppert, S. Fluorescence in situ hybridization (FISH) in the microbiological diagnostic routine laboratory: a review. Crit. Rev. Microbiol. 2017, 43, 263–293. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, K. , Ogawa, M., Taniguchi, H., Saito, M. Molecular approaches to studying microbial communities: Targeting the 16S ribosomal RNA gene. J. UOEH 2016, 38, 223–232. [Google Scholar] [CrossRef]
- García-Hernández, J. , Hernández, M., Moreno, Y. Combination of direct viable count and fluorescent in situ hybridization (Dvc-fish) as a potential method for identifying viable vibrio parahaemolyticus in oysters and mussels. Foods 2021, 10, 1502. [Google Scholar] [CrossRef]
- Gawad, C. , Koh, W., Quake, S.R. Single-cell genome sequencing: current state of the science. Nat. Rev. Genet. 2016, 17, 175–188. [Google Scholar] [CrossRef]
- Glöckner, F.O. , Yilmaz, P., Quast, C., Gerken, J., Beccati, A., Ciuprina, A., Bruns, G., Yarza, P., Peplies, J., Westram, R., Ludwig, W. 25 years of serving the community with ribosomal RNA gene reference databases and tools. J. Biotechnol. 2017, 261, 169–176. [Google Scholar] [CrossRef]
- Gould, A.L. , Zhang, V., Lamberti, L., Jones, E.W., Obadia, B., Korasidis, N., Gavryushkin, A., Carlson, J.M., Beerenwinkel, N., Ludington, W.B. Microbiome interactions shape host fitness. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E11951–E11960. [Google Scholar] [CrossRef]
- Grün, D. , Van Oudenaarden, A. Design and Analysis of Single-Cell Sequencing Experiments. Cell 2015, 163, 799–810. [Google Scholar] [CrossRef] [PubMed]
- Gutleben, J. , Chaib De Mares, M., van Elsas, J.D., Smidt, H., Overmann, J., Sipkema, D. The multi-omics promise in context: from sequence to microbial isolate. Crit. Rev. Microbiol. 2018, 44, 212–229. [Google Scholar] [CrossRef] [PubMed]
- Hafeez, S. , Aslanzadeh, J., 2018. Biochemical Profile-Based Microbial Identification Systems. Adv. Tech. Diagnostic Microbiol. Vol. 1 Tech. Third Ed. 33–67. [CrossRef]
- He, Q. , Niu, X., Qi, R.Q., Liu, M. Advances in microbial metagenomics and artificial intelligence analysis in forensic identification. Front. Microbiol. 2022, 13, 1046733. [Google Scholar] [CrossRef] [PubMed]
- Hengsbach, J.N. , Sabel-Becker, B., Ulber, R., Holtmann, D. Microbial electrosynthesis of methane and acetate—comparison of pure and mixed cultures. Appl. Microbiol. Biotechnol. 2022, 106, 4427–4443. [Google Scholar] [CrossRef]
- Huang, L. , Ma, F., Chapman, A., Lu, S., Xie, X.S. Single-Cell Whole-Genome Amplification and Sequencing: Methodology and Applications. Annu. Rev. Genomics Hum. Genet. 2015, 16, 79–102. [Google Scholar] [CrossRef]
- Huang, Y. , Sheth, R.U., Zhao, S., Cohen, L.A., Dabaghi, K., Moody, T., Sun, Y., Ricaurte, D., Richardson, M., Velez-Cortes, F., Blazejewski, T., Kaufman, A., Ronda, C., Wang, H.H. High-throughput microbial culturomics using automation and machine learning. Nat. Biotechnol. 2023, 41, 1424–1433. [Google Scholar] [CrossRef]
- Hussein, S. , Sulaiman, S., Ali, S., Pirot, R., Qurbani, K., Hamzah, H., Hassan, O., Ismail, T., Ahmed, S.K., Azizi, Z. Synthesis of Silver Nanoparticles from Aeromonas caviae for Antibacterial Activity and In Vivo Effects in Rats. Biol. Trace Elem. Res. 2024, 202, 2764–2775. [Google Scholar] [CrossRef]
- Hussein, S. , Sulaiman, S., Ali, S., Pirot, R., Qurbani, K., Hamzah, H., Hassan, O., Ismail, T., Ahmed, S.K., Azizi, Z., 2023. Synthesis of Silver Nanoparticles from Aeromonas caviae for Antibacterial Activity and In Vivo Effects in Rats. Biol. Trace Elem. Res. 1–12. [CrossRef]
- Jin, Y. , Gan, G., Yu, X., Wu, D., Zhang, Li, Yang, N., Hu, J., Liu, Z., Zhang, Lixin, Hong, H., Yan, X., Liang, Y., Ding, L., Pan, Y. Isolation of Viable but Non-culturable Bacteria from Printing and Dyeing Wastewater Bioreactor Based on Resuscitation Promoting Factor. Curr. Microbiol. 2017, 74, 787–797. [Google Scholar] [CrossRef]
- Ju, F. , Zhang, T. 16S rRNA gene high-throughput sequencing data mining of microbial diversity and interactions. Appl. Microbiol. Biotechnol. 2015, 99, 4119–4129. [Google Scholar] [CrossRef]
- Junca, H. , Venegas, I., 2016. Metagenomics : Theory, Methods and Applications Metagenomics : Theory, Methods and Applications | Book Metagenomics : Theory, Methods and Applications.
- Kader, D.A. , Aziz, D.M., Mohammed, S.J., Maarof, N.N.N., Karim, W.O., Mhamad, S.A., Rashid, R.M., Ayoob, M.M., Kayani, K.F., Qurbani, K. Green synthesis of ZnO/catechin nanocomposite: Comprehensive characterization, optical study, computational analysis, biological applications and molecular docking. Mater. Chem. Phys. 2024, 319, 129408. [Google Scholar]
- Kado, C.I. , Heskett, M.G. Selective media for isolation of Agrobacterium, Corynebacterium, Erwinia, Pseudomonas, and Xanthomonas. Phytopathology 1970, 60, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Kaeberlein, T. , Lewis, K., Epstein, S.S. Isolating “uncultivabte” microorganisms in pure culture in a simulated natural environment. Science 2002, 296, 1127–1129. [Google Scholar] [CrossRef]
- Keegan, K.P. , Glass, E.M., Meyer, F. MG-RAST, a metagenomics service for analysis of microbial community structure and function. Methods Mol. Biol. 2016, 1399, 207–233. [Google Scholar] [CrossRef]
- Keller, M. , Zengler, K. Tapping into microbial diversity. Nat. Rev. Microbiol. 2004, 2, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Kulski, J.K. Next-generation sequencing—an overview of the history, tools, and “Omic” applications. Next Gener. Seq. Appl. challenges 2016, 10, 61964. [Google Scholar]
- Lagier, J.C. , Dubourg, G., Million, M., Cadoret, F., Bilen, M., Fenollar, F., Levasseur, A., Rolain, J.M., Fournier, P.E., Raoult, D. Culturing the human microbiota and culturomics. Nat. Rev. Microbiol. 2018, 16, 540–550. [Google Scholar] [CrossRef]
- Lee, C.-Y. , Chiu, Y.-C., Wang, L.-B., Kuo, Y.-L., Chuang, E.Y., Lai, L.-C., Tsai, M.-H., 2013. Common applications of next-generation sequencing technologies in genomic research. Transl. Cancer Res. 2.
- Levsky, J.M. , Singer, R.H. Fluorescence in situ hybridization: Past, present and future. J. Cell Sci. 2003, 116, 2833–2838. [Google Scholar] [CrossRef]
- Lewis, W.H. , Tahon, G., Geesink, P., Sousa, D.Z., Ettema, T.J.G. Innovations to culturing the uncultured microbial majority. Nat. Rev. Microbiol. 2021, 19, 225–240. [Google Scholar] [CrossRef]
- Li, L. , Rong, S., Wang, R., Yu, S. Recent advances in artificial intelligence and machine learning for nonlinear relationship analysis and process control in drinking water treatment: A review. Chem. Eng. J. 2021, 405, 126673. [Google Scholar] [CrossRef]
- Li, S. , Lian, W.-H., Han, J.-R., Ali, M., Lin, Z.-L., Liu, Y.-H., Li, L., Zhang, D.-Y., Jiang, X.-Z., Li, W.-J. Capturing the microbial dark matter in desert soils using culturomics-based metagenomics and high-resolution analysis. npj Biofilms Microbiomes 2023, 9, 67. [Google Scholar]
- Liu, S. , Moon, C.D., Zheng, N., Huws, S., Zhao, S., Wang, J. Opportunities and challenges of using metagenomic data to bring uncultured microbes into cultivation. Microbiome 2022, 10, 76. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. , Chen, Y., Han, L. Bioinformatics: Advancing biomedical discovery and innovation in the era of big data and artificial intelligence. Innov. Med. 2023, 1, 100012. [Google Scholar] [CrossRef]
- Maghembe, R. , Damian, D., Makaranga, A., Nyandoro, S.S., Lyantagaye, S.L., Kusari, S., Hatti-Kaul, R. Omics for bioprospecting and drug discovery from bacteria and microalgae. Antibiotics 2020, 9, 229. [Google Scholar] [CrossRef] [PubMed]
- Markoulatos, P. , Siafakas, N., Moncany, M. Multiplex polymerase chain reaction: A practical approach. J. Clin. Lab. Anal. 2002, 16, 47–51. [Google Scholar] [CrossRef]
- Martín, R. , Miquel, S., Langella, P., Bermúdez-Humarán, L.G. The role of metagenomics in understanding the human microbiome in health and disease. Virulence 2014, 5, 413–423. [Google Scholar] [CrossRef]
- Maukonen, J. , Mättö, J., Wirtanen, G., Raaska, L., Mattila-Sandholm, T., Saarela, M. Methodologies for the characterization of microbes in industrial environments: A review. J. Ind. Microbiol. Biotechnol. 2003, 30, 327–356. [Google Scholar] [CrossRef]
- Mayo, B. , Rachid, C., Alegria, A., Leite, A., Peixoto, R., Delgado, S. Impact of Next Generation Sequencing Techniques in Food Microbiology. Curr. Genomics 2014, 15, 293–309. [Google Scholar] [CrossRef]
- Mishra, S. , Lin, Z., Pang, S., Zhang, W., Bhatt, P., Chen, S. Recent advanced technologies for the characterization of xenobiotic-degrading microorganisms and microbial communities. Front. Bioeng. Biotechnol. 2021, 9, 632059. [Google Scholar] [CrossRef]
- Müller, S. , Nebe-Von-Caron, G. Functional single-cell analyses: Flow cytometry and cell sorting of microbial populations and communities. FEMS Microbiol. Rev. 2010, 34, 554–587. [Google Scholar] [CrossRef]
- Na, G.N., Kim, S.A., Kwon, O.C., Rhee, M.S. Corrigendum to “Development of selective and differential medium for Shigella sonnei using three carbohydrates (lactose sorbitol xylose) X-Gal” [J. Microbiol. Methods 115 (2015) 35-41]. https://doi.org/10.1016/j.mimet.2015.05.019. J. Microbiol. Methods 2015, 118, 190. [CrossRef]
- Nadarajah, K. , Kumar, I.S., 2019. Molecular Microbial Biodiversity Assessment in the Mycorrhizosphere. Mycorrhizosph. Pedogenes. 401–420. [CrossRef]
- Nagarajan, K. , Loh, K.-C. Molecular biology-based methods for quantification of bacteria in mixed culture: perspectives and limitations. Appl. Microbiol. Biotechnol. 2014, 98, 6907–6919. [Google Scholar] [CrossRef]
- Navarrete-Bolaños, J.L. Improving traditional fermented beverages: How to evolve from spontaneous to directed fermentation. Eng. Life Sci. 2012, 12, 410–418. [Google Scholar] [CrossRef]
- Nural Yaman, B. , Aytar Çelik, P., Enuh, B.M., Çabuk, A., 2022. Molecular Approaches of Microbial Diversity in Agricultural Soil, in: Beneficial Microorganisms in Agriculture. Springer, pp. 1–35. [CrossRef]
- O’Neill, B. , Grossman, J., Tsai, M.T., Gomes, J.E., Lehmann, J., Peterson, J., Neves, E., Thies, J.E. Bacterial community composition in Brazilian Anthrosols and adjacent soils characterized using culturing and molecular identification. Microb. Ecol. 2009, 58, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Oremland, R.S. , Capone, D.G., Stolz, J.F., Fuhrman, J. Whither or wither geomicrobiology in the era of “community metagenomics. ” Nat. Rev. Microbiol. 2005, 3, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Overmann, J. , Abt, B., Sikorski, J. Present and Future of Culturing Bacteria. Annu. Rev. Microbiol. 2017, 71, 711–730. [Google Scholar] [CrossRef]
- Palazzotto, E. , Weber, T. Omics and multi-omics approaches to study the biosynthesis of secondary metabolites in microorganisms. Curr. Opin. Microbiol. 2018, 45, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Pauter, K. , Szultka-Młyńska, M., Buszewski, B. Determination and identification of antibiotic drugs and bacterial strains in biological samples. Molecules 2020, 25, 2556. [Google Scholar] [CrossRef]
- Peccia, J. , Hernandez, M. Incorporating polymerase chain reaction-based identification, population characterization, and quantification of microorganisms into aerosol science: A review. Atmos. Environ. 2006, 40, 3941–3961. [Google Scholar] [CrossRef]
- Pereira, A.C. , Tenreiro, A., Cunha, M. V. When FLOW-FISH met FACS: Combining multiparametric, dynamic approaches for microbial single-cell research in the total environment. Sci. Total Environ. 2022, 806, 150682. [Google Scholar] [CrossRef]
- Pham, V.H.T. , Kim, J. Cultivation of unculturable soil bacteria. Trends Biotechnol. 2012, 30, 475–484. [Google Scholar] [CrossRef]
- Prasad, S. , Malav, L.C., Choudhary, J., Kannojiya, S., Kundu, M., Kumar, S., Yadav, A.N., 2021. Soil Microbiomes for Healthy Nutrient Recycling. Curr. trends Microb. Biotechnol. Sustain. Agric. 1–21. [CrossRef]
- Prodan, A. , Tremaroli, V., Brolin, H., Zwinderman, A.H., Nieuwdorp, M., Levin, E. Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS One 2020, 15, e0227434. [Google Scholar] [CrossRef]
- Qurbani, K. , Ali, S., Hussein, S., Hamzah, H., 2024. Antibiotic resistance in Kurdistan, Iraq: A growing concern. New Microbes New Infect. 57. [CrossRef]
- Qurbani, K. , Hamzah, H. Heavy Metal Tolerant Comamonas species Isolated from Soil Sample in Tanjaro Region of Sulaymaniyah City-Iraq. Ann. Rom. Soc. Cell Biol. 2021, 25, 5068–5074. [Google Scholar]
- Qurbani, K. , Hamzah, H. Intimate communication between Comamonas aquatica and Fusarium solani in remediation of heavy metal-polluted environments. Arch. Microbiol. 2020, 202, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Qurbani, K. , Hussein, S., Hamzah, H., Sulaiman, S., Pirot, R., Motevaseli, E., Azizi, Z. Synthesis of Silver Nanoparticles by Raoultella Planticola and Their Potential Antibacterial Activity Against Multidrug-Resistant Isolates. Iran. J. Biotechnol. 2022, 20, 75–83. [Google Scholar] [CrossRef]
- Qurbani, K. , Khdir, K., Sidiq, A., Hamzah, H., Hussein, S., Hamad, Z., Abdulla, R., Abdulla, B., Azizi, Z. Aeromonas sobria as a potential candidate for bioremediation of heavy metal from contaminated environments. Sci. Rep. 2022, 12, 21235. [Google Scholar] [CrossRef]
- Qurbani, K.A. , Amiri, O., Othman, G.M., Fatah, A.A., Yunis, N.J., Joshaghani, M., Ahmed, S.S., Abdulrahman, N.A. Enhanced antibacterial efficacy through piezo memorial effect of CaTiO3/TiO2 Nano-Composite. Inorg. Chem. Commun. 2024, 165, 112470. [Google Scholar]
- Ranjard, L. , Poly, F., Nazaret, S. Monitoring complex bacterial communities using culture-independent molecular techniques: Application to soil environment. Res. Microbiol. 2000, 151, 167–177. [Google Scholar] [CrossRef]
- Rashid, M. , Stingl, U. Contemporary molecular tools in microbial ecology and their application to advancing biotechnology. Biotechnol. Adv. 2015, 33, 1755–1773. [Google Scholar] [CrossRef]
- Rastogi, G. , Sani, R.K., 2011. Molecular techniques to assess microbial community structure, function, and dynamics in the environment, in: Microbes and Microbial Technology: Agricultural and Environmental Applications. Springer, pp. 29–57. [CrossRef]
- Riesenfeld, C.S. , Schloss, P.D., Handelsman, J. Metagenomics: Genomic analysis of microbial communities. Annu. Rev. Genet. 2004, 38, 525–552. [Google Scholar] [CrossRef]
- Rompré, A. , Servais, P., Baudart, J., De-Roubin, M.R., Laurent, P. Detection and enumeration of coliforms in drinking water: Current methods and emerging approaches. J. Microbiol. Methods 2002, 49, 31–54. [Google Scholar] [CrossRef]
- Rowland, I. , Gibson, G., Heinken, A., Scott, K., Swann, J., Thiele, I., Tuohy, K. Gut microbiota functions: metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef]
- Saravanan, A. , Kumar, P.S., Duc, P.A., Rangasamy, G. Strategies for microbial bioremediation of environmental pollutants from industrial wastewater: A sustainable approach. Chemosphere 2023, 313, 137323. [Google Scholar] [CrossRef]
- Šeligová, B. , Lukáč, Ľ., Bábelová, M., Vávrová, S., Sulo, P. Diagnostic reliability of nested PCR depends on the primer design and threshold abundance of Helicobacter pylori in biopsy, stool, and saliva samples. Helicobacter 2020, 25, e12680. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P. , Pandey, A.K., Kim, S.-H., Singh, S.P., Chaturvedi, P., Varjani, S. Critical review on microbial community during in-situ bioremediation of heavy metals from industrial wastewater. Environ. Technol. Innov. 2021, 24, 101826. [Google Scholar] [CrossRef]
- Sharma, P. , Singh, S.P., Iqbal, H.M.N., Tong, Y.W. Omics approaches in bioremediation of environmental contaminants: An integrated approach for environmental safety and sustainability. Environ. Res. 2022, 211, 113102. [Google Scholar] [CrossRef] [PubMed]
- Singleton, C.M. , Petriglieri, F., Kristensen, J.M., Kirkegaard, R.H., Michaelsen, T.Y., Andersen, M.H., Kondrotaite, Z., Karst, S.M., Dueholm, M.S., Nielsen, P.H., Albertsen, M. Connecting structure to function with the recovery of over 1000 high-quality metagenome-assembled genomes from activated sludge using long-read sequencing. Nat. Commun. 2021, 12, 2009. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.J. , Osborn, A.M. Advantages and limitations of quantitative PCR (Q-PCR)-based approaches in microbial ecology. FEMS Microbiol. Ecol. 2009, 67, 6–20. [Google Scholar] [CrossRef]
- Solís-González, C.J. , Loza-Tavera, H. Alicycliphilus: current knowledge and potential for bioremediation of xenobiotics. J. Appl. Microbiol. 2019, 126, 1643–1656. [Google Scholar] [CrossRef]
- Spieck, E. , Lipski, A., 2011. Cultivation, growth physiology, and chemotaxonomy of nitrite-oxidizing bacteria, in: Methods in Enzymology. Elsevier, pp. 109–130.
- Stackebrandt, E. , Embley, T.M., 2000. Diversity of Uncultured Microorganisms in the Environment, in: Nonculturable Microorganisms in the Environment. Springer, pp. 57–75. [CrossRef]
- Steele, H.L. , Streit, W.R. Metagenomics: Advances in ecology and biotechnology. FEMS Microbiol. Lett. 2005, 247, 105–111. [Google Scholar] [CrossRef]
- Steffan, R.J. , Atlas, R.M. Polymerase chain reaction: Applications in environmental microbiology. Annu. Rev. Microbiol. 1991, 45, 137–161. [Google Scholar] [CrossRef]
- Stepanauskas, R. Single cell genomics: An individual look at microbes. Curr. Opin. Microbiol. 2012, 15, 613–620. [Google Scholar] [CrossRef]
- Streit, W.R. , Schmitz, R.A. Metagenomics - The key to the uncultured microbes. Curr. Opin. Microbiol. 2004, 7, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Struelens, M.J. , Ludden, C., Werner, G., Sintchenko, V., Jokelainen, P., Ip, M. Real-time genomic surveillance for enhanced control of infectious diseases and antimicrobial resistance. Front. Sci. 2024, 2, 1298248. [Google Scholar] [CrossRef]
- Tringe, S.G. , Von Mering, C., Kobayashi, A., Salamov, A.A., Chen, K., Chang, H.W., Podar, M., Short, J.M., Mathur, E.J., Detter, J.C., Bork, P., Hugenholtz, P., Rubin, E.M. Comparative metagenomics of microbial communities. Science 2005, 308, 554–557. [Google Scholar] [CrossRef] [PubMed]
- Truong, D.T. , Franzosa, E.A., Tickle, T.L., Scholz, M., Weingart, G., Pasolli, E., Tett, A., Huttenhower, C., Segata, N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 2015, 12, 902–903. [Google Scholar] [CrossRef]
- Valones, M.A.A. , Guimarães, R.L., Brandão, L.A.C., De Souza, P.R.E., De Albuquerque Tavares Carvalho, A., Crovela, S. Principles and applications of polymerase chain reaction in medical diagnostic fields: A review. Brazilian J. Microbiol. 2009, 40, 1–11. [Google Scholar] [CrossRef]
- Van De Veerdonk, F.L. , Gresnigt, M.S., Romani, L., Netea, M.G., Latgé, J.P. Aspergillus fumigatus morphology and dynamic host interactions. Nat. Rev. Microbiol. 2017, 15, 661–674. [Google Scholar] [CrossRef]
- van Dijk, E.L. , Naquin, D., Gorrichon, K., Jaszczyszyn, Y., Ouazahrou, R., Thermes, C., Hernandez, C. Genomics in the long-read sequencing era. Trends Genet. 2023, 39, 649–671. [Google Scholar] [CrossRef] [PubMed]
- Van Iterson, G. , de Jong, L.E. den den D., Kluyver, A.J., 2013. Martinus Willem Beijerinck: His life and his work.
- Wagner, M. , Hornt, M., Daims, H. Fluorescence in situ hybridisation for the identification and characterisation of prokaryotes. Curr. Opin. Microbiol. 2003, 6, 302–309. [Google Scholar] [CrossRef]
- Wani, A.K. , Roy, P., Kumar, V., Mir, T. ul G. Metagenomics and artificial intelligence in the context of human health. Infect. Genet. Evol. 2022, 100, 105267. [Google Scholar] [CrossRef]
- Wood, J.M. , Singh, N.K., Guan, L., Seuylemezian, A., Benardini, J.N., Venkateswaran, K. Performance of Multiple Metagenomics Pipelines in Understanding Microbial Diversity of a Low-Biomass Spacecraft Assembly Facility. Front. Microbiol. 2021, 12, 2739. [Google Scholar] [CrossRef]
- Yadav, A.N. Microbial biotechnology for bio-prospecting of microbial bioactive compounds and secondary metabolites. J. Appl. Biol. Biotechnol. 2021, 9, 1–6. [Google Scholar] [CrossRef]
- Yamin, D. , Uskoković, V., Wakil, A.M., Goni, M.D., Shamsuddin, S.H., Mustafa, F.H., Alfouzan, W.A., Alissa, M., Alshengeti, A., Almaghrabi, R.H., Fares, M.A.A., Garout, M., Al Kaabi, N.A., Alshehri, A.A., Ali, H.M., Rabaan, A.A., Aldubisi, F.A., Yean, C.Y., Yusof, N.Y. Current and Future Technologies for the Detection of Antibiotic-Resistant Bacteria. Diagnostics 2023, 13, 1–43. [Google Scholar] [CrossRef]
- Yang, S. , Rothman, R.E. PCR-based diagnostics for infectious diseases: Uses, limitations, and future applications in acute-care settings. Lancet Infect. Dis. 2004, 4, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Yasen, A. , Aini, A., Wang, H., Li, W., Zhang, C., Ran, B., Tuxun, T., Maimaitinijiati, Y., Shao, Y., Aji, T., Wen, H. Progress and applications of single-cell sequencing techniques. Infect. Genet. Evol. 2020, 80, 104198. [Google Scholar] [CrossRef] [PubMed]
- Yasir, M. , Al-Zahrani, I.A., Bibi, F., Abd El Ghany, M., Azhar, E.I. New insights of bacterial communities in fermented vegetables from shotgun metagenomics and identification of antibiotic resistance genes and probiotic bacteria. Food Res. Int. 2022, 157, 111190. [Google Scholar] [CrossRef]
- Yu, F.B. , 2016. Characterizing Bacterial Physiology and Diversity Using Microfluidic Systems.
- Yu, Y. , Wen, H., Li, S., Cao, H., Li, X., Ma, Z., She, X., Zhou, L., Huang, S. Emerging microfluidic technologies for microbiome research. Front. Microbiol. 2022, 13, 906979. [Google Scholar] [CrossRef]
- Zhu, H. , Zhang, H., Xu, Y., Laššáková, S., Korabečná, M., Neužil, P. PCR past, present and future. Biotechniques 2020, 69, 317–325. [Google Scholar] [CrossRef]
Figure 1.
Steps of metagenomics approach for non-cultivable microorganisms.
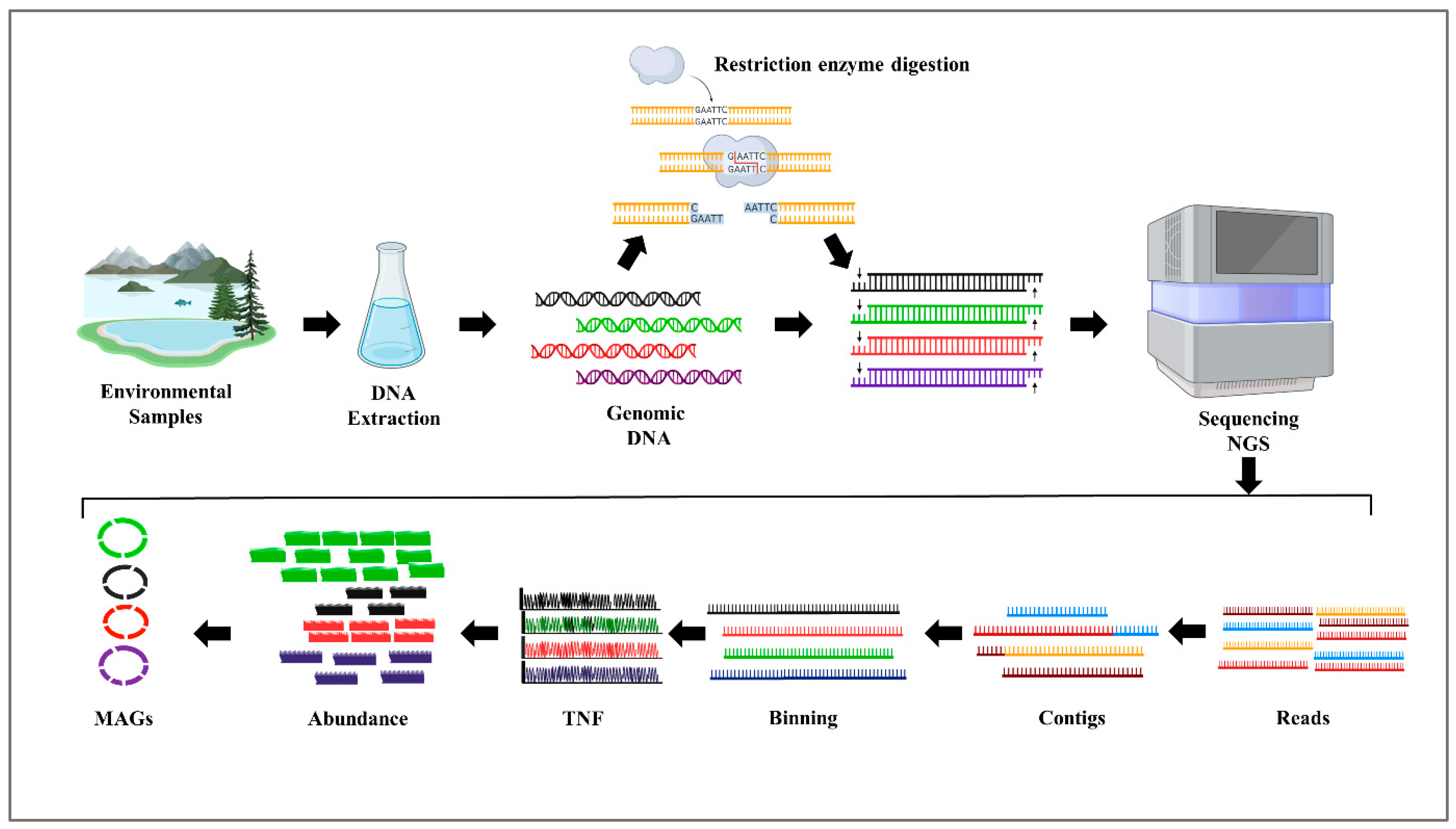
Figure 2.
Single-cell genomic approach.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



