Submitted:
08 October 2024
Posted:
10 October 2024
You are already at the latest version
Abstract
Extremely premature infants are at significant risk for developing bronchopulmonary dysplasia (BPD) and neurodevelopmental impairment (NDI). Although BPD is a predictor of poor neurodevelopmental outcomes, it is currently unknown how BPD contributes to brain injury and long-term NDI in preterm infants. Extracellular vesicles (EVs) are small, membrane-bound structures released from cells into the surrounding environment. EVs are involved in inter-organ communication in diverse pathological processes. Inflammasomes are large, multiprotein complexes that are part of the innate immune system and are responsible for triggering inflammatory responses and cell death. Apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) is pivotal in inflammasome assembly and activating inflammatory caspase-1. Activated caspase-1 cleaves gasdermin D (GSDMD) to release a 30 kD N-terminal domain that can form membrane pores, leading to lytic cell death, also known as proptosis. Activated caspase-1 can also cleave pro-IL-1and pro-IL-18 to their active forms, which can be rapidly released through the GSDMD pores to induce inflammation. Recent evidence has emerged that activation of inflammasomes is associated with neonatal lung and brain injury, and inhibition of inflammasomes reduces hyperoxia-induced neonatal lung and brain injury. Additionally, multiple studies have demonstrated that hyperoxia stimulates the release of lung-derived EVs that contain inflammasome cargos. Adoptive transfer of these EVs into the circulation of normal neonatal mice and rats induces brain inflammatory injury. This review focuses on EV-inflammasomes’ roles in mediating lung-to-brain crosstalk via EV-dependent and EV-independent mechanisms critical in BPD, brain injury, and NDI pathogenesis. EV-inflammasomes will be discussed as potential therapeutic targets for neonatal lung and brain injury.
Keywords:
Preterm
; BPD
; brain injury
; extracellular vesicles
; inflammsome
Introduction
More than 15 million infants are born preterm each year worldwide [1,2,3]. Extremely premature infants born at less than 28 weeks of gestational age (GA) are at significant risk of having multi-organ injury and developmental abnormalities that predominately involve the lung and brain. The lungs of these infants are immature, and they suffer respiratory failure soon after birth and often require supplemental oxygen (O2) [2,3]. However, life-sustaining high fraction inspired O2 (FiO2) can cause lung inflammation, leading to damage of lung structures and, ultimately, bronchopulmonary dysplasia (BPD) [2,3]. These infants also have immature brains prone to injurious stimuli such as oxygen toxicity and inflammation. Consequently, they are at greater risk of developing short-term and long-term neurological complications [4,5,6]. Thus, BPD survivors not only suffer from pulmonary dysfunction but often are also affected by long-term neurodevelopmental impairment (NDI) [6,7]. As such, the cost of treating BPD is $2.4 billion/year [8], and for early intervention is >$1.3 billion/year in the US [6,7,9,10]. There is mounting clinical evidence that severe BPD is an independent risk factor for brain injury and NDI [6,7,11]. Evidence from preclinical studies demonstrates that hyperoxia exposure induces BPD-like pathology and results in brain injury and NDI in newborn rodents [12,13,14,15]. Despite recent advances in neonatal intensive care and extensive research, it is unknown to what extent BPD contributes to NDI, and there is no effective therapy for either of these conditions. Thus, understanding the mechanisms by which BPD contributes to NDI and identifying novel therapeutic targets for these two severe complications of prematurity are paramount. This review focuses on lung-derived extracellular vesicle (EV)-inflammasomes’ roles in mediating lung-to-brain crosstalk via EV-dependent and EV-independent mechanisms critical in BPD and brain injury pathogenesis. EV-inflammasomes will be discussed as potential therapeutic targets for neonatal lung and brain injury.
BPD is Associated with NDI: Clinical Evidence
BPD was first described by Northway, et al. in 1967 as a chronic lung disease of preterm infants who had hyaline membrane disease and required high supplemental oxygen therapy and mechanical ventilation [16]. Over the last 50 years, the incidence of BPD has not decreased because the survival rate of extremely low gestational age newborns (ELGANs) born at <28 weeks GA has increased [17], and the incidence of BPD is inversely related to the GA. BPD occurs in ~35% of ELGANs in the USA, as almost 18,000 out of 50,000 such infants develop BPD was reported in 2015 [18]. However, a wide range of BPD prevalence of 11-50% has been demonstrated by major neonatal research networks worldwide [19,20,21,22,23,24,25], which may be secondary to the non-uniform use of the BPD definitions. The most widely used BPD definition was proposed by the National Institute of Child Health and Human Development (NICHD)/National Heart Lung and Blood Institute (NHLBI) workshop in 2001. This definition was based on the levels of supplemental oxygen and the mode of respiratory support at 36 weeks postmenstrual age (PMA) in preterm infants born at <32 weeks GA who required oxygen therapy at 28 days of life [2]. In 2018, the NICHD workshop proposed a more complex definition that combines oxygen therapy with the mode of respiratory support at 36 weeks PMA. The workshop also proposed to use terms of grades I, II, and III rather than mild, moderate, and severe, as proposed in the 2001 workshop [26]. In 2019, NICHD proposed an updated definition of BPD with severity stratification solely based on the mode of respiratory support at 36 weeks PMA regardless of supplemental oxygen use [27]. The Canadian Neonatal Research Network suggested that evaluating infants at 40 weeks’ PMA may be more predictive of respiratory outcomes [22]. Regardless of what definition is used, the pathological hallmarks of BPD are well known, including alveolar simplification, poor vascularization, variable fibrosis, and chronic inflammation [2,28]. However, the pathogenesis of BPD is poorly understood, and there is no effective therapy for this common complication of prematurity.
There is mounting evidence that preterm infants who develop BPD are at increased risk for brain injury and NDI [29,30,31]. BPD survivors exhibit various degrees of impairment in motor, neurosensory, general cognitive functioning, attention, language, memory and learning, visual-spatial perception, executive skills, academic performance, and psychological problems. Cerebral palsy (CP) occurs in approximately 10% of preterm infants born at <28 weeks GA [32,33]. However, the incidence of CP is even higher at 15% in the BPD survivors [33]. One study showed that BPD was a significant risk factor for CP at 18-22 months corrected age (CA) (odds ratio 1.66, 95% confidence interval 1.01 to 2.74) after adjusting for other confounding factors such as cystic periventricular leukomalacia (PVL) and severe intraventricular hemorrhage (IVH) [34]. At 24 months of age, BPD requiring mechanical ventilation (BPD/MV) at 36 weeks PMA was associated with a nearly sixfold increased risk of quadriparesis and a fourfold increased risk of diparesis [31]. Other non-CP motor impairments also occur more frequently in children who had BPD at 8 years old [35,36,37].
Gallini et al. demonstrated that not only infants with severe BPD but also moderate BPD had an increased risk of overall cognitive impairment at 20 months of CA [38]. In a study of preterm infants at 18 months CA, BPD was an independent risk factor for major neurosensory problems with OR (95% CI) of 2.4 for any of CP, blindness, deafness, or cognitive delay [39]. A similar study showed that at 5 years of age, children with a history of BPD had a higher chance of disability in one or more areas of motor impairment, cognitive impairment, behavior problems, poor general health, deafness, or blindness [40]. A recent study found that increased BPD severity is associated with increased risk of NDI at both 2 years and 5 years CA [41] assessed by Bayley Scales of Infant and Toddler Development (BSID)-II or BSID-III at 2 years of age [41,42,43] and Wechsler Preschool and Primary Scale of Intelligence at 5 years CA [44]. In a study with children at 8 years old, BPD survivors had the worst performance in general intelligence, reading, mathematics, motor performance, memorization, and attention [36]. Children who had more severe BPD compared with children who had mild/moderate BPD performed worse in performance IQ and perceptual organization at 3 and 8 years of age [45]. Some studies have reported language delay in children with BPD. Extremely low birth weight children with a history of BPD had a significant language delay at 18-22 months compared to children without BPD [46]. In a study of 3-year-old very low birth weight preschool children, the group with BPD had lower score in receptive, expressive, and total communicative competence scores [34]. When followed up at 8 years, children with BPD had stepwise decreases in scores in both expressive and receptive language compared to children without BPD [47].
Children with BPD also have impaired visuospatial perception compared to children without BPD at 8-10 years old [48]. Taylor et al. demonstrated that a longer duration of oxygen therapy to treat BPD is associated with difficulties in perceptual-motor tasks in very low birth weight children aged 7 years [49] and also at 16 years of age [49,50]. A recent study in children 5-6 years of age found that BPD was strongly associated with mild, moderate, and severe overall neurodevelopmental disabilities (OR 1.49, 95% CI 1.05 to 2.20; 2.20, 1.41 to 3.42 and 2.71, 1.67 to 4.40) [7]. BPD was also associated with developmental coordination disorders, behavioral difficulties, lower IQ scores, rehospitalization in the last 12 months, and developmental support [7]. A recent study comparing BPD with and without tracheostomy found that BPD with tracheostomy had greater cognitive and motor delays <24 months and more significant cognitive delays >24 months [51]. This study was conducted at 2 years CA in preterm infants with various degrees of BPD, as defined by Jensen et al. [27]. Grade 1 BPD is defined as receiving nasal cannula with a flow rate of 2 L per minute (LPM) or less, grade 2 BPD is defined as receiving nasal cannula of more than 2 LPM flow rate or noninvasive positive pressure support, and grade 3 is defined as receiving invasive respiratory support. Grades 2 and 3, but not Grade 1, BPD were found to be associated with increased odds of a composite adverse neurodevelopmental outcome by 2.7 and 7.2 folds, respectively. A BSID domain-specific analysis showed that higher grades of BPD were associated with lower scores in the cognitive, gross motor, and fine motor domains [9]. Infants with grade 3 BPD had increased odds of CP and developmental delay at 2 years of age, poor academic achievement, and low IQ in adolescence [52].
These data demonstrate that survivors of infants with BPD, particularly those with more severe lung disease in the neonatal period, are at increased risk for NDI throughout childhood. The underlying mechanisms for BPD contributing to NDI remain to be explored.
Extracellular Vesicles in Neonatal Lung and Brain Injury
EVs are lipid-membrane-encircled vesicles secreted by cells into the extracellular environment [53,54,55,56,57] (Figure 1). There are three main subtypes of EVs, exosomes, microvesicles, and apoptotic bodies, named based on their biogenesis, release pathways, size, content, and function [53,57,58]. Exosomes are formed by an endosomal route and are sized 30-150 nm in diameter. The direct outward budding of the cell plasma membrane forms microvesicles 100 nm to 1000 nm in diameter. Apoptotic bodies are released by dying cells into the extracellular environment, and their size ranges from 50 nm to 5000 nm in diameter. EVs carry complex cargoes of proteins, lipids, and nucleic acids, and their cargo composition is highly dependent on the biological function of the parental cells. Being membranous, EVs protect their cargo from the extracellular environment, thus allowing for safe transport and delivery of their intact cargo to target cells, which results in modification of the target cells’ function. In the lungs, it appears that both alveolar epithelial cells (AECs) and alveolar macrophages (AMs) can release bioactive EVs upon inflammatory injury, as AEC- and AM-derived EVs isolated from tracheal aspirate fluid (TAF) have been shown to regulate inflammatory responses in adult lung diseases [59,60,61]. Similarly, EVs have been shown to contribute to several adult central nervous system (CNS) disorders, and it is known that they can bidirectionally cross the blood-brain barrier (BBB) [62,63,64].
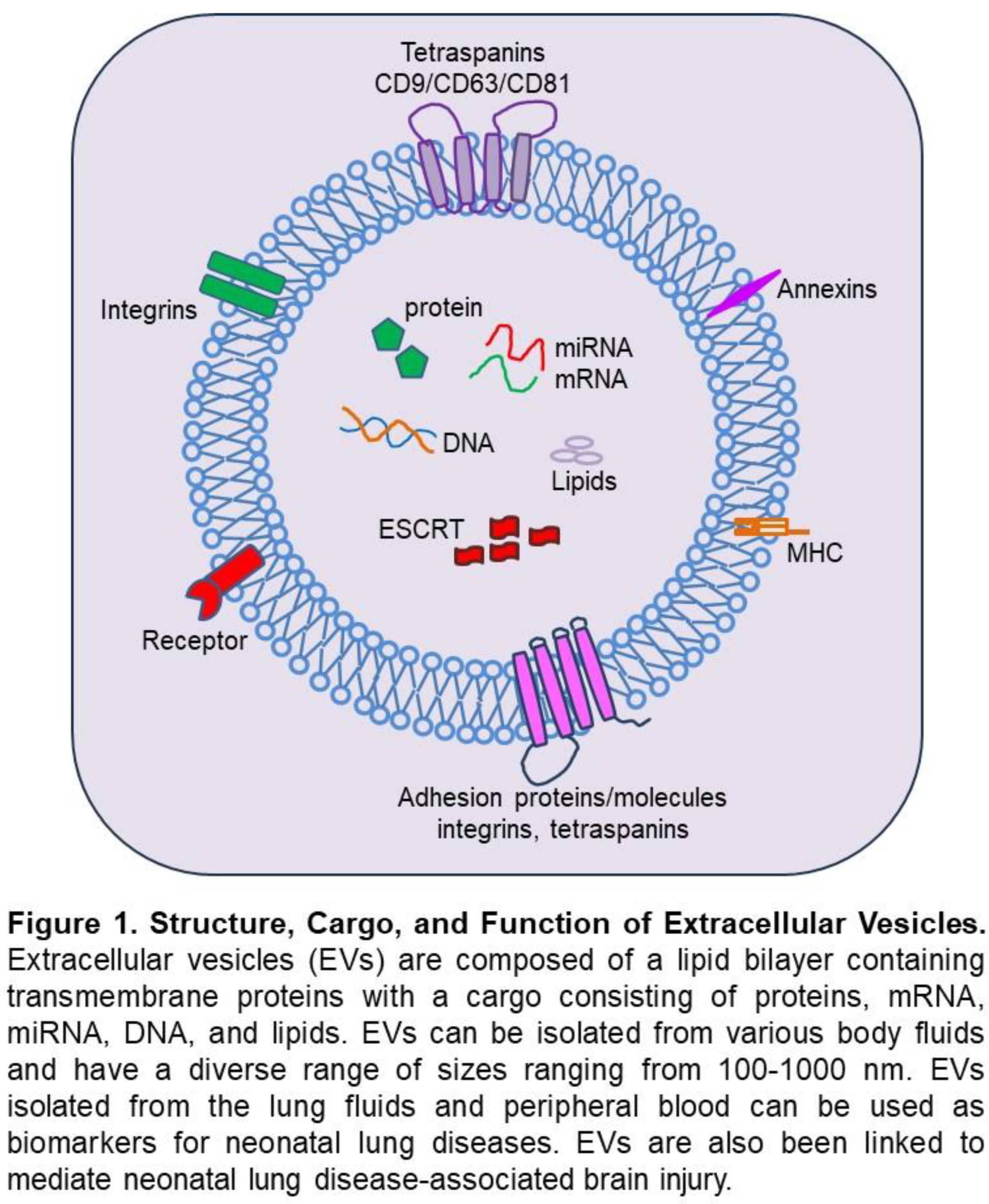
EVs and Neonatal Lung Injury: Although EVs have been characterized in bodily fluids from adults, few studies have identified EVs in the biofluids of neonates [65,66]. Increasing evidence highlights the role of EVs in neonatal lung diseases. EVs have been isolated from a variety of bodily fluids, including TAF [67,68], serum [69], plasma [70], breast milk [71], and amniotic fluid [72] of preterm infants. Increased AEC-derived EVs were detected in TAF from preterm infants with severe BPD compared to infants without BPD [67,68]. To characterize EVs during human lung development, Ransom et al. collected tracheal aspirates from premature neonates between 22 to 35 weeks GA and analyzed the EVs by nanoparticle tracking, electron microscopy, and bead-based flow cytometry [68]. EVs were detectable across late canalicular through saccular stages of lung development, with larger sizes of EVs being detected earlier in gestation. EVs contained an abundance of the EV-enriched tetraspanins CD9, CD63, and CD81, as well as epithelial cell and immune cell markers. EVs had increases in select surface proteins (CD24 and CD14) associated with GA and BPD risk. Finally, the expression data obtained from epithelial cells in a single-cell atlas of murine lung development found that epithelial EV marker expression also changes with developmental time [68]. Together, these data demonstrate an association between EV profile and lung development and provide a foundation for future functional classification of EVs to determine their role in cell signaling during development and harness their potential as new therapeutic targets in BPD [73]. Infants with severe BPD at 36 weeks PMA compared with age-matched full-term controls had a greater number of EVs in TAFs, but as a group, these EVs were smaller than controls [67]. MiRNA-21 was increased in the serum of preterm infants born at <32 weeks GA who developed BPD than without BPD [69]. Increased miRNA was also detected in hyperoxia-exposed mouse lung tissues. EVs isolated from umbilical cord venous blood of neonates revealed differential expression of miRNA-17-5p, miRNA-103-3p, miRNA-185-5p, miRNA-20b-5p, miRNA-2001-3p, and miRNA-765 between BPD and non-BPD infants [74]. Among them, miRNA-103-3p and miRNA-185-5p exhibited the most significant reduction, whereas miRNA-2001-3p showed increased expression. Infants with severe BPD also had decreased expression of EV-miRNA-876-3 in their TAF [67]. Interestingly, treating hyperoxia-exposed mice with a miRNA-876-3p mimic resulted in decreased alveolar hyperplasia and neutrophil infiltration [67]. Utilizing a murine model, Genschmer and collaborators compared the function of EVs derived from bronchoalveolar lavage fluid from BPD and non-BPD infants [75]. Intriguingly, mice that received intranasal BPD-derived EVs had significant alveolar hypoplasia and right ventricular hypertrophy, suggesting a role for EVs in BPD pathogenesis [75]. In another study, Wang et al. demonstrated a total of 317 circRNAs, 104 IncRNAs, and 135 mRNAs that were differentially expressed in umbilical cord blood-derived exosomes of preterm infants with BPD compared to those without BPD [76]. The GO terms and KEGG pathways, mostly involving differentially expressed exosomal RNAs, were closely associated with endothelial or epithelial cell development.
EVs and Neonatal Brain Injury: In the CNS, each cell type is capable of secreting and taking up EVs, which gives them a vital role in health and during disease. Under physiological conditions, brain cells, including astrocytes, endothelial cells, microglia, oligodendrocytes, and neurons, produce EVs [77,78,79,80,81,82,83]. Some of these EVs mediate brain autocrine and paracrine signaling, including neural trophic support, synaptic plasticity, regulation of myelination, and intercellular communication during brain development [84,85,86]. EVs also play an important role in BBB integrity, and one such example demonstrates that pericyte-derived EVs carry neuroprotective cargo [77]. Additional evidence suggests that neural progenitor cell-derived EVs enhance post-ischemic BBB integrity by enhancing pericyte recruitment via down-regulation of ATP binding cassette subfamily member-a (ABCA1) expression and inhibiting the NF-kB pathway and downstream MMP-9 activity [87]. Under physiological conditions, oligodendrocytes are the myelin-forming cells that can release EVs, which microglia can uptake through micropinocytosis that facilitates the clearance of oligodendrocyte-derived EV cargo and occurs in the absence of microglial activation [80].
Over the last two decades, more knowledge has been gained about the role of EVs in neonatal brain injury in human and experimental models. In 2019, Spaull et al. analyzed the CSF EVs of preterm infants with post-hemorrhagic hydrocephalus (PHH) and found a heterogeneous size and concentration of EVs between patients [88]. The pathological placental exosomes have been shown to propagate acute and chronic inflammation, leading to brain injury [89]. A study with exosomes isolated from women with preeclampsia showed that they disrupt the BBB in vitro and in vivo [90]. In a study conducted by Vechetti and the group demonstrated there was a comparison drawn between the circulation of small EVs in terms of concentration and size between individuals affected by CP as well as typically developed (control group) individuals [73]. CP individuals were seen to present an overall lower concentration of small EVs. Also, an interesting observation noted in the study was the upregulation of the skeletal muscle-specific miRNA, miR-486, in small EVs from the CP-affected individuals [73]. In animal models of brain injury, inhibition of EV release from the CNS attenuates systemic responses to CNS inflammation. It also inhibits BBB leukocyte infiltration, suggesting a damaging role of EVs in neuroinflammation [91]. Also, in an IL-1b mouse model of inflammatory brain injury, astrocytes-derived EVs released post-injury can induce a systemic inflammatory response in naïve animals in the absence of injury [91]. Microglia-derived EVs are equally detrimental in promoting a proinflammatory microenvironment response to brain injury. Importantly, recent findings suggest that EVs released from microglia in response to brain injury may represent the major pathway of TNF-a secretion since EV production is markedly induced by activation of the P2X7, P2 receptor X7 by ATP [92]. Activated microglial cells produce EVs containing high amounts of TNF-a and can cause reactive astrocytic conversion and demyelination [93]. Microglia-derived EVs are equally detrimental in promoting a proinflammatory microenvironment response to brain injury [94]. Stimulation of microglia with ATP through activating the P2X7 receptor drastically increases EV release and modifies their proteomics and their effects in increasing IL-1b, IL-6, and TNF-a release that induce a robust inflammatory response [95]. Besides EV’s function in inducing brain injury, EVs also serve as protectors for brain injury. MiRNAs, such as miRNA-182-5p, miRNA-342-3p, and miRNA-92b-3p are present in hypoxia-preconditioned mouse brain EVs, which protect against apoptosis in hypoxia-ischemia-induced mice [96].
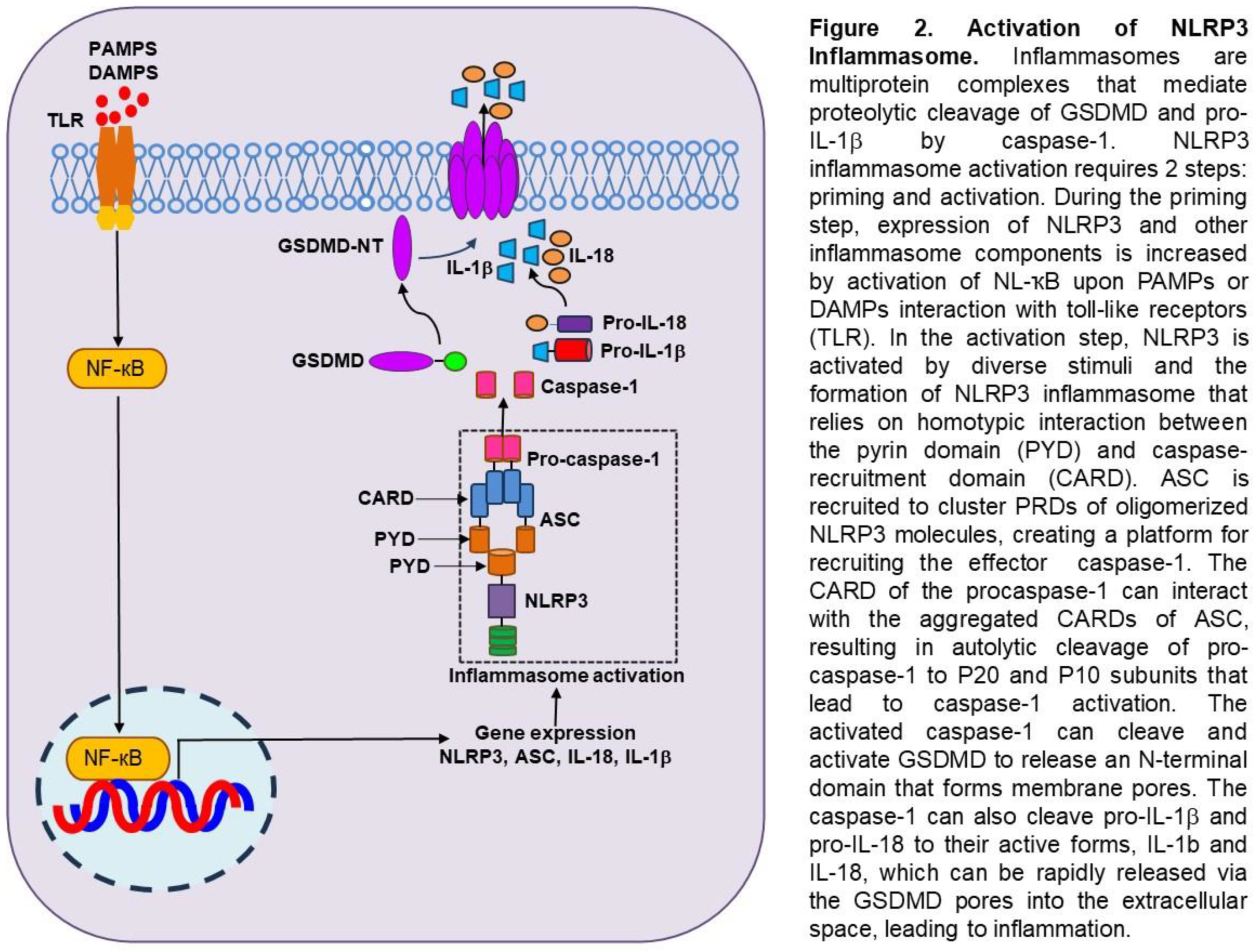
Inflammasomes and Programmed Inflammatory Cell Death in Lung and Brain Injury Inflammasomes are multiprotein complexes that mediate proteolytic cleavage of GSDMD, pro-IL-1b, and pro-IL-18 by caspase-1 [97,98]. One of the most studied inflammasomes is the NLRP3 inflammasome, which belongs to the nucleotide-binding domain (NBD)- and leucine-rich repeat (LRR)-containing protein (NLP) family [99,100,101,102]. NLRP3 activation requires two steps: a priming step and an activation step. First, NLRP3 expression can be primed by germline-encoded pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs), upon recognition of pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) or by cytokines that engage in immune and inflammatory responses. Upon activation of NF-kB or other transcription factors, the expression of NLRP3 and other inflammasome components is transcriptionally upregulated [99,100,101,102]. Posttranslational modifications of NLRP3, including ubiquitination, phosphorylation, and sumoylation, also prime NLRP3 for activation while keeping NLRP3 in an autoinhibited state. In the second step, NLRP3 is activated by diverse microbial and sterile stimuli that often converge to K+ efflux of other ionic changes [99,100,101,102]. The formation of the NLRP3 inflammasome includes three typical components: sensor, adaptor, and effector, and these roles are played by NLRP3, ASC, and caspase-1 [99,100,101,102] (Figure 2). The N-terminal effector domain of NLRP3 consists of a pyrin domain (PYD), 12 leucine-rich repeat (LRR) domains at the C-terminal end, and a central nucleotide-binding oligomerization (NACTH) domain. The ASC comprises an N-terminal PYD domain and a C-terminal caspase-recruitment domain (CARD). Caspase-1 consists of an N-terminal CARD, a large catalytic subunit p20, and a C-terminal small catalytic subunit p10. Assembly of NLRP3 inflammasome relies strictly on homotypic interactions between PYD and CARD. When a ligand is detected, the NLRP3 sensor undergoes a conformational change, releasing itself from its inhibitory state. Through homotypic PYD-PYD interactions, ASC is recruited to cluster PYDs of oligomerized NLRP3 molecules, creating a platform for recruiting effector caspase-1. The CARD of caspase-1 can interact with the aggregated CARDs of ASC, recruiting procaspase-1 and rendering caspase-1 fully proteolytical activity. Once locally recruited, pro-caspase-1 increases and undergoes autolytic cleavage to generate p20 and p10 subunits, which form activated caspase-1. The activated caspase-1 can cleave and activate GSDMD to release an N-terminal domain that can bind to the cell membrane and oligomerize to form transmembrane pores. The caspase-1 simultaneously activates pro-IL-1b and pro-IL-18, converting them into mature proinflammatory cytokines. The GSDMD pores allow water and ions influx, which leads to lytic cell death, a proinflammatory form of programmed cell death known as pyroptosis. The GSDMD pores also allow the release of mature IL-1b and IL-18 into the extracellular space, leading to inflammation [99,100,101,102].
Inflammasomes in Neonatal Lung Diseases: Liao et al. reported in 1995 that activation of NLRP3 inflammasome is associated with the development of BPD [103]. They showed that exposure to 85% hyperoxia in neonatal mice from P3 to P14 increased caspase-1 activation, IL-1b, inflammation, and decreased alveolarization. They further showed that NLRP3 knockout mice have no caspase-1 activity, no IL-1b, no inflammatory response, and undergo normal alveolarization when exposed to 85% oxygen. Treatment of hyperoxia-exposed mice with either IL-1 receptor (IL-1r) antagonist to block IL-1b or glyburide to block the NLRP3 inflammasome resulted in decreased inflammation and increased alveolarization. Notably, the IL-1b:IL-1ra ratio in tracheal aspirates from preterm infants with respiratory failure is predictive of the development of BPD. This important study demonstrates the critical role of NLRP3 in BPD pathogenesis both in experimental models and clinical investigations. While caffeine is commonly used in preterm infants for apnea, in this hyperoxia-induced BPD model, treatment with caffeine significantly reduced oxidative stress, promoted alveolar development, attenuated inflammatory infiltration and lung injury, and these were associated with significant inhibition of NLRP3 inflammasome protein and NF-ҡB pathway [104]. In another newborn mouse model of hyperoxia-induced lung injury, treatment with acetate significantly reduced the expression of TNF-a, IL-1b, IL-18, NLRP3, and caspase-1 [105]. Tert-butylhydroquinone (TBHQ), an inhibitor of Nuclear factor e2-related factor 2 (Nrf2), was shown to reduce NLRP3 inflammasome activation, decrease IL-1b and IL-18 expression and activation, as well as inhibit pyroptosis in the lungs of hyperoxia-induced newborn mice [106]. In neonatal rats exposed to hyperoxia, inhibition of Rac1, a member of the Rho GTPase family with NSC23766, significantly decreased NLRP1 inflammasome activity, reduced lung macrophage infiltration, and improved alveolar and vascular development [107]. Vaidya et al. showed that recombinant cysteine-rich protein 61 (CCN1) reduced macrophage and neutrophil infiltration, decreased NLRP1 inflammasome activation, and improved alveolar and vascular development in hyperoxia-exposed newborn rat lungs [108]. Also, in a hyperoxia-induced BPD rat model, 18b-Glycyrrhetinic acid treatment inhibited the activation of NF-ҡB and NLRP3 inflammasome, decreased ROS level and pulmonary inflammation, improved alveolar development, and increased body weight of neonatal rats exposed to hyperoxia [109]. Simvastatin inhibited NLRP3 inflammasome activation and ameliorated lung injury in hyperoxia-induced BPD via Kruppel-like factor 2 (KLF2)-mediated mechanism [110]. The direct evidence that GSDMD plays an important role in hyperoxia-induced BPD was demonstrated by GSDMD gene knockout protects newborn mice from hyperoxia-induced BPD by reducing macrophage infiltration, improving alveolarization and vascularization, and decreasing cell death [111]. Clearly, there are overwarming data that support the critical role of inflammasomes in experimental BPD.
Inflammasomes in Neonatal Brain Injury: The role of inflammasomes in neonatal brain injury was abundantly studied in experimental models of hypoxic-ischemic encephalopathy (HIE). In 2018, Chen et al. showed that inositol requiring enzyme-1 alpha (IRE1a) expression was significantly increased in the brain after hypoxic-ischemic injury. Intranasal administration of STF-083010, an IRF1a inhibitor, alleviated brain injury, improved neurological behavior, and improved expression of MiR-17-5p. Meanwhile, miR-17-5p mimic administration ameliorated NLRP3 inflammasome activation, caspase-1 cleavage, and IL-1b production, as well as brain infarct volume [112]. Serdar et al. showed that LPS pre-sensitization significantly increases brain area loss and induces microglia activation and neuronal injury after mild hypoxia-ischemia. They also found that microglia upregulate proinflammatory genes involving NLRP3 inflammasome [113]. Lv et al. found that in HIE patients, the elevation levels of pyroptotic pathway tightly correlate with the severity of HIE. Treatment with MCC950, a small molecule inhibiting NLRP3 inflammasome and thus pyroptosis, alleviated pyroptosis and injury severity in rats with neonatal hypoxic-ischemic brain damage [114]. Increasing evidence indicates that miRNAs are involved in the process of HIE, and miR-374a-5p is down-regulated in HIE patients. Further, over-expression of miR-374a-5p significantly attenuated brain injury and inhibited the release of proinflammatory cytokines in neonatal rat HIE models. In vitro, miR-347a-5p inhibited LPS-induced microglial proinflammatory cytokine production by regulating NLRP3 inflammasome [115]. Few studies focused on the roles of inflammasomes in brain white matter injury (WMI). One study tested ZJU-37, a novel dual inhibitor of receptor interacting protein kinase-1 and -3 (RIP1/RIP3) in WMI induced by subjecting postnatal day (P) 3 rat pups to right common carotid artery ligation and hypoxia with or without human neural stem cells (hOPCs). The ZJU-37 combined with hOPCs more effectively decreased the activation of glial cells and NLRP3 inflammasome and improved behavioral function at 12 weeks post-treatment [116]. Caffeine has been shown to inhibit NLRP3 inflammasome activation, reduce expression of Iba-1, an active microglial marker, inhibit microglia M1 polarization, promote microglia M2 polarization, and improve long-term cognitive function in neonatal rats with hypoxic-ischemic WM disease [117]. Diallyl disulfide (DADS) is an allicin extract with detoxifying, antibacterial, and cardiovascular disease protective effects. It was tested in HIE-induced brain damage in rats and showed DADS significantly reduced the cerebral infarct volume, alleviated inflammatory reaction, reduced astrocyte activation, promoted tissue structure recovery, and improved pyroptosis caused by HIE [118]. When GSDMD knockout and wildtype newborn mice were exposed to 85% oxygen, GSDMD knockout mice had considerable resistance to oxygen as they had decreased pyroptosis and increased proliferation compared to wildtype brains. GSDMD knockout also prevented gene expression associated with neuronal and vascular development and differentiation, axonogenesis, glial cell differentiation, hypoxia-induced factor 1 pathway, and neuronal growth factor pathways [119].
EV-inflammasomes Mediate Lung-brain Axis in Neonatal Brain Injury: Recently, our laboratory investigated the critical role of circulating EVs from hyperoxia-exposed and mechanical ventilated newborn rats in inducing brain injury in healthy newborn rats [70,120]. In the hyperoxia model, newborn rats were exposed to room air or 85% oxygen for two weeks, and circulating EVs were isolated from the rats' plasma [70]. The EVs were analyzed using Nanosight tracking, fluorescence-activated cell sorting (FACS), and Western blot. It was found that the EVs from hyperoxia-exposed rats contain increased levels of both surfactant C (SPC), a lung type II epithelial cell marker, and GSDMD, a key executor of inflammasome-induced cell pyroptosis. We performed adoptive transfer experiments by injecting these EVs via the tail veins into healthy newborn rats and found that they were taken up by the lungs and brain. The EVs from the hyperoxia-exposed rats induced lung inflammation, indicated by increased inflammatory cell infiltration in the alveolar airspaces and expression of inflammatory cytokines and chemokines. Furthermore, alveolarization and vascular density were drastically reduced in the lungs that received EVs from hyperoxia-exposed rats. We performed in vitro experiments by treating the pulmonary vascular endothelial cells (PVECs) with EVs from room air and hyperoxia-exposed rats. Interestingly, the PVECs treated with the EVs from hyperoxia-exposed rats had increased cell death and reduced cell survival [70]. Furthermore, EVs from hyperoxia-exposed rats induced brain inflammation by activating microglia and increasing the expression of proinflammatory cytokines. These changes were associated with increased cell death in the cortex, subventricular, and subgranular zones. Additionally, in vitro experiments showed that treating neural stem cells (NSCs) with EVs from hyperoxia-exposed rats decreased cell proliferation and increased cell death [70]. EVs from cultured hyperoxia-exposed lung epithelial cells induced pyroptosis in NSCs [70]. These data revealed a novel lung-brain crosstalk mediated by lung epithelial-derived EVs in both lung and brain injury.
We further investigated this EV-mediated lung-brain crosstalk in mechanical ventilation-associated brain injury in newborn rat models [120]. We demonstrated that injurious mechanical ventilation with higher tidal volumes induced similar markers of inflammation and pyroptosis, such as IL-1β, activated caspase-1, and GSDMD in both lung and brain and induced microglial activation and cell death in the brain [120]. The EVs isolated from neonatal rats with ventilator-induced lung injury had increased caspase-1. We further adoptive transferred these EVs into healthy newborn rats, which led to neuroinflammation with microglial activation and activation of caspase-1 and GSDMD in the brain, similar to that observed in neonatal rats that were mechanically ventilated [120]. Thus, circulating EVs may contribute to brain injury and possibly poor neurodevelopmental outcomes in preterm infants exposed to hyperoxia and mechanical ventilation [120].
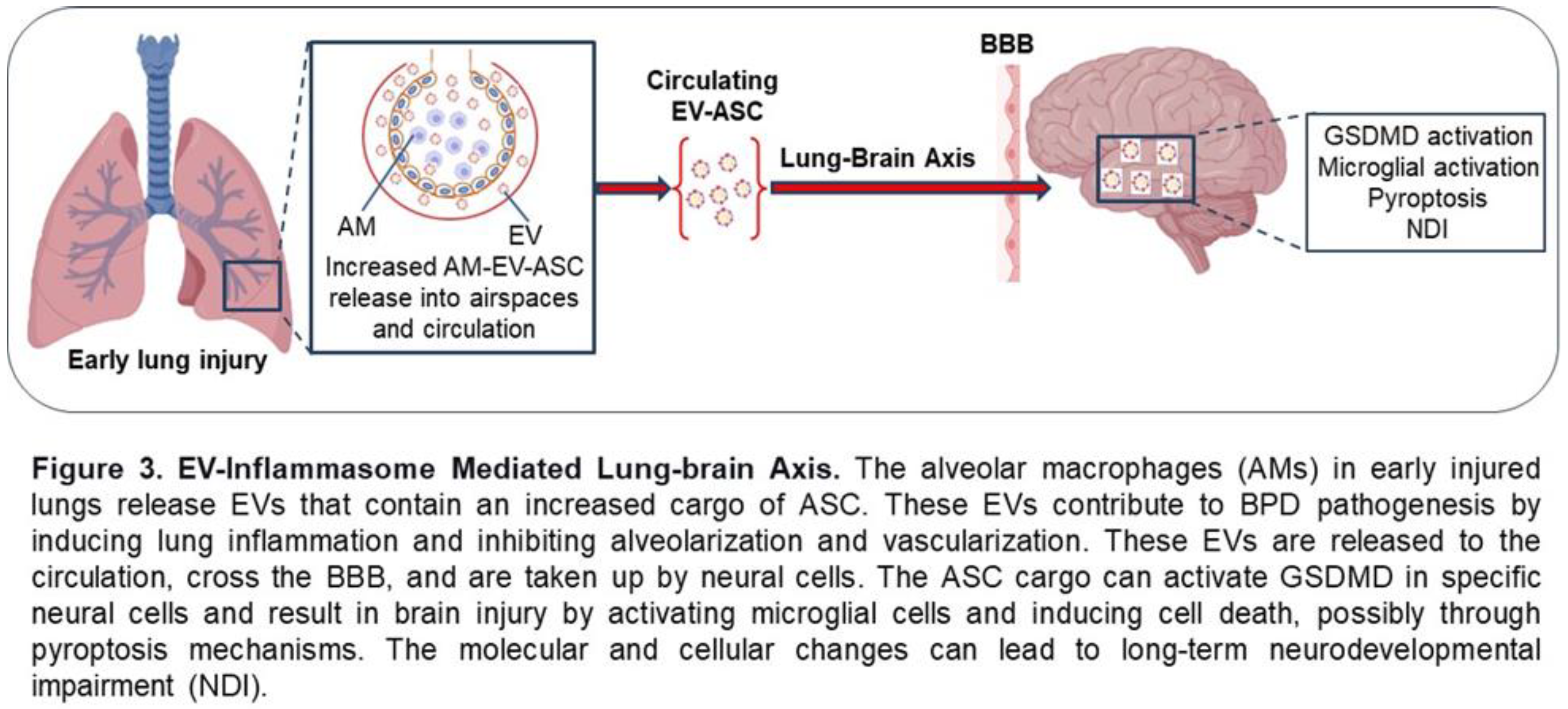
Most recently, we published a study that examined the EVs isolated from the plasma of preterm infants at risk for BPD at 7 days of life [121]. We found that the EVs from infants who were on higher oxygen therapy (≥30%, HO2) had increased levels of alveolar macrophage-derived EV-ASC compared to infants on lower oxygen therapy (<30%, LO2). To assess the function of these EVs, we performed adoptive transfer experiments by injecting them into the circulation of newborn mice and examined the lung and brain on P17. We discovered that mice that received EVs from the HO2 patients had increased lung inflammation, decreased alveolarization, and disrupted vascular development, the hallmarks of BPD. Importantly, these EVs crossed the BBB, and the EVs from infants on HO2 caused inflammation, reduced cell survival, and increased cell death with the feature of pyroptosis in the hippocampus [121]. These data support a novel AM-derived EV-inflammasome model that mediates the lung-brain axis, which leads to brain inflammatory injury (Figure 3). These studies provide experimental and clinical evidence for EV-inflammasomes acting as novel mediators for lung injury-associated brain injury.
Perspective
One important question is whether we can target EVs and inflammasomes to prevent and treat BPD and brain injury in preterm infants. Many pharmacological agents are being investigated on their properties to inhibit EV release as research tools and therapeutic modalities. Some of these agents inhibit EV trafficking, such as calpeptin [122], manumycin A [123], and Y27632 [124]; others inhibit lipid metabolisms, such as D-pantethine [125], imipramine [92], and GW4869 [126]. Additional drugs that inhibit various kinases have also been tested, including bisyndoylmaleimide I (protein kinase C inhibitor) [127], U0126 (inhibitor of MEK 1 and MEK 2, two protein kinases belonging to the mitogen-activated protein kinase family) [128], and clopidogrel (anticoagulant) [129]. Overall, these agents are mainly tested in experimental models of cancer cells [130]. We have a long way to go to identify effective anti-EV agents for preventing and treating BPD and brain injury in preterm infants. In contrast to anti-EV therapy, anti-inflammasome therapy is more promising for preventing and treating BPD and brain injury in preterm infants. Many of the studies that involve anti-inflammasome drugs in experimental models of BPD and brain injury have been reviewed in the above sections. Chiarini et al. summarized an extensive list of specific and non-specific NLRP3 inhibitors tested in brain disorders [131]. VX765 is a specific caspase-1 inhibitor that has been approved by the FDA to be used in clinical trials of adult patients with psoriasis (ClinicalTrials.gov Identifier: NCT00205465) and epilepsy (ClinicalTrials.gov Identifier: NCT01048255 and NCT01501383). Disulfiram inhibits GSDMD pore formation associated with pyroptosis, which the FDA also approves for treating chronic alcoholism [132,133]. GDC-2394 is an NLRP3 inhibitor that was approved by the FDA for the first-in-human phase I trial in normal volunteers [134]. Anakinra, a nonglycosylated recombinant version of human IL-1Ra, is used clinically for rheumatoid arthritis and cryopyrin-associated periodic syndrome [135]. Tadekinig alfa, a recombinant version of human IL18 binding protein, is currently being investigated in a single-arm, open-label phase 3 trial in patients with NLRC4-associated hyperinflammation [136]. Clearly, targeted therapies for EVs and inflammasomes are underdeveloped for BPD and brain injury in preterm infants. It is imperative that some of these therapies be tested in preterm infants with BPD and brain injury in future clinical investigations.
Conclusion
EVs and inflammasomes are emerging targets in clinical and experimental neonatal lung and brain injury. We presented the evidence for BPD as an important risk factor for neonatal brain injury and long-term NDI. We discussed how EV and inflammasome work under physiological conditions and, more importantly, in neonatal lung and brain injury. We presented abundant data demonstrating the critical role of EVs and inflammasomes in regulating lung and brain injury in experimental models, particularly in hyperoxia-induced BPD and brain injury, mimicking clinical scenarios in the neonatal intensive care unit. Intriguingly, lung-derived EVs in rodents and preterm infants carry inflammasome cargo, which can cross the BBB, be taken up by brain cells, and induce brain inflammatory injury. In contrast to many studies investigating the therapeutic role of EVs from various stem cells in neonatal lung and brain injury, this review focused on the novel role of EVs in inducing neonatal lung and brain injury. Targeting EV-inflammasome cascade may have a great potential for treating and preventing preterm infants at risk for developing BPD and brain injury.
References
- Cao, G.; Liu, J.; Liu, M. Global, Regional, and National Incidence and Mortality of Neonatal Preterm Birth, 1990-2019. JAMA Pediatr. 2022, 176, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Jobe, A.H.; Bancalari, E. Bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2001, 163, 1723–1729. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.J.; Jobe, A.H.; Bancalari, E. What is BPD today and in the next 50 years? Am J Physiol Lung Cell Mol Physiol. 2021, 321, L974–L7. [Google Scholar] [CrossRef] [PubMed]
- Volpe, J.J. Brain injury in premature infants: a complex amalgam of destructive and developmental disturbances. Lancet Neurol. 2009, 8, 110–124. [Google Scholar] [CrossRef]
- Faramarzi, R.; Darabi, A.; Emadzadeh, M.; Maamouri, G.; Rezvani, R. Predicting neurodevelopmental outcomes in preterm infants: A comprehensive evaluation of neonatal and maternal risk factors. Early Hum Dev. 2023, 184, 105834. [Google Scholar] [CrossRef]
- Thomas SR, Jain SK, Murthy P, Joseph CJ, Soraisham A, Tang S, et al. Neurodevelopmental Outcomes of Preterm Infants Born <29 Weeks with Bronchopulmonary Dysplasia-Associated Pulmonary Hypertension: A Multicenter Study. Am J Perinatol. 2023.
- Treluyer L, Nuytten A, Guellec I, Jarreau PH, Benhammou V, Cambonie G, et al. Neurodevelopment and healthcare utilisation at age 5-6 years in bronchopulmonary dysplasia: an EPIPAGE-2 cohort study. Arch Dis Child Fetal Neonatal Ed. 2023, 109, 26–33.
- Bhattacharya S, Go D, Krenitsky DL, Huyck HL, Solleti SK, Lunger VA, et al. Genome-wide transcriptional profiling reveals connective tissue mast cell accumulation in bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2012, 186, 349–358.
- Oluwole I, Tan JBC, DeSouza S, Hutchinson M, Leigh RM, Cha M, et al. The association between bronchopulmonary dysplasia grade and risks of adverse neurodevelopmental outcomes among preterm infants born at less than 30 weeks of gestation. J Matern Fetal Neonatal Med. 2023, 36, 2167074.
- Waitzman, N.J.; Jalali, A.; Grosse, S.D. Preterm birth lifetime costs in the United States in 2016: An update. Semin Perinatol. 2021, 45, 151390. [Google Scholar] [CrossRef]
- Malavolti, A.M.; Bassler, D.; Arlettaz-Mieth, R.; Faldella, G.; Latal, B.; Natalucci, G. Bronchopulmonary dysplasia-impact of severity and timing of diagnosis on neurodevelopment of preterm infants: a retrospective cohort study. BMJ Paediatr Open. 2018, 2, e000165. [Google Scholar] [CrossRef]
- Lithopoulos MA, Toussay X, Zhong S, Xu L, Mustafa SB, Ouellette J, et al. Neonatal hyperoxia in mice triggers long-term cognitive deficits via impairments in cerebrovascular function and neurogenesis. J Clin Invest. 2022, 132(22).
- Obst S, Herz J, Alejandre Alcazar MA, Endesfelder S, Mobius MA, Rudiger M, et al. Perinatal Hyperoxia and Developmental Consequences on the Lung-Brain Axis. Oxid Med Cell Longev. 2022, 2022:5784146.
- Kim YE, Park WS, Sung DK, Ahn SY, Sung SI, Yoo HS, et al. Intratracheal transplantation of mesenchymal stem cells simultaneously attenuates both lung and brain injuries in hyperoxic newborn rats. Pediatr Res. 2016, 80, 415–424.
- Dapaah-Siakwan F, Zambrano R, Luo S, Duncan MR, Kerr N, Donda K, et al. Caspase-1 Inhibition Attenuates Hyperoxia-induced Lung and Brain Injury in Neonatal Mice. Am J Respir Cell Mol Biol. 2019, 61, 341–354.
- Northway, W.H.; Jr Rosan, R.C.; Porter, D.Y. Pulmonary disease following respirator therapy of hyaline-membrane disease. Bronchopulmonary dysplasia. N Engl J Med. 1967, 276, 357–368. [Google Scholar] [CrossRef]
- Abman, S.H.; Bancalari, E.; Jobe, A. The Evolution of Bronchopulmonary Dysplasia after 50 Years. Am J Respir Crit Care Med. 2017, 195, 421–424. [Google Scholar] [CrossRef]
- Lapcharoensap W, Gage SC, Kan P, Profit J, Shaw GM, Gould JB, et al. Hospital variation and risk factors for bronchopulmonary dysplasia in a population-based cohort. JAMA Pediatr. 2015, 169, e143676.
- Adams M, Bassler D, Bucher HU, Roth-Kleiner M, Berger TM, Braun J, et al. Variability of Very Low Birth Weight Infant Outcome and Practice in Swiss and US Neonatal Units. Pediatrics. 2018, 141(5).
- Bhunwal, S.; Mukhopadhyay, K.; Bhattacharya, S.; Dey, P.; Dhaliwal, L.K. Bronchopulmonary Dysplasia in Preterm Neonates in a Level III Neonatal Unit in India. Indian Pediatr. 2018, 55, 211–215. [Google Scholar] [CrossRef]
- Bose C, Van Marter LJ, Laughon M, O'Shea TM, Allred EN, Karna P, et al. Fetal growth restriction and chronic lung disease among infants born before the 28th week of gestation. Pediatrics. 2009, 124, e450–8.
- Isayama T, Lee SK, Yang J, Lee D, Daspal S, Dunn M, et al. Revisiting the Definition of Bronchopulmonary Dysplasia: Effect of Changing Panoply of Respiratory Support for Preterm Neonates. JAMA Pediatr. 2017, 171, 271–279.
- Lee, J.H.; Noh, O.K.; Chang, Y.S.; Korean Neonatal, N. Neonatal Outcomes of Very Low Birth Weight Infants in Korean Neonatal Network from 2013 to 2016. J Korean Med Sci. 2019, 34, e40. [Google Scholar] [CrossRef]
- Lin HJ, Du LZ, Ma XL, Shi LP, Pan JH, Tong XM, et al. Mortality and Morbidity of Extremely Low Birth Weight Infants in the Mainland of China: A Multi-center Study. Chin Med J (Engl). 2015, 128, 2743–2750.
- Su BH, Hsieh WS, Hsu CH, Chang JH, Lien R, Lin CH, et al. Neonatal outcomes of extremely preterm infants from taiwan: comparison with Canada, Japan, and the USA. Pediatr Neonatol. 2015, 56, 46–52.
- Higgins RD, Jobe AH, Koso-Thomas M, Bancalari E, Viscardi RM, Hartert TV, et al. Bronchopulmonary Dysplasia: Executive Summary of a Workshop. J Pediatr. 2018, 197:300-8.
- Jensen EA, Dysart K, Gantz MG, McDonald S, Bamat NA, Keszler M, et al. The Diagnosis of Bronchopulmonary Dysplasia in Very Preterm Infants. An Evidence-based Approach. Am J Respir Crit Care Med. 2019, 200, 751–759.
- Thebaud B, Goss KN, Laughon M, Whitsett JA, Abman SH, Steinhorn RH, et al. Bronchopulmonary dysplasia. Nat Rev Dis Primers. 2019, 5, 78.
- Anderson, P.J.; Doyle, L.W. Neurodevelopmental outcome of bronchopulmonary dysplasia. Semin Perinatol. 2006, 30, 227–232. [Google Scholar] [CrossRef]
- Gallini, F.; Arena, R.; Stella, G.; Frezza, S.; Maggio, L. Neurodevelopmental outcomes of premature infants with bronchopulmonary dysplasia. Acta Biomed. 2014, 85, 30–34. [Google Scholar]
- Van Marter LJ, Kuban KC, Allred E, Bose C, Dammann O, O'Shea M, et al. Does bronchopulmonary dysplasia contribute to the occurrence of cerebral palsy among infants born before 28 weeks of gestation? Arch Dis Child Fetal Neonatal Ed. 2011, 96, F20–9.
- Vohr, B.R.; Wright, L.L.; Poole, W.K.; McDonald, S.A. Neurodevelopmental outcomes of extremely low birth weight infants <32 weeks' gestation between 1993 and 1998. Pediatrics. 2005, 116, 635–643. [Google Scholar]
- Skidmore, M.D.; Rivers, A.; Hack, M. Increased risk of cerebral palsy among very low-birthweight infants with chronic lung disease. Dev Med Child Neurol. 1990, 32, 325–332. [Google Scholar] [CrossRef]
- Hintz SR, Kendrick DE, Stoll BJ, Vohr BR, Fanaroff AA, Donovan EF, et al. Neurodevelopmental and growth outcomes of extremely low birth weight infants after necrotizing enterocolitis. Pediatrics. 2005, 115, 696–703.
- Arnaud C, Daubisse-Marliac L, White-Koning M, Pierrat V, Larroque B, Grandjean H, et al. Prevalence and associated factors of minor neuromotor dysfunctions at age 5 years in prematurely born children: the EPIPAGE Study. Arch Pediatr Adolesc Med. 2007, 161, 1053–1061.
- Short EJ, Klein NK, Lewis BA, Fulton S, Eisengart S, Kercsmar C, et al. Cognitive and academic consequences of bronchopulmonary dysplasia and very low birth weight: 8-year-old outcomes. Pediatrics. 2003, 112, e359.
- Davis, N.M.; Ford, G.W.; Anderson, P.J.; Doyle, L.W.; Victorian Infant Collaborative Study, G. Developmental coordination disorder at 8 years of age in a regional cohort of extremely-low-birthweight or very preterm infants. Dev Med Child Neurol. 2007, 49, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Gallini F, Coppola M, De Rose DU, Maggio L, Arena R, Romano V, et al. Neurodevelopmental outcomes in very preterm infants: The role of severity of Bronchopulmonary Dysplasia. Early Hum Dev. 2021, 152:105275.
- Schmidt B, Asztalos EV, Roberts RS, Robertson CM, Sauve RS, Whitfield MF, et al. Impact of bronchopulmonary dysplasia, brain injury, and severe retinopathy on the outcome of extremely low-birth-weight infants at 18 months: results from the trial of indomethacin prophylaxis in preterms. JAMA. 2003, 289, 1124–1129.
- Schmidt B, Anderson PJ, Doyle LW, Dewey D, Grunau RE, Asztalos EV, et al. Survival without disability to age 5 years after neonatal caffeine therapy for apnea of prematurity. JAMA. 2012, 307, 275–282.
- Katz TA, Vliegenthart RJS, Aarnoudse-Moens CSH, Leemhuis AG, Beuger S, Blok GJ, et al. Severity of Bronchopulmonary Dysplasia and Neurodevelopmental Outcome at 2 and 5 Years Corrected Age. J Pediatr. 2022, 243:40-6 e2.
- Reuner, G.; Fields, A.C.; Wittke, A.; Lopprich, M.; Pietz, J. Comparison of the developmental tests Bayley-III and Bayley-II in 7-month-old infants born preterm. Eur J Pediatr. 2013, 172, 393–400. [Google Scholar] [CrossRef]
- Moore, T.; Johnson, S.; Haider, S.; Hennessy, E.; Marlow, N. Relationship between test scores using the second and third editions of the Bayley Scales in extremely preterm children. J Pediatr. 2012, 160, 553–558. [Google Scholar] [CrossRef]
- Rosenberg AA, Lee NR, Vaver KN, Werner D, Fashaw L, Hale K, et al. School-age outcomes of newborns treated for persistent pulmonary hypertension. J Perinatol. 2010, 30, 127–134.
- Short EJ, Kirchner HL, Asaad GR, Fulton SE, Lewis BA, Klein N, et al. Developmental sequelae in preterm infants having a diagnosis of bronchopulmonary dysplasia: analysis using a severity-based classification system. Arch Pediatr Adolesc Med. 2007, 161, 1082–1087.
- Natarajan G, Pappas A, Shankaran S, Kendrick DE, Das A, Higgins RD, et al. Outcomes of extremely low birth weight infants with bronchopulmonary dysplasia: impact of the physiologic definition. Early Hum Dev. 2012, 88, 509–515.
- Lewis BA, Singer LT, Fulton S, Salvator A, Short EJ, Klein N, et al. Speech and language outcomes of children with bronchopulmonary dysplasia. J Commun Disord. 2002, 35, 393–406.
- Majnemer, A.; Riley, P.; Shevell, M.; Birnbaum, R.; Greenstone, H.; Coates, A.L. Severe bronchopulmonary dysplasia increases risk for later neurological and motor sequelae in preterm survivors. Dev Med Child Neurol. 2000, 42, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Taylor, H.G.; Klein, N.; Schatschneider, C.; Hack, M. Predictors of early school age outcomes in very low birth weight children. J Dev Behav Pediatr. 1998, 19, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Taylor, H.G.; Minich, N.; Bangert, B.; Filipek, P.A.; Hack, M. Long-term neuropsychological outcomes of very low birth weight: associations with early risks for periventricular brain insults. J Int Neuropsychol Soc. 2004, 10, 987–1004. [Google Scholar] [CrossRef] [PubMed]
- Annesi, C.A.; Levin, J.C.; Litt, J.S.; Sheils, C.A.; Hayden, L.P. Long-term respiratory and developmental outcomes in children with bronchopulmonary dysplasia and history of tracheostomy. J Perinatol. 2021, 41, 2645–2650. [Google Scholar] [CrossRef]
- DeMauro, S.B. Neurodevelopmental outcomes of infants with bronchopulmonary dysplasia. Pediatr Pulmonol. 2021, 56, 3509–3517. [Google Scholar] [CrossRef]
- Thery, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef]
- Gyorgy B, Szabo TG, Pasztoi M, Pal Z, Misjak P, Aradi B, et al. Membrane vesicles, current state-of-the-art: emerging role of extracellular vesicles. Cell Mol Life Sci. 2011, 68, 2667–2688.
- van der Pol E, Boing AN, Harrison P, Sturk A, Nieuwland, R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol Rev. 2012, 64, 676–705.
- Buzas, E.I. The roles of extracellular vesicles in the immune system. Nat Rev Immunol. 2023, 23, 236–250. [Google Scholar] [CrossRef]
- Thery C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018, 7, 1535750.
- Doyle, L.M.; Wang, M.Z. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells. 2019, 8(7).
- De Lorenzis, E.; Rindone, A.; Di Donato, S.; Del Galdo, F. Circulating extracellular vesicles in the context of interstitial lung disease related to systemic sclerosis: A scoping literature review. Autoimmun Rev. 2023, 22, 103401. [Google Scholar] [CrossRef] [PubMed]
- Xiong, C.; Huang, X.; Chen, S.; Li, Y. Role of Extracellular microRNAs in Sepsis-Induced Acute Lung Injury. J Immunol Res. 2023, 2023:5509652.
- Tine M, Neri T, Biondini D, Bernardinello N, Casara A, Conti M, et al. Do Circulating Extracellular Vesicles Strictly Reflect Bronchoalveolar Lavage Extracellular Vesicles in COPD? Int J Mol Sci. 2023, 24(3).
- Li Z, Wang X, Wang X, Yi X, Wong YK, Wu J, et al. Research progress on the role of extracellular vesicles in neurodegenerative diseases. Transl Neurodegener. 2023, 12, 43.
- Wang, X.; Yang, H.; Liu, C.; Liu, K. A new diagnostic tool for brain disorders: extracellular vesicles derived from neuron, astrocyte, and oligodendrocyte. Front Mol Neurosci. 2023, 16:1194210.
- Kerr NA, de Rivero Vaccari JP, Abbassi S, Kaur H, Zambrano R, Wu S, et al. Traumatic Brain Injury-Induced Acute Lung Injury: Evidence for Activation and Inhibition of a Neural-Respiratory-Inflammasome Axis. J Neurotrauma. 2018, 35, 2067–2076.
- Schiller, E.A.; Cohen, K.; Lin, X.; El-Khawam, R.; Hanna, N. Extracellular Vesicle-microRNAs as Diagnostic Biomarkers in Preterm Neonates. Int J Mol Sci. 2023, 24(3).
- Ransom, M.A.; Blatt, A.M.; Pua, H.H.; Sucre, J.M.S. The emerging role of extracellular vesicles in bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol. 2024, 326, L517–L23. [Google Scholar] [CrossRef]
- Lal CV, Olave N, Travers C, Rezonzew G, Dolma K, Simpson A, et al. Exosomal microRNA predicts and protects against severe bronchopulmonary dysplasia in extremely premature infants. JCI Insight. 2018, 3(5).
- Ransom MA, Bunn KE, Negretti NM, Jetter CS, Bressman ZJ, Sucre JMS, et al. Developmental trajectory of extracellular vesicle characteristics from the lungs of preterm infants. Am J Physiol Lung Cell Mol Physiol. 2023, 324, L385–L92.
- Go H, Maeda H, Miyazaki K, Maeda R, Kume Y, Namba F, et al. Extracellular vesicle miRNA-21 is a potential biomarker for predicting chronic lung disease in premature infants. Am J Physiol Lung Cell Mol Physiol. 2020, 318, L845–L51.
- Ali A, Zambrano R, Duncan MR, Chen S, Luo S, Yuan H, et al. Hyperoxia-activated circulating extracellular vesicles induce lung and brain injury in neonatal rats. Sci Rep. 2021, 11, 8791.
- Zhou Y, Liu Y, Xu G, Liu L, Li H, Li Y, et al. Human breast milk-derived exosomes through inhibiting AT II cell apoptosis to prevent bronchopulmonary dysplasia in rat lung. J Cell Mol Med. 2022, 26, 4169–4182.
- Bellio MA, Young KC, Milberg J, Santos I, Abdullah Z, Stewart D, et al. Amniotic fluid-derived extracellular vesicles: characterization and therapeutic efficacy in an experimental model of bronchopulmonary dysplasia. Cytotherapy. 2021, 23, 1097–1107.
- Vechetti IJ, Norrbom J, Alkner B, Hjalmarsson E, Palmcrantz A, Ponten E, et al. Extracellular vesicle characteristics and microRNA content in cerebral palsy and typically developed individuals at rest and in response to aerobic exercise. Front Physiol. 2022, 13:1072040.
- Zhong XQ, Yan Q, Chen ZG, Jia CH, Li XH, Liang ZY, et al. Umbilical Cord Blood-Derived Exosomes From Very Preterm Infants With Bronchopulmonary Dysplasia Impaired Endothelial Angiogenesis: Roles of Exosomal MicroRNAs. Front Cell Dev Biol. 2021, 9:637248.
- Genschmer KR, Russell DW, Lal C, Szul T, Bratcher PE, Noerager BD, et al. Activated PMN Exosomes: Pathogenic Entities Causing Matrix Destruction and Disease in the Lung. Cell. 2019, 176(1-2):113-26 e15.
- Wang, Y.; Wang, X.; Xu, Q.; Yin, J.; Wang, H.; Zhang, L. CircRNA, lncRNA, and mRNA profiles of umbilical cord blood exosomes from preterm newborns showing bronchopulmonary dysplasia. Eur J Pediatr. 2022, 181, 3345–3365. [Google Scholar] [CrossRef]
- Prada I, Gabrielli M, Turola E, Iorio A, D'Arrigo G, Parolisi R, et al. Glia-to-neuron transfer of miRNAs via extracellular vesicles: a new mechanism underlying inflammation-induced synaptic alterations. Acta Neuropathol. 2018, 135, 529–550.
- Bakhti, M.; Winter, C.; Simons, M. Inhibition of myelin membrane sheath formation by oligodendrocyte-derived exosome-like vesicles. J Biol Chem. 2011, 286, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Fevrier B, Vilette D, Archer F, Loew D, Faigle W, Vidal M, et al. Cells release prions in association with exosomes. Proc Natl Acad Sci U S A. 2004, 101, 9683–9688.
- Fitzner D, Schnaars M, van Rossum D, Krishnamoorthy G, Dibaj P, Bakhti M, et al. Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis. J Cell Sci. 2011, 124(Pt 3):447-58.
- Fruhbeis C, Frohlich D, Kuo WP, Amphornrat J, Thilemann S, Saab AS, et al. Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte-neuron communication. PLoS Biol. 2013, 11, e1001604.
- Lewis, S. Glia: Transporting cargo from A to B. Nat Rev Neurosci. 2013, 14, 589. [Google Scholar] [CrossRef]
- Sharma, P.; Schiapparelli, L.; Cline, H.T. Exosomes function in cell-cell communication during brain circuit development. Curr Opin Neurobiol. 2013, 23, 997–1004. [Google Scholar] [CrossRef]
- Coleman, B.M.; Hill, A.F. Extracellular vesicles--Their role in the packaging and spread of misfolded proteins associated with neurodegenerative diseases. Semin Cell Dev Biol. 2015, 40:89-96.
- Budnik, V.; Ruiz-Canada, C.; Wendler, F. Extracellular vesicles round off communication in the nervous system. Nat Rev Neurosci. 2016, 17, 160–172. [Google Scholar] [CrossRef]
- Thompson AG, Gray E, Heman-Ackah SM, Mager I, Talbot K, Andaloussi SE, et al. Extracellular vesicles in neurodegenerative disease - pathogenesis to biomarkers. Nat Rev Neurol. 2016, 12, 346–357.
- Zhang L, Graf I, Kuang Y, Zheng X, Haupt M, Majid A, et al. Neural Progenitor Cell-Derived Extracellular Vesicles Enhance Blood-Brain Barrier Integrity by NF-kappaB (Nuclear Factor-kappaB)-Dependent Regulation of ABCB1 (ATP-Binding Cassette Transporter B1) in Stroke Mice. Arterioscler Thromb Vasc Biol. 2021, 41, 1127–1145.
- Spaull R, McPherson B, Gialeli A, Clayton A, Uney J, Heep A, et al. Exosomes populate the cerebrospinal fluid of preterm infants with post-haemorrhagic hydrocephalus. Int J Dev Neurosci. 2019, 73:59-65.
- Gall, A.R.; Amoah, S.; Kitase, Y.; Jantzie, L.L. Placental mediated mechanisms of perinatal brain injury: Evolving inflammation and exosomes. Exp Neurol. 2022, 347:113914.
- Leon J, Acurio J, Bergman L, Lopez J, Karin Wikstrom A, Torres-Vergara P, et al. Disruption of the Blood-Brain Barrier by Extracellular Vesicles From Preeclampsia Plasma and Hypoxic Placentae: Attenuation by Magnesium Sulfate. Hypertension. 2021, 78, 1423–1433.
- Dickens AM, Tovar YRLB, Yoo SW, Trout AL, Bae M, Kanmogne M, et al. Astrocyte-shed extracellular vesicles regulate the peripheral leukocyte response to inflammatory brain lesions. Sci Signal. 2017, 10(473).
- Bianco F, Perrotta C, Novellino L, Francolini M, Riganti L, Menna E, et al. Acid sphingomyelinase activity triggers microparticle release from glial cells. EMBO J. 2009, 28, 1043–1054.
- Lombardi M, Parolisi R, Scaroni F, Bonfanti E, Gualerzi A, Gabrielli M, et al. Detrimental and protective action of microglial extracellular vesicles on myelin lesions: astrocyte involvement in remyelination failure. Acta Neuropathol. 2019, 138, 987–1012.
- Wies Mancini, V.S.B.; Mattera, V.S.; Pasquini, J.M.; Pasquini, L.A.; Correale, J.D. Microglia-derived extracellular vesicles in homeostasis and demyelination/remyelination processes. J Neurochem. 2024, 168, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Drago F, Lombardi M, Prada I, Gabrielli M, Joshi P, Cojoc D, et al. ATP Modifies the Proteome of Extracellular Vesicles Released by Microglia and Influences Their Action on Astrocytes. Front Pharmacol. 2017, 8:910.
- Meng, Q.; Yang, P.; Lu, Y. MicroRNA-410 serves as a candidate biomarker in hypoxic-ischemic encephalopathy newborns and provides neuroprotection in oxygen-glucose deprivation-injured PC12 and SH-SY5Y cells. Brain Behav. 2021, 11, e2293. [Google Scholar] [CrossRef]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015, 526, 660–665.
- Li, Z.; Ji, S.; Jiang, M.L.; Xu, Y.; Zhang, C.J. The Regulation and Modification of GSDMD Signaling in Diseases. Front Immunol. 2022, 13:893912.
- Gaidt, M.M.; Hornung, V. The NLRP3 Inflammasome Renders Cell Death Pro-inflammatory. J Mol Biol. 2018, 430, 133–141. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell. 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Fu, J.; Wu, H. Structural Mechanisms of NLRP3 Inflammasome Assembly and Activation. Annu Rev Immunol. 2023, 41:301-16.
- Liao J, Kapadia VS, Brown LS, Cheong N, Longoria C, Mija D, et al. The NLRP3 inflammasome is critically involved in the development of bronchopulmonary dysplasia. Nat Commun. 2015, 6:8977.
- Chen, S.; Wu, Q.; Zhong, D.; Li, C.; Du, L. Caffeine prevents hyperoxia-induced lung injury in neonatal mice through NLRP3 inflammasome and NF-kappaB pathway. Respir Res. 2020, 21, 140. [Google Scholar] [CrossRef]
- Zhang, Q.; Ran, X.; He, Y.; Ai, Q.; Shi, Y. Acetate Downregulates the Activation of NLRP3 Inflammasomes and Attenuates Lung Injury in Neonatal Mice With Bronchopulmonary Dysplasia. Front Pediatr. 2020, 8:595157.
- Wang M, Zhang F, Ning X, Wu C, Zhou Y, Gou Z, et al. Regulating NLRP3 Inflammasome-Induced Pyroptosis via Nrf2: TBHQ Limits Hyperoxia-Induced Lung Injury in a Mouse Model of Bronchopulmonary Dysplasia. Inflammation. 2023, 46, 2386–2401.
- Hummler JK, Dapaah-Siakwan F, Vaidya R, Zambrano R, Luo S, Chen S, et al. Inhibition of Rac1 Signaling Downregulates Inflammasome Activation and Attenuates Lung Injury in Neonatal Rats Exposed to Hyperoxia. Neonatology. 2017, 111, 280–288.
- Vaidya R, Zambrano R, Hummler JK, Luo S, Duncan MR, Young K, et al. Recombinant CCN1 prevents hyperoxia-induced lung injury in neonatal rats. Pediatr Res. 2017, 82, 863–871.
- Qing, C.; Ziyun, L.; Xuefei, Y.; Xinyi, Z.; Xindong, X.; Jianhua, F. Protective Effects of 18beta-Glycyrrhetinic Acid on Neonatal Rats with Hyperoxia Exposure. Inflammation. 2022, 45, 1224–1238. [Google Scholar] [CrossRef]
- Wang X, Huo R, Liang Z, Xu C, Chen T, Lin J, et al. Simvastatin Inhibits NLRP3 Inflammasome Activation and Ameliorates Lung Injury in Hyperoxia-Induced Bronchopulmonary Dysplasia via the KLF2-Mediated Mechanism. Oxid Med Cell Longev. 2022, 2022:8336070.
- Sonny S, Yuan H, Chen S, Duncan MR, Chen P, Benny M, et al. GSDMD deficiency ameliorates hyperoxia-induced BPD and ROP in neonatal mice. Sci Rep. 2023, 13, 143.
- Chen D, Dixon BJ, Doycheva DM, Li B, Zhang Y, Hu Q, et al. IRE1alpha inhibition decreased TXNIP/NLRP3 inflammasome activation through miR-17-5p after neonatal hypoxic-ischemic brain injury in rats. J Neuroinflammation. 2018, 15, 32.
- Serdar M, Kempe K, Rizazad M, Herz J, Bendix I, Felderhoff-Muser U, et al. Early Pro-inflammatory Microglia Activation After Inflammation-Sensitized Hypoxic-Ischemic Brain Injury in Neonatal Rats. Front Cell Neurosci. 2019, 13:237.
- Lv Y, Sun B, Lu XX, Liu YL, Li M, Xu LX, et al. The role of microglia mediated pyroptosis in neonatal hypoxic-ischemic brain damage. Biochem Biophys Res Commun. 2020, 521, 933–938.
- Chen, Z.; Hu, Y.; Lu, R.; Ge, M.; Zhang, L. MicroRNA-374a-5p inhibits neuroinflammation in neonatal hypoxic-ischemic encephalopathy via regulating NLRP3 inflammasome targeted Smad6. Life Sci. 2020, 252:117664.
- Zhang C, Guan Q, Shi H, Cao L, Liu J, Gao Z, et al. A novel RIP1/RIP3 dual inhibitor promoted OPC survival and myelination in a rat neonatal white matter injury model with hOPC graft. Stem Cell Res Ther. 2021, 12, 462.
- Yang, L.; Yu, X.; Zhang, Y.; Liu, N.; Xue, X.; Fu, J. Caffeine treatment started before injury reduces hypoxic-ischemic white-matter damage in neonatal rats by regulating phenotypic microglia polarization. Pediatr Res. 2022, 92, 1543–1554. [Google Scholar] [CrossRef]
- Zheng Y, Zhu T, Chen B, Fang Y, Wu Y, Feng X, et al. Diallyl disulfide attenuates pyroptosis via NLRP3/Caspase-1/IL-1beta signaling pathway to exert a protective effect on hypoxic-ischemic brain damage in neonatal rats. Int Immunopharmacol. 2023, 124(Pt B):111030.
- Challa NVD, Chen S, Yuan H, Duncan MR, Moreno WJ, Bramlett H, et al. GSDMD gene knockout alleviates hyperoxia-induced hippocampal brain injury in neonatal mice. J Neuroinflammation. 2023, 20, 205.
- Chavez L, Meguro J, Chen S, de Paiva VN, Zambrano R, Eterno JM, et al. Circulating extracellular vesicles activate the pyroptosis pathway in the brain following ventilation-induced lung injury. J Neuroinflammation. 2021, 18, 310.
- Starke N, Challa NVD, Yuan H, Chen S, Duncan MR, Cabrera Ranaldi ED, et al. Extracellular Vesicle ASC: A Novel Mediator for Lung-Brain Axis in Preterm Brain Injury. Am J Respir Cell Mol Biol. 2024.
- Fox, J.E.; Austin, C.D.; Reynolds, C.C.; Steffen, P.K. Evidence that agonist-induced activation of calpain causes the shedding of procoagulant-containing microvesicles from the membrane of aggregating platelets. J Biol Chem. 1991, 266, 13289–13295. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.J.; Shin, Y.; Chung, S.; Hwang, D.W.; Lee, D.S. Convective exosome-tracing microfluidics for analysis of cell-non-autonomous neurogenesis. Biomaterials. 2017, 112:82-94.
- Li, B.; Antonyak, M.A.; Zhang, J.; Cerione, R.A. RhoA triggers a specific signaling pathway that generates transforming microvesicles in cancer cells. Oncogene. 2012, 31, 4740–4749. [Google Scholar] [CrossRef]
- Kavian N, Marut W, Servettaz A, Nicco C, Chereau C, Lemarechal H, et al. Pantethine Prevents Murine Systemic Sclerosis Through the Inhibition of Microparticle Shedding. Arthritis Rheumatol. 2015, 67, 1881–1890.
- Menck K, Sonmezer C, Worst TS, Schulz M, Dihazi GH, Streit F, et al. Neutral sphingomyelinases control extracellular vesicles budding from the plasma membrane. J Extracell Vesicles. 2017, 6, 1378056.
- Stratton, D.; Moore, C.; Zheng, L.; Lange, S.; Inal, J. Prostate cancer cells stimulated by calcium-mediated activation of protein kinase C undergo a refractory period before re-releasing calcium-bearing microvesicles. Biochem Biophys Res Commun. 2015, 460, 511–517. [Google Scholar] [CrossRef]
- Li, M.; Yu, D.; Williams, K.J.; Liu, M.L. Tobacco smoke induces the generation of procoagulant microvesicles from human monocytes/macrophages. Arterioscler Thromb Vasc Biol. 2010, 30, 1818–1824. [Google Scholar] [CrossRef]
- Ryu, J.H.; Kim, S.J. Clopidogrel effectively suppresses endothelial microparticle generation induced by indoxyl sulfate via inhibition of the p38 mitogen-activated protein kinase pathway. Blood Purif. 2011, 32, 186–194. [Google Scholar] [CrossRef]
- Catalano, M.; O'Driscoll, L. Inhibiting extracellular vesicles formation and release: a review of EV inhibitors. J Extracell Vesicles. 2020, 9, 1703244. [Google Scholar] [CrossRef]
- Chiarini A, Gui L, Viviani C, Armato U, Dal Pra, I. NLRP3 Inflammasome's Activation in Acute and Chronic Brain Diseases-An Update on Pathogenetic Mechanisms and Therapeutic Perspectives with Respect to Other Inflammasomes. Biomedicines. 2023, 11(4).
- Wang HQ, Song KY, Feng JZ, Huang SY, Guo XM, Zhang L, et al. Caffeine Inhibits Activation of the NLRP3 Inflammasome via Autophagy to Attenuate Microglia-Mediated Neuroinflammation in Experimental Autoimmune Encephalomyelitis. J Mol Neurosci. 2022, 72, 97–112.
- Xu J, Pickard JM, Nunez, G. FDA-approved disulfiram inhibits the NLRP3 inflammasome by regulating NLRP3 palmitoylation. Cell Rep. 2024, 43, 114609.
- McBride C, Trzoss L, Povero D, Lazic M, Ambrus-Aikelin G, Santini A, et al. Overcoming Preclinical Safety Obstacles to Discover (S)-N-((1,2,3,5,6,7-Hexahydro-s-indacen-4-yl)carbamoyl)-6-(methylamino)-6,7-dihydro-5H-pyrazolo [5,1-b][1,3]oxazine-3-sulfonamide (GDC-2394): A Potent and Selective NLRP3 Inhibitor. J Med Chem. 2022, 65, 14721–14739.
- Van Gorp, H.; Van Opdenbosch, N.; Lamkanfi, M. Inflammasome-Dependent Cytokines at the Crossroads of Health and Autoinflammatory Disease. Cold Spring Harb Perspect Biol. 2019, 11(1).
- Canna SW, Girard C, Malle L, de Jesus A, Romberg N, Kelsen J, et al. Life-threatening NLRC4-associated hyperinflammation successfully treated with IL-18 inhibition. J Allergy Clin Immunol. 2017, 139, 1698–1701.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



