Submitted:
09 October 2024
Posted:
11 October 2024
Read the latest preprint version here
Abstract
Recent avian influenza outbreaks have heightened global concern over viral threats with the potential to significantly impact human health. Influenza is particularly alarming due to its history of causing pandemics and zoonotic reservoirs. In response, significant progress has been made toward the development of universal influenza vaccines, largely driven by the discovery of broadly neutralizing antibodies (bnAbs). bnAbs have the potential to neutralize a broad range of influenza viruses, extending beyond the traditional strain-specific response. This could lead to longer-lasting immunity, reducing the need for seasonal vaccinations, and improve preparedness for future pandemics. This review offers a comprehensive analysis of these antibodies, their application in clinical studies, and both their potential and possible shortcomings in managing future influenza outbreaks.
Keywords:
Influenza
; Virus
; Antibodies
; Broadly Neutralizing Antibodies
; Vaccines
; Immunology
1. Introduction
Influenza is a globally endemic respiratory virus typically associated with upper respiratory tract infection, cough, and accompanying fever [1]. While generally not lethal, influenza poses a significant health burden on geriatric, paediatric, or otherwise immunocompromised individuals [2]. The World Health Organization (WHO) estimates around a billion seasonal infections, 3-5 million cases of severe disease outcomes, and up to 650,000 annual deaths can be attributed to influenza each year [3].
Human infections are primarily caused by influenza types A and B; however types C and D are also known. Influenza can be broadly classified by the composition of its major surface glycoproteins; the entry protein, hemagglutinin (HA), and exit protein neuraminidase (NA). The specific combination of HA and NA not only defines the virus's preferred host target and virulence, but also influences its zoonotic potential and pandemic threat [1].
Despite circulating for centuries [4], influenza remains a public health threat. The ability to continue evading existing immune responses is heavily linked to two phenomena; antigenic drift and antigenic shift. Antigenic drift describes the accumulation of glycoprotein mutations in response to selective pressure of acquired immune responses. Antigenic shift describes sudden introductions of new or recombined viral strains. The dramatic rearrangement of the antigenic landscape frequently has a devastating effect on immunologically naïve populations [5]. This has been demonstrated by the four historical flu outbreaks: the 1918 H1N1 Spanish Flu that killed an estimated 40 million people, the 1957 H2N2 Asian Flu, and the 1968 H3N2 Hong Kong Flu affecting 700,000 and 1 million people respectively, and the 2009 H1N1 Swine Flu affecting 16,000 people worldwide [6,7]. All except the 1918 Spanish Flu are attributed to antigenic shift, whereas the Spanish Flu is thought to be a zoonotic avian virus that underwent unusually rapid antigenic drift [6,7].
The fact that mildly antigenically drifted seasonal strains tend to be more immunologically tolerable suggests that a person’s infection history significantly influences disease severity. In the early 1980s, distinctions were made regarding “strain-specific” and “cross-reactive” antibodies, with the latter being mentioned as a possible explanation for the ability to tolerate mildly mutated strains [8]. This was corroborated in 1993, when Okuno and colleagues observed that mice immunized with A/Okuda/57 (H2N2) gained immunity to all H1 and H2 strains through the generation of a singular broadly neutralizing antibody (bnAb), termed C179 [9]. Here we review trends and treatments relating to antibodies capable of neutralizing multiple antigenically drifted, chronologically distinct viruses (intrastrain bnAbs), different viruses within the same Influenza Group (intrasubtypic bnAbs) and viruses within different influenza groups (intergroup bnAbs).
1.1. Hemagglutinin
HA is the primary immunologic target in influenza. Influenza A features 18 different HAs (H1-H18), while Influenza B has two HAs (Yamagata and Victoria). HA is synthesized as an immature HA0 chain, which is proteolytically cleaved by endoplasmic reticulum host proteases into disulphide-linked subunits – HA1 and HA2. HA1 primarily comprises the globular head domain, forming functionally critical structures including the receptor binding site (RBS). HA2, together with a portion of HA1 forms the stem domain. During viral infection, the HA1 RBS binds to sialic acid, inducing viral endocytosis. Upon endosomal acidification, the HA2 subunit undergoes a conformational change leading to the insertion of a hydrophobic fusion peptide into the host membrane. Alongside further conformational changes, this leads to endosomal collapse and the introduction of the viral genome to the host cell (Fig 1) [10].
Antibody potency against HA is influenced by both the mutation frequency of the epitope and its functional significance. As such antibodies targeting the head domain, particularly the RBS tend to be highly immunogenic; however epitopes in the head domain are less stable and tend to drift seasonally [11]. Conversely, antibodies targeting the stem must significantly impede conformational changes during acidification of the endosome or the fusion peptide [11]. Only few such epitopes have been characterised, yet the lower mutational rate of the HA stem means that functional antibodies are more likely to also be broadly neutralising (Fig 2) [12,13,14].
2. bnAbs against the HA Stem
The discovery of C179 in 1993 demonstrated that broadly neutralizing antibodies (bnAbs) targeting the hemagglutinin (HA) stem exist [9]. However, the reduced accessibility of the stem, combined with the immunodominance of the accessible HA head, has been suggested as a barrier to the development of bnAbs targeting the stem [15]. It has been shown that enhancing immune focus away from the head by hyperglycosylating variable regions significantly increases the production of stem-targeting antibodies [16]. To date, only two main immunological stem epitopes have been identified: 1) the Central Stem (CS) Epitope, and 2) the Anchor Epitope/Fusion Peptide (Fig 2). Whether the limited identification of other stem epitopes is due to the functional importance of HA sites, steric constraints, or the dominance of other HA regions remains an open question.
Most stem antibodies characterized to date are IGVH1-69 somatically hypermutated antibodies, that predominantly bind via the heavy-chain complementarity determining region 3 (CDRH3) (Table 2).
Figure 2.
Approximate Locations of Stem Epitopes in Representative Influenza A Group 1 (A/South Carolina/1/1918(H1N1), PDB: 1RUZ), Influenza A Group 2 (A/Hong Kong/1/1968(H3N2), PDB: 4WE4) and Influenza B (B/Hong Kong/8/73, PDB: 3BT6). The central stem (CS) epitope (pink) and fusion peptide (cyan H3N2) or fusion peptide and anchor epitope (cyan H1N1) are in the stem. Conversely the RBS (blue), VE (green), lateral patch (yellow) are situated in the head domain. The occluded epitope and the interface epitope (orange) are marked in orange on a single rotated representative H3N2 monomer.
Figure 2.
Approximate Locations of Stem Epitopes in Representative Influenza A Group 1 (A/South Carolina/1/1918(H1N1), PDB: 1RUZ), Influenza A Group 2 (A/Hong Kong/1/1968(H3N2), PDB: 4WE4) and Influenza B (B/Hong Kong/8/73, PDB: 3BT6). The central stem (CS) epitope (pink) and fusion peptide (cyan H3N2) or fusion peptide and anchor epitope (cyan H1N1) are in the stem. Conversely the RBS (blue), VE (green), lateral patch (yellow) are situated in the head domain. The occluded epitope and the interface epitope (orange) are marked in orange on a single rotated representative H3N2 monomer.
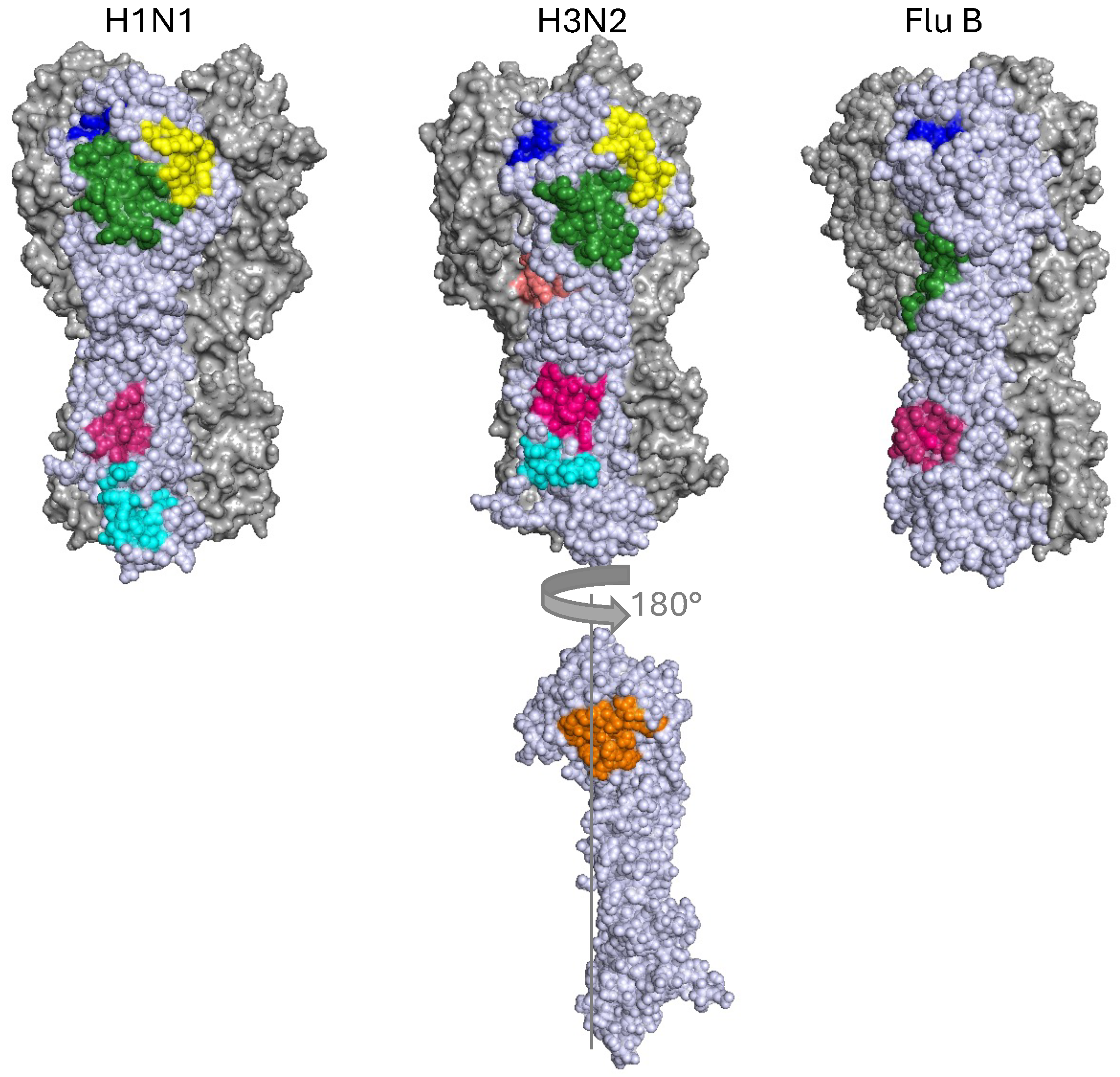
2.1. The Central Stem Epitope
Most stem bnAbs, including C179, target the central stem (CS) epitope (Fig 2, Table 1) [17]. This epitope broadly consists of a conserved hydrophobic pocket, which affects conformational changes related to membrane fusion and HA0 processing [18,19,20,21]. CS antibodies also frequently mediate antibody dependent cellular cytotoxicity (ADCC) (Table 1).
The first human serum-derived bnAb to the CS was discovered in 2008 [12]. This antibody, called CR6261, elicited broad protection in pandemic H5N1 and H1N1 lethally challenged mice [12]. The potency of this site became apparent with F10, a human antibody targeting the CS, capable of neutralising H1N1, H2N2, H5N1, H6N1, H6N2, H8N4 and H9N2 [19]. This was rapidly followed by the discovery of FI6, an antibody that was able to bind to all 16 hemagglutinin subtypes in influenza A, but not influenza B [21]. FI6 has since been shown to elicit in vivo protection in mice, ferrets, and pigs against a panel of Influenza A viruses (Table 1)[21,22,23].
2.2. The Fusion Peptide and Anchor Epitope
A second antigenic site has been identified below the CS epitope and closer to the viral membrane (Fig 2, Table 1). It was initially thought to be unique to Group 2 influenza A viruses, [17,24,25] with CR8020 [24] and CR8043 [25] both eliciting robust protection against H3N2 and H7Nx viruses. However, recently Group 1 influenza has also been found to contain a relevant site near the viral membrane [26]. This site is reported to possess a strong polyclonal response upon H1N1 vaccination or infection, with classified antibodies – 047-09 4F04, 241 IgA 2F04 and 222-1C06 – recognizing a well conserved epitope amongst Group 1 viruses consisting of W343, H354, Q356, S361 and Y363 [26].

Table 1.
Overview of Extensively Studied Stem-Targeting Broadly Neutralizing Antibodies (bnAbs). This table provides details on some of the most thoroughly researched stem-targeting bnAbs, adding to previously published work [17]. It includes information on their in vitro binding affinity, in vitro neutralization capacity, and in vivo protective efficacy. The table also lists the immunoglobulin heavy-chain variable region (IGHV) gene used, the primary complementarity-determining region (CDR) recognition mode, and whether the antibody exhibits antibody-dependent cellular cytotoxicity (ADCC) as a significant protection mechanism. Additionally, it specifies whether the antibody was isolated from mice or humans, any known escape mutations, and the IgG subtype used in generating the findings. A – is shown if the information was not provided.
Table 1.
Overview of Extensively Studied Stem-Targeting Broadly Neutralizing Antibodies (bnAbs). This table provides details on some of the most thoroughly researched stem-targeting bnAbs, adding to previously published work [17]. It includes information on their in vitro binding affinity, in vitro neutralization capacity, and in vivo protective efficacy. The table also lists the immunoglobulin heavy-chain variable region (IGHV) gene used, the primary complementarity-determining region (CDR) recognition mode, and whether the antibody exhibits antibody-dependent cellular cytotoxicity (ADCC) as a significant protection mechanism. Additionally, it specifies whether the antibody was isolated from mice or humans, any known escape mutations, and the IgG subtype used in generating the findings. A – is shown if the information was not provided.
| Name | In vitro Binding | In vitro Neutralisation | In vivo Protection | Germline Encoded IGHV | CDR Recognition Mode | ADCC activity | Source | Escape Mutants | IgG-type in Studies | Ref | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Central Stem | C179 | H1, H2, H5, H6, H9 | H1, H2, H5, H6, H9 | H1, H5 | - | - | Yes | Mouse | T332K, V395E * |
IgG2a | [9,27,28] | ||||||
| 27F3 | H1, H2, H5, H6, H9, H11, H12, H13, H16, H3, H7, H10, FluB | H1, H5, H6, H3, H7, H10 | - | IGHV1–69 | CDRH2 | - | Humans | - | IgG1 | [29,30] | |||||||
| FI6 | H1-H16 | H1, H5, H3, H7 | H1, H5, H3 | IGHV3–30 |
CDRH3 CDRL1 | Yes | Humans | R62K, D239G, R240Q T333K, A388T ° | - | [23] [21,22,31,32] | |||||||
| CR6261 | H1, H2, H5, H6, H8, H9 | H1, H2, H5, H6, H8, H9 | H1, H5 | IGHV1–69 | CDRH2 | Weak | Humans | A388V | IgG1 | [18,30,33,34,35] | |||||||
| CR6323 | H1, H2, H5, H6, H8, H9 | H1, H2, H5, H6, H8, H9 | - | IGHV1–69 | HCDR2 | - | Humans | H357L/T* | IgG1 | [34] | |||||||
| 09-2A06 | H1 | H1 | - | IGHV1–69 | - | - | Humans | - | - | [36] | |||||||
| 09-3A01 | H1 | H1 | - | IGHV4–39 | - | - | Humans | - | - | ||||||||
| 05-2G02 | H1, H3, H5 | H1, H3, H5 | - | IGHV1–18 | - | - | Humans | - | - | ||||||||
| A06 | H1, H5 | H1, H5 | H1 | IGHV1–69 | - | - | Humans | - | IgG1 | [37] | |||||||
| 39.18 | H1, H2 | H1, H2 | - | IGHV1–69 | - | - | Humans | - | - | [38,39] | |||||||
| 39.29 | H1, H2, H3 | H1, H2, H3 | H1, H3 | IGHV3-30 | CDRH3 | - | Humans | G387K, D391Y/G | - | ||||||||
| 81.39 | H1, H2, H3 | H1, H2, H3 | - | IGHV3-15 | - | - | Humans | - | - | ||||||||
| 36.89 | H3 | H3 | - | IGHV1–18 | - | - | Humans | - | - | ||||||||
| FE43 | H1, H5, H6, H9 | H1, H5, H6, H9 | H1, H5, H6 | IGHV1–69 | - | - | Humans | None found | IgG1 | [40] | |||||||
| FB110 | H1, H2, H5 | H1, H2, H5 | - | IGHV3-23 | - | - | Humans | None found | IgG3 | ||||||||
| 3Е1 | H1, H5, H9, H3, H7 | H1, H5, H9, H3, H7 | H1, H5 | IGHV4-4 | Mostly Heavy Chain | - | Humans | - | IgG1 | [41] | |||||||
| CT149 | H1, H5, H9, H3, H7 | H5, H9, H3, H7 | H1, H5, H3, H7 | IGHV1–18 | CDRH3 CDRH2 |
Yes | Humans | - | IgG1 | [42] | |||||||
| 31.a.83 | H1, H2, H5, H9, H3, H7 | H1, H2, H5, H9, H3, H7 | - | IGHV3–23 | Mostly CDRH3 CDRH2 |
- | Humans | - | - | [43] | |||||||
| 56.a.09 | H1, H5, H3, H7 | H1, H5, H3, H7 | - | IGHV6–1 | Mostly CDRH3 CDRH2 |
- | Humans | - | - | ||||||||
| CR9114 | H1, H2, H5, H6, H8, H9, H12, H13, H16, H3, H4, H7, H10, H15, FluB | H1, H2, H5, H6, H8, H9, H12, H3, H4, H7, H10 | H1, H2, H3, H5, H9, FluB | IGHV1–69 | CDRH2 | Weak | Humans | R62K, D239G, R240Q, L335V, D363G, A388T ° | IgG1 | [30,31,33,44,45] | |||||||
| F10 | H1, H2, H5, H6, H8, H9, H11, H13, H16 | H1, H2, H5, H6, H8, H9, H11 | H1, H5 | IGHV1–69 | CDRH2 | Yes | Humans | N460, S123, E190D+G225D, N203VHA + E329KNA * |
IgG1 | [19,30,32,46] | |||||||
| MEDI8852 | H1-H18 | H1, H2, H5, H6, H9, H3, H7 | H1, H5, H3 | IGHV6-1 | CDRH2 CDRH3 CDRL1 |
Yes | Humans | - | IgG1 | [47,48] | |||||||
| CR9117 | Mouse homologue of CR9114, presumed to have similar neutralization capacity | - | Yes | Mouse | - | IgG2a | [33] | ||||||||||
| Anchor Domain | Polyclonal response (FISW84 / 222-1C06 were named) | H1, H2, H5 | H1, H2, H5 | H1 | IGHV3-23 IGHV3-30 IGHV3-30-3 IGHV3-48 |
CDRk3 CDRH2 CDRH3 |
No | Humans | - | IgG1 | [26] | ||||||
| Fusion Peptide | CR8020 | H3, H4, H7, H10, H14, H15 | H3, H7, H10 | H3, H7 | IGHV1–18 | CDRH1 CDRH3 |
Weak | Humans | D372N, G376E * | IgG1 | [20,25,49,50] | ||||||
| CR8043 | H3, H4, H7, H10, H14, H15 | H3, H7, H10 | H3, H7 | IGHV1–3 | CDRH1 CDRH3 |
- | Humans | R378M, Q380R/T * | IgG1 | [25,50] | |||||||
| 9H10 | H3, H9 | H3, H10 | H3 | - | - | - | Mice | R378M T385R Q387R/T G386E * |
- | [50] | |||||||
°=numbering from methionine, *=H3 numbering.
3. bnAbs against the HA Head Domain
The low mutational rate of the HA stem has historically made it more heavily investigated vaccine and bnMab therapy target, yet this usually came with the trade-off of lower potency Mabs [11] and frequently sterically occluded antigenic sites [51]. In contrast, the head domain is mutationally volatile, and immunodominant, yielding a more potent and diverse set of antibodies [15]; both with respect to their germline sequences and their binding mechanisms (Table 2). Many bnAbs against the head have been characterised, but their epitopes can be roughly grouped into four distinct sites; the Receptor Binding Site (RBS), the Lateral Patch, the Vestigial Esterase (VE) and the Occluded Site (Fig 2).
3.1. Receptor Binding Site
Broadly neutralising antibodies against the Receptor Binding Site (RBS) have been described in the literature for almost as long as stem antibodies (Table 2, Figure 2) [52]. These are usually characterized by hemagglutination inhibition (HAI), as antibodies that directly compete with sialic acid for the RBS [52,53,54,55].
Antibodies that neutralize the RBS frequently do so through molecular mimicry, sterically and electrostatically mimicking sialic acid [54,56]. Curiously, while the germline-encoded IGHVs differ widely between different RBS bnAbs, the dominant loop associated with RBS binding tends to be CDRH3. This CDRH3-centric mechanism makes RBS antibodies particularly susceptible to mutations in and around the RBS as small steric or electrostatic constraints in the CDR insertion path can completely abolish antibody binding [30,40,54,57,58,59].
3.2. Lateral Patch
The lateral patch is a region on the HA head that is offset from the RBS (Fig 2). However, reports on HAI activity [60] indicate active or passive inhibition of sialic acid binding. The original study that coined the term “lateral patch” characterized CL6649, an antibody with H1N1 intrastrain activity. However mechanistic insight into the mode of neutralization was lacking [60,61].
A recent study on H7N9 lateral patch antibodies may offer further insights on the mechanism involved with this site. Jia and colleagues found that sialic acid binding is passively inhibited by their antibody, H7.HK1. This antibody inhibits the HA 220-loop (G218-G228 in H7 numbering, or G228-238 in H3 numbering), which makes hydrophobic contacts with sialic acid [62]. It is worth noting, however, that these mechanisms may not translate between influenza subtypes and further research is needed to understand how CL6649 and H7.HK1 compare.
3.3. Vestigial Esterase
The Vestigial Esterase (VE) is a region located in the HA head between the RBS and the start of the HA stem (Fig 2)[63]. Its sequences are highly conserved within subtypes, but not across subtypes [62,63]. VE-specific antibodies tend to lack HAI activity [64,65], but display ADCC through Fc-FcγR responses [62,63,65,66,67] and may be involved in crosslinking and thereby conformationally restricting different HA trimers [64].
3.4. Interface and Occluded Epitope
Finally, the occluded epitope is a name given to an epitope either sitting between two HA monomers, or within the HA core. Despite being characterized as early as 1993 [70], the value of this low variability site for bnAbs had not been realized until three independent research teams demonstrated its ability to provide therapeutic protection against a range of different Influenza A viruses including H1N1, H3N2, H5N1, and/or H7N9 [71,72,73].
Despite its occluded nature, the epitope is available for binding even in intact, trimeric HAs[73], likely through protein dynamics. However, the mechanism by which protection is achieved has been reported to differ widely.
FluA-20 is a bnAb that has been shown to be protect mice against viral challenge with H1N1, H3N2, H5N1 or H7N9 subtypes. It has been found to bind the occluded epitope in uncleaved immature HA0. Trypsin-based treatment destabilizes the FluA-20 contacts, implying that FluA-20 may be disrupting HA trimerization. No HAI activity or significant ADCC has been observed [71].
Conversely, 8H10 bnAb has been shown to bind mature trimeric HAs and elicit protection against various H3N2 strains, and to bind to H4 viruses. Protection is thought to be primarily governed by ADCC [73]. These findings are corroborated in another study that shows a panel of antibodies targeting the occluded epitope to be eliciting protection primarily through ADCC [72].
It is worth noting that broadly neutralising sites have also been reported at an exposed site of the HA multimerization interface (Fig 2)[74]. This bnAb, termed F005-126, lacks HAI activity and protects HA from trypsin-based cleavage against a panel of H3N2 viruses, indicating inhibition of endosomal rearrangements [74].
Table 2.
Overview of Extensively Studied Head-Targeting Broadly Neutralizing Antibodies (bnAbs). This table provides details on some of the most thoroughly researched head-targeting bnAbs, adding to previously published work [17]. It includes information on their in vitro binding affinity, in vitro neutralization capacity, and in vivo protective efficacy. The table also lists the immunoglobulin heavy-chain variable region (IGHV) gene used, the primary complementarity-determining region (CDR) recognition mode, and whether the antibody exhibits antibody-dependent cellular cytotoxicity (ADCC) as a significant protection mechanism. Additionally, it specifies whether the antibody was isolated from mice or humans, any known escape mutations, and the IgG subtype used in generating the findings. A – is shown if the information was not provided.
Table 2.
Overview of Extensively Studied Head-Targeting Broadly Neutralizing Antibodies (bnAbs). This table provides details on some of the most thoroughly researched head-targeting bnAbs, adding to previously published work [17]. It includes information on their in vitro binding affinity, in vitro neutralization capacity, and in vivo protective efficacy. The table also lists the immunoglobulin heavy-chain variable region (IGHV) gene used, the primary complementarity-determining region (CDR) recognition mode, and whether the antibody exhibits antibody-dependent cellular cytotoxicity (ADCC) as a significant protection mechanism. Additionally, it specifies whether the antibody was isolated from mice or humans, any known escape mutations, and the IgG subtype used in generating the findings. A – is shown if the information was not provided.
| Name | In vitro Binding | In vitro Neutralisation | In vivo Protection | Germline Encoded IGHV | CDR Recognition Mode | ADCC activity | Source | Escape Mutants | IgG-type in Studies | Ref | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| RBS | S139/1 | H1, H2, H3, H5, H9, H13 | H1, H2, H3, H5, H9, H13, H16 | H1, H3 | - | CDRH2 | - | Mouse | K156, G158, S193, insertion at 133a * | IgG2a | [53,54,75] |
| C05 | H1, H2, H9, H12, H3 | H1, H2, H3 | H1, H3 | IGHV3-23 | CDRH3 | Weak | Human | insertion at 133a | - | [54,76] | |
| F045-092 | H1, H2, H13, H3 | H1, H2, H3, H13 | - | IGHV1–69 | CDRH3 | - | Human | 133A insertion * | - | [30,57] | |
| K03.12 | H1, H3 | - | - | IGHV1-2 | CDRH3 | - | Human | - | IgG1 | [77] | |
| 2G1 | H2 | H2 | H2 | IGHV1–69 | - | - | Human | - | - | [30,78] | |
| FE17 | H1, H9 | H1, H9 | H1, H5 | IGHV1–69 | - | - | Human | S145N* | IgG1 | [40] | |
| 12H5 | H1, H5 | H1, H5 | H1, H5 | IGHV9-1 alignment | CDRH2, CDRH3 | - | Mouse | Y98A, A137E, H141A, A142E, G143R, A144E, W153A, D190A * | IgG1 | [59] | |
| 1F1 | H1 | H1 | H1 | IGHV3-30 | CDRH3 | - | Human | D190E, D225G * | - | [79] | |
| 5J8 | H1 | H1 | H1 | IGHV4-b | - | - | Human | R(133A)I, K(133A)Q, A137T, D199H, K222Q * | - | [58] | |
| CH65 | H1 | H1 | - | IGHV1-2 | CDRH3 | - | Human | G200D, K/R insertion at 133A * | IgG1 | [56,80] | |
| CH67 | H1 | H1 | - | IGHV1-2 | CDRH3 | - | Human | likely as CH65 | IgG1 | [56,80] | |
| 3D11 | H1 | H1 | H1 | - | - | - | Mouse | K153E, D200E * | IgG1 | [81] | |
| 8M2 | H2 | H2 | H2 | IGHV1–69 | - | - | Human | G142D * | - | [30,78] | |
| 8F8 | H2 | H2 | H2 | IGHV3-33 | - | - | Human | R144Q/M/K, T134K * | - | [78] | |
| A2.91.3 | H3 | H3 | - | - | CDRH3 | - | Mouse | K189N, F193S/K, L194P, Y195A * | IgG1 | [82] | |
| AVFluIgG03 | H5 | H5 | H5 | IGHV3-23 | CDRH3 | - | Human | S159I, R193M/W * | IgG1 | [83,84] | |
| FLD21.140 | H5 | H5 | H5 | IGHV4-31 | CDRH3 | - | Human | S159I, R193M/W * | IgG1 | [84] | |
| 13D4 | H5 | H5 | H5 | Mouse IGHV1-9 | CDRH3 | - | Mouse | K/R193N * | - | [83] | |
| Hab21 | H5 | H5 | - | - | - | - | Mouse | H136A, D197A, A198G, A199G, E200A, N207A, P208A, P225A, N258A | [85] | ||
| H5.3 | H5 | H5 | - | - | CDRH3 | - | Human | - | - | [86] | |
| CR8033 | FluB | FluB | FluB | IGHV3-9 | CDRH Dominant | - | Human | P161Q | - | [55] | |
| VE | H3v-47 | H3 | H3 | H3 | IGHV1–69 | CDRH2, CDRH3, CDRL1, CDRL3 | Yes | Human | None found | IgG1 | [64] |
| F005-126 | H3 | H3 | - | - | CDRH3 | - | Human | N285Y * | IgG1 | [74] | |
| H5M9 | H5 | H5 | H5 | - | CDRH1-3, CDRL1-2 | - | Mouse | D53A/N, E78K, E83aA/K, Y274A * | IgG1 | [87] | |
| 9F4 | H5 | H5 | H5 | - | - | Yes | Mouse | R62G * | IgG2b | [88,89] | |
| 100F4 | H5 | H5 | H5 | - | - | Yes | Human | D72A, E116Q/L * | - | [90,91] | |
| 4F5 | H5 | H5 | H5 | IGHV3-43 | - | Yes | Human | W70, L71, L72, G73, N74, P75 * | - | [92,93] | |
| 1H5 | H7 | - | H7 | - | - | Yes | Mouse | R58K * | IgG2a | [94] | |
| 1H10 | H7 | - | H7 | - | - | Yes | Mouse | R58K * | IgG2a | [94] | |
| 46B8 | FluB | FluB | FluB | - | - | Yes | Human | S301F | IgG1 | [95] | |
| CR8071 | FluB | FluB | FluB | IGHV1-18 | - | Yes | Human | None found | - | [55,95] | |
| Lateral Patch | CL6649 | H1 | H1 | - | IGHV4-39 | CDRH3, CDRL1, CDRL3 |
- | Human | K176Q S175N+K176Q * |
- | [96] |
| H7.HK1 | H7, H10, H15 | H7 | H7 | IGHV4-59 | CDRH1-3, CDRL1, CDRL2 |
- | Human | R57K * | IgG1 | [97,98] | |
| 07-5F01 | H7 | H7 | H7 | IGHV4-31 | - | - | Human | R57K * | IgG2a | [97,98] | |
| HA Multimerization Interface and Occluded Site | FluA-20 | H1-H12, H14, H15 | - | H1, H3, H5, H7 | IGHV4-61 | CDRH3, CDRL2 | Weak | Human | In H1: P103G, R230A, P231G, V233G, R239A * | - | [71] |
| 8H10 | H3, H4 | H3 | H3 | IGHV5-9-1 | CDRH1-3, CDRL1, CDRL3 |
Yes | Human | - | IgG2a, IgG1 | [73] | |
| S5V2-29 | H1, H2, H3, H4, H7, H9, H14 | - | H1, H3 | IGHV4-61 | - | Yes in IgG2c but not IgG1 | Human | - | IgG1 and IgG2c | [72] | |
| H2214 | H1, H2, H3, H4, H14 | - | H1, H3 | IGHV3-23 | - | Yes in IgG2c but not IgG1 | Human | - | IgG1 and IgG2c | [72] | |
| H7-200 | H7, H15 | - | H7 | - | CDRH Dominant, CDRL3 | - | Human | - | - | [99] | |
| H7.5 | H7 | H7 | - | - | CDRH2, CDRL3 | - | Human | - | - | [99,100] |
°=numbering from methionine, *=H3 numbering.
4. bnAbs in Clinical Trials
In the 1970s, Köhler and Milstein pioneered hybridoma technology, facilitating the production of monoclonal antibodies [101]. This breakthrough has significantly advanced the therapeutic use of monoclonal antibodies, particularly in autoimmune diseases, oncology, and infectious diseases. Broadly neutralising monoclonal antibodies (bnMAbs) hold promise as a therapeutic alternative to existing influenza treatments, particularly in combating Influenza A. Ongoing clinical trials aim to evaluate the efficacy and safety of stem-targeting bnMAbs in treating influenza infections (Table 3). Initial findings suggest that bnMAbs may provide significant clinical benefits in managing uncomplicated influenza A, as evidenced by several trials. For instance, in a clinical trial (NCT02071914), patients treated with CT-P27 at doses of 10 mg/kg or 20 mg/kg exhibited a statistically significant reduction in the area under the curve (AUC) of viral load compared to placebo. Another bnMAb, VIS410, administered at doses of 2000 mg or 4000 mg, demonstrated significant improvement in the signs and symptoms of influenza infection on days 3 and 4, along with a reduction in the time required to resolve peak viral load (NCT02989194).
However, when VIS410 was combined with oral oseltamivir at doses of 3600 mg or 8400 mg, it failed to show a statistically significant reduction in the time to oxygen cessation or in viral load in nasopharyngeal samples compared to oseltamivir with a placebo in hospitalized patients with influenza A infection (NCT03040141).
Similarly, the bnMAb MHAA4549A, at a dose of 3600 mg, resulted in a statistically significant reduction in viral AUC in nasopharyngeal samples in a challenge model involving H3N2 influenza (NCT01980966). Despite this reduction in viral load, MHAA4549A, whether at 3600 mg or 8400 mg, did not improve clinical outcomes over oseltamivir alone in patients hospitalized with severe influenza infections (NCT02293863).
Another bnMAb, MEDI8852, was assessed for its efficacy in treating acute, uncomplicated influenza A infections. However, it did not provide statistically significant improvements over oseltamivir alone and was even associated with potentially worsened disease progression compared to oseltamivir (NCT02603952). Consequently, a subsequent clinical trial to evaluate the dosing regimen of MEDI8852 was withdrawn by the sponsoring company (NCT03028909).
The bnMAb CR6261, while able to significantly reduce the percentage of participants exhibiting influenza symptoms following H1N1 challenge, failed to significantly impact viral shedding, AUC, or disease severity (NCT02371668). Additionally, a trial combining CR6261 with CR8020 was withdrawn due to unsatisfactory preliminary efficacy results (NCT01992276). Similarly, a trial involving CT-P27 was terminated due to the inactivation of the bnMAb (NCT03511066).
These findings emphasize the potential of bnMAbs as a therapeutic strategy against influenza, while also underscoring their limitations and the need for further research. Current clinical trials for influenza predominantly utilize IgG1 bnMAbs. IgG1 and IgG3 are the major subclasses generated during viral infections, with distinct functional differences and characteristics. IgG1 has a shorter 15 amino acid hinge region with only 2 disulfide bonds, providing a longer half-life and potentially greater therapeutic efficacy [102,103]. In contrast, IgG3, with its shorter half-life has been overlooked for its therapeutic applications. However, with its longer hinge region of 62 amino acids and 11 disulfide bonds [102,103] IgG3 may be able to overcome steric hindrance, a limitation observed in certain stem-targeting bnMAbs such as CR8020 [104]. Moreover, IgG3 has a higher affinity for the Fc receptors FcγRIIa, FcγRIIIa, and FcγRIIIb in its monomeric form compared to IgG1, while its binding efficiency in complex form exceeds IgG1 for all receptors [102], making it particularly effective at activating CDC and ADCC [103] mechanisms employed by multiple head and stem targeting bnAbs (Table 1 and 2) to indirectly neutralize influenza [105,106,107]. Switching the subclass of MAbs has been seen to have beneficial effects for SARS-CoV-2, with the switch from an IgG1 to an IgG3 enhancing both Fc mediated phagocytosis and the triggering of the classical complement pathway [108]. Additionally, Bolton and colleagues found that IgG3 antibodies exhibited superior binding and neutralisation capacity to antigenically drifted influenza and SARS-CoV-2 viruses relative to other IgG subclasses [109].
To enhance the therapeutic efficacy of IgG1 bnMAbs, modifications to the Fc region could be explored to improve ADCC initiation. Ocaratuzumab is an anti-CD20 mAb which has Fc modifications, P247I/A339Q, which have been shown to increase its binding to lower-affinity FcγRIIIa allowing it to have increased ADCC activity [110,111,112]. Fc modifications can also be used to increase the half life of antibodies to improve their therapeutic efficacy. The MAb therapy Sotrovimab, which was approved for use against SARS-CoV-2, has the Fc modifications M428L/N434S to extend the half life of the antibody [113].
Another promising approach involves the development of chimeric IgG1/IgG3 antibodies, which may enhance ADCC and CDC as well as improving binding to sterically hindered stem regions. Natsume and colleagues generated a chimeric form of rituximab, an anti-CD20 antibody, that consisted of the CH1 and hinge regions from IgG1, with the Fc region of IgG3 with the COOH terminal CH3 domain of IgG1. This chimeric antibody showed enhanced CDC and ADCC activities compared to the wild type [114]. Chimeric antibodies could combine the favourable pharmacokinetics of IgG1 with the functional advantages of IgG3, potentially overcoming the limitations associated with IgG3 monotherapy [114,115]. These strategies highlight avenues for optimizing bnMAb design to improve their utility in influenza treatment. Further research into IgG subclass-specific characteristics and their impact on bnMAb efficacy is warranted.
Table 3.
Table of stem-targeting bnMAb’s currently undergoing clinical trials for use in influenza infection. Information about the type of antibody, target, dose regime and results were taken from the corresponding study record listed on the NIH clinical studies site.
Table 3.
Table of stem-targeting bnMAb’s currently undergoing clinical trials for use in influenza infection. Information about the type of antibody, target, dose regime and results were taken from the corresponding study record listed on the NIH clinical studies site.
| Name | Type and Target | Dosage/ Infection Model | Result | Trial Registry ID/ Reference |
|---|---|---|---|---|
| CT-P27 | CT-120 & CT-149 mAb’s targeting the stem region of group 1 and group 2 influenza hemagglutinin | 10 mg/kg CT-P27, 20 mg/kg CT-P27, or placebo in an influenza challenge model | Reduction of AUC of Viral Load, as measured by Quantitative PCR of Nasopharyngeal Swab for patients who received CT-P27 | NCT02071914, [105] |
| 90 mg/kg CT-P27, 45 mg/kg CT-P27, or placebo | NCT03511066 was terminated due to CT-P27 inactivation | NCT03511066. | ||
| MEDI8852 | Human IgG1 kappa monoclonal antibody (MAb) targeting H1N1 and H3N2 viruses, as well as subtypes such as H2, H5, H6, H7, and H9 via the stem region | 750 mg or 3000mg MEDI8852 given with oseltamivir or 3000 mg MEDI8852 on its own to patients with acute, uncomplicated influenza caused by Type A strains. | MEDI8852 provided no statistically significant improvement over oseltamivir alone, potentially worsened disease in combination compared to oseltamivir alone | NCT02603952,[116] |
| Low dose and high dose of MEDI8852 and oseltamivir in comparison to oseltamivir and placebo | Withdrawn due to company decision | NCT03028909 | ||
| VIS410 | Human immunoglobulin IgG1 monoclonal antibody engineered to bind to the stem region of group 1 and 2 influenza A hemagglutinins | Influenza challenge with H1N1 followed by a single administration of VIS410 or placebo | No results posted | NCT02468115, [117] |
| 2000mg or 4000mg VIS410 was given to patients with uncomplicated influenza A infection and compared to a placebo | Statistically significant improvement in signs and symptoms of influenza infection on day 3 and 4 with VIS410 compared to placebo. Statistically significant reduction in time to resolution of peak viral load when patients were given VIS410. | NCT02989194, [118] | ||
| 3600 mg or 8400 mg VIS410 combined with oral oseltamivir or placebo with oseltamivir in patients hospitalised with influenza A infection | No statistically significant reduction in time to cessation of oxygen, or reduction of viral load in nasopharyngeal samples | NCT03040141 | ||
| MHAA4549A | Human monoclonal antibody, IgG1, targeting the influenza A virus hemagglutinin stem across multiple subtypes | Influenza challenge with H3N2 influenza virus followed by a dose of 400 mg, 1200 mg or 3600 mg | Statistically significant reduction in AUC of virus in nasopharyngeal samples was seen at 3600mg compared to placebo. Influenza symptom scores, mucus weight, and inflammatory biomarkers were also reduced. | NCT01980966 |
| 3600mg or 8400mg given either on its own or with oseltamivir to patients hospitalised with severe influenza infection | MHAA4549A did not improve clinical outcomes over OTV alone. MHAA4549A+OTV did not further reduce viral load versus placebo+OTV.MHAA4549A did not alleviate symptoms quicker than a placebo. | NCT02293863, [119] | ||
| 3600mg or 8400mg given to patients with uncomplicated seasonal influenza A infection | 3600mg dose was able to statistically reduce the number of days to alleviate symptoms compared to the control | NCT02623322 | ||
| CR8020 | A mAb targeting the stem region of group 2 influenza A hemagglutinin |
15 mg/kg CR8020 given before challenge with a H3N2 influenza virus. | No results | NCT01938352 |
| CR6261 | mAb that targets the stem region of group 1 and group 2 influenza hemagglutinin | 50 mg/kg administered one day after challenge with H1N1 | Statistically reduced percentage of participants who experienced influenza symptoms. No statistically significant reduction in AUC or viral shedding. | NCT02371668, [120] |
| CR8020/ CR6261 | Withdrawn due to preliminary efficacy results from an influenza challenge trial | NCT01992276 |
5. Broadly Protective Vaccines in Clinical Trials
bnMAbs represent a promising secondary defence mechanism against influenza, complementing the primary defence provided by vaccination. Current influenza vaccines require annual updates to address antigenic drift and shift. However, the induction of bnAbs via vaccination could offer cross-protection against multiple strains of influenza, irrespective of antigenic variations. Vaccines designed to elicit bnAbs may provide long-term immunity and reduce the need for frequent vaccine reformulations. Several clinical trials are currently investigating novel influenza vaccines aimed at inducing bnAb formation (Table 4). These trials employ various strategies to stimulate bnAb production specifically targeting the hemagglutinin protein of influenza A.
One promising approach involves presenting only the stem region of the influenza virus, aiming to elicit bnAbs against conserved stem epitopes across diverse influenza strains. UFluA, a stabilized stem nanoparticle vaccine currently in Phase 1 trials (NCT05155319), exemplifies this strategy. By stabilizing the HA stem into a nanoparticle format, UFluA aims to induce bnAbs effective against both group 1 and group 2 influenza A viruses, offering broad cross-protection.
Another vaccine candidate, H1ssF, employs the stem domain from Influenza A/New Caledonia/20/1999 (H1N1) genetically fused to the ferritin protein from Helicobacter pylori. This design is intended to enhance the presentation of the stem region to the immune system, thereby inducing bnAbs targeting the stem. Initial results from a Phase 1 trial demonstrated that H1ssF generated an increased IC80 concentration in a pseudoviral neutralization assay against the homologous H1N1 A/New Caledonia/20/99 virus (NCT03814720), indicating promising immunogenicity.
The G1 mHA vaccine utilizes a ‘mini-HA,’ a stabilized form of the HA stem trimer [121]. Previous studies have shown that this design can induce stem-targeting antibodies against various group 1 viruses in non-human primates [122]. Currently, G1 mHA is undergoing a Phase 1/2 trial (NCT05901636) to further evaluate its efficacy and safety in humans.
Chimeric vaccines represent another innovative approach to induce stem-targeting bnAbs. These vaccines employ a prime-boost regimen, using vaccines with different HA head domains but a consistent stem domain to focus the immune response on the stem. GSK3816302A, a chimeric vaccine currently in Phase 1 trials (NCT03275389), incorporates cH8/1 N1, cH5/1 N1, and cH11/1 N1 constructs to elicit bnAbs targeting the H1 stem. Initial results indicate an increase in anti-H1 stem antibodies post-vaccination, with a statistically significant humoral immune response. Notably, increased antibody titres against H2 and H18 subtypes were also observed, suggesting potential cross-reactivity [123].
A specific subset of bnAbs target the HA stem region in its post-fusion form (Table 1). To induce these bnAbs, vaccines must present the stem in a non-native post-fusion conformation. Previous studies have demonstrated that vaccines designed to present this form can induce protective bnAbs in mice, offering cross-protection against mismatched influenza A strains [124]. A clinical trial evaluating a post-fusion hemagglutinin antigen is currently in the recruitment phase (NCT06460064).
In parallel, a universal influenza vaccine, M-001, which incorporates conserved epitopes from the M1 matrix protein, NP, and HA of both influenza A and B, has advanced to Phase 3 trials [125]. Despite initial promise, results from this trial indicated no statistically significant difference between control and vaccine groups regarding the prevention of influenza illness or reduction in symptom severity (NCT03450915). These results show that while inducing the production of bnAb’s against hemagglutinin is an attractive route forward for the formation of a universal influenza vaccine, many challenges exist for these vaccines, and continued research into this area is required.
Table 4.
Table of broadly protective vaccines and vaccine antigens targeting the hemagglutinin currently undergoing clinical trials for use in influenza prevention.
Table 4.
Table of broadly protective vaccines and vaccine antigens targeting the hemagglutinin currently undergoing clinical trials for use in influenza prevention.
| Phase | Name of vaccine | Target/ Type of vaccine | Dosage/ Infection model | Results | Trial Registry ID/ Reference |
|---|---|---|---|---|---|
| Recruiting | fH1/DSP-0546LP | Post-fusion hemagglutinin antigen | Combination of 2 dose levels of fH1 (2 and 8 μg), 3 dose levels of DSP-0546LP (2.5, 5, and 10 μg), and placebo. Each dose level of fH1 will be combined with the low, medium, and high dose level of DSP-0546LP to assess safety, tolerability, and immunogenicity | Active | NCT06460064, [124] |
| Phase 1 | EBS-UFV-001 |
Induction of antibodies against conserved stem antigens across group 1 and 2 via a hemagglutinin stabilized stem nanoparticle vaccine | Testing the safety, tolerability and immunogenicity of 20 µg or 60 µg of UFluA as single dose or as two dose | No results posted | NCT05155319, [126] |
| H1ssF | HA stem domain from Influenza A/New Caledonia/20/1999 (H1N1) genetically fused to the ferritin protein from H. pylori. | 20 mcg was given to group 1, group 2 received 60 mcg on a prime boost schedule. | All regimes generated an increased IC80 concentration when tested in a pseudoviral neutralization assay against the homologous H1N1 A/New Caledonia/20/99 virus | NCT03814720 | |
| GSK3816302A | Chimeric vaccines of D-SUIV cH8/1 N1, D-SUIV cH5/1 N1, and D-SUIV cH11/1 N1 to induce cross reactive stem targeting antibodies against H1 stem | Chimeric H5, H8 and H11 with and without adjuvants AS03 or AS01 were tested for their reactogenicity, safety and immunogenicity. H8 and H5 were given with a placebo second dose, or all three were given. | An increase in anti H1 stem antibodies, as measured by ELISA and MN assay, was seen across all dose schedules with adjuvant AS03 providing a statistically significant increase in humoral immune response for anti-H1 stem antibody by ELISA at Day 29 and Day 85. Increases in antibody titres against H2 and H18 were also identified. | NCT03275389, [123] | |
| Phase 1/2 | G1 mHA | Mini-hemagglutinin stem-derived protein vaccine antigen | Single dose of influenza G1 mHA with or without Al(OH)3 adjuvant at two dose levels to evaluate safety, reactogenicity and immunogenicity | Active | NCT05901636, [121,122] |
| Phase 3 | (M-001) | A recombinant 45 kDa protein produced in Escherichia coli. consisting of three repetitions of nine linear, conserved influenza A and B epitopes to form a single recombinant protein. Epitopes were derived from: M1 matrix protein, NP and HA | Vaccination with 1mg dose of M-001 twice: Once at Day 0, and once at Day 21 then followed for 2 years | No statistical difference in prevention of influenza infection. Did not statistically reduce the number of patients with influenza like symptoms, or a reduction of severity of either qRT-PCR or culture-confirmed influenza illness | NCT03450915, [125] |
6. bnAbs in Current and Future Directions
bnAbs present a promising avenue for the development of universal influenza vaccines and therapeutic interventions for influenza infections. However, bnAbs do have potential downfalls within these areas.
Although several bnAbs have been identified, thus far no bnAb possesses complete universality (Table 1 and 2). This limitation necessitates precise identification of the influenza strain prior to bnAb administration, which is not a common practice in clinical settings, reducing their practicality for routine treatment.
6.1. Escape Mutations
Influenza undergoes antigenic drift and shift over time, resulting in mutations within the HA protein. These mutations can restrict the effectiveness of certain bnAbs. For instance, an amino acid insertion at position 133 induces a localized structural change in the 130-loop of the HA head domain. This insertion is observed in 100% of all H6 and H10, ~ 95% of all H5 and ~ 70% of all H1 sequences. This alteration inhibits the activity of multiple RBS-targeting antibodies (Table 2), such as S139/1 and C05, as demonstrated by Ekiert and colleagues, and Lee and colleagues [127,128]. As a result, these antibodies may have reduced efficacy as therapeutic agents. Furthermore, the antibody FluA-20, which targets an epitope within the HA trimer interface, has already encountered escape mutations (Table 2). A mutation from an arginine to an isoleucine at position 229, one of the five major epitope residues, has already been seen in a H3 strain and rendered FluA-20 ineffective [71].
Although the stem region of HA is less prone to antigenic drift and shift, and therefore more conserved than the head region, escape mutations to stem targeting bnAb’s have also been identified (Table 1). Ekiert and colleagues identified two escape mutations for the fusion peptide targeting antibody CR8020 [24] which had entered clinical trials (Table 3), while Okuno and colleagues identified two escape mutations for the central stem targeting antibody C179 [9]. Although these mutations have not yet been seen in naturally occurring viruses, the potential for escape viruses from stem targeting antibodies is possible.
A further concern is the potential for vaccines and therapeutic bnAb’s that target regions susceptible to escape mutations to drive antigenic drift, leading to the emergence of strains resistant to such therapies. The generation of bnAb’s through universal vaccines, or the therapeutic use of bnAb’s could potentially induce mutations in currently conserved regions. Chai and colleagues determined that although Ser301, a key epitope residue at the centre of the 46B8-binding site, is highly conserved currently, a mutation to a phenylalanine allows viruses to evade neutralisation without compromising their fitness [95]. Park and colleagues were also able to determine that the presence of stem-targeting antibodies may result in mutant virus selection to escape the immune response. Within human challenge models, a mutant virus of A338V H1N1 2009 was preferentially selected for in participants with higher levels of pre-challenge anti-HA stem antibodies [35] .
6.2. Immunogenicity of bnAbs
bnMAbs and universal influenza vaccines in current clinical trials predominantly focus on antibodies targeting the HA stem. However, stem-targeting antibodies have demonstrated reduced immunogenicity compared to head-targeting antibodies [15]. The exact mechanisms behind this difference are not yet fully understood, though several explanations have been proposed.
Andrews and colleagues demonstrated that neutralizing stem antibodies exhibited weaker binding to whole influenza virions compared to head-targeting antibodies, despite showing equivalent binding to recombinant HA [129]. One proposed explanation for this is that stem-targeting antibodies face steric hindrance, as their binding site on the HA stem may be located near the viral membrane, potentially limiting accessibility (Fig 2). Additionally, the negative selection pressures exerted on B-cell clones producing stem-targeting antibodies may also play a role in their reduced immunogenicity. Stem-targeting antibodies are more prone to autoreactivity and polyreactivity, which could lead to immune tolerance or elimination of these B-cell clones. Andrews and colleagues identified that neutralizing stem-targeting antibodies exhibited higher levels of polyreactivity compared to non-neutralizing stem-targeting antibodies or head-targeting antibodies [129]. This observation was further supported by Bajic and colleagues who tested 12 bnAbs—six targeting the HA head and six targeting the stem. Of the stem-targeting antibodies, five displayed some degree of autoreactivity, whereas three head-targeting antibodies exhibited polyreactivity. Notably, CR6261, a stem-targeting bnMAb that had entered clinical trials for use as a therapeutic bnMAb (Table 3), showed both autoreactivity and binding to phospholipids, suggesting potential off-target interactions [130]. The presence of polyreactivity and autoreactivity in stem targeting bnAb’s raises concerns about their safety and efficacy as therapeutic agents. Polyreactivity may induce immune clearance mechanisms, such as B-cell anergy, and increase the risk of adverse effects due to off-target binding. These factors underscore the need for careful consideration of autoreactivity and polyreactivity in the development of bnMAbs, whether for therapeutic applications or as a foundation for universal influenza vaccines.
6.3. Antibody-Dependent Enhancement
The potential for antibody-dependent enhancement (ADE) of infection represents a critical concern in the development of bnAbs as therapeutic agents or as a foundation for universal influenza vaccines. ADE occurs when virus-specific antibodies facilitate, rather than inhibit, viral infection. This phenomenon arises when antibodies are present at subtherapeutic concentrations or function in a non-neutralizing manner, leading to increased disease severity. In such cases, antibodies may promote viral entry into host cells, thereby exacerbating infection, or contribute to the formation of excessive immune complexes, intensifying inflammatory responses [131,132,133,134] .
Non-neutralizing antibodies have been implicated in ADE during various viral infections, including the early stages of HIV-1 infection. Willey and colleagues demonstrated that during the acute phase of HIV-1 infection, non-neutralizing antibodies targeting the viral envelope can initiate complement-mediated ADE, resulting in heightened levels of infection [131]. Similarly, studies on influenza have shown that certain non-neutralizing antibodies can enhance disease outcomes. Winarski and colleagues reported that two non neutralising mAbs that bind to the head domain of HA caused enhanced disease in mice after viral challenge with H3N2. Interactions of these mAb’s with HA led to the destabilization of the HA stem domain, resulting in increased viral load and pathogenicity in mice [135]. Historical data further supports the role of non-neutralizing antibodies in severe influenza outcomes. During the 2009 H1N1 pandemic, middle-aged individuals with preexisting immunity to seasonal influenza exhibited higher rates of severe illness than expected. Monsalvo and colleagues identified the presence of cross-reactive, non-neutralizing antibodies in this population, which were considered to be linked to an increase in immune complex-mediated disease, with this population also having markers for complement activation via immune complexes. A similar mechanism was identified in fatal cases of the 1957 influenza pandemic [132]. These observations highlight the potential risks associated with non-neutralizing antibodies within pandemic influenza cases.
In light of these concerns, the potential for bnAbs to trigger ADE is being carefully studied due to the ongoing clinical trials which are investigating the use of bnAbs as a therapeutic intervention following influenza infection. Rao and colleagues examined the human monoclonal IgG1 bnMAb MHAB5553A, which targets a conserved epitope in the vestigial esterase domain of HA and exhibits strong antiviral activity against multiple influenza B strains in murine models [95]. Notably, MHAB5553A did not enhance pathogenesis in female DBA/2J mice within this study [134]. In conclusion, while bnAbs hold promise for influenza therapy and vaccine development, the risk of ADE must be thoroughly evaluated to ensure the safety and efficacy of these approaches. Ongoing research into the mechanisms of ADE in the context of bnAbs will be crucial for the advancement of universal influenza vaccines and antibody-based therapies.
References
- Krammer, F.; Smith, G.J.D.; Fouchier, R.A.M.; Peiris, M.; Kedzierska, K; Doherty, P.C.; Palese, P; Shaw, M.L; Treanor, J.; Wesbter, R.G.; et al., Influenza. Nature Reviews Disease Primers, 2018. 4(1): p. 3. [CrossRef]
- Collins, J.P., Campbell, A.P.; Openo, K.; Farley, M.; Cummings, C.N.; Hill, M.; Schaffner, W.; Lindegren; M.L.; Thomas, A.; Billing, L.; et al., Outcomes of Immunocompromised Adults Hospitalized With Laboratory-confirmed Influenza in the United States, 2011-2015. Clin Infect Dis, 2020. 70(10): p. 2121-2130. [CrossRef]
- WHO. Influenza seasonal. 2024 [cited 2024 28.08.2024]; Available from: https://www.who.int/health-topics/influenza-seasonal#tab=tab_1.
- Knobler, S.L.; Mack, A.; Mahmoud, A.; Lemon, S.M. Story of Influenza. 2005, Institute of Medicine (US): Forum on Microbial Threats.
- Fierer, J., D. Looney, and J.-C. Pechère, 2 - Nature and Pathogenicity of Micro-organisms, in Infectious Diseases (Fourth Edition), J. Cohen, W.G. Powderly, and S.M. Opal, Editors. 2017, Elsevier. p. 4-25.e1. [CrossRef]
- Ryu, W.-S., Chapter 15 - Influenza Viruses, in Molecular Virology of Human Pathogenic Viruses, W.-S. Ryu, Editor. 2017, Academic Press: Boston. p. 195-211. [CrossRef]
- Fujimura, S.F., Purple Death: The Great Flu of 1918, in Perspectives in Health. 2003: Pan American Health Organization.
- Gerhard, W.; Yedwell, J.; Frankel, M.E.; Webster, R. Antigenic structure of influenza virus haemagglutinin defined by hybridoma antibodies. Nature, 1981. 290(5808): p. 713-717. [CrossRef]
- Okuno, Y.; Isegawa, F.; Sasao, F.; Ueda, S. A common neutralizing epitope conserved between the hemagglutinins of influenza A virus H1 and H2 strains. J Virol, 1993. 67(5): p. 2552-8. [CrossRef]
- Lousa, D.; C.M. Soares, Molecular mechanisms of the influenza fusion peptide: insights from experimental and simulation studies. FEBS Open Bio, 2021. 11(12): p. 3253-3261. [CrossRef]
- Cheung, C.S.-F.; Gorman, J.; Andrews, S.F.; Rawi, R.; Reveiz, M.; Shen, C.-H.; Wang, Y.; Harris, D.R.; Nazzari, A.F.; Olia, A.S.; et al., Structure of an influenza group 2-neutralizing antibody targeting the hemagglutinin stem supersite. Structure, 2022. 30(7): p. 993-1003.e6.
- Throsby, M., van den Brink, E.; Jongeneelen, M.; Poon, L.L.M; Alard, Ph; Cornelissen, L.; Bakker, A.; Cox, F.; van Deventer, E.; Guan, Y.; et al., Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. PLoS One, 2008. 3(12): p. e3942. [CrossRef]
- Nath Neerukonda, S., R. Vassell, C.D. Weiss, Neutralizing Antibodies Targeting the Conserved Stem Region of Influenza Hemagglutinin. Vaccines (Basel), 2020. 8(3). [CrossRef]
- Sun, X., Ling, Z.; Yang, Z.; Sun, B. Broad neutralizing antibody-based strategies to tackle influenza. Curr. Opi. Virol. 2022. 53: p. 101207.
- Tan, H.X., Jegaskanda, S.; Juno, J.A.; Esterbauer, R.; Wong, J.; Kelly, H.G.; Liu, Y.; Tilmanis, D.; Hurt, A.C.; Yedwell, J.W.; e al. Subdominance and poor intrinsic immunogenicity limit humoral immunity targeting influenza HA stem. J Clin Invest, 2019. 129(2): p. 850-862. [CrossRef]
- Eggink, D., P.H. Goff, P. Palese, Guiding the immune response against influenza virus hemagglutinin toward the conserved stalk domain by hyperglycosylation of the globular head domain. J Virol, 2014. 88(1): p. 699-704. [CrossRef]
- Jiao, C., Wang, B; Chen, P.; Jian, Y.; Liu; J. Analysis of the conserved protective epitopes of hemagglutinin on influenza A viruses. Frontiers in Immunology, 2023. 14. [CrossRef]
- Ekiert, D.C., Bhabha, G.; Elsliger, M.-A.; Friesen, R.H.; Jongeneelen, M.; Throsby, M.; Goudsmit, J.; Wilson, I.A. Antibody recognition of a highly conserved influenza virus epitope. Science, 2009. 324(5924): p. 246-51. [CrossRef]
- Sui, J., Hwang, W.C.; Pérez, S .; Wei, G.; Aird, D.; Chen, L.-.m; Santelli, E.; Stec, B.; Cadwell, G.; Ali, M.; et al., Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat. Struct. Mol. Biol. 2009. 16(3): p. 265-273. [CrossRef]
- Tharakaraman, K.; Subramanian, V.; Cain, D.; Sasisekharan, V.; Sasisekharan, R. Broadly neutralizing influenza hemagglutinin stem-specific antibody CR8020 targets residues that are prone to escape due to host selection pressure. Cell Host Microbe, 2014. 15(5): p. 644-51. [CrossRef]
- Corti, D., Voss, J.; Gamblin, S.J.; Codoni, G.; Macagno, A.; Jarrossay, D.; Vachieri, S.G.; Pinna, D.; Minola, A.; Vanzetta, F.; et al., A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science, 2011. 333(6044): p. 850-6. [CrossRef]
- Limberis, M.P., Adam, V.S.; Wong, G.; Gren, J.; Kobasa, D.; Ross, T.M.; Kobinger, G.P.; Tretiakova, A.; Wilson, J.M. Intranasal antibody gene transfer in mice and ferrets elicits broad protection against pandemic influenza. Sci Transl Med, 2013. 5(187): p. 187ra72. [CrossRef]
- Morgan, S.B., Holzer, B.; Hemmink, J.D.; Salguero, F.J.; Schwartz, J.C.; Agatic, G.; Cameroni, E.; Guarino, B.; Porter, E.; Rijal, P.; et al., Therapeutic Administration of Broadly Neutralizing FI6 Antibody Reveals Lack of Interaction Between Human IgG1 and Pig Fc Receptors. Front Immunol, 2018. 9: p. 865. [CrossRef]
- Ekiert, D.C., Friesen, R.H.E.; Bhabha, G.; Kwaks, T.; Jongeneelen, M.; Yu, W.; Ophorst, C.; Cox, F.; Korse, H.J.W.M.; Brandenburg, B.; et al., A highly conserved neutralizing epitope on group 2 influenza A viruses. Science, 2011. 333(6044): p. 843-50. [CrossRef]
- Friesen, R.H., Lee, P.S.; Stoop, E.J.M.; Hoffman, R.M.B.; Ekiert, D.C.; Bhabha, G.; Yu, W.; Juraszek, J.; Koudstaal, W.; Jongeneelen, M.; et al., A common solution to group 2 influenza virus neutralization. Proc Natl Acad Sci U S A, 2014. 111(1): p. 445-50. [CrossRef]
- Guthmiller, J.J., Han, J.; Utset, H.A.; Li, L.; Lan, L.Y.-L.; Henry, C.; Stamper, C.T.; McMahon, M.; O'Dell, G.; Fernández-Quintero, M.L.; et al., Broadly neutralizing antibodies target a haemagglutinin anchor epitope. Nature, 2022. 602(7896): p. 314-320. [CrossRef]
- McCraw, D.M., Myers, M.L.; Gulati, N.M.; Prabhakaran, M.; Brand, J.; Andrews, S.; Gallagher, J.R.; Maldonado-Puga, S.; Kim, A.J.; Torian, U.; et al., Designed nanoparticles elicit cross-reactive antibody responses to conserved influenza virus hemagglutinin stem epitopes. PLoS Pathog, 2023. 19(8): p. e1011514. [CrossRef]
- Sakabe, S., Iwatsuki-Horimoto, K.; Horimoto, T.; Nidom, .C.A; Le, M.t.Q.; Takano, R.; Kubota-Koketsu, R.; Okuno, Y.; Ozawa, M.; Kawaoka, Y. A cross-reactive neutralizing monoclonal antibody protects mice from H5N1 and pandemic (H1N1) 2009 virus infection. Antiviral Res, 2010. 88(3): p. 249-55. [CrossRef]
- Lang, S., Xie, J.; Zhu, X.; Wu, N.C., Lerner, R.A.; Wilson, I.A. Antibody 27F3 Broadly Targets Influenza A Group 1 and 2 Hemagglutinins through a Further Variation in V(H)1-69 Antibody Orientation on the HA Stem. Cell Rep, 2017. 20(12): p. 2935-2943. [CrossRef]
- Chen, F., Tzarum, N.; Wilson, I.A.; Law, M. V(H)1-69 antiviral broadly neutralizing antibodies: genetics, structures, and relevance to rational vaccine design. Curr Opin Virol, 2019. 34: p. 149-159. [CrossRef]
- Roubidoux, E.K., Carreño, J.M; McMahon, M.; Jiang, K.; van Bakel, H.; Wilson, P.; Krammer, F. Mutations in the Hemagglutinin Stalk Domain Do Not Permit Escape from a Protective, Stalk-Based Vaccine-Induced Immune Response in the Mouse Model. mBio, 2021. 12(1). [CrossRef]
- Muralidharan, A., Gravel, C.; Harris, G.; Hashem, A.M.; Zhang, W.; Safronetz, D.; Van Domselaar, G.; Krammer,, F.; Sauve, S.; Rosu-Myles, M.; et al., Universal antibody targeting the highly conserved fusion peptide provides cross-protection in mice. Hum Vaccin Immunother, 2022. 18(5): p. 2083428. [CrossRef]
- Sutton, T.C., Lamirande, E.W.; Bock, K.W.; Moore, I.N.; Koudstaal, W.; Rehman, M.; Weverling, G.J.; Goudsmit, J.; Subbarao, K. In Vitro Neutralization Is Not Predictive of Prophylactic Efficacy of Broadly Neutralizing Monoclonal Antibodies CR6261 and CR9114 against Lethal H2 Influenza Virus Challenge in Mice. J Virol, 2017. 91(24). [CrossRef]
- Throsby, M., van den Brink, E.; Jongeneelen, M.; Poon, L.L.M; Alard, Ph; Cornelissen, L.; Bakker, A.; Cox, F.; van Deventer, E.; Guan, Y.; et al., Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. PLoS One, 2008. 3(12): p. e3942. [CrossRef]
- Park, J.K., Xiao, Y.; Ramuta, M.D.; Rosas, L.A.; Fong, S.; Matthews, A.M.; Freeman, A.D.; Gouzoulis, M.A.; Batchenkova, N.A.; Yang, X.; Pre-existing immunity to influenza virus hemagglutinin stalk might drive selection for antibody-escape mutant viruses in a human challenge model. Nat Med, 2020. 26(8): p. 1240-1246. [CrossRef]
- Li, G.M., Chiu, C.; Wrammert, J.; McCausland, M.; Andrews, S.F.; Zheng, N.-Y.; Lee, J.-H.; Huang, M.; Qu, X.; Edupuganti, S.; et al., Pandemic H1N1 influenza vaccine induces a recall response in humans that favors broadly cross-reactive memory B cells. Proc Natl Acad Sci U S A, 2012. 109(23): p. 9047-52. [CrossRef]
- Kashyap, A.K., Steel, J.; Rubrum, A.; Estelles, A.; Briante, R.; Ilyushina, N.A.; Xu, L.; Swale, R.E.; Faynboym, A.M.; Foreman, P.K.; et al., Protection from the 2009 H1N1 pandemic influenza by an antibody from combinatorial survivor-based libraries. PLoS Pathog, 2010. 6(7): p. e1000990. [CrossRef]
- Nakamura, G., Chai, N.; Park, S.; Chiang, N.; Lin, Z.; Chiu, H.; Fong, R.; Yan, D.; Kim, J.; Zhang, J.; et al., An In Vivo Human-Plasmablast Enrichment Technique Allows Rapid Identification of Therapeutic Influenza A Antibodies. Cell Host Microbe, 2013. 14(1): p. 93-103.
- Chai, N., Swem, L.R.; Reichelt, M.; Chen-Harris, H.; Luis, E.; Park, S.; Fouts, A.; Lupardus, P.; Wu, T.D.; Li, O.; et al., Two Escape Mechanisms of Influenza A Virus to a Broadly Neutralizing Stalk-Binding Antibody. PLoS Pathog, 2016. 12(6): p. e1005702. [CrossRef]
- Corti, D., Suguitan Jr.; A.L.; Pinna, D.; Silacci, C.; Fernandez-Rodriguez, B.M.; Vanzetta, F.; Santos, C.; Luke, C.J.; Torres-Velez, F.J.; Temperton, N.J.; et al., Heterosubtypic neutralizing antibodies are produced by individuals immunized with a seasonal influenza vaccine. J Clin Invest, 2010. 120(5): p. 1663-73. [CrossRef]
- Wang, W., Sun, X.; Li, Y.; Su, J.; Ling, Z.; Zhang, T.; Wang, F.; Zhang, H.; Chen, H.; Ding, J.; et al., Human antibody 3E1 targets the HA stem region of H1N1 and H5N6 influenza A viruses. Nature Communications, 2016. 7(1): p. 13577. [CrossRef]
- Wu, Y.; Cho, M.; Shore, D.; Song, M.; Choi, J.; Jiang, T.; Deng, Y.-Q.; Bourgeois, M.; Almli, L.; Yang, H.; et al., A potent broad-spectrum protective human monoclonal antibody crosslinking two haemagglutinin monomers of influenza A virus. Nature Communications, 2015. 6(1): p. 7708. [CrossRef]
- Joyce, M.G., Wheatley, A.K.; Thomas, P.V.; Chuang, G.-Y.; Soto, C.; Bailer, R.T.; Druz, A.; Georgiev, I.S.; Gillespie, R.A.; Kanekiyo, M.; et al., Vaccine-Induced Antibodies that Neutralize Group 1 and Group 2 Influenza A Viruses. Cell, 2016. 166(3): p. 609-623. [CrossRef]
- Beukenhorst, A.L., Frallicciardi, J.; Koch, C.M.; Klap, J.M.; Phillips, A.; Desai, M.M.; Wichapong, K.; Nicolaes, G.A.F.; Koudstaal, W.; Alter, G.; et al., Corrigendum: The influenza hemagglutinin stem antibody CR9114: Evidence for a narrow evolutionary path towards universal protection. Frontiers in Virology, 2023. 3. [CrossRef]
- Beukenhorst, A.L.; Frallicciardi, J.; Rice, K.L.; Koldijk, M.H.; Moreira de Mello, J.C.; Klap, J.M.; Hadjichrysanthou, C.; Koch, C.M.; da Costa, K.A.S.; Temperton, N.; et al., A pan-influenza monoclonal antibody neutralizes H5 strains and prophylactically protects through intranasal administration. Sci. Rep., 2024. 14(1): p. 3818. [CrossRef]
- Prachanronarong, K.L., Canale, A.S.; Liu, P.; Somasundaran, M.; Hou, S.; Poh, Y.-P.; Han, T.; Zhu, Q.; Renzette, N.; Zeldovich, K.B.; et al., Mutations in Influenza A Virus Neuraminidase and Hemagglutinin Confer Resistance against a Broadly Neutralizing Hemagglutinin Stem Antibody. J Virol, 2019. 93(2). [CrossRef]
- Ali, S.O.; Takas, T.; Nyborg, A.; Shoemaker, K.; Kallewaard, N.L.; Chiong, R.; Dubovsky, F.; Mallory, R.M. Evaluation of MEDI8852, an Anti-Influenza A Monoclonal Antibody, in Treating Acute Uncomplicated Influenza. Antimicrob Agents Chemother, 2018. 62(11). [CrossRef]
- Paules, C.I., Lakdawala, S.; McAuliffe, J.M.; Paskel, M.; Vogel, L.; Kallewaard, N.L.; Zhu, Q.; Subbarao, K. The Hemagglutinin A Stem Antibody MEDI8852 Prevents and Controls Disease and Limits Transmission of Pandemic Influenza Viruses. J Infect Dis, 2017. 216(3): p. 356-365. [CrossRef]
- Mark Throsby, R.H., Edward Friesen, Theodorus Hendrikus, Jacobus Kwaks, Mandy Antonia, Catharina Jongeneelen, Human binding molecules capable of neutralizing influenza virus H3N2 and uses thereof. 2010: United States.
- Tan, G.S., Lee, P.S.; Hoffman, R.M.B.; Mazel-Sanchez, B.; Krammer, F.; Leon, P.E.; Ward, A.B.; Wilson, I.A.; Palese, P. Characterization of a broadly neutralizing monoclonal antibody that targets the fusion domain of group 2 influenza A virus hemagglutinin. J Virol, 2014. 88(23): p. 13580-92. [CrossRef]
- Myers, M.L.; Gallagher, J.R.; Kim, A.J.; Payne, W.H.; Maldonado-Puga, S.; Assimakopoulos, H.; Bock, K.W.; Torian, U.; Moore, I.N.; Harris, A.K. Commercial influenza vaccines vary in HA-complex structure and in induction of cross-reactive HA antibodies. Nature Communications, 2023. 14(1): p. 1763. [CrossRef]
- Yu, X., Tsibane, T.; McGraw, P.A.; House, F.S.; Keefer, C.J.; Hicar, M.D.; Tumpey, T.M.; Pappas, C.; Perrone, L.A.; Martinez, O.; et al, Neutralizing antibodies derived from the B cells of 1918 influenza pandemic survivors. Nature, 2008. 455(7212): p. 532-6. [CrossRef]
- Yoshida, R., Igarashi, M.; Ozaki, H.; Kishida, N.; Tomabechi, D.; Kida, H.; Ito, K.; Takada, A. Cross-protective potential of a novel monoclonal antibody directed against antigenic site B of the hemagglutinin of influenza A viruses. PLoS Pathog, 2009. 5(3): p. e1000350. [CrossRef]
- Ekiert, D.C.; Kashyap, A.K.; Steel, J.; Rubrum, A.; Bhabha, G.; Khayat, R.; Lee, J.H.; Dillon, M.A.; O'Neil, R.E.; Faynboym, A.M.; et al. Cross-neutralization of influenza A viruses mediated by a single antibody loop. Nature, 2012. 489(7417): p. 526-32. [CrossRef]
- Dreyfus, C., Laursen, N.S.; Kwaks, T.; Zuijdgeest, D.; Khayat, R.; Ekiert, D.C.; Lee, J.H.; Metlagel, Z.; Bujny, M.V.; Jongneelen, M.; et al. Highly conserved protective epitopes on influenza B viruses. Science, 2012. 337(6100): p. 1343-8. [CrossRef]
- Whittle, J.R.R., Zhang, R.; Khurana, S.; King, L.R.; Manischewitz, J.; Golding, H.; Dormitzer, P.R.; Haynes, B.F.; Walter, E.B.; Moody, M.A.; et al. Broadly neutralizing human antibody that recognizes the receptor-binding pocket of influenza virus hemagglutinin. Proceedings of the National Academy of Sciences, 2011. 108(34): p. 14216-14221. [CrossRef]
- Lee, P.S., Ohshima, N.; Stanfield, R.L.; Yu, W.; Iba, Y.; Okuno, Y.; Kurosawa, Y.; Wilson, I.A. Receptor mimicry by antibody F045–092 facilitates universal binding to the H3 subtype of influenza virus. Nat. Commun. 2014. 5(1): p. 3614. [CrossRef]
- Krause, J.C., Tsibane, T.; Tumpey, T.M.; Huffman, C.J.; Basler, C.F.; Crowe Jr, J.E. A broadly neutralizing human monoclonal antibody that recognizes a conserved, novel epitope on the globular head of the influenza H1N1 virus hemagglutinin. J Virol, 2011. 85(20): p. 10905-8. [CrossRef]
- Li, T., Chen, J.; Zheng, Q.; Xue, W.; Zhang, L.; Rong, R.; Zhang, S.; Wang, Q.; Hong, M.; Zhang, Y.; et al. Identification of a cross-neutralizing antibody that targets the receptor binding site of H1N1 and H5N1 influenza viruses. Nature Commun, 2022. 13(1): p. 5182. [CrossRef]
- Guthmiller, J.J., Han, J.; Li, L.; Freyn, A.W.; Liu, S.T.H.; Stovicek, O.; Stamper, C.T.; Dugan, H.L.; Tepora, M.E.; Utset, H.A.; et al. First exposure to the pandemic H1N1 virus induced broadly neutralizing antibodies targeting hemagglutinin head epitopes. Sci Transl Med, 2021. 13(596). [CrossRef]
- Raymond, D.D., Bajic, G.; Ferdman, J.; Suphaphiphat, P.; Settembre, E.C.; Moody, M.A.; Schmidt, A.G.; Harrison, S.C. Conserved epitope on influenza-virus hemagglutinin head defined by a vaccine-induced antibody. Proc Natl Acad Sci U S A, 2018. 115(1): p. 168-173. [CrossRef]
- Jia, M.; Zhao, H.; Morano, N.C.; Lu, H.; Lui, Y.-M.; Du, H.; Becker, J.E.; Yuen, K.-Y.; Ho, D.D; Kwong, P.D.; et al. Human neutralizing antibodies target a conserved lateral patch on H7N9 hemagglutinin head. Nature Communications, 2024. 15(1): p. 4505. [CrossRef]
- Zheng, Z., Paul, S.S.; Mo, X.; Yuan, Y.-R.; Tan, Y.J. The Vestigial Esterase Domain of Haemagglutinin of H5N1 Avian Influenza A Virus: Antigenicity and Contribution to Viral Pathogenesis. Vaccines (Basel), 2018. 6(3). [CrossRef]
- Bangaru, S., Zhang, H.; Gilchuk, I.M.; Voss, T.G.; Irving, R.P.; Gilchuk, P.; Matta, P.; Zhu, X.; Lang, S.; Nieusma, T.; et al. A multifunctional human monoclonal neutralizing antibody that targets a unique conserved epitope on influenza HA. Nature Communications, 2018. 9(1): p. 2669. [CrossRef]
- Crowe, J.E., Antibody Determinants of Influenza Immunity. J Infect Dis, 2019. 219(Suppl_1): p. S21-s29. [CrossRef]
- Gao, R.; Sheng, Z.; Sreenivasan, C.C.; Wang, D.; Li, F. Influenza A Virus Antibodies with Antibody-Dependent Cellular Cytotoxicity Function. Viruses, 2020. 12(3): p. 276.
- Chai, N., Swem, L.R.; Park, S.; Nakamura, G.; Chiang, N.; Estevez, A.; Fong, R.; Kamen, L.; Kho, E.; Reichelt, M.; et al. A broadly protective therapeutic antibody against influenza B virus with two mechanisms of action. Nat Commun, 2017. 8: p. 14234. [CrossRef]
- Yan, L., Sun, L.; Guo, C.; Li, L.; Sun, J.; Huang, X.; Zhao, P.; Xie, X.; Hu, J. Neutralizing antibody PR8-23 targets the footprint of the sialoglycan receptor binding site of H1N1 hemagglutinin. J Med Virol, 2021. 93(6): p. 3508-3515. [CrossRef]
- Li, J., Wang, Y.; Liang, Y.; Ni, B.; Wan, Y.; Liao, Z.; Chan, K.-h.; Yuen, K.-y.; Fu, X.; Shang, X.; et al. Fine antigenic variation within H5N1 influenza virus hemagglutinin's antigenic sites defined by yeast cell surface display. Eur J Immunol, 2009. 39(12): p. 3498-510. [CrossRef]
- Yewdell, J.W., Taylor A.; Yellen, A.; Caton, A.; Gerhard, W.; Bächi, T. Mutations in or near the fusion peptide of the influenza virus hemagglutinin affect an antigenic site in the globular region. J Virol, 1993. 67(2): p. 933-42. [CrossRef]
- Bangaru, S., Lang, S.; Schotsaert, M.; Vanderven, H.A.; Zhu, X.; Kose, N.; Bombardi, R.; Finn, J.A.; Kent, S.J.; Gilchuk, P.; et al. A Site of Vulnerability on the Influenza Virus Hemagglutinin Head Domain Trimer Interface. Cell, 2019. 177(5): p. 1136-1152.e18. [CrossRef]
- Watanabe, A., McCarthy, K.R.; Kuraoka, M.; Schmidt, A.G.; Adachi, Y.; Onodera, T.; Tonouchi, K.; Caradonna, T.M.; Bajic, G.; Song, S.; et al. Antibodies to a Conserved Influenza Head Interface Epitope Protect by an IgG Subtype-Dependent Mechanism. Cell, 2019. 177(5): p. 1124-1135.e16. [CrossRef]
- Bajic, G., Maron, M.J.; Adachi, Y.; Onodera, T.; McCarthy, K.R.; McGee, C.E.; Sempowski, G.D.; Takahasi, Y.; Kelsoe, G.; Kuraoka, M.; et al. Influenza Antigen Engineering Focuses Immune Responses to a Subdominant but Broadly Protective Viral Epitope. Cell Host Microbe, 2019. 25(6): p. 827-835.e6. [CrossRef]
- Iba, Y., Fujii, Y.; Ohshima, N.; Sumida, T.; Kubota-Koketsu, R.; Ikeda, M.; Wakiyama, M.; Shirouzu, M.; Okada, J.; Okuno, Y.; et al. Conserved neutralizing epitope at globular head of hemagglutinin in H3N2 influenza viruses. J Virol, 2014. 88(13): p. 7130-44. [CrossRef]
- Lee, P.S.; Yoshida, R.; Ekiert, D.C.; Sakai, N.; Suzuki, Y.; Takada, A.; Wilson, I.A. Heterosubtypic antibody recognition of the influenza virus hemagglutinin receptor binding site enhanced by avidity. Proc Natl Acad Sci U S A, 2012. 109(42): p. 17040-5. [CrossRef]
- He, W.; Tan, G.S.; Mullarkey, C.E.; Lee, A.J.; Lam, M.M.W.; Krammer, F.; Henry, C.; Wilson, P.C.; Ashkar, A.A.; Palese, P. et al. Epitope specificity plays a critical role in regulating antibody-dependent cell-mediated cytotoxicity against influenza A virus. Proc Natl Acad Sci U S A, 2016. 113(42): p. 11931-11936. [CrossRef]
- McCarthy, K.R.; Watanabe, A.; Kuraoka, M.; Do, K.T.; McGee, C.E.; Sempowski, G.D.; Kepler, T.B.; Schmidt, A.G.; Kelsoe, G.; Harrison, S.C. Memory B Cells that Cross-React with Group 1 and Group 2 Influenza A Viruses Are Abundant in Adult Human Repertoires. Immunity, 2018. 48(1): p. 174-184.e9. [CrossRef]
- Krause, J.C.; Tsibane, T.; Tumpey, T.M.; Huffman, C.J.; Albrecht, R.; Blum, D.L.; Ramos, I:; Fernandez-Sesma, A.; Edwards, K.M.; García-Sastre, A.; et al. Human monoclonal antibodies to pandemic 1957 H2N2 and pandemic 1968 H3N2 influenza viruses. J Virol, 2012. 86(11): p. 6334-40. [CrossRef]
- Tsibane, T.; Ekiert, D.C.; Krause, J.C.; Martinez, O.; Crowe Jr., J.E.; Wilson, I.A.; Basler, C.F. Influenza human monoclonal antibody 1F1 interacts with three major antigenic sites and residues mediating human receptor specificity in H1N1 viruses. PLoS Pathog, 2012. 8(12): p. e1003067. [CrossRef]
- Schmidt, A.G.; Therkelsen, M.D.; Stewart, S.; Kepler, T.B.; Liao, H.-X.; Moody, M.A.; Haynes, B.F.; Harrison, S.C. Viral receptor-binding site antibodies with diverse germline origins. Cell, 2015. 161(5): p. 1026-1034. [CrossRef]
- Yang, F.; Yan, S.; Zhu, L.; Wang, F.X.C.; Liu, F.; Cheng, L.; Yao, H.; Wu, N.; Lu, R.; Wu, H. Evaluation of panel of neutralising murine monoclonal antibodies and a humanised bispecific antibody against influenza A(H1N1)pdm09 virus infection in a mouse model. Antiviral Res, 2022. 208: p. 105462.
- Portnoff, A.D.; Patel, N.; Massare, M.J.; Zhou, H.; Tian, J.-H.; Zhou, B.; Shinde, V.; Glenn, G.M.; Smith, G. Influenza Hemagglutinin Nanoparticle Vaccine Elicits Broadly Neutralizing Antibodies against Structurally Distinct Domains of H3N2 HA. Vaccines (Basel), 2020. 8(1). [CrossRef]
- Lin, Q.; Li, T.; Chen, Y.; Lau, S.-Y.; Wei, M.; Zhang, Y.; Zhang, Z.; Yao, Q.; Li, J.; Li, Z.; et al. Structural Basis for the Broad, Antibody-Mediated Neutralization of H5N1 Influenza Virus. J Virol, 2018. 92(17). [CrossRef]
- Zuo, Y., Wang, P.; Sun, J.; Guo, S.; Wang, G.; Zuo, T.; Fan, S.; Zhou, P.; Liang, M.; Shi, X.; et al. Complementary recognition of the receptor-binding site of highly pathogenic H5N1 influenza viruses by two human neutralizing antibodies. J Biol Chem, 2018. 293(42): p. 16503-16517. [CrossRef]
- Wu, R.; Li, X.; Leung, H.-C.; Cao, Z.; Qiu, Z.; Zhou, Y.; Zheng, B.-J.; He, Y A novel neutralizing antibody against diverse clades of H5N1 influenza virus and its mutants capable of airborne transmission. Antiviral Res 2014. 106: p. 13-23.
- Winarski, K.L.; Thornburg, N.J.; Yu, Y.; Sapparapu, G.; Crowe Jr., J.E.; Spiller, B.W. Vaccine-elicited antibody that neutralizes H5N1 influenza and variants binds the receptor site and polymorphic sites. Proceedings of the National Academy of Sciences, 2015. 112(30): p. 9346-9351. [CrossRef]
- Zhu, X.; Guo, Y.-H.; Jiang, T.; Wang, Y.-D.; Chan, K.-H.; Li, X.-F., Yu, W.; McBride, R.; Paulson, J.C.; Yuen, K.-Y., et al. A unique and conserved neutralization epitope in H5N1 influenza viruses identified by an antibody against the A/Goose/Guangdong/1/96 hemagglutinin. J Virol, 2013. 87(23): p. 12619-35. [CrossRef]
- Paul, S.S.; Mok, C.-K.; Mak, T.-M.; Ng, O.-W.;.Aboagye, J.O.; Wohlbold, T.J.; Krammer, F.; Tan, Y.-J. A cross-clade H5N1 influenza A virus neutralizing monoclonal antibody binds to a novel epitope within the vestigial esterase domain of hemagglutinin. Antiviral Res, 2017. 144: p. 299-310.
- Zheng, Z.; Teo, S.H.C.; Arularasu, S.C.; Liu, Z.; Mohd-Ismail, N.K.; Mok, C.K.; Ong, C.B.; Chu, J.J.-h.; Tan, Y.-J. Contribution of Fc-dependent cell-mediated activity of a vestigial esterase-targeting antibody against H5N6 virus infection. Emerg Microbes Infect, 2020. 9: p. 95-110. [CrossRef]
- Qian, M.; Hu, H.; Zuo, T.; Wang, G.; Zhang, L.; Zhou, P. Unraveling of a neutralization mechanism by two human antibodies against conserved epitopes in the globular head of H5 hemagglutinin. J Virol, 2013. 87(6): p. 3571-7. [CrossRef]
- Wang, S.; Ren, H.; Jiang, W.; Chen, H.; Hu, H.; Chen, Z.; Zhou, P. Divergent Requirement of Fc-Fcγ Receptor Interactions for In Vivo Protection against Influenza Viruses by Two Pan-H5 Hemagglutinin Antibodies. J Virol, 2017. 91(11). [CrossRef]
- Zhang, X.; Qi, X.; Zhang, Q.; Zeng, X.; Shi, Z.; Jin, Q:; Zhan, F.; Xu, Y.; Liu, Z.; Feng, Z.; Jiao, Y. Human 4F5 single-chain Fv antibody recognizing a conserved HA1 epitope has broad neutralizing potency against H5N1 influenza A viruses of different clades. Antiviral Res 2013. 99(2): p. 91-99.
- Jin, Q.; Yao, Z.; Liu, F.; Di, Y.; Gao, J.; Zhang, X. The protective effect of a combination of human intracellular and extracellular antibodies against the highly pathogenic avian influenza H5N1 virus. Hum Vaccin Immunother, 2022. 18(1): p. 2035118. [CrossRef]
- Tan, G.S., et al., Broadly-Reactive Neutralizing and Non-neutralizing Antibodies Directed against the H7 Influenza Virus Hemagglutinin Reveal Divergent Mechanisms of Protection. PLOS Pathogens, 2016. 12(4): p. e1005578. [CrossRef]
- Chai, N.; Swem, L.R.; Park, S.; Nakamura, G.; Chiang, N.; Estevez, A.; Fong, R.; Kamen, L.; Kho, E.; Reichelt, M.; et al. A broadly protective therapeutic antibody against influenza B virus with two mechanisms of action. Nat Commun, 2017. 8: p. 14234. [CrossRef]
- Raymond, D.D., Bajic, G.; Ferdman, J.; Suphaphiphat, P.; Settembre, E.C.; Moody, M.A.; Schmidt, A.G.; Harrison, S.C. Conserved epitope on influenza-virus hemagglutinin head defined by a vaccine-induced antibody. Proc Natl Acad Sci U S A, 2018. 115(1): p. 168-173. [CrossRef]
- Henry Dunand, C.J.; Leon, P.E.; Huang, M.; Choi, A.; Chromikova, V.; Ho, I.Y.; Tan, G.S.; Cruz, J.; Hirsh, A.; Zheng, N.-Y.; et al. Both Neutralizing and Non-Neutralizing Human H7N9 Influenza Vaccine-Induced Monoclonal Antibodies Confer Protection. Cell Host Microbe, 2016. 19(6): p. 800-13. [CrossRef]
- Jia, M.; Zhao, H.; Morano, N.C.; Lu, H.; Lui, Y.-M.; Du, H.; Becker, J.E.; Yuen, K.-Y.; Ho, D.D; Kwong, P.D.; et al. Human neutralizing antibodies target a conserved lateral patch on H7N9 hemagglutinin head. Nature Communications, 2024. 15(1): p. 4505. [CrossRef]
- Dong, J.; Gilchuk, I.; Li, S.; Irving, R.; Goff, M.T.; Turner, H.L.; Ward, A.B.; Carnahan, R.H.; Anti-influenza H7 human antibody targets antigenic site in hemagglutinin head domain interface. J Clin Invest, 2020. 130(9): p. 4734-4739. [CrossRef]
- Turner, H.L.; Pallesen, J.; Lang, S.; Bangaru, S.; Urata, S.; Li; S.; Cottrell, C.A.; Bowman, C.A.; Crowe, Jr., J.E.; Wilson, I.A.; et al. Potent anti-influenza H7 human monoclonal antibody induces separation of hemagglutinin receptor-binding head domains. PLoS Biol, 2019. 17(2): p. e3000139. [CrossRef]
- Kohler, G.; C. Milstein. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature, 1975. 256(5517): p. 495-7. [CrossRef]
- Vidarsson, G.; G. Dekkers, T.; Rispens. IgG Subclasses and Allotypes: From Structure to Effector Functions. Frontiers in Immunology, 2014. 5. [CrossRef]
- Brezski, R.J.; G. Georgiou. Immunoglobulin isotype knowledge and application to Fc engineering. Curr Opin Immunol, 2016. 40: p. 62-9. [CrossRef]
- Tharakaraman, K.; Subramanian, V.; Cain, D.; Sasisekharan, V.; Sasisekharan, R. Broadly neutralizing influenza hemagglutinin stem-specific antibody CR8020 targets residues that are prone to escape due to host selection pressure. Cell Host Microbe, 2014. 15(5): p. 644-51. [CrossRef]
- Yi, K.S.; Choi, J.-A.; Kim, P.; Ryu, D.-K.; Yang, E.; Son, D.; Shin, J.; Park, H.; Lee, S.; Lee, H.; et al. Broader neutralization of CT-P27 against influenza A subtypes by combining two human monoclonal antibodies. PLOS ONE, 2020. 15(7): p. e0236172. [CrossRef]
- Kallewaard, N.L.; Corti, D.; Collins, P.J.; Neu, U.; McAuliffe, J.M.; Benjamin, E.; Wachter-Rosati, L.; Palmer-Hill, F.J.; Yuan, A.Q.; Walker, P.A.; et al. Structure and Function Analysis of an Antibody Recognizing All Influenza A Subtypes. Cell, 2016. 166(3): p. 596-608. [CrossRef]
- Tharakaraman, K.; Subramanian, V.; Viswanathan, K.; Sloan, S.; Yen, H.-L.; Barnard, D.L.; Leung, Y.H.C.; Szretter, K.J.; Koch, T.J.; Delaney, J.C.; et al. A broadly neutralizing human monoclonal antibody is effective against H7N9. Proceedings of the National Academy of Sciences, 2015. 112(35): p. 10890-10895. [CrossRef]
- Izadi, A.; Hailu, A.; Godzwon, M.; Wrighton, S.; Olofsson, B.; Schmidt, T.; Söderlund-Strand, A.; Elder, E.; Appelberg, S.; Valsjö, M.; et al. Subclass-switched anti-spike IgG3 oligoclonal cocktails strongly enhance Fc-mediated opsonization. Proceedings of the National Academy of Sciences, 2023. 120(15). [CrossRef]
- Bolton, M.J.; Arevalo, C.P.; Griesman, T.;Li; S.H.; Bates, P.; Wilson, P.C.; Hensley, S.E. IgG3 subclass antibodies recognize antigenically drifted influenza viruses and SARS-CoV-2 variants through efficient bivalent binding. Proc Natl Acad Sci U S A, 2023. 120(35): p. e2216521120. [CrossRef]
- Bowles, J.A.; Wang, S.-Y.; Link, B.K; Allan, B.; Beuerlein, G.; Campbell, M.-A.; Marquis, D.; Ondek, B.; Wooldridge, J.E.; Smith, B.J.; et al. Anti-CD20 monoclonal antibody with enhanced affinity for CD16 activates NK cells at lower concentrations and more effectively than rituximab. Blood, 2006. 108(8): p. 2648-2654. [CrossRef]
- Forero-Torres, A.; de Vos, S.; Pohlman, B.L.; Pashkevich, M.; Cronier, D.M.; Dang, N.H.; Carpenter, S.P.; Allan, B.W.; Nelson, J.G.; Slapak, C.A.; et al. Results of a Phase 1 Study of AME-133v (LY2469298), an Fc-Engineered Humanized Monoclonal Anti-CD20 Antibody, in FcγRIIIa-Genotyped Patients with Previously Treated Follicular Lymphoma. Clinical Cancer Res, 2012. 18(5): p. 1395-1403. [CrossRef]
- Van Der Horst, H.J.; Nijhof, I.S.; Mutis, T.; Chamuleau, M.E.D. Fc-Engineered Antibodies with Enhanced Fc-Effector Function for the Treatment of B-Cell Malignancies. Cancers, 2020. 12(10): p. 3041. [CrossRef]
- Ko, S.; Park, S.; Sohn, M.H.; Jo, M.; Ko, B.J.; Na, J.-H.; Yoo, H.; Jeong, A.L.; Ha, K.; Woo, J.R.; et al. An Fc variant with two mutations confers prolonged serum half-life and enhanced effector functions on IgG antibodies. Exp. Mol. Med., 2022. 54(11): p. 1850-1861. [CrossRef]
- Natsume, A.; In, M.; Takamura, H.; Nakagawa, T.; Shimizu, Y.; Kitajima, K.; Wakitani, M.; Ohta, S.; Satoh, M.; Shitara, K.; et al. Engineered antibodies of IgG1/IgG3 mixed isotype with enhanced cytotoxic activities. Cancer Res, 2008. 68(10): p. 3863-72. [CrossRef]
- Chu, T.H.; E.F. Patz; M.E. Ackerman. Coming together at the hinges: Therapeutic prospects of IgG3. mAbs, 2021. 13(1): p. 1882028. [CrossRef]
- Ali, S.O., et al., Takas, T.; Nyborg, A.; Shoemaker, K.; Kallewaard, N.L.; Chiong, R.; Dubovsky, F.; Mallory, R.M. Evaluation of MEDI8852, an Anti-Influenza A Monoclonal Antibody, in Treating Acute Uncomplicated Influenza. Antimicrob Agents Chemother, 2018. 62(11). [CrossRef]
- Sloan, S.E.; Szretter, K.J.; Sundaresh, B.; Narayan, K.M.; Smith, P.F.; Skurnik, D.; Bedard, S.; Trevejo, J.M.; Oldach, D.; Shriver, Z. Clinical and virological responses to a broad-spectrum human monoclonal antibody in an influenza virus challenge study. Antiviral Res, 2020. 184: p. 104763. [CrossRef]
- Hershberger, E.; Sloan, S.; Narayan, K.; Hay, C.A.; Smith, P.; Engler, F.; Jeeninga, R.; Smits, S.; Trevejo, J.; Shriver, Z.; et al. Safety and efficacy of monoclonal antibody VIS410 in adults with uncomplicated influenza A infection: Results from a randomized, double-blind, phase-2, placebo-controlled study. EBioMedicine, 2019. 40: p. 574-582. [CrossRef]
- Lim, J.J.; Nilsson, A.C.; Silverman, M.; Assy, N.; Kulkarni, P.; McBride, J.M.; Deng, R.; Li, C.; Yang, X.; Nguyen, A.; et al. A Phase 2 Randomized, Double-Blind, Placebo-Controlled Trial of MHAA4549A, a Monoclonal Antibody, plus Oseltamivir in Patients Hospitalized with Severe Influenza A Virus Infection. Antimicrob Agents Chemother, 2020. 64(7). [CrossRef]
- Han, A.; Czajkowski, L; Rosas, L.A.; Cervantes-Medina, A.; Xiao, Y.; Gouzoulis, M.; Lumbard, K.; Hunsberger, S.; Reed, S.; Athota, R.; et al. Safety and Efficacy of CR6261 in an Influenza A H1N1 Healthy Human Challenge Model. Clinical Infectious Diseases, 2021. 73(11): p. e4260-e4268. [CrossRef]
- Impagliazzo, A.; Milder, F.; Kuipers, H.; Wagner, M.V.; Zhu, X.; Hoffman, R.M.B.; van Meersbergen, R.; Huizingh, J.; Wanningen, P.; Verspuij, J.; et al. A stable trimeric influenza hemagglutinin stem as a broadly protective immunogen. Science, 2015. 349(6254): p. 1301-1306. [CrossRef]
- Van Der Lubbe, J.E.M.; Huizingh, J.; Verspuij, J.W.A.; Tettero, L.; Schmit-Tillemans, S.P.R.; Mooij, P.; Mortier, D.; Koopman, G.; Bogers, W.M.J.M.; et al., Mini-hemagglutinin vaccination induces cross-reactive antibodies in pre-exposed NHP that protect mice against lethal influenza challenge. NPJ Vaccines, 2018. 3(1). [CrossRef]
- Folschweiller, N.; Abeele, C.V.; Chu, L.; Van Damme, P.; García-Sastre, A.; Krammer, F.; Nachbagauer, R.; Palese, P; Solórzano, A.; Bi, D.; et al. Reactogenicity, safety, and immunogenicity of chimeric haemagglutinin influenza split-virion vaccines, adjuvanted with AS01 or AS03 or non-adjuvanted: a phase 1-2 randomised controlled trial. Lancet Infect Dis, 2022. 22(7): p. 1062-1075. [CrossRef]
- Nishiyama, A.; Adachi, Y.; Tonouchi, K.; Moriyama, S.; Sun, L.; Aoki, M.; Asanuma, H.; Shirakura, M.; Fukushima, A.; Yamamoto, T.; et al. Post-fusion influenza vaccine adjuvanted with SA-2 confers heterologous protection via Th1-polarized, non-neutralizing antibody responses. Vaccine, 2023. 41(31): p. 4525-4533. [CrossRef]
- Atmar, R.L.; Bernstein, D.I.; Winokur, P.; Frey, S.E.; Angelo, L.S.; Bryant, C.; Ben-Yedida-T.; Roberts, P.C.; El Sahly, H.M.; Keitel, W.A. Safety and immunogenicity of Multimeric-001 (M-001) followed by seasonal quadrivalent inactivated influenza vaccine in young adults - A randomized clinical trial. Vaccine, 2023. 41(16): p. 2716-2722. [CrossRef]
- Corbett, K.S.; Moin, S.M.; Yassine, H.M.; Cagigi, A.; Kanekiyo, M.; Boyoglu-Barnum, S.; Myers, S.I.; Tsybovsky, Y.; Wheatley, A.K.; Schramm, C.A.; et al. Design of Nanoparticulate Group 2 Influenza Virus Hemagglutinin Stem Antigens That Activate Unmutated Ancestor B Cell Receptors of Broadly Neutralizing Antibody Lineages. mBio, 2019. 10(1). [CrossRef]
- Lee, P.S.; Yoshida, R.; Ekiert, D.C.; Sakai, N.; Suzuki, Y.; Takada, A.; Wilson, I.A. Heterosubtypic antibody recognition of the influenza virus hemagglutinin receptor binding site enhanced by avidity. Proc Natl Acad Sci U S A, 2012. 109(42): p. 17040-5. [CrossRef]
- Ekiert, D.C.; Kashyap, A.K.; Steel, J.; Rubrum, A.; Bhabha, G.; Khayat, R.; Lee, J.H.; Dillon, M.A.; O'Neil, R.E.; Faynboym, A.M.; et al. Cross-neutralization of influenza A viruses mediated by a single antibody loop. Nature, 2012. 489(7417): p. 526-32. [CrossRef]
- Sarah F. Andrews, Y.H., Kaval Kaur, Lyubov I. Popova, Irvin Y. Ho, Noel T. Pauli, Carole J. Henry Dunand, William M Taylor, Samuel Lim, Min Huang, Xinyan Qu, Jane-Hwei Lee, Marlene Salgado-Ferrer, Florian Krammer, Peter Palese, Jens Wrammert, Rafi Ahmed, and Patrick C. Wilson. Immune history profoundly affects broadly protective B cell responses to influenza. Science Translational Medicine, 2015. 7(316): p. 316ra192.
- Bajic, G.; van der Poel, C.E.; Kuraoka, M.; Schmidt, A.G.; Carroll, M.C.; Kelsoe, G.; Harrison, S.C. Autoreactivity profiles of influenza hemagglutinin broadly neutralizing antibodies. Scientific Reports, 2019. 9(1). [CrossRef]
- Willey, S.; Aasa-Chapman, M.M.I.; O'Farrell, S.; Pellegrino, P.; Williams, I.; Weiss, R.A.; Neil, S.J.D. Extensive complement-dependent enhancement of HIV-1 by autologous non-neutralising antibodies at early stages of infection. Retrovirology, 2011. 8(1): p. 16. [CrossRef]
- Monsalvo, A.C.; Batalle, J.P.; Lopez, M.F.; Krause, J.C.; Klemenc, J.; Hernandez, J.Z.; Maskin, B.; Bugna, J.; Rubinstein, C.; Aguilar, L.; et al. Severe pandemic 2009 H1N1 influenza disease due to pathogenic immune complexes. Nature Medicine, 2011. 17(2): p. 195-199. [CrossRef]
- Lee, W.S.; Wheatley, A.K.; Kent, S.J.; DeKosky, B.J. Antibody-dependent enhancement and SARS-CoV-2 vaccines and therapies. Nat. Microbiol., 2020. 5(10): p. 1185-1191. [CrossRef]
- Rao, G.K.; Prell, R.A.; Laing, S.T.; Burleson, S.C.M.; Nguyen, A.; McBride, J.M.; Zhang, C.; Sheinson, D.; Halpern, W.G. In Vivo Assessment of Antibody-Dependent Enhancement of Influenza B Infection. Toxicol Sci, 2019. 169(2): p. 409-421. [CrossRef]
- Winarski, K.L.; Tang, J.; Klenow, L.; Lee, J.; Coyle, E.M.; Manischewitz, J.; Turner, H.L.; Takeda, K.; Ward, A.B.; Golding, H.; et al. Antibody-dependent enhancement of influenza disease promoted by increase in hemagglutinin stem flexibility and virus fusion kinetics. Proc Natl Acad Sci U S A, 2019. 116(30): p. 15194-15199. [CrossRef]
Figure 1.
Steps in Hemagglutinin Endosomal Collapse. The process begins when hemagglutinin (HA) binds to sialic acid on the host cell surface, facilitating viral endocytosis. In the endosome, HA initially exists in its a) prefusion state and is bound to sialic acid (not shown). Upon endosomal acidification, HA releases the bound sialic acid and undergoes a conformational change to its b) extended intermediate state, allowing the fusion peptide (shown in red) to insert into the endosomal membrane. The c) extended intermediate state collapses, and HA undergoes a "jackknife" motion (c1 → c2), drawing the viral and endosomal membranes together. As membrane fusion occurs, HA adopts its d) post-fusion state, forming a fusion pore that facilitates the release of viral genetic material into the cytoplasm.
Figure 1.
Steps in Hemagglutinin Endosomal Collapse. The process begins when hemagglutinin (HA) binds to sialic acid on the host cell surface, facilitating viral endocytosis. In the endosome, HA initially exists in its a) prefusion state and is bound to sialic acid (not shown). Upon endosomal acidification, HA releases the bound sialic acid and undergoes a conformational change to its b) extended intermediate state, allowing the fusion peptide (shown in red) to insert into the endosomal membrane. The c) extended intermediate state collapses, and HA undergoes a "jackknife" motion (c1 → c2), drawing the viral and endosomal membranes together. As membrane fusion occurs, HA adopts its d) post-fusion state, forming a fusion pore that facilitates the release of viral genetic material into the cytoplasm.
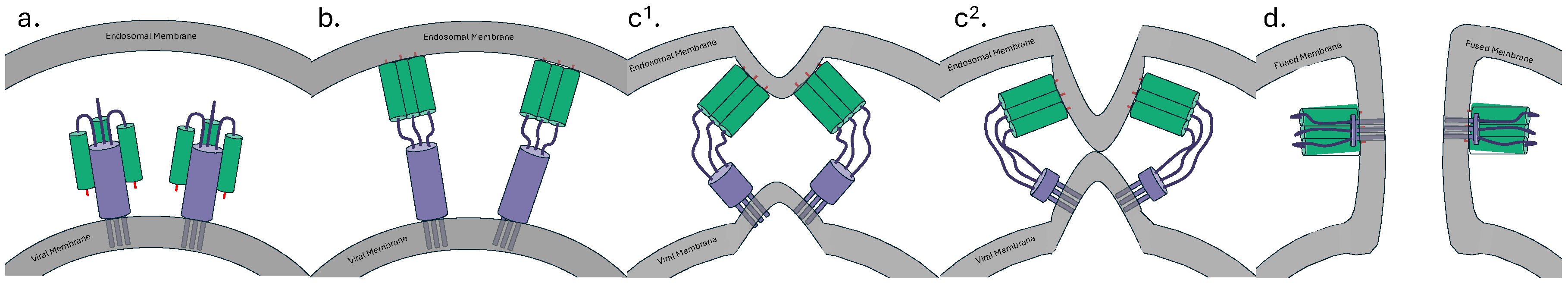
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



