
Submitted:
12 October 2024
Posted:
15 October 2024
You are already at the latest version
Abstract
L-(+)-Furanomycin 1 is a miniature antibacterial natural product that contains an -amino acid core. This non-proteinogenic α-amino acid was first isolated in 1967 by Katagiri and co-workers from the fermentation broth of Streptomyces threomyceticus L-803 (ATCC 15795). It is a substrate of isoleucyl aminoacyl tRNA synthetase that replaces isoleucine in the protein translation process and exhibits antibacterial properties in vitro. It effectively acts as an antibacterial agent against M. tuberculosis, E. coli, B. subtilis, and some Shigella and Salmonella bacterial species at concentrations as low as the micromolar range. Consequently, synthetic chemists have garnered considerable interest from their specific structure-activity profile, distinctive chemical compositions, and distinct biological profile. This review comprehensively describes cutting-edge synthetic methodologies for synthesizing furanomycin and its analogs reported to date. Therefore, this review will offer an initial perspective on synthesizing furanomycin and its customized compounds.
Keywords:
Furanomycin
; non-proteinogenic amino acids
; antibiotics natural products
; total synthesis
1. Introduction
Amino acids are the essential building blocks of proteins, the most abundant biological macromolecules in all cell parts. [1,2,3] They are crucial in converting genetic information into essential biological products. These include enzymes, hormones, antibodies, muscle fibers, and other vital biological materials.[4,5,6] Besides the 20 proteinogenic amino acids, there are about 500 naturally occurring non-proteinogenic amino acids, many of which are building blocks for small bioactive peptide structures. The non-proteinogenic amino acids possess unique biological functional properties that are not found in the 20 standard amino acid building blocks. [7,8] The existence of natural peptides containing non-proteinogenic amino acids explicitly highlights the biological processes involved in their integration into peptides and their significant potential for use in protein engineering and medicinal chemistry.
The natural metabolites and peptides that contain non-proteinogenic amino acid residues highlight the process used to select and include these non-proteinogenic building blocks into nonribosomal peptides. Additionally, artificial amino acids have been created to be recognized by tRNAs and aminoacyl-tRNA synthetases, enabling their integration into proteins within living cells. This advancement holds potential for utilizing internally produced non-proteinogenic amino acids in protein engineering. [9,10] This compels researchers to synthesize alloproteins containing novel nonprotein amino acid residues, significantly expanding the potential for designing specific proteins with unique functions.
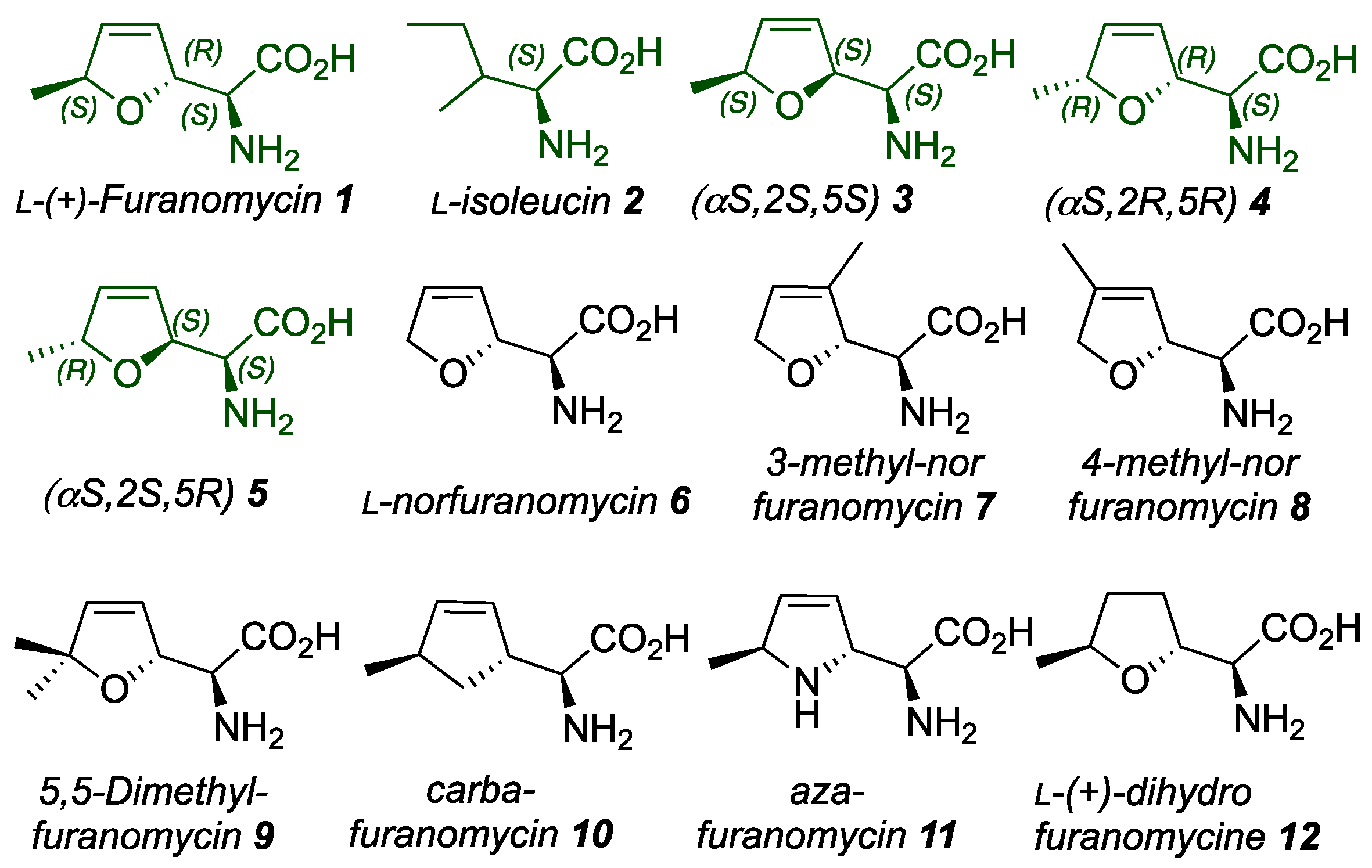
The infrequent and the most petite antibacterial natural product L-(+)-furanomycin 1 (Figure 1), with a molecular weight of 157 g/mol, was first reported by Katagiri and co-workers in 1967 (Figure 1).[11] After that, in 2013, Banowetz et al. identified the ninhydrin-active compound L-furanomycin 1 being produced and released with selective antimicrobial properties by the plant root-associated γ–proteobacterium Pseudomonas fluorescens SBW25.[12] Very recently, Fariq et al. documented the production of furanomycin 1 from halophilic bacteria Halomonas elongata for the first time.[13] The absolute configuration of the isolated product has been conclusively established as αS,2R,5S by X-ray structural analysis of the N-acetyl derivative of 1 [14] and chemical methods. [15] The biological profile of (+)-L-furanomycin 1 exhibited antibacterial activity against numerous bacteria at low concentrations in the micromolar range (MIC 1 – 5 μg/mL).[11,16,17,18] Even though the chemical structure of (+)-L-furanomycin 1 differs from L-isoleucine 2 (Figure 1), it surprisingly preferred isoleucyl aminoacyl-tRNA synthetase.[17] In the protein translation processes, the conformation of enzyme-bound amino acids determines the affinity of an amino acid to aminoacyl-tRNA synthetase. These structural observations of compound 1 have been further supported by NMR studies that showed that the conformation of the enzyme-bound furanomycin is similar to the isoleucine.[17,19,20] Furthermore, standard amino acids featuring a methyl group, such as leucine (Leu) and valine (Val) substrates, are effectively recognized and differentiated from isoleucine by the enzymatic machinery. This essential function of the methyl group underscores the importance of understanding its influence on substrate recognition. It can be inferred that the C5-methyl group in compound 1 likely occupies the same binding pocket of the enzyme IleRS. While furanomycin 1 is recognized and activated by the enzyme IleRS in place of L-isoleucine 2, its conformation does not precisely match that of isoleucine 2. Thus, the research indicated that the larger side chain of furanomycin may interfere with the natural conformations and functions of essential proteins in cellular physiology. This alteration in protein structure accounts for its antibacterial effect in bacteria. In this scenario, although L-furanomycin 1 inhibits isoleucyl-tRNA formation in E. coli, other aminoacyl-tRNAs remain unaffected.[17,20,21]
The study aimed to understand the biological significance of compound 1 and its derivatives through structural analysis. As illustrated in Figure 1, the basic set of structures of furanomycin used to explore the SAR was expanded to include diastereomers of furanomycin 1 (3, 4, and 5), norfuranomycin 6, 3-methyl-norfuranomycin 7, 4-methyl-norfuranomycin 8, furanomycin derivative 9, which contains two methyl groups, carba-analogs 10, aza-furanomycin 11, and dihydrofuranomycin 12. The structural prerequisites of isoleucyl aminoacyl-tRNA synthetase for furanomycin-like substrates were quite stringent. A very constricted SAR study was concluded, and all synthetic isomers and derivatives of the natural antibiotic 1 demonstrated different MICs against a panel of selected Gram-positive and Gram-negative wild-type pathogens, including S. aureus and E. coli. [21,22] Additionally, biological studies showed that changing the position of the methyl group in the central furan ring influenced the antibacterial properties of furanomycin 1, including nor-furanomycin 6, 3-methyl-norfuranomycin 7, and 4-methyl-norfuranomycin 8, against certain Gram-negative bacteria. (Figure 1). In this context, many other L-(+)-furanomycin congeners have been chemically synthesized in enantiomerically pure or racemic forms and scrutinized biologically. For example, L-(+)-dihydrofuranomycin 12 exhibited borderline MIC (32-64 μg/mL) against S. aureus.
In contrast, the chiral carba-analogue 10 exhibited weak antibacterial activity (4 μg/mL) against an efflux pump-deficient E. coli and its antibacterial properties in vitro and in vivo. Likewise, L-(+)-furanomycin displays drug-like features, such as polarity, sufficient aqueous solubility, and moderate structural complexity, which render it an attractive synthetic target to medicinal chemists. To this end, several synthetic approaches targeting 1 have been developed, consisting of up to 20 steps. Some syntheses start from chiral substrates such as carbohydrates, amino acids, etc., whereas some approaches use achiral starting materials like furans, cyclopentadiene, etc. The approaches starting from amino acids are primarily attractive because they involve only six to seven steps, making them short and flexible enough to access furanomycin 1 and their analogs rapidly. Therefore, we briefly discuss in detail the synthetic studies of the synthesis of furanomycin 1 and its functionalized derivatives. The present review offers comprehensive information and motivation for developing new antibacterial targets. Investigating the intricate mechanisms of newly developed antibacterial agents would also be beneficial in identifying the best antibacterial candidates effectively.
2. Biosynthesis of Furanomycin:
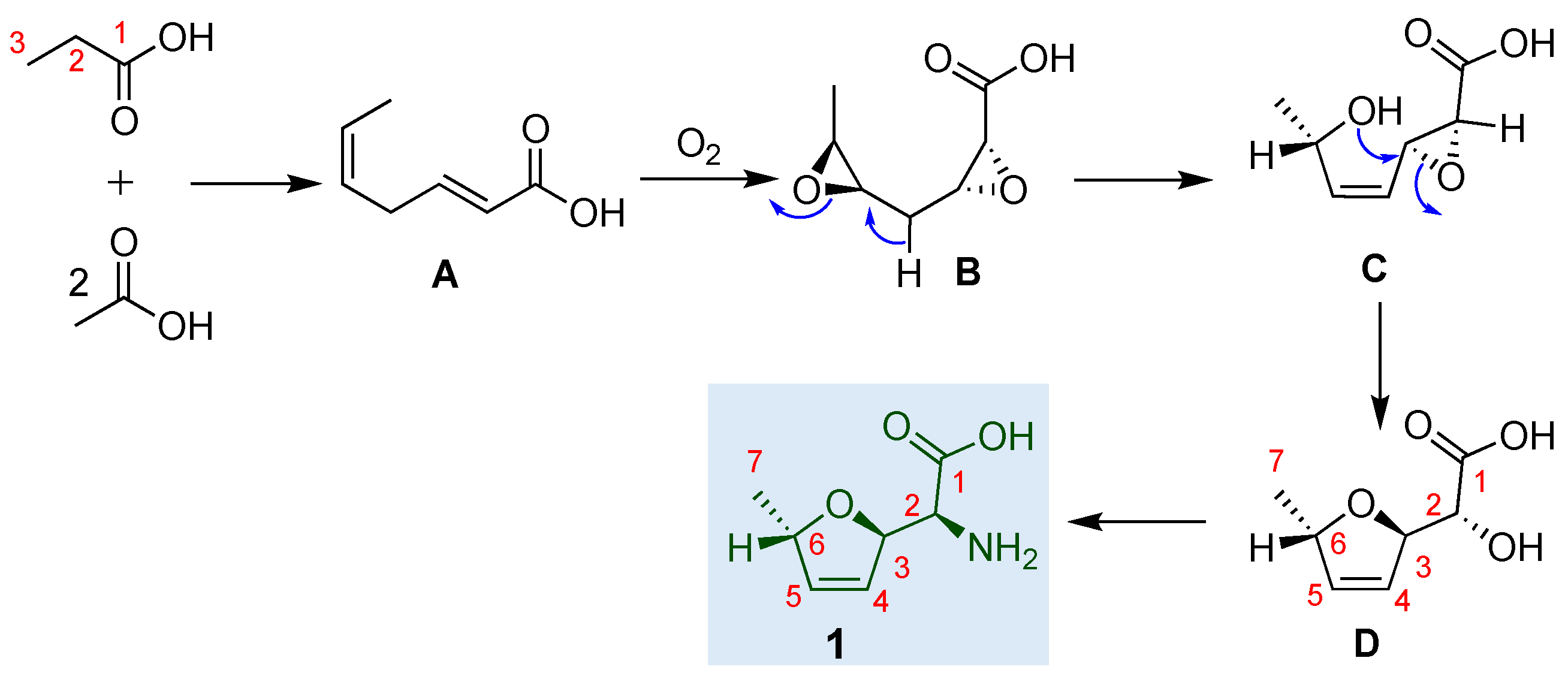
Parry et al. proposed the biosynthetic pathway for furanomycin 1 (Scheme 1). [23,24] In biogenesis, the carbon skeleton of the inquisitive amino acid furanomycin D is formed via a polyketide pathway starting from one propionate and two acetate units. The carbon atom 6 in D could come from C-2 of propionate. During the oxidation at C-2, one of the propionate protons is lost from this site without forming a lactate intermediate. Later, the study strongly supported this observation by confirming that the 2-pro-R proton from the propionate unit. Since the configuration at C-6 of D is S, the introduction of the ether oxygen at C-2 of propionate occurs with an overall inversion of configuration. Overall, analogous to the polyether antibiotics, the substrate A could undergo epoxidation to form the epoxide intermediate B, the 5,6-epoxide ring could open, and the resulting hydroxyl group could attack the adjacent epoxide, producing the cyclic ether D. It can then easily be converted into furanomycin 1 (Scheme 1). [24] Labeling experiments with [l-13C, 18O2] acetate and 18O2 gas proved that the ether oxygen originates from aerial oxygen, not acetate.
3. Chemical Syntheses of L-(+)-Furanomycin and Its Analogues
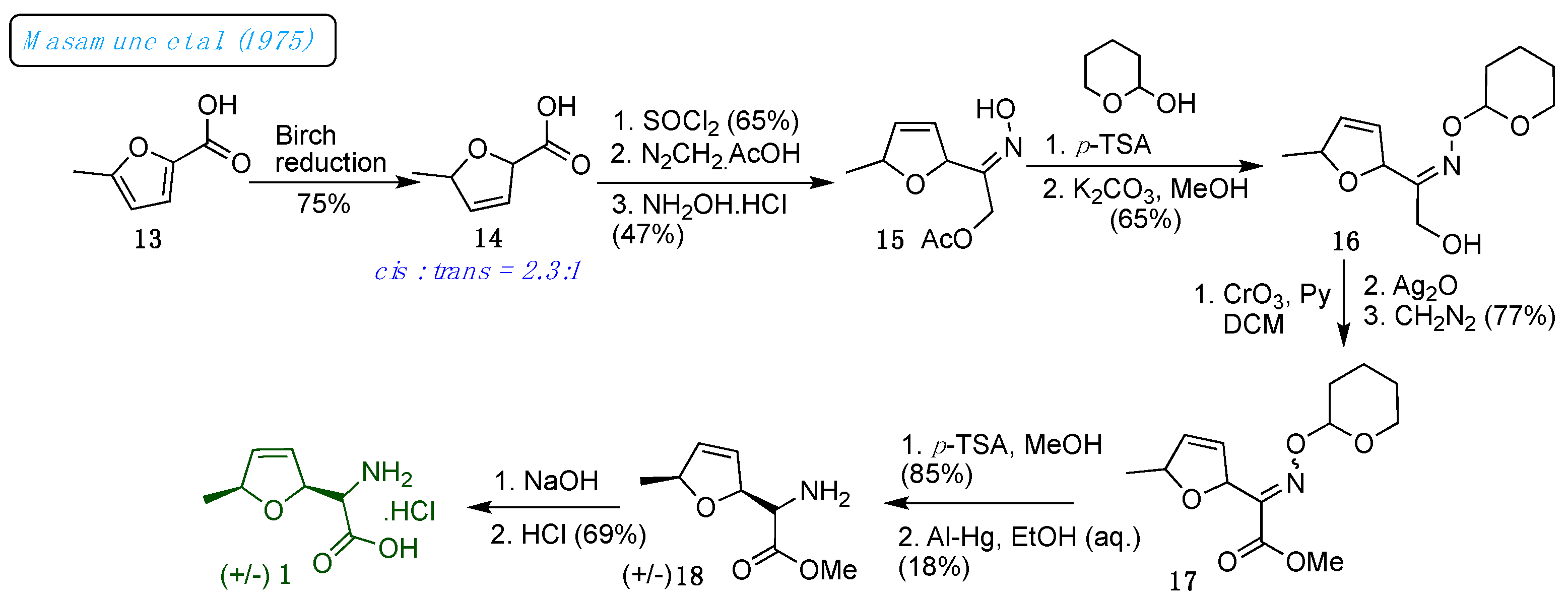
Furanomycin 1 possesses a unique trans-2,5-substituted, five-membered unsaturated cyclic ring system with an S-configured amino acid side chain, setting them apart as crucial components in biological processes. Thus, the synthetic challenge is assembling trans-2,5-dihydrofuran ring and (S)-amino carboxylic acid units. Over the past few years, many approaches to synthesizing L-(+)-furanomycin have been developed. In this direction, At first, in 1975, Masamune and Ono reported the total synthesis of the racemic (±)-furanomycin 1 (Scheme 2) in 16 steps, with an overall yield of <0.02%.[19]
The 5-methyl-2-furoic acid 13 was utilized to access the dl-furanomycin 1. The partial Birch reduction of 13 produced a crystalline mixture of diastereoisomeric 5-methyl-2,5-dihydrofuroic acids 14, from which a trans-isomer was isolated in 23% yield as a minor isomer. The remaining acid mixture contained a major cis-isomer in 53% yield. The amino and acid groups in the structure were installed in the consecutive steps. Thus, acid 14 was converted into keto-acetates to oxime acetates 15 in a three-step reaction sequence. Next, the oxime protection introduced the acid group, followed by acetate deprotection, which produced alcohol 16. Oxidation of 16 employing a modified Colline oxidation protocol followed by silver oxide treatment furnished acids; subsequently, acids were esterified with diazomethane to esters 17. The oxime acetates 17 on a chemo-selective reduction of oximino-esters with aluminum amalgam in aqueous ethanol resulted in diastereoisomeric amino esters 18 in 18% yield. Diastereomeric amino esters 18 were converted into hydrochloride salts; the data obtained was identical to isolated furanomycin 1.
Until 1979, the configurations at the asymmetric centers of (+)-furanomycin were not confirmed by X-ray studies or stereospecific synthesis. Based on a combination of spectroscopic and chemical degradation techniques, the first structural assignment of asymmetric centers of furanomycin 1 was established into (αR,2R,5R) configuration.[11] Due to structural ambiguity, Joullie et al. first developed the chirality transfer strategy to synthesize the furanomycin and its configurational isomers.[15] As shown in Scheme 3 (A), the synthesis proceeded with nine steps from the D-glucose. Thus, furanose derivative 19 was treated with excess sodium phenylselenide to afford diselenide 20. Reductive removal of phenylseleno groups with the Raney nickel produced alcohol 21. Tosylation of 21, followed by base-catalyzed elimination, afforded 2,5-dihydrofuran 22. The acid hydrolysis of 22 provided the aldehyde intermediate, which, upon being subjected to Ugi four-component reaction with (R)-(+)-α-methyl benzylamine, benzoic acid, and tert-butyl isocyanide afforded the diastereomeric adducts 23a and 23b (1:1). Debenzylation of 23a and 23b in acidic condition (95% formic acid) followed by hydrolysis with 6N HCl gave (α-1R,2R,5S)-furanomycin ent-5 and 5S,2R-α−S-furanomycin 1, respectively. In the synthetic study, the absolute and relative stereochemistry of 1 was accurately established for the first time.
Later, as shown in Scheme 3 (B), the same group confirmed the configuration of naturally occurring antibiotic furanomycin as (αS,2R,5S) by synthesizing a series of four 2,5-cis stereoisomers of α-amino-2,5-dihydro-5-methylfuranacetic acid and two trans isomers from the D-glucose. [25] Five years later, they reported the synthesis of two new trans-isomers of furanomycin from D-glucosamine hydrochloride (Scheme 4). In this strategy, the compound 30 was efficiently prepared from D-glucosamine and stereo-selectively transformed into enantiomer (α−R,2S,5R)-furanomycin and its isomer with the opposite configuration at the amino acid functionality (α−S,2S,5R)-furanomycin 5 in good yields. [26]
1982, Parker et al. developed a synthetic strategy for furanomycin 1 and its diastereoisomer from D-ribose (Scheme 5).[27] A known sequence prepared the 1,3-diphenylimidazolidine derivative 33.[28] It was further transformed into appropriately protected seven-carbon 3,6-anhydrous compounds by benzylating the primary alcohol 33. Deprotection of the aldehyde followed Strecker-type side chain synthesis and subsequent acetylation of the resulting epimeric 2-hydroxyl groups provided 34a and 34, benzyl deprotection and mesylation followed by iodide replacement on 34a/b afforded iodo derivatives 35a/b. The catalytic hydrogenolysis of 35a/b with Pd/C afforded a 36a/b. The amine group was mounted on the ring's core structure following the deprotection of the 2-hydroxyl group and subsequent mesylation and azidation reactions. The 2,3-dihydrofurane ring in 37a/b was created by methanolysis of the amide, removal of the isopropylidene to form a cyclic thiono-carbonate group, and then reductive decomposition of the cyclic thiono-carbonate group under Corey-Winter reaction conditions. This process also resulted in the reduction of azide and the formation of amine intermediates. Finally, hydrolysis of the amide gave ent-3 and 4 diastereomers of antibiotic 1.
As shown in Scheme 6, Kang et al. reported a highly enantioselective total synthesis of (+)-furanomycin 1 utilizing the mercury cation-mediated stereoselective cyclizations of γ-hydroxy alkene 40 as a critical step.[29] The sequential mercury-promoted cyclization postulated the required trans-2,5-disubstituted tetrahydrofuran and the (S)-amino acid side unit from homoallylic compounds 40. The silyl ether 38 was prepared from dimethyl L-tartrate by a known literature method and followed by PPh3 mediated iodination, which afforded iodide 39, transformed into an essential diene diol 40 via substitution of the vinyl group followed by hydrolysis of acetonide. The formation of trans- and cis-2,5-disubstituted tetrahydrofurans 41a/b was achieved with Hg(OCOCF3)2 and K2CO3 followed by triethyl borane/LiBH4 mediated demercuration. The tetrahydrofurans 41a/b transformed to the corresponding trichloroacetimidate separated as 42a and 42b. The Hg(OCOCF3)2 mediated intramolecular amination produced the desired mercuric bromide in 95% yield, sequentially converted to the dihydro-1,3-oxazine 43 via demercuration followed by protection. The bicyclic compound 43 was transformed into dihydrofurane intermediate 44 by acid-catalyzed hydrolysis and iodination reactions. Elimination in 44 established an endocyclic double bond, and Zn promoted reductive N-O bond cleavage that smoothly afforded compound 45. Oxidation of primary hydroxyl group in Swern-oxidation condition, successive Pinnick oxidation of the aldehyde to acid, and removal of Cbz group provided unmasked 1 (Scheme 6).
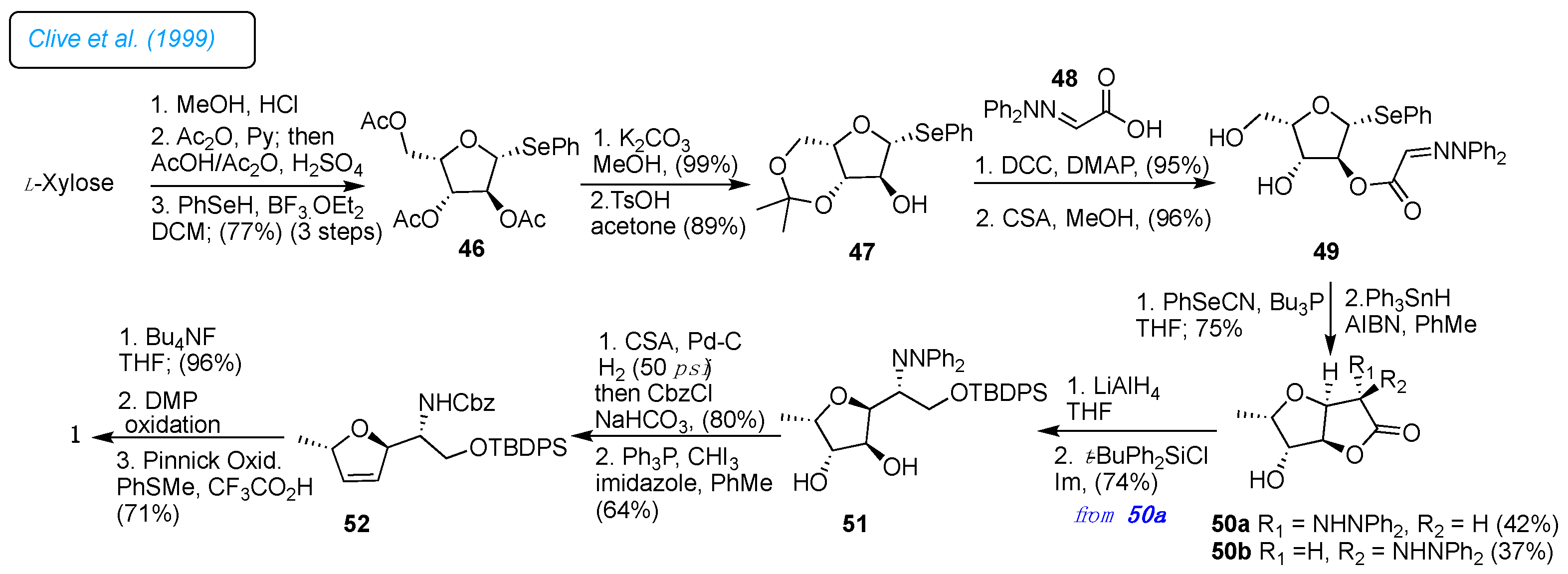
In 1999, Clive and a co-worker developed the chiral pool strategy for synthesizing 1, which involved radical cyclization as a key step.[30] The synthesis started from L-xylose, which was converted into its methyl glycoside 46 via a three-step sequence with an overall yield of 77% without isolation of the intermediates (Scheme 7). Compound 46 is used for mild hydrolysis, followed by acetonide protection provided by ketal 47. Next, DCC-mediated coupling 47 with (2,2-diphenylhydrazono)acetic acid 48 gave the hydrazone ester 49 in 95% yield. The critical one-step radical cyclization and deoxygenation of the C5-OH group of 49 were accomplished with AIBN and Ph3SnH reagent, which provided the isomeric mixture of chromatographically separable hydrazino lactones 50a (43%) and 50b (37%) epimeric at C2.
Further, compound 50a was converted into compound 51 by reducing the ester and protecting the primary hydroxyl group. The dihydrofuran ring 52 was created by removing the remaining two hydroxyl groups with PPh3 and CHI3. Extensive bis-dehydration was also noted in the corresponding furan system. Under optimum conditions, 51 was obtained in 64% yield. Finally, desilyation and the oxidation of the primary hydroxyl group to acid were achieved. In the oxidation steps to avoid the epimerization at the C2 position, the Dess-Martin conditions were used instead of the Swern oxidation, and treatment with TFA in the presence of PhSMe observed free amino group and liberated furanomycin 1 of 98% purity.
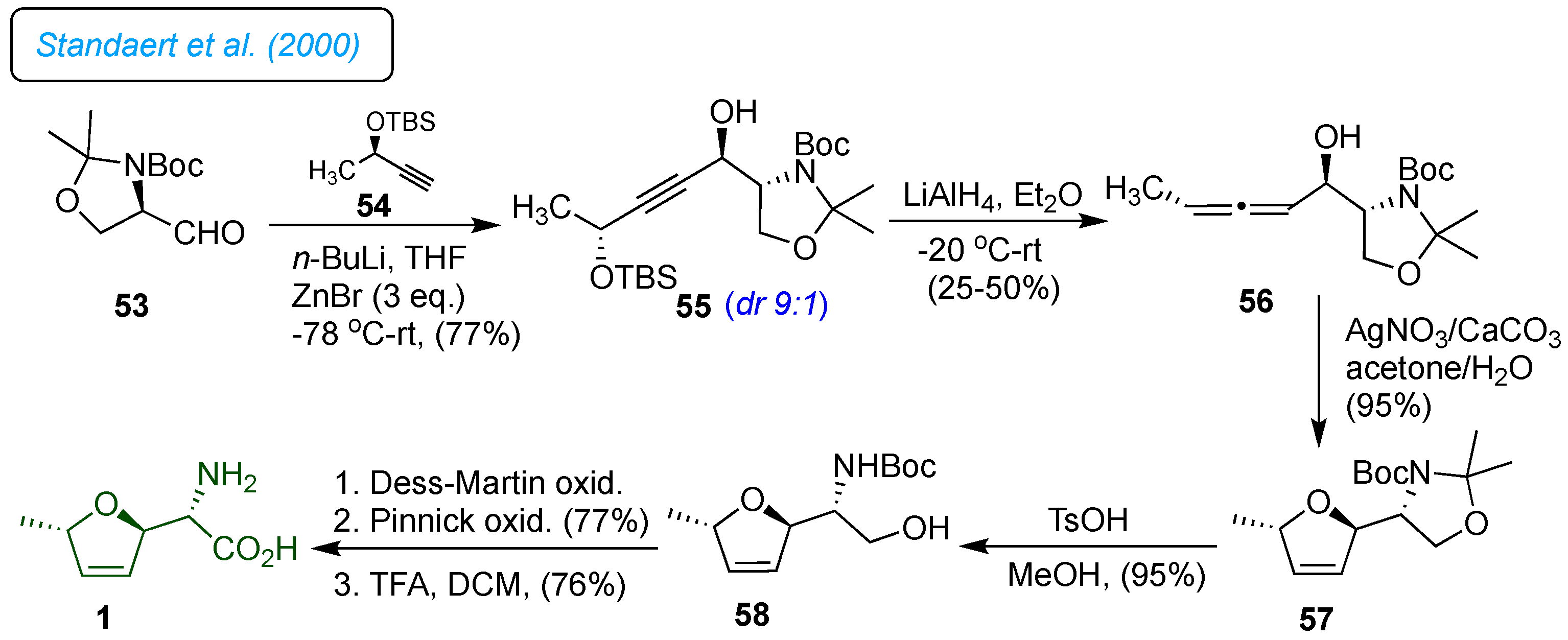
Standaert et al. reported concise and efficient synthesis of L-(+)-furanomycin 1 in seven total steps starting from the (R)-Garner aldehyde 53 (Scheme 8).[31] The first step in the synthesis was the stereoselective addition of acetylide 54 to the (R)-Garner aldehyde 53 to afford a 9:1 mixture of diastereomers 55, which was stereoselectively reduced with LAH to give allenic alcohol 56. This transformation proceeded via trans-hydrometallation followed by anti-elimination of the alkoxide to allenic alcohol. This approach showed the usefulness of constructing a dihydrofuran ring system directly propargyl alcohols, simplifying the synthetic problem with a stereoselective addition of acetylene to L-serine. Next, the trans-2,5 dihydrofuran ring 57 was formed by Ag-assisted cyclization of the allenicalcohol 56 in the dark, affording the dihydrofuran 57 in 97% yield. Base catalyzed selective deprotection of the O, N-acetonide in 57 afforded furanomycinol 58. The sequential oxidation using the Dess-Martin reagent followed by NaClO2, as a procedure reported by Clive for oxidation of N-Cbz furanomycinol [30] and further TFA-mediated deprotection of the amino group yielded the desired (+)-furanomycin 1 in 76% yield.
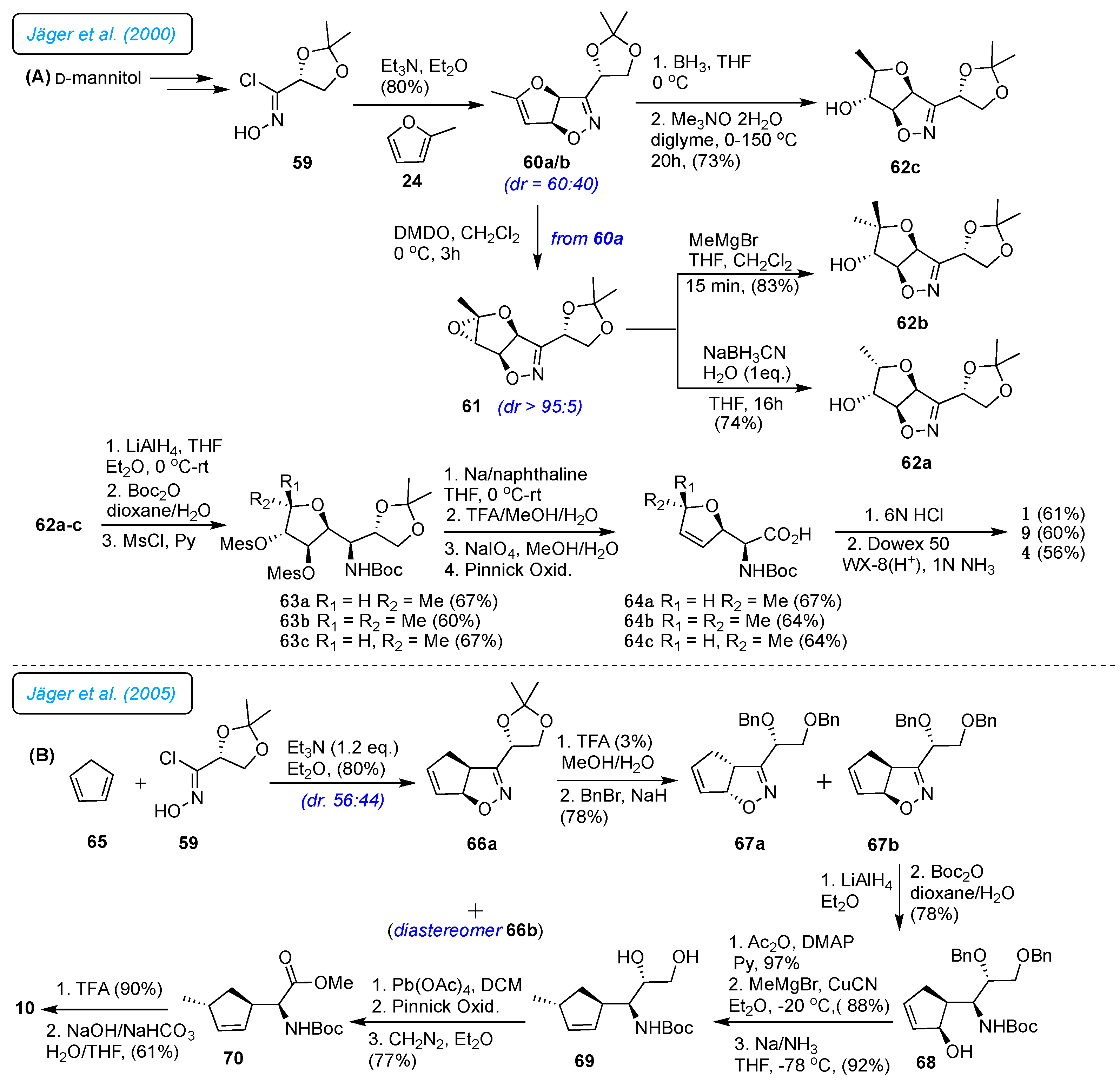
Jäger et al. presented a novel stereoselective 1,3-dipolar cycloaddition of nitrile oxides to furan for the synthesis of L-(+)-furanomycin 1, its C5-epimer analog 4 and 5,5-dimethyl analog furanomycin 9 based on previous work (Scheme 9A).[32] The new synthesis method has some unique features compared to previous ones. It can determine the relative arrangement of the three stereocenters in intermediate tetrahydrofuran-3,4-diols. It used a chiral glycine equivalent called glyceronitrile oxide as a starting material, providing a direct route to four of the eight stereoisomers of 1. Different nucleophiles (such as Grignard reagents) can produce structural analogs 1 from the main epoxide 61. As outlined in Scheme 9A, diisopropylidene mannitol was transformed to known hydroxamic acid chloride 59 in three steps.[33] The 1,3-dipolar cycloaddition of 59 with 2-methylfuran 24 in TEA gave a mixture of the diastereomeric furoisoxazolines 60a/b (dr 60:40). The major dihydrofuran 60a exposed to epoxidation with DMDO afforded stable epoxide 61 with high selectivity (dr >95:5). The endo-stereoselective hydride transfer with sodium cyanoborohydride on 61, as well as regioselective epoxide opening with MeMgBr, led to alcohols 62a and 62b, respectively. Subsequently, the hydroboration-oxidation reaction of the compound 60a led to high exo-selectivity of the alcohol 62c, which is C5-epimer of 62a. The 3,4 dihydrofuran ring 63a-c was effortlessly obtained from isoxazolines 62a-c by successive LAH reduction followed by a twofold mesylation and demesylation procedure. Finally, in 63a-c on acetonide hydrolysis, subsequent diol cleavage to an aldehyde, and further oxidation to an acid using Pinnick oxidation conditions provided 64a-c. Cleavage of the carbamate in 64a-c provided 1, 5,5-dimethyl analog 9 and 5-epimer 4 (Scheme 9A).
The same group in 2005 reported the synthesis of L-carba-furanomycin 10. As shown in Scheme 9B, the 1,3-dipolar cycloaddition of cyclopentadiene 65 and the chiral nitrile oxide 59 gave a mixture of 66a/b in 80% yield (dr 56:44).[34] Worthy of note, the compound cyclopentadiene 65 serves as the cyclopentene core of carba-furanomycin 10, and the chiral cyclopentaisoxazoline 67 contains three stereogenic centers that provide stereogenic carba-furanomycin 10. Thus, the mix of 66a/b was treated with dilute TFA in aqueous methanol to provide diols, which were benzylated to give 67a/b in 78% yield. The stereoselective reduction of the cyclopentaisoxazoline 67a was followed by amino alcohol protection to the amino group with Boc2O in dioxane and water (3:1), providing the significant product cyclopentenol 68. The acetylation was trailed by introducing the methyl group and subsequent debenzylation (Na, liq.NH3) to give diol 69. The intermediate aldehyde was prepared by cleavage of diol 29 with Pb(OAc)4, which was directly oxidized to the carboxylic acid in Pinnick oxidation, followed by esterification with diazomethane providing methyl ester 70. Finally, the NH-Boc deprotection in 70 with 90% TFA and hydrolysis of ester gave L-carba-furanomycin 10.
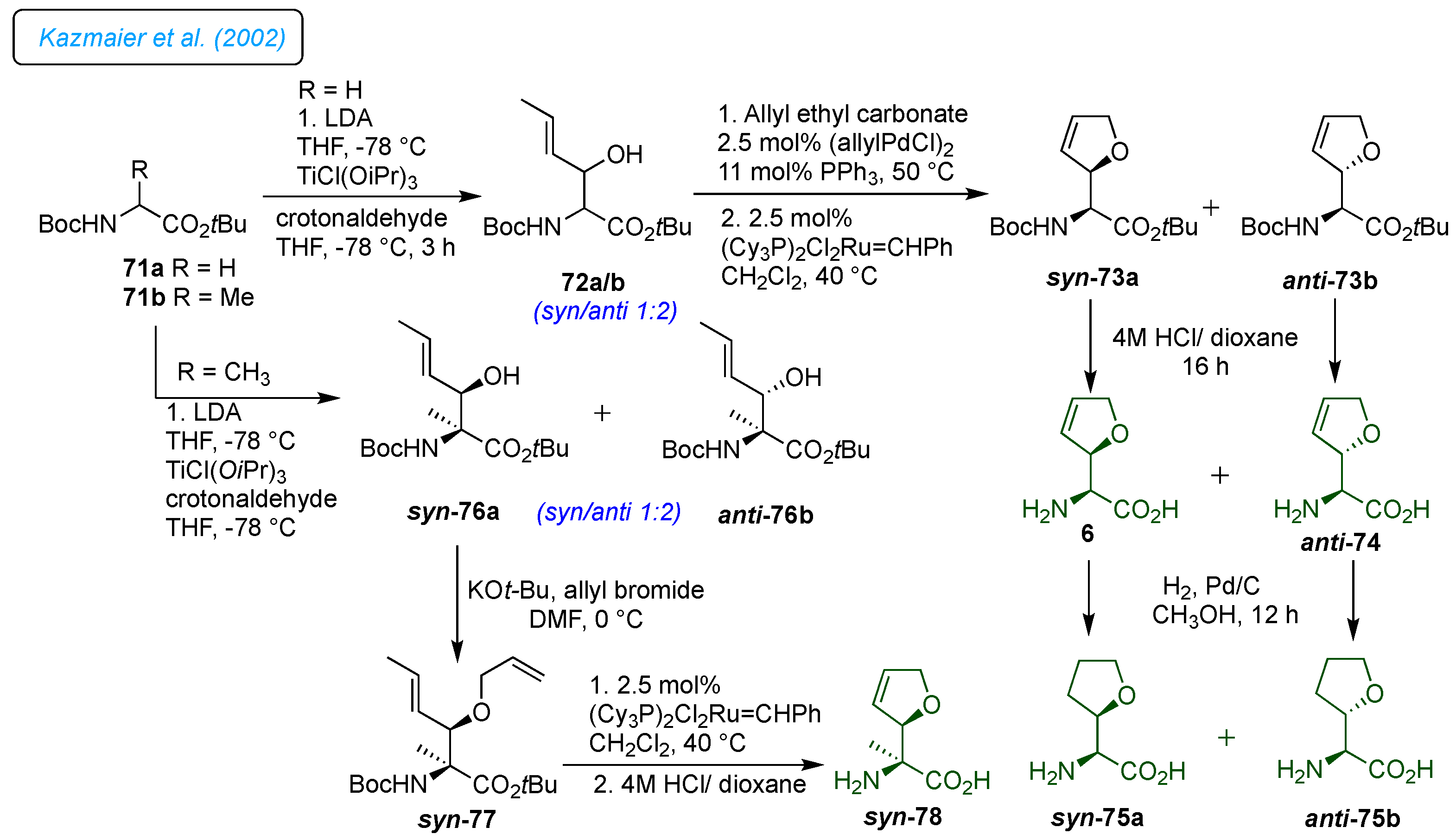
Kazmaier et al. developed straightforward protocols for the 3- and 4-methylated derivatives, corresponding to hydrogenated analogs of furanomycin (Scheme 10) and carba-furanomycin (Scheme 11). [22] They wanted to determine the structural parameters required for biological activity and identify those that can be altered or removed. Their goal was to create simple procedures for producing derivatives without the 5-methyl group (norfuranomycin 6), also found to be biologically active. Thus, the glycine esters 71a/b were used to synthesize norfuranomycin 6 and its α-methylated derivative 78. Primarily, the synthesis of norfuranomycin 6 started with a titanium-assisted aldol reaction of crotonaldehyde with glycine ester 71a, which resulted in a preferentially anti-product 72a/b (syn/anti 1:2) in good yields (Scheme 10). The palladium-catalyzed O-allylation followed by Grubbs cyclization on 72a/b yielded a separable mixture of diastereomers syn-73a and anti-73b. After removing the Boc groups, they could obtain diastereomerically pure norfuranomycin 6 and anti-74. Consequently, independent catalytic hydrogenation of 73a/b provided the saturated derivatives syn-75a and anti-75b. On the other hand, the synthesis of α-methylated derivatives syn-78 was achieved following the same synthetic protocol from 71b.
The 3-methylated derivatives 7 and anti-81 and 4-methylated derivatives 8 and anti-84 were synthesized from methacrolein and acrolein by a similar protocol described in Scheme 10 (Scheme 11).
For synthesizing syn-89a and anti-89b, the precursor ester 85 was obtained by coupling cyclopentenol with Boc-glycine (Scheme 12). [35] The protected cyclopentenyl glycine diastereomers (syn/anti 8:2) 86a/b were obtained through a base-promoted Claisen rearrangement of ester 85 with zinc chloride and iodolactonation of a diastereomeric mixture of 86 provided separable syn-87a and anti-87b in good yields. Further, cleavage of iodolactones and subsequent deprotection afforded the desired diastereomerically pure cyclopentenyl glycine syn-89a and anti-89b. In addition, fully saturated cyclopentane glycine derivative 90 was directly obtained from 86 after hydrogenation and deprotection.
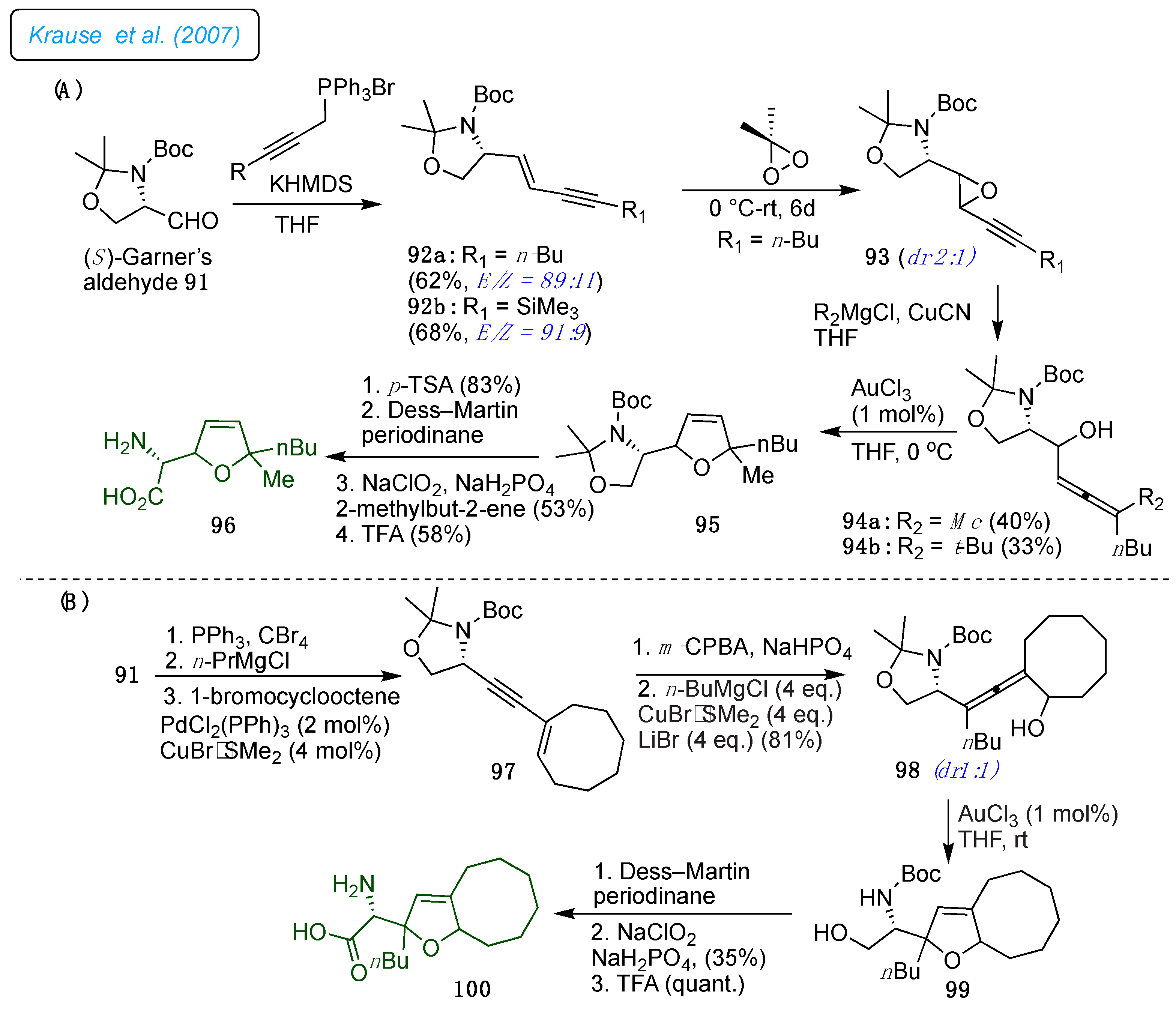
In 2007, Krause and co-workers developed a strategy to prepare novel furanomycin analogs 96 and 100 employing the gold-catalyzed cycloisomerization of α-of hydroxyallenes 94a/b as the foremost step (Scheme 13 A and B).[36] Notably, the present gold-catalyzed strategy is more efficient for cyclizing highly functionalized substrates than the traditional approach, where stoichiometric amounts of silver salts are used. As shown in Scheme 13, the reactive hydroxy allenes 94a/b were favorably synthesized by the copper-mediated SN2, substituting propargyl oxiranes 93, obtained from Garner's aldehyde (S)-91 via conjugated enynes 92a/b Wittig olefination with the propargyl phosphonium salts. Further, Au(III)-catalyzed cycloisomerization of the α-hydroxyallene 94a/b retained the formation of the 2,5-dihydrofuran by 95 in good yield. Here, the sterically challenging allene reacted sluggishly and gave the dihydrofuran a lower yield, accompanied by several side products. The low reactivity of alkene 92 led to the utilization of the electron-rich alkyne (Scheme 13B) for the epoxidation reaction, producing oxirane 97 (1:1 mixture of diastereomers) with a 79% yield. Later, a stereoselective hydroxyl allene scaffold 98 was generated and subjected to a cycloisomerization reaction with 1 mol% of AuCl3, which ended with 89% of bicyclic dihydrofuran 99 with concomitant acetal cleavage. Next, the oxidation of the primary hydroxyl in the Pinnick oxidation condition gave a better carboxylic acid yield (35% over 2 steps). Finally, acidic cleavage of the Boc group 99 afforded the bicyclic furanomycin derivative 100 as a 1:1 mixture of diastereomers.
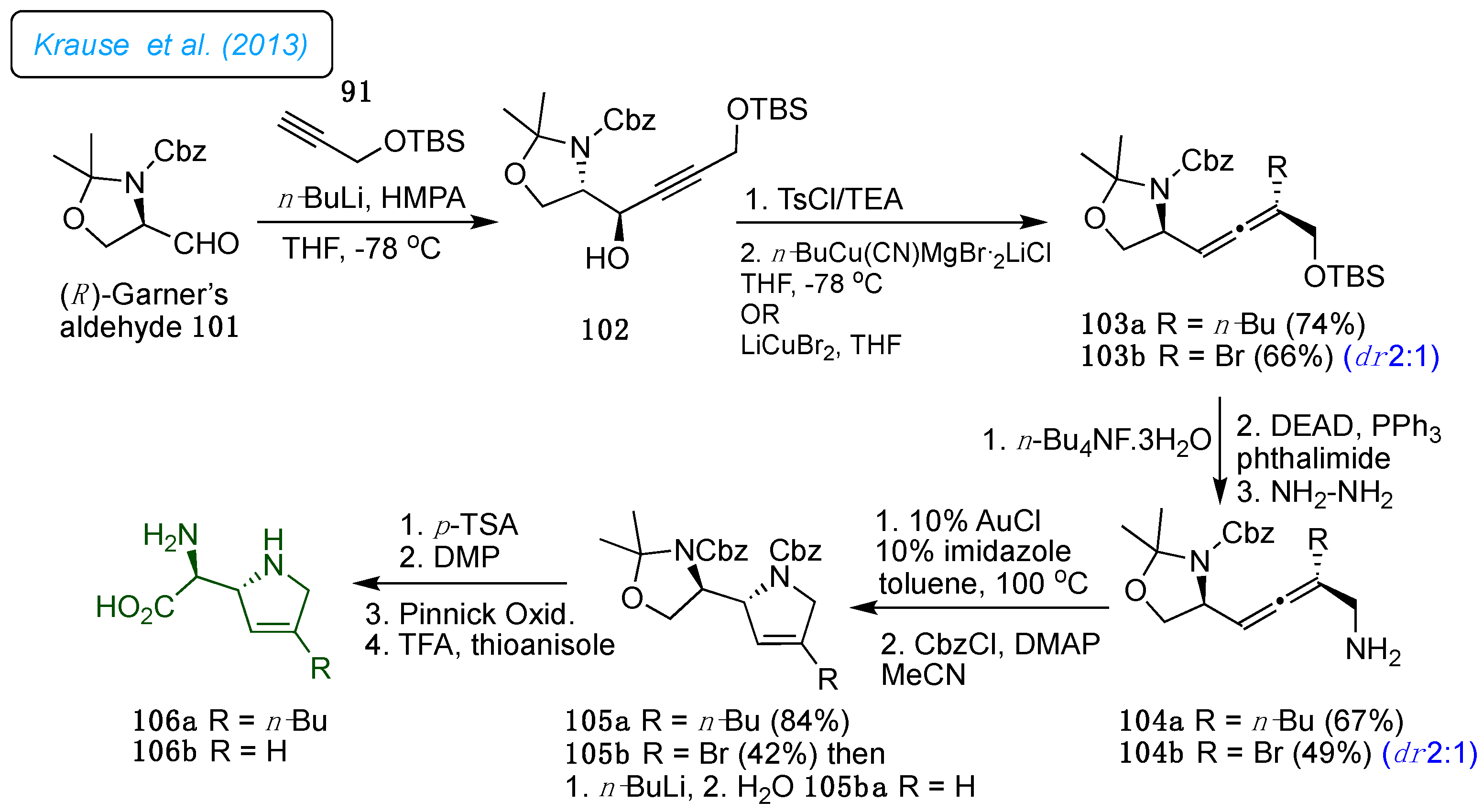
Later, in 2013, the same group expanded the gold-catalyzed cycloisomerization of α-hydroxyallene strategy towards the first synthesis of the azafuranomycin analogs (αS, 2R)-(2,5-dihydro-1H-pyrrol-2-yl)glycine 106a/b in 13 linear steps with an overall yield of 2.4% starting from the Cbz-protected(R)-Garner aldehyde 101 (Scheme 14). [37] The vital step, gold-catalyzed cycloisomerization of α-amino allenes 104a/b to dihydropyrroles 105a/b, gave a good yield. As described in Scheme 13, dihydropyrroles 105a/b were converted into respective aza-furanomycin 106a/b with a higher yield.
Chattopadhyay et al. have demonstrated the synthesis of optically pure nor-furanomycin from Garner's aldehyde 53 (Scheme 15).[38] Thus, the alcohols 107a and 107b obtained by Grignard reaction from Garner's aldehyde 53 yielded the diastereomers in a 3:7 ratio, unfavorable for the natural furanomycin 1. The treatment of allyl bromide with syn- and anti-diastereomers provided a corresponding separable mixture of allyl ethers. Distinctly, the critical ring-closing metathesis reaction of related allyl ethers worked well, resulting in the dihydrofuran derivatives 108a and 108b, respectively, in good yields. The oxazolidine unit in both isomers was removed to N-Boc-protected amino alcohols. Jones' oxidation of alcohols was carried out under biphasic conditions, which was afforded N-Boc-protected norfuranomycin derivatives 109a and 109b. This methodology further extended for the synthesis of dihydro furanylglycine, furanyl glycine, furanyl alanine, and homo-furanyl alanine derivatives from enantiomerically pure aldehydes derived from the natural amino acids such as serine, aspartic acid, and glutamic acid.
On a similar line, in 2008, the same group demonstrated the synthesis of N-protected derivatives of L-(+)-furanomycin 1 and its 5’-epi-furanomycin 4 and 4-methyl norfuranomycin 112 from D-serine as a chiral pool through a critical ring-closing metathesis (RCM).[39] The linear concise eight-step synthesis of the N-Boc L-(+)-furanomycin 115a was achieved in an overall yield of 9% from Garner's aldehyde 53. The simple and short methodology was developed from a common intermediate for the syntheses of the N-protected derivatives of 5’-epi-furanomycin 115b and 4-methyl norfuranomycin 112 (Scheme 15). Adding lithium trimethylsilyl acetylide to Garner's aldehyde 53 made the 110 a separable mixture in syn: anti 9:1. After deprotection of syn-propargyl alcohol 110 and partial hydrogenation of the acetylene group afforded a common intermediate, allyl alcohol 111. Ensuing alkylating of the 111 with 3-chloro-1-butene with Ag2O afforded a mixture of dienes 113 in moderate yield. Then, employing RCM protocol on mixture 113 delivered the desired oxazolidine rings as an inseparable diastereomeric mixture (1:1.2) 114. Further, deprotection and oxidation of corresponding alcohols to acids afforded 115a and 115b in a combined 74% yields. The HPLC separation provided the N-Boc derivative of L-(+)-furanomycin 115a as a major isomer, reflecting that the diastereoselectivity in the RCM reaction favored the trans-isomer. Correspondingly, allyl alcohol 111 was also utilized to synthesize the N-Boc derivative of 4-methyl norfuranomycin 112—alkylation with methallyl bromide, followed by RCM, sequential deprotection, and oxidation procedures.
Vedejs et al. developed the palladium-catalyzed coupling of an aziridinylzinc chloride intermediate with alkenyl and aryl halides with the retention of aziridine stereochemistry (Scheme 16).[40] The synthesis of protected 5,5-dimethyl furanomycin 122 was achieved to demonstrate the protocol's potential. The stannylated aziridine 116 and iodoacrylate 117 afforded coupled product 118 under standard conditions of the Negeshi coupling. The Grignard addition on 118 afforded the tertiary alcohol 119, followed by acid-catalyzed aziridine cleavage, providing dihydrofuran 120. The deprotection of the trityl group, subsequent protection of amine as Boc derivative, and benzyl deprotection under Na/NH3 reduction furnished primary alcohol 121. The primary alcohol 121 was treated with Dess-Martin periodinane, followed by Pinnick oxidation, which produced protected 5,5-dimethyl furanomycin 122.
Koskinen et al. utilized Garner's aldehyde as a chiral precursor for synthesizing Boc-protected epi-norfuranomycin 109a and 109b isomers.[41] The alcohols 124a and 124b were prepared by coupling 91 with vinyl iodide 123 (Scheme 18). Deprotection of the TBS ethers separately with tetrabutylammonium fluoride in THF provided the diols 125a and 125b. Then, In the critical step, both diols were subjected independently to the cyclization reaction through the in-situ formation of sulfonate 126a/b, which led to the 2,5-dihydrofuran 127a and 127b, respectively. In the next step, the N, O-acetal was removed with FeCl3 adsorbed on silica in CHCl3 to provide primary alcohols 128a and 128b in excellent yields. Finally, oxidation of amino alcohols with TEMPO/BAIB and further oxidation to acids gave target molecules 109a and 109b.
Carbery et al. utilized the Ireland-Claisen [3,3]-sigmatropic rearrangement of an allylic glycinate 135 bearing a remote chiral enol ether to synthesize natural antibiotic 1 (Scheme 19).[42] The synthesis started from the oxy-Michael reaction of chiral allylic alcohol 132 with methyl propiolate 130 catalyzed by DABCO to provide vinylogous carbonate 133. DIBAL-H mediated reduction of the ester and consequent rapid EDCl mediated coupling of 134 with di-Boc-glycine afforded appropriate allylic glycinate 135. The Ireland-Claisen [3,3]-sigmatropic rearrangement of an allylic glycinate 135 bearing a remote chiral enol ether afforded syn-β-alkoxy α-amino ester 136 as two inseparable diastereomers in a ratio of anti, syn/syn, syn = 72:28. Subsequent ring-closing metathesis reaction of 136 with Grubbs Ist catalyst gave the cyclic olefin 137. NOE experiments confirmed the formation of dihydrofuran 137 in a stereo-controlled manner. The mono-Boc deprotection of 137 was achieved with TFA to afford 138. The diastereomeric mixture of 139a/b was achieved through LAH-mediated reduction of ester 138. The formal total synthesis of furanomycin 1 has been reported.
Ha et al. utilized chiral aziridine to synthesize L-(+)-furanomycin 1 and its analogs (Scheme 20).[43] The synthesis started from the stereoselective Pd-catalyzed etherification of 3-hydroxyalkyl aziridine 140, which provided a diastereomeric mixture of 141a/b (dr 39:61). The RCM on 141a/b and chromatography separation gave 142. The identification of the configuration of two diastereomers was confirmed by NOE experiments of corresponding 5′-methyldihydrofuran fused oxazolidin-2-one 143 as a major product. The α-methylbenzyl group of the oxazolidine-2-one 143 was removed by treatment with Na and liq.NH3. Subsequent hydrolysis of the oxazolidine-2-one, followed by Boc-protection, gave N-Boc-amino alcohol. Further, alcohol oxidation provided 144, and N-Boc deprotection provided furanomycin 1. For the synthesis of norfuranomycin 6, the allylic alcohol 140 was protected as allyl ether 145. Following the similar reaction sequences described for the synthesis of furanomycin 1, the synthesis of N-Boc-protected norfuranomycin 109a was accomplished.
Furanomycin 1 is an important amino acid antibiotic that has led to the synthesis of numerous analogs; on the same line, cyclobutane α-amino acids are crucial targets in organic synthesis. In this context, Avenoza et al. established a Michael-Dieckmann process and a stereoselective Michael-aldol methodology for synthesizing cyclobutane amino acid analogs. [44,45] The same strategy was extended for the synthesis of 2-oxabicyclo[3.2.0]heptane as distinctive conformationally restricted dihydronorfuranomycin analogs via stereoselective Michael-aldol process with 2,3-dihydrofuran and 2-acylaminoacrylates derivatives (Scheme 20). [46] The protocol was primarily examined with [2+2] cycloaddition reaction of the 2-methoxy-propene 146 as a donor alkene and methyl 2-acetamidoacrylate 147 as an acceptor alkene in the presence of Lewis acid methyl aluminoxane (MAO) in methylene chloride at 20 °C which led to a substituted single isomer of cyclobutane 148 in 60% yield. The bulky aluminum-derived Lewis acid MAO obtained this exclusive selectivity in the cycloaddition reaction. In support, the 2D NMR experiments determined the stereochemistry of the newly generated structure 148. To extend the scope of the reactivity of alkenes with different cyclic vinyl ether, 2,3-dihydrofuran 149 and 3,4-dihydro-2H-pyran 153 were elaborated under the optimized reaction conditions. Therefore, the methyl 2-acetamidoacrylate 150a reacted with 2,3-dihydrofuran 149 and 3,4-dihydro-2H-pyran 153, resulting in a diastereomeric mixture of 2-oxabicyclo[3.2.0]heptanes core 151a/b (dr 25:75) and 2-oxabicyclo-[4.2.0]octane core 154b as a significant isomer. The same reaction applied to alkene acceptor trifluoroacetamidoacrylate 150b with 2,3-dihydrofuran 153 using MAO enhanced the selectivity and afforded 152a/b (dr 19:81) in 75% yield.
Meanwhile, the addition of 153 made 155b a significant product. Conformational studies of cyclobutane dihydronorfuranomycin analogs 151a and 152b were conducted to understand the interesting dynamic properties in the solution. Both compounds were found to have relatively rigid conformations.
Pilli et al. developed the stereoselective addition of chiral titanium enolates to 5-substituted five-membered oxocarbenium ions, as outlined in Scheme 21.[47] The methodology application was elaborated to synthesize key intermediate 158 in a diastereoselective manner from γ-lactols 157 derived from (S)-glutamic acid. After desilylation of 158a/b with aqueous HF/CH3CN and LiBH4 reduction, it allowed the efficient preparation of the corresponding trans-2,5-disubstituted tetrahydrofuran diols with the recovery of the chiral auxiliary. The bromo derivative trans-158a is a critical intermediate for the preparation of (-)-furanomycin 1, the enantiomeric form of the isoleucine antagonist (+)-furanomycin 1 (Scheme 21). Therefore, employing the protocol developed by Jager et al. could synthesize both isomers of 1.[18]
Bartoli and co-workers reported a new method for synthesizing the (R)-Garner-type aldehyde 161 and adding a vinyl organocerium compound to provide intermediate syn-162a for (+)-furanomycin synthesis.[48] Thus, the hydroxyl group of L-serine was protected with silyl ether (Scheme 22). Reduction of the ester to the corresponding amino alcohol and N-Boc protection of this amino alcohol afforded the desired intermediate 160 in high yield. The cyclohexylidine protection of amino alcohol 160 and cleavage of the silyl ether moiety, followed by Parikh–Doering oxidation of primary alcohol to optically pure stable aldehyde 161. A vinyl cerium reagent was diastereoselectively added to aldehyde 161 to get the carbon framework in a mixture of 162a/b (dr 82:18). Selective deprotection of cyclic ketal of 163a/b served the valuable building blocks 163a/b in the syntheses of naturally occurring and pharmacologically exciting compounds. [16,49]
5. Conclusions and Outlooks:
Medicinal chemistry employs powerful tools such as total synthesis, semi-synthesis, and de novo synthesis to iteratively optimize natural products and create patentable drug molecules with enhanced pharmacokinetic, physicochemical, and toxicological properties. Medicinal chemistry programs can uncover pathways to future therapies like reverse genetics by carefully selecting natural antibiotic lead structures and valid targets. In silico modeling of the binding of these privileged structures to their targets enables the conception of less complex molecules with improved properties, which can serve as scaffolds for medicinal and combinatorial chemistry. Medicinal and combinatorial organic chemists have devised numerous effective methods to produce innumerable novel and diverse molecules for human health care. The chiral pool, primarily α-amino acids, remains crucial for targeting molecules in chiral synthesis. The efforts outlined in this review on synthesizing natural products should further fortify and accelerate translational research using chiral building blocks, offering new biomedical breakthroughs and disease treatments.
The present review outlines synthetic studies on the antibacterial natural product furanomycin 1 and its analogs. Furanomycin 1 prevents the formation of isoleucyl–tRNA in E. coli while sparing other aminoacyl–tRNAs. Aminoacyl–tRNA synthetases are vital in all organisms and are being explored as new targets in bacterial protein synthesis. Combined drug-like features of furanomycin 1, such as polarity, sufficient solubility, and moderate structural complexity, make it attractive to medicinal chemists, in addition to its antibacterial activity in vitro and in vivo. Several synthetic approaches towards 1 have been developed, some of which consist of up to 20 steps. Many syntheses have started from chiral templates such as D-glucose, D-ribose, D-glucosamine, L-xylose, D-mannitol, and dimethyl-L-tartrate. Other approaches have used amino acids like serine, glycine, or furanes as starting points. Approaches starting from amino acids were captivating; with six to seven steps, they were short and flexible enough to access isomers and close congeners to evaluate the SAR rapidly. First, it was necessary to learn which structural parts in furanomycin were essential for activity and which parts could be removed or modified. Since norfuranomycin had been reported to exhibit antibacterial activity (MIC) against some Gram-negative bacteria, straightforward protocols toward derivatives of 1 were envisaged. To gain rapid insight into SARs, control of diastereoselectivity was considered more important than controlling the absolute configuration. A ring-closing metathesis was used to construct the dihydrofuran ring. Only L-(+)-dihydrofuranomycin 6 showed borderline MIC (32-64 mg/mL) against S. aureus, and the chiral carbon-analog 10 exhibited weak antibacterial activity (4 mg/mL) against an efflux-pump-deficient E. coli.
In summary, the chemical synthesis of both natural and non-natural versions of furanomycin could offer valuable lead compounds for studying protein translation processes and modification. Creating modified non-proteinogenic amino acids could provide new insights into protein function, expand the genetic code in organisms, and produce diverse peptide libraries for drug discovery.[50,51] This review will provide an initial perspective on synthesizing furanomycin and its modified compounds.
Author Contributions
"Conceptualization, R.R; visualization, R.R.; writing—original draft preparation, R.R., and R.M.; writing—review and editing, R.R. and R.M.; supervision, R.R. and R.M.; All authors have read and agreed to the published version of the manuscript."
Funding
This research received no external funding.
Data Availability Statement
Not applicable.
Acknowledgments
Rajendra Rohokale reports administrative support or supplies from the University of Florida. Rajendra Mane was supported by SERB-SIRE, New Delhi (SIR/2022/000426).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Chemistry and Biochemistry of the Amino Acids, 1 ed.; Barrett, G.C., Ed.; Springer Dordrecht: London, 1985; pp. X, 684.
- O'Donnel, M.J. Preface. Tetrahedron 1988, 44, xiii. [Google Scholar] [CrossRef]
- Wagner, I.; Musso, H. New Naturally Occurring Amino Acids. Angewandte Chemie International Edition in English 1983, 22, 816–828. [Google Scholar] [CrossRef]
- Nelson, D.L.D.L.-.; Cox, M.; Lehninger, A.L. Lehninger principles of biochemistry, 4th ed. ed.; New York : W.H. Freeman ; Houndmills, Basingstoke : Macmillan Learning 2017: 2017; pp. pp 71-103.
- Herbert, R.B. The Biosynthesis of Secondary Metabolites, 1 ed.; Springer Dordrecht: 1981; Volume.
- Izumi, Y.; Chibata, I.; Itoh, T. Production and Utilization of Amino Acids. Angewandte Chemie International Edition in English 1978, 17, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Finking, R.; Marahiel, M.A. Biosynthesis of Nonribosomal Peptides1. Annual Review of Microbiology 2004, 58, 453–488. [Google Scholar] [CrossRef]
- Caboche, S.; Pupin, M.; Leclère, V.; Fontaine, A.; Jacques, P.; Kucherov, G. NORINE: a database of nonribosomal peptides. Nucleic Acids Res 2008, 36, D326–331. [Google Scholar] [CrossRef]
- Johnson, J.A.; Lu, Y.Y.; Van Deventer, J.A.; Tirrell, D.A. Residue-specific incorporation of non-canonical amino acids into proteins: recent developments and applications. Current Opinion in Chemical Biology 2010, 14, 774–780. [Google Scholar] [CrossRef]
- Liu, C.C.; Schultz, P.G. Adding New Chemistries to the Genetic Code. Annual Review of Biochemistry 2010, 79, 413–444. [Google Scholar] [CrossRef]
- Katagiri, K.; Tori, K.; Kimura, Y.; Yoshida, T.; Nagasaki, T.; Minato, H. A New Antibiotic. Furanomycin, an Isoleucine Antagonist. Journal of Medicinal Chemistry 1967, 10, 1149–1154. [Google Scholar] [CrossRef]
- Trippe, K.; McPhail, K.; Armstrong, D.; Azevedo, M.; Banowetz, G. Pseudomonas fluorescens SBW25 produces furanomycin, a non-proteinogenic amino acid with selective antimicrobial properties. BMC Microbiology 2013, 13, 111. [Google Scholar] [CrossRef]
- Fariq, A.; Yasmin, A. Production, characterization and bioactivities of biosurfactants from newly isolated strictly halophilic bacteria. Process Biochemistry 2020, 98, 1–10. [Google Scholar] [CrossRef]
- Shiro, M.; Nakai, H.; Tori, K.; Nishikawa, J.; Yoshimura, Y.; Katagiri, K. X-Ray crystal structure determination of furanomycin. Journal of the Chemical Society, Chemical Communications 1980, 375a-375a. [CrossRef]
- Joullie, M.M.; Wang, P.C.; Semple, J.E. Total synthesis and revised structural assignment of (+)-furanomycin. Journal of the American Chemical Society 1980, 102, 887–889. [Google Scholar] [CrossRef]
- von Nussbaum, F.; Brands, M.; Hinzen, B.; Weigand, S.; Häbich, D. Antibacterial Natural Products in Medicinal Chemistry—Exodus or Revival? Angewandte Chemie International Edition 2006, 45, 5072–5129. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Kohda, D.; Haruki, M.; Yokoyama, S.; Miyazawa, T. Nonprotein amino acid furanomycin, unlike isoleucine in chemical structure, is charged to isoleucine tRNA by isoleucyl-tRNA synthetase and incorporated into protein. Journal of Biological Chemistry 1990, 265, 6931–6935. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, P.J.; Lee, J.Y.; Hlobilova, I.; Endermann, R.; Häbich, D.; Jäger, V. Synthesis of L-Furanomycin and Its Analogues via Furoisoxazolines. European Journal of Organic Chemistry 2005, 2005, 3450–3460. [Google Scholar] [CrossRef]
- Masamune, T.; Ono, M. The synthesis of dl-furanomycin. Chemistry Letters 1975, 625–626. [Google Scholar] [CrossRef]
- Masamune, T.; Ono, M. THE SYNTHESIS OF dl-FURANOMYCIN. Chemistry Letters 2006, 4, 625–626. [Google Scholar] [CrossRef]
- Tanaka, K.; Tamaki, M.; Watanabe, S. Effect of furanomycin on the synthesis of isoleucyl-tRNA. Biochimica et Biophysica Acta (BBA) - Nucleic Acids and Protein Synthesis 1969, 195, 244–245. [Google Scholar] [CrossRef]
- Kazmaier, U.; Pähler, S.; Endermann, R.; Häbich, D.; Kroll, H.-P.; Riedl, B. Straightforward syntheses of furanomycin derivatives and their biological evaluation. Bioorganic & medicinal chemistry 2002, 10, 3905–3913. [Google Scholar]
- Parry, R.J.; Buu, H.P. Investigations of the biosynthesis of furanomycin. Unexpected derivation from acetate and propionate. Journal of the American Chemical Society 1983, 105, 7446–7447. [Google Scholar] [CrossRef]
- Parry, R.J.; Turakhia, R.; Phuoc, B.H. The biosynthesis of furanomycin. Mechanism of formation of the ether linkage. Journal of the American Chemical Society 1988, 110, 4035–4036. [Google Scholar] [CrossRef]
- Semple, J.E.; Wang, P.C.; Lysenko, Z.; Joullie, M.M. Total synthesis of (+)-furanomycin and stereoisomers. Journal of the American Chemical Society 1980, 102, 7505–7510. [Google Scholar] [CrossRef]
- Chen, S.Y.; Joullie, M.M. oTal synthesis of two furanomycin stereoisomers. The Journal of Organic Chemistry 1984, 49, 1769–1772. [Google Scholar] [CrossRef]
- Robins, M.J.; Parker, J.R. Chiral transformations of D-ribose to 2 (S)-amino-2-[2, 5-dihydro-5 (R)-methylfuran-2 (R)-yl] ethanoic acid, a diastereomer of the antibiotic (+)-furanomycin. Canadian Journal of Chemistry 1983, 61, 317–322. [Google Scholar] [CrossRef]
- Trummlitz, G.; Moffatt, J.G. C-Glycosyl nucleosides. III. Facile synthesis of the nucleoside antibiotic showdomycin. The Journal of Organic Chemistry 1973, 38, 1841–1845. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Lee, S. Total synthesis of (+)-furanomycin. Chemical Communications 1998, 761-762.
- Zhang, J.; Clive, D.L. Synthesis of (+)-Furanomycin: Use of Radical Cyclization. The Journal of organic chemistry 1999, 64, 1754–1757. [Google Scholar] [CrossRef]
- VanBrunt, M.P.; Standaert, R.F. A short total synthesis of (+)-Furanomycin. Organic Letters 2000, 2, 705–708. [Google Scholar] [CrossRef]
- Zimmermann, P.J.; Blanarikova, I.; Jäger, V. A General Approach to l-(+)-Furanomycin and Some Stereoisomers and Analogues Using Furoisoxazoline Intermediates. Angewandte Chemie International Edition 2000, 39, 910–912. [Google Scholar] [CrossRef]
- Müller, R.; Leibold, T.; Pätzel, M.; Jäger, V. A New Synthesis of 1,3,4-Trideoxy-1,4-iminoglycitols of Varying Chain Length by (C3 + Cn)-Coupling of Allyl Halides with Glycononitrile Oxides. Angewandte Chemie International Edition in English 1994, 33, 1295–1298. [Google Scholar] [CrossRef]
- Lee, J.Y.; Schiffer, G.; Jäger, V. Synthesis of L-carbafuranomycin, an unnatural analogue of the antibiotic amino acid furanomycin. Organic Letters 2005, 7, 2317–2320. [Google Scholar] [CrossRef]
- Uli, K. Stereoselective synthesis of 2-(2′-cycloalkenyl) glycinates via [3,3] sigmatropic rearrangement of chelated ester-enolates. Tetrahedron 1994, 50, 12895–12902. [Google Scholar] [CrossRef]
- Erdsack, J.; Krause, N. Synthesis of furanomycin derivatives by Gold-catalyzed cycloisomerization of α-hydroxyallenes. Synthesis 2007, 2007, 3741–3750. [Google Scholar]
- Erdsack, J.; Krause, N. An approach towards azafuranomycin analogs by gold-catalyzed cycloisomerization of allenes: synthesis of (αS,2R)-(2,5-dihydro-1H-pyrrol-2-yl)glycine. Beilstein Journal of Organic Chemistry 2013, 9, 1936–1942. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, S.K.; Sarkar, K.; Karmakar, S. Enantioselective synthesis of dihydrofuranylglycine, furanylglycine, furanylalanine and homo-furanylalanine derivatives. Synlett 2005, 2005, 2083–2085. [Google Scholar] [CrossRef]
- Bandyopadhyay, A.; Pal, B.K.; Chattopadhyay, S.K. A short route to N-protected furanomycin, 5′-epi-furanomycin and isofuranomycin derivatives. Tetrahedron: Asymmetry 2008, 19, 1875–1877. [Google Scholar] [CrossRef]
- Nelson, J.M.; Vedejs, E. Metalated aziridines for cross-coupling with aryl and alkenyl halides via palladium catalysis. Organic letters 2010, 12, 5085–5087. [Google Scholar] [CrossRef]
- Passiniemi, M.; Koskinen, A.M.P. Enantioselective synthesis of Boc-protected norfuranomycins. Tetrahedron Letters 2011, 52, 6736–6738. [Google Scholar] [CrossRef]
- Tellam, J.P.; Carbery, D.R. Ireland-Claisen rearrangement of substrates bearing chiral enol ether units. Tetrahedron letters 2011, 52, 6027–6029. [Google Scholar] [CrossRef]
- Jung, J.-H.; Yoon, D.-H.; Lee, K.; Shin, H.; Lee, W.K.; Yook, C.-M.; Ha, H.-J. Stereoselective Pd-catalyzed etherification and asymmetric synthesis of furanomycin and its analogues from a chiral aziridine. Organic & Biomolecular Chemistry 2015, 13, 8187–8195. [Google Scholar]
- Avenoza, A.; Busto, J.H.; Canal, N.; García, J.I.; Jiménez-Osés, G.; Peregrina, J.M.; Pérez-Fernández, M. Mechanistic study of the ring-size modulation in Michael–Dieckmann type reactions of 2-acylaminoacrylates with ketene diethyl acetal. New Journal of Chemistry 2007, 31, 224–229. [Google Scholar] [CrossRef]
- Avenoza, A.; Busto, J.H.; Canal, N.; Peregrina, J.M.; Pérez-Fernández, M. Selective Michael−Aldol Reaction by Use of Sterically Hindered Aluminum Aryloxides as Lewis Acids: An Easy Approach to Cyclobutane Amino Acids. Organic Letters 2005, 7, 3597–3600. [Google Scholar] [CrossRef]
- Avenoza, A.; Busto, J.H.; Canal, N.; Corzana, F.; Peregrina, J.M.; Pérez-Fernández, M.; Rodriguez, F. Cyclobutane amino acid analogues of furanomycin obtained by a formal [2+ 2] cycloaddition strategy promoted by methylaluminoxane. The Journal of Organic Chemistry 2010, 75, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Pilli, R.A.; Riatto, V.B. Stereoselective addition of chiral titanium enolates to 5-substituted five-membered oxocarbenium ions. Journal of the Brazilian Chemical Society 2008, 19. [Google Scholar] [CrossRef]
- Bartoli, G.; Di Antonio, G.; Fiocchi, R.; Giuli, S.; Marcantoni, E.; Marcolini, M. Synthesis of an (R)-Garner-type Aldehyde from l-Serine: Useful Building Block for a (+)-Furanomycin Derivative. Synthesis 2009, 2009, 951–956. [Google Scholar] [CrossRef]
- Herold, P. Synthesis of D-Erythro- and D-Threo-Sphingosine Derivatives From L-Serine. Helvetica Chimica Acta 1988, 71, 354–362. [Google Scholar] [CrossRef]
- Chhibber-Goel, J.; Yogavel, M.; Sharma, A. Structural analyses of the malaria parasite aminoacyl-tRNA synthetases provide new avenues for antimalarial drug discovery. Protein Science 2021, 30, 1793–1803. [Google Scholar] [CrossRef]
- Hartman, M.C.T. Non-canonical Amino Acid Substrates of E. coli Aminoacyl-tRNA Synthetases. ChemBioChem 2022, 23, e202100299. [Google Scholar] [CrossRef]
Figure 1.
The chemical structure of furanomycin 1 and its isomers and analogs, 3-12 and L-isoleucine 2.
Figure 1.
The chemical structure of furanomycin 1 and its isomers and analogs, 3-12 and L-isoleucine 2.
Scheme 1.
The biosynthetic pathway of furanomycin 1.
Scheme 2.
The synthesis of dl-furanomycin 1 from 5-methyl-2-furoic acid.
Scheme 3.
(A) The synthesis of L-furoanomycin 1 from D-glucose, and (B) its 3, ent-3, 4, ent-4, ent-5 from 2-methyl furan.
Scheme 3.
(A) The synthesis of L-furoanomycin 1 from D-glucose, and (B) its 3, ent-3, 4, ent-4, ent-5 from 2-methyl furan.
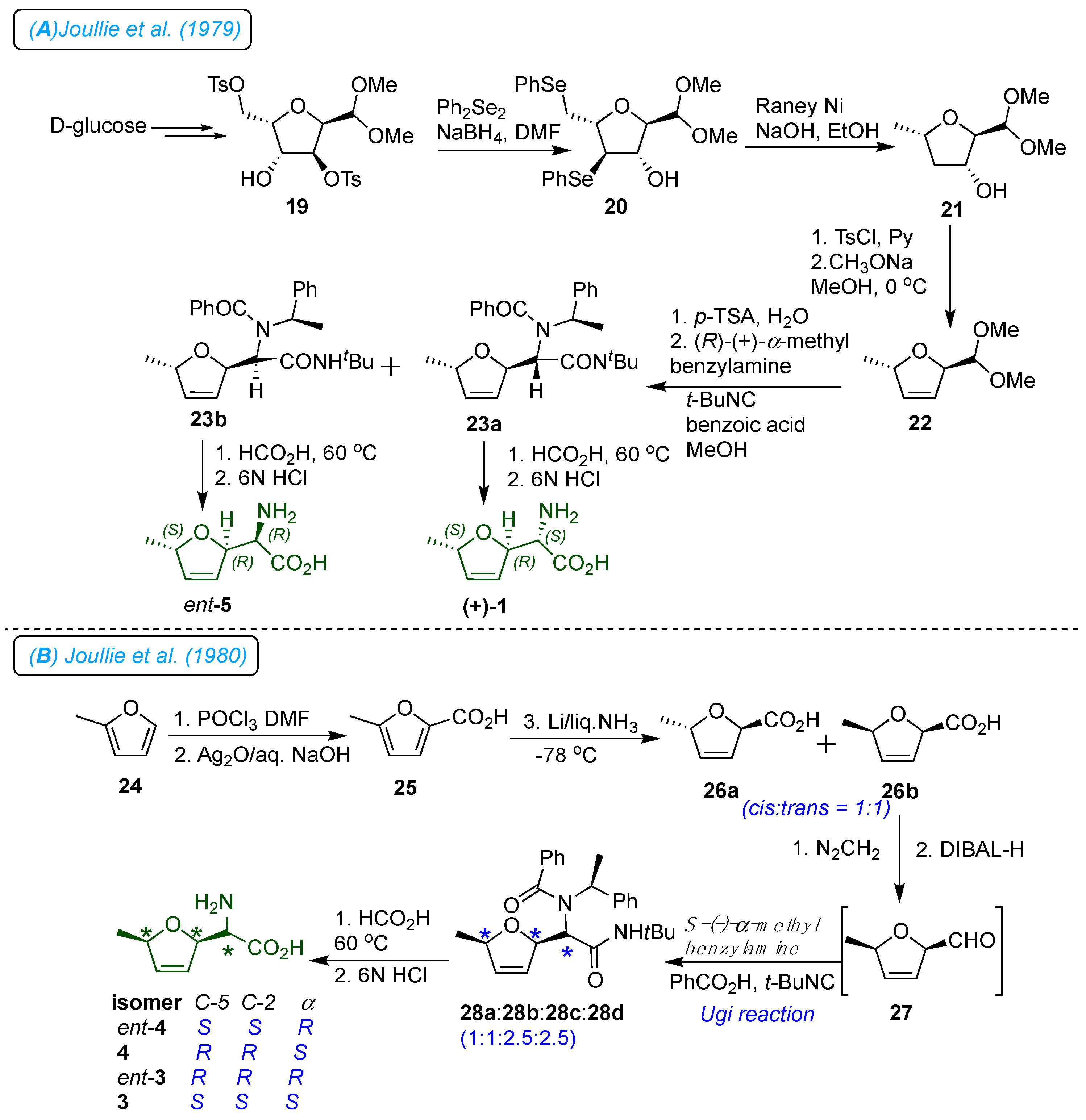
Scheme 4.
The furoanomycin (-)-1 synthesis and its isomer 5 from D-glucosamine.
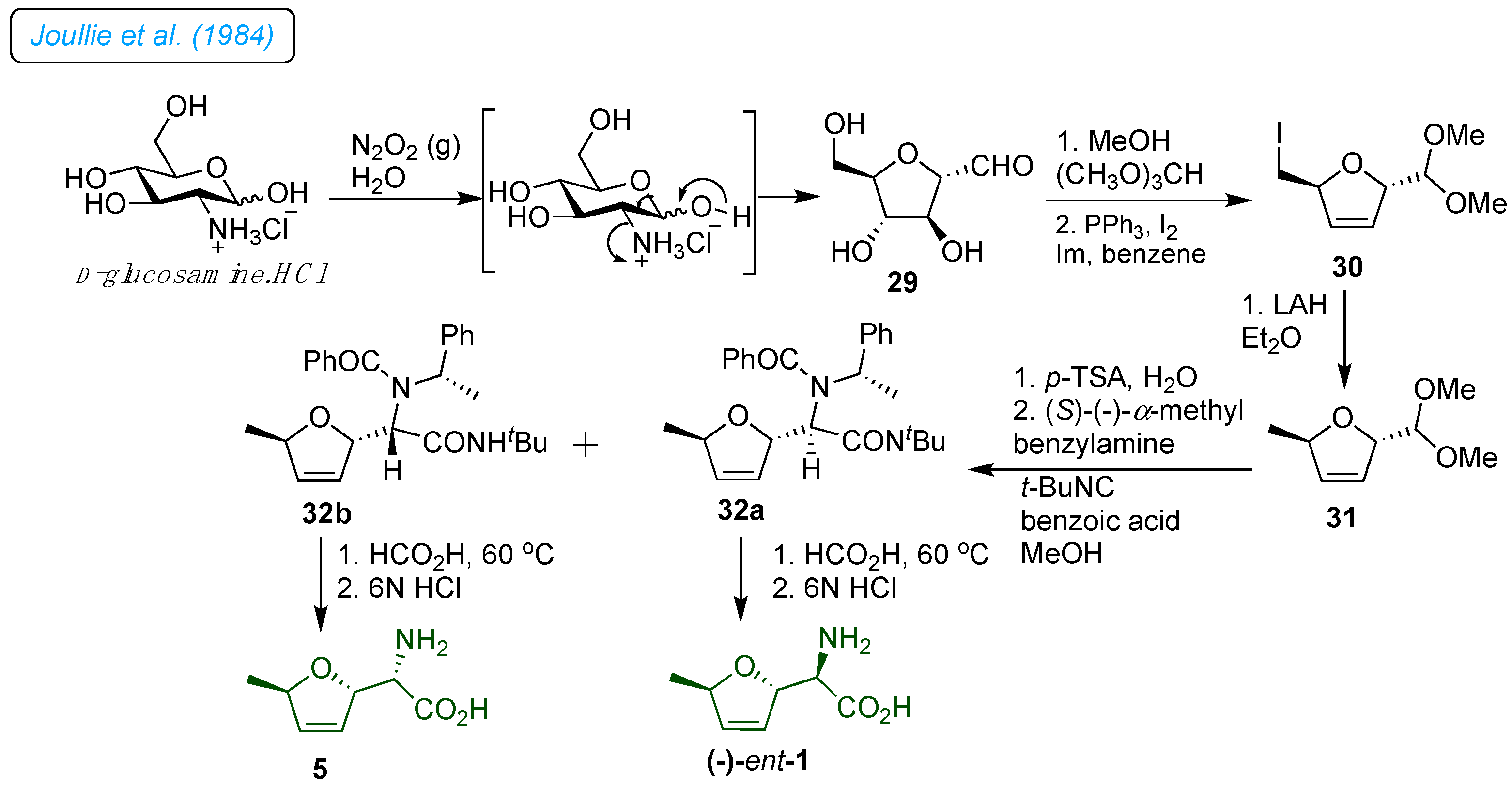
Scheme 5.
The synthesis of ent-3 and 4 from D-ribose.
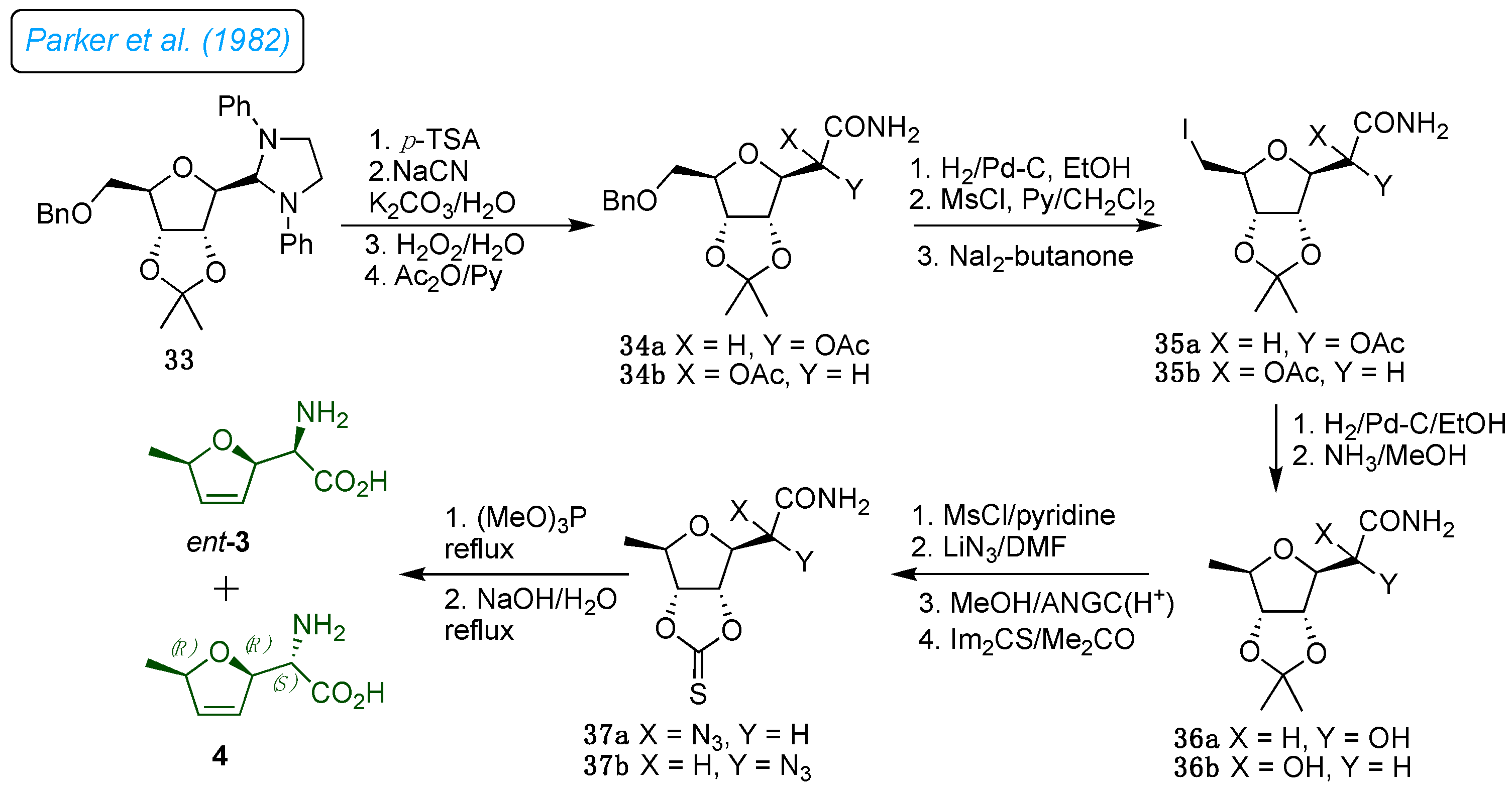
Scheme 6.
The enantioselective synthesis of furanomycin 1 from L-tartaric acid.
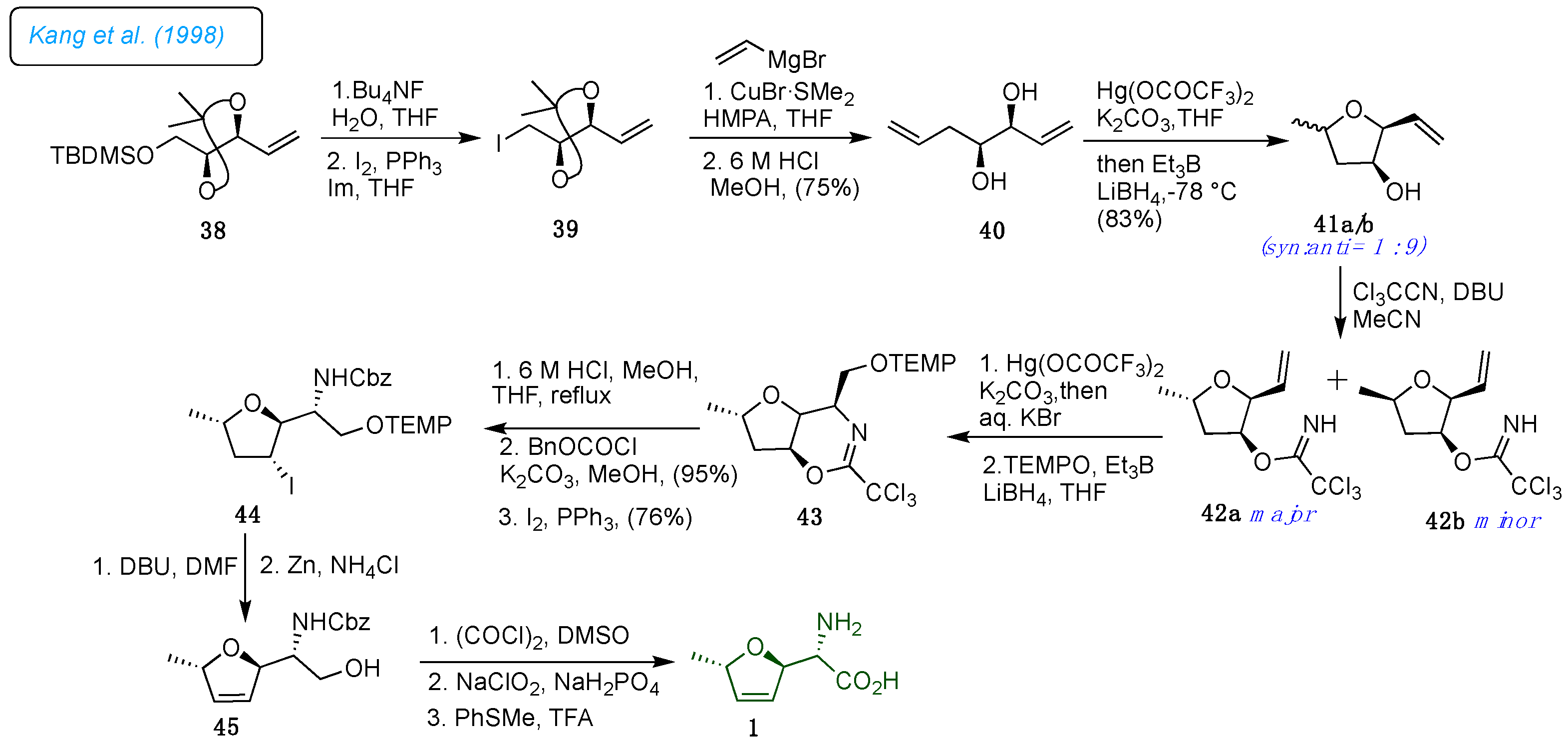
Scheme 7.
The synthesis of furanomycin 1 from L-xylose.
Scheme 8.
The synthesis of L-(+)-furanomycin 1 from Garner aldehyde.
Scheme 9.
(A) The synthesis of L-furanomycin 1 and its analogs 6, 7 from 2-methyl furan and D-mannitol; (B) A synthesis of compound 6 from cyclopentadiene and D-mannitol.
Scheme 9.
(A) The synthesis of L-furanomycin 1 and its analogs 6, 7 from 2-methyl furan and D-mannitol; (B) A synthesis of compound 6 from cyclopentadiene and D-mannitol.
Scheme 10.
The synthesis of norfuranomycin 6 and its analogs from glycine ester.
Scheme 11.
The synthesis of 3-methyl-norfuranomycin 7 and 4-methyl-norfuranomycin 8 from glycine ester.
Scheme 11.
The synthesis of 3-methyl-norfuranomycin 7 and 4-methyl-norfuranomycin 8 from glycine ester.
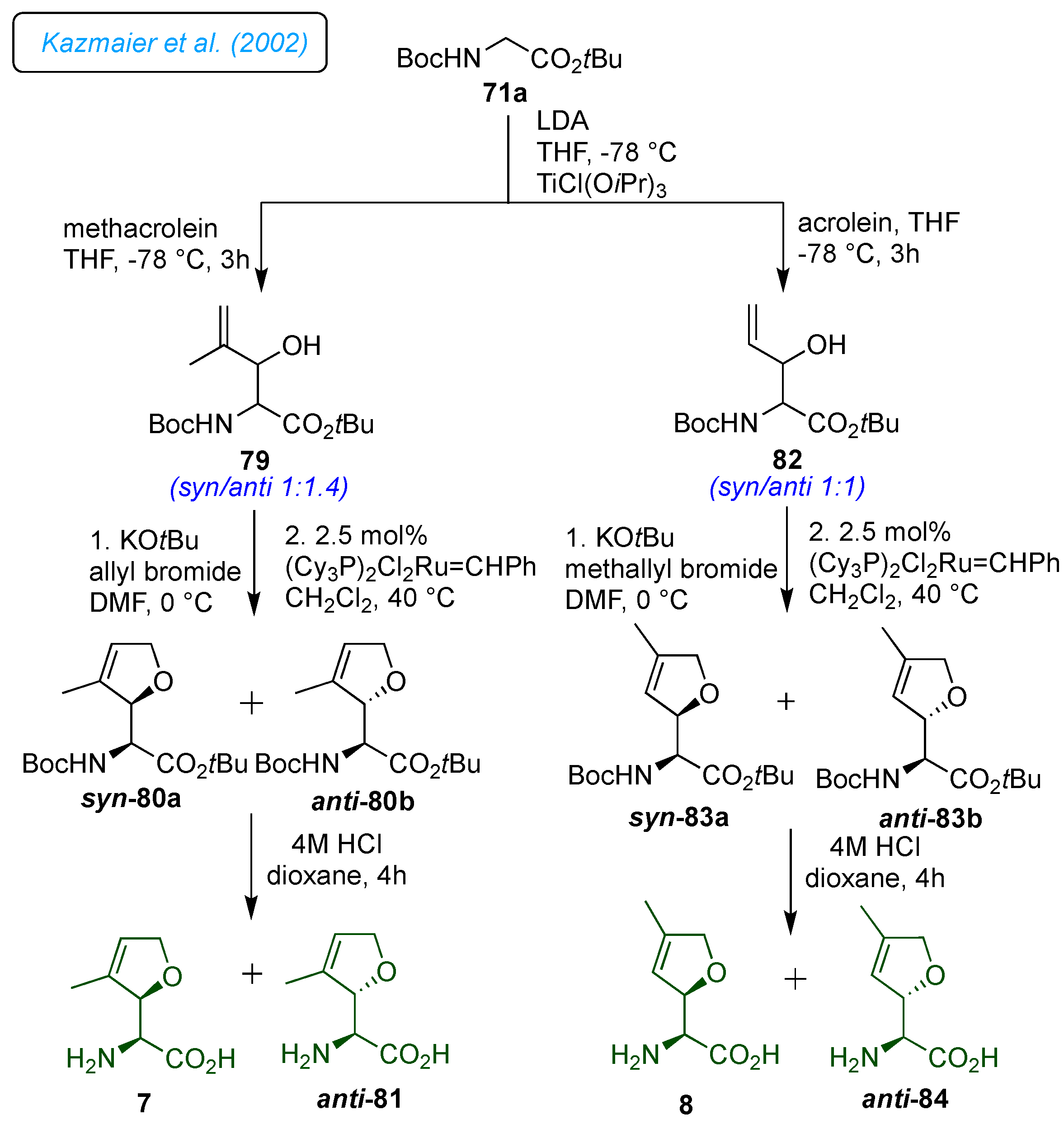
Scheme 12.
The synthesis of carba-norfuranomycin 89a/b from cyclopentenol with glycine ester.
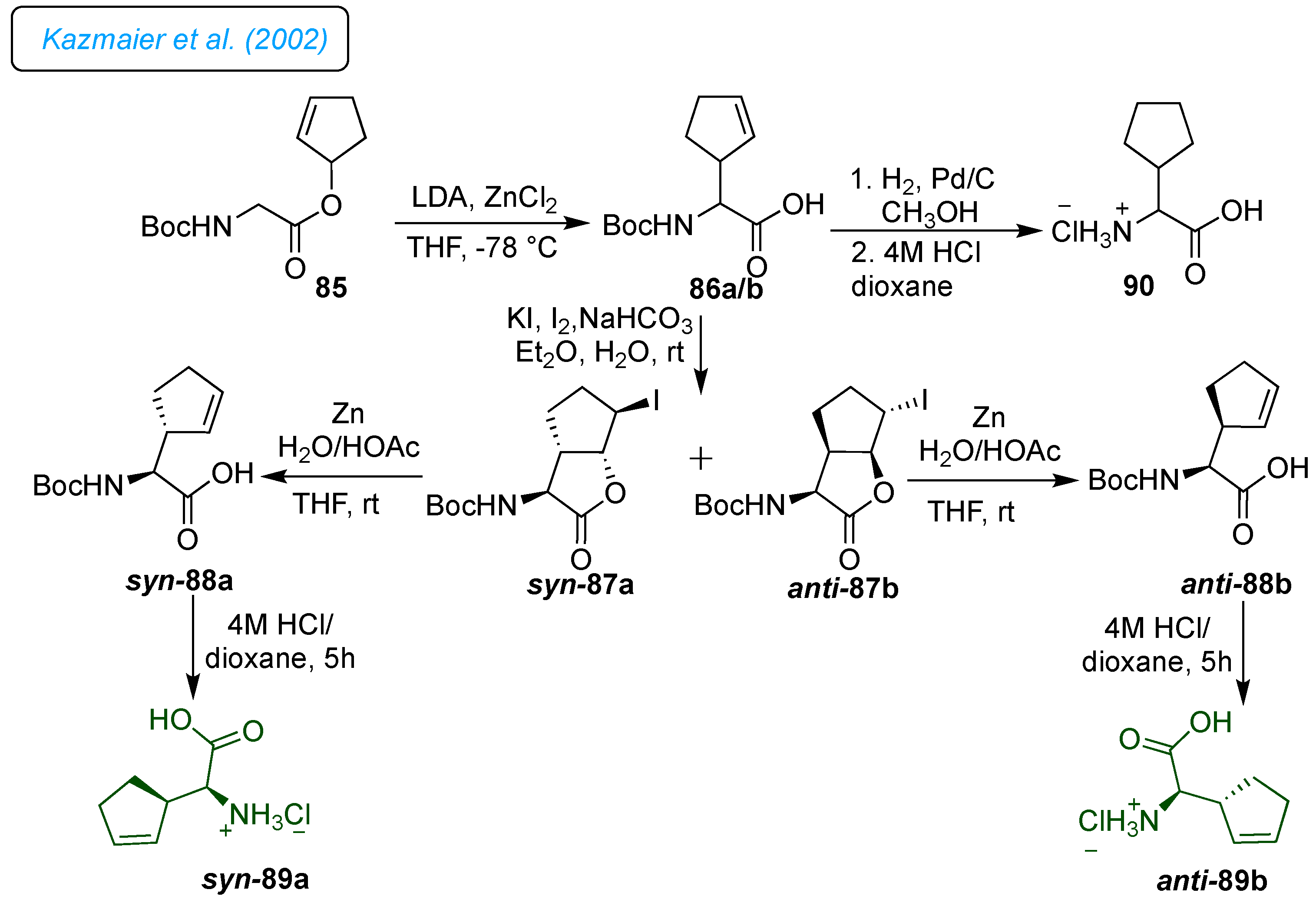
Scheme 13.
Synthesis of furanomycin 96 and 100 analogs from S-Garnaldehyde 91.
Scheme 14.
Synthesis of aza-furanomycin 106a and 106b analogs from R-Garnaldehyde.
Scheme 15.
The synthesis of NHBoc-protected furanomycine 115a/b and norfuroanomycin 109a/b from R-Garner's aldehyde.
Scheme 15.
The synthesis of NHBoc-protected furanomycine 115a/b and norfuroanomycin 109a/b from R-Garner's aldehyde.
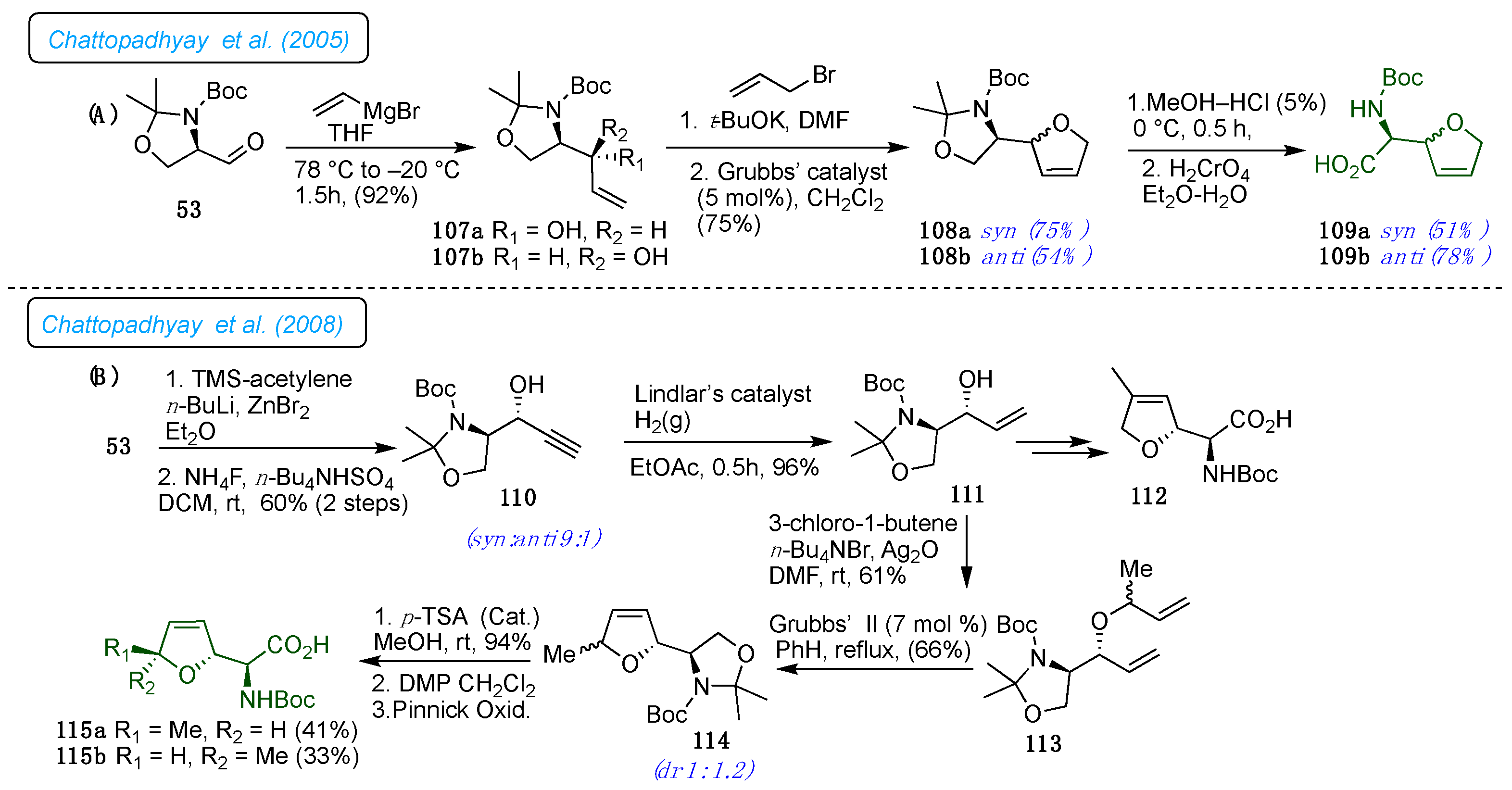
Scheme 16.
The synthesis of analogue of furoanomycin 122 from chiral aziridine 116.
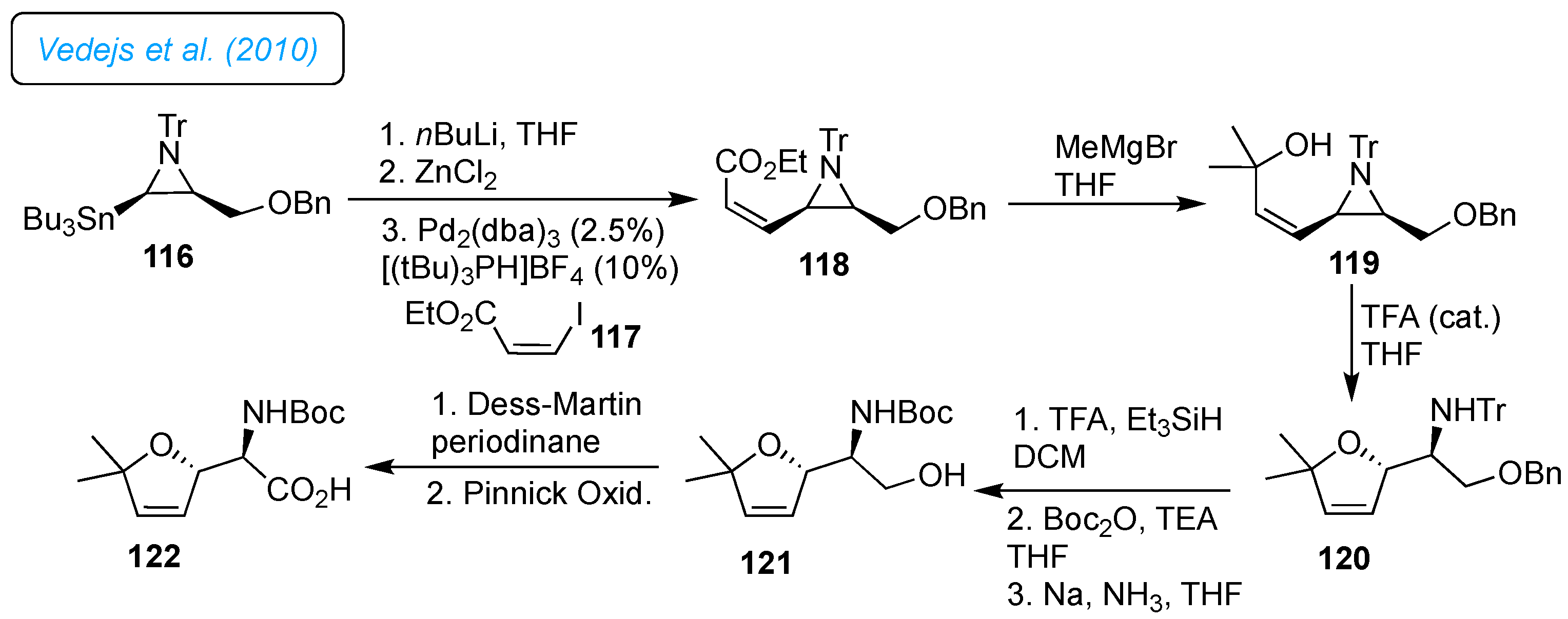
Scheme 17.
The synthesis of NH-Boc-protected norfuroanomycin 109a/b from S-Garnaldehyde 91.
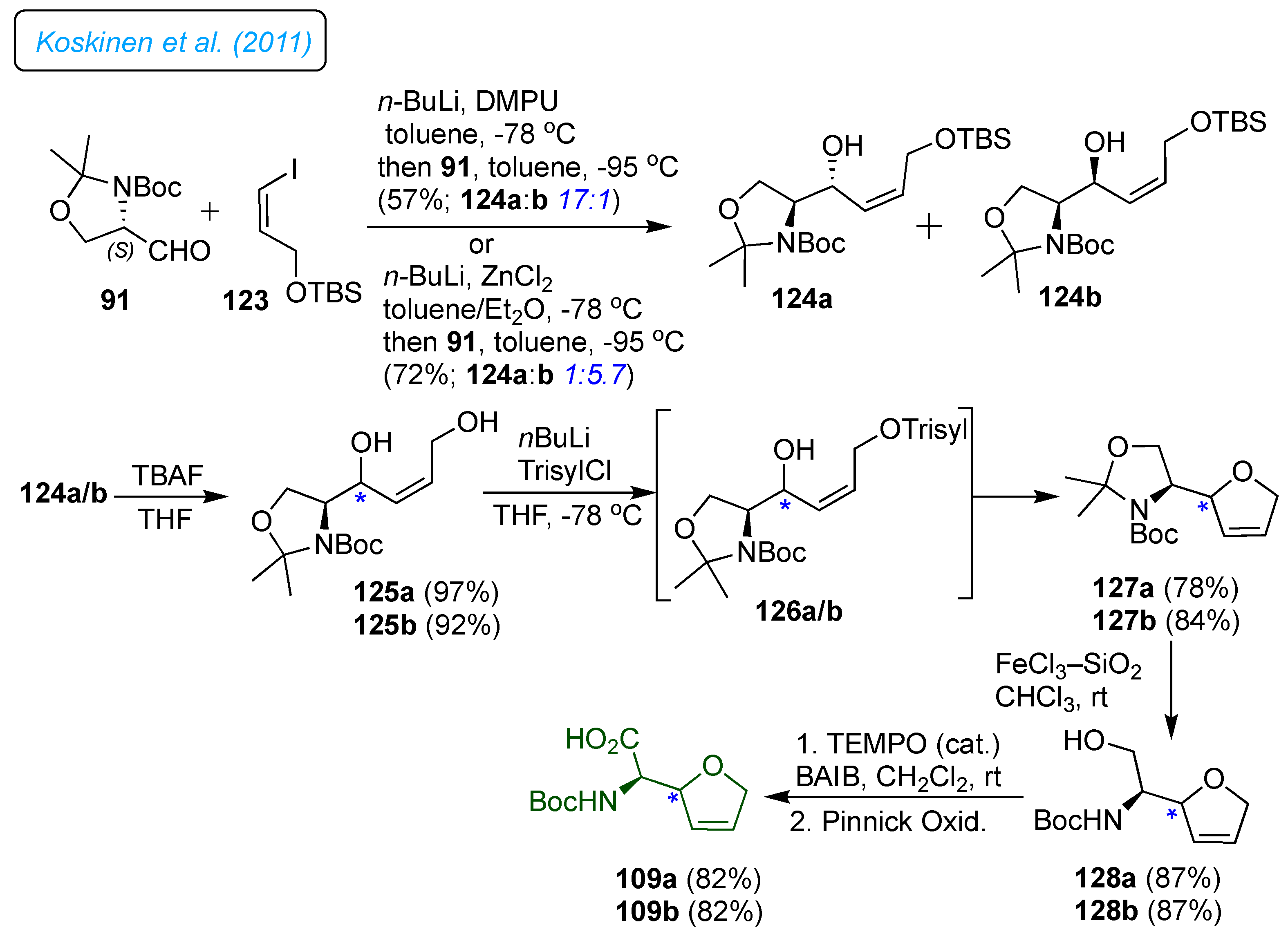
Scheme 18.
The synthesis of intermediate of furanomycin 139a/b from (S)-but-3-en-2-ol.
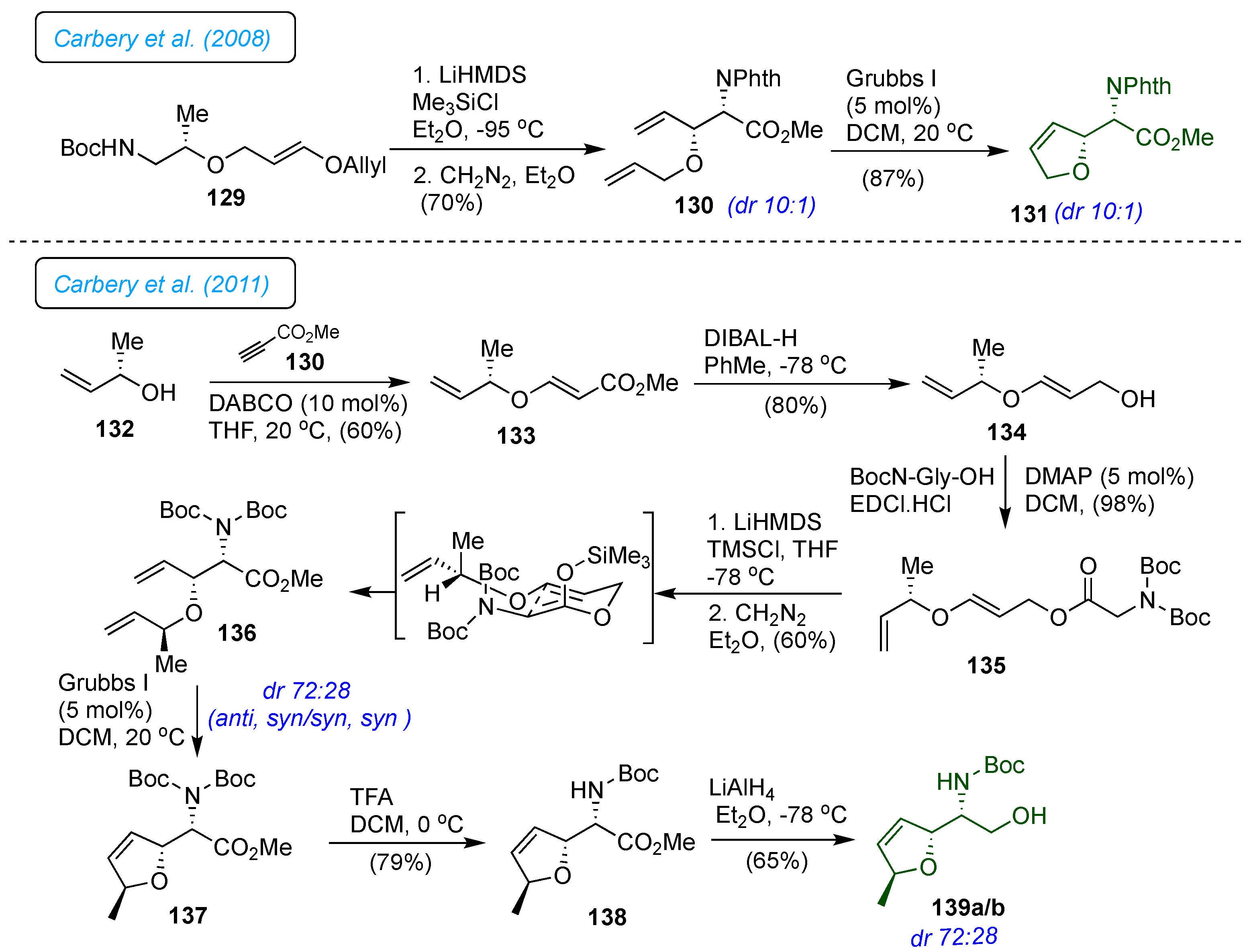
Scheme 19.
The synthesis of analogue of furoanomycin 122 from chiral aziridine 116.
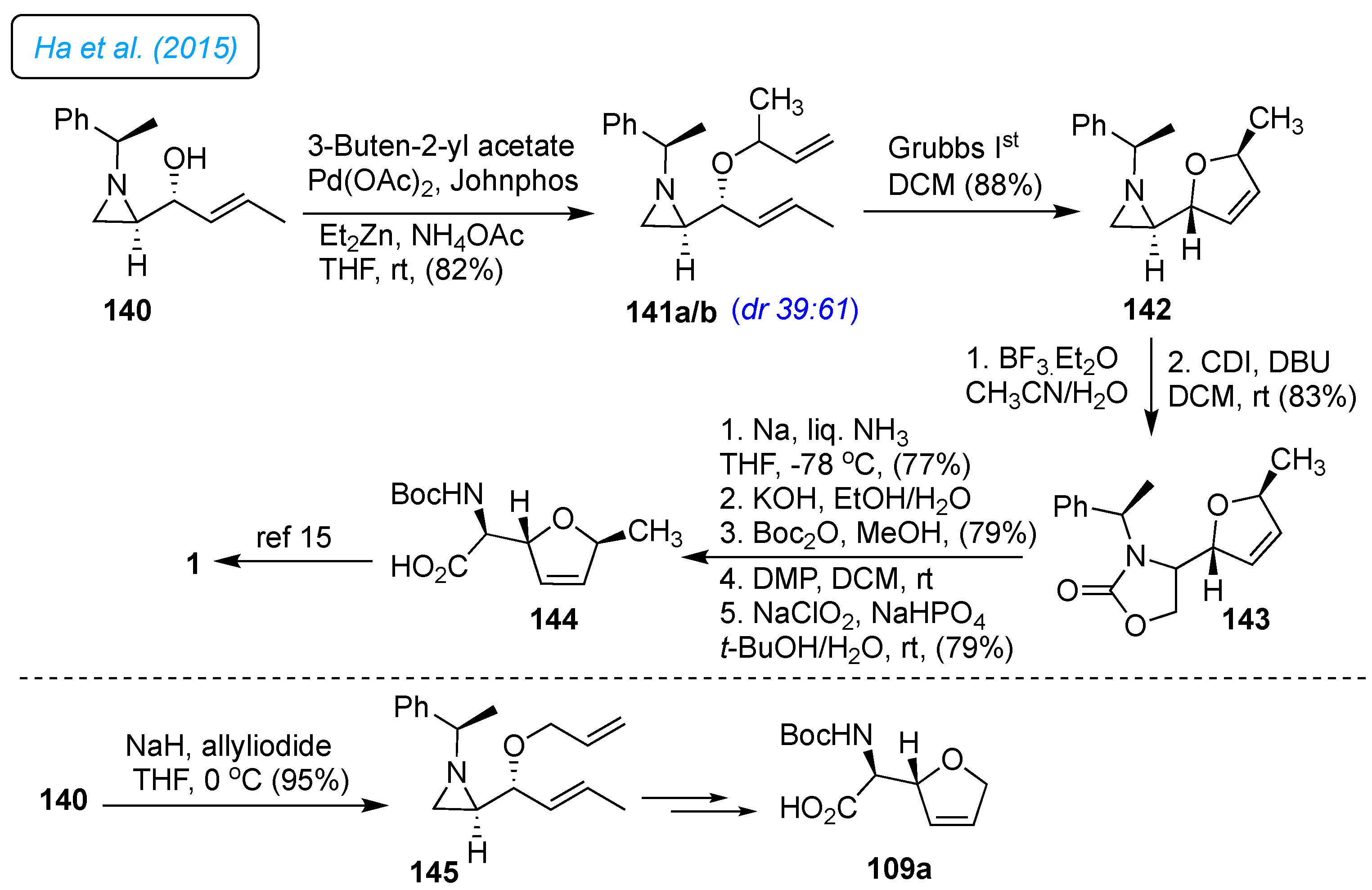
Scheme 20.
The synthesis of cyclobutane a-amino acid derivative analogs of furanomycin from acylaminoacrylates and cyclic vinyl ethers.
Scheme 20.
The synthesis of cyclobutane a-amino acid derivative analogs of furanomycin from acylaminoacrylates and cyclic vinyl ethers.
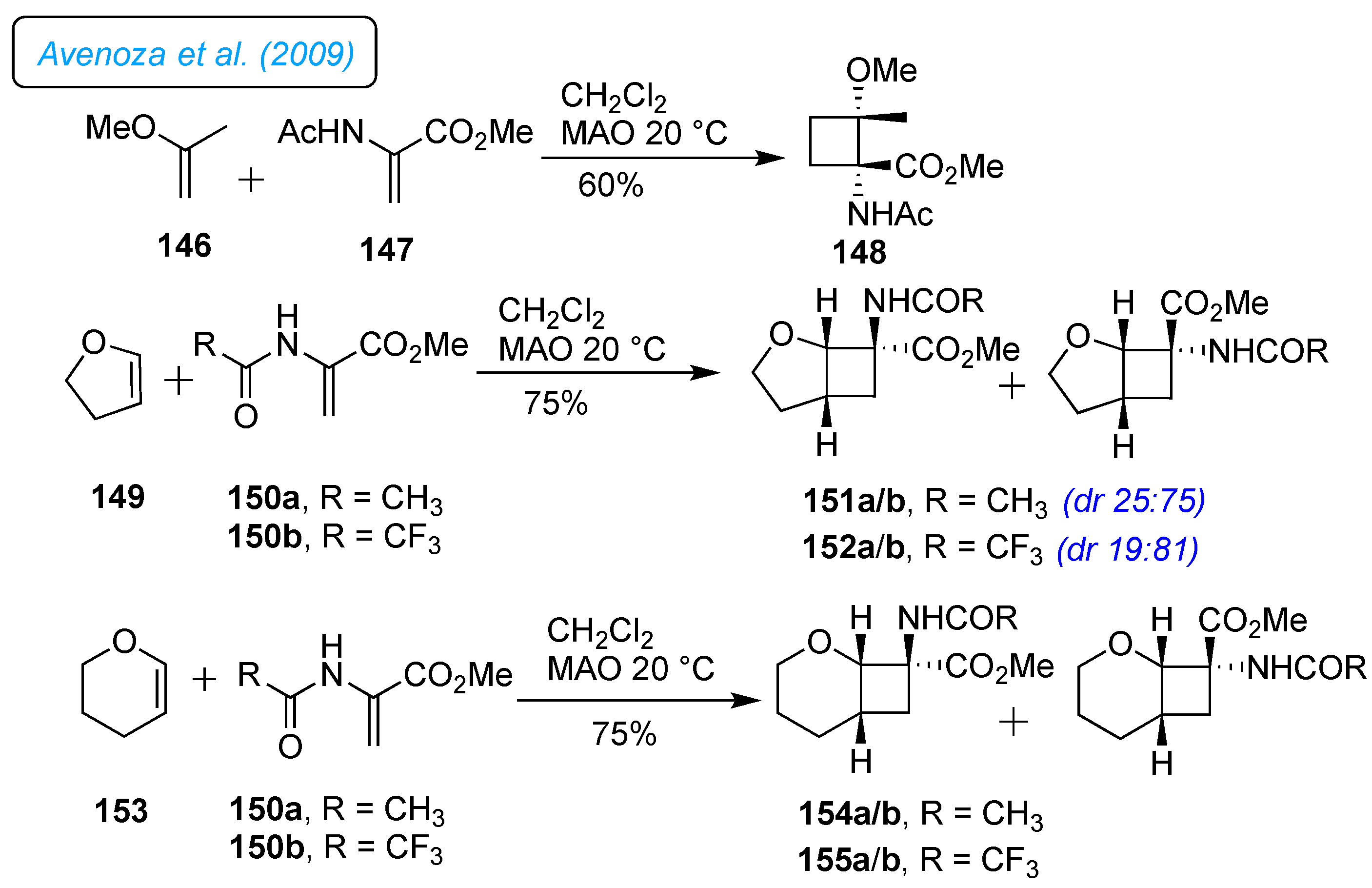
Scheme 21.
The synthesis of (-)-furoanomycin 1 from chiral auxiliary 158a/b.
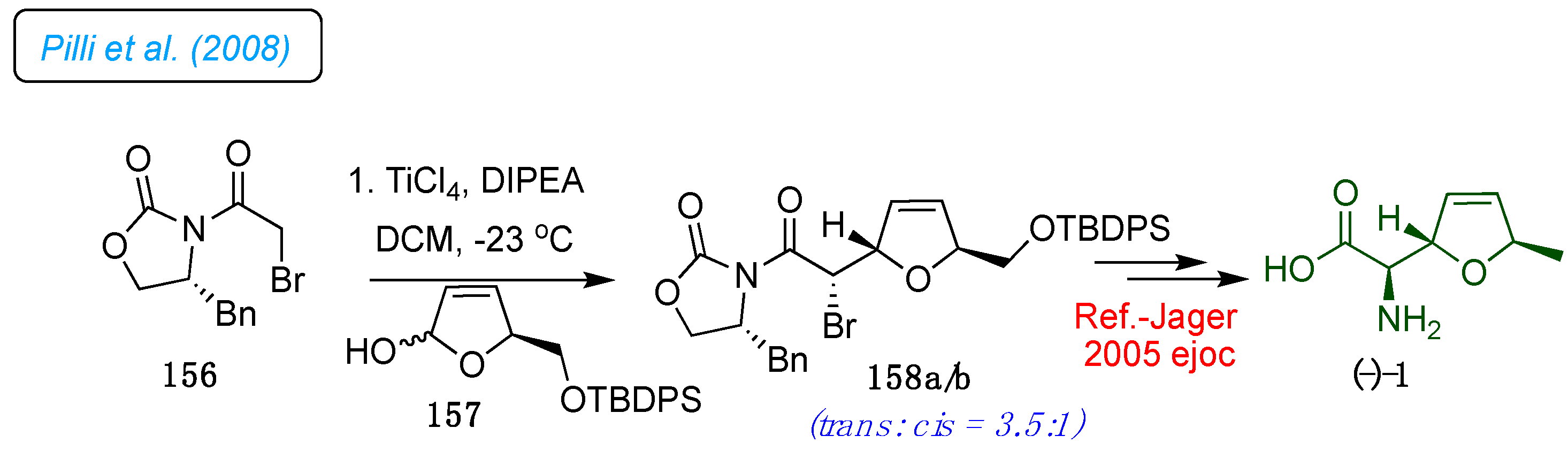
Scheme 22.
The synthesis of essential precursor 161 for the synthesis of furanomycin.
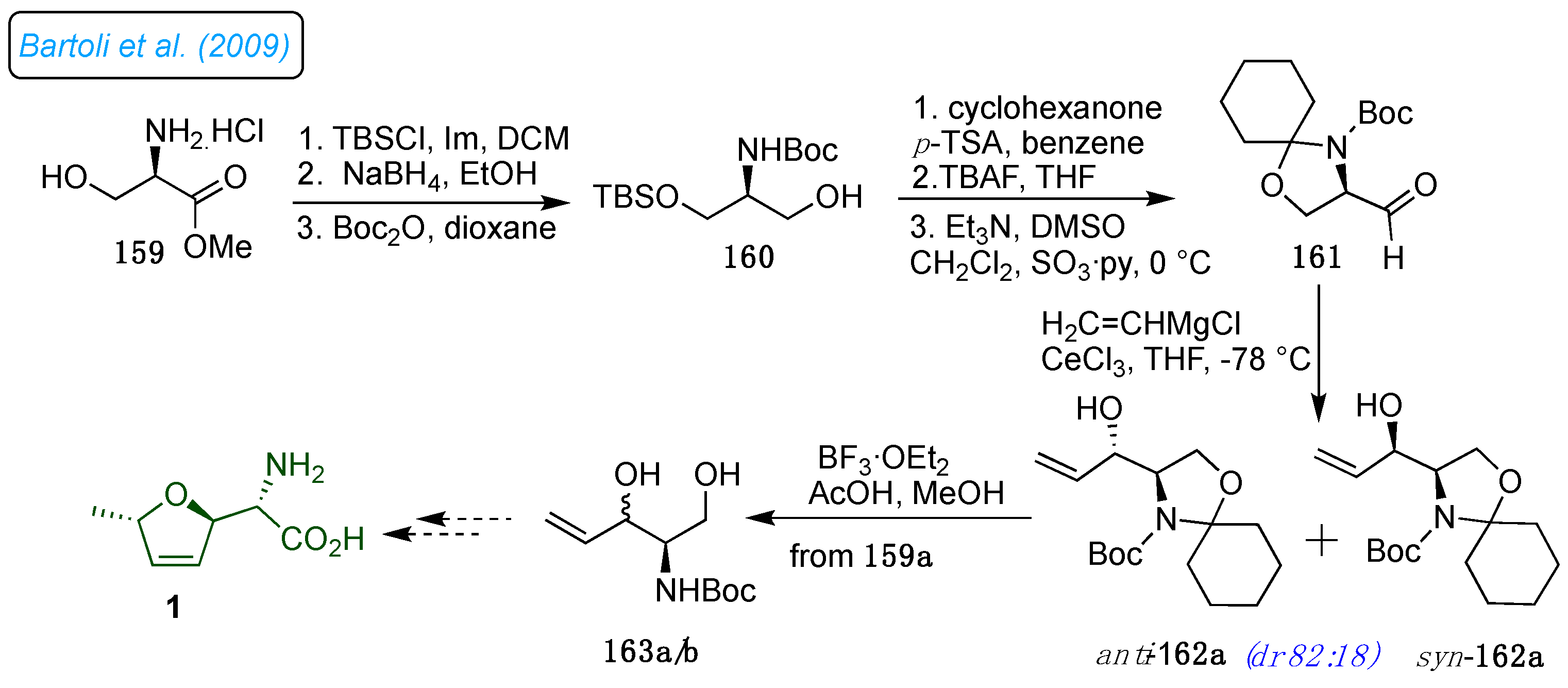
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



