
Submitted:
13 October 2024
Posted:
14 October 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Obesity is among the most prevalent risk factors in the severe forms of Coronavirus disease 2019 (COVID-19) infection. COVID-19 patients with obesity often face severe complications that might be associated with overexpression of adiponectin, inflammatory cytokines, and angi-otensin-converting enzyme 2 (ACE2) receptors in visceral fat. The pre-existing subclinical in-flammation associated with obesity can also lead to severe inflammatory responses. Elevation of pro-inflammatory cytokines considerably activates coagulation cascades, including the tissue factor (TF) pathway. The hypercoagulable state in COVID-19 is presented by severe pulmonary complications such as venous thromboembolism (VTE), disseminated intravascular coagulation (DIC), and disruption of vascular endothelial cells which can lead to severe complications and death. The interaction between inflammatory response and coagulation mechanism in COVID-19 patients with obesity warrants a further understanding of prognosis and potential therapeutic approaches. This review discussed the crosstalk between inflammation and coagulopathy in obesity-related severe COVID-19 infection.
Keywords:
COVID-19
; coagulation
; inflammation
; obesity
; cytokine storm
1. Introduction
Coronavirus disease 2019 (COVID-19) has emerged as one of the most highly contagious disease outbreaks in history, caused by the severe acute respiratory syndrome coronavirus (SARS-CoV)-2 virus. This disease has given rise to the most consequential global economic and health crisis, infecting over 700 million people and causing more than 6.99 million deaths worldwide as of December 2023 [1]. The most common clinical symptoms of COVID-19 are severe cough, fever, fatigue, and dyspnea [2]. However, the disease can lead to severe complications including pneumonia-induced acute respiratory distress syndrome (ARDS), sepsis, organ failure, and eventually death. Many clinical findings have associated severe COVID-19 with obesity, hypertension, diabetes, cardiovascular disease, asthma, and aging [3]. In Malaysia, the crucial determinants of severe COVID-19 include old age, chronic kidney disease, chronic pulmonary diseases, shortness of breath, and high serum C-reactive protein (CRP) (≥5 mg/dL) [4]. Obesity, in particular, has been linked to severe complications of COVID-19, where most patients require intensive care unit (ICU) hospitalization [5]. In most cases, obesity is accompanied by other diseases, such as dyslipidemia, hypertension, and diabetes mellitus, providing an increased risk of developing severe COVID-19 [6].
Obesity is a low degree of chronic inflammation in which adipose tissue expresses a broad spectrum of Toll-like receptors and produces proinflammatory cytokines and chemokines [7]. The prevalence of severe COVID-19 cases among patients with obesity and the associated mortality varies in different countries. However, the consensus is these patients face a higher risk of death than non-obese patients [8]. Previous reports had associated patients with obesity and severe COVID-19, where most of them suffered from ARDS and often required ventilators compared to their normal-weight counterparts [6,9]. In the United States of America (USA), obesity was reported as the most significant association with COVID-19 mortality in patients under 60 years old [10]. In France, approximately 85% of COVID-19 patients with a body mass index (BMI) >40 needed ventilators upon hospitalization [11]. Meanwhile, in Malaysia, comorbidities contribute to the highest incidence (75.4%) of mortality, with 5.8% being obesity [12].
Coagulopathy, or bleeding disorder is seen more in severe COVID-19 patients associated with impairment in the coagulation cascade and clot formation giving rise to disseminated intravascular coagulation (DIC). Importantly, patients with obesity are more susceptible to coagulopathy and cytokine storms (elevation of prothrombin factors and pro-inflammatory cytokines, respectively) when they are affected by COVID-19, and hence the mortality rate in these patients is high [13]. Understanding the mechanisms of inflammation and coagulopathy in obesity-related severe COVID-19 is essential to developing treatment approaches for managing the severity and mortality. Thus, this review comprehensively discussed the association of coagulopathy and inflammation with the development of severe COVID-19 in patients with obesity.
2. Materials and Methods
A comprehensive search on Google Scholar and PubMed databases was conducted for articles published between May 2010 to January 2024. The keywords include “inflammation, coagulation, obesity, SARS-CoV-2, COVID-19, coagulopathy, and cytokine storms”. We also include COVID-19 data from the World Health Organization (WHO) 2023.
3. Mechanisms of Inflammation and Coagulopathy in Severe COVID-19
3.1. Entry and Life Cycle of SARS-CoV-2 Virus
SARS-CoV-2 viral entry occurs via a host cell receptor known as angiotensin-converting enzyme 2 (ACE2), previously recognized as a SARS-CoV-2 receptor; a process mediated via its spike (S) protein, which consists of S1 and S2 subunits [14]. ACE2 is a functional receptor that is highly expressed in pulmonary epithelial cells. This receptor is also present in cells of other vital organs, such as gastrointestinal tracts, heart, endothelial cells, and kidneys [15]. During SARS-CoV-2 infection, the S1 protein binds to ACE2, leading to exposure of another cleavage site, called the ‘S2 site’ on S2 subunit, which is subsequently cleaved by host proteases [16]. These host proteases, namely type II transmembrane serine protease (TMPRSS2) and lysosomal cathepsins are critical for S2 subunit activation, resulting in dissociation of S1 and drastic structural change in S2, exposing fusion peptide, promoting membrane fusion [17]. S2 subunit activation at the plasma membrane surface is stimulated by TMPRSS2, while that in endosomes is induced by lysosomal cathepsin, which can also compensate for viral invasion into cells lacking TMPRSS2 [17]. Following membrane fusion, the viral RNA genome is released into the cytosol of the host cell via fusion pore, inducing viral polymerase translation and RNA replication by host cell mechanisms, which subsequently leads to virion assembly and exocytosis of the newly synthesized virion, which can infect new host cells and continue the replication cycle [18].
3.2. SARS-CoV-2-Induced Coagulopathy
Coagulation is a crucial mechanism to prevent excessive bleeding in the damaged vessel walls. It consists of two main phases, namely primary and secondary hemostasis. During primary hemostasis, following a vascular injury, platelets in circulation begin to adhere to the exposed collagen fibers at the site of injury, facilitated by von Willebrand factor (vWF) (Figure 1). The platelets become activated and secrete their granules, such as adenosine diphosphate (ADP), and produce substances such as thromboxane A2 (TXA2) which promote further platelet aggregation. The coagulation cascades begin in secondary hemostasis when the platelets have formed a temporary plug at the site of injury. Activation of Tissue Factor (TF) subsequently triggers the intrinsic (Factor XII) and/or extrinsic (Factor XII) pathway activation and eventually converges at the activation of factor X, which then combines with other factors to activate prothrombin into thrombin by the prothrombinase complex. Thrombin mediates the conversion of soluble fibrinogen into insoluble fibrin which polymerizes and is cross-linked by factor XIIIa to create a stable blood clot [19,20]. Coagulation is kept in check by two other systems, namely the anti-coagulant, mediated by Thrombomodulin (TM), Protein C and Protein S; and the fibrinolytic involving factors such as plasminogen and its activator [21,22]. The latter system converges on the formation of plasmin which then breaks down fibrin. An imbalance of any of these systems results in coagulopathy, leading to uncontrolled thrombosis or bleeding.
3.3. SARS-CoV-2-Induced Inflammation
Following viral infection alveolar epithelial cells undergo programmed cell death induced by pathogen-associated molecular patterns (PAMPs) such as viral RNA the subsequent release of damage-associated molecular patterns (DAMPs) such as RNA or histone, triggers activation of macrophages that lead to high cytokines secretion, particularly interleukin (IL)-6 and reactive oxygen species (ROS) [23]. Circulation of IL-6 receptor complexes and cytokines indirectly activates a series of cells, including endothelial cells, platelets, and neutrophils, culminating in an exaggerated production of pro-inflammatory cytokines, which ultimately results in a cytokine storm. Cytokine storms occur due to significant overexpression of inflammatory cytokines that initiate lung inflammation and damage, followed by pneumonia, ARDS, septic shock, loss of respiratory functions, and ultimately death [24]. The critical nuclear factor-κB (NF-ĸB) pathway is activated in a cytokine storm, releasing excessive inflammatory cytokines, such as IL-1β, IL-2, IL-7, IL-10, granulocyte-colony stimulating factor (G-CSF), Interferon-γ-inducible protein 10 (IP10), monocyte chemoattractant protein-1 (MCP-1), macrophage inflammatory protein (MIP) 1-A, and tumor necrosis factor (TNF)-α in severe COVID-19 patients and the resulting inflammation may lead to the development and progression of ARDS. COVID-19 patients admitted to the ICU had a higher concentration of these cytokines than healthy individuals, and has been reported in secondary hemophagocytic lymphohistiocytosis (HLH) associated with severe COVID-19 [25,26].
3.4. The Crosstalk Between Inflammation and Coagulation
Crosstalk between factor XIIa in the coagulation cascade and kallikrein and kinin system (KKS) is crucial in the cytokine storm and coagulopathy in COVID-19 patients, activated by SARS-CoV-2 RNA (Figure 2) [27]. Loss of inactivation of the pro-inflammatory des-Arg9 bradykinin by ACE2 increases the production of inflammatory cytokines and oxidative stress by activating the kinin B1 receptor (KB1R) [28]. Increased inflammatory cytokines in COVID-19 patients also induce TF and P-selectin in vascular endothelium cells, promoting vascular thrombosis and neutrophil extracellular traps (NETs) formation, respectively [29]. Moreover, inhibition of anticoagulants by IL-1 elevates thrombin production and aggravates vascular thrombosis [30]. Overexpression of P-selectin directly correlates with higher TF levels in monocytes, promoting DIC occurrence [31]. This condition is common in severe COVID-19 patients, where D-dimer induces P-selectin and, subsequently, TF overexpression [32]. Therefore, stimulating P-selectin production via D-dimer and high TF expression level is a mechanism for coagulopathy in severe COVID-19 patients.
Overexpression of both angiotensin II (AngII) and angiotensin II receptor type 1 (AT1R) can also lead to overproduction of TF by monocytes and vascularized endothelial cells [33]. Activated platelets have also been shown to stimulate TF expression by monocytes, via P-selectin and integrin αIIb/β3 [34], thus promoting a TF-dependent coagulation pathway. AngII/AT1R axis also inhibits the fibrinolytic pathway by increasing plasminogen activator inhibitor 1 (PAI-1) expression, further promoting a hypercoagulable state [35]. Moreover, overexpression of AngII and AT1R leads to the upregulation of both intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) in vascular endothelium, promoting leucocyte adhesion, and therefore associated with the severity of COVID-19 disease [36,37]. The resultant activation of endothelial cells is reflected by elevated levels of D-dimer, which is considered a biomarker for coagulopathy and associated with poor prognosis in COVID-19 patients. Although D-dimer represents the degradation of clots by the fibrinolytic pathway hence generated in the late stage of the coagulation pathway, the mechanisms of D-dimer elevation in COVID-19 patients are not fully understood.
Systemic inflammation in COVID-19 resulting from the severe elevation of inflammatory cytokines has been observed in association with the activation of coagulation cascades, thrombosis formation, and endothelial cell dysfunction [38]. The resultant hyperinflammatory and hypercoagulability condition, characterized by cytokine storm and overstimulated coagulation cascade with impaired fibrinolytic system, respectively, is aptly coined as immuno-thrombosis [37]. Generation of vascular microthrombi eventually leads to systemic DIC, followed eventually by multi-organ failure [37]. In addition, the formation of platelet-neutrophil complexes (PNCs) and toxic NETs promote coagulopathy in several ways, including further cellular recruitment, activation of platelet and coagulation cascade pathways; and inhibition of anti-coagulant and fibrinolytic pathways which could confer detrimental effects in endothelial injury, lung damage, and thrombosis [39,40].
Disseminated intravascular coagulopathy is a condition where the formation of blood clots and thrombosis in vessels obstruct blood flow due to inflammation-mediated activation of the blood coagulation cascade [41]. DIC, accompanied by fibrin formation in blood vessels, leads to organ dysfunction and increased patient mortality [42]. Coagulopathy occurs when the balance between coagulation and anticoagulant factors is disturbed [43]. On one hand, pro-inflammatory cytokines and CRP can induce blood coagulation cascade through TF/factor VIIa complex activation, primarily driven by the vascular endothelium cells. On the other hand, when these cells encounter pathogens or inflammatory cytokines, they lose their antithrombin (AT) properties, resulting in overall favorable conditions for coagulation [44,45]. In addition, hypoxic conditions due to lung damage in COVID-19 patients exacerbate the stimulation of TF production by the endothelium; thus, high TF levels are predictors of increased morbidity and mortality [46].
The TF pathway activation by cytokines IL-1, IL-6, and TNF-α also downregulates anticoagulant factors such as AT, thrombomodulin (TM), and endothelial protein C receptor (EPCR), thus preventing fibrinolysis and promoting DIC [47,48]. Nevertheless, the relationship between inflammation and coagulation is not unidirectional. Coagulation can lead to inflammatory responses by protease-activated receptors (PARs) located on the endothelium of vessels and platelets. PARs bind to factor VIIa upon activation and increase inflammatory cytokines production, including IL-1β and TNF-α and ROS-induced oxidative stress [49,50]. Furthermore, TM/activated protein C (APC) and its EPCR are essential in eliminating coagulation factors, such as factor Va and VIIIa [51]. The APC downregulation leads to endotheliopathy and vascular thrombosis[52].
High levels of coagulation factors, such as von Willebrand factor (vWF), factor VIII, and soluble TM indicate endothelial damage in severe COVID-19 found in patients’ lung samples [53]. The TNF-α and IL-1 cytokines play an important role in activating the endothelial damage biomarkers in this process, indicating the activation of coagulation cascades by inflammatory cytokines [54]. In addition, downregulated AT levels have been reported in hospitalized COVID-19 patients, which led to increased ventilator requirement and mortality [55]. Therefore, AT administration is recommended for critical COVID-19 patients hospitalized in the ICU, but discrepancies have been highlighted between studies [56].
Coagulopathy caused by venous thromboembolism (VTE) events and unresponsiveness to prophylactic anticoagulants is common in severe COVID-19 patients in the ICU [57]. This condition promotes heart and brain ischemia, which increases mortality among COVID-19 patients [58]. D-dimer, which is a coagulopathy biomarker, exceeding 1000 ng/mL predicts VTE incidence and mortality in COVID-19 [59]. Furthermore, the upregulation of TF, factor VIII, and vWF and the downregulation of anticoagulant factors, such as protein C and AT III, have been reported in COVID-19 cases [60].
4. Hyperinflammation and Coagulation in COVID-19 Patients with Obesity
The prevalence of obesity has continued to rise in recent years, and approximately a quarter of the world’s population is obese or overweight. Obesity-associated complications and mortality are relatively high due to co-morbidities such as cardiovascular disease (CVD), hypertension, and diabetes mellitus [61]. In light of the recent global pandemic, obesity has been identified as one of the risk factors for severe COVID-19 [62]. Clinical reports have shown that almost half of COVID-19 patients hospitalized in the ICU had a BMI > 30, and the high severity of COVID-19 among young patients was also correlated with obesity [63]. The most critically ill COVID-19 patients admitted to the ICU exhibited hypercoagulopathy symptoms, primarily caused by severe inflammatory responses and endotheliopathy, which activate the coagulation cascade through the TF pathway [64]. These patients also demonstrate high D-dimer levels and long prothrombin time (PT). Therefore, increased inflammation and activation of coagulation pathways lead to coagulopathy, known as thromboinflammation.
A meta-analysis demonstrated that obese subjects are at higher risk of severe disease and mortality due to COVID-19 [65]. Meanwhile, Gao et al. (2020) reported high plasma CRP levels and low lymphocyte counts in COVID-19 patients with obesity [66]. Consequently, these patients had a longer hospital stay (median = 23 days) compared to non-obese COVID-19 patients (median = 18 days) [66]. Therefore, COVID-19 patients with obesity face a higher risk of severe COVID-19, ICU admission, and death compared to non-obese patients. ACE2 overexpression, leptin production, and pre-existing inflammation are reported to contribute to severe COVID-19 in patients with obesity and further reduce their chances of survival.
4.1. Overexpression of ACE2
SARS-CoV-2 utilizes the ACE2 receptor to initiate infection, and the high expression of this receptor has been associated with severe COVID-19 [67]. One of the contributing factors to the high probability of severe COVID-19 among patients with obesity is the high visceral fat tissue, which is associated with the high expression of the ACE2 receptor [68]. Overexpression of the ACE2 gene in adipose tissue has been associated with increased ICU admission, ventilator requirement, and mortality rate in severe patients with obesity (BMI > 40) affected by COVID-19 [68]. Furthermore, high expression of the ACE2 receptor is also found particularly in vital organs such as the lung, heart, kidney, gastrointestinal tract, and visceral tissue could lead to further dissemination and high viral load in COVID-19 patients with obesity [69].
4.1. Leptin
The adipose tissue is a source of energy and essential in regulating inflammatory responses to infections [70]. Adipocytes are responsible for secreting various adiponectins, including leptin, which is abundantly detected in individuals with obesity [71]. High serum leptin in individuals with obesity is an indicator of cell resistance to leptin, impacting the signal transduction pathways Janus kinase-2 (JAK2) and signal transducer and activator of transcription 3 (STAT3), besides promoting inflammatory cytokines including IL-6 and TNF-α production by monocytes [72,73].
Leptin also causes CRP overexpression in hepatocytes, indicating the pro-inflammatory role of this adipokine [74]. Precisely, leptin regulates the inflammatory responses via leptin receptor modulation and upregulates multiple inflammatory cytokines (i.e., TNF-α and IL-6) [72]. Likewise, a surge in leptin levels leads to the activation and proliferation of immune cells, increasing TNF-α and IL-6 levels, thus aggravating the inflammatory responses of obesity with high leptin levels [75]. A previous report highlighted that severe COVID-19 patients hospitalized in ICU and under mechanical ventilation have high leptin levels, providing the basis for ARDS development [76]. High leptin levels render patients with obesity susceptible to inflammation and increase their risk of highly severe COVID-19. High leptin levels positively correlate with neutrophilic lung inflammation, lung damage, and ARDS in COVID-19 patients [77]. Thus, obesity is a significant risk factor for the high severity of COVID-19 due to the high leptin and inflammatory factors production, particularly IL-6, by excessive visceral fat.
4.1. Pre-Existing Inflammation
Visceral tissue composition is correlated with metabolic syndrome diseases, such as type 2 diabetes mellitus (T2DM) [78]. High levels of inflammatory factors such as CRP, IL-6, and TNF-α have been detected in obesity, which is attributed to the excessive visceral fat tissue that regulates the body’s metabolism by producing adipokines, chemokines, and various growth factors [79,80]. Thus, subclinical inflammation exists in obesity, creating a suitable environment for other diseases to develop in the long term, such as T2DM [81].
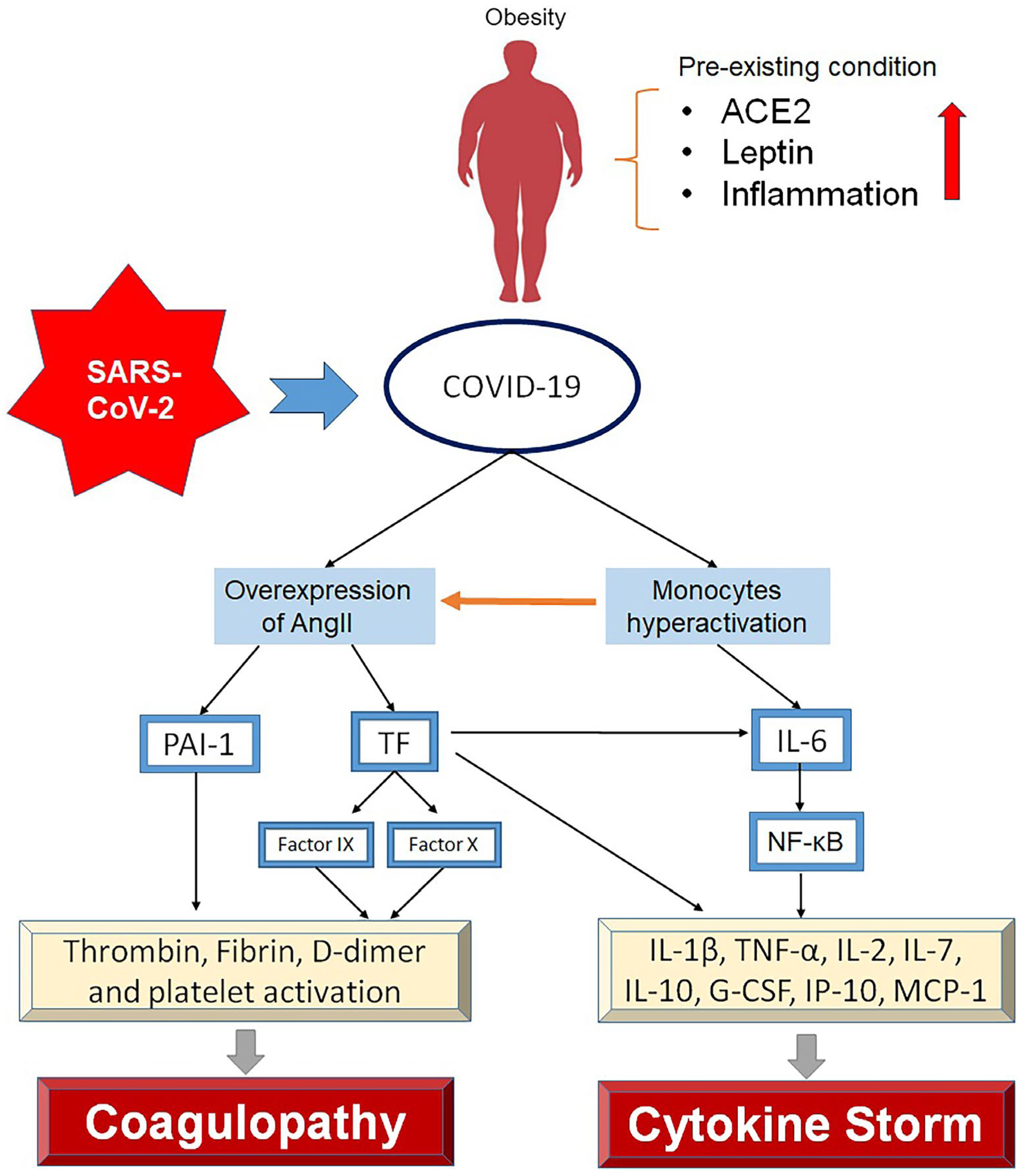
Acute IL-6 upregulation in visceral tissue has been reported in infectious diseases associated with the cytokine storm and various tissue damage [82]. High IL-6 levels often lead to a cytokine storm in COVID-19 patients, particularly those who are obese [83]. The relationship between IL-6 and leptin has been linked to the increased mortality of COVID-19 patients with obesity. High leptin, IL-6, and TNF-α levels have been reported in obesity, which provides a favorable environment for cytokine storms (Figure 3) [84]. Leptin and IL-6 receptors activate the JAK/STAT pathway and increase IL-6 production, causing the phosphorylation of the Akt/PI3K STAT3 signaling pathway [85]. This process triggers the activation of the NF-κB pathway and IL-6 overexpression, giving rise to TNF-α and IL-1 production [86]. Moreover, IL-6 overproduces to increase vascular permeability, resulting in a high viral load [87], intense inflammatory responses, and the overproduction of inflammatory cytokines. This condition activates the TF coagulation pathway, thrombosis, and coagulopathy. This process elucidates the sensitivity of patients with obesity to SARS-CoV-2 infection and their potential vulnerability to the disease severity and mortality. When IL-6 receptor inhibitor (i.e., Tocilizumab) was administered to patients, a reduction in ARDS was observed [88]. In COVID-19 patients with obesity, the dosage of neutralizing antibodies is indiscernible compared to autoimmune antibodies, causing a weak response to infection and high-grade fevers [89].
5. Therapeutic Approach in COVID-19 Patients with Obesity
The development of COVID-19 vaccines has significantly reduced the death toll of this disease worldwide. In general, COVID-19 vaccines can be divided into three types; mRNA, vector, and protein vaccines, all of which target the S protein of the virus[90]. However, there are inter-individual differences in vaccine responses. For example, individuals with obesity were administered with double doses of the BNT162b2 or CoronaVac (Sinovac) vaccine mostly displayed a significantly lower antibody titer than their non-obese counterparts, indicating the poor response of this group to the vaccine [91]. Gender differences in response to the vaccine have also been reported; men with obesity had lower antibody titers compared to women; thus, men are more susceptible to the SARS-CoV-2 infection [92].
Alternatively, inhibition of inflammatory factors, particularly IL-6 and TNF-α, may reduce the severity of COVID-19 in obesity [93]. Lonicera japonica and Astragalus membranaceus have demonstrated inhibitory effects against IL-6 and TNF-α through the upregulation of let-7a, miR-148b, and miR-146a, preventing the cytokine storm in COVID-19 patients [94]. Other IL-6 inhibitors include baricitinib, which reduces the recovery time of COVID-19 patients compared to remdesivir (95% confidence interval [CI], 6 to 8) [95]. In addition, sarilumab binds to the IL-6 receptor and was found to improve the survival of hospitalized patients [96]. Tocilizumab, another IL-6 receptor inhibitor, reduced the mortality of critically ill COVID-19 patients [97]. Administration of IL-6 receptor inhibitors in COVID-19 patients with obesity potentially prevents cytokine storms and improves survival. Therefore, specific treatment strategies for patients with obesity should be established to avoid future complications in this group.
6. Conclusions
Obesity is a risk factor for the highly severe SARS-CoV-2 infection and increases mortality. The primary contributing factors to this condition are pre-subclinical inflammation and high IL-6 levels, which increase one’s risk of severe inflammatory responses by activating inflammatory pathways. These pro-inflammatory factors activate coagulation pathways, such as TF, and provide the basis for thrombosis, DIC, and hypercoagulopathy. Therefore, there is a synergistic interaction between inflammation and coagulation in SARS-CoV-2 infection, whereby inflammation leads to the activation of coagulation cascades. This crosstalk is more severe in COVID-19 patients with obesity, given their background pro-inflammatory state, thus rendering them susceptible to high disease severity and mortality. Inhibiting the inflammatory cytokines IL-6/TNF-α could be a promising treatment option for this group of patients and should be further explored in future studies.
Author Contributions
NT: MH, and ERMT conceived and designed the manuscript with input and advice from MA, SYZS, AMG, NFZZ, IZI, and SAN. NT and MH wrote the manuscript and NT, MA, ERMT, SYZS, AMG, and MH revised the manuscript.
Funding
This review is part of research funded by the Fundamental Research Grant Scheme (grant number 5540518) from the Ministry of Education, Malaysia.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Organization, W.H. Number of COVID-19 deaths reported to WHO. Available online: https://data.who.int/dashboards/covid19/deaths?n=c (accessed on 1 September 2024).
- Alimohamadi, Y.; Sepandi, M.; Taghdir, M.; Hosamirudsari, H. Determine the most common clinical symptoms in COVID-19 patients: a systematic review and meta-analysis. J. Prev. Med. Hyg. 2020, 61, E304–E312. [Google Scholar] [CrossRef] [PubMed]
- Castilla, J.; Guevara, M.; Miqueleiz, A.; Baigorria, F.; Ibero-Esparza, C.; Navascués, A.; Trobajo-Sanmartín, C.; Martínez-Baz, I.; Casado, I.; Burgui, C.; et al. Risk Factors of Infection, Hospitalization and Death from SARS-CoV-2: A Population-Based Cohort Study. J. Clin. Med. 2021, 10, 2608. [Google Scholar] [CrossRef] [PubMed]
- Sim, B.L.H.; Chidambaram, S.K.; Wong, X.C.; Pathmanathan, M.D.; Peariasamy, K.M.; Hor, C.P.; Chua, H.J.; Goh, P.P. Clinical characteristics and risk factors for severe COVID-19 infections in Malaysia: A nationwide observational study. Lancet Reg. Heal. - West. Pac. 2020, 4, 100055–100055. [Google Scholar] [CrossRef] [PubMed]
- Fericean, R.M.; Citu, C.; Manolescu, D.; Rosca, O.; Bratosin, F.; Tudorache, E.; Oancea, C. Characterization and Outcomes of SARS-CoV-2 Infection in Overweight and Obese Patients: A Dynamic Comparison of COVID-19 Pandemic Waves. J. Clin. Med. 2022, 11, 2916. [Google Scholar] [CrossRef]
- Aghili, S.M.M.; Ebrahimpur, M.; Arjmand, B.; Shadman, Z.; Sani, M.P.; Qorbani, M.; Larijani, B.; Payab, M. Obesity in COVID-19 era, implications for mechanisms, comorbidities, and prognosis: a review and meta-analysis. Int. J. Obes. 2021, 45, 998–1016. [Google Scholar] [CrossRef] [PubMed]
- Renovato-Martins, M.; Moreira-Nunes, C.; Atella, G.C.; Barja-Fidalgo, C.; de Moraes, J.A. Obese Adipose Tissue Secretion Induces Inflammation in Preadipocytes: Role of Toll-Like Receptor-4. Nutrients 2020, 12, 2828. [Google Scholar] [CrossRef]
- Zhou, Y.; Chi, J.; Lv, W.; Wang, Y. Obesity and diabetes as high-risk factors for severe coronavirus disease 2019 (Covid-19). Diabetes/Metab. Res. Rev. 2020, 37, e3377. [Google Scholar] [CrossRef]
- Petrilli, C.M.; Jones, S.A.; Yang, J.; Rajagopalan, H.; O’donnell, L.; Chernyak, Y.; Tobin, K.A.; Cerfolio, R.J.; Francois, F.; Horwitz, L.I. Factors associated with hospital admission and critical illness among 5279 people with coronavirus disease 2019 in New York City: prospective cohort study. BMJ 2020, 369. [Google Scholar] [CrossRef]
- Lighter, J.; Phillips, M.; Hochman, S.; Sterling, S.; Johnson, D.; Francois, F.; Stachel, A. Obesity in Patients Younger Than 60 Years Is a Risk Factor for COVID-19 Hospital Admission. Clin. Infect. Dis. 2020, 71, 896–897. [Google Scholar] [CrossRef]
- Simonnet, A.; Chetboun, M.; Poissy, J.; Raverdy, V.; Noulette, J.; Duhamel, A.; Labreuche, J.; Mathieu, D.; Pattou, F.; Jourdain, M. High prevalence of obesity in severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) requiring invasive mechanical ventilation. Obesity 2020, 28, 1195–1199. [Google Scholar] [CrossRef]
- Danial, M.; Arulappen, A.L.; Soelar, S.A.; Ch’ng, A.S.H.; Looi, I. Clinical Characteristics of Individuals Died with COVID-19 in Malaysia. Malays. J. Pharm. Sci. 2022, 20, 105–120. [Google Scholar] [CrossRef]
- Jiang, Y.; Rubin, L.; Peng, T.; Liu, L.; Xing, X.; Lazarovici, P.; Zheng, W. Cytokine storm in COVID-19: from viral infection to immune responses, diagnosis and therapy. Int. J. Biol. Sci. 2022, 18, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, W.; Yang, L.; You, R. Physiological and pathological regulation of ACE2, the SARS-CoV-2 receptor. Pharmacol. Res. 2020, 157, 104833–104833. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Beyerstedt, S.; Casaro, E.B.; Rangel, É.B. COVID-19: angiotensin-converting enzyme 2 (ACE2) expression and tissue susceptibility to SARS-CoV-2 infection. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 905–919. [Google Scholar] [CrossRef]
- Periayah, M.H.; Halim, A.S.; Mat Saad, A.Z. Mechanism Action of Platelets and Crucial Blood Coagulation Pathways in Hemostasis. Int. J. Hematol. Oncol. Stem Cell Res. 2017, 11, 319–327. [Google Scholar]
- Palta, S.; Saroa, R.; Palta, A. Overview of the coagulation system. Indian J. Anaesth. 2014, 58, 515–523. [Google Scholar] [CrossRef]
- Amiral, J.; Seghatchian, J. Revisiting the activated protein C-protein S-thrombomodulin ternary pathway: Impact of new understanding on its laboratory investigation. Transfus. Apher. Sci. 2019, 58, 538–544. [Google Scholar] [CrossRef]
- Wang, L.; Zhong, J.; Xiao, D.; Huang, W.; Zheng, Z.; Jiang, Y. Thrombomodulin (TM), thrombin-antithrombin complex (TAT), plasmin-α2-plasmininhibitor complex (PIC), and tissue plasminogen activator-inhibitor complex (t-PAIC) assessment of fibrinolytic activity in postpartum hemorrhage: a retrospective comparative cohort study. Ann. Transl. Med. 2022, 10, 1273–1273. [Google Scholar] [CrossRef] [PubMed]
- Subbarao, K.; Mahanty, S. Respiratory Virus Infections: Understanding COVID-19. Immunity 2020, 52, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Prompetchara, E.; Kettoy, C.; Palaga, T. Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic. Asian Pac. J. Allergy Immunol. 2020, 38, 1–9. [Google Scholar] [CrossRef]
- Claudia, N.-T.; Ana, F.-G.; Esther Moreno, M.; Belen, P.-M.; Jesús, V.; Sandra, C.; Javier, L.-J.; Palacios, J.; Miguel, P.-V.; Mónica, G.-C. Secondary haemophagocytic lymphohistiocytosis in COVID-19: correlation of the autopsy findings of bone marrow haemophagocytosis with HScore. J. Clin. Pathol. 2022, 75, 383. [Google Scholar]
- Abdin, S.M.; Elgendy, S.M.; Alyammahi, S.K.; Alhamad, D.W.; Omar, H.A. Tackling the cytokine storm in COVID-19, challenges and hopes. Life Sci. 2020, 257, 118054–118054. [Google Scholar] [CrossRef]
- Mackman, N.; Antoniak, S.; Wolberg, A.S.; Kasthuri, R.; Key, N.S. Coagulation abnormalities and thrombosis in patients infected with SARS-CoV-2 and other pandemic viruses. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2033–2044. [Google Scholar] [CrossRef]
- Zhang, X.; Lowry, J.L.; Brovkovych, V.; Skidgel, R.A. Characterization of dual agonists for kinin B1 and B2 receptors and their biased activation of B2 receptors. Cell. Signal. 2012, 24, 1619–1631. [Google Scholar] [CrossRef]
- Agrati, C.; Sacchi, A.; Tartaglia, E.; Vergori, A.; Gagliardini, R.; Scarabello, A.; Bibas, M. The Role of P-Selectin in COVID-19 Coagulopathy: An Updated Review. Int. J. Mol. Sci. 2021, 22, 7942. [Google Scholar] [CrossRef]
- Burzynski, L.C.; Morales-Maldonado, A.; Rodgers, A.; Kitt, L.A.; Humphry, M.; Figg, N.; Bennett, M.R.; Clarke, M.C.H. Thrombin-activated interleukin-1α drives atherogenesis, but also promotes vascular smooth muscle cell proliferation and collagen production. Cardiovasc. Res. 2023, 119, 2179–2189. [Google Scholar] [CrossRef]
- Mosad, E.; Elsayh, K.I.; Eltayeb, A.A. Tissue Factor Pathway Inhibitor and P-Selectin as Markers of Sepsis-Induced Non-overt Disseminated Intravascular Coagulopathy. Clin. Appl. Thromb. 2009, 17, 80–87. [Google Scholar] [CrossRef]
- Hottz, E.D.; Azevedo-Quintanilha, I.G.; Palhinha, L.; Teixeira, L.; Barreto, E.A.; Pão, C.R.R.; Righy, C.; Franco, S.; Souza, T.M.L.; Kurtz, P.; et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood 2020, 136, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Ekholm, M.; Kahan, T. The Impact of the Renin-Angiotensin-Aldosterone System on Inflammation, Coagulation, and Atherothrombotic Complications, and to Aggravated COVID-19. Front. Pharmacol. 2021, 12, 640185. [Google Scholar] [CrossRef] [PubMed]
- Hottz, E.D.; Azevedo-Quintanilha, I.G.; Palhinha, L.; Teixeira, L.; Barreto, E.A.; Pão, C.R.R.; Righy, C.; Franco, S.; Souza, T.M.L.; Kurtz, P.; et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood 2020, 136, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Daher, J. Endothelial dysfunction and COVID-19 (Review). Biomed. Rep. 2021, 15, 1–6. [Google Scholar] [CrossRef]
- Tong, M.; Jiang, Y.; Xia, D.; Xiong, Y.; Zheng, Q.; Chen, F.; Zou, L.; Xiao, W.; Zhu, Y. Elevated Expression of Serum Endothelial Cell Adhesion Molecules in COVID-19 Patients. J. Infect. Dis. 2020, 222, 894–898. [Google Scholar] [CrossRef]
- Gando, S.; Wada, T. Thromboplasminflammation in COVID-19 Coagulopathy: Three Viewpoints for Diagnostic and Therapeutic Strategies. Front. Immunol. 2021, 12, 649122. [Google Scholar] [CrossRef]
- Darif, D.; Hammi, I.; Kihel, A.; El Idrissi Saik, I.; Guessous, F.; Akarid, K. The pro-inflammatory cytokines in COVID-19 pathogenesis: What goes wrong? Microb. Pathog. 2021, 153, 104799. [Google Scholar] [CrossRef] [PubMed]
- González-Jiménez, P.; Méndez, R.; Latorre, A.; Piqueras, M.; Balaguer-Cartagena, M.N.; Moscardó, A.; Alonso, R.; Hervás, D.; Reyes, S.; Menéndez, R. Neutrophil Extracellular Traps and Platelet Activation for Identifying Severe Episodes and Clinical Trajectories in COVID-19. Int. J. Mol. Sci. 2023, 24, 6690. [Google Scholar] [CrossRef]
- Morris, G.; Bortolasci, C.C.; Puri, B.K.; Olive, L.; Marx, W.; O’Neil, A.; Athan, E.; Carvalho, A.F.; Maes, M.; Walder, K.; et al. The pathophysiology of SARS-CoV-2: A suggested model and therapeutic approach. Life Sci. 2020, 258, 118166. [Google Scholar] [CrossRef]
- Gando, S.; Levi, M.; Toh, C.-H. Disseminated intravascular coagulation. Nat. Rev. Dis. Primers 2016, 2, 16037. [Google Scholar] [CrossRef]
- Gando, S.; Fujishima, S.; Saitoh, D.; Shiraishi, A.; Yamakawa, K.; Kushimoto, S.; Ogura, H.; Abe, T.; Mayumi, T.; Sasaki, J.; et al. The significance of disseminated intravascular coagulation on multiple organ dysfunction during the early stage of acute respiratory distress syndrome. Thromb. Res. 2020, 191, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Hadid, T.; Kafri, Z.; Al-Katib, A. Coagulation and anticoagulation in COVID-19. Blood Rev. 2020, 47, 100761. [Google Scholar] [CrossRef] [PubMed]
- Norooznezhad, A.H.; Mansouri, K. Endothelial cell dysfunction, coagulation, and angiogenesis in coronavirus disease 2019 (COVID-19). Microvasc. Res. 2021, 137, 104188–104188. [Google Scholar] [CrossRef] [PubMed]
- Grover, S.P.; Mackman, N. Tissue factor in atherosclerosis and atherothrombosis. Atherosclerosis 2020, 307, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Rosell, A.; Havervall, S.; Von Meijenfeldt, F.; Hisada, Y.; Aguilera, K.; Grover, S.P.; Lisman, T.; Mackman, N.; Thålin, C. Patients With COVID-19 Have Elevated Levels of Circulating Extracellular Vesicle Tissue Factor Activity That Is Associated With Severity and Mortality—Brief Report. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 878–882. [Google Scholar] [CrossRef] [PubMed]
- Popescu, N.I.; Lupu, C.; Lupu, F. Disseminated intravascular coagulation and its immune mechanisms. Blood 2021, 139, 1973–1986. [Google Scholar] [CrossRef]
- Heidari, Z.; Naeimzadeh, Y.; Fallahi, J.; Savardashtaki, A.; Razban, V.; Khajehا, S. The Role of Tissue Factor In Signaling Pathways of Pathological Conditions and Angiogenesis. Curr. Mol. Med. 2024, 24, 1135–1151. [Google Scholar] [CrossRef]
- Barra, A.; Brasil, A.F.; Ferreira, T.L.; Fernandes-Braga, W.; Marconato, D.G.; Faria-Pinto, P.; Alvarez-Leite, J.I.; Capettini, L.d.S.A.; Klein, A. Protease-activated receptor 2 enhances innate and inflammatory mechanisms induced by lipopolysaccharide in macrophages from C57BL/6 mice. Inflamm. Res. 2022, 71, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Posma, J.J.; Grover, S.P.; Hisada, Y.; Owens III, A.P.; Antoniak, S.; Spronk, H.M.; Mackman, N. Roles of coagulation proteases and PARs (protease-activated receptors) in mouse models of inflammatory diseases. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 13–24. [Google Scholar] [CrossRef]
- Ikezoe, T. Thrombomodulin/activated protein C system in septic disseminated intravascular coagulation. J. Intensiv. Care 2015, 3, 1–8. [Google Scholar] [CrossRef]
- Agarwal, S.; Cohen, C.T.; Zobeck, M.; Jacobi, P.M.; Sartain, S.E. Downregulation of thrombomodulin-thrombin-activated protein C pathway as a mechanism for SARS-CoV-2 induced endotheliopathy and microvascular thrombosis. Thromb. Update 2022, 8, 100116. [Google Scholar] [CrossRef]
- Cugno, M.; Meroni, P.L.; Gualtierotti, R.; Griffini, S.; Grovetti, E.; Torri, A.; Lonati, P.; Grossi, C.; Borghi, M.O.; Novembrino, C.; et al. Complement activation and endothelial perturbation parallel COVID-19 severity and activity. J. Autoimmun. 2020, 116, 102560. [Google Scholar] [CrossRef] [PubMed]
- Cacciola, R.; Cacciola, E.G.; Vecchio, V.; Cacciola, E. Cellular and molecular mechanisms in COVID-19 coagulopathy: role of inflammation and endotheliopathy. J. Thromb. Thrombolysis 2021, 53, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Gazzaruso, C.; Paolozzi, E.; Valenti, C.; Brocchetta, M.; Naldani, D.; Grignani, C.; Salvucci, F.; Marino, F.; Coppola, A.; Gallotti, P. Association between antithrombin and mortality in patients with COVID-19. A possible link with obesity. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 1914–1919. [Google Scholar] [CrossRef]
- Anaklı. ; Özcan, P.E.; Polat,.; Orhun, G.; Alay, G.H.; Tuna, V.; Çeliksoy, E.; Kılıç, M.; Mercan, M.; Ali, A.; et al. Prognostic Value of Antithrombin Levels in COVID-19 Patients and Impact of Fresh Frozen Plasma Treatment: A Retrospective Study. Turk. J. Hematol. 2021, 38, 15–21. [Google Scholar] [CrossRef]
- Klok, F.A.; Kruip, M.J.H.A.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.A.M.P.J.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020, 191, 145–147. [Google Scholar] [CrossRef]
- Stein, L.K.; Mayman, N.A.; Dhamoon, M.S.; Fifi, J.T. The emerging association between COVID-19 and acute stroke. Trends Neurosci. 2021, 44, 527–537. [Google Scholar] [CrossRef]
- Elkhalifa, A.M.E. D-dimer as a predictive and prognostic marker among COVID-19 patients. SciVee 2022, 43, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Mesa, J.E.; Galindo-Coral, S.; Montes, M.C.; Martin, A.J.M. Thrombosis and Coagulopathy in COVID-19. Curr. Probl. Cardiol. 2021, 46, 100742–100742. [Google Scholar] [CrossRef]
- Blüher, M. Obesity: global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef]
- Drucker, D.J. Diabetes, obesity, metabolism, and SARS-CoV-2 infection: the end of the beginning. Cell Metabolism 2021, 33, 479–498. [Google Scholar] [CrossRef]
- Kass, D.A.; Duggal, P.; Cingolani, O. Obesity could shift severe COVID-19 disease to younger ages. Lancet 2020, 395, 1544–1545. [Google Scholar] [CrossRef] [PubMed]
- Joly, B.S.; Siguret, V.; Veyradier, A. Understanding pathophysiology of hemostasis disorders in critically ill patients with COVID-19. Intensiv. Care Med. 2020, 46, 1603–1606. [Google Scholar] [CrossRef]
- Singh, R.; Rathore, S.S.; Khan, H.; Karale, S.; Chawla, Y.; Iqbal, K.; Bhurwal, A.; Tekin, A.; Jain, N.; Mehra, I.; et al. Association of Obesity With COVID-19 Severity and Mortality: An Updated Systemic Review, Meta-Analysis, and Meta-Regression. Front. Endocrinol. 2022, 13, 780872. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Zheng, K.I.; Wang, X.-B.; Sun, Q.-F.; Pan, K.-H.; Wang, T.-Y.; Chen, Y.-P.; Targher, G.; Byrne, C.D.; George, J.; et al. Obesity Is a Risk Factor for Greater COVID-19 Severity. Diabetes Care 2020, 43, e72–e74. [Google Scholar] [CrossRef]
- Krishnamurthy, S.; Lockey, R.F.; Kolliputi, N. Soluble ACE2 as a potential therapy for COVID-19. Am. J. Physiol. Physiol. 2021, 320, C279–C281. [Google Scholar] [CrossRef]
- Al-Benna, S. Association of high level gene expression of ACE2 in adipose tissue with mortality of COVID-19 infection in obese patients. Obes. Med. 2020, 19, 100283–100283. [Google Scholar] [CrossRef]
- Bourgonje, A.R.; Abdulle, A.E.; Timens, W.; Hillebrands, J.L.; Navis, G.J.; Gordijn, S.J.; Bolling, M.C.; Dijkstra, G.; Voors, A.A.; Osterhaus, A.D. Angiotensin-converting enzyme 2 (ACE2), SARS-CoV-2 and the pathophysiology of coronavirus disease 2019 (COVID-19). J. Pathol. 2020, 251, 228–248. [Google Scholar] [CrossRef]
- Sun, S.; Ji, Y.; Kersten, S.; Qi, L. Mechanisms of Inflammatory Responses in Obese Adipose Tissue. Annu. Rev. Nutr. 2012, 32, 261–286. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.I.; Mittendorfer, B.; Klein, S. Metabolically healthy obesity: facts and fantasies. J. Clin. Investig. 2019, 129, 3978–3989. [Google Scholar] [CrossRef]
- Agrawal, S.; Gollapudi, S.; Su, H.; Gupta, S. Leptin activates human B cells to secrete TNF-α, IL-6, and IL-10 via JAK2/STAT3 and p38MAPK/ERK1/2 signaling pathway. J. Clin. Immunol. 2011, 31, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, A.G.; Crujeiras, A.B.; Casanueva, F.F.; Carreira, M.C. Leptin, Obesity, and Leptin Resistance: Where Are We 25 Years Later? Nutrients 2019, 11, 2704. [Google Scholar] [CrossRef] [PubMed]
- Hribal, M.L.; Fiorentino, T.V.; Sesti, G. Role of C Reactive Protein (CRP) in Leptin Resistance. Curr. Pharm. Des. 2014, 20, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Acedo, S.C.; Gambero, S.; Cunha, F.G.P.; Lorand-Metze, I.; Gambero, A. Participation of leptin in the determination of the macrophage phenotype: an additional role in adipocyte and macrophage crosstalk. Vitr. Cell. Dev. Biol. - Anim. 2013, 49, 473–478. [Google Scholar] [CrossRef]
- van der Voort, P.H.; Moser, J.; Zandstra, D.F.; Kobold, A.C.M.; Knoester, M.; Calkhoven, C.F.; Hamming, I.; van Meurs, M. Leptin levels in SARS-CoV-2 infection related respiratory failure: A cross-sectional study and a pathophysiological framework on the role of fat tissue. Heliyon 2020, 6, 04696. [Google Scholar] [CrossRef]
- Rosa, B.A.; Ahmed, M.; Singh, D.K.; Choreño-Parra, J.A.; Cole, J.; Jiménez-Álvarez, L.A.; Rodríguez-Reyna, T.S.; Singh, B.; Gonzalez, O.; Carrion, R.; et al. IFN signaling and neutrophil degranulation transcriptional signatures are induced during SARS-CoV-2 infection. Commun. Biol. 2021, 4, 290. [Google Scholar] [CrossRef]
- Samaras, K.; Botelho, N.K.; Chisholm, D.J.; Lord, R.V. Subcutaneous and Visceral Adipose Tissue Gene Expression of Serum Adipokines That Predict Type 2 Diabetes. Obesity 2010, 18, 884–889. [Google Scholar] [CrossRef]
- Al-Mansoori, L.; Al-Jaber, H.; Prince, M.S.; Elrayess, M.A. Role of Inflammatory Cytokines, Growth Factors and Adipokines in Adipogenesis and Insulin Resistance. Inflammation 2021, 45, 31–44. [Google Scholar] [CrossRef]
- Illán-Gómez, F.; Gonzálvez-Ortega, M.; Orea-Soler, I.; Alcaraz-Tafalla, M.S.; Aragón-Alonso, A.; Pascual-Díaz, M.; Pérez-Paredes, M.; Lozano-Almela, M.L. Obesity and Inflammation: Change in Adiponectin, C-Reactive Protein, Tumour Necrosis Factor-Alpha and Interleukin-6 After Bariatric Surgery. Obes. Surg. 2012, 22, 950–955. [Google Scholar] [CrossRef]
- Esser, N.; Legrand-Poels, S.; Piette, J.; Scheen, A.J.; Paquot, N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res. Clin. Pract. 2014, 105, 141–150. [Google Scholar] [CrossRef]
- Michalakis, K.; Ilias, I. SARS-CoV-2 infection and obesity: Common inflammatory and metabolic aspects. Diabetes Metab. Syndr. : Clin. Res. Rev. 2020, 14, 469–471. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M. Cytokine storm and immunomodulatory therapy in COVID-19: Role of chloroquine and anti-IL-6 monoclonal antibodies. Int. J. Antimicrob. Agents 2020, 55, 105982–105982. [Google Scholar] [CrossRef] [PubMed]
- Muniz, M.G.R.; Palfreeman, M.; Setzu, N.; Sanchez, M.A.; Portillo, P.S.; Garza, K.M.; Gosselink, K.L.; Spencer, C.T. Obesity Exacerbates the Cytokine Storm Elicited by Francisella tularensis Infection of Females and Is Associated with Increased Mortality. BioMed Res. Int. 2018, 2018, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Malanga, D.; De Marco, C.; Guerriero, I.; Colelli, F.; Rinaldo, N.; Scrima, M.; Mirante, T.; De Vitis, C.; Zoppoli, P.; Ceccarelli, M.; et al. The Akt1/IL-6/STAT3 pathway regulates growth of lung tumor initiating cells. Oncotarget 2015, 6, 42667–42686. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Li, J.; Che, Y.-Q. CXCL8 gene silencing promotes neuroglial cells activation while inhibiting neuroinflammation through the PI3K/Akt/NF-κB-signaling pathway in mice with ischemic stroke. J. Cell. Physiol. 2019, 234, 7341–7355. [Google Scholar] [CrossRef]
- Terroni, B.; Lopes, J.R.; Chin, C.M.; Dos Santos, J.L. Pleiotropic Effects of Nitric Oxide on SARS-CoV-2 Infections. Coronaviruses 2021, 2, 10–17. [Google Scholar] [CrossRef]
- Sánchez-Montalvá, A.; Sellarés-Nadal, J.; Espinosa-Pereiro, J.; Fernández-Hidalgo, N.; Pérez-Hoyos, S.; Salvador, F.; Durà, X.; Miarons, M.; Antón, A.; Eremiev-Eremiev, S. Early outcomes in adults hospitalized with severe SARS-CoV-2 infection receiving tocilizumab. Med. Clínica (Engl. Ed.) 2022, 158, 509–518. [Google Scholar] [CrossRef]
- Frasca, D.; Reidy, L.; Romero, M.; Diaz, A.; Cray, C.; Kahl, K.; Blomberg, B.B. The majority of SARS-CoV-2-specific antibodies in COVID-19 patients with obesity are autoimmune and not neutralizing. Int. J. Obes. 2022, 46, 427–432. [Google Scholar] [CrossRef]
- Ndwandwe, D.; Wiysonge, C.S. COVID-19 vaccines. Curr. Opin. Immunol. 2021, 71, 111–116. [Google Scholar] [CrossRef]
- Kara, Z.; Akçin, R.; Demir, A.N.; Dinç, H.Ö.; Taşkın, H.E.; Kocazeybek, B.; Yumuk, V.D. Antibody Response to SARS-CoV-2 Vaccines in People with Severe Obesity. Obes. Surg. 2022, 32, 2987–2993. [Google Scholar] [CrossRef]
- Yamamoto, S.; Mizoue, T.; Tanaka, A.; Oshiro, Y.; Inamura, N.; Konishi, M.; Ozeki, M.; Miyo, K.; Sugiura, W.; Sugiyama, H.; et al. Sex-associated differences between BMI and SARS-CoV-2 antibody titers following the BNT162b2 vaccine. Obesity 2022, 30, 999–1003. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Hybertson, B.M.; Cota-Gomez, A.; Gao, B. Nrf2 activator PB125® as a carnosic acid-based therapeutic agent against respiratory viral diseases, including COVID-19. Free. Radic. Biol. Med. 2021, 175, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.-C.; Doan, L.H.; Huang, Z.-Y.; Chu, L.-W.; Shi, T.-H.; Lee, Y.-R.; Wu, C.-T.; Lin, C.-H.; Chiang, S.-T.; Liu, H.-K.; et al. Honeysuckle (Lonicera japonica) and Huangqi (Astragalus membranaceus) Suppress SARS-CoV-2 Entry and COVID-19 Related Cytokine Storm in Vitro. Front. Pharmacol. 2022, 12, 765553. [Google Scholar] [CrossRef] [PubMed]
- Kalil, A.C.; Patterson, T.F.; Mehta, A.K.; Tomashek, K.M.; Wolfe, C.R.; Ghazaryan, V.; Marconi, V.C.; Ruiz-Palacios, G.M.; Hsieh, L.; Kline, S.; et al. Baricitinib plus Remdesivir for Hospitalized Adults with COVID-19. N. Engl. J. Med. 2021, 384, 795–807. [Google Scholar] [CrossRef]
- Marino, A.; Munafò, A.; Augello, E.; Bellanca, C.M.; Bonomo, C.; Ceccarelli, M.; Musso, N.; Cantarella, G.; Cacopardo, B.; Bernardini, R. Sarilumab Administration in COVID-19 Patients: Literature Review and Considerations. Infect. Dis. Rep. 2022, 14, 360–371. [Google Scholar] [CrossRef]
- Tleyjeh, I.M.; Kashour, Z.; Damlaj, M.; Riaz, M.; Tlayjeh, H.; Altannir, M.; Altannir, Y.; Al-Tannir, M.; Tleyjeh, R.; Hassett, L.; et al. Efficacy and safety of tocilizumab in COVID-19 patients: a living systematic review and meta-analysis. Clin. Microbiol. Infect. 2020, 27, 215–227. [Google Scholar] [CrossRef]
Figure 1.
The schematic representation of primary and secondary hemostasis pathways results in clot formation. In primary hemostasis, the initial response to vascular injury involves a series of processes including the roles of von Willebrand factor (vWF), thromboxane A2 (TXA2) production, and platelet aggregation. During secondary hemostasis, activation of Tissue Factor (TF) initiates the activation of intrinsic (Factor XII) and/or extrinsic (Factor VIII) pathways leading to subsequent activation of Factor X. Thrombin produced from prothrombin and converts fibrinogen into fibrin and form a stable clot.
Figure 1.
The schematic representation of primary and secondary hemostasis pathways results in clot formation. In primary hemostasis, the initial response to vascular injury involves a series of processes including the roles of von Willebrand factor (vWF), thromboxane A2 (TXA2) production, and platelet aggregation. During secondary hemostasis, activation of Tissue Factor (TF) initiates the activation of intrinsic (Factor XII) and/or extrinsic (Factor VIII) pathways leading to subsequent activation of Factor X. Thrombin produced from prothrombin and converts fibrinogen into fibrin and form a stable clot.
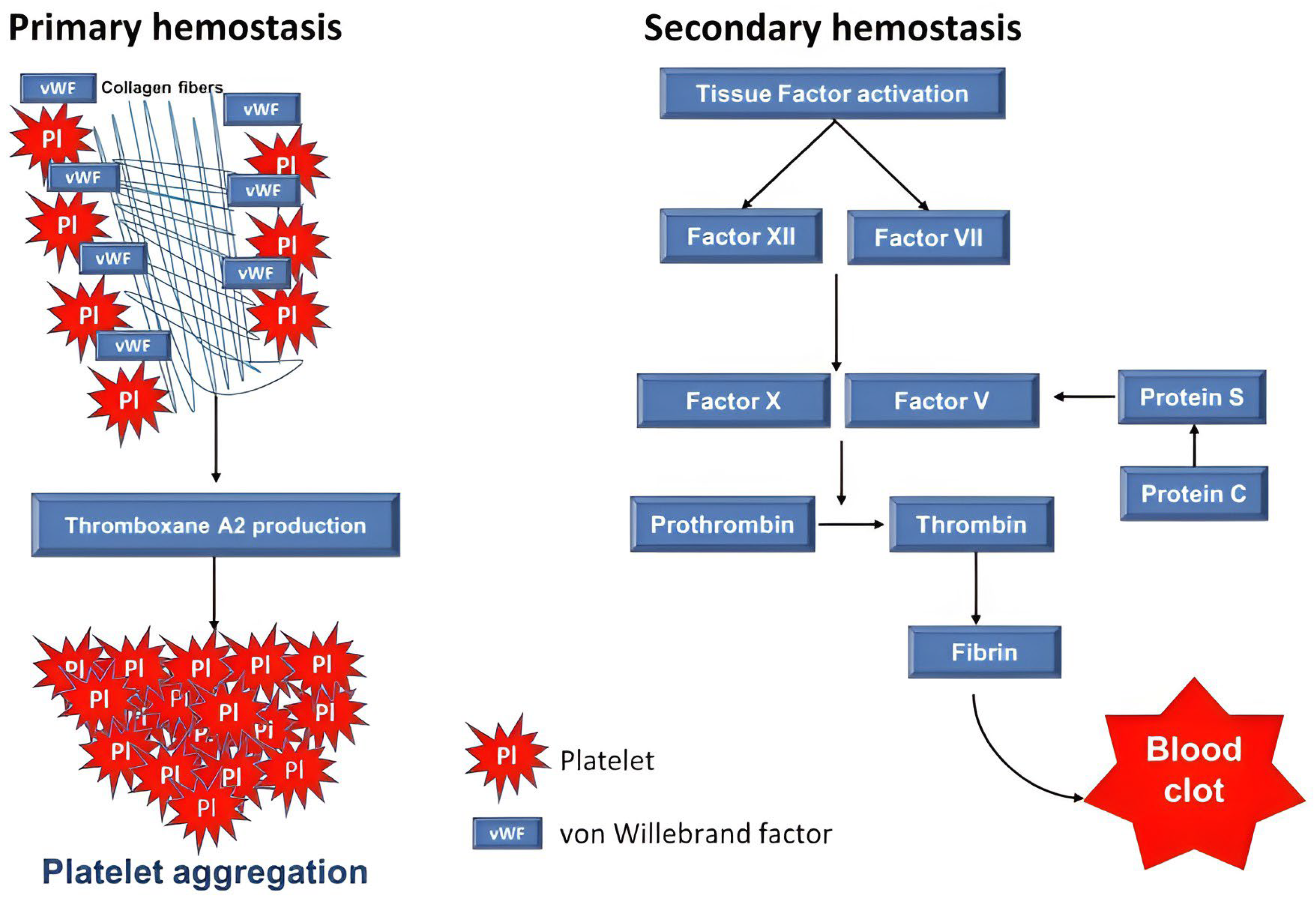
Figure 2.
The crosstalk between inflammation and coagulation in COVID-19 infection. SARS-CoV-2 through the activation of kallikrein and kinin system (KKS) and angiotensin-converting enzyme 2 (ACE2), angiotensin II (AngII), and the angiotensin II receptor type 1 (AT1R) (ACE2-AngII-AR1R) axis overexpresses the IL-1β, TNF-α, and IL-6 cytokines. The overexpression of tissue factor (TF) and P-selectin mediated by cytokines downregulates the anti-coagulants including antithrombin (AT), thrombomodulin (TM), endothelial protein C receptor (EPCR), and neutrophil extracellular trap (NET) formation. Overexpression of P-selectin and TF results in the activation of von Willebrand factor (vWF), Factor VIII, and soluble TM which subsequently increase the levels of coagulants including thrombin, fibrin, and D-dimer that eventually leads to SARS-CoV-2-induced coagulopathy. The overexpression of inflammatory cytokine increases the platelets aggregation by αIIb/β3 integrin resulting in coagulopathy.
Figure 2.
The crosstalk between inflammation and coagulation in COVID-19 infection. SARS-CoV-2 through the activation of kallikrein and kinin system (KKS) and angiotensin-converting enzyme 2 (ACE2), angiotensin II (AngII), and the angiotensin II receptor type 1 (AT1R) (ACE2-AngII-AR1R) axis overexpresses the IL-1β, TNF-α, and IL-6 cytokines. The overexpression of tissue factor (TF) and P-selectin mediated by cytokines downregulates the anti-coagulants including antithrombin (AT), thrombomodulin (TM), endothelial protein C receptor (EPCR), and neutrophil extracellular trap (NET) formation. Overexpression of P-selectin and TF results in the activation of von Willebrand factor (vWF), Factor VIII, and soluble TM which subsequently increase the levels of coagulants including thrombin, fibrin, and D-dimer that eventually leads to SARS-CoV-2-induced coagulopathy. The overexpression of inflammatory cytokine increases the platelets aggregation by αIIb/β3 integrin resulting in coagulopathy.
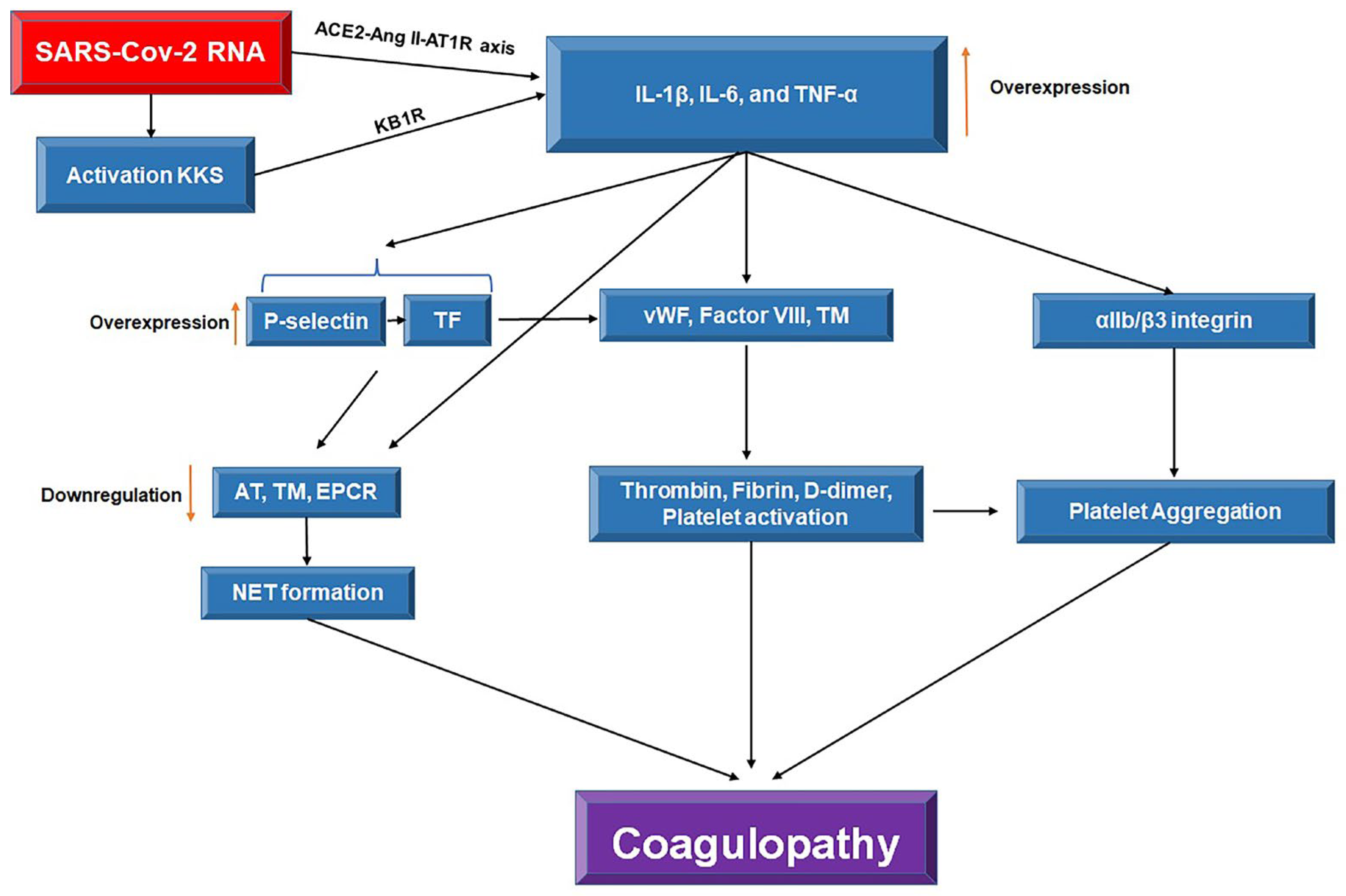
Figure 3.
The pathophysiological mechanism of SARS-CoV-2-induced coagulopathy and cytokine storm in obese patients. Pre-existing conditions include a high frequency of ACE2 receptors, high leptin, and inflammation contributing to severe COVID-19 mediated by cytokine storm and coagulopathy. SARS-CoV-2 infection induces overexpression of angiotensin II (AngII) and hyperactivation of monocytes. AngII overexpression resulted in both tissue factor (TF) and plasminogen activator inhibitor 1 (PAI-1) upregulations, which the former increases the factors IX and X. PAI-1 also upregulates thrombin, fibrin, D-dimer, and platelets leading to coagulopathy in obese COVID-19. Hyperactivation of monocytes and AngII-induced TF overexpression results in the upregulation of IL-6 and the activation of NF-κB pathway leading to the overproduction of inflammatory cytokines.
Figure 3.
The pathophysiological mechanism of SARS-CoV-2-induced coagulopathy and cytokine storm in obese patients. Pre-existing conditions include a high frequency of ACE2 receptors, high leptin, and inflammation contributing to severe COVID-19 mediated by cytokine storm and coagulopathy. SARS-CoV-2 infection induces overexpression of angiotensin II (AngII) and hyperactivation of monocytes. AngII overexpression resulted in both tissue factor (TF) and plasminogen activator inhibitor 1 (PAI-1) upregulations, which the former increases the factors IX and X. PAI-1 also upregulates thrombin, fibrin, D-dimer, and platelets leading to coagulopathy in obese COVID-19. Hyperactivation of monocytes and AngII-induced TF overexpression results in the upregulation of IL-6 and the activation of NF-κB pathway leading to the overproduction of inflammatory cytokines.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



