Submitted:
17 October 2024
Posted:
18 October 2024
You are already at the latest version
Abstract
Alzheimer's disease (AD) and Parkinson's disease (PD) are the most prevalent and irreversible neurological disorders that primarily affect the aged population. These conditions lead to disability and mortality as there is currently no medication for a complete cure. Existing drugs only provide symptomatic relief of the disease. Inhibition of Acetylcholinesterase (AChE) and Butyrylcholinesterase (BuChE) will reduce the hydroxylation of Acetylcholine, a molecule possessing a significant role in the neurotransmission pathways and is accounted as a remedial objective for the treatment of AD and PD. This study aimed to identify potent lead molecules from plants that could inhibit AChE and BuChE. We employed molecular docking, molecular dynamic simulation, and MMPBSA approaches to screen and evaluate potential compounds. The docking results were analysed based on binding affinity and hydrogen bond interactions at the active-site residues of the targets. From the analysis, four phytocompounds - Tricetin, Luteolin, Withasomniferol C, and Withanolide were identified as potential hits. These compounds demonstrated strong binding affinities of -8.49, -9.85, -25.26, and -24.1 kcal/mol, respectively. The significant binding affinity suggests that these compounds may effectively inhibit the activity of AChE and BuChE, potentially leading to therapeutic benefits for AD and PD. However, further in-vitro analysis is recommended to validate the activity of these compounds. In-vitro experiments will provide a better understanding of their efficacy, safety, and potential as drug candidates for treating AD and PD.
Keywords:
ayurinformatics
; neurodegenerative diseases
; Alzheimer’s disease
; Parkinson’s disease
; acetylcholinesterase
; butyrylcholinesterase
1. Introduction
Neurodegenerative diseases (NDD), are a globally distributed category of diseases that threaten the physical and mental lifestyles of a significant percentage of the aged population. NDDs are characterized by the progressive degeneration of the structure and function of cells and the central or peripheral nervous system that are crucial for coordination, sensation, mobility, and cognition (Marcos-Rabal et al., 2021; Baumard et al., 2021; H Ferreira-Vieira et al., 2016). Alzheimer’s disease and Parkinson’s disease are the most commonly reported condition among NDDs. Alzheimer’s disease (AD) is generally distinguished by the presence of neuron apoptosis, neurofibrillary tangles (NFTs), and amyloid plaques (Aβ) (Hyman et al., 1989), Parkinson’s disease (PD) is a progressive multifactorial NDD that leads to the disablement of voluntary motor control. These disorders are debilitating and incurable, affecting an individual’s speech, movement, intelligence, and memory leading to movement disorders, moodiness, memory loss, agitation, depression, and anxiety(Balestrino and Schapira, 2020).
Acetylcholine (ACh), is the chief neurotransmitter that blocks and stimulates a response, escalates bodily secretions, dilates blood vessels, and contracts muscles, proving the significance of the molecule in neurotransmission pathways. Healthy individuals have balanced dopamine and ACh levels (Gu and Wang, 2021). Studies reported that decreased levels of ACh further develop to cause AD and PD. In PD, the increase in the level of ACh gradually leads to the death of the dopaminergic neurons, thus a decrease in dopamine levels which are considered a crucial neurochemical impairment (Ha, Mathew and Yeong, 2020). Homologous enzymes, Acetylcholinesterase (AChE), and Butyrylcholinesterase (BuChE) were reported for the hydrolyzation of Ach (Alves et al., 2022). AChE, the primary cholinesterase member of the serine hydrolase family, plays a significant part in the CNS and PNS (peripheral nervous system) by catalyzing the hydrolysis of ACh into acetate ions and choline under normal circumstances (Vanessa and Mah, 2021). BuChE is a non-specific serine cholinesterase enzyme that can hydrolyze choline-based esters. The potential significance of BuChE recompense the deficit of AChE in AChE knocked out mice model by sustaining the usual cholinergic pathways. Another study confirms these by substituting AChE with BuChE in nullizygous animals. Thus, BuChE can surrogate AChE and hydrolyze ACh in deficient AChE conditions (Pagano et al., 2015), (Emamzadeh and Surguchov, 2018). The inhibited enzymatic activity of AChE and BuChE can lead to increased ACh levels, a major therapeutic strategy for treating AD and PD. On this account, AChE and BuChE can be considered potential therapeutic targets. Several medications and drugs are adopted for the treatment of the diseases but the main challenge is the absence of a completely curing drug. Current drugs focus on the symptomatic relief of the disease and were reported for side effects (Breijyeh and Karaman, 2020). Hence, it is necessary to discover and design effective AChE and BuChE inhibitors without/less side effects in treating Alzheimer’s and Parkinson’s diseases.
Diverse herbal medicines have been used for centuries to treat severe illnesses and medical conditions worldwide. An integrative approach of traditional and western medicine has improved the treatment of NDDs (Tanaka and Kashiwada, 2021). A study demonstrated that Western medicine combined with Chinese western medicine had reduced non-motor disorders (Chen and Pan, 2014). Withania somnifera, Bacopa monnieri, Cannabis sativa, Ginkgo bilobo, and Clitoria ternatea are reported to have potential activities and are used in treating NDDs. Withania somnifera was used to treat loss of memory, nervous exhaustion, insomnia, and general disability (Kuboyama, Tohda and Komatsu, 2014). Bacopa monnieri had reported the cholinergic and neuroprotective effects of the secondary metabolites on AD, similar to the mechanism of the current medications (Basheer et al., 2022). Phytocannabinoids from Cannabis sativa were nonpsychoactive, having high anti-inflammatory and antioxidant activity and anxiolytic, neuroprotective, and anticonvulsant properties (de Barros Viana et al., 2022). Ginkgo bilobo (a tree native to China) has exhibited protective effects against amyloidogenesis and Aβ aggregation and is studied for its anti-inflammatory, anti-apoptotic and antioxidant activities (Arunima, Julia and Prasobh, 2021). The roots of Clitoria ternatea had been proven to enhance memory (by escalating memory retention and memory power) and reduce psychotic stress ( Murugesan et al., 2022).
The study attempts an ayurinformatics approach in which the traditional medicine system - Ayurveda incorporates bioinformatics to identify the capacity of natural compounds with innumerable therapeutic properties. Herein, the neuroprotective and regenerating potential phytocompounds from the five herbal plants extensively used in traditional medicines were evaluated in silico to inhibit AChE and BuChE.
2. Materials and Methods
2.1. Target
AChE and BuChE are the targets considered for the study. The crystal 3D structures of Recombinant Human Acetylcholinesterase in Complex with (-)-galantamine (PDB ID: 4EY6) (Cheung et al., 2013) and Human butyrylcholinesterase (PDB ID:1P0I) (Nicolet et al., 2003) were downloaded from PDB (http://www.rcsb.org). The A chains of the structure 4EY6 with 542 amino acid residues and 1P0I with 529 amino acid residues were considered for analysis. The secondary structure information (Table 1) and the physicochemical properties (Table 2) of the protein 4EY6 and 1P0I were calculated using SOPMA (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html) (Geourjon and Deleage, 1995) and Protparam 4(web.expasy.org/protparam/) ( Johansson et al., 2012), respectively. Protein purification was performed using discovery studio visualizer software (2021 Client). The missing residues were added using the Swiss PDB viewer v4.1.0 (Laskowski, 2001). The active sites residues identified from pdbsum ligplots (Kim et al., 2021) and literature survey - Tyr 72, Asp74, Trp86, Tyr 124, Ser 203, Trp 286, Glu 334, Tyr 337, Phe338, Tyr 341, His447 and Asn68, Asp70, Trp82, Gln119, Ser198, Ala277, Leu286, Glu 325, Ala328, Phe329, His438, Val288 were defined for the protein 4EY6 and 1P0I respectively.
2.2. Ligands
The phytocompounds from Clitoria ternatea, Cannabis Sativa, Bacopa monnieri, Withania Somnifera, and Ginkgo Biloba were listed and subjected to property calculation. The phytocompounds were screened based on Lipinski’s rule of five and ADMET (Absorption, Distribution, Metabolism & Excretion) properties. The 2D structure of the selected phytocompounds was downloaded from the small molecule database PubChem (https://pubchem.ncbi.nlm.nih.gov/) () in .sdf format. Ligands were converted into. pdbqt format using OpenBabel (http://openbabel.org) (O’Boyle et al., 2011).
2.3. Molecular Docking
Molecular docking of the selected phytocompounds against the targets 4EY6 and 1P0I was executed in the Autodock Vina software (v.1.2.0.) (Trott and Olson, 2010; Vanessa and Mah, 2021). Pre-docking steps, like the addition of polar hydrogen, the addition of Kolmann charges, and defining the binding site, were performed in the purified protein structure using Autodock Tools 1.5.7 and the prepared structure was saved in. pdbqt format. Autodock vina calculates and results, in the top 10 poses of protein-ligand interaction based on the binding affinity. The poses with the least binding affinity and the greatest number of H bonds were considered favourable interactions. The molecular interaction between protein and ligands complex was analyzed using Discovery Studio Visualizer 2017. The selected poses of protein-ligand complexes were further subjected to molecular dynamics simulation.
2.4 Molecular Dynamics Simulation
MD simulations of selected complexes were performed using GROMACS version 2020.1 (Valdés-Tresanco et al., 2021; Bauer, Hess, and Lindahl, 2022). MD package in Ubuntu 20.04.2 (AMD® Ryzen 9 3900x 12-core processor × 24). The topology file of selected ligands was created using the CGenFf server, followed by solvation, the addition of ions, Energy minimization, and Equilibration using NVT (300 K), and NPT (1 atm) ensemble. Protein-ligand complexes were subjected to MDS for 100 ns. Trajectory analyses, including the RMSD, RMSF, SASA, Rg, and Hbond interactions were performed using the GROMACS version 2020.1 package. Visualization of the complexes after MD simulation was performed using DS Visualizer, Visual Molecular Dynamics, and Pymol. The trajectory plot analysis was performed using Xmgrace.
2.4.1. MMPBSA
The poses of docked complexes considered for Molecular Dynamics simulation were also subjected to MMPBSA, to estimate the binding free energy calculations. gmx_mmpbsa tool (Valdés-Tresanco et al., 2021) developed for GROMACS was employed for MMPBSA calculations. The binding energy was further decomposed on the per-residue basis to estimate the binding free energy of individual residue to the MGAM-TW/WA interaction. The estimation of binding free energy of ligand-protein complexes in a solvent can be calculated by
∆G bind = ∆G complex − [∆G protein + ∆G lig],
∆G complex - overall free energy of the complex, ∆G protein - free energy of isolate protein, ∆G lig - free energy of isolate ligand,
Conceptually, the MM-PBSA approach can be explained as
∆G bind = ∆E gas + ∆G sol = ∆E vdw + ∆E ele + ∆G polar + ∆G nonpolar
where ∆E gas (gas-free energy) = average molecular mechanic potential energy in a vacuum (van der Waals (∆E vdw ) + electrostatic ( ∆E ele ) interactions),
∆G sol = polar solvation (∆G polar) energy + nonpolar solvation (∆G nonpolar) energy.
3. Result and Discussion
In this study, the neuroprotective and regenerative potentiality of phytocompounds from the plants Clitoria ternatea, Cannabis Sativa, Bacopa monnieri, Withania Somnifera, and Ginkgo Biloba against targets Acetylcholinesterase (AChE) and Butyrylcholinesterase (BuChE) from Alzheimer’s disease and Parkinson’s disease has been investigated through insilico approaches. The research protocol includes Data retrieval, Molecular property calculation, ADMET prediction, Molecular docking, Interaction analysis, Molecular Dynamic Simulation, and MMPBSA calculation.
The selected targets, AChE and BuChE play an important role in cholinergic mediation and were also found to be the targets of many clinically approved drugs (Kandiah et al., 2017). The Recombinant Human Acetylcholinesterase in Complex with (-)-galantamine (PDB ID: 4EY6) and Human butyrylcholinesterase (PDB ID:1P0I) were the considered target structures. A total of 196 phytocompounds from five plants were listed and subjected to molecular property and ADMET prediction. Among that 190 phytocompounds, satisfied Lipinski’s rule of 5 and ADMET properties. The selected target was also subjected to physiochemical properties and secondary structure prediction. Table 1 shows the estimated physiochemical properties, and Table 2 shows the secondary structure prediction values of target proteins to conclude the feasibility of these protein structures. The screening/training dataset is composed of 2 targets and 190 ligand molecules.
Molecular docking of the selected targets and ligands was carried out. The complexes with 1870 poses were obtained separately for each target and 187 phytocompounds out of 190 were docked with the targets. Interaction analysis of the docked complexes was carried out and the poses were ranked based on the most number of Hydrogen bond interactions at active sites and binding affinity (supplementary data). Top-ranked poses from each target were screened and three ligands from each target were shortlisted. A total of 6 poses (Acetyl-4ey6_490802, Acetyl-4ey6_5281701, Acetyl_5280445, Butryl-1P0I_44259428, Butryl-1P0I_101710597, and Butryl-1P0I_118701104) were selected for the molecular dynamics simulation to validate the stability of interactions.
From the 6 poses, Acetyl-4ey6_490802 shows H bond interaction at the active site TYR 72, ASP 74, ASN 87 with a binding energy of -10.9, Acetyl_5280445 form Hydrogen bond with the active site residues GLN 71, TRP 86, GLU 202, HIS 447, TYR 72, and ASP 74 possessing binding energy of -10.6 were illustrated in Figure 1. Whereas Butryl-1P0I_44259428 interacts with the active sites SER 198, GLY 116, PRO 285, and ASN 289 with a binding score of -11, and Butryl-1P0I_101710597 acquire binding energy of -11 which interacts with ASN 289, SER 198, GLY 117, and GLY 116 were shown in Figure 2. Comparatively, the binding energy of ligands in the present study with acetylcholinesterase with the binding energy of approved drugs like Donepezil (-5.1), Rivastigmine (-3.3), and Chlorzoxazone (-6.0) calculated using Autodock in other studies, it is found that Tricetin and Luteolin have good potency against the Acetylcholinesterase (Baskaran et al., 2020). Similarly, Withasomniferol C and Withanolide show similar and satisfiable binding energy with Butyrylcholinesterase when compared with docked results in other studies (Khare, Maheshwari, and Jha, 2021).
After the MD simulation, the interaction analysis of the complexes and the trajectory plot analysis (RMSD, RMSF, RoG, SASA, Hbond distribution) were performed (Khare, Maheshwari, and Jha, 2021). RMSD provides inference on the extent of deviation for a group of atoms from the corresponding initial reference structure (Schreiner et al., 2012). Thus high RMSD deviation points out the instability of the structure. The ligands with high RMSD for their corresponding protein-ligand complex indicates the inadequate accommodation of ligand in the binding pocket of protein over the given Molecular Dynamics simulation time frames (Al-Karmalawy et al., 2021). The RMSF plot determines the average deviation of each residue over time from its reference position (Benson and Daggett, 2012). The notable changes within structural movements could be estimated using an RMSF cut-off value of 0.30 Å, where residues with values >0.30 were considered to have decreased mobility (de Souza et al., 2019). RoG is used to determine the compactness of a protein. An increase in RoG values implies a decrease in protein structure compactness, thereby suggesting increased flexibility and less stability. When a protein is very compact, it tends not to fold easily. SASA often correlates to the molecular surface area that can be measured by solvent molecules, providing a quantitative assessment of the degree of protein/solvent interaction (Pirolli et al., 2014). The result includes both stable (interaction in amino residues remains the same as in the docking result) and unstable interactions (bond breakage, variation in residue, movement to alternate sites, etc). The ligands that exhibit better interaction and stability with the target protein were selected were illustrated in Table 3. The relative binding affinity of ligands towards the protein was also calculated using the MMPBSA approach. The binding energy for each molecule with its corresponding targets is tabulated in Table 4.
From the in silico results, the phytocompounds Tricetin, Luteolin, and Withasomniferol C, Withanolide are found to be the potential molecules that exhibit better interaction and good binding energy with AChE and BuChE respectively were shown in Table 3. The calculated molecular properties summarise the potentiality of these phytocompounds as drug molecules (Rivera-Pérez, Yépes-Pérez, and Martínez-Pabón, 2019). From Table 4 and Table 5, it is evident that these phytocompounds obey Lipinski’s Rule of Five and satisfy ADMET properties.
In the trajectory analysis of Tricetin (Acetyl-4ey6_5281701), the average RMSD was calculated as 0.20333 nm. The total RMSD varied between 0.099378 nm and 0.245959 nm at 0.2 ns and 97.9 ns respectively exhibiting the stability of the complex (Figure 3(a)). The RMSF graph implies the fluctuation of the protein residues, with residue 198 having the lowest RMSF value of 0.0331 nm, residue 4 having the highest RMSF value of 0.5093 nm, and residue 542 having the second highest RMSF value of 0.4966 nm. Unlike the previous complex, residues from 250-400 showed fluctuations between 0.1nm and 0.3 nm (Figure 3(b)). The lowest RoG value was 2.29377 nm at 800 ps, the highest value was 2.33971 nm at 81.6 ns, and an average of 2.321226 nm were calculated for the whole simulation period (Figure 3(c)). The Acetylcholinesterase-Tricetin complex showed an ascent in the SASA up to 40 ns and continued at a stable progression till 100 ns. An average SASA value of 226.5834 nm2 ranging from 207.686 nm2 to 238.012 nm2 is calculated from the trajectory. Residue TYR 72 and ASN 87 broke H bonds with the receptor while ASP 74 remained stable in the active site (Figure 3(d)).
In the trajectory analysis of Luteolin (Acetyl-4ey6_5280445), the average RMSD was calculated as 0.180523 nm. The total RMSD varied between 0.095933 nm and 0.219514 nm at the beginning of the simulation and 35.1 ns respectively exhibiting the stability of the complex (Figure 4(a)). The RMSF graph implies the fluctuation of the protein residues, with residue 147 having the lowest RMSF value of 0.033 nm, residue 76 having the highest RMSF value of 0.4422 nm, and residue 4 having the second highest RMSF value of 0.5697 nm. Residues from 250-400 showed fluctuations between 0.1nm and 0.25 nm (Figure 4(b)). The lowest RoG value, 2.29549 nm at 100 ps, the highest value, 2.33899 nm at 47.4 ns, and an average of 2.31743 nm were calculated for the whole simulation period (Figure 4(c)). The Acetyl-Luteolin complex has an average SASA value of 221.2316 nm2 and ranges from 207.32 nm2 to 232.759 nm2. Apart from HIS 447 all other H bonds were either shifted or broken from the active site(Figure 4(d)).
In the trajectory analysis of Withanosomniferol C (Butryl-1P0I_101710597), the plot shifted after the first half of the simulation time with an average RMSD calculated as 0.173959 nm. The total RMSD varied between 0.086082 nm and 0.244917 nm at 200 ps and 95.3 ns respectively exhibiting the stability of the complex (Figure 5(a)). The RMSF graph implies the fluctuation of the protein residues, with residue 142 having the lowest RMSF value of 0.0349 nm, residue 376 having the highest RMSF value of 0.2762 nm, and residue 529 having the second highest RMSF value of 0.4223 nm (Figure 5(b)). As in the RMSD plot, the RoG plot too exhibited a sight upwards after 50 ns. The lowest RoG value 2.29755 nm at 45.9 ns, the highest value was 2.37108 nm at 58.6 ns and an average of 2.327914 nm were calculated for the whole simulation period(Figure 5(c)). The Butryl-Withanosomniferol C complex has an average SASA value of 221.0537 nm2 and ranges from 212.376 nm2 to 231.462 nm2 showing an unfluctuating plot over the simulation time(Figure 5(d)).
In the trajectory analysis of Withanolide (Butryl-1P0I_118701104), The RMSD plot had a slanting nature, and the average RMSD was calculated as 0.17939 nm. The total RMSD varied between 0.109934 nm and 0.242825 nm at 100 ps and 96.8 ns respectively exhibiting the stability of the complex (Figure 6(a)). The RMSF graph implies the fluctuation of the protein residues, with residue 142 having the lowest RMSF value of 0.035 nm, residue 73 having the highest RMSF value of 0.3084 nm, and residue 74 having the second highest RMSF value of 0.3055 nm. Residues between 200 and 500 were more fluctuating in comparison with the other complexes (Figure 6(b)). The lowest RoG value was 2.30033 nm at 0.8 ns, the highest value was 2.34204 nm at 99.3 ns, and an average of 2.323457 nm were calculated for the whole simulation period(Figure 6(c)). The Butryl- Withanolide complex has an average SASA value of 224.917 nm2 and ranges from 216.28 nm2 to 234.712 nm2 (Figure 6(d)).
The computational approaches carried out in this study for protein-ligand interaction prediction led to the identification of novel interactions. Based on the binding affinity, stability, and molecular properties of four phytocompounds Tricetin, Luteolin, and Withasomniferol C, Withanolide can be suggested as lead molecules from this analysis for the inhibition of therapeutic targets of Alzheimer’s and Parkinson’s disease.
4. Conclusion
The current work is an ayurinformatics study to identify potential lead molecules against the therapeutic targets (AChE and BuChE) of Alzheimer’s disease and Parkinson’s disease. Phytocompounds from five plants reported for neuroprotective and regenerative activity were considered ligands. Molecular interaction analysis, Molecular Dynamic simulation, and MMPBSA calculations were performed using Autodock Vina and GROMACS respectively. Four phytocompounds; Tricetin, Luteolin, Withasomniferol C, and Withanolide exhibited stable interaction even after molecular dynamics simulation. They possess favorable binding affinity in molecular docking as well as MMPBSA calculations. The molecular properties and descriptors of these molecules point to the qualification of these molecules as lead molecules by obeying the Lipinski rule of 5. ADMET prediction supports the above statement. The insilico analysis supports and suggests these lead molecules for the inhibition of therapeutic targets Acetylcholinesterase and butyrylcholinesterase of Alzheimer’s and Parkinson’s disease. Future wet lab experiments have to be performed to validate the activity of these lead molecules.
Author Contributions
This research was supported by Dr. Nidhin Sreekumar, Director, Accubits Invent Pvt. Ltd. and supervised by Shahanas Naisam, Research Associate. We thank all our colleagues who provided insight and expertise that greatly assisted the research. Shahanas Naisam conceived of the presented idea and designed the work plan and computational framework. Aswin Mohan, and Gayathri S S, performed the data collection and computations. Viji V. S. And Vidhya V. S. has provided support in data collection. Shahanas Naisam verified the result. We are immensely grateful to Dr. Suvanish Kumar, Research Scientist of Accubits Invent Pvt. Ltd. for his assistance with constructive criticism and comments that significantly improved the manuscript.
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Availability of Data and Material
The datasets generated during and/or analyzed during the current study are not publicly available due to the confidential nature of the work but are available from the corresponding author on reasonable request.
Acknowledgments
This research was supported by Accubits Invent Pvt Ltd. and supervised by Dr. Nidhin Sreekumar, Chief Research Scientist. We thank all our colleagues who provided insight and expertise that greatly assisted the research.
Conflicts of Inter
The authors have no relevant financial or non-financial interests to disclose.
References
- Al-Karmalawy, A. A., Dahab, M. A., Metwaly, A. M., Elhady, S. S., Elkaeed, E. B., Eissa, I. H., & Darwish, K. M. (2021). Molecular Docking and Dynamics Simulation Revealed the Potential Inhibitory Activity of ACEIs Against SARS-CoV-2 Targeting the hACE2 Receptor. Frontiers in Chemistry, 9. [CrossRef]
- Luana Severo Alves, Valdir Cechinel Filho, Pires, R., & Furtado-Alle, L. (2022). BChE inhibitors from marine organisms – A review. 367, 110136–110136. [CrossRef]
- Arunima, C., Julia, J. and Prasobh, G. (2021) ‘A review on role of ginkgo biloba in treating alzheimer’s disease’.
- Balestrino, R. and Schapira, A.H. V (2020) ‘Parkinson disease’, European journal of neurology, 27(1), pp. 27–42.
- de Barros Viana, M. et al. (2022) ‘Cannabis sativa and Cannabidiol: A Therapeutic Strategy for the Treatment of Neurodegenerative Diseases?’, Medical Cannabis and Cannabinoids, 5(1), pp. 207–219. [CrossRef]
- Basheer, A. et al. (2022) ‘Use of Bacopa monnieri in the Treatment of Dementia Due to Alzheimer Disease: Systematic Review of Randomized Controlled Trials’, Interactive journal of medical research, 11(2), p. e38542. [CrossRef]
- Baskaran, K.P. et al. (2020) ‘Insilico method for prediction of maximum binding affinity and ligand—protein interaction studies on Alzheimer’s disease’, Int J Res Granthaalayah, 8(11), pp. 362–370.
- Bauer, P., Hess, B. and Lindahl, E. (2022) ‘GROMACS 2022 manual’.
- Baumard, J. et al. (2021) ‘Physical understanding in neurodegenerative diseases’, Cognitive Neuropsychology, 38(7–8), pp. 490–514.
- Benson, N.C. and Daggett, V. (2012) ‘A comparison of multiscale methods for the analysis of molecular dynamics simulations’, The Journal of Physical Chemistry B, 116(29), pp. 8722–8731. [CrossRef]
- Breijyeh, Z. and Karaman, R. (2020) ‘Comprehensive review on Alzheimer’s disease: causes and treatment’, Molecules, 25(24), p. 5789. [CrossRef]
- Chen, X. and Pan, W. (2014) ‘The treatment strategies for neurodegenerative diseases by integrative medicine’, Integrative Medicine International, 1(4), pp. 223–225. [CrossRef]
- Cheung, J. et al. (2013) ‘Structures of human acetylcholinesterase bound to dihydrotanshinone I and territrem B show peripheral site flexibility’, ACS medicinal chemistry letters, 4(11), pp. 1091–1096. [CrossRef]
- Emamzadeh, F.N. and Surguchov, A. (2018) ‘Parkinson’s disease: biomarkers, treatment, and risk factors. Front Neurosci 12: 612’.
- Geourjon, C. and Deleage, G. (1995) ‘SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments’, Bioinformatics, 11(6), pp. 681–684. [CrossRef]
- Gu, X. and Wang, X. (2021) ‘An overview of recent analysis and detection of acetylcholine’, Analytical Biochemistry, 632, p. 114381. [CrossRef]
- H Ferreira-Vieira, T. et al. (2016) ‘Alzheimer’s disease: targeting the cholinergic system’, Current neuropharmacology, 14(1), pp. 101–115.
- Ha, Z.Y., Mathew, S. and Yeong, K.Y. (2020) ‘Butyrylcholinesterase: a multifaceted pharmacological target and tool’, Current Protein and Peptide Science, 21(1), pp. 99–109. [CrossRef]
- Hyman, B.T. et al. (1989) ‘Alzheimer’s disease’, Annual review of public health, 10(1), pp. 115–140.
- Johansson, M.U. et al. (2012) ‘Defining and searching for structural motifs using DeepView/Swiss-PdbViewer’, BMC bioinformatics, 13(1), pp. 1–11.
- Kandiah, N. et al. (2017) ‘Rivastigmine: the advantages of dual inhibition of acetylcholinesterase and butyrylcholinesterase and its role in subcortical vascular dementia and Parkinson’s disease dementia’, Clinical interventions in aging, 12, p. 697. [CrossRef]
- Khare, N., Maheshwari, S.K. and Jha, A.K. (2021) ‘Screening and identification of secondary metabolites in the bark of Bauhinia variegata to treat Alzheimer’s disease by using molecular docking and molecular dynamics simulations’, Journal of Biomolecular Structure and Dynamics, 39(16), pp. 5988–5998.
- Kim, S. et al. (2021) ‘PubChem in 2021: new data content and improved web interfaces’, Nucleic Acids Research, 49(D1), pp. D1388–D1395. [CrossRef]
- Kuboyama, T., Tohda, C. and Komatsu, K. (2014) ‘Effects of Ashwagandha (roots of Withania somnifera) on neurodegenerative diseases’, Biological and Pharmaceutical Bulletin, 37(6), pp. 892–897. [CrossRef]
- Laskowski, R.A. (2001) ‘PDBsum: summaries and analyses of PDB structures’, Nucleic acids research, 29(1), pp. 221–222.
- Marcos-Rabal, P. et al. (2021) ‘Neurodegenerative Diseases: A Multidisciplinary Approach’, Current Pharmaceutical Design, 27(30), pp. 3305–3336.
- Murugesan, V. et al. (2022) ‘Phytonutrients in Neurological Disorders’, in Role of Nutrients in Neurological Disorders. Springer, pp. 3–15.
- Nicolet, Y. et al. (2003) ‘Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products’, Journal of Biological Chemistry, 278(42), pp. 41141–41147. [CrossRef]
- O’Boyle, N.M. et al. (2011) ‘Open Babel: An open chemical toolbox’, Journal of cheminformatics, 3(1), pp. 1–14.
- Pagano, G. et al. (2015) ‘Cholinesterase inhibitors for Parkinson’s disease: a systematic review and meta-analysis’, Journal of Neurology, Neurosurgery & Psychiatry, 86(7), pp. 767–773. [CrossRef]
- Pirolli, D. et al. (2014) ‘Insights from molecular dynamics simulations: structural basis for the V567D mutation-induced instability of zebrafish alpha-dystroglycan and comparison with the murine model’, PLoS One, 9(7), p. e103866. [CrossRef]
- Rivera-Pérez, W.A., Yépes-Pérez, A.F. and Martínez-Pabón, M.C. (2019) ‘Molecular docking and in silico studies of the physicochemical properties of potential inhibitors for the phosphotransferase system of Streptococcus mutans’, Archives of Oral Biology, 98, pp. 164–175. [CrossRef]
- Schreiner, W. et al. (2012) ‘Relaxation estimation of RMSD in molecular dynamics immunosimulations’, Computational and mathematical methods in medicine, 2012. [CrossRef]
- de Souza, A.S. et al. (2019) ‘3-Acyltetramic acids as a novel class of inhibitors for human kallikreins 5 and 7’, Bioorganic & medicinal chemistry letters, 29(9), pp. 1094–1098.
- Tanaka, N. and Kashiwada, Y. (2021) ‘Phytochemical studies on traditional herbal medicines based on the ethnopharmacological information obtained by field studies’, Journal of natural medicines, 75(4), pp. 762–783. [CrossRef]
- Trott, O. and Olson, A.J. (2010) ‘AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading’, Journal of computational chemistry, 31(2), pp. 455–461.
- Valdés-Tresanco, M.S. et al. (2021) ‘gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS’, Journal of Chemical Theory and Computation, 17(10), pp. 6281–6291. Available at: . [CrossRef]
- Vanessa, V. V and Mah, S.H. (2021) ‘Xanthone: Potential Acetylcholinesterase Inhibitor for Alzheimer’s Disease Treatment’, Mini reviews in medicinal chemistry, 21(17), pp. 2507–2529. [CrossRef]
Figure 1.
Molecular interaction between ligands and Acetylcholinesterase (4EY6). (a) Interaction of Tricetin - 4EY6 complex after molecular docking. (B) Interaction of Luteolin - 4EY6 complex after molecular docking.
Figure 1.
Molecular interaction between ligands and Acetylcholinesterase (4EY6). (a) Interaction of Tricetin - 4EY6 complex after molecular docking. (B) Interaction of Luteolin - 4EY6 complex after molecular docking.
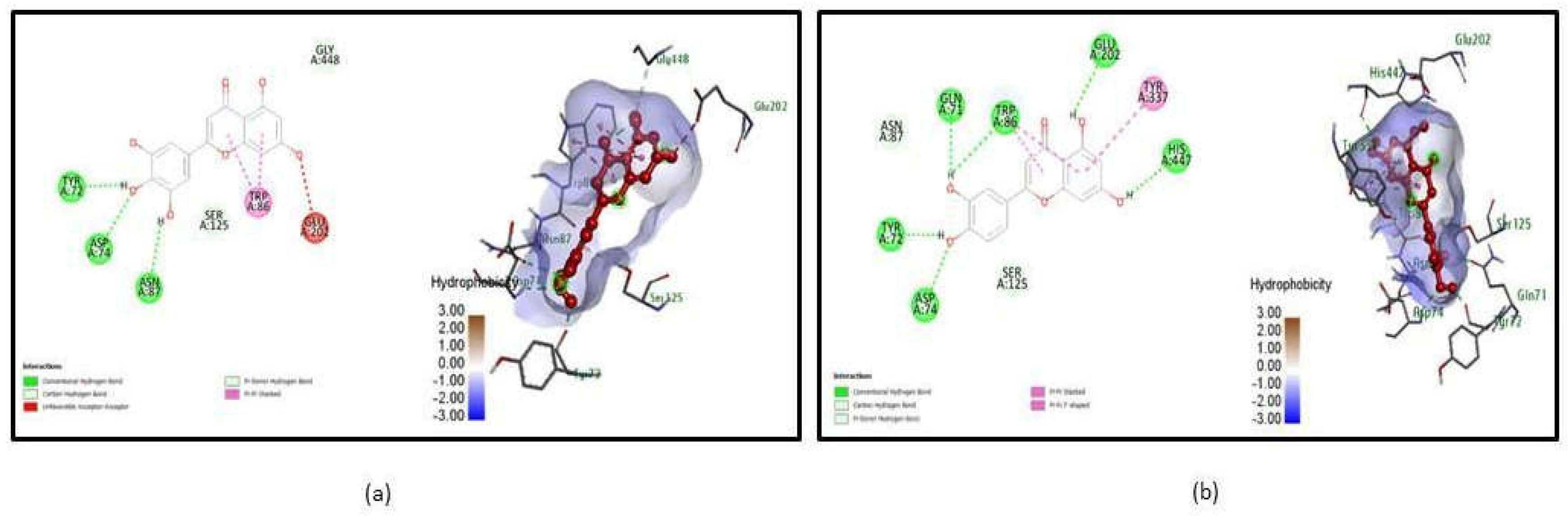
Figure 2.
Molecular interaction between ligands and Butyrylcholinesterase (1P0I). (a) Interaction of Withasomniferol C - 1P0I complex after molecular docking. (b) Interaction of Withanolide-1P0I complex after molecular docking.
Figure 2.
Molecular interaction between ligands and Butyrylcholinesterase (1P0I). (a) Interaction of Withasomniferol C - 1P0I complex after molecular docking. (b) Interaction of Withanolide-1P0I complex after molecular docking.
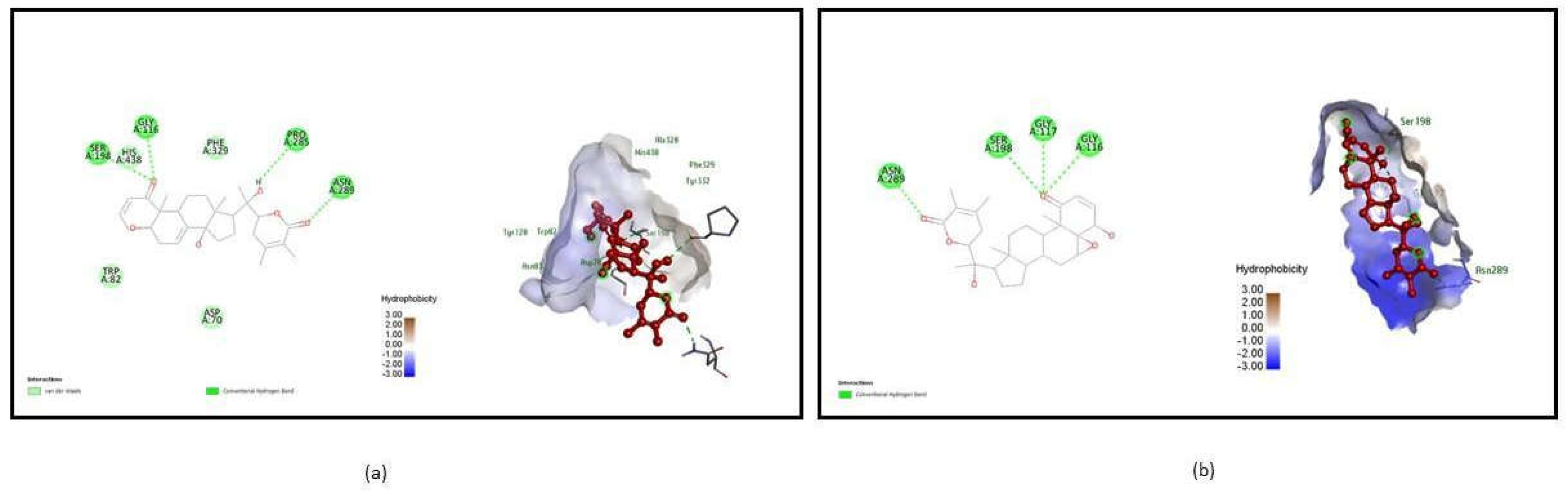
Figure 3.
Trajectory plots of Tricetin - 4EY6 MD simulation. (a) RMSD plot of Tricetin - 4EY6 MD simulation. (b) RMSF plot Tricetin - 4EY6 MD simulation. (c) Radius of Gyration plot of Tricetin - 4EY6 MD simulation. (d) H-bond distribution plot of Tricetin - 4EY6 MD simulation.
Figure 3.
Trajectory plots of Tricetin - 4EY6 MD simulation. (a) RMSD plot of Tricetin - 4EY6 MD simulation. (b) RMSF plot Tricetin - 4EY6 MD simulation. (c) Radius of Gyration plot of Tricetin - 4EY6 MD simulation. (d) H-bond distribution plot of Tricetin - 4EY6 MD simulation.
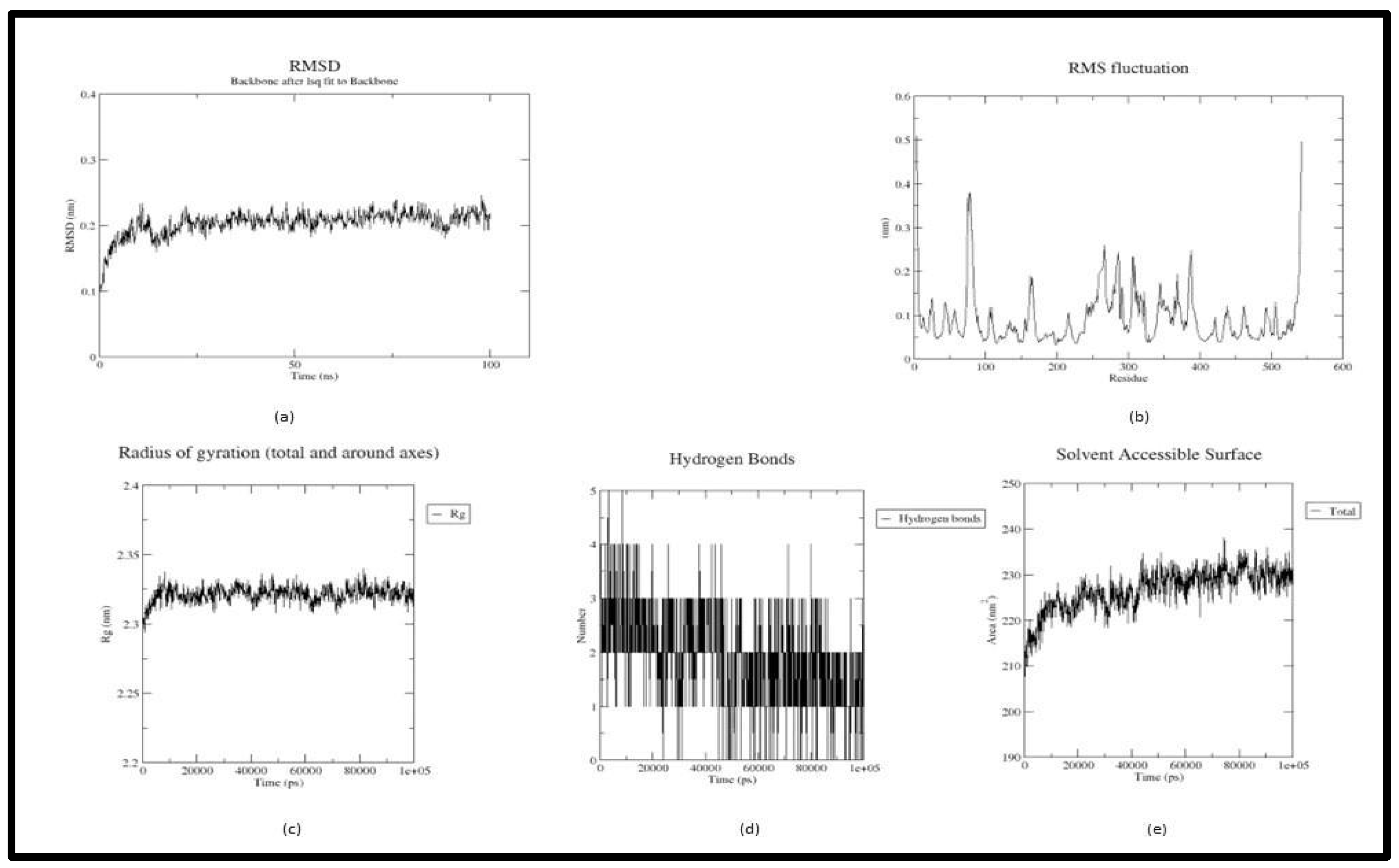
Figure 4.
Trajectory plots of Luteolin - 4EY6 MD simulation. (a) RMSD plot of Luteolin - 4EY6 MD simulation. (b) RMSF plot Luteolin - 4EY6 MD simulation. (c) Radius of Gyration plot of Luteolin - 4EY6 MD simulation. (d) H-bond distribution plot of Luteolin - 4EY6 MD simulation.
Figure 4.
Trajectory plots of Luteolin - 4EY6 MD simulation. (a) RMSD plot of Luteolin - 4EY6 MD simulation. (b) RMSF plot Luteolin - 4EY6 MD simulation. (c) Radius of Gyration plot of Luteolin - 4EY6 MD simulation. (d) H-bond distribution plot of Luteolin - 4EY6 MD simulation.
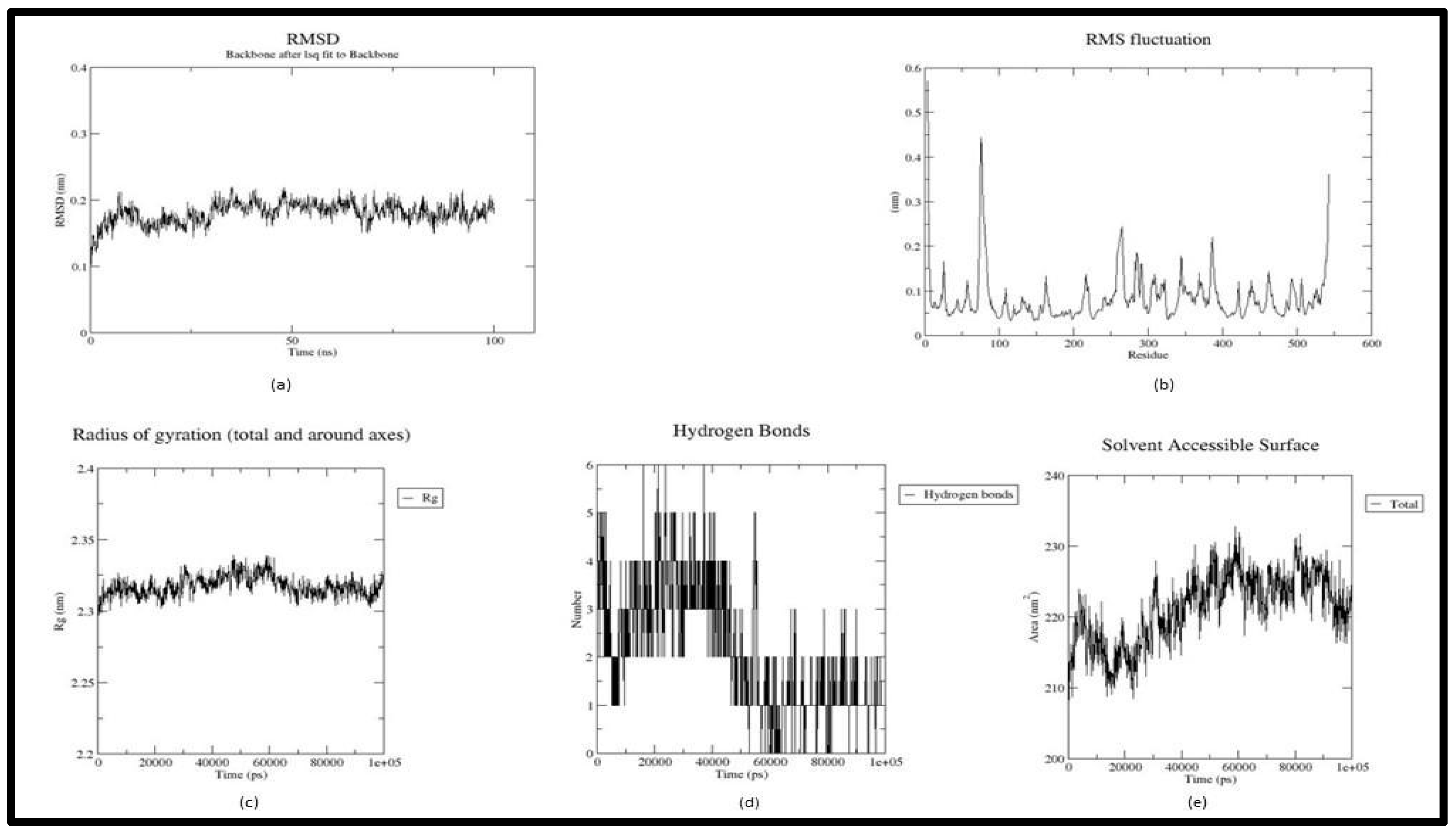
Figure 5.
Trajectory plots of 1P0I- Withasomniferol C MD simulation. (a) RMSD plot of 1P0I- Withasomniferol C MD simulation. (b) RMSF plot 1P0I- Withasomniferol C MD simulation. (c) Radius of Gyration plot of 1P0I- Withasomniferol C MD simulation. (d) H-bond distribution plot of 1P0I- Withasomniferol C MD simulation.
Figure 5.
Trajectory plots of 1P0I- Withasomniferol C MD simulation. (a) RMSD plot of 1P0I- Withasomniferol C MD simulation. (b) RMSF plot 1P0I- Withasomniferol C MD simulation. (c) Radius of Gyration plot of 1P0I- Withasomniferol C MD simulation. (d) H-bond distribution plot of 1P0I- Withasomniferol C MD simulation.
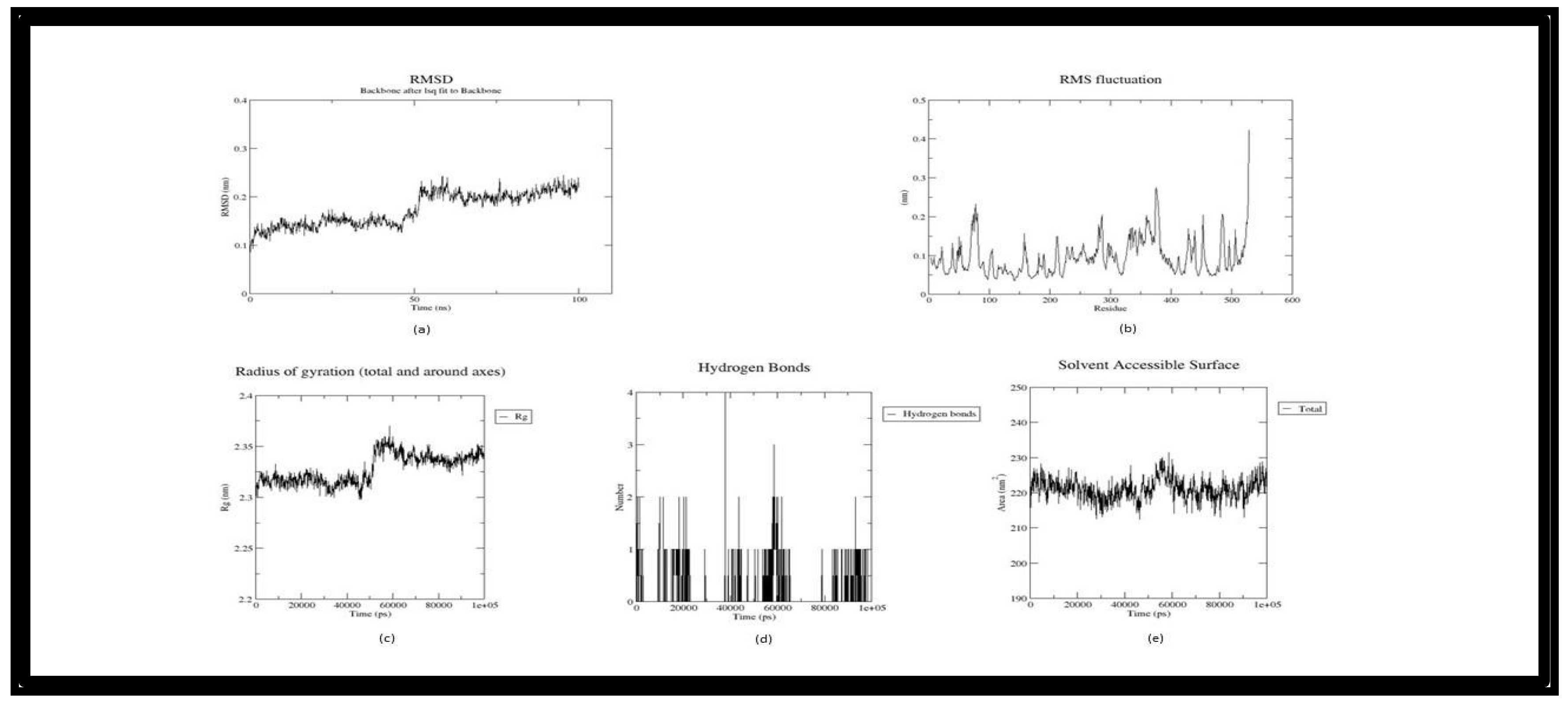
Figure 6.
Trajectory plots of 1P0I- Withanolide MD simulation. (a) RMSD plot of 1P0I- Withanolide MD simulation. (b) RMSF plot 1P0I- Withanolide MD simulation. (c) Radius of Gyration plot of 1P0I- Withanolide MD simulation. (d) H-bond distribution plot of 1P0I- Withanolide MD simulation.
Figure 6.
Trajectory plots of 1P0I- Withanolide MD simulation. (a) RMSD plot of 1P0I- Withanolide MD simulation. (b) RMSF plot 1P0I- Withanolide MD simulation. (c) Radius of Gyration plot of 1P0I- Withanolide MD simulation. (d) H-bond distribution plot of 1P0I- Withanolide MD simulation.
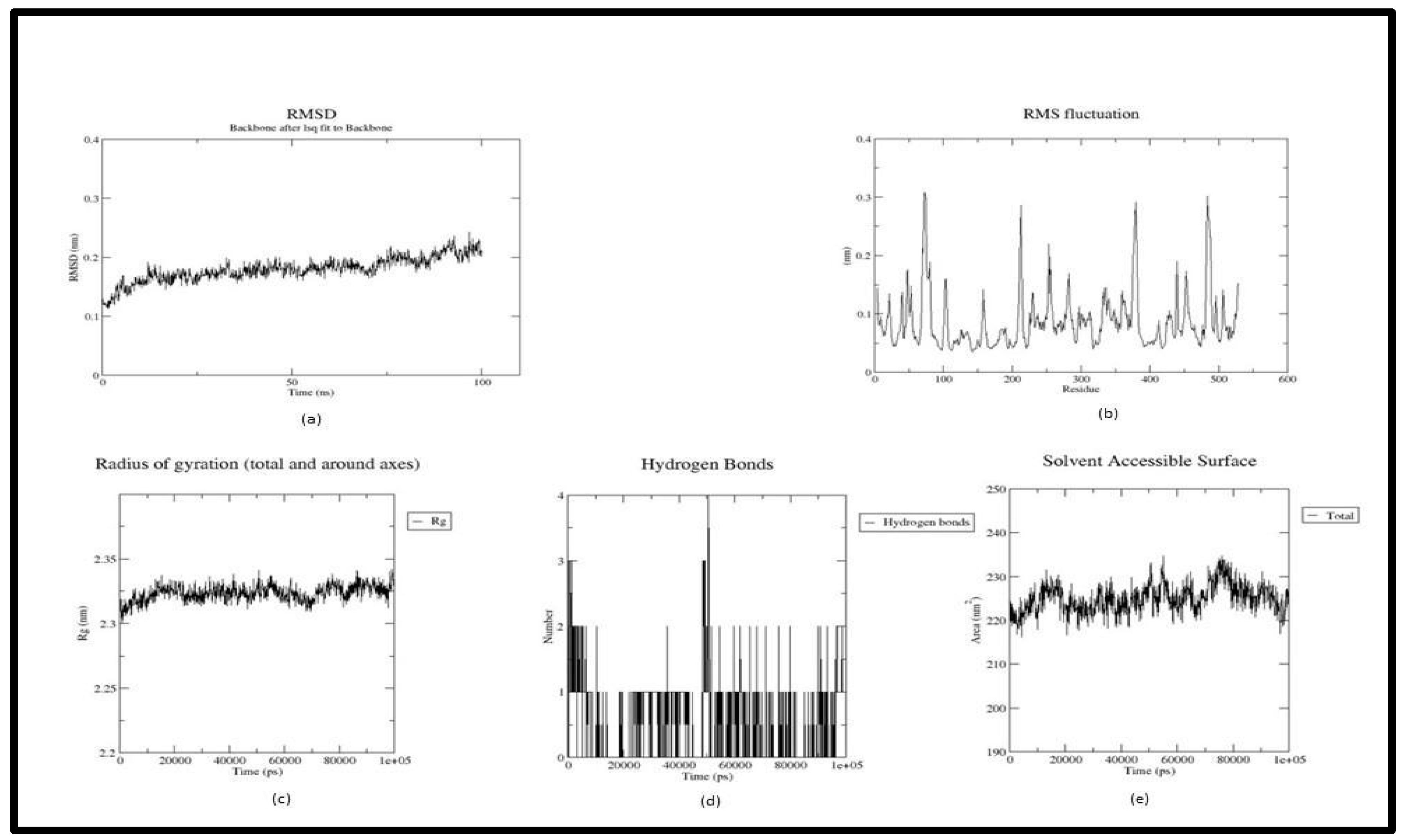

Table 1.
Physicochemical properties of the targets.
| PDB_ID | T- PI | Al | Mol. Weight |
No. of amino acids |
GV |
II |
Half Life | Extinction Coefficient | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mammalian | Yeast | E-coli | Cysteines | Cys reduced | |||||||
| 4EY6 | 5.83 | 84.00 | 59390.46 | 542 | -0.110 | 40.07 | 30 hours | >20 hours | >10 hours | 100185 |
99810 |
| 1P0I | 7.22 | 77.60 | 59653.87 | 529 | -0.275 | 38.32 | 1 hours | 30 min | >10 hours | 104195 | 103820 |
*T-PI – Theoretical PI, *AI- Aliphatic sIndex, *MW- Molecular Weight, *GV- Gravy Value, *II – Instability index.
Table 2.
Secondary structure prediction of targets.
| PDB _ID | Alpha helix percent |
Beta turn Percent |
Random Coil |
Extended strand |
|---|---|---|---|---|
| 4EY6 | 29.11% | 4.73% | 48.02% | 18.15% |
| 1P0I | 28.04% | 5.35% | 48.15% | 18.45% |
Table 3.
Interaction of selected compounds with targets.
| Target | Ligand_Name | H-Bond Interaction |
No. of H-Bonds | Binding Affinity (kcal/mol) |
H-bond interaction after MD Simulation | Binding energy (kcal/mol) |
|---|---|---|---|---|---|---|
| 4EY6 | Tricetin | TYR 72, ASP 74, ASN 87 | 3 | -10.9 | ASP 74 | -8.49 |
| 4EY6 |
Luteolin | GLN 71, TRP 86, GLU 202, HIS 447, TYR 72, ASP 74 | 6 | -10.6 | TYR 341, HIS 447 |
-9.85 |
| 1P0I | Withasomniferol C | SER 198, GLY 116, PRO 285, ASN 289 | 4 | -11 | TRP 231 | -25.26 |
| 1P0I |
Withanolide | ASN 289, SER 198, GLY 117, GLY 116 | 4 | -11 | TYR 332 | -24.1 |
Table 4.
Lipinski’s Rule of Five.
| Ligand_Name | MW | H-Bond Donor | H-Bond Acceptor | Log P | MR |
|---|---|---|---|---|---|
| Tricetin | 302.24 | 5 | 7 | 1.33 | 78.04 |
| Luteolin | 286.24 | 4 | 6 | 1.86 | 76.01 |
| Withasomniferol C | 470.6 | 3 | 6 | 3.49 | 129.28 |
| Withanolide | 470.6 | 2 | 6 | 3.78 | 127.53 |
Table 5.
ADMET Properties.
| Property | Model Name | Tricetin | Luteolin | Withasomniferol C | Withanolide |
|---|---|---|---|---|---|
| Absorbtion | Water solubility | -3.028 | -3.094 | -5.269 | -5.127 |
| Caco2 permeability | -0.272 | 0.096 | 0.769 | 0.831 | |
| Intestinal absorption (human) | 78.366 | 81.13 | 97.917 | 99.2 | |
| Skin Permeability | -2.735 | -2.735 | -3.764 | -3.267 | |
| P-glycoprotein substrate | Yes | Yes | Yes | Yes | |
| P-glycoprotein I inhibitor | No | No | No | Yes | |
| P-glycoprotein II inhibitor | No | No | No | Yes | |
| Distribution | VDss (human) | 0.932 | 1.153 | 0.142 | -0.048 |
| Fraction unbound (human) | 0.208 | 0.168 | 0.23 | 0.093 | |
| BBB permeability | -1.38 | -0.907 | -0.774 | -0.315 | |
| CNS permeability | -3.557 | -2.251 | -3.008 | -2.696 | |
| Metabolilsm | CYP2D6 substrate | No | No | No | No |
| CYP3A4 substrate | No | No | Yes | Yes | |
| CYP1A2 inhibitior | Yes | Yes | No | No | |
| CYP2C19 inhibitior | No | No | No | No | |
| CYP2C9 inhibitior | No | Yes | No | No | |
| CYP2D6 inhibitior | No | No | No | No | |
| CYP3A4 inhibitior | No | No | No | No | |
| Excretion | Total Clearance | 0.513 | 0.495 | 0.43 | 0.347 |
| Renal OCT2 substrate | No | No | No | No | |
| Toxicity | AMES toxicity | No | No | No | No |
| Max. tolerated dose (human) | 0.545 | 0.499 | -0.722 | -0.867 | |
| hERG I inhibitor | No | No | No | No | |
| hERG II inhibitor | No | No | No | No | |
| Hepatotoxicity | No | No | No | No | |
| Skin Sensitisation | No | No | No | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



