
Submitted:
15 October 2024
Posted:
17 October 2024
You are already at the latest version
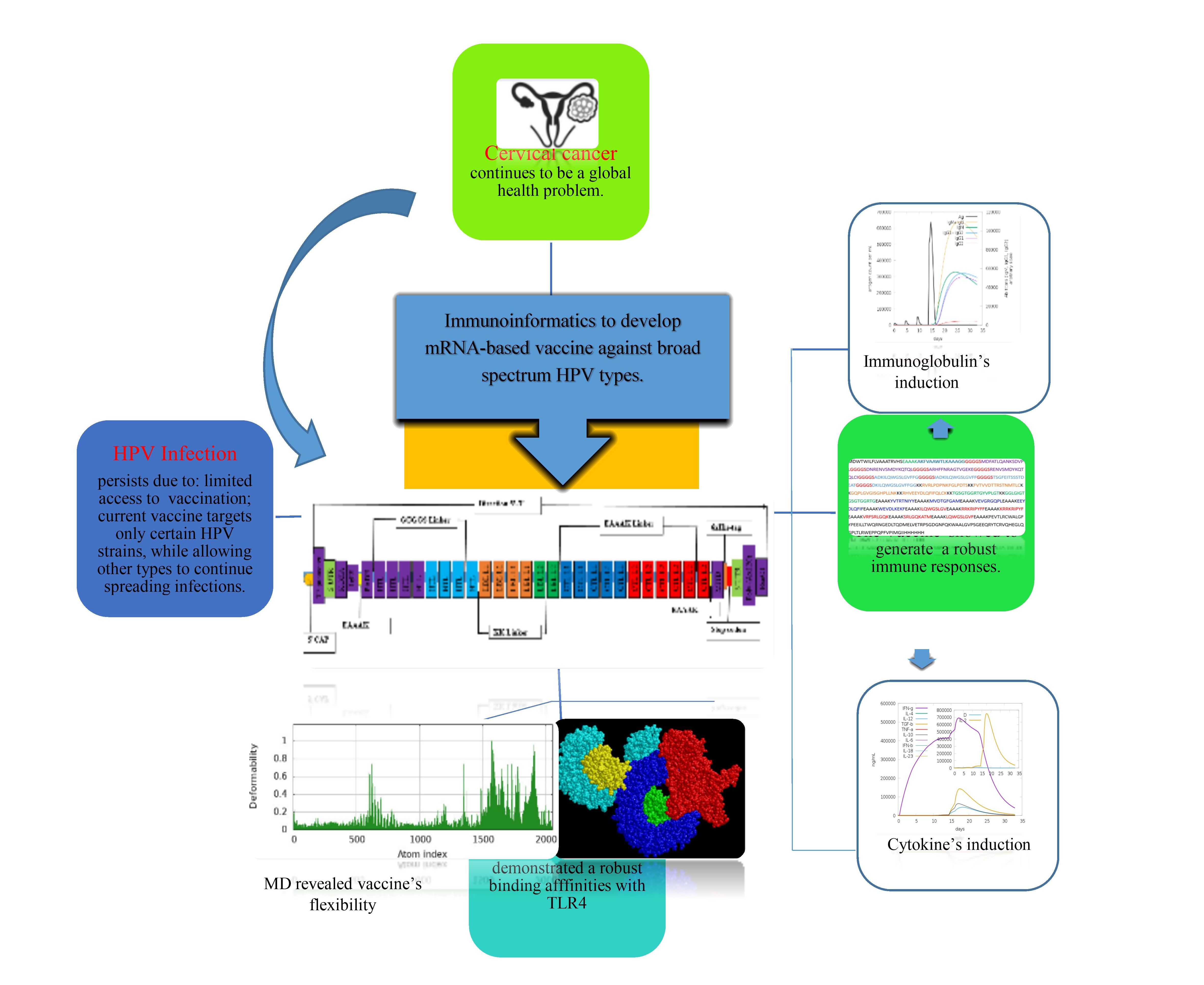
Abstract
(1) Background: Human papillomaviruses (HPV) are primarily related to cervical cancer around the world. This study presented an in silico design of a chimeric L1/L2 mRNA-based vaccine candidate against a diverse spectrum of human papillomaviruses; (2) Methods: We employed a computational strategy to predict and screen for antigenicity, allergenicity, toxicity, and conservation of immunodominant cytotoxic T cells, helper T cells, and linear B-cell epitopes from the most conserved HPV L1 and L2 proteins. The selected epitope peptides were coupled together with appropriate linkers to create a multi-epitope chimeric peptides. The mRNA-based vaccine was developed by combining an optimized open reading frame with co-translation regulation and co-transcription components; (3) Results: Twenty five (25) non-toxic, non-allergenic, highly antigenic, and conserved epitopes were identified. The final vaccine had a molecular weight of 60161.29 kDa, a theoretical pI of 9.44, and a solubility of 0.451. The mRNA-based vaccine had a stable mRNA structure with a minimum free energy of -731.10 kcal/mol and an estimated molecular weight of 644.98 kDa. The suggested vaccine had no cross-affinity for the human genome and achieved a global population coverage rate of 86.24%. The vaccine demonstrated a stable docking complex and robust interactions with Toll-like receptor 4 (TLR4) with binding affinities of -1377.5 kcal/mol and was capable of stimulating immunoglobulin IgG, IgM, T and B cells lymphocytes, and cytokines by in silico simulation; (4) Conclusion: The vaccine is considered a preventive and therapeutic candidate; however, further in vitro and in vivo tests are needed to prove its safety and efficacy.
Keywords:
HPV
; IVT
; mRNA
; multi-epitope vaccine
; molecular docking
; molecular dynamics simulation
; immune simulation
1. Introduction
Human papillomaviruses are circular, non-enveloped double stranded DNA viruses responsible for 99% of cervical cancer, 93% of anus cancer, 40% of vaginal, 40% of penile, 63% of oropharyngeal, and 51% of vulva cancer [1]. Cervical cancer is a significant global health challenge, ranking as the fourth most common cancer among women worldwide, with an approximation of 604 127 new cases and 341 831 deaths reported in 2020 due to this disease [2]. Africa has the highest cervical cancer burden globally, with 119,000 new cases and 69,000 deaths in 2018 [3]. This is due to limited access to screening and treatment, lack of awareness, and a global high-risk HPV infection rates in Sub-Saharan Africa , averaging 24% [4]; whereas South-East Asia (14%) and Eastern Europe (14%) next, followed by Latin America and the Caribbean (16%) [5]. Fourteen (14) human papillomaviruses—namely HPV 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66, 68, have been associated with cervical cancer [6], while 23 low-risk (LR-HPV) variants, such as 6, 11, 26, 40, 42, 53, 54, 55, 61, 62, 64, 67, 69, 70, 71, 72, 73, 81, 82, 83, 84, IS39, and CP6108 cause around 2% of cervical cancer cases or benign genital warts, and eight (8) are benign skin-driving-warts, or cutaneous-associated HPV genotypes: 1, 2, 3, 4, 27, 29, 57, and 63 have been reported [7,8].
A recent vaccination that is in use is a three FDA-approved prophylactic vaccines against human papillomavirus, Gardasil4®, Gardasil9®, and CervarixTM [9,10]. However, these vaccine target a limited number of HPV genotypes and display a narrow range of neutralization antibodies [11], indicating that some untargeted HPV genotypes continue spreading infection. Progress in HPV L2-based vaccination, aiming at providing type-specific and broad range protection against a variety of Human Papillomavirus genotypes and associated illnesses, has been reported as a means of overcoming these barriers [7,12]. It has been established that the L2 vaccine can neutralize antibodies produced during several types of HPV infections because L2 proteins include highly cross-conserved epitopes [13]; nevertheless, L2 immunization itself stimulates minimal immunological responses. To circumvent these drawbacks, multivalent L2 epitopes (peptide vaccines), fusion, or chimeric procedures between L1/L2 [8,14,16], or fusion of L1 with other immunogenic proteins [17], and multi-epitope approaches [8], have emerged as feasible options. Furthermore, fusing L2 with early viral (L1) antigens has the potential to stimulate both preventive and therapeutic immunity [7]. Epitope prediction is a computational method used to design multi-subunit vaccines for emerging infections, triggering cellular and humoral immunity through Th cell activation cytokine secretion, Tc-cell production, and B-cell induction [1,18,19].
The mRNA-based technology is increasingly used for preventative vaccinations due to its quick manufacturing process and assumed to be non-infectious due to their lack of genomic integration and replication with the exception of rare instances of recombination between single-stranded RNA molecules [20], as well as, promoting humoral and cell-mediated immunity [21].
In this work, an in silico technique was used to predict epitopes from five HPV cutaneous-related HPV, six HPV low risks (6 LR HPV), and fourteen HPV high risks (14HR-HPV) of major capsids (L1) and minor capsids (L2) proteins, with the aim of designing chimeric L1/L2 mRNA-based vaccine against a broad range of HPV genotypes. All successful epitopes were connected together with the necessary linkers, and this construct was used to build mRNA structure to create a multi-epitope chimeric L1/L2 mRNA-based vaccine. This study found that the proposed mRNA-based vaccine construct is highly efficient and induced immune responses by in silico immune simulation. Furthermore, the physiochemical properties of the vaccine demonstrated that the vaccine is stable and soluble, along with the affinity for innate immune receptors (TLRs), indicating its ability to promote both innate and adaptive immunity against targeted viral infection. Therefore, the proposed vaccine design is a promising contender for producing mRNA vaccines for global use. The design of a possibly universal vaccine is a significant step forward in vaccine research and pandemic preparedness.
2. Materials and Methods
2.1. Retrieval of protein sequence, identification of conserved regions and generating consensus sequences
This research involved 25 amino acid sequences derived from 14 high risks (HR), 6 low risks (LR) and 5 cutaneous or skin warts–related human papillomavirus (HPV). All these sequences were derived from major (L1) and minor (L2) capsids protein of human papillomavirus downloaded from human papillomaepisteim (PaVE) data-base (https:// pave.niaid.nih.gov/) and were used as input for future bioinformatics analysis. In each category, the selected HPV types were picked from highly carcinogenic (HR), and low risks (LR)-associated with benign vaginal warts or epithelial lesions, and those mainly affecting skin as stated in the literature review [6]. Multiple sequence alignment was conducted by (BioEdit software v7.0.0) [22]. Subsequently, alignment result was uploaded into the Jalview software v2.11.3.2) (https://www .jalview.org/download/) [23] to generate consensus sequence and then these consensus sequences were employed to predict potential immunogenic T cells and B cells epitopes for constructing a chimeric L1/L2 mRNA-based vaccine candidate. Figure 1 displays an overview of all the operations.
2.2. Prediction of immune cell epitopes
The epitopes for MHC-class I or the cytotoxic T lymphocytes (CTL) were predicted using NetCTLpan-1.1 online tools available at https://services.healthtech.dtu.dk in which prediction parameters were set to default [24]. The prediction was done by selecting 12 alleles as default parameters. The Immune Epitope Database (IEDB) analysis tools (http://tools.iedb.org/mhcii/) was utilized to predict the epitopes for MHC-class II, or Helper T lymphocytes (HTL) [25]. The parameters were set as default by picking species/locus as (Human, HLA-DR), selecting whole HLA reference set with the panel of 27 alleles, and using netMHCIIpan 4.1 EL (recommended epitope predictor 2023.09) as the prediction method [26]. The ABC Pred server was used to screen Linear B Cell Lymphocytes (LBL), setting threshold score of more than ≥ 0.81[27]. This service predicts LBL epitopes with a length of 15mer amino acids with 65.93% accuracy using a recurrent neural network [28].
2.3. Screening the best epitopes for toxicity, antigenicity, allergenicity, and evaluation of vaccine physiochemical characteristics and solubility
Non-toxic epitopes were evaluated using ToxPred2 webserver available (https:// webs.iiitd.edu. in/raghava/toxinPred2/) using parameters, such as batch submission and protein scanning. The VaxiJen v2.0 and Aller-TOP servers (https://www.ddg-pharmfac .net/vaxijen/VaxiJen/VaxiJen. html) were used to calculate the antigenicity and allergenicity of the predicted epitopes by selecting viral organisms as target species. All of these predictions were performed as reported by Sanami and Oladipo [18,19]. Similarly, the VaxiJen v2.0, the Aller-TOP, and ToxPred2 webservers were used for antigenicity, allergenicity and toxicity analysis of the constructed vaccine. Solubility of the vaccine was predicted using Protein-Sol server (https://protein-sol.Manchester.ac.uk/cgi-bin/solubility/ sequence-prediction.php,) while physiochemical properties were verified by the Expasy-ProtParam tool (https://web.Expasy.org/cgi-bin/protparam/protparam) [29].
2.4. Epitope human homology and conservancy analysis
Epitope human homology analysis is an important bioinformatics tool used in the rational design of epitope-based vaccines because it aids in identifying the most immunogenic and pathogen-specific epitopes while avoiding those that are similar to self-proteins, thereby improving both the efficacy and safety of vaccine candidates [30]. The cross-reactivity of epitopes to human proteome was assessed by uploading individual epitope to the Multiple Peptide Match tool available in PI (https://research.bioinformatics.udel. edu/peptidematch/batchpeptidematch.jsp)UniProtKB, restricting Homo sapiens [taxi: 9606] as the target organism [31]. The epitope conservancy analysis (conservation across antigens) was performed using (IEDB server (http://tools.iedb. org/conservancy/). Other variable were maintained at its initial configuration, with the exception of the sequence identity threshold, which was set to 100% [32].
2.5. Population coverage analysis of CTL and HTL epitopes
The distribution of human leucocyte (HLA) alleles (class I and II) varies by geographic region around the world. As a result, when developing an effective vaccine, population coverage must be considered in order to reach the greatest number of people possible [33]. The IEDB server's population coverage tool (http://tools.iedb.org/population/) was used to calculate the coverage of chosen CTL and HTL epitopes.
2.6. Designing of mRNA-based vaccine
The final selected top ranked epitopes, such as CTL, HTL, and LBL, were combined using amino acid linkers to build a chimeric L1/L2 vaccine model, following the methods described by Oladipo, Kakakhel and Mohammadi, 2023 [19,34-35] with modifications. The KK linker was used to join intra-B cells epitopes, while EAAAK, and GGGGS were used to combine intra-CTL and HTL epitopes, respectively. Furthermore, EAAAK linker was used to link IgE signal peptide and PADRE sequence. The N-terminal of this vaccine design had Pan-(PADRE) [36] and the IgE leader signal peptide (MDWTWILFLV- AAATRVHS), whereas the C-terminal contained MITD (UniProt ID: Q5S1P3) and 6xHis-tag. All of these linkers were used to ensure that the proteins remained stable, and flexible [26]. To build the mRNA structure, additional regulatory components were introduced to the construct, including T7 promoter, UTRs (5’UTR: α-human globin), (3’UTR: β-human globin), and transcription initiator (Kozak sequence, GCCGCCACCATGGCG), Poly (A) tail, translation stop codon, and a plasmid DNA linearization enzyme (BspQI) [37]. UTRs were provided by GenScript.
2.7. Predicting 2D and 3D structures, refining 3D structure, and validating the vaccine candidates
SOPMA tool (https://npsa-prabi.ibcp.fr/cgi-bin/npsaautomat.pl?page=/NPSA/npsa sopma.html) and PSIPRED (http://bioinf.cs. ucl.ac.uk/psipred/) were run to predict 2D structure’s conformation states (beta-sheet, coil, and alpha-helix) of the vaccine design [32], while Rosetta (https://yanglab.qd.sdu.edu.cn/trRosetta/) [38] webservers were utilized to build the 3D structure of the vaccine. Moreover, the 3D predicted structure was refined to enhance structural quality by using the Galaxyrefine server (http://galax .seoklab.org). The quality assessment of the projected model was validated by the use of the Pro-SA online platform (https://prosa.services.came.sbg.ac.at/prosa.php),and PROCHECK and ERRAT (https:saves.mbi.ucla.edu/), which were considered for the stereochemistry analysis using the Ramachandran plot [18].
2.8. Identification of discontinuous B-cells epitopes
The non-linear B-cell epitopes were detected from the improved vaccine 3D structure using the Elli-Pro online tool, which is available on the IEDB server (https: //www.iedb.org). Discontinuous B cells are essential components of antibody epitopes [39].
2.9. Molecular docking between the vaccine and TRL4
Toll-like receptor TLR4 is protein that interact with the antigens of various viral and microbial organisms, promoting the stimulation of immune responses including cytokines and Type I interferon’s [40]. The molecular docking was performed using protein-protein docking servers, including the HawkDock (http://cadd.zju.edu.cn/hawkdock/) and ClusPro server (https://cluspro.bu.edu/). The HawkDock was ran to analyze binding interactions, evaluate binding free energy, and perform mm-GBSA analysis. The human Toll-like receptors TLR4 with the PDB ID code of (3FXI), which was retrieved from Protein Data Bank (RCSB PDB) (https://www.rcsb.org/ ) was used. The Dock Prep, which is integrated in the UCSF chimera v1.17.3 software, was used to prepare receptors. The interaction between the vaccine and the receptors were resolved using the LigPlot+ DIMPLOT software [40].
2.10. Simulating molecular dynamics of docking complex
Molecular dynamics simulation (MD) approach plays the role in determining atomic behavior and other biomolecules based on interatomic interactions [41]. The internal coordinate’s normal mode analysis server (iMODS), a tool that can be obtained at https://imods.iqf.csic.es/ was employed to analyze the interatomic and motions between the vaccine and receptors complexes. This tool predicts torsional angles of the complex, investigates the collective motions of proteins and nucleic acids utilizing the normal mode analysis (NMA) in internal coordinates, as well as structure deformation, eigenvalues of interacting residues, root-mean square deviation (RMSD) values, covariance among individual residues, and complex stability [42].
2.11. In Silico immune response simulation
Immune modeling predicts how the immune system recognizes epitope peptides, which is critical for assessing vaccine immunogenicity [43].The plots relative to the simulation and the outcome of the epitopes were produced by the C-IMMSIM Online server available at (https://kraken.Iac.rm. cnr.it/C-IMMSIM/index.php ). The simulation volume was set to [10-100]. Three injections were set as at time step for injection scheduled on days 1, 14, 28, and 42 (The first dose was given at time = 0, and each time-step equals to 8 h of real life) with a number of antigen to inject optimized to 20, 50, 100, and 1000. Other parameters were set to default, such as simulation steps set to 100, random seed set to 12345, host HLA selection, vaccine no LPS, and adjuvant set to 100.
2.12. Codon optimization, secondary structure prediction for mRNA vaccine, and cloning simulation
Codon optimization of the multi-epitope chimeric L1/L2 vaccine was performed using the Vector-Builder's Codon Optimization tool (https://en.vectorbuilder.com/tool/ codon-optimization. html), which was adapted for human (Homo sapiens) [44]. The generated sequence was then cloned into a standard cloning vector provided by GenScript (pUC57) using SnapGene software, franking between EcoRI and SacI restriction enzymes. The final mRNA construct was created using the optimized DNA from the Vector Builder's Codon Optimization tool, which was then uploaded into the RNAfold websever (http://rna.tbi. univie.ac.at/cgibin/RNAWebSuite/ RNAfold.cgi) to predict the secondary structure of the mRNA vaccine construct. The RNAfold server predicts mRNA structures using thermodynamics and assigns a minimal free energy (MFE) score to examine the MFE of secondary and centroid structures [45].
3. Results
3.1. Retrieval of protein sequences, multiple sequence alignment and generating consensus sequence
Twenty-five (25) HPV amino acids sequences were chosen from high-risks, low-risks, or genital wart-related and benign skin wart types in both major (L1) and minor (L2) proteins. Sequences were retrieved from (PaVE). These HPV strains have been demonstrated to share highly conserved regions [7]. The consensus sequence was produced from the highly conserved portions at each place in the sequence to promote the creation of an efficient vaccine against multi-types. The MSA and consensus sequence were generated by using BioEdit software v7.0.0 and Jalview v2.11.3.2, respectively.
3.2. Prediction and evaluation of MHC class I and MHC class II and B CELL (LBL) epitopes based on L1 protein
To identify potential T cell, or CD8+ T cell epitopes, we used the NetCTLpan 1.1 servers to classify immunogenic epitopes based on MHC class I, whereas the IEDB was served to predict MHC class II binding epitopes. Out of 45 potential CTL epitopes predicted, only 11.11% (5 out of 45) passed screening for toxicity, antigenicity, allergenicity, conservancy analysis, and MHCI-binding affinity score at percentile rank value ≤ 2.0, indicating a good binder (Table 1). The IEDB server identified 53 MHC II binding epitope candidates. After screening for antigenicity, allergenicity, and toxicity, only 16.98% (9 of 53) epitopes were found to be non-toxic, non-allergenic, and probable antigens. Based on conservation analysis, 44.4% (4 out of 9) epitopes demonstrated conservation at threshold ≤ 100% (Table 1). Using the threshold of (0.81), 31 potential Linear B cell lymphocytes (LBL) were identified. Out of thirty-one (31), 25.8% (8 out of 31) were non-toxic, probable antigen, and non-allergen. Among the eight (8) screened epitopes, 5 were found to be conserved in the sequences and were retained for further investigation.
3.3. Prediction and evaluation of CTL and HTL) and B CELL (LBL) based on L2 protein
NetCTLpan 1.1 server identified 48 possible MHC ligands or epitopes with MHCI-binding affinity scores of ≤ 2.0 percentile rank. Only 26.7% (13 out 48) epitopes successfully passed screening for toxicity, antigenicity and allergenicity. Out of these, 46.15% (6 out of 13) were highly conserved (Table 2). For HTL, a total of 38 potential epitopes were discovered utilizing the IEDB server. After screening for toxicity, antigenicity, and allergenicity, only 10.5% (4 out of 38) epitopes were non-toxic, non-allergenic, and most likely antigens. B cell epitopes play an important role in antigenic determinants and are detected by B lymphocyte receptors. Thirty-seven Linear B cell epitopes were found using the ABCpred server with a threshold of 0.81. The AllerTOP 2.0, Toxinred, and the VaxiJen servers determined 48.64% (18 out of 37) of LBL epitopes as non-toxic, non-allergen and probable antigen, respectively. Only two epitopes that showed conservation utilizing the conservancy analysis method were retained for further examination (Table 2).
3.4. Epitope human homology
Epitope human homology analysis is an important bioinformatics tool used in the rational design of epitope-based vaccines because it aids in identifying the most immunogenic and pathogen-specific epitopes while avoiding those that are similar to self-proteins, thereby improving both the efficacy and safety of vaccine candidates [30]. Individual epitope was checked by restricting organism from UniProt complete proteomes for (Homo sapiens taxi: 9606). All predicted epitopes were not detected in the human genome, indicating that they have no cross-reactivity with the human proteome.
1. Population coverage analysis
In this study, the IEDB population coverage approach was used. The population coverage analysis of combined epitopes in both L1 and L2 proteins (CTL L1 and CTL L2) and (HTL L1 and HTL L2) revealed to cover the majority of Europe (92.93%), followed by North America (88.33%), East Asia (76.73%), South Asia (75.51%), and North Africa (75.59 %). However, the lowest population coverage rates was observed in Central America (21.92%), Central Africa (44.53%), South America (51.7%), and East Africa (53.67%). Overall, the vaccine achieved a coverage rate of 86.24% globally (Figure 2)
3.5. Chimeric mRNA–based vaccine assembly
A chimeric L1/L2 mRNA-based vaccine model with 25 epitopes was generated by conjugating various combinations of shortlisted CTL, HTL, and B-cell (LBL) epitopes with particular linkers. The chimeric mRNA- based vaccine composed of 4 HTL L1 and 4 HTL L2 joined together using GGGGS, 4 LBL L1 and 2 LBL L2 linked with KK flexible linker, and 5 CTL L1 and 6 CTL L2 combined with EAAAK rigid linker. Other regulatory elements added to the primary construct included signal peptide (IgE leader sequence and Pan DR-binding epitope (PADRE) as a human Toll-like receptor agonist [36], MITD (UniProt ID: Q5S1P3) to direct MHC class I epitopes to the endoplasmic reticulum [19]. In addition, a 6xHis-tag was added to the C- terminal for purification purposes. To design a complete mRNA structure, a T7 promoter RNA polymerase (TAATACGACTCACTA TA GGG), 5’UTR (human alpha globin gene: HBA), Kozak sequence were added in the N-terminal, while a translation stop signal (a stop codon), 3’UTR (human beta globin gene: HBB), and Poly (Ax120) tail were added to the construct. Moreover, the restriction enzyme (BspQI: GCTCTTC) was added after the Poly (A) tail to facilitate the plasmid DNA linearization for in vitro transcription. UTRs for α-human globin (5'UTR) and β-human globin (3'UTR) were introduced to maintain mRNA stability. Figure 3 represents graphical representation of the multi-epitope chimeric L1/L2 mRNA-based vaccine against HPV species
3.5. Assessment of antigenicity, allergenicity, and solubility profiling and physiochemical properties of the vaccine design
The ProtParam server projected the multi-epitope chimeric peptides vaccine to have a molecular weight of 60161.29~60kDa and a theoretical pI of 9.44, indicating it is basic in nature. Fifty-six (56) of the 562 amino acid residues were negatively charged (Asp+Glu), while 73 were positively charged (Arg+Lys). The vaccine demonstrated thermostability (aliphatic index = 68.49) and stability (instability index = 36.89). A protein with an instability greater than 40 is considered unstable [35]. Besides, the grand average of hydropathicity (GRAVY) was predicted to be -0.366, depicting hydrophilic in nature, and demonstrating its ability to interact better with the adjacent water molecules [40]. The Protein-Sol server demonstrated a protein solubility of 0.451 for the vaccine, indicating an acceptable profile (≥ 45.0) compared to the average solubility of E. coli protein in the experimental solubility dataset. Furthermore, the multi-epitope chimeric L1/L2 had an average half-life of 30 hours in vivo (in mammalian reticulocytes), > 20 hours (yeast), and >10 hours (Escherichia coli). The vaccine had an antigenicity score of 0.9178 at the threshold of ≥ 0.4 and no suspected allergens, as validated by VaxiJen 2.0 and AllerTOP v.2.0 servers. Table 3 summarizes the physicochemical properties of the vaccine design, demonstrating the protein's stability and potential to induce a strong defensive response in the body. It is also expected to be safe, as allergenicity testing revealed no danger of allergenicity in the protein sequence.
3.5. Secondary (2D) structure prediction of the chimeric L1/L2 mRNA–based vaccine
2D structure of the produced vaccine was predicted and validated by applying SOPMA and PSIPRED servers (Figure 4). SOPMA servers calculate the fraction of 2D configurations using 4 conformational states, such as alpha helix, random coil, and beta-turn with an accuracy of 80% [33]. As predicted by the SOPMA server, the secondary structure result revealed that the proposed vaccine had 129 (22.95%) alpha helixes, 158 (28.11%) extended strands, and 275 (48.93%) random coils. The outcomes revealed that the designed chimeric L1/L2 vaccine had acceptable configurations states of protein characterized by flexibility, stability, which indicates that epitopes are located in accessible region of the protein.
3.5. Modelling and validation of tertiary (3D) structure of the vaccine construct
Rosetta online server showed that overall assessment of numerous protein quality parameters to be on high quality as assessed by PROCHECK and ProSA web servers before and after 3 D structure refinement. As depicted in Figure 5B of the refined model, 96.1%, 3.5%, 0.4 %, and 0.2% of the residues are located in most favoured, additional allowed, generously allowed, and disallowed regions, respectively. In the improved model, the vaccine construct had a Z-score of -7.2, indicating good quality (Z< 0) (Figure C) and an ERRAT's quality factor of 88.3721. GalayxRefine2 webserver demonstrated that the refined 3D structure had GDT-HA scores (0.9644), RMSD (0.372), MolProbity (1.390), clash score (7.1), poor rotamers (0.2), and ram favored score (98.8). A high resolution quality model should have over 90% of amino acid residues in its most favored region [46]. This finding indicated that the modeled protein had good overall stereo-chemical quality and that the majority of its residues was in energetically favorable conformations.
3.10. Identification of discontinuous B-cell epitopes
ElliPro: Antibody epitope prediction was used to identify non-linear B cell epitopes on the 3D structure of the vaccine setting the threshold of minimum score greater than (≥ 0.5) and a maximum distance of 6 angstrom (6Å) as default.
Figure 6.
Discontinuous B cells epitopes in the chimeric L1/L2 mRNA-based vaccine 3D structure (Figure 6 (1-12). The vaccine design is showed by gray sticks, whereas the discontinuous B cell epitopes are depicted by cyan spheres. The number (1-12) demonstrate residues and their corresponding scores as are shown in Table 4.
Figure 6.
Discontinuous B cells epitopes in the chimeric L1/L2 mRNA-based vaccine 3D structure (Figure 6 (1-12). The vaccine design is showed by gray sticks, whereas the discontinuous B cell epitopes are depicted by cyan spheres. The number (1-12) demonstrate residues and their corresponding scores as are shown in Table 4.
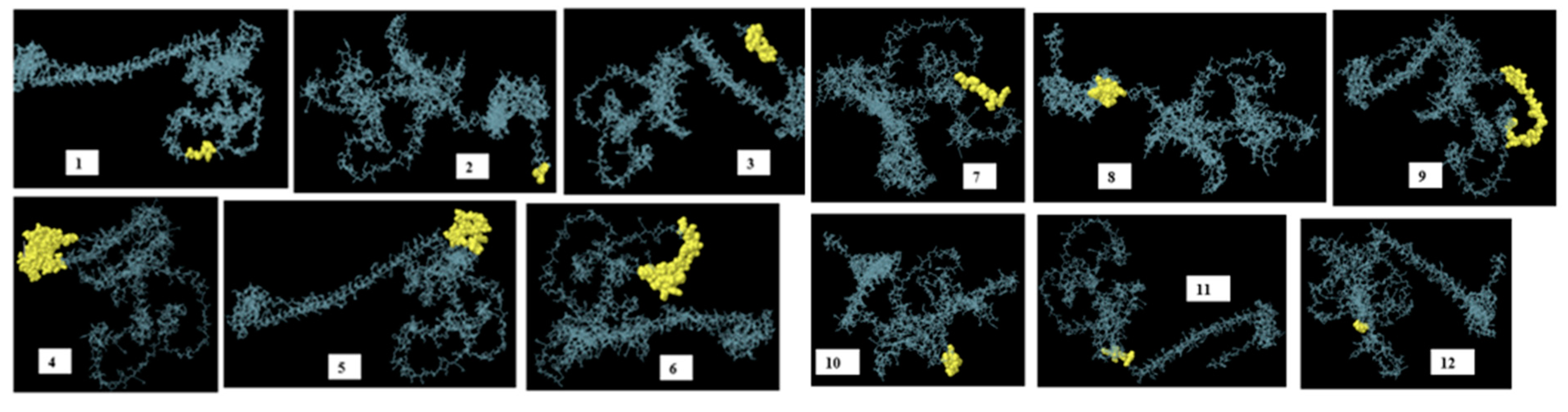

Table 4.
A list of predicted discontinuous B-Cell epitopes by the IEDB: ElliPPro server.
| No. | Residues | Number of residues | score |
| 1 | A:N106, A:V107, A:S108, A:M109, A:D110, A:Y111 | 6 | 0.979 |
| 2 | A:H560, A:H561, A:H562 | 3 | 0.969 |
| 3 | A:F549, A:V550, A:P551, A:I552, A:M553, A:G554, A:I555, A:I556, A:H557, A:H558, A:H559 | 11 | 0.895 |
| 4 | A:S438, A:R439, A:L440, A:G441, A:Q442, A:K443, A:A444, A:T445, A:M446, A:E447, A:A448, A:A449, A:A450, A:K451, A:L452, A:Q453, A:W454, A:G455, A:S456, A:L457, A:G458, A:V459, A:F460, A:E461, A:A462, A:A463, A:A464, A:K465, A:P466, A:E467, A:V468, A:T469, A:L470, A:R471, A:C472, A:W473, A:A474, A:L475, A:G476, A:F477, A:Y478, A:P479, A:E480, A:E481, A:I482, A:L483, A:L484, A:T485, A:W486, A:Q487, A:R488, A:N489, A:G490, A:E491, A:D492, A:L493, A:T494, A:Q495, A:D496, A:M497, A:E498, A:L499, A:V500, A:E501, A:T502, A:R503, A:P504, A:S505, A:G506, A:D507, A:G508, A:N509, A:F510, A:Q511, A:K512, A:W513, A:A514, A:A515, A:L516, A:G517, A:V518, A:P519, A:S520, A:G521, A:E522, A:E523, A:Q524, A:R525, A:Y526, A:T527, A:C528, A:R529, A:V530, A:Q531, A:H532, A:E533, A:G534, A:L535, A:Q536, A:G537, A:P538, A:L539, A:T540, A:L541, A:R542, A:W543, A:E544, A:P545, A:P546, A:Q547, A:P548 | 111 | 0.776 |
| 5 | A:T232, A:L233, A:C234, A:K235, A:K236, A:G237, A:Q238, A:P239, A:L240, A:G241, A:V242, A:G243, A:I244, A:S245, A:G246, A:H247, A:P248, A:L249, A:L250, A:N251, A:K252, A:K253, A:K254, A:R255 | 24 | 0.741 |
| 6 | A:D64, A:N65, A:E67, A:N68, A:V69, A:S70, A:M71, A:D72, A:Y73, A:K74, A:Q75, A:T76, A:Q77, A:L78, A:G79, A:G82, A:S83, A:A84, A:H86, A:F87, A:F88, A:N89, A:R90, A:A91, A:G92, A:T93, A:V94, A:G95, A:E96, A:K97, A:G99, A:G100, A:G101, A:G102, A:S103, A:R104, A:E105 | 37 | 0.74 |
| 7 | A:K112, A:Q113, A:T114, A:Q115, A:L116, A:C117, A:I118, A:G119 | 8 | 0.724 |
| 8 | A:G345, A:Q346, A:P347, A:L348, A:E349, A:A350, A:A351, A:A352, A:K353, A:E354, A:E355, A:Y356, A:D357, A:L358, A:Q359, A:F360, A:I361, A:E363, A:A364, A:K367 | 20 | 0.701 |
| 9 | A:W151, A:G152, A:S153, A:L154, A:G155, A:V156, A:F157, A:F158, A:G159, A:G160, A:G161, A:G162, A:S163, A:T164, A:S165, A:G166, A:F167, A:E168, A:I169, A:T170, A:S171, A:S172, A:S173, A:T174, A:D175, A:E176, A:A177, A:T178, A:G179, A:G180, A:G181 | 31 | 0.682 |
| 10 | A:G282, A:Y283, A:V284, A:P285, A:L286, A:G287, A:T288, A:K289, A:G291, A:G292, A:L293, A:G294, A:I295, A:G296 | 14 | 0.594 |
| 11 | A:A338, A:K339, A:V340, A:E341, A:V342, A:G343, A:R344 | 7 | 0.528 |
| 12 | A:A36, A:G37, A:G38, A:G39, A:G40 | 5 | 0.511 |
3.11. Molecular docking of vaccine construct with TLR4
To determine the modes and energies of the proposed vaccine with human Toll-like receptor 4 (TLR4), molecular docking techniques were applied. The ClusPro 2.0 server produced top five ranked models of docking complex between vaccine and TLR4. The best model had 52 members, demonstrated the lowest energy value of -1377.5kcal/mol and a representative center with a weighted score value of -1363.1. The docking complex vaccine and TLR4 was simulated using ClusPro 2.0 server (Figure 7I), whereas the interactions between residues were resolved by LigPlot+/DIMPLOT (Figure 7II, III, and IV). The hydrogen bonds that had formed between residues of vaccine and receptor are: ASN 365 (B):LYS 388 (A); ASN 365 (B):ASN 365 (A); LYS 388 (B):ASN 365 (A); GLN 507 (B):GLN 507 ( A); ASP 101 (C):ARG 764 (A); ASP 101 (C):SER 317 (A); TYR 102 (C):ARG 764 (A); SER 103 (C):ASN 266 (A); THR 116 (C):ASN 265 (A); GLN 111 (C):HIS 159 (A); THR 112 (C):GLU 135 (A); THR 112 (C):ARG 87 (A); GLY 110 (C):ARG 87 (A); ARG 68 (C):GLU 42 (A); LYS 109 (C):SER 62 (A); ASP 100 (C):ARG 234 (A); ASP 99 (C):ARG 289 (A); ARG 106 (C):SER 18(A); ARG 106 (C):SER 183 (A); ARG 90 (D):GLU 439 (A); GLY 123 (D):SER 416 (A); LYS125 (D):SER 445 (A); LYS 125 (D):ASN 417 (A); LYS 125 (D):GLU 422 (A)
3.12. Molecular dynamics simulation (MD)
Molecular dynamics simulation (iMODs) performs structural critical analysis by altering the complex's force field over time intervals. The resulting of the complex between vaccine andTLR4 showed moderate protein deformability or fluctuations at each residue capacity level because the complex contained some regions with lower and higher hinges or peaks (Figure 8b). The location of the chain ‘hinges’ can be derived from high deformability regions. The complex exhibited an Eigenvalue of 5.599239e -08 (Figure 8c). The Eigenvalue for each normal mode represents the motion stiffness. Its value is directly proportional to the energy required to distort the structure. The smaller the eigenvalue, the softer the mode and the simpler the deformation, indicating flexibility; however, it is unstable vice versa.
The graph in Figure 8 C displays the relationship between the complex's NMA and PDB fields, along with a B-factor, which represents how much atoms or residues in a molecule move around their average positions during simulation. The results showed a few disordered regions with a larger B-factor, which corresponds to increased atomic displacement, indicating flexible loops that facilitate antigen presentation and immune recognition. The elastic network model (Figure 8F), describes which pairs of atoms are connected by a spring or represents interactions between residues in which each dot on the graph represents a spring between the corresponding atoms. Dots are colored according on stiffness, with darker grays signifying stiffer springs and vice versa.
The covariance matrix or heat map shows whether pairs of residues experience correlated (red), uncorrelated (white), or anti-correlated (blue) motions (Figure 8G). The result indicated highly co-related regions in heat maps, revealing greater correlations between individual residues. Overall, the iMODS analysis demonstrated that the vaccine-TLR4 complexes remained stable and flexible, with specific places exhibiting compactness and deformability within the complex's coordinate system.
3.13. Immune simulation of the chimeric vaccine
The C-IMMSIM tool simulates B-cell epitope binding, class I and II HLA epitope binding, and T-cell receptor binding to HLA peptide complexes [47]. As a result, (Figure 9A,B) show that high immunoglobulin titers were formed following the fourth injection of the vaccination. The first response was indicated by an elevated level of IgM+IgG, followed by IgM. During the simulation period, antibody levels (IgM+IgG, IgG1+IgG2, IgM, IgG1) increased as the antigen injection volume increased during secondary responses. Following vaccination, there was a significant B-cell population, including memory B cells and Plasma B lymphocytes count sub-divided per isotype (IgM, IgG1 and IgG2), as well as antigen presentation. As shown in (Figure 9D), there was a significant rise in the population of TH cells, including Th memory. Moreover, cytotoxic T lymphocytes and NK cells were found. Figure 9C, shows an increase in macrophage activity and dendritic cells, which are key antigen presentation components of the immune system, during vaccine delivery, as well as helper T cells (Figure 9D). The immunization with this vaccine significantly increased IFN-γ titers and IL-2 levels (Figure 9F). This proposed vaccine demonstrated to generate an effective immune response that could protect against the targeted illness, as evidenced by these data.
3.14. Codon-optimization and in Silico cloning
The final constructed chimeric vaccine was codon-optimized to human (Homosapiens) using VectorBuilder online tool. The optimized ORF consisted of 1686 nucleotides. The results showed that the vaccine's DNA sequence had a CAI of 0.91 and a GC content of 59.92%, indicating a stable DNA sequence and robust protein expression. The complete mRNA vaccine was then inserted into the standard vector pUC57, provided by GenSript, franked between two restriction sites of EcoRI (GAATTC) and SacI (GAGCTC) using SnapGene software in the opposite direction to the lac promoter. In addition, BspQI (GCTCTTC) restriction enzyme sequence was added to the constructed vaccine after poly (A) tail to facilitate plasmid DNA linearization during IVT mRNA synthesis. The resulting plasmid vector construct designed “chimeric L1/L2_pUC57” consisted of 4725 bp, comprising 2027 bp of the inserted sequence (Figure 10).
3.14. Prediction of secondary structure and minimum free enery of the chimeric vaccine
The RNAfold webserver was used to compute the MFE of the mRNA vaccine, resulting in an MFE of -731.10 kcal/mol, while that of the centroid secondary structure was-527.51 kcal/mol. Furthermore, the thermodynamic ensemble has a free energy of -757.80 kcal/mol, a frequency of 0.00 % for the MFE structure, and an ensemble diversity of 560.43.These findings indicate that the mRNA may remain stable after production (Figure 11). The structure below is colored by base-pairing probabilities. For unpaired regions the color denotes the probability of being unpaired.
4. Discussion
This study developed a chimeric L1/L2 mRNA-based vaccine against human papillomavirus (HPVs), which is a major health concern, including cervical cancer [1]. Currently, prophylactic vaccinations, such as Gardasil-9, Gardasil-4, and CervarixTM, only target a limited number of HPV species [10], highlighting the need for broad protective vaccinations. Fusion vaccines can be created by co-expressing major and minor proteins, using immuno-informatics and in silico approaches [14,48]. These vaccines can be designed against various viral and pathogenic infections, including HPV, using multi-epitope peptides and mRNA technology [27]. Epitope-based peptide vaccine creation is easy, economical, and quick, resulting in stable proteins and high immune responses [33]. In silico techniques have been used to create universal peptide vaccines from HPV proteins, and prophylactic multi-epitopic DNA vaccines have been developed [8,49]. DNA vaccines are less preferred due to high costs, difficulty, and limited immunogenicity, and there is a possibility of genome mutation [20]. Because of its safety qualities, mRNA-based vaccines are required for both preventive and therapeutic applications.
In this study, as in many others, the GGGGS linker has demonstrated exceptional flexibility and solubility, making it a great candidate for fusion protein domains that require particular movements or interactions [50]. The KK linker improves epitope processing and presentation, enhancing immunogenicity, whereas the EAAAK linker provides structural stability and promotes epitope attachment to adjuvants, resulting in better immune activation [29].
The vaccine's chimeric L1/L2 mRNA structure was optimized for efficient human expression, incorporating untranslated regions, Poly (A) tail, and Kozak sequence for stability, translation efficiency, and immunogenicity, for enhancing its efficacy [51]. This vaccine showed no cross-reactivity with the human genome, confirming safety and lack of insertional mutagenesis.
In the context of vaccine physiochemical characteristics, the constructed vaccine had an antigenicity score of 0.9178 and a solubility of 0.451, indicating its potential to generate immunological responses and influencing the efficacy and stability of the vaccine during production and storage, respectively. In addition, the vaccine found to be non-toxic and non-allergenic, which confirms its safety in the body. The Expasy-ProtParam projected the vaccine to have a molecular weight of 60161.29~60 kDa for the chimeric peptide vaccine construct, indicating an appropriate vaccine candidate as the protein falls within the approved range of less than 110kDa, making it easier to express and purify than heavier proteins [18,40]. The molecular weight of the chimeric mRNA-based vaccine was 644.98, as estimated using the RNA Molecular Weight Calculator online tool. The molecular weight of mRNA-based vaccine can vary greatly depending on its specific design and components, as numerous research found a varying size of 285.3 kDa [19], 50.417 kDa [47], and 71.897.13kDa [29]. The vaccine's solubility exceeded the average solubility of E.coli protein in the experimental solubility dataset (0.45), demonstrating good soluble protein [52]. The mRNA-based vaccine comprised multi-epitope peptides with an estimated a theoretical pI of 9.44, instability index of 36.89 and aliphatic index of 68.49. The higher aliphatic index indicates that the created vaccine is heat-stable in nature because the higher the aliphatic index of a protein, the greater its thermal stability [53]. A protein with an instability greater than 40 is considered unstable [35]. Besides, the grand average of hydropathicity (GRAVY) was predicted to be -0.366, depicting hydrophilic in nature, and demonstrating its ability to interact better with the adjacent water molecules [40]. The proposed vaccine construct's half-life was calculated to be 30 h in mammalian reticulocytes, more than 20 h in yeast, and more than 10 h in E. coli, suggesting that the vaccine lasts longer when exposed to the immune system [53]. The findings revealed that the produced vaccine is a promising contender for future use because its physicochemical properties coincided with previous investigations [29,47].
The global distribution of HLA alleles, class I and II, necessitates a focus on population coverage when developing effective vaccines to reach the maximum number of people [33]. This vaccine achieved a global coverage rate of 86.24%, with a higher population coverage rate of 92.93% observed in Europe.
The vaccine's 2D structure modeling identified stable, flexible, and globular secondary structural features in α-helices, β-sheets, and coils. 3D structure modeling revealed the vaccine had the most amino acid residues in the most preferred region, with a coverage of 96.1% and a Z-score value of -7.2, indicating good quality (Z< 0). A high resolution quality model should have over 90% of amino acid residues in its most favored region [46]. A larger negative value for Z-score indicates that the structure is of higher quality [54].
Based on molecular dynamics simulation, the vaccine demonstrated good binding to TLR4 receptor, with binding affinities of (-1377.5 kcal/mol). A lower ΔG values indicate a more favorable interaction [55], implying that the vaccine interacted more strongly to the receptor. The proposed vaccine efficiently interacts with Toll-like receptor 4 (TLR4), producing hydrogen bonds, salt bridges, and hydrophobic interactions. The iMODS server results revealed the vaccine's deformability at each residue, moderate motion stiffness, high correlations between residue pairings, and few regions with a higher B-factor. The developed vaccine was successfully demonstrated to generate long-term IgG, IgM, B lymphocytes, and cytotoxic T responses, as well as cytokine responses when administered four times over a 42-day period with a 2-week interval. The release of cytokines that are capable of eliminating the virus-infected cells indicates that the T cells are activated [40].
In the context of mRNA vaccine design, the secondary structure of the mRNA plays a crucial role in the stability, translation efficiency, and immunogenicity of the vaccine [55-56]. The RNAfold server predicted the mRNA structure with a negative MFE value of -731.10 kcal/mol, whereas its centroid secondary structure produced -527.51 kcal/mol, indicating that the expected vaccine may be very stable following transcription in vivo. Studies suggested mRNA secondary structure stability with negative MFE; however, no universally accepted MFE value for mRNA vaccines, as it depends on specific sequence and desired properties [19]. VectoBuilder's codon optimization tool yielded satisfactory results with CAI (0.91) and GC content (59.92%), meeting optimal expression criteria for target organism [18]. Regarding the facts of this research presenting a good future outcomes, it is recommended to validate this vaccine candidate in vitro and in vivo to assure safety and efficacy.
5. Conclusions
HPV infections persist due to limited preventative vaccinations targeting specific species. Cross-protective vaccinations are effective for targeting diverse HPV genotypes. We used immuno-informatics approach to develop a chimeric mRNA-based vaccine against a broad spectrum HPV strains. This study aimed to create a universal chimeric mRNA-based vaccine that generates cellular and humoral immunity by incorporating T-cells and B-cell epitopes and promoting innate and adaptive immunity against targeted viral infections. The immunosimulation results supported the vaccine's ability to produce immune responses, whereas molecular dynamics simulation indicated that the vaccine engaged and triggered TLR4 on immune responses through a robust vaccine-receptor interaction. The proposed research is promising for future outcomes; nevertheless, it is an in silico study that requires laboratory validation; hence, we urge that this vaccine design be validated by in vitro and vivo testing.
Author Contributions
Conceptualization, N.S. (Nshimiyimana Sylvere), J.K. (James Kimotho) and C.W.N. (Caroline Wangari Ngugi); methodology, N.S. (Nshimiyimana Sylvere), J.K. (James Kimotho), and C.W.N. (Caroline Wangari Ngugi); formal analysis, N.S. ( Nshimiyimana Sylvere), J.K. (James Kimotho) and C.W.N. ( Caroline Wangari Ngugi); investigation, N.S. (Nshimiyimana Sylvere).; resources, N.S. ( Nshimiyimana Sylvere); data curation, N.S. (Nshimiyimana Sylvere), J.K. (James Kimotho) and C.W.N. ( Caroline Wangari Ngugi); validation, N.S. (Nshimiyimana Sylvere), J.K. (James Kimotho) and C.W.N (Caroline Wangari Ngugi); writing—original draft preparation, N.S. (Nshimiyimana Sylvere); writing—review and editing; N.S. (Nshimiyimana Sylvere), J.K. ( James Kimotho), C.W.N. ( Caroline Wangari Ngugi); visualization, N.S. (Nshimiyimana Sylvere), J.K. (James Kimotho) and C.W.N. (Caroline Wangari Ngugi); supervision, J.K. (James Kimotho) and C.W.N. ( Caroline Wangari Ngugi); project administration, J.K. (James Kimotho) ; funding acquisition, N.S. (Nshimiyimana Sylvere). All authors have read and agreed to the published version of the manuscript. All authors contributed equally. All authors have read and agreed to the published version of the manuscript.
Funding
Please add: This work was supported by the African Union Commission through the Pan African University Institute for Basic Sciences, Technology, and Innovation (PAUSTI).
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable
Data Availability Statement
All data are provided in this manuscript.
Acknowledgments
The authors extend their gratitude for the support provided by the African Union Commission through the Pan African Union through Pan African University Institute for Basic Sciences, Technology and Innovation (PAUSTI).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Mahmoudvand, S.; Shokri, S.; Makvandi, M.; Taherkhani, R.; Rashno, M.; Jalilian, F. A.; Angali, K. A. In Silico Prediction of T-Cell and B-Cell Epitopes of Human Papillomavirus Type 16 L1 Protein. Biotechnol. Appl. Biochem. 2022, 69, 514–525. [Google Scholar] [CrossRef]
- Singh, D.; Vignat, J.; Lorenzoni, V.; Eslahi, M.; Ginsburg, O.; Lauby-Secretan, B.; Arbyn, M.; Basu, P.; Bray, F.; Vaccarella, S. Global Estimates of Incidence and Mortality of Cervical Cancer in 2020: A Baseline Analysis of the WHO Global Cervical Cancer Elimination Initiative. Lancet Glob. Heal. 2023, 11, e197–e206. [Google Scholar] [CrossRef] [PubMed]
- Jedy-Agba, E.; Joko, W. Y.; Liu, B.; Buziba, N. G.; Borok, M.; Korir, A.; Masamba, L.; Manraj, S. S.; Finesse, A.; Wabinga, H.; Somdyala, N.; Parkin, D. M. Trends in Cervical Cancer Incidence in Sub-Saharan Africa. Br. J. Cancer 2020, 123, 148–154. [Google Scholar] [CrossRef]
- Drokow, E. K.; Fangninou, F. F.; Effah, C. Y.; Agboyibor, C.; Zhang, Y.; Arboh, F.; Deku, M. A.; Xinyin, W.; Wang, Y.; Sun, K. Cervical Cancer Survival Times in Africa. Front. Public Heal. 2022, 10. [Google Scholar] [CrossRef]
- Bruni, L.; Diaz, M.; Castellsagué, X.; Ferrer, E.; Bosch, F. X.; De Sanjosé, S. Cervical Human Papillomavirus Prevalence in 5 Continents: Meta-Analysis of 1 Million Women with Normal Cytological Findings. J. Infect. Dis. 2010, 202, 1789–1799. [Google Scholar] [CrossRef] [PubMed]
- Haręża, D. A.; Wilczyński, J. R.; Paradowska, E. Human Papillomaviruses as Infectious Agents in Gynecological Cancers―Oncogenic Properties of Viral Proteins. Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Olczak, P.; Roden, R. B. S. Progress in L2-Based Prophylactic Vaccine Development for Protection against Diverse Human Papillomavirus Genotypes and Associated Diseases. Vaccines 2020, 8, 1–22. [Google Scholar] [CrossRef]
- Namvar, A.; Bolhassani, A.; Javadi, G.; Noormohammadi, Z. In Silico/In Vivo Analysis of High-Risk Papillomavirus L1 and L2 Conserved Sequences for Development of Cross-Subtype Prophylactic Vaccine. Sci. Rep. 2019, 9, 1–22. [Google Scholar] [CrossRef]
- Boxus, M.; Fochesato, M.; Miseur, A.; Mertens, E.; Dendouga, N.; Brendle, S.; Balogh, K. K.; Christensen, N. D.; Giannini, S. L. Broad Cross-Protection Is Induced in Preclinical Models by a Human Papillomavirus Vaccine Composed of L1/L2 Chimeric Virus-Like Particles. J. Virol. 2016, 90, 6314–6325. [Google Scholar] [CrossRef]
- Huber, B.; Wang, J. W.; Roden, R. B. S.; Kirnbauer, R. Rg1-Vlp and Other L2-Based, Broad-Spectrum Hpv Vaccine Candidates. J. Clin. Med. 2021, 10, 1–21. [Google Scholar] [CrossRef]
- Begum Akuzum1,†, Sinae Kim2, 3,†, Tam Thanh Nguyen2, 3,†, J. H.; Lee3, S.;, Eunhye Kim2, 3, J. K.; Choi1, Y.;, Hyunjhung Jhun3, 4; Lee5, Y.; Kim6, H.; Sohn7, D. H.;, Soohyun Kim1, 2. L1 Recombinant Proteins of HPV Tested for Antibody Forming Using Sera of HPV Quadrivalent Vaccine. Immune Netw. 2018, 18, 1–13. [Google Scholar]
- Huber, B.; Schellenbacher, C.; Shafti-Keramat, S.; Jindra, C.; Christensen, N.; Kirnbauer, R. Chimeric L2-Based Virus-like Particle (VLP) Vaccines Targeting Cutaneous Human Papillomaviruses (HPV). PLoS One 2017, 12, 1–27. [Google Scholar] [CrossRef]
- Schellenbacher, C.; Roden, R. B. S.; Kirnbauer, R. Developments in L2-Based Human Papillomavirus (HPV) Vaccines. Virus Res. 2017, 231, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Tumban, E.; Peabody, J.; Peabody, D. S.; Chackerian, B. A Pan-HPV Vaccine Based on Bacteriophage PP7 VLPs Displaying Broadly Cross-Neutralizing Epitopes from the HPV Minor Capsid Protein, L2. PLoS One 2011, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Varsani, A.; Williamson, A.-L.; de Villiers, D.; Becker, I.; Christensen, N. D.; Rybicki, E. P. Chimeric Human Papillomavirus Type 16 (HPV-16) L1 Particles Presenting the Common Neutralizing Epitope for the L2 Minor Capsid Protein of HPV-6 and HPV-16. J. Virol. 2003, 77, 8386–8393. [Google Scholar] [CrossRef] [PubMed]
- McGrath, M.; de Villiers, G. K.; Shephard, E.; Hitzeroth, I. I.; Rybicki, E. P. Development of Human Papillomavirus Chimaeric L1/L2 Candidate Vaccines. Arch. Virol. 2013, 158, 2079–2088. [Google Scholar] [CrossRef]
- Wang, J. W.; Roden, R. B. S. L2, the Minor Capsid Protein of Papillomavirus. Virology 2013, 445, 175–186. [Google Scholar] [CrossRef]
- Sanami, S.; Nazarian, S.; Ahmad, S.; Raeisi, E.; ul Qamar, M. T.; Tahmasebian, S.; Pazoki-Toroudi, H.; Fazeli, M.; Ghatreh Samani, M. In Silico Design and Immunoinformatics Analysis of a Universal Multi-Epitope Vaccine against Monkeypox Virus. PLoS One 2023, 18. [Google Scholar] [CrossRef]
- Oladipo, E. K.; Adeniyi, M. O.; Ogunlowo, M. T.; Irewolede, B. A.; Adekanola, V. O.; Oluseyi, G. S.; Omilola, J. A.; Udoh, A. F.; Olufemi, S. E.; Adediran, D. A.; Olonade, A.; Idowu, U. A.; Kolawole, O. M.; Oloke, J. K.; Onyeaka, H. Bioinformatics Designing and Molecular Modelling of a Universal MRNA Vaccine for SARS-CoV-2 Infection. Vaccines 2022, 10. [Google Scholar] [CrossRef]
- Maruggi, G.; Zhang, C.; Li, J.; Ulmer, J. B.; Yu, D. MRNA as a Transformative Technology for Vaccine Development to Control Infectious Diseases. Mol. Ther. 2019, 27. [Google Scholar] [CrossRef]
- Rcheulishvili, N.; Mao, J.; Papukashvili, D.; Feng, S.; Liu, C.; Yang, X. Candidate for Monkeypox, Smallpox, and Vaccinia Viruses : 2023.
- Hall, T. Bio Edit. Pdf. 2004. [Google Scholar]
- Waterhouse, A. M.; Procter, J. B.; Martin, D. M. A.; Clamp, M.; Barton, G. J. Jalview Version 2-A Multiple Sequence Alignment Editor and Analysis Workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Stranzl, T.; Larsen, M. V.; Lundegaard, C.; Nielsen, M. NetCTLpan: Pan-Specific MHC Class I Pathway Epitope Predictions. Immunogenetics 2010, 62, 357–368. [Google Scholar] [CrossRef]
- Jensen, K. K.; Andreatta, M.; Marcatili, P.; Buus, S.; Greenbaum, J. A.; Yan, Z.; Sette, A.; Peters, B.; Nielsen, M. Improved Methods for Predicting Peptide Binding Affinity to MHC Class II Molecules. Immunology 2018, 154, 394–406. [Google Scholar] [CrossRef]
- Dehghani, A.; Mamizadeh, M.; Karimi, A.; Hosseini, S. A.; Siamian, D.; Shams, M.; Ghiabi, S.; Basati, G.; Abaszadeh, A. Multi-Epitope Vaccine Design against Leishmaniasis Using IFN-γ Inducing Epitopes from Immunodominant Gp46 and G63 Proteins. J. Genet. Eng. Biotechnol. 2024, 22, 100355. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sahu, U.; Kumari, P.; Dixit, A.; Khare, P. Designing of Multi-Epitope Chimeric Vaccine Using Immunoinformatic Platform by Targeting Oncogenic Strain HPV 16 and 18 against Cervical Cancer. Sci. Rep. 2022, 12, 1–16. [Google Scholar] [CrossRef]
- Sanchez, G. Las Instituciones de Ciencia y Tecnología En Los Procesos de Aprendizaje de La Producción Agroalimentaria En Argentina. El Sist. argentino innovación Inst. Empres. y redes. El desafío la creación y apropiación Conoc. 2013, 14, 659–664. [Google Scholar] [CrossRef]
- Naveed, M.; Hassan, J. U.; Ahmad, M.; Naeem, N.; Mughal, M. S.; Rabaan, A. A.; Aljeldah, M.; Shammari, B. R. A.; Alissa, M.; Sabour, A. A.; Alaeq, R. A.; Alshiekheid, M. A.; Turkistani, S. A.; Elmi, A. H.; Ahmed, N. Designing MRNA- and Peptide-Based Vaccine Construct against Emerging Multidrug-Resistant Citrobacter Freundii: A Computational-Based Subtractive Proteomics Approach. Med. 2022, 58. [Google Scholar] [CrossRef]
- De Groot, A. S.; Moise, L.; Terry, F.; Gutierrez, A. H.; Hindocha, P.; Richard, G.; Hoft, D. F.; Ross, T. M.; Noe, A. R.; Takahashi, Y.; Kotraiah, V.; Silk, S. E.; Nielsen, C. M.; Minassian, A. M.; Ashfield, R.; Ardito, M.; Draper, S. J.; Martin, W. D. Better Epitope Discovery, Precision Immune Engineering, and Accelerated Vaccine Design Using Immunoinformatics Tools. Front. Immunol. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Chen, C.; Li, Z.; Huang, H.; Suzek, B. E.; Wu, C. H. A Fast Peptide Match Service for UniProt Knowledgebase. Bioinformatics 2013, 29, 2808–2809. [Google Scholar] [CrossRef]
- Hasan, M.; Ahmed, S.; Imranuzzaman, M.; Bari, R.; Roy, S.; Hasan, M. M.; Mia, M. M. Designing and Development of Efficient Multi-Epitope-Based Peptide Vaccine Candidate against Emerging Avian Rotavirus Strains: A Vaccinomic Approach. J. Genet. Eng. Biotechnol. 2024, 22, 100398. [Google Scholar] [CrossRef]
- Shawan, M. M. A. K.; Sharma, A. R.; Halder, S. K.; Arian, T. Al; Shuvo, M. N.; Sarker, S. R.; Hasan, M. A. Advances in Computational and Bioinformatics Tools and Databases for Designing and Developing a Multi-Epitope-Based Peptide Vaccine. Int. J. Pept. Res. Ther. 2023, 29, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Kakakhel, S.; Ahmad, A.; Mahdi, W. A.; Alshehri, S.; Aiman, S.; Begum, S.; Shams, S.; Kamal, M.; Imran, M.; Shakeel, F.; Khan, A. Annotation of Potential Vaccine Targets and Designing of MRNA-Based Multi-Epitope Vaccine against Lumpy Skin Disease Virus via Reverse Vaccinology and Agent-Based Modeling. Bioengineering 2023, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, Y.; Nezafat, N.; Negahdaripour, M.; Eskandari, S.; Zamani, M. In Silico Design and Evaluation of a Novel MRNA Vaccine against BK Virus: A Reverse Vaccinology Approach. Immunol. Res. 2023, 71, 422–441. [Google Scholar] [CrossRef] [PubMed]
- Ghaffari-Nazari, H.; Tavakkol-Afshari, J.; Jaafari, M. R.; Tahaghoghi-Hajghorbani, S.; Masoumi, E.; Jalali, S. A. Improving Multi-Epitope Long Peptide Vaccine Potency by Using a Strategy That Enhances CD4+ T Help in BALB/c Mice. PLoS One 2015, 10, 1–12. [Google Scholar] [CrossRef]
- Pichon, C.; Perche, F. Design and Delivery of Messenger RNA-Based Vaccines. Biochem. (Lond). 2021, 43, 4–7. [Google Scholar] [CrossRef]
- Du, Z.; Su, H.; Wang, W.; Ye, L.; Wei, H.; Peng, Z.; Anishchenko, I.; Baker, D.; Yang, J. The TrRosetta Server for Fast and Accurate Protein Structure Prediction. Nat. Protoc. 2021, 16, 5634–5651. [Google Scholar] [CrossRef]
- Ponomarenko, J.; Bui, H. H.; Li, W.; Fusseder, N.; Bourne, P. E.; Sette, A.; Peters, B. ElliPro: A New Structure-Based Tool for the Prediction of Antibody Epitopes. BMC Bioinformatics 2008, 9, 1–8. [Google Scholar] [CrossRef]
- Akbay, B.; Abidi, S. H.; Ibrahim, M. A. A.; Mukhatayev, Z.; Ali, S. Multi-Subunit Sars-Cov-2 Vaccine Design Using Evolutionarily Conserved t-and b-Cell Epitopes. Vaccines 2021, 9, 1–27. [Google Scholar] [CrossRef]
- Hollingsworth, S. A.; Dror, R. O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef]
- López-Blanco, J. R.; Aliaga, J. I.; Quintana-Ortí, E. S.; Chacón, P. IMODS: Internal Coordinates Normal Mode Analysis Server. Nucleic Acids Res. 2014, 42, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Rapin, N.; Lund, O.; Castiglione, F. Immune System Simulation Online. Bioinformatics 2011, 27, 2013–2014. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, Q.; Zhao, F. Synonymous but Not Silent: The Codon Usage Code for Gene Expression and Protein Folding. Annu. Rev. Biochem. 2021, 90, 375–401. [Google Scholar] [CrossRef]
- Gruber, A. R.; Lorenz, R.; Bernhart, S. H.; Neuböck, R.; Hofacker, I. L. The Vienna RNA Websuite. Nucleic Acids Res. 2008, 36, 70–74. [Google Scholar] [CrossRef]
- Morris, A. L.; MacArthur, M. W.; Hutchinson, E. G.; Thornton, J. M. Stereochemical Quality of Protein Structure Coordinates. Proteins Struct. Funct. Bioinforma. 1992, 12, 345–364. [Google Scholar] [CrossRef]
- Adam, K. M. Immunoinformatics Approach for Multi-Epitope Vaccine Design against Structural Proteins and ORF1a Polyprotein of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2). Trop. Dis. Travel Med. Vaccines 2021, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Manuscript, A. Capsomeres to Broaden Protection against HPV Infection. Vaccination 2011, 28, 4478–4486. [Google Scholar]
- Gupta, S. K.; Singh, A.; Srivastava, M.; Gupta, S. K.; Akhoon, B. A. In Silico DNA Vaccine Designing against Human Papillomavirus (HPV) Causing Cervical Cancer. Vaccine 2009, 28, 120–131. [Google Scholar] [CrossRef]
- Chen, X.; Zaro, J. L.; Shen, W. C. Fusion Protein Linkers: Property, Design and Functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef]
- Tossberg, J. T.; Esmond, T. M.; Aune, T. M. A Simplified Method to Produce MRNAs and Functional Proteins from Synthetic Double-Stranded DNA Templates. Biotechniques 2020, 69, 281–288. [Google Scholar] [CrossRef]
- Niwa, T.; Ying, B. W.; Saito, K.; Jin, W.; Takada, S.; Ueda, T.; Taguchi, H. Bimodal Protein Solubility Distribution Revealed by an Aggregation Analysis of the Entire Ensemble of Escherichia Coli Proteins. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 4201–4206. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M. R.; Appel, R. D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server; The Proteomics Protocols Handbook. Humana Press 2005, 112, 531–52. [Google Scholar]
- Wiederstein, M.; Sippl, M. J. ProSA-Web: Interactive Web Service for the Recognition of Errors in Three-Dimensional Structures of Proteins. Nucleic Acids Res. 2007, 35, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Sarvmeili, J.; Baghban Kohnehrouz, B.; Gholizadeh, A.; Shanehbandi, D.; Ofoghi, H. Immunoinformatics Design of a Structural Proteins Driven Multi-Epitope Candidate Vaccine against Different SARS-CoV-2 Variants Based on Fynomer. Sci. Rep. 2024, 14, 1–26. [Google Scholar] [CrossRef]
- Lu, Q.; Wu, H.; Meng, J.; Wang, J.; Wu, J.; Liu, S.; Tong, J.; Nie, J.; Huang, W. Multi-Epitope Vaccine Design for Hepatitis E Virus Based on Protein ORF2 and ORF3. Front. Microbiol. 2024, 15, 1–10. [Google Scholar] [CrossRef]
Figure 1.
AB. The immuno-informatics flow chart for developing a chimeric L1/L2 mRNA- vaccine against several HPV species.
Figure 1.
AB. The immuno-informatics flow chart for developing a chimeric L1/L2 mRNA- vaccine against several HPV species.
Figure 2.
Population coverage of combined epitopes in both L1 and L2 proteins.
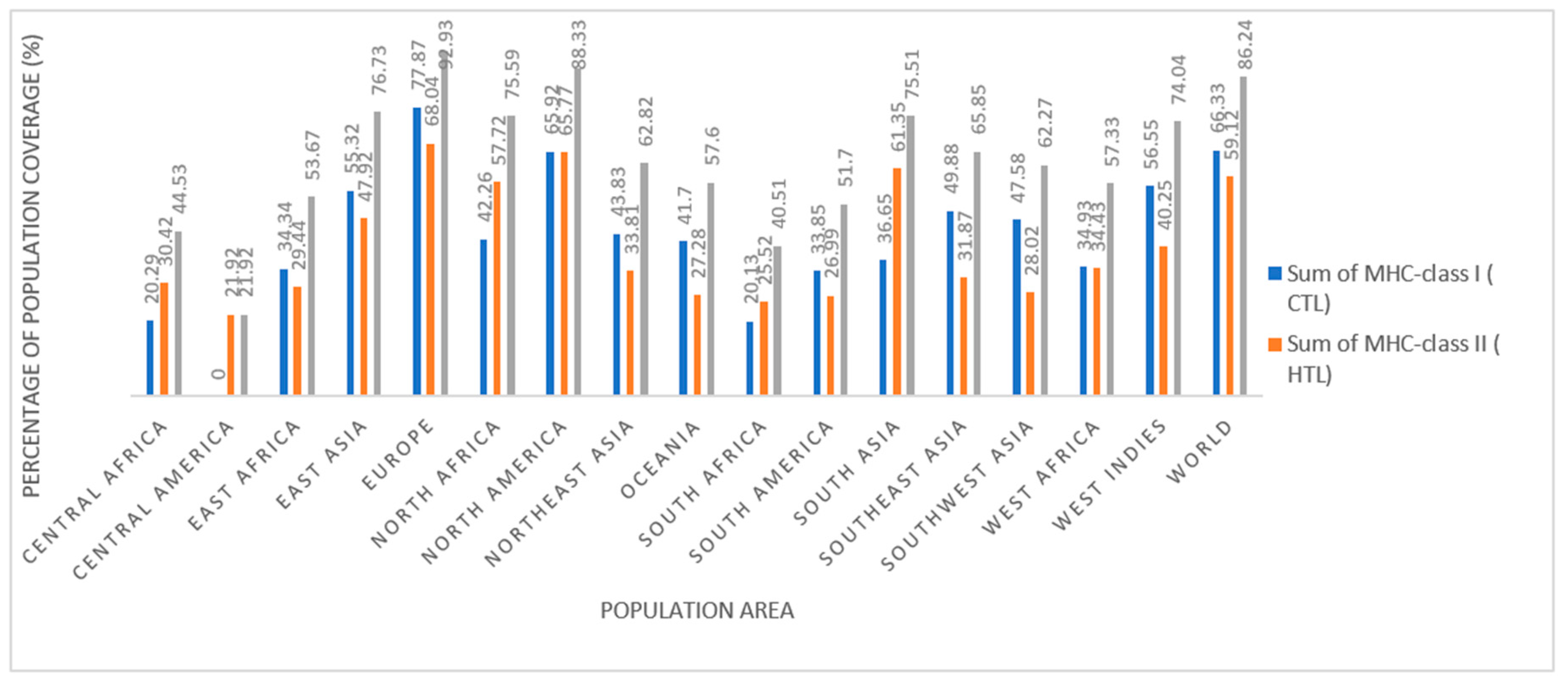
Figure 3.
Figure 3. Graphical representation of chimeric L1/L2 mRNA-based vaccine construct against HPV species. The construct consists of 25 epitopes, a T7 promoter, α-human globin (5'UTR), β-human globin (3'UTR), a Kozak sequence, an IgESP (signal peptide), a stop codon, a Poly (Ax120) tail, a 6xHis-Tag sequence, a plasmid linearization restriction enzyme sequence BspQI, and various linkers, such as GGGGS, EAAAK, and KK.
Figure 3.
Figure 3. Graphical representation of chimeric L1/L2 mRNA-based vaccine construct against HPV species. The construct consists of 25 epitopes, a T7 promoter, α-human globin (5'UTR), β-human globin (3'UTR), a Kozak sequence, an IgESP (signal peptide), a stop codon, a Poly (Ax120) tail, a 6xHis-Tag sequence, a plasmid linearization restriction enzyme sequence BspQI, and various linkers, such as GGGGS, EAAAK, and KK.
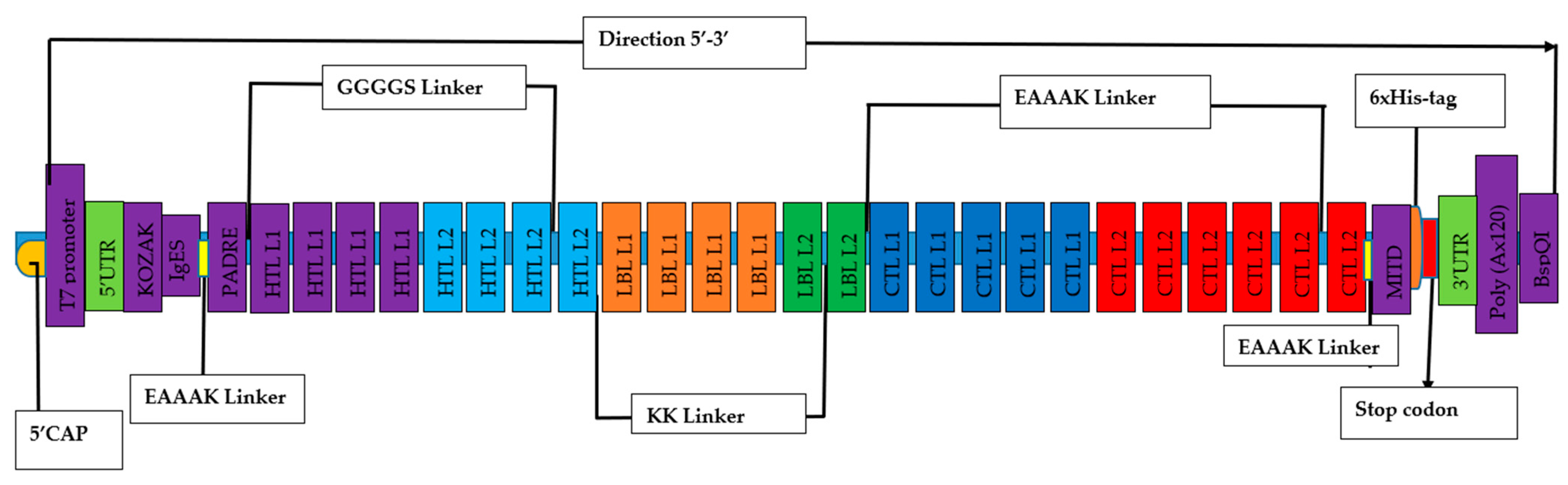
Figure 4.
Prediction of secondary structure (2D) of chimeric L1/L2 mRNA-based vaccine. The legend within the picture explains how the different sections (helixes, strands, coils) are colored. The 2D structure was constructed with PSIPRED, while conformation states was generated with the NPSA/SOPMA webserver.
Figure 4.
Prediction of secondary structure (2D) of chimeric L1/L2 mRNA-based vaccine. The legend within the picture explains how the different sections (helixes, strands, coils) are colored. The 2D structure was constructed with PSIPRED, while conformation states was generated with the NPSA/SOPMA webserver.

Figure 5.
Validation of the vaccine design's 3D structure with a Ramachandran plot and the ProSA web server. Figure A illustrates 3D structure of the vaccine visiluazed using the UCSF chimera v1.17.3 software, (B Protein sequence of the vaccine construction, PADRE sequence is lighted in brown, HTL epitopes are lighted in purple, CTL epitopes in blue and B epitopes are lighted in green, MITD sequence is lighted in light black, IgE signal sequence is lighted in red, 6xHistidine tag is lighted in gold darker, all the linkers are high- lightened in yellow color, (C) validation of the model's 3D structure using a Ramachandran plot (96.1 %,) of residues were located in the most favoured regions, (D) overall model quality with a Z-Score value (a Z-score value of -7.2) determined by x-ray crystallography using ProSA.
Figure 5.
Validation of the vaccine design's 3D structure with a Ramachandran plot and the ProSA web server. Figure A illustrates 3D structure of the vaccine visiluazed using the UCSF chimera v1.17.3 software, (B Protein sequence of the vaccine construction, PADRE sequence is lighted in brown, HTL epitopes are lighted in purple, CTL epitopes in blue and B epitopes are lighted in green, MITD sequence is lighted in light black, IgE signal sequence is lighted in red, 6xHistidine tag is lighted in gold darker, all the linkers are high- lightened in yellow color, (C) validation of the model's 3D structure using a Ramachandran plot (96.1 %,) of residues were located in the most favoured regions, (D) overall model quality with a Z-Score value (a Z-score value of -7.2) determined by x-ray crystallography using ProSA.
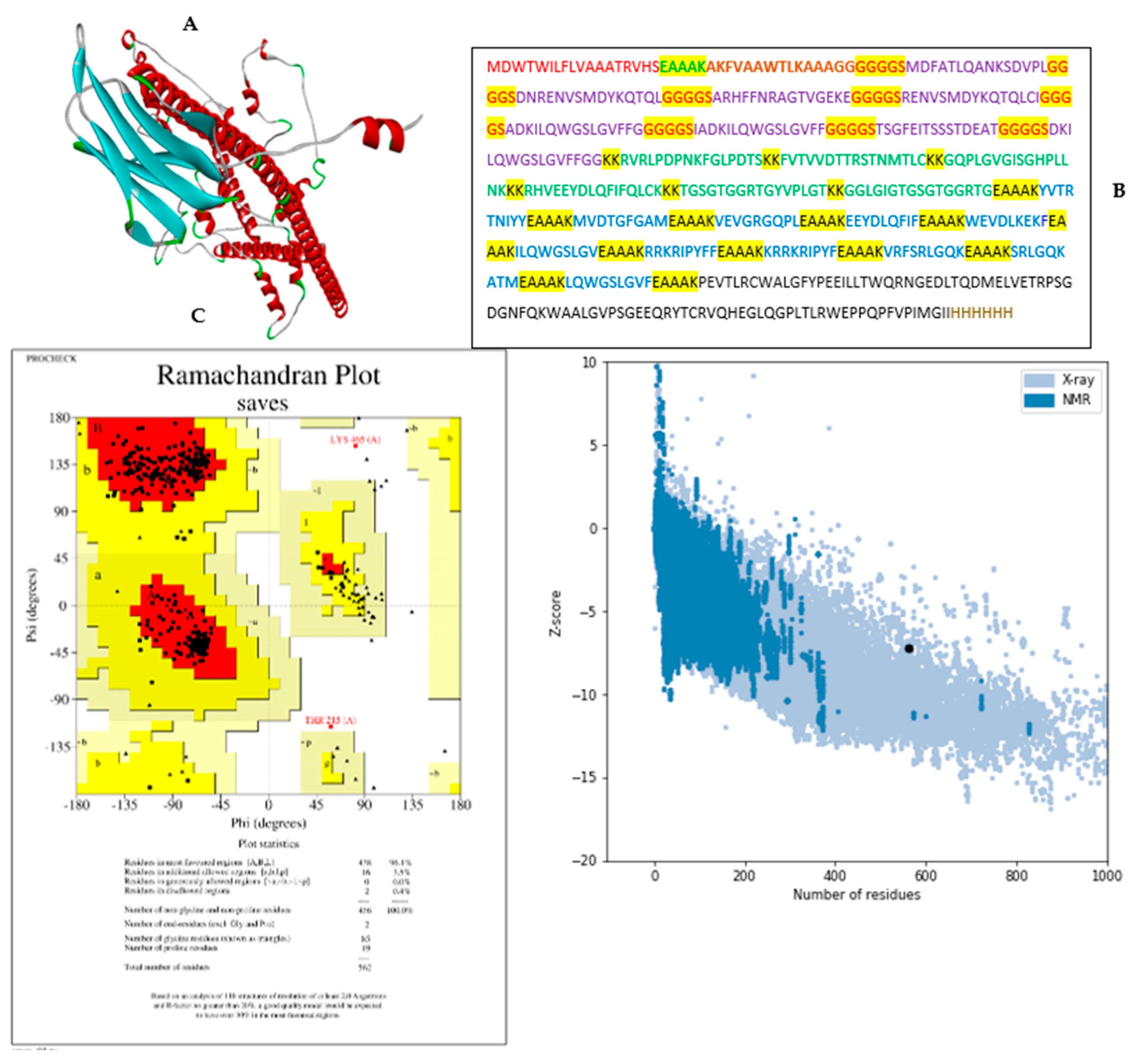
Figure 7.
The docked complex between vaccine and human Toll-like receptor 4 (TLR4), and residue interactions analysis. (I) Docked complex between vaccine and TLR 4 and its interactions (II, III, IV). In the LIGPLOT+/DIMPLOT, hydrogen bonds are depicted by dashed (green) lines with the bond length displayed inside, while hydrophobic interactions are denoted by red dashed lines. The interactions of residues between vaccine and receptor are denoted as (A: B; A: C; A: D), with residues in vaccine denoted in blue, and receptor residues in green.
Figure 7.
The docked complex between vaccine and human Toll-like receptor 4 (TLR4), and residue interactions analysis. (I) Docked complex between vaccine and TLR 4 and its interactions (II, III, IV). In the LIGPLOT+/DIMPLOT, hydrogen bonds are depicted by dashed (green) lines with the bond length displayed inside, while hydrophobic interactions are denoted by red dashed lines. The interactions of residues between vaccine and receptor are denoted as (A: B; A: C; A: D), with residues in vaccine denoted in blue, and receptor residues in green.
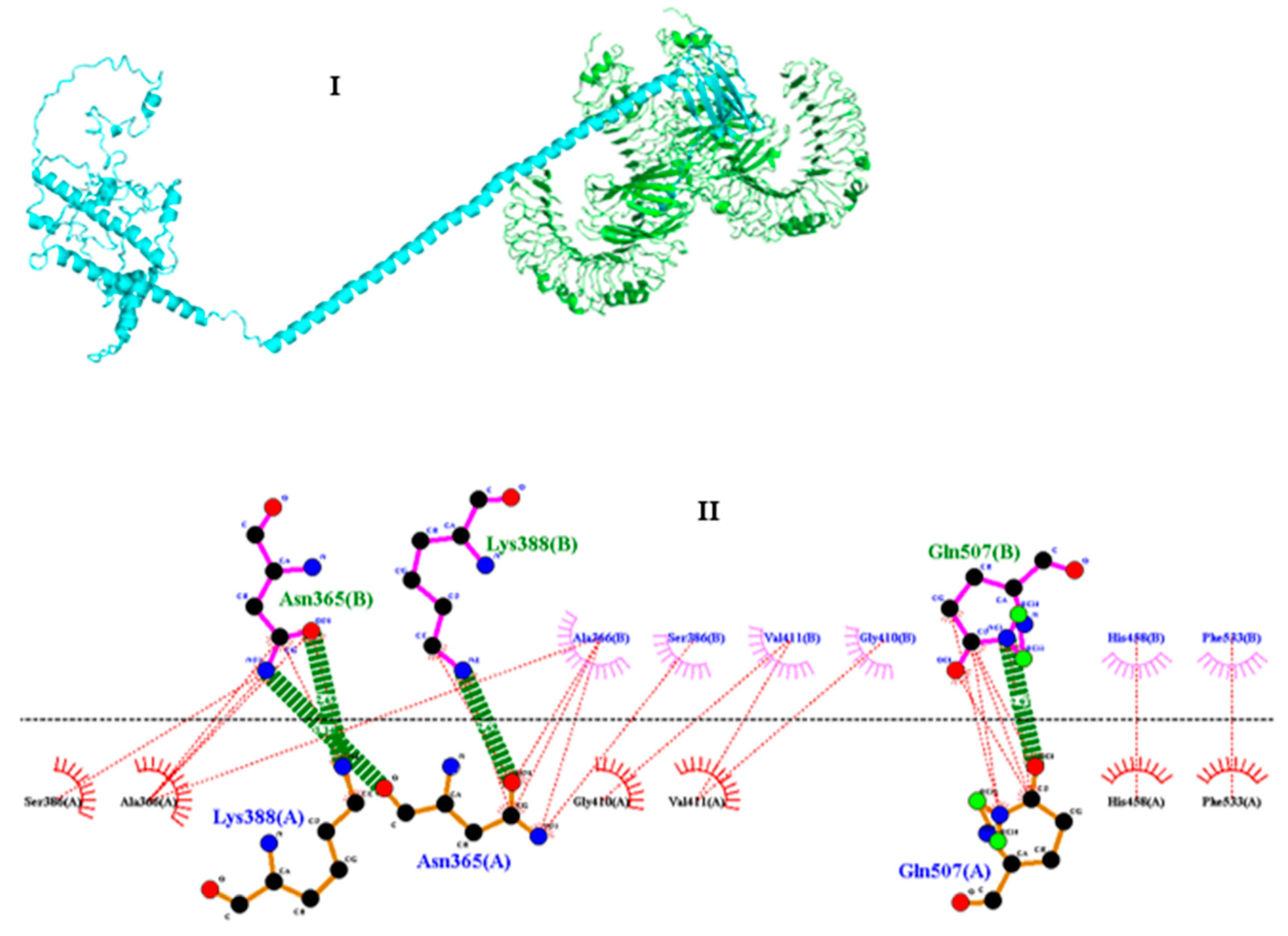
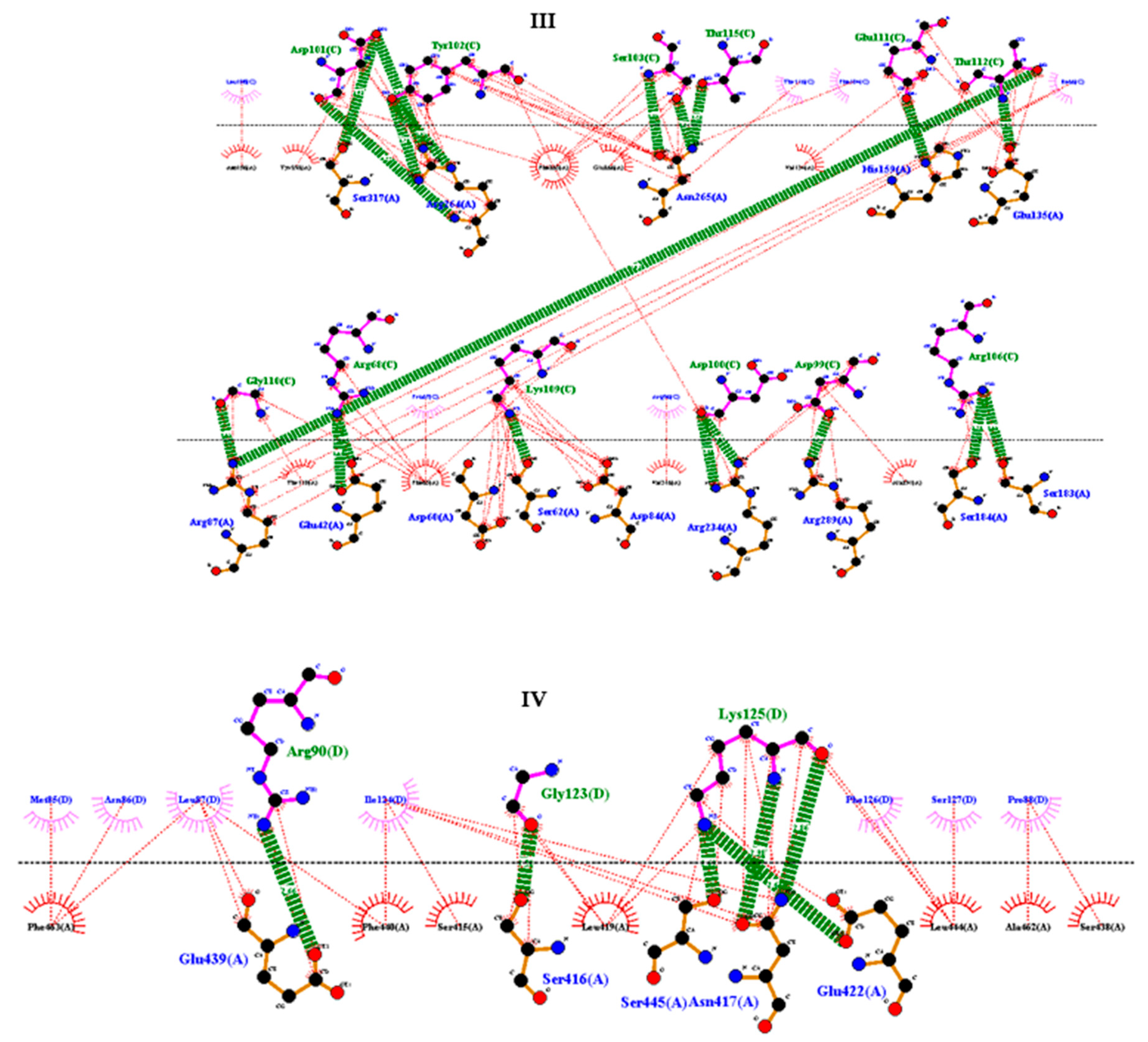
Figure 8.
Molecular dynamic simulation analysis of the vaccine design and the docked complexes. Figure (A) represents vaccine-TLR4 complexes in its NMA mobility visualized in the JSmol, (B) Deformability, (C) relationship between the complex's NMA and PDB fields, along with a B-factor, (D) Cumulative variance %, (E) the NMA Eigenvalues, (F) Elastic network, and (G) Covariance map shows correlated (red),uncorrelated ( white), and anti-correlated (blue) motions of paired residues.
Figure 8.
Molecular dynamic simulation analysis of the vaccine design and the docked complexes. Figure (A) represents vaccine-TLR4 complexes in its NMA mobility visualized in the JSmol, (B) Deformability, (C) relationship between the complex's NMA and PDB fields, along with a B-factor, (D) Cumulative variance %, (E) the NMA Eigenvalues, (F) Elastic network, and (G) Covariance map shows correlated (red),uncorrelated ( white), and anti-correlated (blue) motions of paired residues.
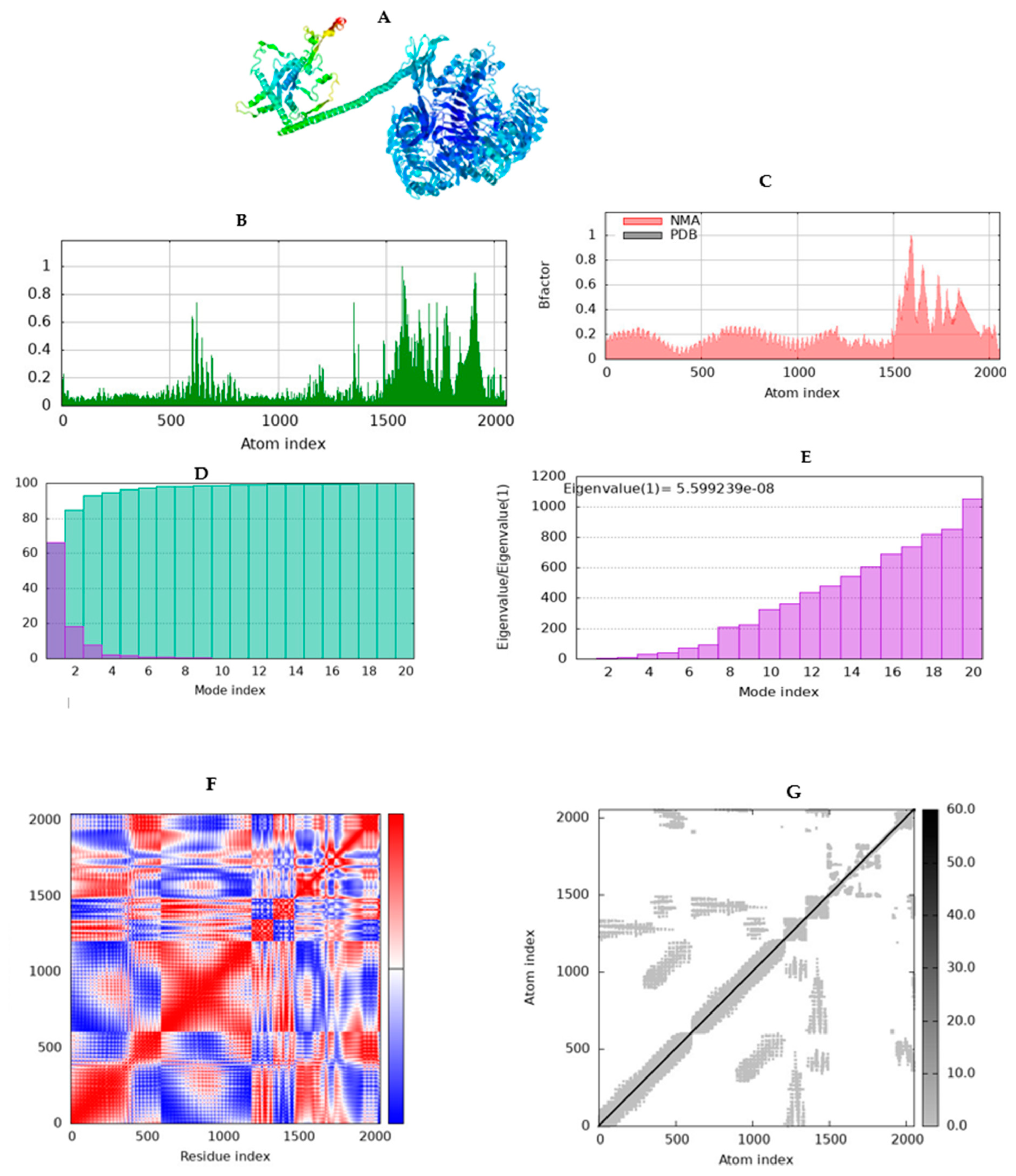
Figure 9.
Various immune responses generated by C-ImmSim server. (A) The produced immunoglobulins in response to antigen injection. Various subtypes of immunoglobulin are represented as colored peaks, (B) Shows B cell population after injections, (C) Macrophage population by state, (E) Population per state of T-helper cell, (E) DC population by state, (F) Production of cytokines and interleukins (increased production of IFN-γ and IL-2) after immunization.
Figure 9.
Various immune responses generated by C-ImmSim server. (A) The produced immunoglobulins in response to antigen injection. Various subtypes of immunoglobulin are represented as colored peaks, (B) Shows B cell population after injections, (C) Macrophage population by state, (E) Population per state of T-helper cell, (E) DC population by state, (F) Production of cytokines and interleukins (increased production of IFN-γ and IL-2) after immunization.
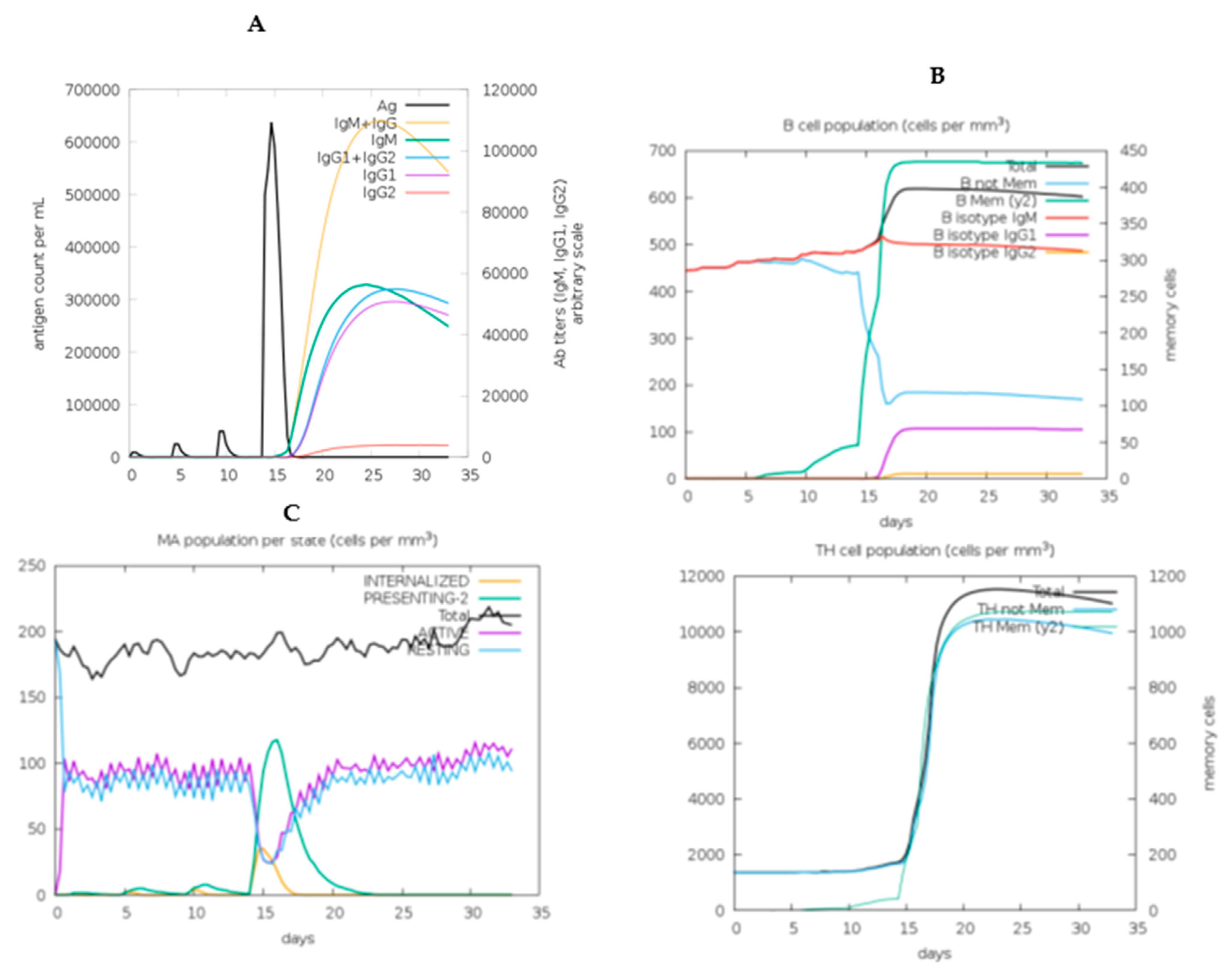
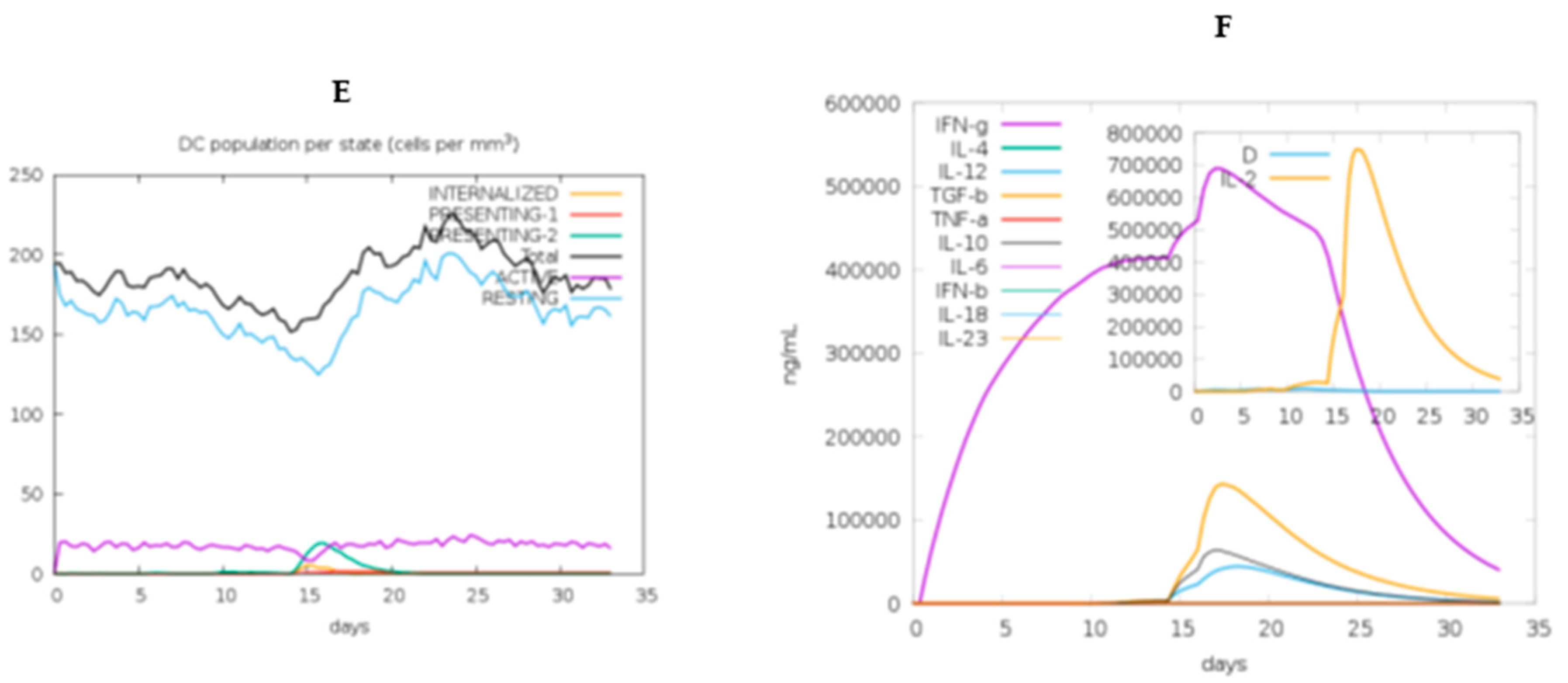
Figure 10.
Cloning simulation of the final vaccine design against HPV species into the standard vector pUC57 (size of 4725 bp) between the EcoRI (at a position of 396) and SacI (at a position of 2421). The red spots represent BspQI restriction sites for plasmid DNA linearization, whereas the longer orange arrow represents a multi-epitope chimeric L1/L2 mRNA vaccine of 2027 bp length.
Figure 10.
Cloning simulation of the final vaccine design against HPV species into the standard vector pUC57 (size of 4725 bp) between the EcoRI (at a position of 396) and SacI (at a position of 2421). The red spots represent BspQI restriction sites for plasmid DNA linearization, whereas the longer orange arrow represents a multi-epitope chimeric L1/L2 mRNA vaccine of 2027 bp length.
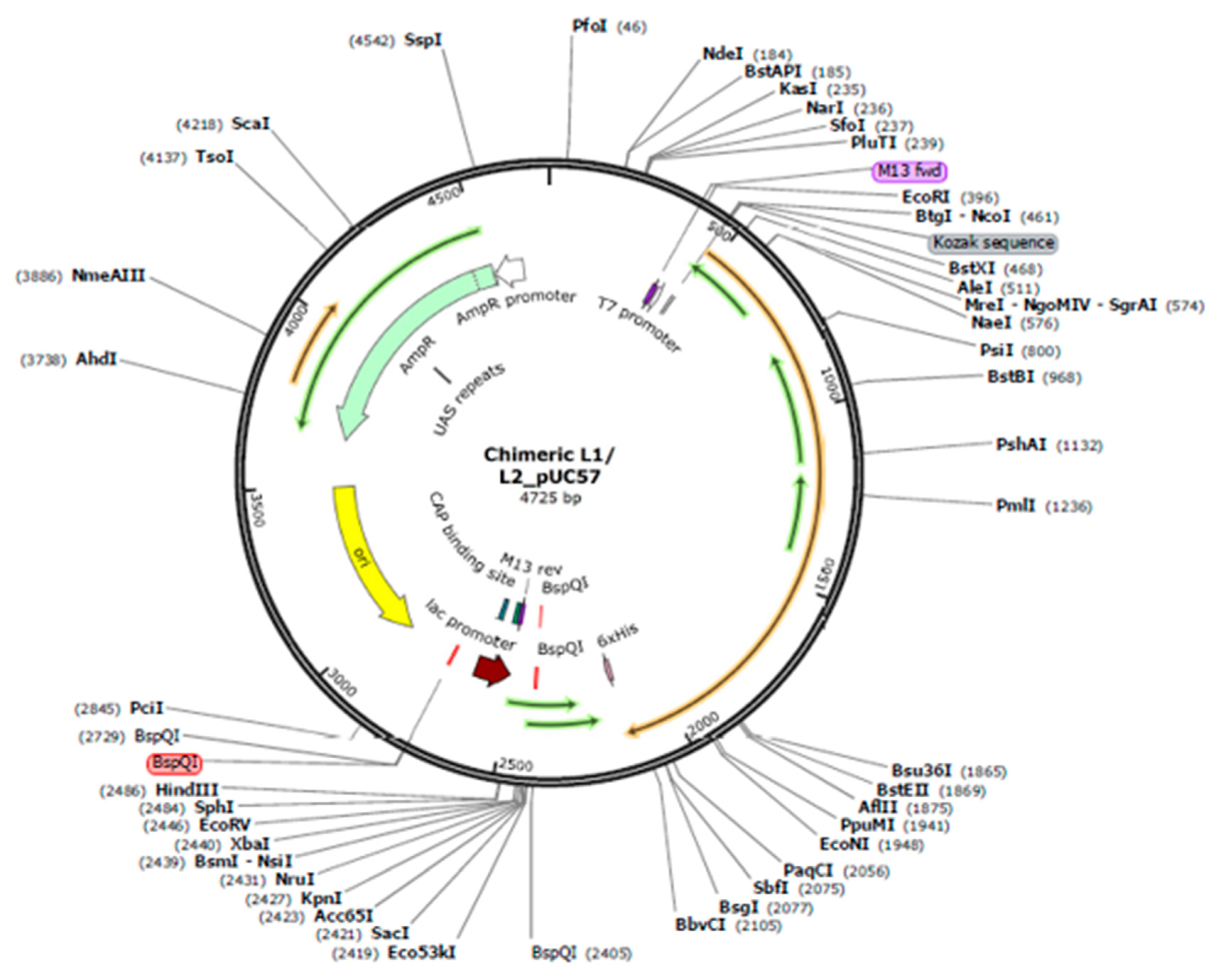
Figure 11.
Secondary structure prediction of the final constructed mRNA-based vaccine using RNAfold server. (A) Model representation of MFE secondary structure of the vaccine, (B) predicted model of Centroid secondary structure of the vaccine, (C) A mountain plot representation of the MFE structure, the thermodynamic ensemble of mRNA structures, and the centroid structure and the positional entropy plot for each position; where red: MFE, green: PF, blue: centroid.
Figure 11.
Secondary structure prediction of the final constructed mRNA-based vaccine using RNAfold server. (A) Model representation of MFE secondary structure of the vaccine, (B) predicted model of Centroid secondary structure of the vaccine, (C) A mountain plot representation of the MFE structure, the thermodynamic ensemble of mRNA structures, and the centroid structure and the positional entropy plot for each position; where red: MFE, green: PF, blue: centroid.
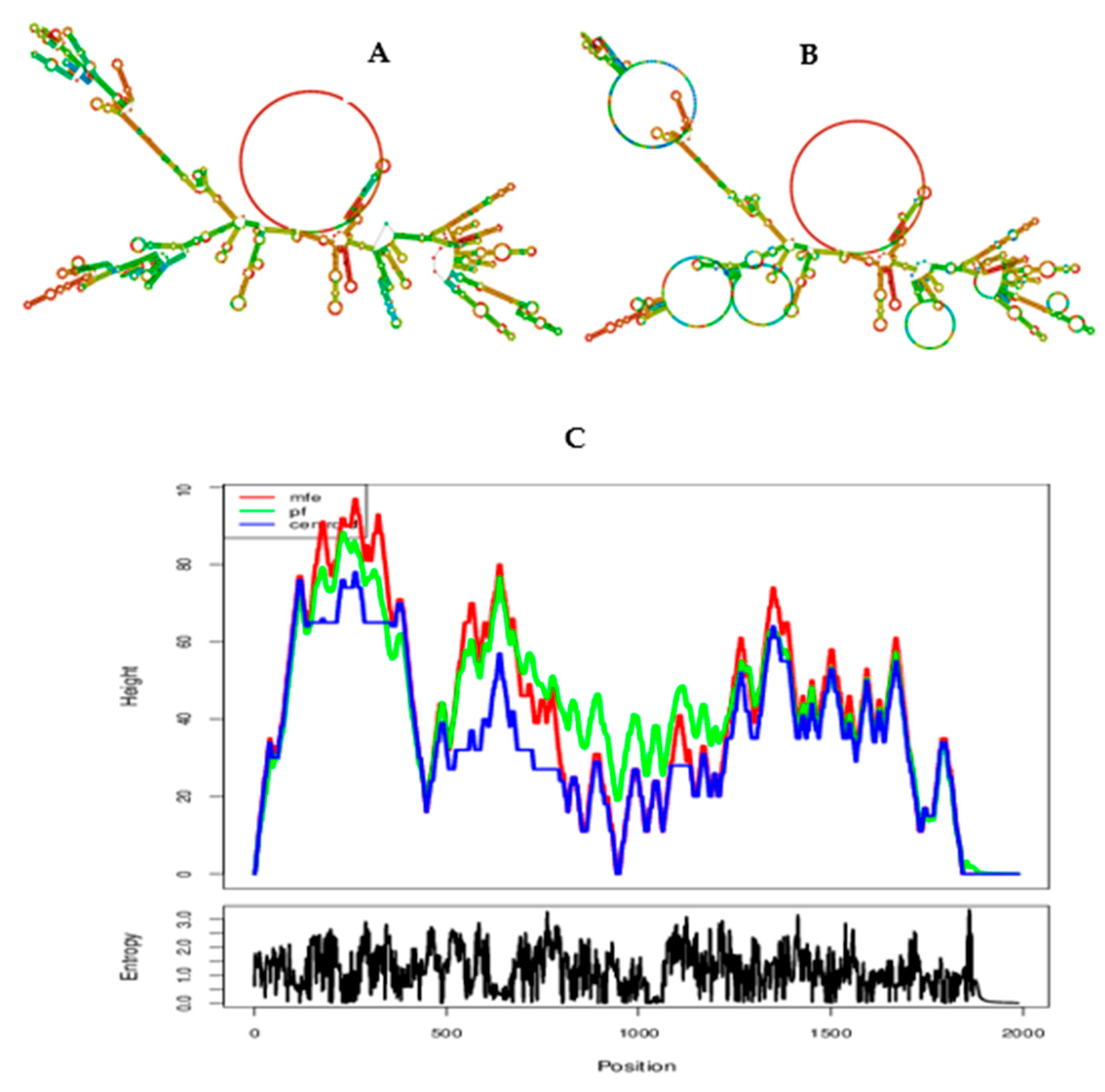
Table 1.
The L1 CTL, HTL and LBL (B CELL) epitopes successfully passed screening based on antigenicity, toxicity, allergenicity and conservancy analysis.
Table 1.
The L1 CTL, HTL and LBL (B CELL) epitopes successfully passed screening based on antigenicity, toxicity, allergenicity and conservancy analysis.
| Type of epitopes | Allele | Epitope sequence | Antigenicity score | Toxicity | Conservancy analysis at the max. identity ≤ 100% |
| CTL (5) | HLA-A*01:01,HLA-A*26:01 | YVTRTNIYY | 0.6843 | Non-toxin | 100.00%: HPV31,HPV35,HPV29 |
| HLA-A*01:01,HLA-A*26:01 | MVDTGFGAM | 1.6337 | Non-toxin | 100.00%: HPV31, HPV35, HPV66, HPV6, HPV11, HPV40, HPV43, HPV44 | |
| HLA-B*39:01,HLA-B*40:01 | VEVGRGQPL | 1.2418 | Non-toxin | 100.00%: HPV16, HPV35, HPV39, HPV51, HPV40, HPV27, HPV29, HPV57 | |
| HLA-B*40:01 | EEYDLQFIF | 1.7384 | Non-toxin | 100.00%:HPV16,HPV18,HPV35,HPV39,HPV45,HPV45,HPV68, HPV6, HPV43,HPV27, HPV29, HPV57 | |
| HLA-B*40:01 | WEVDLKEKF | 1.2621 | Non-toxin | 100.00%: HPV35, HPV52, HPV33 | |
| HTL (4) | HLA-DRB1*01:01 | MDFATLQANKSDVPL | 0.7698 | Non-toxin | 93.33% |
| HLA-DRB1*03:01 | DNRENVSMDYKQTQL | 1.4352 | Non-toxin | 100%: HPV42 | |
| HLA-DRB1*04:05 | ARHFFNRAGTVGEKE | 0.7295 | Non-toxin | 86.67% | |
| HLA-DRB1*03:01 | RENVSMDYKQTQLCI | 1.6266 | Non-toxin | 93.33% | |
| FVTVVDTTRSTNMTLC | 1.0558 | Non-toxin | 100.00%: HPV52, HPV58, HPV6, HPV11, HPV33 | ||
| GQPLGVGISGHPLLNK | 0.5668 | Non-toxin | 100.00%: HPV16, HPV31, HPV35, HPV52, HPV42, HPV44, HPV33 | ||
| LBL (4) | RHVEEYDLQFIFQLCK | 0.4410 | Non-toxin | 93.75% | |
| RVRLPDPNKFGLPDTS | 0.4553 | Non-toxin | 100.00%: HPV16, HPV31, HPV35, HPV52, HPV42, HPV44, HPV33 |
CTL (5): Cytotoxic T lymphocytes epitopes, HTL (4): Helper T lymphocytes, LBL (4): Linear B Cell lymphocytes, HPV: Human papillomavirus.
Table 2.
The L2 CTL, HTL and LBL (B CELL) epitopes successfully passed screening based on antigenicity, toxicity, allergenicity and conservancy analysis.
Table 2.
The L2 CTL, HTL and LBL (B CELL) epitopes successfully passed screening based on antigenicity, toxicity, allergenicity and conservancy analysis.
| Type of epitopes | Allele | Epitope sequence | Antigenicity score at the threshold (≥ 0.4) | Toxicity | Conservancy analysis at the max. identity ≥ ≤ 100% |
| CTL (6) | HLA-A*02:01 | ILQWGSLGV | 1.8703 | Non-toxin | 100.00%: HPV42, HPV29 |
| HLA-B* 15:01 | LQWGSLGVF | 2.2096 | Non-toxin | 100%: HPV42 | |
| HLA-B*27:05 | RRKRIPYFF | 1.5931 | Non-toxin | 100.00%:HPV35,HPV56,HPV66, HPV51 | |
| HLA-B*27:05 | KRRKRIPYF | 1.7636 | Non-toxin | 100.00%: HPV51 | |
| HLA-B*27:05 | VRFSRLGQK | 2.1048 | Non-toxin | 100.00%:HPV51 | |
| HLA-B*27:05 | SRLGQKATM | 1.2853 | Non-toxin | 100.00%: HPV51 | |
| HTL (4) | HLA-DRB1*15:01 | ADKILQWGSLGVFFG | 0.8722 | Non-toxin | 100.00%: HPV42 |
| HLA-DRB1*15:01 | IADKILQWGSLGVFF | 0.9813 | Non-toxin | 93.33% | |
| HLA-DRB1*07:01 | TSGFEITSSSTDEAT | 0.6108 | Non-toxin | 73.33% | |
| HLA-DRB1*15:01 | DKILQWGSLGVFFGG | 0.6126 | Non-toxin | 100.00%: HPV42 | |
| LBL (2) | TGSGTGGRTGYVPLGT | 1.3789 | Non-toxin | 93.75% | |
| GGLGIGTGSGTGGRTG | 1.4744 | Non-toxin | 100.00%: HPV16, HPV18, HPV58, HPV59, HPV6, HPV43, HPV44, HPV27, HPV29,HPV 57 |
CTL (6): Cytotoxic T lymphocytes epitopes, HTL (4): Helper T lymphocytes, LBL (2): Linear B Cell lymphocytes, HPV: Human papillomavirus.
Table 3.
The physiochemical properties of the multi-epitope chimeric vaccine construct.
| Features | Value |
| Number of amino acids | 562aa |
| Molecular weigh | 60161.29~ 60kDa |
| Theoretical pI Solubility (≥ 0.45) Antigenicity score (≥ 0.4) Allergenicity Instability index Aliphatic index Grand average of hydropath city GRAVY) Formula Total number of atoms Estimated half-life |
9.44 0.451 0.9178 Non-allergen 36.89 68.49 -0.366 C2683H4187N761O786S15 8432 30 hours (mammalian reticulocytes, in vitro) > 20 hours (yeast, in vivo) > 10 hours (Escherichia coli, in vivo) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



