
Submitted:
21 October 2024
Posted:
22 October 2024
You are already at the latest version
Abstract
Purpose:
Report of a novel genetic variant in a family with an unusual clinical presentation of posterior polymorphous corneal dystrophy (PPCD).
Methods:
Documentation of clinical findings of the anterior and posterior segments using slit lamp analysis and imaging tools (Scheimpflug imaging, optical coherence tomography), along with documentation of best spectacle corrected visual acuity (BSCVA; LogMAR). Next-generation sequencing (NGS) of the genes of interest was performed by the IDTxGen® sytem (Inherited Disease Panel v1.0 and sequencing on an Illumina NextSeq® 500) based on DNA extracted from peripheral blood lymphocytes. Data analysis was performed using the SeqNext module of the SEQUENCE Pilot software (version 5.1.0, JSI Medical Systems). A histological examination of four corneas in both patients (father and son) was carried out by hematoxylin-eosin (HE) staining.
Results:
A novel heterozygous variant in the ZEB1 gene was identified in a 41-year-old man and his 13-year-old son. The 2-bp duplication in exon 8 results in a frameshift and a premature stop codon (p.(Asp883Argfs*39)), predicted to result in nonsense-mediated decay (NMD) and to lead to functional impairment of the ZEB1 protein. The variant was classified as likely pathogenic applying the American College of Medical Genetics and Genomics (ACMG) criteria. The variant is located in the penultimate 3´base pairs of the ZEB1 gene. The C-terminus of the ZEB1 gene encodes the C2H2-type protein domains of the ZEB1 zinc finger transcription factor protein which are relevant in epithelial-mesenchymal transition (EMT) by mediating the transition between cell lineages. The patients presented with an unusual symmetrical crescent-shaped bullous edema with a temporal focus with retrocorneal snail-track-like changes. Histological examination of all hematoxylin-eosin (HE) stained samples revealed a peripheral multilayer of endothelial cells adjacent to the trabecular meshwork (TMW), along with a central reduction of corneal endothelial cells and a pathological thickening of Descemet's membrane of up to 18.7 m.
Conclusions:
We report a novel variant of dominantly inherited PPCD3. Pathological production of a protein with an effect on the transition between cell lineages could explain the pathogenic epithelial transformation of the corneal endothelium, characterized by snail-track-like retrocorneal changes with a symmetric temporal crescent-shaped corneal edema.
Keywords:
Posterior Polymorphous Corneal Dystrophy (PPCD)
; ZEB1 mutation
; Zinc finger transcription factor
Introduction
Corneal dystrophies (CDs) are genetic disorders that affect one or more layers of the cornea. In most cases, these diseases follow an autosomal dominant inheritance pattern, although autosomal recessive and X-chromosomal inheritance also exists. In contrast to dysgenesis of the anterior segment, CDs appear mostly bilaterally and show a slow progression. Patients suffer from progressive visual loss and often photophobia. A foreign body sensation and ocular surface pain may also occur. There are usually no systemic manifestations in CDs (except e.g. in macular corneal dystrophy).
Posterior polymorphous corneal dystrophy (PPCD) is an endothelial corneal dystrophy that had been first described by Koeppe in 1916.1 It often occurs asymmetrically. Visual impairment at birth is rare, and its progression is slow over many decades. Slit lamp examination findings include retrocorneal geographic deposits of Descemet's membrane with vesicular, snail-track-like, or diffuse changes with sometimes gray-white plaques.1,2 Histologically, abnormal thickening of Descemet’s basement membrane and multilayered epithelium transformation can be observed.
The polymorphic lesions described can be detected via slit lamp biomicroscopy at moderate to high magnification, preferably using the retro-illumination technique. Vesicular lesions occur in clustered groups and are characterized by a cystic appearance and may vary greatly in size, often showing a translucent center surrounded by a darker rim. The band-like lesions are usually easily recognized by two parallel bands with serrated edges. These bands can occur in any orientation, but in the vast majority of cases, they are horizontal and range in length from 2 to 10 mm. In comparison to Descemet’s findings in trauma or congenital glaucoma (Haab striae), the bands in PPCD do not thin out towards the ends. Diffuse polymorphic opacities are less common in PPCD but are most disturbing to the affected patient, often causing photophobia due to abnormal refraction. Geographic clusters of small opacities are usually distributed over part of Descemet’s membrane and blur the posterior stroma, causing a kind of peau d’orange appearance under retro-illumination. Therefore, the clinical manifestations of this disease vary widely. Corneal edema associated with PPCD can occur at a younger age. However, most patients with PPCD are asymptomatic for a long time, and the progression is very slow, often leading to a diagnosis years later during a routine eye examination.1-3
PPCD may also affect the iris and iridocorneal angle in rare cases. Abnormal migration of endothelial cells, which might not be limited to the posterior surface of the cornea but extend uncontrollably across the chamber angle and onto the peripheral iris surface. Peripheral iridocorneal adhesions, which can induce glaucoma, may be detected by gonioscopy. These adhesions may cause corectopia and pupillary ectropion.
To date, four genes (OVOL2, COL8A2, ZEB1, GRHL2) have been associated with monogenic forms of PPCD, all displaying autosomal dominant inheritance pattern. Additional genetic heterogeneity is assumed.4-6 Thus far, in the literature four different single nucleotide substitutions in OVOL2 have been described, along with a 23 bp insertion in a highly conserved region of the OVOL2 promoter, and an approximately 49 kb microduplication encompassing the entire OVOL2 gene. All variants were classified as (likely) pathogenic.7,8,9,10 No OVOL2 expression has been detected in human corneal endothelium, but multiple binding sites for transcription factors expressed in corneal endothelium are located in the OVOL2 promoter. Two single-base pair deletions and one single-base pair substitution as well as stop mutation were identified in a non-coding intronic regulatory region of GRHL2.2,6,11-14
Numerous ZEB1 loss-of-function variants have been identified, including nonsense variants and single nucleotide substitutions that alter conserved splice site motifs. Additionally, small deletions, small duplications, structural and missense variants with deleterious effect of ZEB1 have also been described. Furthermore, twenty-three frameshift variants, listed in the Human Gene Mutation Database (HGMD), have been documented.5
The ZEB1 gene, also known as Zinc Finger E-Box Binding Homeobox 1, is a crucial regulator of various cellular processes, including embryonic development, tissue differentiation, and tumor progression. It plays a significant role in the epithelial-to-mesenchymal transition (EMT), a fundamental process in development and disease.15 In normal physiological conditions, ZEB1 regulates the transition of epithelial cells into mesenchymal cells, a process essential for embryogenesis and wound healing. However, dysregulation of ZEB1 expression or activity can contribute to pathological conditions and can influence other cellular processes, such as cell proliferation, survival, and resistance to therapy. Understanding the role of the ZEB1 gene in cancer and other diseases holds significant implications for the development of targeted therapies aimed at modulating its activity or expression. Additionally, ZEB1 expression levels may serve as prognostic markers for disease progression and patient outcomes in various diseases.15 De novo occurrence of heterozygous ZEB1, GRHL2 and OLOV2 variants has been reported and should be considered if the parents of an index case are unaffected.11,14,16
Presentation of the Case
The index patient is a 41-year-old man who presented first at the Department of Ophthalmology at the University Hospital Cologne in 2019 with bilateral pronounced bullous keratopathy. Due to the severity of the disease and permanently recurrent corneal decompensations with distinct temporal focus, the patient had undergone three Descemet membrane endothelial keratoplasties (DMEK) of the right eye between 2019 and 2021 and two DMEK surgeries of the left eye at the University Hospital Cologne in 2020 and 2021.
In 2022, a fourth lamellar keratoplasty was performed on the right eye due to rapid deterioration. In the context of PPCD, the index patient also presented with secondary glaucoma with a cup-disc ratio (CDR) of 0.6 in both eyes and a reduction of the retinal nerve fiber layer (RNFL). The patient was using multiple antiglaucomatous eye drops at the time of presentation but had well-controlled intraocular pressure. Best corrected visual acuity (BSCVA) was 0.9 logMAR on the right eye and 0.6 logMAR on the left before his first DMEK procedure in 2019. The index patient has been wearing glasses since the age of 13.
The 13-year-old son of the index patient was diagnosed with bullous keratopathy bilaterally in 2019 with temporal focus as well. The son had been wearing glasses since the age of three and received his first lamellar keratoplasty on the left eye at the University Hospital Cologne in 2020, also in the form of a DMEK. Before the DMEK procedure in 2020, the best corrected visual acuity of the 13-year-old son was 0.2 logMAR on his right eye and 0.4 logMAR on his left eye. Measurement of the endothelial cell count (ECC) and a gonioscopy could not be performed because of a temporally emphasized crescent bullous edema up to central cornea combined with a high degree of photophobia (compare Figure 1a). Retinal examination of the son showed a CDR of 0.4 bilaterally. The RNFL was intact. In both patients, the macular OCT (Optical Coherence Tomography) was unremarkable.
Before first DMEK surgery, the slit-lamp microscopy results were similar in the index patient and the son in respect to the clinical presentation:
Both patients showed a temporally pronounced bullous edema up to complete cornea with a stromal haze (Figure 1a,b). In the regressed light with dilated pupil snail-track-like changes of the cornea over the entire posterior corneal surface were detectable (Figure 1c). There were no typical vesicles, parallel bands or gray-white plaques typical for PPCD and less peau d'orange. In both patients, strongly progressive and symmetrical findings were present with marked visual impairment and photophobia caused by PPCD. There is no history of further ocular or other relevant diseases in both patients and the extended family. There is no consanguinity.
The findings described above suggest a genetic cause with autosomal dominant inheritance pattern for CD in father and son.
Figure 1a:
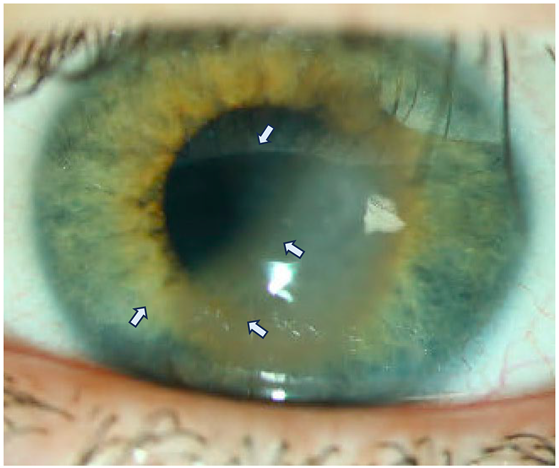
Slit lamp microscopy of the left eye of the son at the first presentation in the clinic:
Clinical slit lamp findings at the first presentation of the younger patient (13 years old son) with temporally emphasized crescent bullous edema up to central cornea (white arrows). The surrounding cornea is rather clear, the anterior chamber is deep and there is no sign of infection. The epithelium is closed. The BSCVA (logMAR) is 0.4.
Figure 1b:
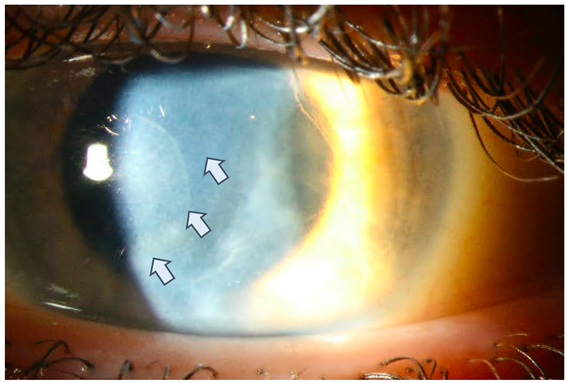
Preoperative slit lamp microscopy of the left eye of the son:
Distinct corneal edema throughout the whole cornea with a central corneal thickness of 717 µm and a BSCVA (logMAR) of 0.4. Temporal: the pronounced corneal edema leads to an incipient fibrosis of the stroma (white arrows).
Figure 1c:

Preoperative slit lamp microscopy of the left eye with dilated pupil and regredient light (13-year-old son) Clearly visible snail-track-like changes (white arrows) of the cornea over the entire posterior corneal surface in regredient light.
Methods
Clinical parameters of the anterior and posterior segments were constantly monitored in both patients by using the slit lamp and imaging techniques, including Pentacam (Oculus®, Wetzlar, Germany), spectral domain optical coherence tomography (OCT) and swept-source OCT, as well as a documentation of best-corrected visual acuity (BSCVA) in LogMAR and the intraocular pressure.
Descemet membrane endothelial keratoplasty (DMEK) was performed in a standardized procedure as described previously.17-21 The surgically obtained endothelial Descemet remnants from both patients in their respectively first DMEK procedure of each eye were histologically stained by hematoxylin-eosin (HE) staining and analyzed with regard to morphology.
A next-generation sequencing (NGS) panel analysis for 10 genes associated with hereditary corneal dystrophy (CHST6, COL17A1, COL8A2, DCN, GSN, KRT3, SCLA411, TGFB1, VSX1 and ZEB1) was performed in the index patient, followed by segregation analysis for the detected ZEB1 variant in the son using genomic DNA from EDTA blood samples in both patients.
NGS analysis was performed by enrichment of the genes of interest’s coding sequences using the IDTxGen®, Inherited Disease Panel v1.0., sequencing on an Illumina NextSeq 500 (Illumina, San Diego, Cal.) system, and analysis using the SeqNext module of the Sequence Pilot software (version 5.1.0, JSI Medical Systems). Copy number variants affecting one or more exons, structural rearrangements, changes in repeat/homopolymer and regulatory domains, as well as deep intronic variants cannot be excluded with this methodology.
Minor allele frequency (MAF) describes the frequency with which the second most common allele appears in a determined population. MAFs have a crucial role in heritability, because if they occur only once, the so-called "singletons" cause pronounced selection.
MAF is commonly used in population genetic studies because it offers information to distinguish between common and rare variants in the population. Detected variants with minor allele frequencies (MAF) of < 1% were subjected to further scientific evaluation according to the latest scientific knowledge and classified according to the recommendations of the American College for Medical Genetics and Genomics (ACMG).22
Results
A heterozygous variant c.2645_2646dupAG, p.(Asp883Argfs*39) in the ZEB1 gene was identified in both patients – father and son – and was classified as likely pathogenic (class 4).22 This variant has not yet been described in the literature nor in disease variant (ClinVar, HGMD, UniProt, LOVD) or population-specific (genomAD ex.) databases.
This novel variant describes a 2 bp duplication and leads to a frameshift resulting in the formation of a premature stop codon (p.(Asp883Argfs*39)), null variant). It is located in the penultimate exon within the last 50 bp. It can therefore be expected to cause nonsense-mediated mRNA decay (NMD) and to result in a truncation of the ZEB1 protein and impairment of its function. Monoallelic pathogenic ZEB1 variants, especially null variants, cause PPCD3, OMIM #609141.
Histologic analysis of HE stainings in both patients indicates a complete and therefore non-measurable loss of corneal endothelial cells centrally, with simultaneous presence of a peripheral multi-row epithelial-like cell layer in the region of the trabecular meshwork, displaying visible pigmentation. This finding is consistent across multiple sections of the samples (compare Figure 2a and b).
In addition to those findings, an abnormal and pronounced thickening of Descemet’s membrane of up to 18.7 μm and multiple blurred lacerations centrally is evident. (compare Figure 2a and 2c).
Discussion
Hereditary corneal dystrophies are a genetically very heterogeneous group of mostly non-syndromic eye diseases. Heterozygous, mostly truncating, variants in the ZEB1 gene can lead to different autosomal dominantly inherited corneal diseases – Fuchs endothelial dystrophy (FECD) type 6 (OMIM 613270) or posterior polymorphic corneal dystrophy (PPCD) type 3 (OMIM 609141). Disease expression can be very variable, with incomplete penetrance also having been described.12
ZEB1 is a transcription factor that modulates the process of epithelial-mesenchymal transition (EMT), which is complementary to mesenchymal-epithelial transition (MET). EMT is a transition that can occur at various stages of development, wound healing, fibrosis, and disease, in which epithelial cells lose their characteristic properties, such as their gene expression profile that includes suppression of epithelial genes and activation of mesenchymal genes.15,23 In addition, changes in cell polarity and cell-cell adhesion occur and an increased migratory capacity is evident.
A key event in this crossover is the conversion of E-cadherin gene expression to N-cadherin gene expression, resulting in changes of cell adhesion properties.23 Considering both, clinical and genetic findings, the newly identified ZEB1 variant in our patients is very likely causative for the autosomal dominant PPCD phenotype in both affected family members. In view of the clinically unaffected other family members, the ZEB1 variant could have occurred de novo in the index patient; incomplete penetrance or variable expressivity are another possible explanation. The parents of the 41-year-old patient were not available for genetic testing.
This novel mutation variant in ZEB1 is located within the last 50 bp of the penultimate exon of the gene, which could result in the production of a truncated protein instead of NMD. The 3´-terminal part of the ZEB1 gene encodes transcription factor C2H2-type domains of the ZEB1 zinc finger protein, which regulates the expression of other genes and plays a role in EMT by mediating the transition between cell lineages.23
A zinc cation (Zn2+) attaches to multiple conserved amino acids in C2H2-type zinc finger proteins (generally Cysteine and Histidine). Two histidine pairs at the C-terminus of the α-helix and the two cysteines at the terminus of the β-strand bind Zn2+ to form a tetrahedral structure. Zn2+ is located within between an α-helix and two antiparallel β-strands; the functionality of the ββα-structure system is kept constant by the interlocked bond supplied by Zn2+. Zn2+ maintains the stability of the overall zinc finger structure and the regular helical structure. Therefore, a truncated protein with loss of function may provide a pathogenic explanation for epithelialization of the corneal endothelium, which is a hallmark of PPCD.23,24
As a result of ZEB1 insufficiency, several epithelial-like features are noted in PPCD corneal endothelium, including stratified cell organization, desmosomal intracellular junctions, and exposure to an epithelial-like transcriptome profile, which includes increased/ectopic expression of epithelial-associated keratins and cadherins as well as a migration. On top of this, decreased ZEB1 expression leads to elevated cell death and cell barrier function. 16,23,24 In synopsis of the current literature, all this may explain the abnormal migration of corneal endothelial cells, which are not restricted to the posterior surface of the cornea but also irregularly migrate across the chamber angle and onto the peripheral iris surface and trabecular meshwork.
There is only sparse long-term follow-up data on patients with PPCD3 in the literature. In a study by Dudakova et al., 19 affected patients from 12 different families were examined.25 Seven of the affected patients aged 20 to 59 years had moderate visual impairment, uni- or bilaterally. Four of these patients underwent a single corneal transplantation, in contrast to our patients who already required multiple corneal transplants. Moreover, in Dudakova's study, no patient had secondary glaucoma in the setting of PPCD.
Due to the singularity of the c.2645_2646dupAG mutation and the variability of pathogenic variants in both patients, the expression and severity of the disease and the course cannot be predicted reliably. Because of the autosomal dominant inheritance pattern of ZEB1-associated CD, the recurrence probability is 50% although it is not known if the disease is fully penetrant. The clinical expression and severity of the disease may obviously require multiple corneal transplants because of recurrent corneal decompensations in patients affected by this novel detected mutation. Predictive genetic testing can be offered to at-risk family members after genetic counseling, facilitating a special screening and early detection program.
Conclusions
We identified a novel mutation in the ZEB1 gene (OMIM #609141), which leads to a frameshift and a premature stop codon. This mutation variant is predicted to result in significant functional impairment, which is clinically expressed in a snail-track-like phenotype and irregular changes in regressed light with pronounced temporal focus. Genetic counseling for predictive testing could be offered to family members regarding screening and early detection programs, also with regard to further scientific workup of different pathogenic variants in the ZEB1 gene and their clinical manifestations.
Conflicts of Interest statement
S. B. Zwingelberg, C. Velmans, J. Busse, M. Becker, P. Gehle, M. Mestanoglu, A. V. Schilcher, N.Luft, S. Priglinger, C. Netzer and H. J. Bolz and C. Cursiefen state that there is no conflict of interest. It is assured that there are no links with a company whose product is mentioned in the article or with a company that sells a competitive product. The presentation of the topic is independent and the presentation of the content is product-neutral.
Acknowledgments
We would like to thank Prof. Dr. med. Björn Bachmann and Dr. med. Prof. Mario Matthaei for their clinical care of the patients.
References
- Koeppe, L. : Klinische Beobachtungen mit der Nernstspaltlampe und dem Hornhautmikroskop. In: Albrecht von Graefes Archiv für Klinische und Experimentelle Ophthalmologie Bd. 91, 1916, S. 363–379.
- Vanathi M, Raj N, Kusumesh R, Aron N, Gupta N, Tandon R. Update on pediatric corneal diseases and keratoplasty. Surv Ophthalmol. 2022 Nov-Dec;67(6):1647-1684. [CrossRef]
- Weiss JS, Rapuano CJ, Seitz B, Busin M, Kivelä TT, Bouheraoua N, Bredrup C, Nischal KK, Chawla H, Borderie V, Kenyon KR, Kim EK, Møller HU, Munier FL, Berger T, Lisch W. IC3D Classification of Corneal Dystrophies-Edition 3. Cornea. 2024 Apr 1;43(4):466-527. [CrossRef]
- Krafchak CM, Pawar H, Moroi SE, Sugar A, Lichter PR, Mackey DA, et al. Mutations in TCF8 cause posterior polymorphous corneal dystrophy and ectopic expression of COL4A3 by corneal endothelial cells. Am J Hum Genet. 2005; 77:694-708.
- Davidson AE, Liskova P, Evans CJ, Dudakova L, Noskova L, Pontikos N, et al. Autosomal-dominant corneal endothelial dystrophies CHED1 and PPCD1 are allelic disorders caused by noncoding mutations in the promoter of OVOL2. Am J Hum Genet. 2016;98:75-89.
- Liskova P, Dudakova L, Evans CJ, Rojas Lopez KE, Pontikos N, Athanasiou D, et al. Ectopic GRHL2 expression due to non-coding mutations promotes cell state transition and causes posterior polymorphous corneal dystrophy.
- Chung DD, Frausto RF, Cervantes AE, Gee KM, Zakharevich M, Hanser EM, Stone EM, Heon E, Aldave AJ. Confirmation of the OVOL2 promoter mutation c.-307T>C in Posterior Polymorphous Corneal Dystrophy 1. PloS One. 2017 Jan 3;12(1):e0169215.
- Janeschitz-Kriegl L, Kamdar D, Quinodoz M, Kaminska K, Folcher M, György B, Meyer P, Wild A, Escher P, Scholl HPN, Rivolta C, Goldblum D. c.-61G>A in OVOL2 is a Pathogenic 5’ Untranslated Region Variant Causing Posterior Polymorphous Corneal Dystrophy 1. Cornea. 2022 Jan 1;41(1):89-94. [CrossRef]
- Muijzer MB, Kroes HY, van Hasselt PM, Wisse RPL. Bilateral posterior lamellar corneal transplant surgery in an infant of 17 weeks old: Surgical challenges and the added value of intraoperative optical coherence tomography. Clin Case Rep. 2022 Apr 4;10(4):e05637. [CrossRef]
- Davidson AE, Liskova P, et al.. Autosomal-Dominant Corneal Endothelial Dystrophies CHED1 and PPCD1 Are Allelic Disorders Caused by Non-coding Mutations in the Promoter of OVOL2. Am J Hum Genet. 2016 Jan 7;98(1):75-89. [CrossRef]
- Nischal, KK. Genetics of Congenital Corneal Opacification—Impact on Diagnosis and Treatment. Cornea. 2015 Oct;34 Suppl 10:S24-34. [CrossRef]
- Lisch W, Weiss JS. Clinical and genetic update of corneal dystrophies. Exp Eye Res. 2019 Sep; 186:107715.
- Nischal, KK. A new approach to the classification of neonatal corneal opacities. Curr Opin Ophthalmol. 2012;23(5):344-354.
- Seitz B, Lisch W, Weiss J. Die revidierte neueste IC³D-Klassifikation der Hornhautdystrophien [The revised newest IC³D classification of corneal dystrophies]. Klin Monbl Augenheilkd. 2015 Mar;232(3):283-94. German. [CrossRef]
- Zhang Y, Liu X, Liang W, Dean DC, Zhang L, Liu Y. Expression and Function of ZEB1 in the Cornea. Cells. 2021 Apr 16;10(4):925. [CrossRef]
- Liskova P, Evans CJ, Davidson AE, Zaliova M, Dudakova L, Trkova M, et al. Heterozygous deletions at the ZEB1 locus verify haploinsufficiency as the mechanism of disease for posterior polymorphous corneal dystrophy type 3. Eur J Hum Genet. 2016;24:985-91.
- Matthaei, M. , Schrittenlocher S., Cursiefen C. et al. 10 years of Descemet membrane endothelial keratoplasty in Fuchs endothelial corneal dystrophy: What have we learned. Ophthalmologe. (2019); 116(3):236-242.
- Schaub, F. , Collmer M., Hos D. et al. Outcome of Descemet Membrane Endothelial Keratoplasty Using Corneas from Donors ≥80 Years of Age. Am J Ophthalmol. (2020) 211:200-206.
- Dapena I, Moutsouris K, Melles GR, et al. Standardized "no-touch" technique for descemet membrane endothelial keratoplasty. Arch Ophthalmol. (2011) 129(1):88-94.
- Kruse FE, Schrehardt US, Tourtas T. Optimizing outcomes with Descemet's membrane endothelial keratoplasty. Curr Opin Ophthalmol. (2014) 25(4):325-334.
- Schrittenlocher S, Schaub F, Hos D, et al. Evolution of Consecutive Descemet Membrane Endothelial Keratoplasty Outcomes Throughout a 5-Year Period Performed by Two Experienced Surgeons. Am J Ophthalmol. (2018) 190:171-178.
- Masson E, Zou WB, Génin E, Cooper DN, Le Gac G, Fichou Y, Pu N, Rebours V, Férec C, Liao Z, Chen JM. Expanding ACMG variant classification guidelines into a general framework. Hum Genomics. 2022 Aug 16;16(1):31. [CrossRef]
- Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–96.
- Chiambaretta F, De Graeve F, Turet G, Marceau G, Gain P, Dastugue, B, Rigal D, Sapin V. Cell and tissue specific expression of human Krüppel-like transcription factors in human ocular surface. Mol Vis. 2004 Nov 23;10:901-9. [PubMed]
- Dudakova L, Evans CJ, Pontikos N, Hafford-Tear NJ, Malinka F, Skalicka P, Horinek A, Munier FL, Voide N, Studeny P, Vanikova L, Kubena T, Rojas Lopez KE, Davidson AE, Hardcastle AJ, Tuft SJ, Liskova P. The utility of massively parallel sequencing for posterior polymorphous corneal dystrophy type 3 molecular diagnosis. Exp Eye Res. 2019 May;182:160-166. [CrossRef]
Figure 2.
Histological patient sample obtained during first DMEK in the index patient (A&B) and the son (C) with the novel mutation in PPCD: a multilayer epithelium-like endothelium is here a typical histological feature: A: Histologic evaluation by using hematoxylin-eosin staining shows a loss of corneal endothelial cells centrally and blurred lacerations at the level of Descemet's membrane (see blue arrow) A/B: Simultaneous peripheral multi-row corneal endothelial cell layer next to the trabecular meshwork (see red arrow) with visible pigmentation (see brown arrows). C: Abnormal thickening of the Descemet's membrane up to 18.7m in the hematoxylin-eosin staining (see green arrows). Cave: During the paraffinization process, there is a regular shrinkage of the tissue, i.e. a reduction in Descemet's thickness.
Figure 2.
Histological patient sample obtained during first DMEK in the index patient (A&B) and the son (C) with the novel mutation in PPCD: a multilayer epithelium-like endothelium is here a typical histological feature: A: Histologic evaluation by using hematoxylin-eosin staining shows a loss of corneal endothelial cells centrally and blurred lacerations at the level of Descemet's membrane (see blue arrow) A/B: Simultaneous peripheral multi-row corneal endothelial cell layer next to the trabecular meshwork (see red arrow) with visible pigmentation (see brown arrows). C: Abnormal thickening of the Descemet's membrane up to 18.7m in the hematoxylin-eosin staining (see green arrows). Cave: During the paraffinization process, there is a regular shrinkage of the tissue, i.e. a reduction in Descemet's thickness.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



