
Submitted:
21 October 2024
Posted:
22 October 2024
You are already at the latest version
Abstract
Gastrointestinal (GI) cancers are the most common causes of global cancer burden and cancer-related deaths. Chronic inflammation is a hallmark feature of GI tumors and plays an important role in tumor initiation, progression, and therapeutic response. The genetic makeup of cancer cell, as well as epigenetic alterations, and biological processes constitute an inflammatory tumor microenvironment (TME) that provides an essential nich for cancer initiation and progression. Cancer-associated inflammation, a key component of TME, displays protumoral or antitumoral activity depending on the composition, spatial distribution, and functional status of the immune cells involved, thus adversely affecting response to cancer treatment and clinical outcomes, whereas acute inflammation supports T-cell priming, activation, and infiltration into malignant tissues, thereby conferring anti-tumoral immunity. Inflammatory cells and proinflammatory molecules within the TME act as a bridge between inflammation and cancer. The majority of cells involved in the inflammatory process in the TME are innate immune cells, which play a critical role in shapping both the cellular composition of the TME and the plasticity of cancer cells and stromal cells. Activation of innate immune cells in the TME initiates the production of inflammatory, regenerative, and anti-inflammatory soluble molecules, resulting in adaptive immune response to the cancer. Additionally, aberrant activation of intracellular signaling pathways, inflammatory molecules, and metabolites have critical roles in the development and healing of inflammation during carcinogenesis. Cancer-associated inflammation may inhibit the effectiveness of anti-cancer agents by causing enzymatic degradation of drugs and modulating the immun activity, thus affecting survival of patient. In this review article, we highlight the genetic and cellular mechanisms involved in the formation of the inflammatory component of the TME in GI cancers, the soluble molecules, the cancer cell-immune cell interactions, and the implications of all these for clinical outcomes. We also comprehensively address the mechanisms linking chronic inflammation to cancer, with a focus on the most frequent GI cancers.
Keywords:
tumor microenvironment
; cancer-associated inflammation
; cancer-associated fibroblasts
; gastrointestinal cancer
; innate immunity
; adaptive immunity
Introduction
Gastrointestinal (GI) cancers, such as colorectal cancer (CRC), stomach cancer, pancreatic cancer and liver cancer, are a major cause of global cancer burden and cancer-related deaths (1). Although unprecedented advances have been made in cancer research and treatment in the last two decades, cancer-associated death remains one of the most common causes of death globally (1). During this period, the role of the immune system and chronic inflammation on carcinogenesis, tumor progression, and treatment efficacy has been extensively investigated (1,2,3). Approximately 30% of patients develop treatment resistance to molecular targeted therapies, especially immunotherapies, which are widely used today, and chronic inflammation constitute one of the important mechanisms of treatment resistance (4,5). Today, we observe a paradigm shift in the concept of tumor biology; we understand tumor microenvironment (TME)-centered biology rather than cancer cell-centered biology. TME is an intricate and dynamic ecosystem, which contains cellular and molecular components, including tumor cells, stromal cells, inflammatory cells, and soluble inflammatory molecules (3,4,6). Preclinical trials have demonstrated that chronic inflammation recruits and activates innate and adaptive immune cells within the TME (7,8). Inflammation, which contains sufficient innate and adaptive immune cells, is considered a protective immune response that maintains tissue homeostasis by eliminating harmful stimuli (8,9,10). Cancer-associated inflammation, which can be considered as the chronic inflammatory component of the TME, which begins to develop at the beginning of carcinogenesis and continues to develop at every stage of the tumor, is non-resolving, unlike wound healing and infection (8,9,10,11). The molecular and cellular mechanisms involved in the pathogenesis of cancer-associated inflammation and inflammation resulting from infection and tissue injury show functional differences and similarities (3,8,10). Damage to epithelial tissue by injury and infection triggers the activation of myeloid cells expressing inflammatory cytokines, activating innate and adaptive immunity, thus providing homeostasis in the damaged tissue (8,9,10). However, if damage to epithelial tissue results from an oncogenic stimulus, the emerging immunity will predispose to carcinogenesis through inflammation and cytokines rather than improving initial injury (8,10).
Chronic inflammation has long been recognized to contribute to cancer development, particularly across colorectal cancer, gastric cancer and liver cancer (8,10,11). The existence of a link between chronic inflammation and cancer was first suggested by the German pathologist Rudolph Virchow in 1863, and inflammatory cells were abundant in tumor tissues (12). 15-20% of solid tumors originate from tissues and organs with long-term chronic inflammation. The well-known examples are inflammatory bowel disease (IBD), chronic hepatitis, and H. pylori-associated gastritis, with an increased risk of colorectal cancer (CRC), liver cancer, and stomach cancer, respectively (13,14,15,16,17). The inflammatory TME formed by the genetics of cancer cell and other mechanisms such as epigenetic alterations, deregulation of intracellular signaling pathways and biological processes of cancer cell is a key determinant in tumor progression and response to anticancer treatments (18,19). Today, the mechanisms that govern the development of tumor-associated inflammation, and its interaction with and modulation of anti-tumor immunity have been largely elucidated (8,9,10,15,18,19). Single-cell technologies have shown increased expression of various inflammatory cytokines and chemokines by cancer cells in primary tumors or metastatic lesions (3,8,10,20,21). Another important type of inflammation is therapy-induced inflammation, which occurs in response to various anti-cancer therapies, including chemotherapy, radiotherapy, and immunotherapy (8,9,10,11). Cancer-associated inflammation enhances immunosuppression by recruiting immunosuppressive cells such as myeloid-derived immunosuppressive cells (MDSCs) and regulatory T (Tregs) cells, thus contributing to the formation of the immunosuppressive TME, which promotes cancer development and metastasis (4,8,14,15,21). While treatment-induced chronic inflammation promotes drug resistance and cancer progression, acute inflammation accentuates the eradication of cancer cells by triggering the activation of CD8+ T cells (7,8,11). Studies have shown that acute inflammation caused by exogenous factors foster anti-tumor function by triggering the activation of dendritic cells (DCs) and CD8+ T cells (1,4,7,8,10). While various environmental factors, such as obesity, smoking, and excessive alcohol consumption predispose carcinogenesis and promote tumor progression by causing low-grade local or systemic inflammation, cancer-associated inflammation contributes to cancer progression by promoting the accumulation and activation of inflammatory cells (8,10). Cancer-associated inflammation has a critical function in shaping the cellular composition of the TME and the plasticity of tumor cells, as well as contributing to cancer progression by blocking anti-tumor immunity and directly transmitting tumor-promoting signals to cancer cells (3,8,9,10). The future and utility of cancer immunotherapy depend on a better understanding of these interactions and the ability to orchestrate them beneficially.
Molecular Mechanisms Linking Chronic Inflammation to Cancer
Acute inflammation is thought to be a protective immune response that occurs during infection and tissue injury, the main function of which is to mitigate tissue damage and infection (8,9,10). Innate immune cells, including macrophages, monocytes, neutrophils, dendritic cells (DCs), and Kupffer cells (KCs) initiate inflammation in damaged tissue (3,8,10,22). Inflammatory mediators expressed by innate immune cells, such as cytokines, chemokines, leukotrienes, and prostaglandins have been demonstrated to promote the recruitment of immune cells and stimulate surrounding cells to produce cytokines, forming a potent barrier against the microorganisms (3,8,23,24,25,26). Acute inflammation is typically triggered during times of infection via an interaction between pattern recognition receptors (PRRs) on innate immune cells, particularly tissue-resident macrophages, and pathogen-associated molecular patterns (PAMPs) (7,8,10). Granulocytes constitute the majority of cells involved in acute inflammatory reactions (27,28). In the next step, inflammatory cells that undergo apoptosis-induced cell death are phagocyted by macrophages (11,27,30). Tissue-resident macrophages, particularly KCs, respond to tissue changes by several mechanisms, such as eliminating dying cells, expressing chemotactic molecules, orchestrating immune mechanisms, and contributing to the recruitment of stem cells (8,10,29). In the initial and amplification phases, inflammation exerts a significant immunostimulatory effects (1,7,8,10). The resolution phase is characterized by the secretion of anti-inflammatory lipid mediators, such as SMPs, cytokines such as IL-10 and growth factors including TGFβ and epidermal growth factor (EGF) (3,8,9). SMPs play an important role in the resolution of inflammation by hindering neutrophil recruitment in damaged tissue, attenuating the secretion of inflammatory cytokines, and fostering the capacity of macrophages to phagocyte apoptotic neutrophils (30,31,32,33). These mediators augment the production of Tregs and B cells to suppress excessive activation of adaptive immunity (30,32). When acute inflammation becomes chronic inflammation, adaptive immune cells recruit in the inflammation field to eliminate detrimental factors (27,29). Although inflammatory mechanisms are necessary for the regeneration of normal tissue and the maintenance of tissue homeostasis, they can also contribute to the initiation of cancer and tumor progression (29). Nowadays, it has become clear that the inflammatory process comprises many cell types and molecules governing cell chemotaxis, migration, and proliferation (11,31,32).
Chronic inflammation is considered to be one of the main drivers in the development of cancer and the anti-cancer response and may show pro-tumoral or anti-tumoral activity depending on the type of immune cells it involves and the functional status of the cells (8,10,22,34) (Figure 1). In some situations, the resolution of acute inflammatory reactions can be hampered by biological and environmental factors, leading to chronic inflammation, the immune cell composition of which and the molecular mediators produced are quite different from acute inflammation (1,7,8,10,35). In contrast to the acute inflammatory response, chronic inflammation is typically activated by DAMPs in the absence of activation of PAMPs (1,8). The clinical consequences of chronic inflammation can be severe and its key role in carcinogenesis and tumor progression has been proven (1,7,8,29). Approximately 25% of cancers arise from a chronic inflammatory microenvironment (1). Helicobacter pylori infection causes chronic gastritis that is associated with the generation of reactive oxygen species (ROS) and nitric oxide metabolites and a reduction of vitamin C levels, which can lead to peptic ulcer, gastric cancer, and gastric mucosa-associated lymphoid tissue lymphoma (36). In Asian and Eastern European countries, where gastric cancer incidence and mortality are high, the lifetime risk of developing gastric cancer in H. pylori-positive individuals is 1-5% (17). Additionally, in these geographic regions, high salt intake, smoking habits, low iron levels, and pickled foods contribute to the development of gastric cancer (17). Chronic inflammation damages parietal cells and these cells lose their acid-producing properties, which results in hypochlorhydria or achlorhydria, which pave the way for the colonization of proinflammatory gastric microorganisms, thus producing more genotoxic and pro-inflammatory metabolites and carcinogens, which are directly effective on malignant epithelial cell transformation (37,38).
Another well-known example of chronic inflammation-derived cancer is colitis-associated colorectal cancer (CAC) (13,14). Persistent chronic inflammation of the colon caused by inflammatory bowel diseases (IBDs), such as Crohn's colitis and ulcerative colitis, is associated with an increased incidence of CAC, which is considered the most serious complication of IBD (39,40). CAC represents an important model of inflammation-associated carcinogenesis: the risk of CAC increases as the duration of colitis increases and the anatomical extent of colitis expands and in the presence of concomitant inflammatory diseases, such as primary sclerosing cholangitis (13,40). Chronic inflammation itself is the major driver in the development of CAC in IBD (13,14). Chronic inflammation-associated CAC is thought to arise as a result of the expansion of pro-tumorigenic clones (14). Somatic driver mutations can be detected in non-dysplastic inflamed colon years before the diagnosis of CAC (13). Multiple studies using whole exome sequencing have identified TCGA point mutation in the KRAS, BRAF, ERBB2, ERBB3, TP53, and FBXW7 genes in non-dysplastic colon mucosa (41). Chronic inflammation present in the microenvironment paves the way for driver mutations to form tumors (13,41). Local tissue damage leads to inflammation, which cooperates with driver mutations in the KRAS and p53 genes for malignant transformation of the cell (13,41). Many teams of researchers have demonstrated a distinct genomic landscape CAC in terms of driver gene mutations, copy number alterations, germline changes, and the functional effects of these genomic alterations (13,41). Mutations may not be the initial event that triggers carcinogenesis in IBD (41). While p53 mutation is not detected in the colon mucosa, it is detected in approximately 50% of the dysplastic mucosa, and the frequency of p53 mutations gradually increases towards the dysplastic-carcinoma cascade (13,41). Additionally, copy number alterations accumulate progressively from low-grade dysplasia to high-grade dysplasia and cancer (13,43,44). Chatila et al. showed that cancer development in IBD predominantly arises from independent genetic events and that there is no field effect of genetic changes that predispose to cancer development (14). Two important differences have been detected between CAC and sporadic CRC in terms of genomic landscapes (14). The first is that p53 alterations are early and highly recurrent events in CACs, which occur in half of dysplasia, while it is a late event in sporadic CRC (14). Secondly, while APC mutations are detected at a rate of 81% in sporadic microsatellite stable (MSS) CRC, they are detected at a rate of 11-22% in CAC (14). Studies in mouse models have demonstrated that SMAD4 is also an important player in the regulation of inflammation and loss of SMAD4 leads to the upregulation of some inflammatory signaling pathways, including IL-6/STAT3 and NF-κB (45). Significant differences have been identified between CAC and sporadic CRC in terms of driver mutations in oncogenes and tumor suppressor genes, copy number changes, and functional effects of these genomic alterations (13,14,45). One of the most prominent examples in this field is the absence of Wnt signaling activity in CAC, which is one of the fundamental genomic landscapes of sporadic CRC (13,45).
Chronic liver diseases such as viral hepatitis or nonalcoholic fatty liver disease (NAFLD) are characterized by persistent inflammation and subsequent liver fibrosis, leading to liver cirrhosis and hepatocellular carcinoma (HCC) (46,47). HCC represents the most common type of liver cancer, which usually arises in the inflamed liver microenvironment caused by HBV and HCV infection, alcohol abuse and NAFLD (46). NAFLD is a spectrum of chronic liver disease that ranges from simple steatosis to non-alcoholic steatohepatitis (NASH) and is strongly associated with metabolic syndrome (46,47). NASH is an emerging risk factor for HCC (46). Accumulating evidence has indicated that pre-cirrhotic NAFLD might provide an increased risk of HCC, independent of cirrhosis (48). A range of single nucleotide polymorphisms, such as patatin-like phospholipase domain containing 3 (PNPLA3; rs 738409) and transmembrane superfamily member 2 (TM6SF2; rs 58542926) have been associated with the presence of NAFLD and disease progression to advanced fibrosis and HCC (46,49,50). Many research groups from different countries have shown that HCC is 3 times higher in patients carrying the PNPLA3 polymorphism (46,49). Recently, the combined use of single-cell RNA sequencing and single-cell spatial analysis in tumor tissue samples has largely elucidated the molecular and cellular characteristics of NASH-associated HCC (51). Genomic analyses have indicated key pathways altered in HCC, including Wnt/β-catenin, P13K/Ras, and cell-cycle pathways (46,52). The most frequently mutated genes in NASH-associated HCCs are TERT, CTNNB1, TP53 and ACVR2A genes (46,52). NASH-associated HCC samples have significantly higher rates of ACVR2A mutations than samples of other aetiologies (52). In vitro observations revealed that ACVR2A mutations act as a tumor suppressor in HCC (52). The Wnt/TGF-β proliferation subclass is more frequent in NASH-driven HCC than in HCCs of other etiologies (53). Another molecular landscape of NASH-driven HCC is that it has an immunosuppressive pro-carcinogenic and inflammatory tumor microenvironment (52,53).
In homeostasis, immune cells, particularly KCs are densely populated in the liver, which rapidly sense hepatocyte stress and injury signals, and lead to the activation of pro-inflammatory pathways (51). Metabolic stress induced by several factors causes metabolic disturbance in hepatocytes, increasing reactive oxygen species (ROS), endoplasmic reticulum (ER) stress and oxidative stress and resulting in hepatic metabolic reprogramming (47,48,51). These processes result in hepatocyte death of apoptotic or necroptotic type (48,51). In the liver, dying or damaged hepatocytes release soluble mediators that act as damage-associated molecular patterns (DAMPs) (51). Preclinical studies have revealed that dying hepatocytes release P2Y14 ligands, such as uridine 5’-diphosphate (UDP)-glucose and UDP-galactose, that bind to the P2Y14 receptor on HSC and induce activation in both mouse and human HSCs (54). Liver parenchymal and non-parenchymal cells, including HSCs, KCs, and liver sinusoidal endothelial cells (LSECs) perceive these dangerous signals released from the dying hepatocytes via PRRs and form inflammasome as the first response (55). Inflammasome, a protein complex, initiates the inflammatory response by producing IL-1β, releasing IL-18, and ultimately promoting inflammatory cell death (55). Pro-inflammatory cytokine-producing Innate and adaptive immune cells rapidly accumulate in the inflammatory microenvironment and disrupt hepatocyte metabolism by promoting hepatic metabolic reprogramming, thereby promoting hepatocyte injury and death, DNA damage, and hepatocyte proliferation (56). Inflammatory molecules promote the activation of hepatic stellate cells (HSCs), and inflammatory mediators secreted by activated HSCs contribute to the expansion of chronic inflammation and hepatocarcinogenesis (49,51). Mounting evidence indicates that chronic liver inflammation damages hepatic epithelial cells, including hepatocytes and biliary epithelial cells (51). This damage leads to a significant increase in liver cell proliferation (46,51). Simultaneously, chronic inflammation induces ROS production and DNA damage, increasing the frequency of genomic alterations (49). Furthermore, chronic liver inflammation induces phenotypic changes in hepatocytes and hepatic immune cells, especially macrophages (16,49,51). Chronic inflammation initiates hepatocarcinogenesis through the transformation of hepatocytes into liver progenitor cells and the differentiation of macrophages into tumor-associated macrophages (M2 phenotype) (16). TNFα and IL-6 produced by macrophages in the cirrhotic liver, as well as TNF receptor 1 (TNFR1) signaling expressed by hepatocytes play critical roles in the development and progression of HCC (16). TNFα is one of the protumorigenic cytokines, that activates both NF-κB and JNK signaling pathways (16,51). Many studies demonstrate that activation of innate immune receptors, such as Toll-like 4 receptors (TLRs), particularly TLR4, plays a role in HCC development because IL-6 is induced through MyD88, a universal adaptor molecule for TLRs (16,57).
The gut microbiome, which harbors more than 100 trillion microorganisms including bacteria, viruses, fungi and archaea, plays a critical role in the development and progression of HCC by contributing to the establishment and growth of chronic liver inflammation (58). The liver and gut interact via the anatomically (portal venous system) and the gut microbiome (59). The liver regulates the intestinal microbiota with bile containing bile acids, IgA and antibacterial metabolites (60). Environmental factors, such as high-fat diets and alcohol consumption, can disrupt microbial compositions, leading to gut dysbiosis, which induces intestinal inflammation, which contributes to intestinal barrier dysfunction and translocation of microorganism-associated molecular patterns (MAMPs), such as lipopolysaccharide (LPS), to the liver and systemic circulation (61). In diet-induced NASH mouse models, a positive correlation between serum LPS levels and liver injury has been demonstrated (62). Cholestasis due to liver tissue remodeling in cirrhosis leads to intestinal dysbiosis (58). Cirrhosis patients display increased bacterial abundance in hepatic tissue, which induces pronounced transcriptional changes, including activation of fibro-inflammatory pathways as well as circuits mediating cancer immunosuppression (63). Increased intestinal permeability in cirrhotic patients allows the translocation of MAMPs and contributes to increased systemic and local inflammation in the liver (63). Dysbiosis changes the metabolism of intestinal bile acids, with less conversion of primary to secondary bile acids (64). Several studies have indicated that fecal microbial diversity is decreased in patients with cirrhosis compared with healthy controls, however, diversity increases as one progresses from cirrhosis to HCC (65). Preclinical studies have shown that myc-transgenic mice capable of developing HCC arise lower numbers and sizes of HCC when given antibiotics (66). Another key finding of this study is that primary bile acids increase the accumulation of hepatic NK cells, whereas secondary bile acids reverse this situation (66). Several mouse model trials have revealed a link between the activation of inflammatory signals caused by intestinal permeability, the translocation of MAMPs, and the development of HCC (67). Rats with diethylnitrosamine-induced HCC have increased serum LPS, and administration of antibiotics decreased the tumor size and numbers (67). Another remarkable finding of this study was that tumor size was significantly reduced in TLR4-knock-out mice treated with diethylnitrosamine compared to wild-type mice (67).
The microenvironment of chronic inflammatory disease consists of immune cells, stromal cells, and soluble molecules such as cytokines and chemokines, which lead to genetic and epigenetic alterations in healthy cells to promote tumorigenesis (1,4,8,11,39,40). In chronic inflammatory diseases, oncogenes such as KRAS and some signaling pathways such as NF-κB, JAK/STAT, MAPK, Wnt/β-catenin, and P13K are induced, resulting in the synthesis of chemokines and adhesion molecules (11,39,40). In chronic inflammatory disorders, the activation of oncogenes and some signaling pathways and the production of some mediators lead to the accumulation of immune cells, the expression of cytokines (IL-1β, IL-17, TNF-α) and growth factors (GM-CSF) and COX-2 that stimulate cancer cell proliferation and survival, and the synthesis of proteases that induce cancer progression (1,4,7,8,9,10,11). One of the most characteristic features of cancer is inactivation of tumor suppressor genes (10,18,19,21). P53 protein has multifaced roles to orchestrate cellular biological processes and exerts transcriptional antagonism to NF-κB, a key regulator of inflammation, in cancer (68,69). Considering the presence of NF-κB signals almost always within the TME, loss of p53 functions leads to increased production of NF-κB-related genes (68,69). Studies conducted in patients with CRCs have demonstrated that this inflammatory signal is involved in tumor progression and metastasis (70). Many environmental stimuli can promote carcinogenesis by inducing chronic inflammation (1,8,10,18). In this situation, inflammation may precede or accompany carcinogenesis (8,10). In humans, these noxious stimuli may exert local or systemic effects (8,10). For instance, cigarette smoke and asbestos primarily promote the accumulation of immune cells, such as macrophages, neutrophils, and monocytes in the airway, which causes lung and airway inflammation, promoting carcinogenesis (71,72). Inhalable risk factors promote lung carcinogenesis through causing oxidative stress, that stimulates the production of pulmonary inflammation mediators and activation of MAPK and NF-κB pathways (73).
In chronic inflammation, tissue destruction and healing occur simultaneously and numerous immune cells, mainly macrophages, neutrophils, and monocytes are accumulated in the field of inflammation (8,10,11,18,21). The recruitment of immune cells has a pivotal role in carcinogenesis and tumor progression (74,75). In the acute inflammatory responses, the chemokine monocyte chemotactic protein (MCP)1/CCL2 initiates the accumulation of pro-inflammatory monocytes that produce the chemokine receptor CCR2 (76). If the acute inflammation does not resolve and chronic inflammation occurs, the M-CSF, which induces the differentiation of monocytes to macrophages, is overexpressed in the inflammation field (77,78). Macrophages exert multiple roles in cancer, a reflection of their plasticity in response to environmental stimuli (79). These cells have the potential to kill tumor cells, mediate cellular cytotoxicity and phagocytosis, elicit vascular damage and tumor necrosis, and activate immune cell-mediated mechanisms of therapy resistance (80,81). M1 phenotype macrophages exert a pro-inflammatory effect and play a key role in hepatocarcinogenesis by expressing pro-inflammatory mediators, such as IL-6 and TNF-α (8,10,79,80). Conversely, macrophages contribute to tumor progression by various mechanisms, including the promotion of cancer cell proliferation and survival, angiogenesis, and inhibition of immune responses (79,81). Tumor-associated macrophages (TAMs), M2 macrophages, are an essential component of the TME and have a role in the regulation of angiogenesis, extracellular remodeling, immunosuppression, and therapy resistance (82). Tissue-resident macrophages (TRMs) may play divergent roles in the inflammatory processes of tissues and the development of cancer (81,82). For example, in pancreatitis, TRMs, on the one hand, exert a protective effect by triggering the accumulation and activation of fibroblasts, while on the other hand, TRM-induced fibrosis drives the pathogenesis and progression of cancer (82). Furthermore, SPM downregulates the translation of inflammatory mediators by upregulating microRNAs influencing inflammatory genes in macrophages (83). Neutrophils are the first responders to acute inflammation and contribute to the resolution of inflammation. However, in chronic inflammation, the role of neutrophils has been described as either beneficial or detrimental, causing tissue damage and accentuating immune responses (84). These cells have the potential to establish neutrophil extracellular traps (NETs) (85). Mast cells are considered to exert critical proinflammatory functions and potential immunoregulatory roles through the secretion of mediators such as histamine, cytokines, proteases, and chemokines (86,87).
T cells are vital for immune functions to maintain health and restrain disease (1,7,8). Effector CD8+ T cells release pro-inflammatory cytokines such as IFN-γ and TNF to inhibit viral replication (7,8). During inflammation, naive CD8+ T cells develop into CD8+ CTLs expressing various mediators, whereas naïve CD4+ T cells develop into Th1, Th2, Th17, Th22, and CTL subsets with indicated phenotypes to exert protective functions (1,7,8,14). CD4+ CTLs contribute to pathogen clearance through direct cytolytic activity or indirectly by enabling help to other immune cells (1,3,4,5,7,8,9,11). T-cell dysfunction is a hallmark of many cancers, but the mechanisms are not fully understood (7,8). Progenitor or stem-like exhausted CD8+ T cells can be defined by intermediate expression of PD-1 and the expression of the chemokine receptor CXCR5 (3). Terminally exhausted T cells co-express high levels of PD-1, Tim-3 and other co-inhibitory receptors (3,21). These two subsets have significant functional distinctions, and only progenitor-exhausted cells proliferate after checkpoint-blockade therapy (3,21). Regulatory T (Treg) cells are unique in terms of their immune and metabolic features, which have a distinctive metabolism and immunosuppressive ability (88,89). Tregs represent about 10% of the CD4+ T cells and promote the immunotolerance of malignant cells, resulting in unfavorable clinical consequences for multiple cancers (3,8,21). Dendritic cells (DCs) play pivotal roles in both the promotion of immune defense and the maintenance of immune tolerance (88,89). DCs exert different immune functions, including antigen recognition and capture, inflammatory molecule expression, chemotactic migration and regulation (88,89,90).
Main Drivers Involved in The Generation of Cancer-Association Inflammation
We discussed in the previous section that some solid tumors originate from a long-term chronic inflammatory background and the cellular and molecular mechanisms that initiate and promote carcinogenesis. Inflammation may exit before carcinogenesis or both processes may begin simultaneously (7,8,9,10,14). Epidemiological studies have revealed that a very small proportion of patients with long-standing inflammatory disease develop cancer from an inflamed field (8,10,11). For example, approximately 2% of CRC is linked to pre-existing inflammation known as CAC, but most develop in patients without underlying IBD (8,10,13). Cancer-associated inflammation, inflammatory component of the TME, is a new concept; the molecular and cellular mechanisms involved in its pathogenesis, and its prognostic and therapeutic implications are only just beginning to be understood (8,9,10,13,14,21). The hallmarks of cancer-associated inflammation include the presence of inflammatory cells and inflammatory mediators, such as chemokines, cytokines and prostaglandins, in tumor tissues, tissue remodeling and angiogenesis similar to that seen in the chronic inflammatory response, and tissue repair (8,9,10,11,20,22,34). Regardless of the risk factors that initiate carcinogenesis, the vast majority of tumors contain inflammatory cells and soluble inflammatory and immunosuppressive molecules in the tumor tissue (8,9,10,14,20). Nowadays, it has been established that there is a causal connection between inflammation and cancer and that the composition and functional status of the immune infiltrate in the tumor tissue have prognostic and predictive value for molecularly targeted therapies (8,9,10,13,21,33). Furthermore, the role of the genetic makeup of the cancer cell, such as the driver mutations in oncogenes and loss of function of TSGs, tumor mutational burden, in the pathogenesis of cancer-related inflammation, as well as the interaction between the genetic landscape of the cancer cell and the composition and activation of the immune infiltrate in the inflammatory TME, has been largely elucidated (18,20,21).
The genetic landscape of the cancer cell promotes the secretion of proinflammatory molecules from the cancer cell, leading to recruitment of inflammatory cells within the tumor tissue, playing a critical role in the formation of cancer-associated inflammation (8,9,10,18,21) (Figure 2). Studies in experimental models have demonstrated that oncogenes and tumor suppressor genes regulate essential cellular biological processes such as cell cycle, apoptosis, senescence, autophagia, migration, and survival and that genetic alterations leading to dysregulation or loss of function in these genes can result in chronic inflammation and malignant transformation (9,11,18,20,21,93,94,95). Solid tumors generate inflammatory and immunosuppressive microenvironment (TME) to promote cancer cell growth and metastasis (18,21,92,93). The mechanisms by which oncogenes and TSGs regulate tumor-associated inflammation are beginning to be elucidated (94). Oncogenes and TSGs alter the inflammatory cell composition of the TME by modulating cancer cell-intrinsic programs as well as changing cancer cell inflammatory mediator secretion (8,18,21,92,93). The p53 pathway can modulate the immunological composition of the tumor tissue by regulating NF-κB signaling, which is generally activated by the loss of p53 (8,18,21,33,93). One remarkable example is NF-κB which orchestrates cell survival and proliferation, but also the production of inflammatory cytokines (8,9,18,21,93). The activation of the NF-κB signaling pathway induced carcinogenesis in the KrasLSL-G12D/+; Trp53F/F lung adenocarcinoma model (21,93,96). On the contrary, NF-κB inactivation accelerates intratumoral immune cell migration and prevents carcinogenesis in KrasLSL-G12D/+; Trp53F/F mice, indicating a link between loss of p53, NF-κB activation, and an inflammatory TME (18,21,93). The tumor suppressor p53 gene can drive immune infiltration and inflammation in a wide range of tumor types by controlling the pro-inflammatory NF-κB pathway (8,10,11,14,15,18,21,93). In experimental models, concomitant loss of E-cadherin and p53 promotes NF-κB activation, which is accompanied by increased macrophage recruitment and proinflammatory mediator production (21,93). Experimental studies have shown that NF-κB is controlled by p53 in the tumor tissue and that loss of p53 activates NF-κB, inducing the production of the inflammatory soluble molecules from cancer cells, which in turn alters the immune context through paracrine interactions (18,93,97). Furthermore, studies in p53 knockout mouse models have revealed that NF-κB-mediated inflammatory response is a driving force of carcinogenesis (18,93,97). In cancer, p53 missense mutations may exhibit tumor suppressor or tumor promoter activity. In addition to causing loss of p53 function, these mutations have gain-of-function activity and contribute to the malignant properties of cancer cells (93,98,99). Accumulating evidence suggests that in cancer, mutant p53 leads to the release of miR-1246-rich exomes, causing M1 macrophages to polarize into tumor-promoting M2 phenotype, thereby contributing to the establishment of an immunosuppressive and inflammatory TME (100,101).
Because of its critical effects on cellular biological processes, the MYC oncogene is a key player in generating cancer-related inflammation (95). The effect of MYC signaling on cancer-associated inflammation has been investigated in many tumors. It orchestrates several cancer cell-intrinsic and host-dependent pathways suh as proliferation, metabolism, invasiveness, autophagy, and protein and ribosomal biosynthesis to promote cancer cell growth and survival (102,103). Experimental models have shown that MYC inactivation can affect tumor cell growth and host immunity, leading to tumor regression (104). In mouse models of pancreatic cancer, MYC inactivation results in significant reductions in the number of M2 macrophages and tumor-associated neutrophils (TANs) in the TME as well as tumor regression (104,105). The cellular biological processes and clinical consequences of MYC inactivation vary depending on the tissue origin of the cancer, for example, the cellular biological processes including cellular differentiation, senescence and apoptosis, differ in MYC-deriven haematological cancers and MYC-deriven mesenchymal tumors (105,106).
Activation of the MYC pathway promotes the secretion of proinflammatory cytokines such as CCL5 and IL-1β from cancer cells, leading to the proliferation of inflammatory cells and amplification of inflammation in the TME (95,107,108). MYC signaling can control the infiltration of immune cells into tumor tissue, but this ability can be inhibited by other aberrant expressed genes (18,95). MYC can also drive the immune phenotype of the tumor by altering the expression levels of immune checkpoint molecules by cancer cells, thus affecting T cell response (109,110). Experimental studies have revealed that MYC increases the production of both PDL1 and CD47 on cancer cells by binding to their respective promoters (110). Overexpression of PDL1 and CD47 limits the accumulation of CD4+ T cells and macrophages in the TME (110). Furthermore, MYC inactivation decreases CD47 and PDL1 production (110). MYC may contribute to cancer cell evasion of immune surveillance by promoting the production of immune checkpoint molecules by cancer cells (111). Activation of MYC pathway impedes the accumulation of adaptive immune cells such as B, T, NK cells, in the TME by promoting IL-23 expression form cancer cells, while inducing the production of CCL9 from cancer cells and allowing macrophages to accumulate in the TME (109). Considering all the findings, targeting MYC in cancer may be a reasonable strategy to enhance anti-tumor immunity (95).
Apart from the genomic alterations in oncogenes and TSGs mentioned above, many genetic events affect the immune landscape of the tumor and contribute to the formation of chronic inflammation within the TME. Members of the Ras family are the most frequently mutated oncogenes in cancers and activated oncogenic components of the Ras-Raf signaling pathway, in turn, promote the expression of tumor-promoting inflammatory chemokines and cytokines (94). The effect of the Kras oncogene on tumor-associated myeloid cells has been demonstrated in many studies (18,94). In experimental models, the mutant Kras oncogene potently promotes the accumulation of myeloid cells in the TME and the production of the cytokines IL-6 and IL-8, which induce tumor progression (112). In addition, many studies have shown that Kras-derived pancreatic ductal adenocarcinoma (PDAC) expresses high levels of the growth factor GM-CSF, which is accompanied by the accumulation of immunosuppressive tumor-associated myeloid cells within the tumor tissue (113). In preclinical models, genetic ablation or neutralization of GM-CSF reduces myeloid infiltration, increases CD8+ T cell accumulation in the TME and leads to significant reduction in tumor size (113). These findings suggest a causal link between Ras oncogenic signaling pathways and the inflammatory landscape of tumors (112,113). Notch signaling can activate monocytes and macrophages by driving CCL2 and IL-1b production (114). In addition, Notch signaling also orchestrates TGF-β -receptor and uPA production, thereby promoting tumor growth (115). Notch can mitigate the antitumor immune response by inhibiting c/EBPb and thus limiting IL-1, IL-6, and IL-8 expression (115). Loss of tumor suppressor gene LKB1 can promote the production of G-CSF, CXCL7, and IL-6, which induces neutrophil accumulation and can prevent the recruitment of immune cells that exhibit antitumor activity (116). PTEN can inhibit NF-κB signaling, as such, loss of PTEN increases NF-κB-mediated expression of soluble molecules, which promotes the recruitment of inflammatory and immunosuppressive cells in the TME, such as neutrophil, macrophage, and Treg (117). Beyond the roles of cancer-derived cytokines, chemokines, and nonimmune cells in shaping inflammatory characteristics of tumors, the mutational landscape of cancer cells, which directly reflects the immunogenicity of the tumor, may determine the extent and phenotype of the immune infiltrate, i.e. inflammation, in the TME (93,94,95).
One of the main inducers of tumor-associated inflammation is the high metabolic stress of tumor, which is characterized by low glucose levels, amino acid deprivation, impaired lipid metabolism and lipotoxicity, and severe hypoxia (118,119). In the tumor tissue, immune cells and other stromal cells may be exposed to all of these metabolic stress components (118).
Tumor hypoxia develops as a result of the rapid consumption of oxygen by tumor cells and quick angiogenesis within the tumor (118,119). Metabolic stress, particularly hypoxia can contribute to generating cancer-related inflammation (120). Hypoxia leads to hypoxia-inducible factor (HIF)-1α induction, CAF and TAM activation, and the secretion of numerous chemokines, which in turn result in the accumulation of proinflammatory myeloid cells, particularly macrophages, in the TME. Hypoxia inhibits glycolysis and may promote the angiogenic activity of TAMs (118). In response to hypoxia, HIF-1α potentiates the polarization and suppression of the effect of M2 (121). Hypoxia also promotes the progression of T cells to exhaustion status (122). It promotes the production of immune checkpoint molecules, such as PD1, PD-L1, CTLA-4, LAG-3, and TIM-3, contributing to the TME becoming more inflammatory and immunosuppressive (123). Although hypoxia affects all cells within the tumor tissue, two recent studies have shown that it specifically affects cells with an inflammatory phenotype (119,120). Macrophages are effector immune cells that undergo significant changes when entering tumors or infected wounds (118,119,120). Hypoxia generates distinct responses from macrophages depending on the activation state. Hypoxia induces transcriptome turnover in macrophages, but inflammatory macrophages exhibit significantly increased mRNA destabilization compared to resting macrophages (119,124,125). In another study investigating the relationship between hypoxia and inflammation, Mello et al. found that hypoxia promotes the induction of inflammatory phenotype cancer-associated fibroblasts (iCAFs) by modulating their interaction with tumor cells and that hypoxic regulation of the iCAF phenotype is independent of tumor HIF1α or HIF2α (120). The drivers of tumor-associated inflammation are different in microbial-rich tumors and sterile tumors (8,20). For example, in CRC, disruption of the intestinal barrier by oncogene in the mucosa where cancer originates leads to translocation of commensal bacteria and their metabolites, which are sensed by tumor-associated myeloid cells, such as TAM, and TAN, to promote IL-23 production and IL-23-mediated cancer-related inflammation (126,127). In contrast, in tumors not originating in the mucosa genomic and metabolic changes, cell death and hypoxia may be initial inflammatory stimuli (126,127))
Stromal cells, such as CAFs, TAMs, TANs, are pivotal players in the generation and expansion of tumor-associated inflammation. CAFs are major component of the tumor tissue, and critically modulate cancer progression through various mechanisms, including production of growth factors, inflammatory ligands and exosomes as cell as ECM remodeling, angiogenesis, tumor mechanics and treatment responses (128). CAF-secreted inflammatory ligands and chemokines promote inflammation and tumor cell proliferation (8,18,128). Interaction between CAFs and cancer cells is mediated with a complex signaling network that consists of signaling pathways for TGFβ, mitogen-activated protein kinase (MAPK), Wnt/β-catenin pathway, JAK/STAT pathway, epidermal growth factor receptor (EGFR), and NF-κB (128). During carcinogenesis, experimental trials have reported that cancer-derived CAFs modulate immune system through recruiting immune cells, such as neutrophils, monocytes and dendritic cells, and promote these cells to acquire immunosuppressive phenotypes that boost immune evasion (128). Tumor-associated macrophages (TAMs) are an essential component of the TME and have an important role in the regulating of angiogenesis, extracellular matrix remodeling, cancer cell proliferation, metastasis and immunosuppression and as well as resistance to cancer therapy (2,7,18,21). Additionally, TAMs promote tumor-associated inflammation by producing pro-inflammatory mediators (2,7,8,11,18,19). TAMs inhibit T cell functions by secreting certain mediators, contributing to the maintenance of immunosuppressive TME (2,4,7,8,10,14,129,130). Neutrophils dominate the early phase of inflammation and pave the way for tissue damage to be repair by macrophages (129,131). These functions are regulated by various cytokines and the production of their receptors (129,130,131). In chronic inflammation, neutrophils exert dual role; beneficial or detrimental, driving tissue damage or accentuating immune response (2,3,4). Tumor-associated neutrophils (TANs) have been reported to be associated with tumor growth, metastasis and worse prognosis (2,129,131). N2 TAN can form NETs, which can promote carcinogenesis in the context of chronic inflammation (2,3,4,129). However, growing evidence suggests that TANs could impair the development of tumors by killing tumor cells or activating innate and adaptive immunity (1,2,3,129,131). Myeloid-derived suppressor cells (MDSCs) are one of the immunosuppressive cells, that drive tumor development and progression (1,7). Several studies have reported that tumor-associated tissue eosinophils (TATE) are associated with better overall survival in many hematological and solid cancers, such as breast, and CRC, indicating the contribution of eosinophils to the anti-tumor activity (3,7,10,132) (Figure 3).
Cancer-Inhibitory Inflammation
Although many experimental and clinical studies have reported that chronic inflammation has a pro-tumor activity, it is well known that chronic inflammation is not always associated with the risk of cancer development. As mentioned in previous sections, immune cells can kill tumor cells by expressing various molecules, playing a critical role in hampering tumor development and progression (1,3,4,8,9,10). Specific immune cells involved in chronic inflammation and cytokines can exert anti-tumor function (3,7,8,14). Although the innate immune system is thought to contribute to tumor-related inflammation by promoting the release of inflammatory mediators, DCs, an important member of the innate immune system, have been reported to control the development of CAC through the production of IL-22BP, which neutralizes IL-22 (3,7,8,9,13,14). However, cancer cells can hijack DCs to promote chronic inflammation and mitigate TAA presentation, thus accelerating carcinogenesis (13,14,133,134). The functions of DCs in inflammation and immune response are largely determined by their movement in lymphoid and nonlymphoid tissues (3,7,8,133,134). Different DC subsets have distinct mobilization capacities and exhibit different immunological functions (7,8,18,133,134). CCR7 is the most important chemotactic mediator of DC migration which recognizes the chemokine ligands CCL19 and CCL21 and it is the main guide in the migration of DCs to lymphoid tissue (134). The presence of inflammatory cells in some tumors, such as eosinophils in CRC and TAMs in breast and pancreatic cancer, is associated with a favorable prognosis (135). These observations may be an indication that inflammatory cells destroy cancer cells as well as normal tissue cells (134,135).
Macrophages, another basic cell of the innate immune system, exert significant antitumor activity by mediating cancer cell phagocytosis and cytotoxic cancer killing (57,81,82,136). As previously described, macrophages are classified into M1 and M2 phenotype (81,82). M1 macrophages produce high levels of proinflammatory mediators, such as TNF-α, IL-1β, IL-6, IL-12, CXCL9, and major histocompatability complex class II (MHC-II), but low levels of IL-10 and arginase (57,136,137). By releasing pro-inflammatory soluble molecules, M1 macrophages promote the polarization and accumulation of tumor cells, mediating the phagocytosis of tumor cells (136). The stimuli that polarize the macrophages towards the M1 phenotype may determine the type of function of the M1 phenotype, for example, M1 macrophages polarized with IFN-γ display anti-tumor activity, while repolarization of macrophages from the M2 phenotype to the M1 phenotype leads to a significant reduction in tumor size (82,136). M2 macrophages (TAMs) are polarized by IL-4, IL-10, and IL-13 and display immunosuppressive activity to trigger tissue repair (82,136,137). TAMs exhibit a role in regulating of angiogenesis, ECM remodeling, cancer cell proliferation, metastasis, and resistance to cancer treatments (81,82,129,130,138). TAMs are recruited and set in tumor-promoting mode by signals by tumor cells, B cells, CAFs and macrophages themselves (82,129,130,138). Molecules involved in the recruitment and education of TAMs include TGF-β, macrophage colony-stimulating factor 1 (CSF1), chemokines such as CCL2, cytokines such as IL-4 and IL-2 (82,129,130). Many studies have revealed that high TAM infiltration within the tumor or particularly in peritumor area are associated with worse clinical outcomes (11,57,81,82,136). Cancer cells educate TAMs to become immunosuppressive through the secretion of CCL2, SPP1 and extracellular vesicles (11,18,138). Macrophage-derived CCL2 promotes the production of PD-L1 and fosters the immunosuppressive landscape of the TME (81,82,138). Additionally, M2 macrophages secrete mediators such as IL-10, IL-6, IL-8 and VEGF, resulting in T cell and NK cell inhibition (11,14). As such, repolarization of TAM to the M1 phenotype may modulate the immunosuppressive feature of the TME and accentuate antitumor immunity (7,11,14,139). TANs are polarized to a pro-tumorigenic (N2) phenotype by tumor, which exert potent immunosuppressive functions including production of PD-L1, secretion of immunosuppressive molecules (IL-8, CCL2), NETosis (131,139). Conversely, N1-TANs are characterized by expressing several pro-inflammatory cytokines, including CCL2, CCL3, CXCL8, IL-6, and TNF-a and recruiting cytotoxic anti-tumor effectors within the TME (1,2,3,4,7,8,14). Additionally, N1-TANs promote the activation of cytotoxic anti-tumor effectors through the high production of co-stimulatory molecules (CD86, ICAM-1), subsequently accentuating anti-tumor immune function (131). Innate lymphoid cells (ILCs) can exert antitumor function in some tumors by recruiting DCs and CD8+ T cells within the TME via CCL5 (140). NK cells are cytotoxic innate-like lymphocytes that recognize stressed cells, such as cancer cells, and mediate the killing of cancer cells (141). Natural Killer T (NKT) cells possess potent tumor-killing activity and can infiltrate tumors and they bridge innate and adaptive immunity (142).
The core function of innate immune system is to recognize and present tumor-associated antigens (TAAs) to cytotoxic anti-tumor effectors (5,6). Anti-tumor effector cells can kill cancer cells directly or eliminate them by sensitizing them to biological molecules such as Fas ligand, perforin or granzyme (143,144,145). CD8+ T cells are pivotal mediators in the elimination of cancer cells, which harbor distinct T cell receptors (TCRs) (21,146). Growing evidence indicates that several key transcriptional factors (T-bet vs Bcl-6, STAT4 vs STAT3), epigenetic mechanisms (DNA methylation and histone modification) and metabolic reprogramming are involved in the differentiation of naïve T cells into effector cells (146,147,148). During cancer, naive CD8+ T cells differentiate into CD8+ CTLs producing a range of chemokine receptors and effector molecules (146). Factors within the TME can drive CD8+ T cells to exhausted T cells, which account for unique cellular phenotype, heterogeneity, and functional capacity (21,147,148). During exhaustion, CD8+ T cells gradually lose expression of IL-2 and TNF-α, and cytotoxic function (149). Terminal exhausted CD8+ T cells also lose IFN-γ expression (149). Experimental trials have reported that terminal exhausted CD8+ T cells can maintain their capacity to produce molecules such as MIP1α, MIP1β, RANTES, and IL-10 (149). Immune checkpoint molecule expression of terminal exhausted CD8+ T cells are high and response to immunotherapies is quite low in these patients (143,144,145). B cells can inhibit carcinogenesis by expressing tumor-reactive antibodies, promoting tumor killing by NK cells, phagocytosis by macrophages and priming cytotoxic effector cells (144,145). Several studies have reported that distinct B-cell types, such as tumor-infiltrating B-lymphocytes (TIBs) display pro-tumor effect during some cancer progression (144,150). Activated B cells can destroy tumor cells (150). Furthermore, TIBs were detected to have cytotoxicity toward hepatocellular carcinoma cells by the production of granzyme and TRAIL (150). Approximately 50% of the numerous studies investigating the functions of TIL-B cells report that CD20+ TIL-B cells have positive prognostic effects (150). It has been shown in many studies that the prognostic significance of TIB cells is compatible with CD3+ and CD8+ T cells and that T cells show stronger anti-tumor activity in the presence of TIB cells (7,8,14,150). Briefly, growing evidence suggests that TIB cells exert anti-tumor activity (7,8,9).
Cancer Therapy-Induced Inflammation
Anti-cancer therapy is associated with an inflammatory response in tumor tissue, which either drives an anti-tumor immune response or, conversly, promotes tumor growth (8,10). Recently, the effects of cancer therapy-induced inflammation on tumor recurrence and clinical outcomes have been largely understood, with the elucidation that cancer cell death elicits an anti-tumor immune response and that lipid mediators such as prostaglandin 2 (PG2) and platelet activation factor (PAF), play a role in the clearance of dead cells in the TME and in tumor growth (151). Many cancer treatments are very effective in destroying cancer cells, but the main difficulty in cancer treatment is that some cancer cells cannot be eliminated and these cells proliferate, leading to tumor recuurrence (151). Chemotherapy and radiotherapy often cause apoptotic cell death (8,92,151). In fact, apoptosis is a physiological process in which apoptotic cells are cleared by phagocytes such as macrophages and DCs, which prevents the emergence of inflammation by inhibiting the release of proinflammatory cytokines such as IL-10 and TGF-β, from phagocytes (82). However, during the anticancer therapy, delayed clearance of apoptotic cells can lead to secondary necrosis, which can result in the release of proinflammatory cytokines and thus inflammation (8,82,151).
Another type of cell death caused by anti-cancer treatments, including chemotherapy and radiotherapy, is necrosis (8,9,10,82,92). The main landscape of necrotic cell death is the rupture of the cancer cell membrane and the release of damage-associated molecular pattern molecules (DAMPs), which engage distinct receptors present on the innate immune cells (8,9,10,92,151,152). Upon recognition, DAMPs activate DCs and promotes the engulfment of dying cells, thereby improving anti-tumor response and clinical consequences (82,92,152). Conversely, DAMPs released from dying cancer cells accumulate macrophages to promote cancer cell clearence and polarize tham to a M2 phenotype, which may contribute to immunosuppression and cancer cell resistance to anti-cancer therapy (82). Dying cancer cells also express lipid mediators, such as PGE2 and PAF, promoting the proliferation and survival of remaning cancer cells (8,9,10,152). Experimental trials have revealed that DAMPs are not only released from necrotic cells, but certain forms of programmed cell death can induce DAMPs relase and lead to immunogenic cell death (152). Distinct chemotherapeutic agents, such as anthracyclines, oxaliplatin, cyclophosphamide, mitochontrone, bortezomid and radiotherapy result in immunogenic cell death (ICD) of cancer cells, and stimulate the release of DAMPs from dying cells (153). Following ICD, DAMPs released as a result of ICD promote the activation of cytotoxic effectors, which play a crucial role in the therapy response (153). Chemotherapeutic agents exert anti-cancer influences by influencing main cellular biological processes, which are indispensable for the robust proliferation of tumor cells (153). In addition, anti-cancer drugs modulate the immune cell profile of the TME, by triggering immune reactions and promoting cytotoxic effectors (154,155). A few anti-cancer agents, such as cisplatin, can cause chronic inflammation through the release of some pro-inflammatory mediators, leading to angiogenesis, tumor progression and resistance to treatment (156,157).
Currently, radiotherapy has established an important part of conventional cancer treatments, as high dose of radiation kills cancer cells and reduces tumor size (158). Radiotherapy activates transcription factors, including NF-κB and STAT, which generate multiple radioresistance signals through modulating anti-apoptotic pathways (159). Radiotherapy-induced NF-κB activation contributes to the prevention of apoptosis and cell cycle arrest (159). Additionally, NF-κB activation also orchestrate the transcription of a myriad of genes regulating immunity, proliferation, invasion, and angiogenesis, which favor radiotherapy resistance (8,10,152). Experimental trials in some types of cancer have reported that drugs that suppress the biological effects of NF-κB, such as indomethacin and curcimin, enhance the radiosensitivity of cancer cells by accentuating radiotherapy-induced apoptosis (158). Irradiated tumor cells secrete many soluble molecules, including proinflammatory cytokines, that also have biological effects on non-irradiated cells (158,159). In addition, radiotherapy promotes the formation of a proinflammatory TME, by inducing the network between inflammatory pathways, ultimately leading to tumor cell death (158,160). Radiation-induced inflammation accentuates the adaptive antigen-specific immune response and tumor-host interaction is reshaped during radiotherapy, which contributes to the favorable clinical outcomes (159). Additionally, chronic inflammation induced by radiotherapy in the TME promotes to recruitment of immunosuppressive cells (TAMs, myeloid-derived cells, and regulatory T cells) (159). Radiotherapy can foster proliferation of cancer cell and tumor progression, thus paving the way for treatment resistance (152,161,162).
In some solid tumors, such as HCC, immune checkpoint inhibitors (ICIs) have been shown to significantly improve overall survival (151,163,164). However, immune-related tissue injury is common in patients with cancer receiving ICIs, adversely affecting clinical outcomes (165,166). Although immune-related tissue injury can affect any organ, it is most commonly observed in the skin, endocrine system, gastrointestinal system and liver. In randomized controlled trials (RCTs), the incidence of immune-related tissue injury has been reported to be between 1% and 15% (165,166,167). In particular, in patients with HCC receiving ICIs, the incidence of immune-related liver damage is directly related to the degree of underlying liver disease and the combined administration of molecularly targeted drugs (165,166). ICIs may cause inflammation due to lysis of tumor cells, especially in patients who achieve an objective radiological response (166,168). ICIs also led to increased T cell infiltration within the TME, resulting in overproduction of proinflammatory cytokines by these cells, thereby enhancing the inflammatory response (165,166). Cytokine release syndrome (CRS) is one of the most serious clinical toxicity of immunotherapies (168). Many studies have shown that CRS can develop not only during the use of immunotherapies in patients with cancer but also during the treatment of other diseases, where CRS is accompanied by severe inflammation (168). Recently, two prospective studies of patients with advanced HCC who received ICIs reported that patients with high baseline AFP levels had a higher frequency of immune-related liver injury and significantly higher levels of inflammartion markers than patients with normal AFP levels (166). Although ICIs have provided significant progress in cancer treatment, they fail in a significant proportion of cancer patients due to their side effects and the development of treatment resistance (165,166,167,168). The main reason for resistance to immunotherapies is the inflammatory TME, where chronic inflammation is the fundamental driver of cancer cell proliferation, angiogenesis, and recruitment of immunosuppressive cell populations (151,152,165,166). The fact that immunotherapies have fatal side effects such as cytokine storm and coagulopathy limit the indication field of these treatments (169,170).
Conclusions and Future Perspectives
Emerging evidence indicates that chronic inflammation is an important driver in carcinogenesis and tumor progression, as well as response to treatment. Recent molecular trials have reported that pro-tumorigenic inflammatory pathways are orchestrated by intrinsic mechanisms of tumor cells. In addition to the genetic makeup of the cancer cell, epigenetic alterations and activation of intracellular signaling pathways create an inflammatory TME, which usually emerges before carcinogenesis but may develop late in carcinogenesis in some tumors. The TME consists of tumor cells, immune cells, stromal cells and pro-inflammatory cytokines and chemokines. Chronic inflammation acts as a tumor promoter by boosting cell survival, proliferation, and angiogenesis during tumorigenesis and tumor progression. In addition, chronic inflammation is involved in carcinogenesis by activating NF-κB and STAT signaling pathways. Changes in driver oncogenes can contribute to establishing an inflammatory microenvironment to promote the initiation of carcinogenesis. Currently, the molecular mechanisms, immune cells and soluble molecules involved in the pathogenesis of tumor-associated inflammation have been largely elucidated. In the vast majority of tumors, inflammation exerts dual effects; normally, immune cells inhibit tumor growth by eradicating tumor cells, on the other hand, in inflammatory tumors, some immune cells, inflammatory cells and molecules promote cancer growth by boosting cancer cell proliferation and prolonging its survival
The TME, of which chronic inflammation is the substantial component, plays a critical role in orchestrating cancer cell progression, metastasis, immune evasion and drug ressitance. A significant number of cancer patients receiving ICIs emerge primary or acquired drug resistance. The interaction between cancer cells and stromal cells in the TME can alter the cellular composition and soluble molecule landscape of the TME and may predispose to the development of drug resistance. Therefore, it is necessary to elucidate this complex crosstalk between cancer cells and immune cells, particularly inflammatory cells to develop effective anti-cancer drugs and improve the overall survival of cancer patients. Considering all of these developments, it can be suggested that treatment of cancer-associated inflammation, elimination of immunosuppressive landscape of the TME and repolarization of terminal exhousted CD8+ T cells into effector CD8+ T cells are necessary for the anti-cancer therapies to be succesful in GI cancers.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| GI | gastrointestinal |
| TME | tumor microenvironment |
| CRC | colorectal cancer |
| IBD | inflammatory bowel disease |
| MDSCs | myeloid-derived immunosuppressive cells |
| DCs | dendritic cells |
| KCs | Kupffer cells |
| PRRs | pattern recognition receptors |
| PAMPs | pathogen-associated molecular patterns |
| EGF | epidermal growth factor |
| ROS | reactive oxygen species |
| CAC | colitis-associated colorectal cancer |
| NAFLD | nonalcoholic fatty liver disease |
| HCC | hepatocellular carcinoma |
| ER | endoplasmic reticulum |
| DAMPs | damage-associated molecular patterns |
| UDP | uridine 5’-diphosphate |
| LSECs | liver sinusoidal endothelial cells |
| HSCS | hepatic stellate cells |
| MAMPs | microorganism-associated molecular patterns |
| LPS | lipopolysaccharide |
| TAMs | Tumor-associated macrophages |
| TRMs | Tissue-resident macrophages |
| NETs | neutrophil extracellular traps |
| HIF | hypoxia-inducible factor |
| ICAFs | inflammatory phenotype cancer-associated fibroblasts |
| MAPK | mitogen-activated protein kinase |
| MDSCs | Myeloid-derived suppressor cells |
| TATE | tumor-associated tissue eosinophils |
| CSF1 | macrophage colony-stimulating factor 1 |
| NKT | Natural Killer T |
| TAAs | tumor-associated antigens |
| TCRs | T cell receptors |
| PAF | platelet activation factor |
| DAMPs | damage-associated molecular pattern molecules |
| ICD | immunogenic cell death |
| ICIs | immune checkpoint inhibitors |
| RCT | randomized controlled trials |
| CRS | Cytokine release syndrome |
References
- Liu, X.; Yin, L.; Shen, S.; Hou, Y. Inflammation and cancer: paradoxical roles in tumorigenesis and implications in immunotherapies. Genes Dis. 2023, 10, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Sohrab, S.S.; Raj, R.; Nagar, A.; Hawthorne, S.; Paiva-Santos, A.C.; Kamal, M.A.; El-Daly, M.M.; Azhar, E.I.; Sharma, A. Chronic Inflammation’s Transformation to Cancer: A Nanotherapeutic Paradigm. Molecules 2023, 28, 4413. [Google Scholar] [CrossRef] [PubMed]
- Afify, S.M.; Hassan, G.; Seno, A.; Seno, M. Cancer-inducing niche: the force of chronic inflammation. Br. J. Cancer 2022, 127, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Zhuo, Y.; Zhang, X.; Li, Y.; Li, Y.; Duan, X.; Shi, J.; Xu, C.; Gao, Y.; Yu, Z. Tumor Microenvironment in Hepatocellular Carcinoma: Key Players for Immunotherapy. J. Hepatocell. Carcinoma 2022, 9, 1109–1125. [Google Scholar] [CrossRef]
- Baker, D.J.; Arany, Z.; Baur, J.A.; Epstein, J.A.; June, C.H. CAR T therapy beyond cancer: the evolution of a living drug. Nature 2023, 619, 707–715. [Google Scholar] [CrossRef]
- Albelda, S.M. CAR T cell therapy for patients with solid tumours: key lessons to learn and unlearn. Nat. Rev. Clin. Oncol. 2024, 21, 47–66. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Zhu, Y.; Zhang, C.; Yang, X.; Gao, Y.; Li, M.; Yang, H.; Liu, T.; Tang, H. Chronic inflammation, cancer development and immunotherapy. Front. Pharmacol. 2022, 13, 1040163. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Diakos, C.I.; Charles, K.A.; McMillan, D.C.; Clarke, S.J. Cancer-related inflammation and treatment effectiveness. Lancet Oncol. 2014, 15, e493–e503. [Google Scholar] [CrossRef]
- Shalapour, S.; Karin, M. Pas de Deux: Control of Anti-tumor Immunity by Cancer-Associated Inflammation. Immunity 2019, 51, 15–26. [Google Scholar] [CrossRef]
- Zhao H, Wu, Yan G, Chen Y, Zhou M, Wu Y and Li Y. Inflammation and tumor progression: signaling pathways and targeted intervention.
- Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet 2001;357(9255):539-545.
- Zhou, R.W.; Harpaz, N.; Itzkowitz, S.H.; Parsons, R.E. Molecular mechanisms in colitis-associated colorectal cancer. Oncogenesis 2023, 12, 48. [Google Scholar] [CrossRef] [PubMed]
- Chatila, W.K.; Walch, H.; Hechtman, J.F.; Moyer, S.M.; Sgambati, V.; Faleck, D.M.; Srivastava, A.; Tang, L.; Benhamida, J.; Ismailgeci, D.; et al. Integrated clinical and genomic analysis identifies driver events and molecular evolution of colitis-associated cancers. Nat. Commun. 2023, 14, 110. [Google Scholar] [CrossRef] [PubMed]
- Yu L-X, Ling Y and Wang H-Y. Role of unresolving inflammation in hepatocellular carcinoma development and progression. Precision Oncology 2018;2: 1-12.
- Galun, E. Liver inflammation and cancer: The role of tissue microenvironment in generating the tumor-promoting niche (TPN) in the development of hepatocellular carcinoma. Hepatology 2015, 63, 354–356. [Google Scholar] [CrossRef] [PubMed]
- Malfertheiner P, Camorgo MC, El-Omar E, Lio J-M, Peek R, Schulz C, Smith SI, and Suerbaum S. Helicobacter pylori infection Nature Reviews disease primers 2023;9: 19.
- Wellenstein, M.D.; de Visser, K.E. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 2018, 48, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, C.F.A.; Taranto, D.; Ando-Kuri, M.; de Groot, M.H.P.; Tsouri, E.; Huang, Z.; de Groot, D.; Kluin, R.J.C.; Kloosterman, D.J.; Verheij, J.; et al. Cancer cell genetics shaping of the tumor microenvironment reveals myeloid cell-centric exploitable vulnerabilities in hepatocellular carcinoma. Nat. Commun. 2024, 15, 2581. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Nigam, M.; Mishra, A.P.; Deb, V.K.; Dimri, D.B.; Tiwari, V.; Bungau, S.G.; Bungau, A.F.; Radu, A.-F. Evaluation of the association of chronic inflammation and cancer: Insights and implications. Biomed. Pharmacother. 2023, 164, 115015. [Google Scholar] [CrossRef]
- Hu, W.; Pasare, C. Location, location, location: tissue-specific regulation of immune responses. J. Leukoc. Biol. 2013, 94, 409–421. [Google Scholar] [CrossRef]
- Rivera, A.; Siracusa, M.C.; Yap, G.S.; Gause, W.C. Innate cell communication kick-starts pathogen-specific immunity. Nat. Immunol. 2016, 17, 356–363. [Google Scholar] [CrossRef]
- Watanabe, S.; Alexander, M.; Misharin, A.V.; Budinger, G.S. The role of macrophages in the resolution of inflammation. J. Clin. Investig. 2019, 129, 2619–2628. [Google Scholar] [CrossRef] [PubMed]
- Bulfone-Paus, S.; Nilsson, G.; Draber, P.; Blank, U.; Levi-Schaffer, F. Positive and Negative Signals in Mast Cell Activation. Trends Immunol. 2017, 38, 657–667. [Google Scholar] [CrossRef]
- Wright, H.L.; Moots, R.J.; Bucknall, R.C.; Edwards, S.W. Neutrophil function in inflammation and inflammatory diseases. Rheumatology 2010, 49, 1618–1631. [Google Scholar] [CrossRef]
- Kumar, R.; Clermont, G.; Vodovotz, Y.; Chow, C.C. The dynamics of acute inflammation. J. Theor. Biol. 2004, 230, 145–155. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Savill, J.S.; Wyllie, A.H.; Henson, J.E.; Walport, M.J.; Henson, P.M.; Haslett, C. Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J. Clin. Investig. 1989, 83, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.G.; Sawatzky, D.A.; Walker, A.; Ward, C.; Sheldrake, T.A.; Riley, N.A.; Caldicott, A.; Martinez-Losa, M.; Walker, T.R.; Duffin, R.; et al. Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat. Med. 2006, 12, 1056–1064. [Google Scholar] [CrossRef]
- Gilroy, D.W.; Lawrence, T.; Perretti, M.; Rossi, A.G. Inflammatory Resolution: new opportunities for drug discovery. Nat. Rev. Drug Discov. 2004, 3, 401–416. [Google Scholar] [CrossRef]
- Serhan, C.N. The resolution of inflammation: the devil in the flask and in the details. FASEB J. 2011, 25, 1441–1448. [Google Scholar] [CrossRef]
- Yang YM, Kim SY & Seki E. Inflammation and liver cancer: Molecular mechanisms and thearpeutic targets. Semin Liver Dis 2019;39: 26-42.
- van Weverwijk’ A & de Visser, E. Mechanisms driving the immunoregulatory function of cancer cells. Nature reviews cancer 2023;23: 193-215.
- Pacifico L, Anania C, Osborn JF, F Ferraro, C Chiesa. Consequences of Helicobacter pylori infection in children. World J Gastroenterol 2010;16(41): 5181-5194.
- Gonzalez, C.A. & Lopez-Carrillo, L. Helicobacter pylori, nutrition and smoking interaction: their impact in gastric carcinogenesis. Scand J. Gastroenterol 2010;45: 6-14.
- Kumar, S.; Metz, D.C.; Ellenberg, S.; Kaplan, D.E.; Goldberg, D.S. Risk Factors and Incidence of Gastric Cancer After Detection of Helicobacter pylori Infection: A Large Cohort Study. Gastroenterology 2020, 158, 527–536.e7. [Google Scholar] [CrossRef]
- Huang, L.C.; Merchea, A. Dysplasia and Cancer in Inflammatory Bowel Disease. Surg. Clin. North Am. 2017, 97, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Brindley PJ, Bachini M, Ilyas SI, Khan SA, Loukos A, Sirica AE, Bin Tean Teh, Sopit Wongkham & Gregory J. Gores. Cholangiocarcinoma. Nat Rev Dis Primers 2021;65: 1-17.
- Olafsson S, Mcintyre RE, Coorens T, Butler T, Jung H, Robinson PS, Henry Lee-Six, Mathijs A. Sanders, Kenneth Arestang, Claire Dawson. Somatic Evoluation in Non-neoplastic IBD-affected Colon. Cell 2020;182: 672-684 e611.
- de Krijger, M.; Carvalho, B.; Rausch, C.; Bolijn, A.S.; Diemen, P.M.D.-V.; Tijssen, M.; van Engeland, M.; Mostafavi, N.; Bogie, R.M.M.; Dekker, E.; et al. Genetic Profiling of Colorectal Carcinomas of Patients with Primary Sclerosing Cholangitis and Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2022, 28, 1309–1320. [Google Scholar] [CrossRef]
- Robles, A.I. , G Traverso, M Zhang, NJ Roberts, MA Khan, C Joseph, GY Lauwers, FM Selaru, Maria Popoli, Meredith E. P. et al. Whole-exome sequencing analysis of inflammatory bowel disease-associated colorectal cancers. Gastroenterology 2016;150: 931-943.
- Yaeger, R. , MA Shah, VA Miller, JR Kelsen, K Wang, ZJ Heins, JS Ross, Y He, E Sanford, Rhonda K. Y., et al. Genomic alterations observed in colitis-associated colorectal cancers and vary by type of inflammatory bowel disease. Gastroenterology 2016;151: 278-287, e6.
- Hanna DN, Smith PM, Novitskly SV, Washington MK, Zi J, Weaver CJ, Jalal A. Hamaamen, Keeli B. Lewis, Jing Zhu, Jing Yang, et al. SMAD4 Suppresses Colitis-associated Carcinoma Through Inhibition of CCL20/CCR6-mediated Inflammation. Gastroenterology 2022;163: 1334-1350.
- Liovet, J.M. Robin Kate Kelley, Augusto Villanueva, Amit G. Singal, Eli Pikarsky, Sasan Roayaie, Riccardo Lencioni, Kazuhiko Koike, Jessica Zucman-Rossi & Richard S. Finn. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2021;7: 6.
- Ringelhan, M.; Pfister, D.; O’connor, T.; Pikarsky, E.; Heikenwalder, M. The immunology of hepatocellular carcinoma. Nat. Immunol. 2018, 19, 222–232. [Google Scholar] [CrossRef]
- Sharma, S.; Le Guillou, D.; Chen, J.Y. Cellular stress in the pathogenesis of nonalcoholic steatohepatitis and liver fibrosis. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 662–678. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Willoughby, C.E.; Singal, A.G.; Greten, T.F.; Heikenwälder, M.; El-Serag, H.B.; Finn, R.S.; Friedman, S.L. Nonalcoholic steatohepatitis-related hepatocellular carcinoma: pathogenesis and treatment. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 487–503. [Google Scholar] [CrossRef]
- Akkiz, H.; Taskin, E.; Karaogullarindan, U.; Delik, A.; Kuran, S.; Kutlu, O. The influence of RS738409 I148M polymorphism of patatin-like phospholipase domain containing 3 gene on the susceptibility of non-alcoholic fatty liver disease. Medicine 2021, 100, e25893. [Google Scholar] [CrossRef] [PubMed]
- Hammerich, L.; Tacke, F. Hepatic inflammatory responses in liver fibrosis. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 633–646. [Google Scholar] [CrossRef]
- Pinyol R, Torrecilla S, Wang H, Montironi C, Pique-Gilli M, Torres-Martin M, Wei-Qiang L, Willoughby CE, Ramadori P, Andreu-Oller C, et al. Molecular characterization of hepatocellular carcinoma in patients with non-alcoholic steatohepatitis. J Hepatol 2021;75: 865-878.
- Foerster, F.; Gairing, S.J.; Müller, L.; Galle, P.R. NAFLD-driven HCC: Safety and efficacy of current and emerging treatment options. J. Hepatol. 2021, 76, 446–457. [Google Scholar] [CrossRef]
- Maderacke I, A Filliol, S Affo, A Nair, C Hernandez, Q Sun, F Hamberger, F Brundu, Y Chen, Aashreya Ravichandra, et al. the purinergic P2Y14 receptorlinks hepatocyte death to hepatic stellate cell activation and fibrogenesis in the liver. Sci. Transl Med 2022;14: eab5795.
- Knorr, J.; Wree, A.; Tacke, F.; Feldstein, A.E. The NLRP3 Inflammasome in Alcoholic and Nonalcoholic Steatohepatitis. Semin. Liver Dis. 2020, 40, 298–306. [Google Scholar] [CrossRef]
- Mchedlidze, T.; Waldner, M.; Zopf, S.; Walker, J.; Rankin, A.L.; Schuchmann, M.; Voehringer, D.; McKenzie, A.N.J.; Neurath, M.F.; Pflanz, S.; et al. Interleukin-33-Dependent Innate Lymphoid Cells Mediate Hepatic Fibrosis. Immunity 2013, 39, 357–371. [Google Scholar] [CrossRef]
- Sica A & Lazzeri, M. Specialization determines outcomes in inflammation and cancer. Nat Immunol 2023;24: 1399-1401.
- Hsu CL & Schnabi, B. The gut-liver axis and gut microbiota in health and liver disease. Nat. Rev. Microbiol 2023;21: 719-733.
- Fan Y & Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2021;19: 56-71.
- Schulthess J, S Pandey, M Capitani, KC Rue-Albrecht, I Arnold, F Franchini, A Chomka, Nicholas E. Ilott, Daniel G.W. Johnston, Elisabete Pires, et al. The short chain fatty acid butyrate imprints an antimicrobialprogram in macrophages. Immunity 2019;50: 432-445.e7.
- Schwabe RF & Greten, TF. Gut microbiome in HCC – Mechansisms, diagnosis and therapy. J Hepatol 2020;72: 230-238.
- Luther, J.; Garber, J.J.; Khalili, H.; Dave, M.; Bale, S.S.; Jindal, R.; Motola, D.L.; Luther, S.; Bohr, S.; Jeoung, S.W.; et al. Hepatic Injury in Nonalcoholic Steatohepatitis Contributes to Altered Intestinal Permeability. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 222–232.e2. [Google Scholar] [CrossRef] [PubMed]
- Chang CS, Chen GH, Lien HC & Yeh HZ. Small intestine dysmotility and bacterial overgrowth in cirrhotic patients with spontenous bacterial peritonitis. Hepatology 1998;28: 1187-1190.
- Kakiyama, G.; Pandak, W.M.; Gillevet, P.M.; Hylemon, P.B.; Heuman, D.M.; Daita, K.; Takei, H.; Muto, A.; Nittono, H.; Ridlon, J.M.; et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J. Hepatol. 2013, 58, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Li, A.; Jiang, J.; Zhou, L.; Yu, Z.; Lu, H.; Xie, H.; Chen, X.; Shao, L.; Zhang, R.; et al. Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut 2018, 68, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Han, M.; Heinrich, B.; Fu, Q.; Zhang, Q.; Sandhu, M.; Agdashian, D.; Terabe, M.; Berzofsky, J.A.; Fako, V.; et al. Gut microbiome–mediated bile acid metabolism regulates liver cancer via NKT cells. Science 2018, 360. [Google Scholar] [CrossRef]
- Yu, L.-X.; Yan, H.-X.; Liu, Q.; Yang, W.; Wu, H.-P.; Dong, W.; Tang, L.; Lin, Y.; He, Y.-Q.; Zou, S.-S.; et al. Endotoxin Accumulation Prevents Carcinogen-Induced Apoptosis and Promotes Liver Tumorigenesis in Rodents. Hepatology 2010, 52, 1322–1333. [Google Scholar] [CrossRef]
- Komarova, E.A.; Krivokrysenko, V.; Wang, K.; Neznanov, N.; Chernov, M.V.; Komarov, P.G.; Brennan, M.-L.; Golovkina, T.V.; Rokhlin, O.W.; Kuprash, D.V.; et al. p53 is a suppressor of inflammatory response in mice. FASEB J. 2005, 19, 1030–1032. [Google Scholar] [CrossRef] [PubMed]
- Schwitalla S, Ziegler PK, Horst D, Becker V, Kerle I, Begus-Nahrmann Y, Lechel A, Rudolph KL, Langer R, Slotta-Huspenina J, et al. loss of p53 in eneterocytes generates an inflammatory microenvironment enabling invasion and lymph node metastasis of carcinogen-induced colorectal tumors. Cancer Cell 2013;23: 93-106.
- Elyada E, Pribluda A, Goldstein RE, Morgenstern Y, Brachya G, Cojocaru G, Snir-Alkalay I, Burstain I, Haffner-Knausz R, Jung S, et al. CKIα ablation highlights a critical role for p53 in invasiveness control. Nature 2011;470: 409-413.
- Kadariya Y, Menges CW, Talarcheck J, Cai KQ, Klein-Szanto AJ, Pletrotesa RA, Christofidou-Solomidou M, Cheung M, Mossman BT, Shuida A and Testa JR. Inflammation-Related IL1β/IL1RSignalin Promotes the Development of Asbestos-induced Malignant Mesothelioma. Cancer Prev Res 2016;9: 406-414.
- Takahashi H, Ogata H, Nishigaki R, Broide H and Karin M. Tobacco smoke promotes lung tumorigenesis by triggering IKKbeta- and JNK1-dependent inflammation. Cancer Cell 2010;17: 89-97.
- Ma QY, Huang DY, Zhang HJ, S Wang, XF Chen. Exposure to particulate matter 2.5 (PM2.5) induced by increased macrophage-dependent inflammation, characterized by increased Th1/Th2 cytokine secretion and cytotoxicity. Int Immunopharm 2017;50: 139-145.
- Ploeger HE, Takken T, de Greef MH & Timmons BW. The feccets of acute and chronic exercise on inflammatory markers on in children and adults with a chronic inflammatory disease: a systematic review. Exerc Immunol Rev 2009;15: 6-41.
- Boyle M, C Chun, C Strojny, R Narayanan, A Bartholomew, P Sundivakkam, S Alapatie. Chronic inflammation and angiogenic signaling axis impairs differentiation of dental-pump stem cells. PLoS ONE 2014;9: e113419.
- Dewald O, P Zymek, K Winkelmann, A Koerting, G Ren, T Abou-Khamis, LH Michael, Barrett J. Rollins, Mark L. Entman, and Nikolaos G. Frangogiannis. CCL2/monocyte chemoattractant protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res 2005;96: 881-889.
- Frantz, S.; Hofmann, U.; Fraccarollo, D.; Schäfer, A.; Kranepuhl, S.; Hagedorn, I.; Nieswandt, B.; Nahrendorf, M.; Wagner, H.; Bayer, B.; et al. Monocytes/macrophages prevent healing defects and left ventricular thrombus formation after myocardial infarction. FASEB J. 2012, 27, 871–881. [Google Scholar] [CrossRef]
- Zhao Y, Zou W, Du J & Zhao Y. the origins and homeostasis of monocytes and tissue-resident macrophages in physiological situation. J Cell Physiol 2018;233: 6425-6439.
- Bied M, Ho WW & Bieriot C. Roles of macrophages in tumor development: a spatiotemporal perspective. Cellular & Molecular Immunology 2023;20: 983-992.
- Kennedy, L. Tilting the Scales: Sirtuin 1 Favors Proinflammatory Macrophage Response Via Inflammasome Signaling and Metabolic Reprogramming. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 1261–1262. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Marchesi, F.; Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 2022, 21, 799–820. [Google Scholar] [CrossRef]
- Bleriot C, Dunsmore G, Alanso-Curbela D & Ginhoux F. A temporal perpective for tumor-associated macrophage identities and functions. Cancer Cell 2024;42: 747-758.
- Fredman, G.; Serhan, C.N. Specialized proresolving mediator targets for RvE1 and RvD1 in peripheral blood and mechanisms of resolution. Biochem. J. 2011, 437, 185–197. [Google Scholar] [CrossRef]
- Williams, M.R.; Azcutia, V.; Newton, G.; Alcaide, P.; Luscinskas, F.W. Emerging mechanisms of neutrophil recruitment across endothelium. Trends Immunol. 2011, 32, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C. Neutrophils and immunity: challenges and opportunities. Nat. Rev. Immunol. 2006, 6, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Blokhuis, B.R.; Garssen, J.; Redegeld, F.A. Non-IgE mediated mast cell activation. Eur. J. Pharmacol. 2016, 778, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Dudeck, A.; Dudeck, J.; Scholten, J.; Petzold, A.; Surianarayanan, S.; Köhler, A.; Peschke, K.; Vöhringer, D.; Waskow, C.; Krieg, T.; et al. Mast Cells Are Key Promoters of Contact Allergy that Mediate the Adjuvant Effects of Haptens. Immunity 2011, 34, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Schuster S, Hurrel B & Tacchini-Cottler F. Crosstalk between neutrophils and dendritic cells: a context-dependent process. J Leukoc Biol 2013;94: 671-675.
- Angeli V & Rudolph, GJ. Inflammation, lymphatic function, and dendritic cell migration. Lymphat Res Biol 2006;4: 217-228.
- Wu, Y.; Antony, S.; Meitzler, J.L.; Doroshow, J.H. Molecular mechanisms underlying chronic inflammation-associated cancers. Cancer Lett. 2013, 345, 164–173. [Google Scholar] [CrossRef]
- Crusz, S.M.; Balkwill, F.R. Inflammation and cancer: advances and new agents. Nat. Rev. Clin. Oncol. 2015, 12, 584–596. [Google Scholar] [CrossRef]
- Ghorani, E.; Swanton, C.; Quezada, S.A. Cancer cell-intrinsic mechanisms driving acquired immune tolerance. Immunity 2023, 56, 2270–2295. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, T.; Su, W.; Dou, Z.; Zhao, D.; Jin, X.; Lei, H.; Wang, J.; Xie, X.; Cheng, B.; et al. Mutant p53 in cancer: from molecular mechanism to therapeutic modulation. Cell Death Dis. 2022, 13, 1–14. [Google Scholar] [CrossRef]
- Singhal, A.; Li, B.T.; O’reilly, E.M. Targeting KRAS in cancer. Nat. Med. 2024, 30, 969–983. [Google Scholar] [CrossRef]
- Dhanasekaran, R.; Deutzmann, A.; Mahauad-Fernandez, W.D.; Hansen, A.S.; Gouw, A.M.; Felsher, D.W. The MYC oncogene — the grand orchestrator of cancer growth and immune evasion. Nat. Rev. Clin. Oncol. 2021, 19, 23–36. [Google Scholar] [CrossRef]
- Karin, M. Nuclear factor-kappaB in cancer development and progression. Nature 2006;441: 431-436.
- Szlosarek PW & Balkwill, FR. Tumor necrosis factor alpha: a potential target for the therapy of solid tumors. Lancet Oncol 2003;4: 565-573.
- Parrales, A.; Iwakuma, T. Targeting Oncogenic Mutant p53 for Cancer Therapy. Front. Oncol. 2015, 5, 288. [Google Scholar] [CrossRef]
- Dittmer, D.; Pati, S.; Zambetti, G.; Chu, S.; Teresky, A.K.; Moore, M.; Finlay, C.; Levine, A.J. Gain of function mutations in p53. Nat. Genet. 1993, 4, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Akkari, L.; Simon, J.; Grace, D.; Tschaharganeh, D.F.; Bolden, J.E.; Zhao, Z.; Thapar, V.; Joyce, J.A.; Krizhanovsky, V.; et al. Non-Cell-Autonomous Tumor Suppression by p53. Cell 2013, 153, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Cooks, T.; Pateras, I.S.; Jenkins, L.M.; Patel, K.M.; Robles, A.I.; Morris, J.; Forshew, T.; Appella, E.; Gorgoulis, V.G.; Harris, C.C. Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR-1246. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, L.; Hyland, J.K.; Watt, R.; Rosenberg, M.; Baserga, R. Microinjected c- myc as a Competence Factor. Science 1985, 228, 1313–1315. [Google Scholar] [CrossRef]
- Karn, J.; Watson, J.; Lowe, A.; Green, S.; Vedeckis, W. Regulation of cell-cycle duration by c-myc levels. 1989, 4, 773–787.
- Jain, M.; Arvanitis, C.; Chu, K.; Dewey, W.; Leonhardt, E.; Trinh, M.; Sundberg, C.D.; Bishop, J.M.; Felsher, D.W. Sustained Loss of a Neoplastic Phenotype by Brief Inactivation of MYC. Science 2002, 297, 102–104. [Google Scholar] [CrossRef]
- Feisher DW & Bishop, JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol. Cell. 1999;4: 199-207.
- Wu, C.-H.; van Riggelen, J.; Yetil, A.; Fan, A.C.; Bachireddy, P.; Felsher, D.W. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc. Natl. Acad. Sci. 2007, 104, 13028–13033. [Google Scholar] [CrossRef]
- Baudino, T.A.; McKay, C.; Pendeville-Samain, H.; Nilsson, J.A.; Maclean, K.H.; White, E.L.; Davis, A.C.; Ihle, J.N.; Cleveland, J.L. c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev. 2002, 16, 2530–2543. [Google Scholar] [CrossRef]
- Dews M, Jamie L Fox, Stacy Hultine, Prema Sundaram, Wenge Wang, Yingqiu Y Liu, Emma Furth, Gregory H Enders, Wafik El-Deiry, Janell M Schelter. The Myc-miR-17-92 axis blunts TGFβ-dependent antiangiogenic factors. Cancer Res 2010;70: 8233-8246.
- Kortlever, R.; Sodir, N.; Wilson, C.; Burkhart, D.L.; Pellegrinet, L.; Swigart, L.B.; Littlewood, T.; Evan, G. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 2017;171: 1301-1315.e14. [CrossRef]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gütgemann, I.; Eilers, M.; et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef]
- Swaminathan, S.; Hansen, A.S.; Heftdal, L.D.; Dhanasekaran, R.; Deutzmann, A.; Fernandez, W.D.M.; Liefwalker, D.F.; Horton, C.; Mosley, A.; Liebersbach, M.; et al. MYC functions as a switch for natural killer cell-mediated immune surveillance of lymphoid malignancies. Nat. Commun. 2020, 11, 2860. [Google Scholar] [CrossRef]
- Ancrile, B.; Lim, K.-H.; Counter, C.M. Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev. 2007, 21, 1714–1719. [Google Scholar] [CrossRef] [PubMed]
- Pylayeva-Gupta, Y.; Lee, K.E.; Hajdu, C.H.; Miller, G.; Bar-Sagi, D. Oncogenic Kras-Induced GM-CSF Production Promotes the Development of Pancreatic Neoplasia. Cancer Cell 2012, 21, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Cohen, B.; Zheng, W.; Rahbar, R.; Martin, B.; Murakami, K.; Lamorte, S.; Thompson, P.; Berman, H.; Zúñiga-Pflücker, J.C.; et al. Notch Shapes the Innate Immunophenotype in Breast Cancer. Cancer Discov. 2017, 7, 1320–1335. [Google Scholar] [CrossRef] [PubMed]
- Hoare M, Ito Y, Kang TW, Weekes MP, Matheson NJ, Patten DA, Shetty S, Parry AJ, Menon S, Salama R, et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat. Cell. Biol. 2016;18: 979-992.
- Koyama S, Akbay EA, Li YY, Aref AR, Skoulidis F, Herter-Sprie GS, Buczkowski KA, liu Y, Awad MM, Denning WL, et al. STK11/LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-cell Activity in the Lung Microenvironment. Cancer Res. 2016;76: 3928-3937.
- Ying H, Elpek KG, Vinjamoori A, Zimmerman SM, Chu GC, Yan H, Fietcher-Sananikone E, Zhang H, Liu Y, Wang W, et al. PTEN is a major tumor suppressor gene in pancreatic ductal adenocarcinoma and regulates an NF-κB cytokine network. Cancer Discov 2011;1: 158-169.
- Kao K-C, Vilbois S, Tsai C-H & Ho P-C. Metabolic communication in the tumor-immune microenvironment. Nat. Microbiol. 2022;24: 1574-1583.
- Courvan EMC & Parker, RR. Hypoxia and inflammation induce synergistic transcriptome turnover in macropgahes. Cell Reports 2024;43: 114452.
- Mello, A.M.; Ngodup, T.; Lee, Y.; Donahue, K.L.; Li, J.; Rao, A.; Carpenter, E.S.; Crawford, H.C.; di Magliano, M.P.; Lee, K.E. Hypoxia promotes an inflammatory phenotype of fibroblasts in pancreatic cancer. Oncogenesis 2022, 11, 56. [Google Scholar] [CrossRef]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef]
- Yu, Y.-R.; Imrichova, H.; Wang, H.; Chao, T.; Xiao, Z.; Gao, M.; Rincon-Restrepo, M.; Franco, F.; Genolet, R.; Cheng, W.-C.; et al. Disturbed mitochondrial dynamics in CD8+ TILs reinforce T cell exhaustion. Nat. Immunol. 2020, 21, 1540–1551. [Google Scholar] [CrossRef]
- Doedens, A.L.; Phan, A.T.; Stradner, M.H.; Fujimoto, J.K.; Nguyen, J.V.; Yang, E.; Johnson, R.S.; Goldrath, A.W. Hypoxia-inducible factors enhance the effector responses of CD8+ T cells to persistent antigen. Nat. Immunol. 2013, 14, 1173–1182. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef]
- Bauer, R.; Meyer, S.P.; Kloss, K.A.; Ruiz, V.M.G.; Reuscher, S.; Zhou, Y.; Fuhrmann, D.C.; Zarnack, K.; Schmid, T.; Brüne, B. Functional RNA Dynamics Are Progressively Governed by RNA Destabilization during the Adaptation to Chronic Hypoxia. Int. J. Mol. Sci. 2022, 23, 5824. [Google Scholar] [CrossRef]
- Grivennikov SI, Greten FR and Karin M. Immunity, inflammation, and cancer. Cell 2010;140: 883-899.
- Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabi B, Jauch D, Taniguchi K, Yu GY, Christoph H Osterreicher, Kenneth E Hung, et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumor growth. Nature 2012;491: 254-258.
- Akkız, H. Emerging role of cancer-associated fibroblast in the pathogenesis and treatment of hepatocellular carcinoma. Int J Mol Science 2023;24(4): 3941.
- Kim J & Bae, JS. Tumor-associated macrophages and neutrophils in tumor microenvironment. Mediators Inflamm 2016;2016: 6058147.
- Shu Y, Qin M, Song Y, Tang Q, Huang Y, Shen F, Yan Lu. M2 polarization of tumor-associated macrophages is dependent on integrin β3 via peroxisome proliferator-activated receptor-γ up-regulation in breast cancer. Immunology 2020;160: 345-356.
- Nicolás-Ávila, J.Á.; Adrover, J.M.; Hidalgo, A. Neutrophils in Homeostasis, Immunity, and Cancer. Immunity 2017, 46, 15–28. [Google Scholar] [CrossRef]
- Gleich GJ, Adolphson CR & Leiferman KM. The biology of the eosinophilic leukocyte. Annu Rev Med 1993;44: 85-101.
- Del Prete A, Salvi V, Sariani A, Lattranchi M, Sozio F, Bosisşo D & Sozzani S. Dendritic cell subsets in cancer immunity and tumor antigen sensing. Cellular and Molecular Immunology 2023;20: 432-447.
- López, L.; Morosi, L.G.; La Terza, F.; Bourdely, P.; Rospo, G.; Amadio, R.; Piperno, G.M.; Russo, V.; Volponi, C.; Vodret, S.; et al. Dendritic cell-targeted therapy expands CD8 T cell responses to bona-fide neoantigens in lung tumors. Nat. Commun. 2024, 15, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.E.; Pollard, J.W. Distinct Role of Macrophages in Different Tumor Microenvironments. Cancer Res. 2006, 66, 605–612. [Google Scholar] [CrossRef]
- Baer, J.M.; Zuo, C.; Kang, L.-I.; de la Lastra, A.A.; Borcherding, N.C.; Knolhoff, B.L.; Bogner, S.J.; Zhu, Y.; Yang, L.; Laurent, J.; et al. Fibrosis induced by resident macrophages has divergent roles in pancreas inflammatory injury and PDAC. Nat. Immunol. 2023, 24, 1443–1457. [Google Scholar] [CrossRef]
- Vonderlin J, Chavakis T, Sieweke M & Tackle F. The Multifaced Roles of Macrophages in NAFLD Pathogenesis. Cell Mol Gastroenterol Hepatol 2023;15: 1311-1324.
- Kumar V & Gabrilowich DIJI. Hypoxia-inducible factors in regulation of immune responses in tumour microenvironment. Immunology 2014;143: 512-519.
- Smith HA & Kang YJJ. The metastasis-promoting roles of tumor-associated immune cells. J Mol Med 2013;91: 411-429.
- Lopes, N.; Vivier, E.; Narni-Mancinelli, E. Natural killer cells and type 1 innate lymphoid cells in cancer. Semin. Immunol. 2023, 66, 101709. [Google Scholar] [CrossRef]
- Coënon, L.; Geindreau, M.; Ghiringhelli, F.; Villalba, M.; Bruchard, M. Natural Killer cells at the frontline in the fight against cancer. Cell Death Dis. 2024, 15, 614. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Wang, Y.; Su, M.; Huang, Y.; Zhang, Y.; Dou, J.; Zhao, C.; Cai, Y.; Pan, J.; Bai, S.; et al. Motility and tumor infiltration are key aspects of invariant natural killer T cell anti-tumor function. Nat. Commun. 2024, 15, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Thommen, D.S.; Schumacher, T.N. T Cell Dysfunction in Cancer. Cancer Cell 2018, 33, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Crespo, J.; Sun, H.; Welling, T.H.; Tian, Z.; Zou, W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 214–221. [Google Scholar] [CrossRef]
- Knutson, K.L.; Disis, M.L. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol. Immunother. 2005, 54, 721–728. [Google Scholar] [CrossRef]
- Koh C-H, Lee S, Kwak M, Kim B-S & Chung Y. CD8 T-cell subsets: heterogeneity, functions, and therapeutic potential. Experimental & Molecular Medicine 2023;55: 2287-2299.
- Seo, W.; Jerin, C.; Nishikawa, H. Transcriptional regulatory network for the establishment of CD8+ T cell exhaustion. Exp. Mol. Med. 2021, 53, 202–209. [Google Scholar] [CrossRef]
- Vignali, P.D.A.; DePeaux, K.; Watson, M.J.; Ye, C.; Ford, B.R.; Lontos, K.; McGaa, N.K.; Scharping, N.E.; Menk, A.V.; Robson, S.C.; et al. Hypoxia drives CD39-dependent suppressor function in exhausted T cells to limit antitumor immunity. Nat. Immunol. 2022, 24, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Chow, A.; Perica, K.; Klebanoff, C.A.; Wolchok, J.D. Clinical implications of T cell exhaustion for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2022, 19, 775–790. [Google Scholar] [CrossRef]
- Wouters MCA & Nelson, BH. Prognostic significance of tumor-infiltrating B cells and plasma cells in human cancer. Clin Cancer Res 2018;24: 6125-6135.
- Rauth, S.; Malafa, M.; Ponnusamy, M.P.; Batra, S.K. Emerging Trends in Gastrointestinal Cancer Targeted Therapies: Harnessing Tumor Microenvironment, Immune Factors, and Metabolomics Insights. Gastroenterology 2024, 167, 867–884. [Google Scholar] [CrossRef]
- Wang, M.; Chen, S.; He, X.; Yuan, Y.; Wei, X. Targeting inflammation as cancer therapy. J. Hematol. Oncol. 2024, 17, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic Cell Death in Cancer Therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Hirata, E.; Sahai, E. Tumor Microenvironment and Differential Responses to Therapy. Cold Spring Harb. Perspect. Med. 2017, 7, a026781. [Google Scholar] [CrossRef]
- Li H, Zhou L, Zhou J, Li Q & Ji Q. Underlying mechanisms and drug intervention strategies for the tumor microenvironment. J Exp Clin Cancer Res 2021;40: 97.
- Rivera G & Wakelee, HA. Resistance to therapy. Cancer Treat Res 2016;170: 183-202.
- Yao, X.; Panichpisal, K.; Kurtzman, N.; Nugent, K. Cisplatin nephrotoxicity: A review. Am. J. Med. Sci. 2007, 334, 115–124. [Google Scholar] [CrossRef]
- Jaffray, DA. Image-guided radiotherapy: from current concept to future perpectives. Nat Rev Clin Oncol 2012;9: 688-699.
- McLoughlin M, Emmanuel C Patin, Malin Pedersen, Anna Wilkins, Magnus T Dillon, Alan A Melcher, Kevin J Harrington. Inflammatory microenvironment remodeling by tumor cells after radiotherapy. Nat Rev Cancer 2020;20: 203-217.
- Schaue, D.; Kachikwu, E.L.; McBride, W.H. Cytokines in Radiobiological Responses: A Review. Radiat. Res. 2012, 178, 505–523. [Google Scholar] [CrossRef]
- Wu, C.-T.; Chen, M.-F.; Chen, W.-C.; Hsieh, C.-C. The role of IL-6 in the radiation response of prostate cancer. Radiat. Oncol. 2013, 8, 159–159. [Google Scholar] [CrossRef]
- Culig Z & Puhr, M. Interleukin-6: multifunctional targetable cytokine in human prostate cancer. Mol Cell Endocrinol 2012;360: 52-58.
- Li M, Bhoori S, Mehta N, Mazzaferro V. Immunotherapy for hepatocellular carcinoma: The next evoluation in expanding access to liver transplantation. J Hepatol 2024;81: 743-755.
- Rimassa, L.; Finn, R.S.; Sangro, B. Combination immunotherapy for hepatocellular carcinoma. J. Hepatol. 2023, 79, 506–515. [Google Scholar] [CrossRef]
- Sangro, B.; Melero, I.; Wadhawan, S.; Finn, R.S.; Abou-Alfa, G.K.; Cheng, A.-L.; Yau, T.; Furuse, J.; Park, J.-W.; Boyd, Z.; et al. Association of inflammatory biomarkers with clinical outcomes in nivolumab-treated patients with advanced hepatocellular carcinoma. J. Hepatol. 2020, 73, 1460–1469. [Google Scholar] [CrossRef] [PubMed]
- Celsa C, Cabibbo G, Fulgenzi CCA, Camma C, Cortellini A, Pinato DJ. Characteristics and outcomes of immunotherapy-related liver injury in patients with hepatocellular carcinoma versus other advanced solid tumors. H Hepatol 2024;80: 431-442.
- Min H-Y & Lee H-Y. Molecular targeted therapy for anticancer treatment. Experimental & Molecular Medicine 2022;54: 1670-1694.
- Meng L, Wu H, Wu J, Ding P, He J, Sang M, Liu L. Mechanims of immune checkpoint inhibitors: insights into the regulation of circular RNAs involved in cancer hallmarks. Cell Death & Disease 2024;15: 3.
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Tabrizian, P.; Abdelrahim, M.; Schwartz, M. Immunotherapy and transplantation for hepatocellular carcinoma. J. Hepatol. 2024, 80, 822–825. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Mechanisms connecting inflammation to cancer. During acute inflammatory process, upon uptake of tumor-associated antigens (TAAs), dendritic cells (DCs) can orchestrate the anti-tumor immunity by several mechanisms, such as presentation of TAAs and priming tumor-specific CD8+ T cells, polarization of immune cells towards tumor suppression, accumulating NK cells. In chronic inflammatory microenvironment, cancer cells inhibit TAA presentation by DCs as well as leading to accumulation of large numbers of immunosuppressive cells, such as MDSCs, Tregs, M2-TAMs, N2-TANs, and Th2 cells, through releasing various soluble molecules and inflammatory mediators. Subsequently, these immunosuppressive cells generate pro-angiogenic and pro-tumoral microenvironment, and mitigate anti-tumor activity.
Figure 1.
Mechanisms connecting inflammation to cancer. During acute inflammatory process, upon uptake of tumor-associated antigens (TAAs), dendritic cells (DCs) can orchestrate the anti-tumor immunity by several mechanisms, such as presentation of TAAs and priming tumor-specific CD8+ T cells, polarization of immune cells towards tumor suppression, accumulating NK cells. In chronic inflammatory microenvironment, cancer cells inhibit TAA presentation by DCs as well as leading to accumulation of large numbers of immunosuppressive cells, such as MDSCs, Tregs, M2-TAMs, N2-TANs, and Th2 cells, through releasing various soluble molecules and inflammatory mediators. Subsequently, these immunosuppressive cells generate pro-angiogenic and pro-tumoral microenvironment, and mitigate anti-tumor activity.
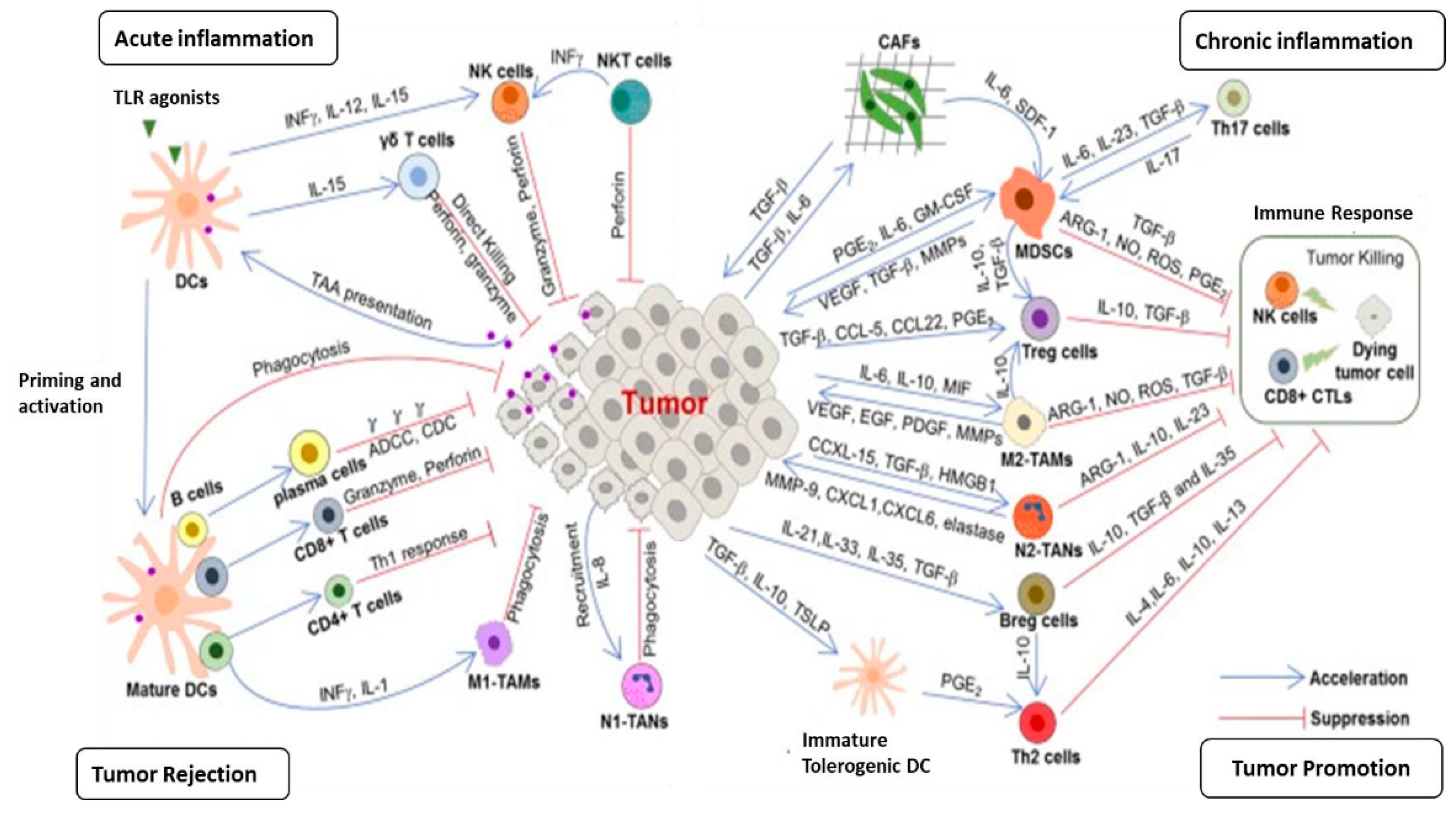
Figure 2.
Inflammation in cancer: mechanisms and effects on tumor biology. Stress, cell death, obesity and bacterial infections trigger the activation of innate immune cells and increase the secretion of inflammatory mediators, which promote the recruitment of adaptive immune cells to damaged tissue. DCs and other myeloid cells take up antigens and present them to CD8+ T cells, activating these cells. On the other hand, cell death has an immunosuppressive effect, thus inhibiting CD8+ T cell activation. Similarly, macrophages can impede the anti-tumor activity of CD8+ T cells by secreting IL-10 and TGF-β.
Figure 2.
Inflammation in cancer: mechanisms and effects on tumor biology. Stress, cell death, obesity and bacterial infections trigger the activation of innate immune cells and increase the secretion of inflammatory mediators, which promote the recruitment of adaptive immune cells to damaged tissue. DCs and other myeloid cells take up antigens and present them to CD8+ T cells, activating these cells. On the other hand, cell death has an immunosuppressive effect, thus inhibiting CD8+ T cell activation. Similarly, macrophages can impede the anti-tumor activity of CD8+ T cells by secreting IL-10 and TGF-β.
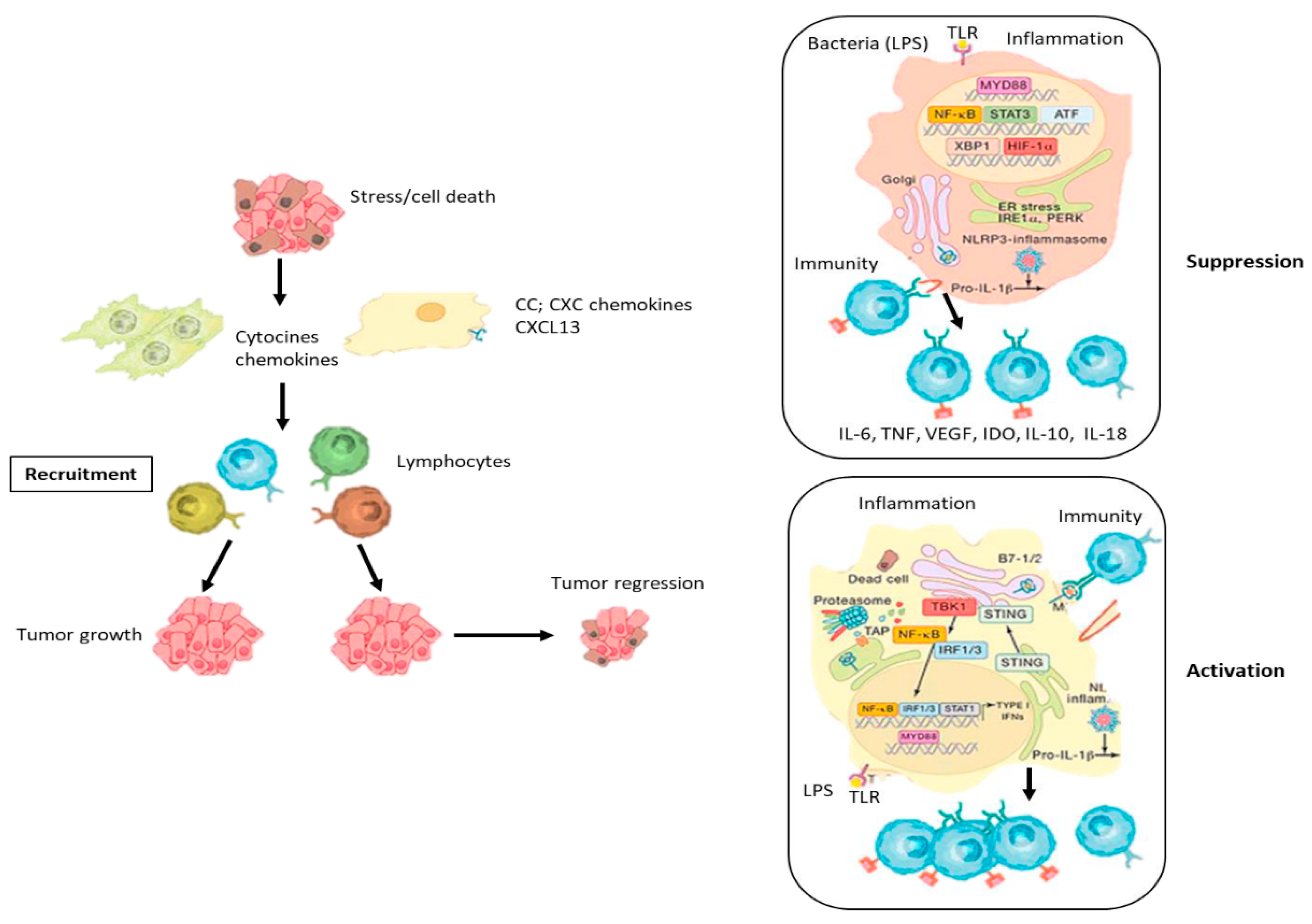
Figure 3.
Complex interaction between cancer cell and immune cells. Tumor and stromal cells secrete soluble molecules to recruit immunosuppressive cells, including TAMs and neutrophils, MDSCs, Dcs and T cells. These soluble molecules induce polarization of immune cells, such as M1 macrophages and N1 neutrophils, into cancer-promoting cells, including M2 macrophages and N2 neutrophils. Immunosuppressive TME impedes effector functions and proliferation capacity of CD8+ T cells, leading to terminal exhausted T cells, which produce high levels of immune checkpoint molecules, inculding PD-1 and PD-L1. Tumor-associated cells also promote the growth of tumor cells by expressing a number of growth factors, such as VEGF, EGF, and HGF.
Figure 3.
Complex interaction between cancer cell and immune cells. Tumor and stromal cells secrete soluble molecules to recruit immunosuppressive cells, including TAMs and neutrophils, MDSCs, Dcs and T cells. These soluble molecules induce polarization of immune cells, such as M1 macrophages and N1 neutrophils, into cancer-promoting cells, including M2 macrophages and N2 neutrophils. Immunosuppressive TME impedes effector functions and proliferation capacity of CD8+ T cells, leading to terminal exhausted T cells, which produce high levels of immune checkpoint molecules, inculding PD-1 and PD-L1. Tumor-associated cells also promote the growth of tumor cells by expressing a number of growth factors, such as VEGF, EGF, and HGF.
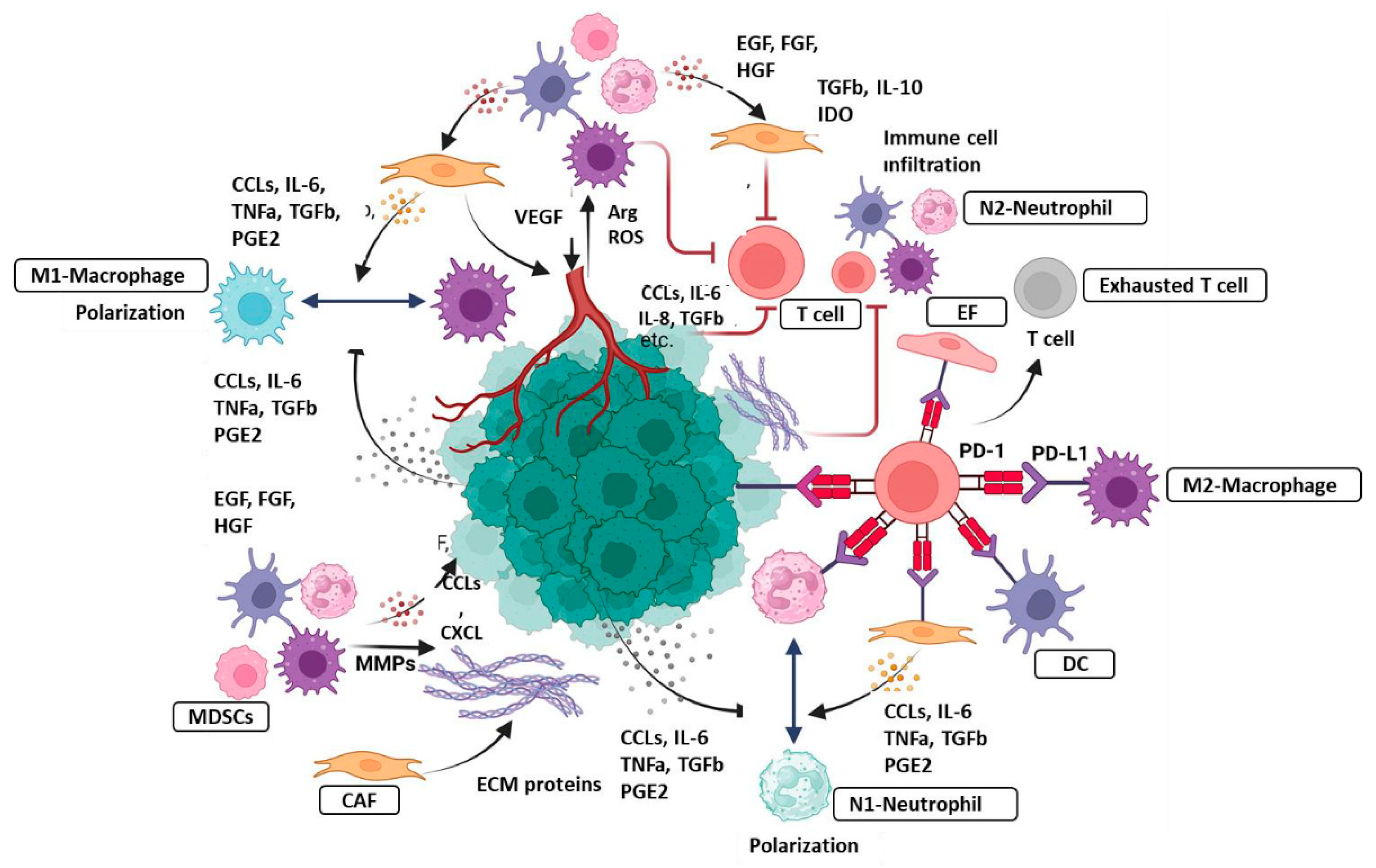
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



