
Submitted:
21 October 2024
Posted:
24 October 2024
Read the latest preprint version here
Abstract
A Gram-positive, aerobic, rod-shaped and spore forming bacterium strain designation B190/17, was isolated from an air monitoring sample of a Brazilian immunobiological production facility in 2017. The strain was not identifiable by biochemical methodology VITEK® 2 or by MALDI-TOF MS with VITEK® MS RUO and MALDI Biotyper®. The 16S rRNA gene sequencing showed 98.51% similarity with Bacillus wudalianchiensis FJAT 27215T, 98.28% with ‘Bacillus aerolatus’ CX 253T, 97.96% with Bacillus badius MTCC 1458T, 97.63% with Bacillus xiapuensis FJAT 46582T and 97.21% with Bacillus thermotolerans SGZ8T. Biochemical data showed the strain was alanine arylamidase, Ala-Phe-Pro arylamidase, ELLMAN, leucine arylamidase, phenyalanine arylamidase and tyrosine arylamidase positive. The genomic DNA G+C% content of B190/17 was 41.6 mol%. The phylogenetic, genomic taxonomy and biochemical tests suggested that B190/17 represents a novel species and should be classified as the type strain of a novel Bacillus species. The name Bacillus lumedeirus sp. nov. is proposed. After characterization, B190/17 was added to the MALDI Biotyper® database as Bacillus lumedeirus sp. nov.
Keywords:
Bacillus lumedeirus
; bacterial identification
; pharmaceutical industry
; genomic taxonomy
; MALDI-TOF MS
; 16S rRNA
1. Introduction
Microbial contamination is one of the main problems in the pharmaceutical industry. It is of particular concern during the manufacture of thermosensitive sterile products such as vaccines and other immunobiologicals, as these cannot be subject to terminal sterilization. Consequently, these products need to be produced aseptically according to strict quality assurance requirements [1,2]. In 2022, the European Medicines Agency established the Contamination Control Strategy (CCS) approach. This aims to identify microorganisms isolated from clean areas and to evaluate the risk of their presence in the environmental production. Subsequently, appropriate measures can be put in place to eliminate high-risk contamination and ensure safe and quality products [3]. To achieve this goal, the identification of microorganisms isolated in pharmaceutical production plants is essential [4,5].
Various methodologies can be employed for microbial identification in the pharmaceutical industry, ranging from biochemical methods to genomics. However these are laborious and expensive to implement [1,2,6]. Although biochemical methods are cheaper and easier to perform than DNA sequence-based methods, their databases are often limited to the more frequently isolated species. In addition, microorganisms isolated from nutrient-limited clean production areas which are subject to the frequent use of disinfectants may not express their standard characteristics in biochemical tests, resulting in inaccurate, or even incorrect results [1,4,7]. Previous studies have shown that matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) is a significantly faster and more accurate identification system than conventional biochemical methods. The method is based on the extraction of proteins from whole microbial cells. Although MALDI-TOF MS databases have improved over the years, some environmental species cannot be identified. This limitation applies to Bacillus and related genera, that are among the main bacteria isolated in pharmaceutical industries [1,4,7,8]. In these cases, DNA sequencing of the 16S rRNA gene and other housekeeping genes like rpoB (encoding the beta subunit of RNA polymerase) and gyrB, (encoding the beta subunit of DNA gyrase) can be performed [1,9,10].
Until 2020 the Bacillus genus was composed of more than 280 species. However studies by Gupta et al. [11] and Patel and Gupta [12] demonstrated how taxonomic and phylogenetic varied the genus was. Consequently, they proposed the reclassification of more than a hundred species into new genera [11,12]. Currently, the genus Bacillus comprises of 111 species with validly published and correct names [13].
Pharmaceutical industry facilities possess potential environments for the discovery of new species of microorganisms which have not been well studied and due to commercial reasons may not have been publicized [1,7,8]. In two previous studies, Costa et al. [1,8] characterized 97 Bacillus and related genera strains isolated from an immunobiological pharmaceutical facility by MALDI-TOF MS and 16S rRNA gene full-length sequencing. These earlier studies include the potential isolation of new bacterial species however, further investigation was required before their formal recognition could be proposed.
The aim of this study was the taxonomic characterization of the novel Bacillus strain B190/17 and to introduce its spectrum into two MALDI-TOF MS system databases. The strain was firstly analyzed by Costa et al [8] and is proposed, in this study, as Bacillus lumedeirus sp. nov., with the type strain B190/17T (= CBAS1225T=CCGBXXXT). In this study, the genome sequence of B190/17 was analyzed to determine genomic and phenotypic features of the new species.
2. Materials and Methods
2.1. Bacterial Strains and Culture Conditions
The strain B190/17 was isolated from an air environmental monitoring sample at a Brazilian immunobiological production facility in 2017 [8]. The strain was streaked on Sheep Blood Agar 5% (SBA) plates and incubated at 30–35 ◦C for 24–48 h. Stock cultures were prepared with Brain Heart Infusion Broth (Merck KGaA, Darmstadt, Germany), containing 20% glycerol (Merck KGaA, Darmstadt, Germany). The strain was deposited at the Coleção de Bactérias do Ambiente e Saúde (CBAS) hosted at Fundação Oswaldo Cruz (Fiocruz), Rio de Janeiro, Brazil (www.cbas.fiocruz.br). The deposit number was 1225T. CBAS is affiliated with the World Federation for Culture Collections (WFCC) and registered as World Data Centre for Microorganisms (WDCM) 958. B190/17 was also deposited at the Coleção de Culturas do Gênero Bacillus e Gêneros Correlatos (CCGB) hosted at Fiocruz, Rio de Janeiro, Brazil (www.ccgb.fiocruz.br) and the deposit number was XXXXT. CCGB is affiliated with the WFCC and registered as WDCM 574.
2.2. Phenotypic Tests
The B190/17 strain was streaked on Tryptic Soy Agar (TSA) plates (BioCen do Brasil, Campinas, Brasil), incubated at 30–35ºC for 24–48 h and submitted to the commercial test API® 50 CH (bioMérieux, Craponne, France), according to the manufacturer`s guidelines and to the motility test in Sulphide Indol Motility (SIM) medium (Merck KGaA, Darmstadt, Germany). The strain was also analyzed by VITEK® 2 Compact System (bioMérieux, Craponne, France) with the BCL card, which was developed for the automated identification of aerobic endospore forming bacteria. Results obtained by VITEK® 2 with a percentage of probability <85% were considered unidentified. Proteomic analysis with MALDI-TOF MS was performed with two different systems: VITEK® MS RUO (bioMérieux, Craponne, France) and MALDI Biotyper® (Bruker Daltonics, Massachusetts, EUA), according to the manufacturer’s instructions. For both systems, a portion of the colony was applied to the slide in duplicate together with one microlitre of formic acid 70% and, after drying, one microlitre of alpha-cyano-4-hydroxycinnamic acid matrix solution (VITEK MS-CHCA, bioMérieux, Craponne, France and Bruker HCCA, Bruker Corporation, Billerica, USA) was also applied. Calibration of VITEK® MS RUO was performed with Escherichia coli ATCC 8739 whereas MALDI Biotyper® was calibrated with the Bacterial Test Standard (BTS) (Bruker Corporation, Billerica, USA). The results were analyzed by SARAMIS Premium software v.4.0.0.14 and MBT Compass HT v.5.1.100, respectively. VITEK® MS RUO and MALDI Biotyper® isolate results with identity percentage ≤75% and scores ≤1.70 were considered unidentified, respectively.
2.3. Genotypic Identification by 16S rRNA Gene SEquencing
Full 16S rRNA gene sequencing was performed using MicroSEQ™ Full Gene 16S rDNA kit (Thermo Fisher Scientific, Waltham, USA), according to the manufacturer’s instructions. The prepared plates were analyzed on the 3500 Series Genetic Analyzer (Applied Biosystems, Waltham, USA) and the sequences were assembled using DNA Star LaserGene SeqMan software v. 7.0.0. An identification was obtained from the website https://www.ezbiocloud.net/ [14] and the sequence was deposited at the Genbank/NCBI (https://www.ncbi.nlm.nih.gov/) with the access number OK586830.1 [1]. Results retrieved with ≥97.0% identity with EzBioCloud were considered valid for identification at genus level, and ≥98.7% for identification at species level [15]. The 16S rRNA gene sequences of related taxa were obtained from EzBioCloud and from the GenBank Database (https://www.ncbi.nlm.nih.gov/nucleotide). The sequences were aligned with PhyloSuite v.1.2.2 [16,17] with MAFFT v.7 plugin default parameters [18]. Maximum likelihood phylogenetic tree was built using PhyloSuite v.1.2.2 [19] and IQ-TREE v. 2.2.0 plugin [20]. The general time-reversible (GTR) substitution model was used according to ModelFinder. FigTree v.1.4.4 was used to pre-edit the phylogenetic tree and the final image was obtained with Inkscape v.1.0.1.
2.4. Genome Sequencing, Assembly, and Annotation
Genomic DNA was extracted and purified using the DNeasy Blood & Tissue Genomic DNA Isolation kit (Qiagen, Hilden, Germany). Next-generation sequencing was performed at Fiocruz Platform Network (Oswaldo Cruz Institute, Brazil, Rio de Janeiro) using Illumina HiSeq 2500 sequencer (Illumina Inc, San Diego, USA). A library was constructed with the Nextera XT DNA Library Preparation Kit (Illumina Inc, San Diego, USA). The reads were assembled de novo using the SPAdes v. 3.15.4 [21]. The genomes were annotated using Rapid Annotation using System Technology (RAST) [22].
2.5. Phylogenetic Analysis of 16S rRNA, rpoB and gyrB Genes
A concatenated tree was built with complete sequences of the housekeeping genes: 16S rRNA (previously obtained), rpoB and gyrB (extracted from the genome with RAST). The 16S rRNA sequences of the type strains for the closest related species were downloaded from EzBioCloud or GenBank, whereas the rpoB and gyrB gene sequences of the closest species were downloaded from GenBank. These three genes were individually aligned with PhyloSuite v.1.2.2 and MAFFT v.7 plugin, concatenated with PhyloSuite and exported in FASTA format. Maximum likelihood phylogenetic tree was built using PhyloSuite and IQ-TREE v.2.2.0 plugin. The GTR substitution model was used, according to Modelfinder. FigTree v.1.4.4 was used to pre-edit the phylogenetic tree and the final image was obtained in Inkscape v.1.0.1.
2.6. Genomic Taxonomy Analysis
To carry out the genomic taxonomy analysis, whole-genome sequences of type strains ‘Bacillus aerolatus’ CX 253T (WEIO01000001.1), Bacillus badius NBRC15713T (NZ_BCVF01000001.1), Bacillus thermotolerans SGZ8T (JWJE02000001.1), Bacillus wudalianchiensis FJAT27215T (NZ_MAYT01000001.1), and Bacillus xiapuensis FJAT46582T (NJAY01000001.1) was downloaded from the GenBank Database (https://www.ncbi.nlm.nih.gov/genome). The Average Nucleotide Identity (ANI) was calculated according to the OrthoANI algorithm using the OrthoANI tool v. 0.93.1 [23]. The DNA-DNA hybridization (DDH) value was determined in silico for these genomes using Genome-to-Genome Distance Calculator (GGDC) v.3.0 by the Basic Local Alignment Search Tool (BLAST) method. Genome-wide super tree building was performed by Type Strain Genome Server (TYGS) (https://tygs.dsmz.de) [24].
2.7. Genome Sequence Deposit
The whole-genome shotgun sequence of B190/17 was deposited at GenBank under the accession number JAUIYO000000000.
2.8. Addition of B190/17 spectra to the VITEK® MS RUO and MALDI Biotyper® database
The SuperSpectrum of B190/17 was created and added to the Saramis Premium software v.4.0.0.14 database of VITEK® MS RUO, as previously described by Costa et al. [1]. For addition to the MALDI Biotyper® database, B190/17 was streaked on SBA and incubated at 30-35ºC for 24-48 h. Then, a portion of the colonies was suspended in 300 μL of sterile water and after vigorous homogenization, 900 μL of absolute ethanol (Merck KGaA, Darmstadt, Germany) was added. After another vigorous homogenization, the sample was centrifugated at 13000-15000 rpm for 2 min. The supernatant was discarded and the centrifugation was performed again to ensure its complete removal. Then, the pellet was dried at room temperature for 3-5 min with the tube cap open. Next, 25-100 μL of 70% formic acid (Merck KGaA, Darmstadt, Germany) was added and, after homogenization, the same volume of acetonitrile (Merck KGaA, Darmstadt, Germany) was added to the pellet. After further centrifugation under the same conditions, one microlitre of supernatant was applied to eight spots of the slide and one microlitre of BTS was applied to only one. After drying, one microlitre of Bruker HCCA was added to the spots. The calibration was performed in BTS spot with Flex Control v.3.4, according to manufacturer`s instructions. For spectra acquisition, the eight sample spots were measured three times with Flex Control. The selection of the most homogeneous spectra of the sample and the elimination of the discrepant ones was carried out in Flex Analysis v.3.4, according to the manufacturer's instructions, so that the Main Spectrum Profile (MSP) could be created with at least 20 spectra. In MBP Compass Explorer v. 4.1, the new MSP was compared with all the existing MSP libraries and then inserted into the customized library. Finally, the new version of this library was imported into the MBT Compass HT v. 5.1.100. Afterwards, B190/17 was subjected once again to proteomic analysis by MALDI Biotyper® in duplicate, as previously described, so that the new MSP could be challenged as to their suitability for identifying the desired strain. Results with score values <1.70 were considered not identified; between 1.70 and 1.99 were considered with low-confidence for species identification, and scores ≥2.00 were considered with high-confidence for species identification.
3. Results and Discussion
Costa et al [1,8] previously isolated the strain B190/17 from an air monitoring sample at an immunobiological production facility and proposed that it could be designated as a new bacterial species. B190/17 was not identified by VITEK® 2 [8], but was given the bionumber 0303101000000000 for its biochemical profile. It was only positive for the biochemical tests: alanine arylamidase, Ala-Phe-Pro arylamidase, ELLMAN, leucine arylamidase, phenyalanine arylamidase and tyrosine arylamidase. The strain was negative for all the biochemical tests of API® 50 CH and was not motile. The VITEK® 2 biochemical test results are present in Table 1. A comparison of phenotypic characteristics of B190/17 with reference strains is given in Table 2. Biochemical methods are not suitable for a reliable identification of Bacillus and related genera species.
Due to its limited and outdated database the BCL card of VITEK® 2 claims to identify 21 Bacillus species [4,7,25] (Table 3). However, currently the number of identifiable Bacillus species is only seven since 14 species are now classified in other genera; for example Bacillus megaterium to Priestia megaterium and Bacillus circulans to Niallia circulans [11,13,25].
In the pharmaceutical industry, accurate identification of microorganisms is very important, as a misidentification could lead to product release based on a false negative, or product withdrawal due to a false positive result [5]. Furthermore according to EU Annex 1, microorganisms detected in the grades A and B areas (cleanrooms used for high-risk activities) should be identified to species level, as well as spore-forming microorganisms isolated from grade C and grade D areas (cleanrooms used for low-risk activities) should to enable a robust risk assessment to reach and eliminate the source of contamination [3]. Therefore, there is a need to apply rapid, yet accurate identification methods.
MALDI-TOF MS is an appropriate methodology for use by microbiological control laboratories in the pharmaceutical industry. It is not laborious and the results can be quickly obtained. However, its database needs to be regularly expanded and updated [1,2,4]. In this study, MALDI Biotyper® and VITEK® MS RUO from Bruker and bioMérieux were used as they are the only companies that sell MALDI-TOF MS in Brazil. At the time these studies, VITEK® MS prime had not been launched. Neither of these MALDI-TOF MS systems were able to identify the B190/17 strain. Figure 1 shows the comparison between the spectra provided by MALDI Biotyper® for the B190/17 strain and the two closest strains of the database: B. badius strains DSM 23T and DSM 30822. It was apparent that the spectrum of the B190/17 strain matched just four and seven peaks, respectively, with B. badius DSM 23T and B. badius DSM 30822. These matches were not enough for a reliable identification.
Most of the studies related to Bacillus species taxonomy are based on the 16S rRNA gene sequences [12]. If the 16S rRNA sequence of the target strain is compared to sequences in EzBioCloud or GenBank and there are no species with similarity >98.7%, then it is an indication that it may be a new species. Therefore further genomic analysis is recommended [26]. 16S rRNA gene sequence analysis showed that all the Bacillus strains, including the strains without validly published nomenclatural status, and Domibacillus strains compared to the B190/17 strain showed similarity that was <98.7%. This supported the earlier proposal by Costa et al. [1,8] that B190/17 was a new bacterial species. By convention, B190/17 would be the type strain of the new species.
The phylogenetic tree based on 16S rRNA gene sequences revealed that the designated type strain B190/17 formed a separate branch closest to B. badius MTCC 1458T and B. wudalianchiensis FJAT 27215T strains (Figure 2), whose similarity were 97.96 and 98.51%, respectively. B. xiapuensis FJAT 46582T, ‘B. aerolatus’ CX253T, B. ectoiniformans NU-14T and B. thermotolerans SGZ8T were placed in different clusters and showed similarities of 97.63, 98.28, 96.49 and 97.21% with the 16S rRNA gene sequence from B190/17 strain, respectively. All others type strains shared less than 96.49% 16S rRNA gene similarities in comparison with B190/17 strain. ‘Bacillus aerolatus’ was described in 2020 by Chen et al [27]. However, this species name has not been validated by the International Code of Nomenclature of Prokaryotes (ICNP). Although the species name is not validly published, its actual taxonomic status is referred to as ‘preferred name’ according to the List of Prokaryotic names with Standing in Nomenclature [13]. As the species ‘Bacillus aerolatus’ was one of the most closely related species to B190/17 strain, it was decided to retain it in this study for comparative purposes. Bacillus smithii was used as an outgroup. This species was chosen because according to a maximum-likelihood phylogenetic tree for 303 genome-sequenced Bacillaceae species (based on concatenated sequences for 650 core proteins), it was the closest species to the genera Domibacillus and Pseudobacillus [11]. At the time of the Gupta et al. [11] study, B. badius and B. wudalianchensis were classified as Pseudobacillus badius and Pseudobacillus wudalianchensis [28].
Phylogenetic analysis of Bacillus species has also been performed using other housekeeping genes [1,27]. Therefore, to confirm the 16S rRNA gene analysis, further analysis was carried out using the concatenated sequences of 16S rRNA (obtained by Sanger sequencing), rpoB and gyrB genes from the whole genome sequences of B190/17 strain. There is no cut-off percentage suggested in the literature for species and genus identification based on rpoB and gyrB genes analysis as there is for 16S rRNA gene. Therefore, it should be analyzed on a case by case basis by considering the phylogenetic analysis of the genes [1]. The concatenated phylogenetic tree also showed the B190/17 strain was in a separate cluster from ‘B. aerolatus’ CX253, B. badius MTCC 1458 and NBRC 15713 and B. wudalianchiensis FJAT 27215 type strains (Figure 3). The percentage of similarity of rpoB and gyrB sequences of B190/17 strain and their closest neighbors, respectively, were ‘B. aerolatus’ CX253T (87.50%, 85.43%), B. badius NBRC 15713T (86.91%, 80.90%) and B. wudalianchiensis FJAT 27215T (87.38%, 81.17%).
The genomic taxonomy results of B190/17 are shown in Table 4 and Table 5. The average size of the B190/17 genome was 3.43 Mb. The DNA G+C % content was 41.6 mol% and the coverage was 73x. The assembly produced 89 contigs with a total of 3,434,160 bp, N50 of 219,177 bp and 3,544 coding sequences. Other sequenced genome metrics provided by RAST are shown in the Table 5. The 16S rRNA gene sequence obtained with Sanger sequencing (OK586830.1; 1,499 bp) was also compared with the 16S sequence of B190/17 genome (JAUIYO000000000; 1,355 bp) and the percentage identity was 99.92%.
The ANI values of B190/17 strain and the related species ‘B. aerolatus’ CX253T, B. badius NBRC 15713T, B. wudalianchiensis FJAT 27215T were 79.55 %, 76.47% and 77.64%, respectively, which are lower than the cut-off value (95-96%) established to consider as belonging to the same species [29]. Moreover, estimation of in silico DNA–DNA hybridization (in silico DDH) by using the GGDC in comparison to the same type species above were 24.00, 21.60 and 22.50, respectively. All values are also lower than the cut-off value (70%) proposed for delineation of novel species, indicating that are distinct species [29].
The phylotaxonomic tree constructed on the TYGS server provided further evidence for the distinct taxonomic status of the B190/17 strain within the genus Bacillus. Figure 4 shows the position of B190/17 strain in comparison with the most closely related type strains based on whole-genome sequences. B190/17 is in a different cluster from their closer relatives ‘B. aerolatus’, B. badius and B. wuadalianchiensis. These results supported the earlier conclusion that B190/17 strain represents a novel species of the genus Bacillus. The new species is proposed as Bacillus lumedeirus sp. nov., with the type strain B190/17T.
In the past, the criteria used by taxonomists to classify a Bacillus species was its ability to produce spores in the presence of oxygen. However whole genome studies of the Bacillus genus have resulted in the reclassification of many of former Bacillus species into new genera. Gupta et al [11] and Patel and Gupta [12] proposed that the Bacillus genus should be composed only of two clades and that strains not in these clades should be transferred to new genera. The two clades being “Subtilis clade” composed of Bacillus sensu stricto, and the “Cereus clade” containing a variety of important pathogenic species. However, some species that do not form part of these two clades are still part of the genus Bacillus. These include species close to the B190/17 strain: B. badius, B. thermotolerans, B. wudalianchiensis and B. xiapuensis [13]. Verma et al. [28] proposed the reclassification of B. badius and B. wudalianchiensis as the new genus Pseudobacillus, as mentioned before. Currently it remains for further studies to justify the reclassification of ‘B. aerolatus’, B. badius, B. wudalianchiensis, and Bacillus lumedeirus sp. nov. into a new genus.
Costa et al. [1] added the B190/17 strain in VITEK® MS RUO simply as Bacillus spp. However, at that time the genomic taxonomy analysis had not been concluded. Based on the conclusion that the B190/17 strain was a novel species of the genus Bacillus, it was added in MALDI Biotyper® database. The strain was again submitted to proteomic analysis and was identified as Bacillus lumedeirus sp. nov. with the higher score = 2.32. Therefore the addition of B190/17 to the MALDI Biotyper® database will facilitate the identification of any further isolates.
According to 16S rRNA gene and whole genome analysis, the closest valid species to Bacillus lumedeirus sp. nov. are B. badius, B. thermotolerans, B. wudalianchiensis and B. xiapuensis. With the exception of B. badius, these species were not present in MALDI Biotyper® database. The database represents only 32 of the 110 species already described [30]. This shows how the database can be further expanded to improve the identification of Bacillus and related genera in microbiological control laboratories. Consequently, MALDI-TOF MS analysis can be used to ensure greater safety in the release of pharmaceutical products, and for tracing sources of contamination within a pharmaceutical facility.
Description of Bacillus lumedeirus sp. nov.
Bacillus lumedeirus (lu.me.dei.rus. N.L. gen. fem. n. medeirus) of Luciane Martins Medeiros, in memoriam, a Brazilian scientist at the Institute of Immunobiological Technology (Bio-Manguinhos) of Fiocruz in Rio de Janeiro who made significant contributions to the Laboratory Microbiological Control of Bio-Manguinhos, including introducing new microorganism identification systems.
Cells are Gram-positive, spore-forming, non-motile, growth in aerobic conditions (24–48 h) at 30-37ºC (optimum temperature: 37ºC). Cream, non-mucoid, circular, irregular edge, smooth, brilliant and medium colonies (up to 6 mm in diameter) were observed after 24 h of incubation on TSA, at 37°C. The type strain was found to be positive for alanine arylamidase, Ala-Phe-Pro arylamidase, ELLMAN, leucine arylamidase, phenyalanine arylamidase and tyrosine arylamidase positive. Activities of α and β-glucosidase and β-manosidase were not detected. Regarding the acid production capacity, this was not observed for galactose, glycogen glucose, mannitol and ribose.
The genome size of B190/17 strain is estimated at 3.43 Mb with 41.6 mol % of G+C content. The genome is deposited in GenBank under access number JAUIYO000000000. The type strain is B190/17 (= CBAS 1225T = CCBG XXXXT), isolated in 2017 from air environmental monitoring in an Immunobiological production facility in Brazil.
Author Contributions
Conceptualization, L.C.; J.R. and V.V.; methodology, L.C.; J.R. and V.V.; software, J.R. and J.V.; formal analysis, L.C.; L.A.; R.M.; T.V. and E.V.; investigation, L.C.; J.R. and V.V.; resources, M.B. and V.V.; writing—original draft preparation, L.C.; writing—review and editing, J.N.; S.F. and M.B; visualization, J.N.; S.F. and M.B; supervision, V.V.; project administration, L.C. and V.V.; funding acquisition, L.C. and V.V. All authors have read and agreed to the published version of the manuscript.
Funding
Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (Faperj/RJ) provided financial support to Leticia Albuquerque (Processo E-26/260.341/2022) and to Juliana Ramos (Processo E-26/202.088/2020).
Data Availability Statement
Data are contained within the article.
Acknowledgments
The authors are grateful to the Instituto de Tecnologia em Imunobiológicos (Bio-Manguinhos)/Fundação Oswaldo Cruz (Fiocruz), to the Instituto Oswaldo Cruz (IOC/Fiocruz) and to the Faperj/RJ.
Conflicts of Interest
Author Stephen James Forsythe is the maintainer of Foodmicrobe.com Ltd. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Costa, L.V.D.; Miranda, R.V.D.S.L.; Reis, C.M.F.; Andrade, J.M.; Cruz, F.V.; Frazão, A.M.; Fonseca, E.L.; Ramos, J.N.; Brandão, M.L.L.; Vieira, V.V. MALDI-TOF MS database expansion for identification of Bacillus and related genera isolated from a pharmaceutical facility. J. Microbiol. Methods 2022, 203, 106625. DOI: 10.1016/j.mimet.2022.106625. [CrossRef]
- Song, M.; Li, Q.; Liu, C.; Wang, P.; Qin, F.; Zhang, L.; Fan, Y.; Shao, H.; Chen, G.; Yang, M. A comprehensive technology strategy for microbial identification and contamination investigation in the sterile drug manufacturing facility—a case study. Front. Microbiol. 2024, 15, 1327175. DOI: 10.3389/fmicb.2024.1327175. [CrossRef]
- European Medicines Agency. European Medicines Agency. European Union guidelines for good manufacturing practice for medicinal products for human and veterinary use. In The Rules Governing Medicinal Products in the European Union; Annex 1: Manufacture of Sterile Medicinal Products; European Medicines Agency: Brussels, Belgium, 2022; Volume 4.
- Miranda, R.V.D.S.L.; da Costa, L.V.; Albuquerque, L.S.; Dos Reis, C.M.F.; Braga, L.M.P.D.S.; de Andrade, J.M.; Ramos, J.N.; Mattoso,J.M.V.; Forsythe, S.J.; Brandão, M.L.L. Identification of Sutcliffiella horikoshii strains in an immunobiological pharmaceutical industry facility. Lett. Appl. Microbiol. 2023, 76, ovad056. DOI: 10.1093/lambio/ovad056. [CrossRef]
- Mattoso, J.M.V.; Costa, L.V.C.; Vale, B.A.; Reis, C.M.F.; Andrade, J.M.; Braga, L.M.P.S.; Conceição, G.M.S.; Costa, P.B.M.; Silva, I.B.; Rodrigues, L.A.P.; Anjos, J.P.; Brandão, M.L.L. Quantitative and qualitative evaluation of microorganism profile identified in bioburden analysis in a biopharmaceutical facility in Brazil: Criteria for classification and management of results. PDA J. Pharm. Sci. Technol. 2024, 78, 3, 1. DOI: 10.5731/pdajpst.2023.012883. [CrossRef]
- Stamatoski, B.; Ilievska, M.; Babunovska, H.; Sekulovski, N.; Panov, S. Optimized genotyping method for identification of bacterial contaminants in pharmaceutical industry. Acta Pharm. 2020, 66, 2, 289-295. DOI: 10.1515/acph-2016-0011. [CrossRef]
- Caldeira, N.G.S.; de Souza, M.L.S.; de Miranda, R.V.D.S.L.; da Costa, L.V.; Forsythe, S.J.; Zahner, V.; Brandão, M.L.L. Characterization by MALDI-TOF MS and 16S rRNA gene sequencing of aerobic endospore-forming bacteria isolated from pharmaceutical facility in Rio de Janeiro, Brazil. Microorganisms 2024, 12, 4, 724. DOI: 10.3390/microorganisms12040724. [CrossRef]
- Costa, L.V.D.; Miranda, R.V.D.S.L.; Fonseca, E.L.; Gonçalves, N.P.; Reis, C.M.F.; Frazão, A.M.; Cruz, F.V.; Brandão, M.L.L.; Ramos, J.N.; Vieira, V.V. Assessment of VITEK® 2, MALDI-TOF MS and full gene 16S rRNA sequencing for aerobic endospore-forming bacteria isolated from a pharmaceutical facility. J. Microbiol. Methods 2022, 194, 106419. DOI: 10.1016/j.mimet.2022.106419. [CrossRef]
- Husni, A.A.A.; Ismail, S.I.; Jaafar, N.M.; Zulperi, D. Current classification of the Bacillus pumilus group species, the rubber-pathogenic bacteria causing trunk bulges disease in Malaysia as assessed by MLSA and multi rep-PCR approaches. Plant. Pathol. J. 2021, 37, 3, 243. DOI: 10.5423/PPJ.OA.02.2021.0017. [CrossRef]
- Qi, H.Y.; Wang, D.; Han, D.; Song, J.; Ali, M.; Dai, X.; Zhang, X.; Chen, J. Unlocking antagonistic potential of Bacillus amyloliquefaciens KRS005 to control gray mold. Front. Microbiol. 2023, 14, 1189354. DOI 10.3389/fmicb.2023.1189354.
- Gupta, R.S.; Patel, S.; Saini, N.; Chen, S. Robust demarcation of 17 distinct Bacillus species clades, proposed as novel Bacillaceae genera, by phylogenomics and comparative genomic analyses: Description of Robertmurraya kyonggiensis sp. nov. and proposal for an emended genus Bacillus limiting it only to the members of the Subtilis and Cereus clades of species. Int. J. Syst. Evol. Microbiol. 2020, 70, 5753–5798. DOI: 10.1099/ijsem.0.004475. [CrossRef]
- Patel, S.; Gupta, R.S. A phylogenomic and comparative genomic framework for resolving the polyphyly of the genus Bacillus: Proposal for six new genera of Bacillus species, Peribacillus gen. nov., Cytobacillus gen. nov., Mesobacillus gen. nov., Neobacillus gen. nov., Metabacillus gen. nov. and Alkalihalobacillus gen. nov. Int. J. Syst. Evol. Microbiol. 2020, 70, 406–438. DOI: 10.1099/ijsem.0.003775. [CrossRef]
- Parte, A.C.; Sardà Carbasse, J.; Meier-Kolthoff, J.P.; Reimer, L.C.; Göker, M. List of Prokaryotic names with Standing in Nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 2020, 70, 5607–5612. DOI: 10.1099/ijsem.0.004332. [CrossRef]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617.. DOI: 10.1099/ijsem.0.001755. [CrossRef]
- Woo, P.C.Y.; Lau, S.K.P.; Teng, J.L.L.; Tse, H.; Yuen, K-Y. Then and now: use of 16S rDNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories. Clin. Microbiol. Infec. 2008, 14, 10, 908-934. DOI: 10.1111/j.1469-0691.2008.02070.x. [CrossRef]
- Xiang, C-Y.; Gao, F.; Jakovlic, I.; Lei, H-P.; Hu, Y.; Zhang, H.; Zou, H.; Wang, G-T.; Zhang, D. Using PhyloSuite for molecular phylogeny and tree-based analyses. iMeta 2023, e87. DOI: 10.1002/imt2.87. [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Res. 2020, 20, 1, 348–355. DOI: 10.1111/1755-0998.13096. [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 4, 772–780. DOI: 10.1093/molbev/mst010. [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 6, 587–589. DOI: 10.1038/nmeth.4285. [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268-274. DOI: 10.1093/molbev/msu300. [CrossRef]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinformatics 2020, 70, 1, e102. DOI: 10.1002/cpbi.102. [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; Meyer, F.; Olsen, G.J.; Olson, R.; Osterman, A.L.; Overbeek, R.A.; McNeil, L.K.; Paarmann, D.; Paczian, T.; Parrello, B.; Pusch, G.D.; Reich, C.; Stevens, R.; Vassieva, O.; Vonstein, V.; Wilke, A.; Zagnitko, O. The RAST Server: rapid annotations using subsystems technology. BMC Genomics 2008, 9, 1-15. DOI: 10.1186/1471-2164-9-75. [CrossRef]
- Lee, I.; Kim, Y.O.; Park, S.C.; Chun, J. OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–3. DOI: 10.1099/ijsem.0.000760. [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 2019, 10, 1, 2182. DOI 10.1038/s41467-019-10210-3.
- bioMerieux. Guide for VITEK® 2 BCL card, v. 045519- 02 - 2019-03. 2019, 1-24.
- Chun, J.; Oren, A.; Ventosa, A.; Christensen, H.; Arahal, D.R.; Costa, M.S.; Rooney, A.P.; Yi, H.; Xu, X.; Meyer, S.D.; Trujillo, M.E. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int J Syst Evol Microbiol 2018, 68, 1, 461-466. DOI: 10.1099/ijsem.0.002516. [CrossRef]
- Chen, P.; Wang, D. ; Ren, Q. ; Wu, J.; Jiang, Y.; Wu, Z.; Pan, Y.; Zhong, Y.; Guan, Y.; Chen, K.; Zhang, G. Bacillus aerolatus sp. nov., a novel member of the genus Bacillus, isolated from bioaerosols in a school playground. Arch Microbiol 2020, 202, 2373-2378. DOI: 10.1007/s00203-020-01955-3. [CrossRef]
- Verma, A.; Pal, Y.; Ojha, A.K.; Kumari, M.; Khatri, I.; Rameshkumar, N.; Schumann, P.; Dastager, S.G.; Mayilraj, S.; Subramanian, S.; Krishnamurthi, S. Taxonomic insights into the phylogeny of Bacillus badius and proposal for its reclassification to the genus Pseudobacillus as Pseudobacillus badius comb. nov. and reclassification of Bacillus wudalianchiensis Liu et al., 2017 as Pseudobacillus wudalianchiensis comb. nov. Syst. Appl. Microbiol. 2019, 42, 3, 360-372. DOI: 10.1016/j.syapm.2019.03.003. [CrossRef]
- Riesco, R.; Trujillo, M.E. Update on the proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 2024, 74, 3, 006300. DOI: 10.1099/ijsem.0.006300. [CrossRef]
- Bruker. Species/Entry List MBT Compass Library Revision K. 2022, 1-75.
Figure 1.
Comparison between spectra provided by MALDI Biotyper® for B190/17 strain and B. badius. a) Comparison between spectra of B190/17 and B. badius DSM 23T. b) Comparison between spectra of B190/17 and B. badius DSM 30822. The ten closest matching classification results and their score values are displayed in the box. Traffic-light color-coding markers indicate the closeness of the match (green = full match, yellow = partial match, red = no match). The higher half of the graphic displays the peak list of B190/17 and their colour reflects the degree of matching with the reference main spectrum profile (MSP) whereas the lower half displays the peak list of the selected MSP in blue using an inverted scale.
Figure 1.
Comparison between spectra provided by MALDI Biotyper® for B190/17 strain and B. badius. a) Comparison between spectra of B190/17 and B. badius DSM 23T. b) Comparison between spectra of B190/17 and B. badius DSM 30822. The ten closest matching classification results and their score values are displayed in the box. Traffic-light color-coding markers indicate the closeness of the match (green = full match, yellow = partial match, red = no match). The higher half of the graphic displays the peak list of B190/17 and their colour reflects the degree of matching with the reference main spectrum profile (MSP) whereas the lower half displays the peak list of the selected MSP in blue using an inverted scale.
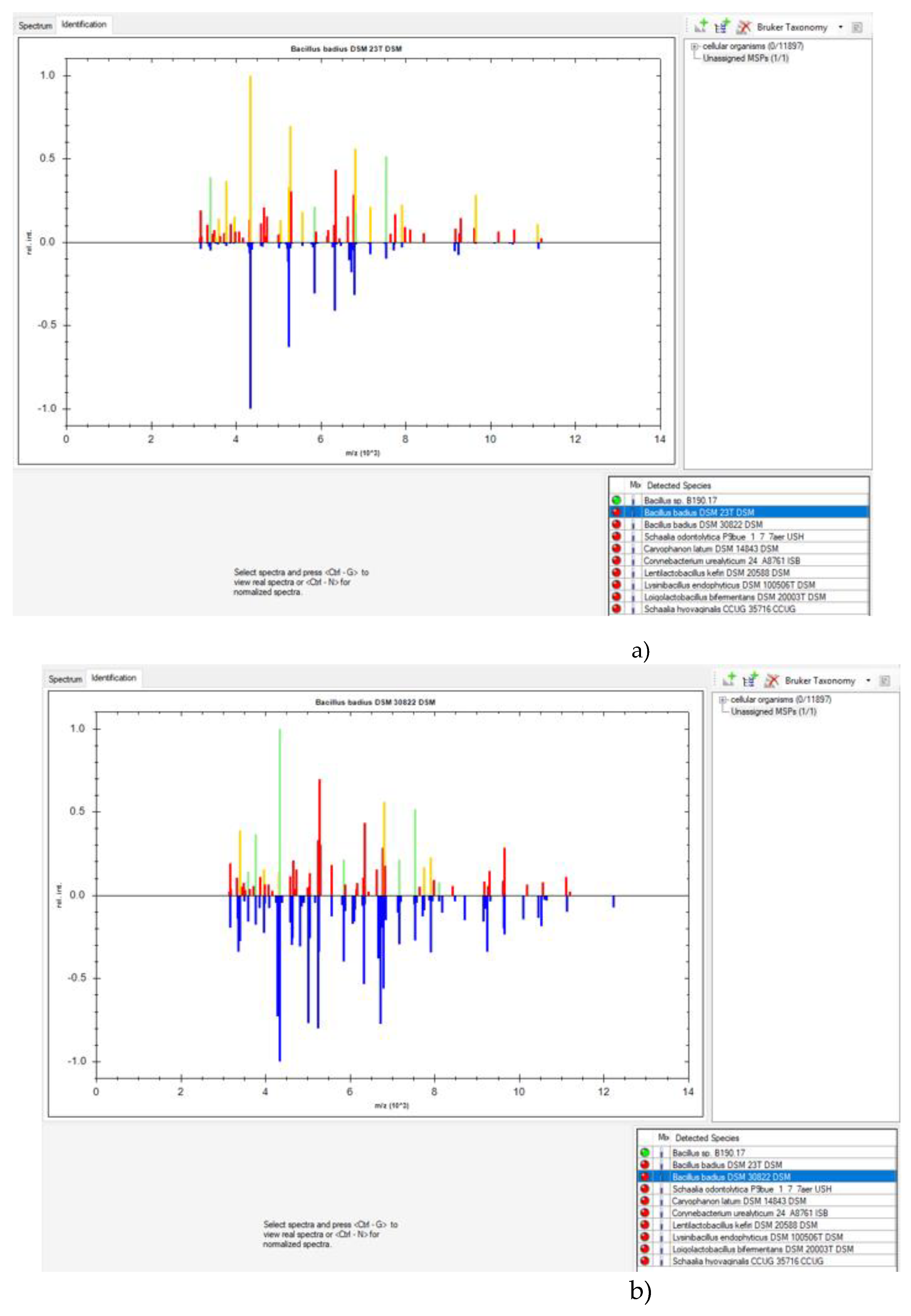
Figure 2.
Maximum likelihood phylogenetic tree showing the position of B190/17 strain based on 16S rRNA gene sequences (22 strains with 1,487 bp). SH-arLT/bootstrap values (>50 %) based on 5,000 repetitions are shown. The sequence of Bacillus smithii DSM4216 was used as outgroup. Bar 0.01% estimated sequence divergence. GenBank accession number is given and type strains are indicated (T). SH-aLRT = Shimodaira-Hasegawa approximate likelihood ratio test.
Figure 2.
Maximum likelihood phylogenetic tree showing the position of B190/17 strain based on 16S rRNA gene sequences (22 strains with 1,487 bp). SH-arLT/bootstrap values (>50 %) based on 5,000 repetitions are shown. The sequence of Bacillus smithii DSM4216 was used as outgroup. Bar 0.01% estimated sequence divergence. GenBank accession number is given and type strains are indicated (T). SH-aLRT = Shimodaira-Hasegawa approximate likelihood ratio test.
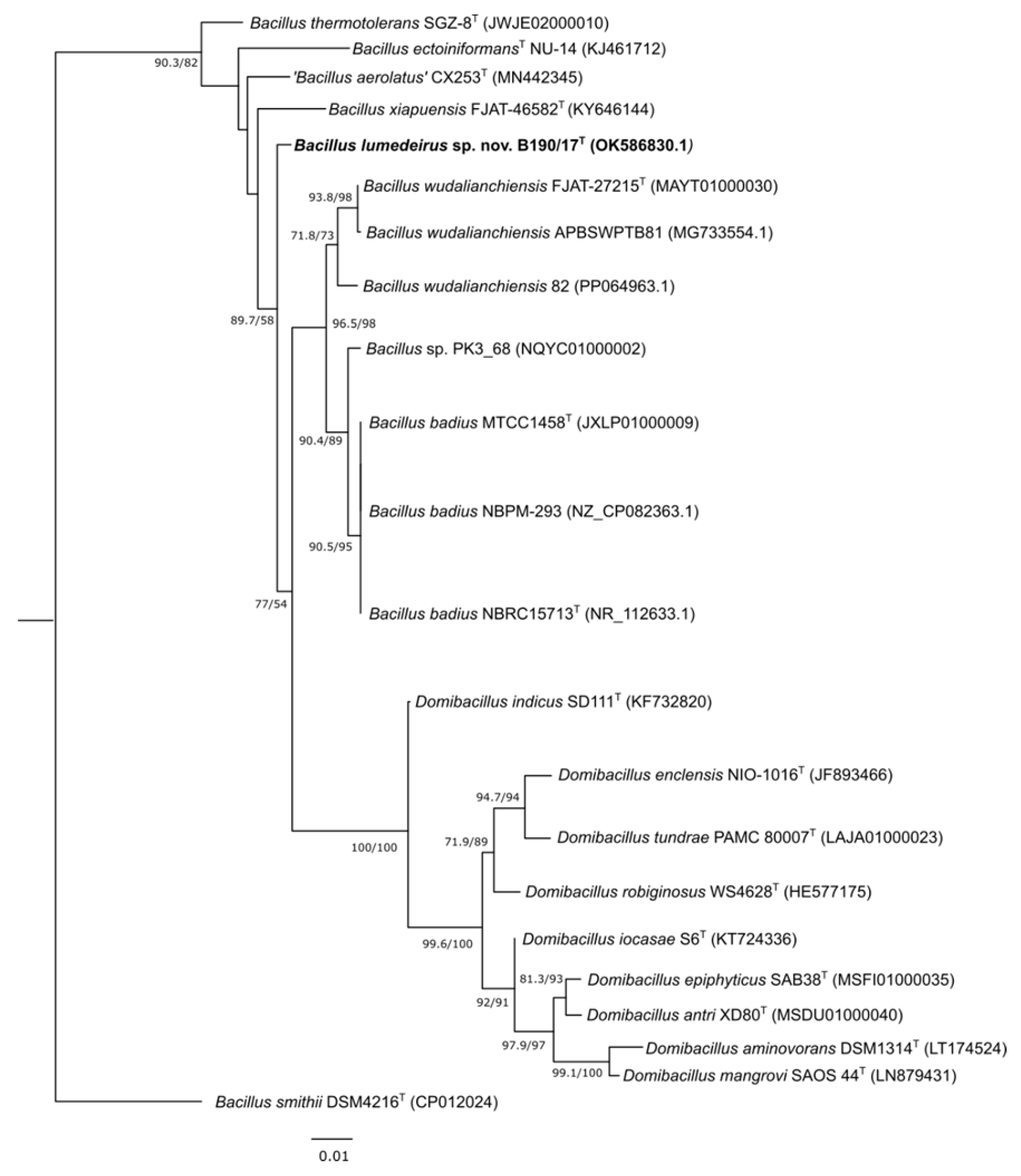
Figure 3.
Maximum likelihood phylogenetic tree based on the sequences of 16S rRNA, rpoB and gyrB showing the position of B190/17 strain against the most closely related strains (18 strains with 6,973 nucleotides). SH-arLT/bootstrap values (>50 %) based on 5,000 repetitions are shown. The strain Bacillus smithii DSM4216 was used as outgroup. Bar 0.06% estimated sequence divergence. Type strains are indicated (T). SH-aLRT = Shimodaira-Hasegawa approximate likelihood ratio test.
Figure 3.
Maximum likelihood phylogenetic tree based on the sequences of 16S rRNA, rpoB and gyrB showing the position of B190/17 strain against the most closely related strains (18 strains with 6,973 nucleotides). SH-arLT/bootstrap values (>50 %) based on 5,000 repetitions are shown. The strain Bacillus smithii DSM4216 was used as outgroup. Bar 0.06% estimated sequence divergence. Type strains are indicated (T). SH-aLRT = Shimodaira-Hasegawa approximate likelihood ratio test.
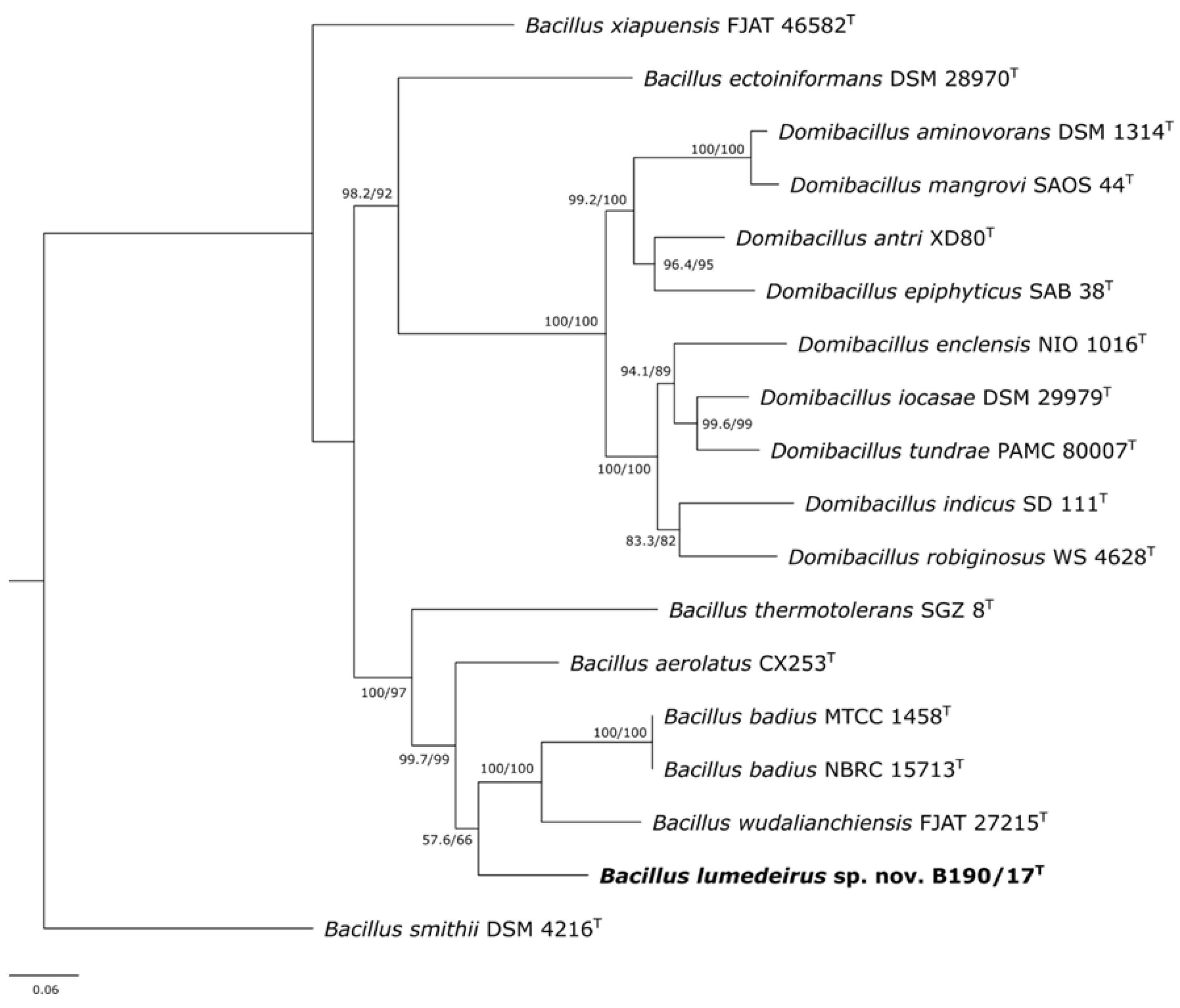
Figure 4.
Phylotaxonomic genome tree showing the position of B190/17 strain. against the most closely related type strains based on whole-genome sequences. The genomic sequences were analyzed by type (strain) genome server (TYGS) with standard parameters. Tree inferred with FastME 2.1.6.1 from GBDP distances calculated from genome sequences. The branch lengths are scaled in terms of GBDP distance formula d5. The numbers above branches are GBDP pseudo-bootstrap support values (> 60%) from 100 replications, with an average branch support of 36.3%. Bacillus encimensis is synonym of Bacillus badius and Bacillus aerolatus is not validly published [13].
Figure 4.
Phylotaxonomic genome tree showing the position of B190/17 strain. against the most closely related type strains based on whole-genome sequences. The genomic sequences were analyzed by type (strain) genome server (TYGS) with standard parameters. Tree inferred with FastME 2.1.6.1 from GBDP distances calculated from genome sequences. The branch lengths are scaled in terms of GBDP distance formula d5. The numbers above branches are GBDP pseudo-bootstrap support values (> 60%) from 100 replications, with an average branch support of 36.3%. Bacillus encimensis is synonym of Bacillus badius and Bacillus aerolatus is not validly published [13].
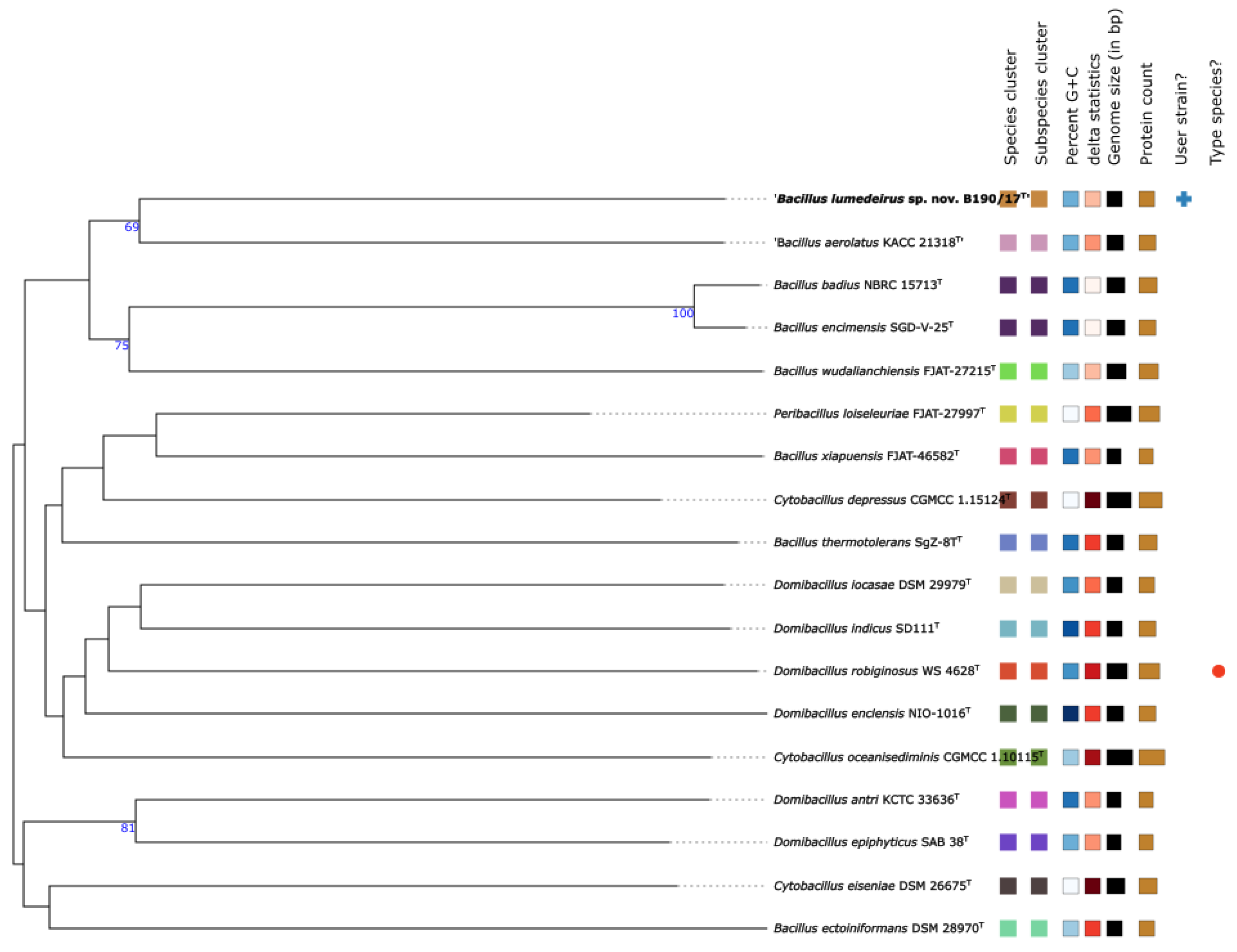

Table 1.
Biochemical profile (bionumber 0303101000000000) of B190/17 strain identified by VITEK® 2.
| Biochemical test | Result | Biochemical test | Result | Biochemical test | Result | Biochemical test | Result | Biochemical test | Result | Biochemical test | Result |
|---|---|---|---|---|---|---|---|---|---|---|---|
| BXYL | - | LysA | - | AspA | - | LeuA | + | PheA | + | ProA | - |
| BGAL | - | PyrA | - | AGAL | - | AlaA | + | TyrA | + | BNAG | - |
| APPA | + | CDEX | - | dGAL | - | GLYG | - | INO | - | MdG | - |
| ELLM | + | MdX | - | AMAN | - | MTE | - | GlyA | - | dMAN | - |
| dMNE | - | dMLZ | - | NAG | - | PLE | - | IRHA | - | BGLU | - |
| BMAN | - | PHC | - | PVATE | - | AGLU | - | dTAG | - | dTRE | - |
| INU | - | dGLU | - | dRIB | - | PSCNa | - | NaCI 6.5% | - | KAN | - |
| OLD | - | ESC | - | TTZ | - | POLYB_R | - |
BXYL = Beta-xylosidase; LysA = L-lysine arylamidase; AspA = L-aspartate arylamidase; LeuA = Leucine arylamidase; PheA = Phenilalanine arylamidase; ProA = L-Proline arylamidase; BGAL = Beta galactosidase; PyrA = L-Pyrrolidonyl arylamidase; AGAL = Alpha galactosidase; AlaA = Alanine arylamidase; TyrA = Tyrosine arylamidase; BNAG = Beta-N-acetyl-glucosaminidase; APPA = Ala-Phe-Pro arylamidase; CDEX = Cyclodextrine; dGAL = D-galactose; GLYG = glycogen; INO = Myo-inositol; MdG = Acidification methyl-A-D-glucopiranoside; ELLM = Ellman; MdX = Methyl-D-xyloside; AMAN = alpha-mannosidase; MTE = Maltotriosis; GlyA = Glycine arilamidase; dMAN = D-mannitol; dMNE = D-mannose; dMLZ = D-melezitosis; NAG N-acetyl-D-glucosamine; PLE = Palatinose; IRHA = Ramnose; BGLU = Beta-glucosidase; BMAN = Beta-manosidase; PHC = Phosphoryl choline; PVATE = Pyruvate; AGLU = Alpha-glucosidase; dTAG = D-tagatose; dTRE = D-trealose; INU = Inulina; dGLU = D-glucose; dRIB = D-ribose; PSCNa = Putrescine assimilation; NaCl 6.5% = Growth in NaCl 6.5%; KAN = Kanamycin resistance; OLD = Oleandomycin resistance; ESC = Sulin hydrolysis; TTZ = Tetrazolium red; POLYB_R = Polymyxin_B resistance; ‘–‘ = 0% to 5% positive; ‘+’ = 95 to 100% positive.
Table 2.
Phenotypic characteristics of strain B190/17 and reference strains.
| Characteristics | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Cell shape | rod | rod | rod | ovoid | rod | rod |
| Motility | - | + | + | - | + | + |
| Optimum temperature for growth (ºC) | 37 | 37 | 30 | 50 | 37 | 25 |
| Catalase | + | + | + | + | + | + |
| Starch | - | - | - | - | - | - |
| L-arabinose | - | - | - | - | - | - |
| L-rhamnose | - | - | - | - | - | ND |
| Lactose | - | - | - | - | - | - |
| Glucose | - | + | - | - | - | + |
| L-sorbose | - | - | - | + | - | ND |
| Mannitol | - | + | - | - | - | - |
| Sucrose | - | + | - | - | - | - |
| Amygdalin | - | + | - | - | - | ND |
| Inositol | - | + | - | - | - | - |
| G + C content (%) | 41.6 | 42.3 | 41.2 | 44.4 | 44.0 | 44.2 |
Strain: 1, Bacillus lumedeirus sp. nov. B190/17T; 2, ‘B. aerolatus’ CX253T; 3, B. wudalianchiensis CCTCC AB 2015266T; 4, B. thermotolerans CCTCC AB 2012108T; 5, B. badius JCM 12228T; 6, B. xiapuensis FJAT-46582T. Five strains were oxidase-negative, and do not hydrolyse casein, sodium thiosulfate, tryptophan and pyruvate. +, Positive; −, negative. ND, not described. Data for B190/17 were taken from this study. Data for strains 2-5 were taken from Chen et al., 2020 and for strain 6 were taken from Liu et al., 2019.
Table 3.
Comparison between the number of Bacillus and related genera species described in literature [13] (last access: 08/09/24) and the species present in the VITEK® 2 database for the BCL card [25].
| Genera | Number of species/group of species in VITEK® 2 database | Number of species described |
|---|---|---|
| Alicyclobacillus | 1 | 29 |
| Aneurinibacillus | 1 | 9 |
| Bacillus | 21 | 111 |
| Brevibacillus | 8 | 33 |
| Geobacillus | 4 | 12 |
| Lysinibacillus | 1 | 22 |
| Paenibacillus | 14 | 310 |
| Virgibacillus | 2 | 34 |
Table 4.
Genomic taxonomy results of B190/17 strain compared to five closely related taxa.
| Strains | 16S rRNA (%) | rpoB (%) | gyrB 的(%) | Ortho ANI (%) | GGDC (%) | Mol GC distance (%) |
|---|---|---|---|---|---|---|
| ‘Bacillus aerolatus’ CX253T | 98.28 | 87.50 | 85.43 | 80.01 | 24.00 | 0.70 |
| Bacillus badius NBRC 15713T | 97.96 | 86.91 | 80.90 | 76.97 | 21.60 | 2.33 |
| Bacillus thermotolerans SGZ8T | 97.21 | 84.09 | NSSF | 73.73 | 20.10 | 2.81 |
| Bacillus wudalianchiensis FJAT 27215T | 98.51 | 87.38 | 81.17 | 78.22 | 22.50 | 0.39 |
| Bacillus xiapuensis FJAT 46582T | 97.63 | 82.91 | NSSF | 72.82 | 21.10 | 2.63 |
ANI, Average Nucleotide Identity; AAI, Average Amino Acid Identity; GGDC, Genome-to-genome Distance Calculator. ‘Species not validly published, according to the List of Procaryotic names with Standing in Nomenclature (Parte et al., 2020). T, Type strain; NSSF, no significant similarity found.
Table 5.
General features of genome sequences of Bacillus lumedeirus sp. nov.
| Characteristics | B190/17 |
|---|---|
| Estimated genome size (pb) | 3,434,160 |
| Cover | 73x |
| G+C content (%) | 41.6 |
| N50 | 219177 |
| L50 | 4 |
| Number of contigs | 89 |
| Number of Subsystems | 305 |
| Number of Coding Sequences | 3544 |
| Number of RNA genes | 159 |
bp, base pair; RNA, ribonucleic acid.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



