Submitted:
25 October 2024
Posted:
25 October 2024
Read the latest preprint version here
Abstract
The volatility of global energy markets, particularly electricity prices, plays a crucial role in influencing international economic activities. In the era of big data, machine learning has revolutionized the field of cancer research, particularly in analyzing gene expression data. This study explores the application of machine learning models to the GSE45827 dataset, which contains breast cancer gene expression profiles. With over 54,000 genes and 151 samples categorized into six classes, the dataset presents a high-dimensional challenge that is addressed using dimensionality reduction techniques such as Principal Component Analysis (PCA) and t-distributed Stochastic Neighbor Embedding (t-SNE). The PCA method proved most effective in retaining the critical features of the data in lower dimensions, allowing for clearer visualization and enhanced model performance.The reduced dataset was then classified using the eXtreme Gradient Boosting (XGBoost) model, achieving promising multi-class classification results. The model demonstrated high precision, recall, and F1-scores across several classes, particularly excelling in classes 1, 2, and 5. However, certain classes, such as 0 and 4, exhibited lower recall, highlighting areas for further refinement. The integration of PCA and XGBoost not only improved the interpretability and computational efficiency of the model but also contributed to the accurate identification of breast cancer subtypes, emphasizing the importance of machine learning in cancer diagnosis and treatment.
Keywords:
XGBoost
; PCA
; t-SNE
; machine learning
; cancer gene expression
1. Introduction
In the era of big data, the fields of genomics and medical research are experiencing a transformative shift thanks to the advent of machine learning techniques. Among the most critical applications of these techniques is the analysis of gene expression data for cancer research. Gene expression data, which captures the activity levels of thousands of genes across various conditions or tissues, holds the key to understanding the complex biological mechanisms underlying cancer development and progression. This understanding is essential not only for diagnosing and predicting the onset of cancer but also for identifying potential therapeutic targets.
One of the significant challenges in utilizing gene expression data effectively is its high dimensionality, which can obscure meaningful biological insights when not handled correctly. Techniques such as Principal Component Analysis (PCA) and t-distributed Stochastic Neighbor Embedding (t-SNE) have been pivotal in reducing this complexity by transforming the high-dimensional data into lower-dimensional spaces that are more interpretable while preserving critical information. These dimensionality reduction techniques not only facilitate a clearer visualization of the data but also enhance the performance of predictive models by eliminating irrelevant features.
Another layer of complexity in cancer research is the heterogeneity of the disease, with many subtypes and stages that can drastically differ in their genetic expressions. Addressing this, advanced machine learning models like eXtreme Gradient Boosting (XGBoost) offer robust multi-class classification capabilities. These models can leverage the intricate patterns hidden in gene expression data to distinguish between different cancer types and their respective stages, providing a powerful tool for precision medicine.
The dataset GSE45827 from the Curated Microarray Database (CuMiDa) focused on breast cancer gene expression, stands as a quintessential example of the application of these sophisticated machine learning tools. Comprising 151 samples across 6 classes and involving 54,676 genes, this dataset provides a rich resource for exploring the genetic underpinnings of breast cancer.
This thesis aims to dive deep into the analysis of the GSE45827 dataset using machine learning models. By employing PCA and t-SNE for data preprocessing and XGBoost for classification, this work seeks to predict disease risk and identify significant biomarkers that are crucial for the diagnosis and treatment of breast cancer. Through this approach, the thesis endeavors to contribute to the broader goal of enhancing the accuracy and efficacy of cancer diagnosis and treatment, harnessing the power of machine learning to unlock new frontiers in medical research.
2. Literature Review
In recent years, machine learning techniques, particularly ensemble models like XGBoost, have played a critical role in cancer classification and gene expression analysis. This literature review explores key contributions to the field, focusing on dimensionality reduction, feature selection, and classification models in breast cancer research.
Zelli et al. demonstrated the effectiveness of XGBoost in classifying tumor types by transforming genomic alterations into a vector space [1]. Their study highlighted how XGBoost outperformed other classifiers, showing high accuracy in distinguishing between different cancer types by capturing genomic variance. Similarly, Hoque et al. employed XGBoost in breast cancer classification, reporting strong performance metrics, particularly in precision and recall [2]. Both studies underscore the robustness of XGBoost in dealing with high-dimensional genomic data, making it a favored choice in cancer diagnosis.
The application of dimensionality reduction techniques, such as Principal Component Analysis (PCA), has been essential in handling large genomic datasets. Song et al. applied PCA to binary genomic data, revealing its utility in reducing the dimensionality while preserving key data characteristics [3]. Laghmati et al. combined PCA with machine learning models for breast cancer prediction, finding that PCA enhanced the model's ability to identify significant patterns in the data [4]. These findings highlight PCA’s critical role in simplifying complex datasets, which is particularly beneficial when using models like XGBoost for classification.
Feature selection is another vital aspect in improving classification performance. Sharma and Mishra performed an extensive analysis of machine learning-based feature selection approaches, emphasizing that optimized feature selection significantly enhances diagnostic accuracy in breast cancer [5]. Nguyen et al. (2019) explored ensemble voting and feature selection techniques in breast cancer prediction, revealing that a combination of methods can lead to more accurate and stable predictions [6]. These studies suggest that integrating feature selection with models like XGBoost can further improve performance by focusing on the most informative features.
Additionally, XGBoost's application is not limited to cancer. Song et al. utilized XGBoost in mining diagnostic markers for COVID-19, demonstrating its versatility in healthcare [7]. This further reinforces its potential in identifying critical biomarkers in diverse diseases, including cancer. Meanwhile, Liew et al. focused specifically on XGBoost-based algorithms for breast cancer, corroborating its effectiveness in classification tasks with complex, high-dimensional data [8].
Other researchers have explored multi-omics data integration. Meng et al. examined dimension reduction techniques for multi-omics data, stressing the importance of PCA and other methods for extracting meaningful insights from large datasets [9]. Zhang et al. discussed the discovery of multi-dimensional modules through integrative analysis of cancer genomic data, further highlighting the potential of combining PCA with machine learning to uncover new patterns in cancer research [10].
Finally, Chiu et al. extended the scope to prostate cancer diagnosis, showing that machine learning techniques, including XGBoost, can enhance diagnostic accuracy when combined with advanced algorithms [11]. This broader application suggests that the findings in breast cancer research may be transferable to other forms of cancer, further validating the role of XGBoost in the medical domain.
The reviewed literature consistently demonstrates that XGBoost, when combined with dimensionality reduction (PCA), feature selection, and integrative multi-omics approaches, offers significant improvements in cancer classification. These studies provide a solid foundation for further exploration into breast cancer gene expression classification and diagnosis, particularly through multi-class and multi-omics data integration.
3. Data and Methods
3.1. Data Introduction
The dataset GSE45827 from the Curated Microarray Database (CuMiDa) specifically focuses on breast cancer gene expression. This dataset comprises 151 samples, which are categorized into 6 distinct classes, potentially representing different subtypes or stages of breast cancer. With an extensive collection of 54,676 genes, this dataset provides a comprehensive basis for in-depth genetic analysis.
CuMiDa is a meticulously curated resource, drawing from over 30,000 studies available in the Gene Expression Omnibus (GEO). The primary objective of CuMiDa is to provide datasets that are uniformly preprocessed and standardized, making them highly suitable for machine learning applications in cancer research. By focusing on quality control measures such as sample quality assessment, removal of unwanted probes, and appropriate background correction and normalization, CuMiDa ensures that the datasets are of high reliability for computational studies.
For research on predicting disease risk or identifying important biomarkers using machine learning techniques, the GSE45827 dataset serves as a valuable resource. By leveraging the homogeneously preprocessed data, you can focus on fine-tuning your models and experimenting with different algorithms to derive meaningful insights into breast cancer gene expression patterns. The ability to download various data representations (such as PCA and t-SNE results) further aids in exploratory data analysis and model interpretation.
In summary, the dataset GSE45827 provides a rich resource for thesis on machine learning applications in cancer gene expression analysis. Its comprehensive gene coverage, coupled with rigorous preprocessing and validation tools from CuMiDa, sets a solid foundation for developing predictive models and identifying biomarkers critical in the context of breast cancer.
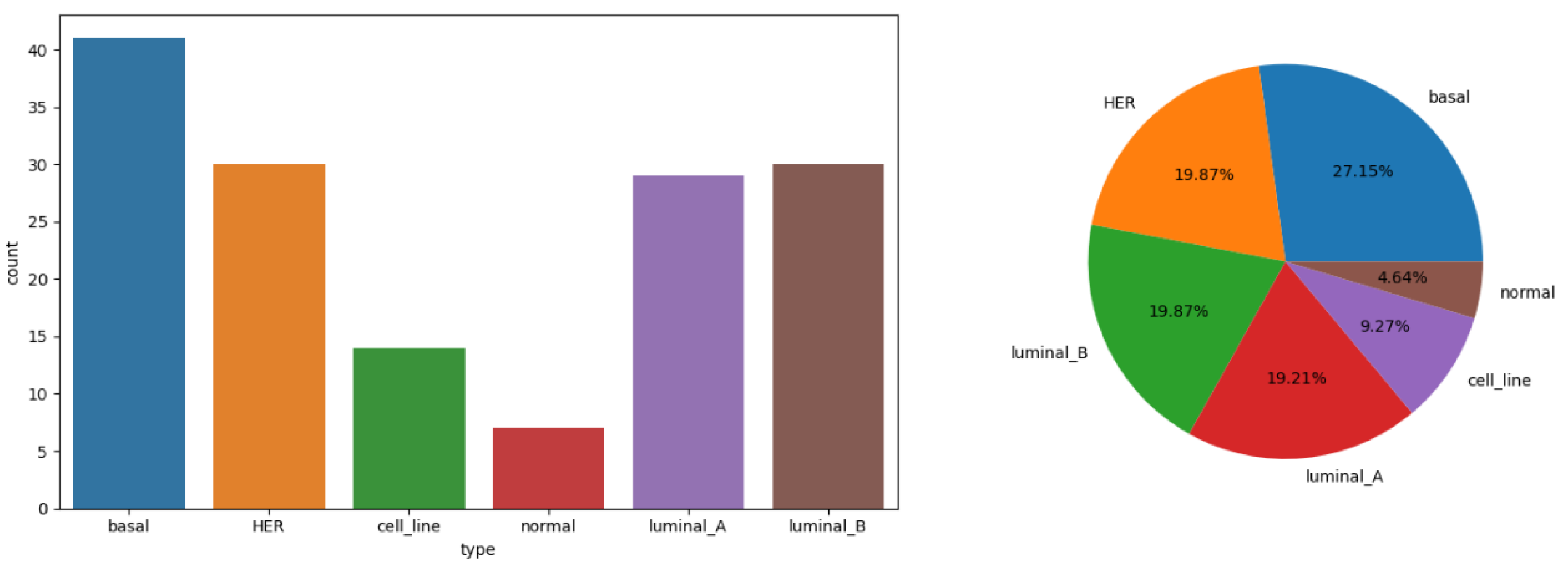
Figure 1.
Statistical chart of explained variable distribution.
3.2. T-Distributed Stochastic Neighbor Embedding
T-SNE is a very powerful technology, which is mainly used for the visualization of high-dimensional data. It reduces the dimension of data by clustering similar data points in high-dimensional space and separating dissimilar data points. The working mechanism of t-SNE is to calculate the similarity between data points in high-dimensional space, which is usually realized by Gaussian joint probability. Then, this similarity is reconstructed in the low-dimensional space, but the T distribution is used. This method is especially suitable for displaying complex multi-class data sets in two or three dimensions. T-SNE is very suitable for exploratory data analysis, because it can reveal the structure in the data, such as which points are closely connected.
3.3. Principal Component Analysis
Principal component analysis (PCA) is a statistical technique, which is used to transform data from possibly highly correlated variables into a set of variables with fewer values through linear transformation. These variables are called principal components. The principle of PCA is to find the direction with the largest variance in data, and then use it as first principal component. Next, the direction orthogonal to first principal component (i.e. uncorrelated) with the largest variance is found as the second principal component, and so on. As a new axis, these principal components can simplify the data structure with the least loss of information, and are often used for data compression and preprocessing.
3.4. Extreme Gradient Boosting
XGBoost is an optimized distributed gradient lifting library, which is designed to realize machine learning algorithm efficiently, flexibly and portable. Although it was originally designed for binary classification problems, XGBoost can also be used for multi-classification problems. In the setting of multi-classification, XGBoost uses the softmax function to extend the output to multiple categories and predict the categories. It uses the gradient lifting framework and adds a new weak prediction model (such as decision tree) in each step to try to correct the prediction error of the previous step. In this way, XGBoost gradually improves the accuracy of its prediction. The advantages of XGBoost include processing various types of data, automatically processing missing values, supporting regularization to prevent over-fitting, and high customization performance and optimization ability.
4. Model Analysis
Each data point in Figure 2 represents the position of a high-dimensional data projected onto these three principal components in 3D space. The data information of different dimensions is condensed into fewer dimensions through these three principal components, thus retaining the main features of the data. The colors of the dots in the picture are different, which means that you may have classified or grouped the data in some way. The distribution of dots with different colors in space indicates the different positions of these groups or categories in the principal component space. Some data points are closely clustered, which may be clusters of similar categories, while others are scattered, showing the difference of data. The fractional data points in the middle of the figure are clustered together, especially in the area near the origin. This shows that these data have strong similarity in the reduced principal component space. There are also a few points far away from other clusters, showing possible outliers or characteristics different from other data. Although the three principal components are all involved in forming the graph, from the graph, PCA-one and PCA-three have a large range of changes, while PCA-two has a relatively small range, which may mean that PCA-one and PCA-three retain more important information in the process of dimensionality reduction, while PCA-two's contribution may be low. To sum up, this 3D scatter plot reveals the data structure after dimensionality reduction by PCA, which helps us to observe the clustering and outliers in the data.
Figure 3 shows the 3D scatter plot of T-SNE dimension reduction, but from the data distribution results in the graph, it is not as good as PCA dimension reduction results, so the subsequent analysis is based on PCA dimension reduction results.
The multi-classification results using the XGBoost model on the dataset provide a detailed insight into the model's performance across different classes of breast cancer gene expression.
Micro Average: Reflects an overall precision of 85%, recall of 72%, and an F1-score of 78%. This average accounts for the total number of true positives, false negatives, and false positives.
Macro Average: Offers an average precision of 88%, recall of 79%, and an F1-score of 81%, without weighting for class imbalance. It provides a general idea of how the model performs across all classes.
Weighted Average: Shows a precision of 89%, recall of 72%, and F1-score of 78%, where each class contribution is weighted by its size. This score is more informative when class imbalance exists.
Samples Average: Records average scores of 69% precision, 72% recall, and 70% F1-score, calculated per sample.
Class 0: Shows moderate precision (50%) but higher recall (67%), indicating that while only half of the predicted positive instances were correct, the model successfully identified 67% of all actual positives. The F1-score, which balances precision and recall, stands at 57%, suggesting room for improvement.
Class 1: Exhibits high precision (93%) and decent recall (78%), leading to a strong F1-score of 85%. This indicates a robust performance in this class, with most positive predictions being correct and a good rate of actual positive identification.
Class 2: This class has a high precision (83%) and perfect recall (100%), resulting in an F1-score of 91%. The model excellently identifies all actual positives in this class, although there might be some over-prediction.
Class 3: Achieves perfect precision (100%) but lower recall (77%), culminating in an F1-score of 87%. The model is precise with no false positives, though it misses some actual positives.
Class 4: Has perfect precision (100%) but low recall (50%), with an F1-score of 67%. While all predictions are accurate, the model fails to identify half of the actual positives.
Class 5: Displays perfect scores in precision, recall, and F1-score (100%), indicating excellent model performance in this class with all positive instances correctly identified and predicted.
5. Conclusions
The application of machine learning models, particularly XGBoost, has shown promising results in the multi-class classification of breast cancer gene expression data. The model's overall performance across six distinct classes reveals its strength in identifying genetic patterns associated with different cancer subtypes. Notably, the model excels in certain classes, such as Class 1, Class 2, and Class 5, with high precision and recall scores, indicating that the model effectively distinguishes these cancer types with minimal error. However, the relatively lower recall in Class 0 and Class 4 suggests the need for further model optimization to avoid missing critical positive cases in these subtypes. Enhancing recall in these classes is essential for developing a robust diagnostic tool that can accurately classify all breast cancer subtypes, especially in real-world clinical applications where missed diagnoses can have significant consequences.
In addition to the classification results, this analysis also highlights the importance of dimensionality reduction techniques like Principal Component Analysis (PCA) in gene expression studies. PCA was employed to tackle the high-dimensional nature of gene expression data, transforming the original dataset into a lower-dimensional space while preserving the most important variance. This step was crucial in improving the interpretability of the data and in reducing computational complexity. By focusing on the most significant principal components, the model could concentrate on key features that contribute most to distinguishing between cancer subtypes, rather than being overwhelmed by irrelevant noise present in the full set of over 54,000 genes.
PCA not only facilitated a more efficient preprocessing workflow but also enhanced the model’s performance by eliminating collinearity and redundant features, which often degrade predictive accuracy. The dimensionality reduction effectively filtered out noise, ensuring that the XGBoost model could work with the most meaningful patterns in the dataset. As a result, PCA was instrumental in improving the stability and generalization ability of the model across various cancer subtypes.
Overall, the combination of PCA for dimensionality reduction and XGBoost for multi-class classification demonstrates a powerful approach to handling the complexity of breast cancer gene expression data. However, future work could explore other advanced dimensionality reduction methods or fine-tune PCA by selecting the optimal number of principal components to further improve classification accuracy. Additionally, techniques such as hyper-parameter tuning, model ensemble methods, or adjusting class weights could help address the recall issues observed in some classes. With these refinements, the predictive model could become an even more reliable tool for precision medicine, aiding in the early detection and targeted treatment of breast cancer based on genetic profiles.
References
- Zelli, V.; Manno, A.; Compagnoni, C. , et al. Classification of tumor types using XGBoost machine learning model: a vector space transformation of genomic alterations. Journal of Translational Medicine 2023, 21, 836. [Google Scholar] [CrossRef] [PubMed]
- Hoque, R.; Das, S.; Hoque, M. , et al. Breast Cancer Classification using XGBoost. World Journal of Advanced Research and Reviews 2024, 21, 1985–1994. [Google Scholar] [CrossRef]
- Song, Y.; Westerhuis, J.A.; Aben, N. , et al. Principal component analysis of binary genomics data. Briefings in bioinformatics 2019, 20, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Laghmati, S.; Hamida, S.; Hicham, K. , et al. An improved breast cancer disease prediction system using ML and PCA. Multimedia Tools and Applications 2024, 83, 33785–33821. [Google Scholar] [CrossRef]
- Sharma, A.; Mishra, P.K. Performance analysis of machine learning based optimized feature selection approaches for breast cancer diagnosis. International Journal of Information Technology 2022, 14, 1949–1960. [Google Scholar] [CrossRef]
- Nguyen, Q.H.; Do, T.T.T.; Wang, Y. , et al. In Breast cancer prediction using feature selection and ensemble voting. In Proceedings of the 2019 International Conference on System Science and Engineering (ICSSE). IEEE; 2019; pp. 250–254. [Google Scholar]
- Song, X.; Zhu, J.; Tan, X. , et al. XGBoost-based feature learning method for mining COVID-19 novel diagnostic markers. Frontiers in Public Health 2022, 10, 926069. [Google Scholar] [CrossRef] [PubMed]
- Liew, X.Y.; Hameed, N.; Clos, J. An investigation of XGBoost-based algorithm for breast cancer classification. Machine Learning with Applications 2021, 6, 100154. [Google Scholar] [CrossRef]
- Meng, C.; Zeleznik, O.A.; Thallinger, G.G. , et al. Dimension reduction techniques for the integrative analysis of multi-omics data. Briefings in bioinformatics 2016, 17, 628–641. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, C.C.; Li, W. , et al. Discovery of multi-dimensional modules by integrative analysis of cancer genomic data. Nucleic acids research 2012, 40, 9379–9391. [Google Scholar] [CrossRef] [PubMed]
- Chiu PK, F.; Shen, X.; Wang, G. , et al. Enhancement of prostate cancer diagnosis by machine learning techniques: an algorithm development and validation study. Prostate cancer and prostatic diseases 2022, 25, 672–676. [Google Scholar] [CrossRef] [PubMed]
Figure 2.
PCA dimension reduction 3D scatter plot.
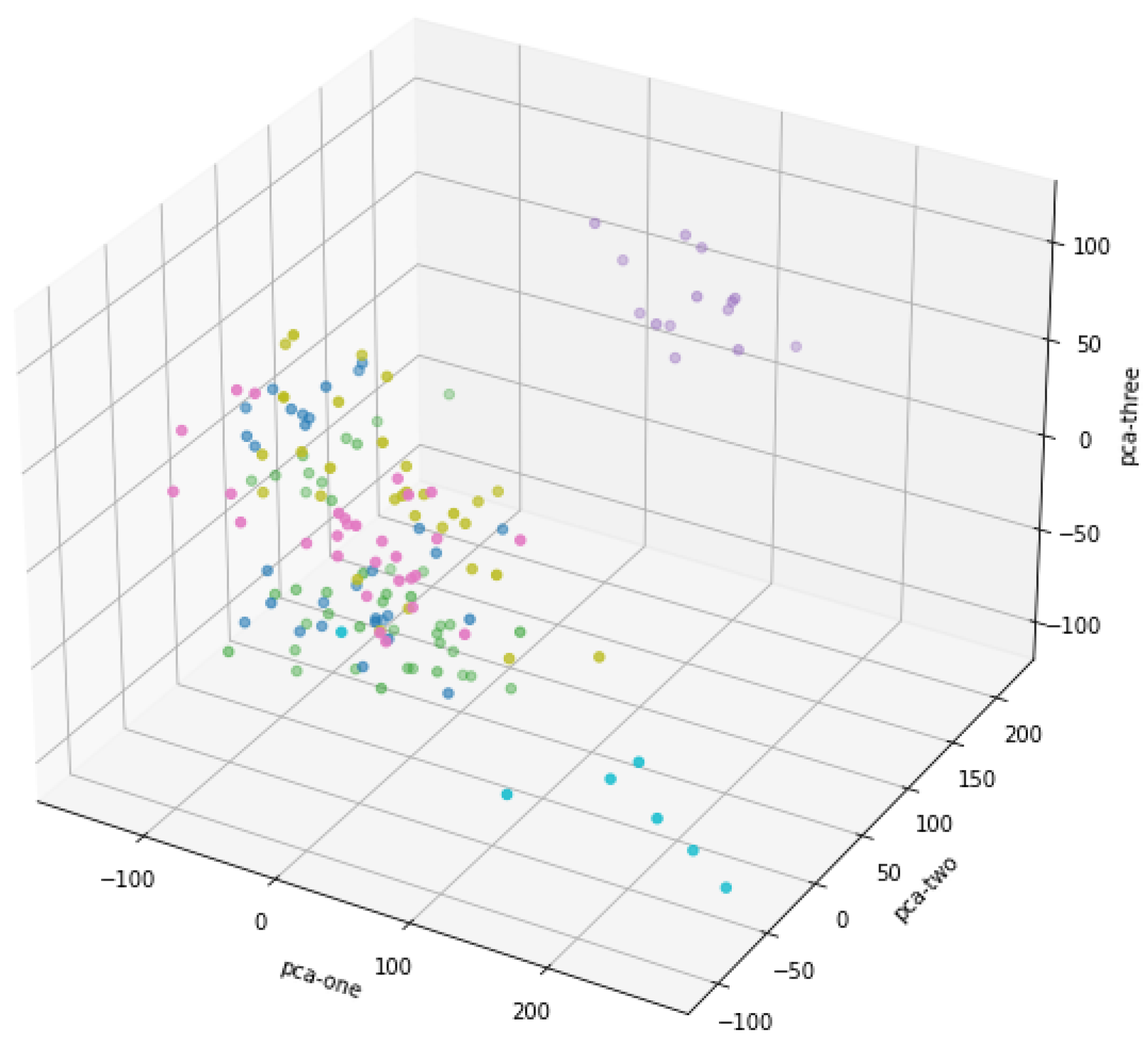
Figure 3.
TSE dimension reduction 3D scatter plot.
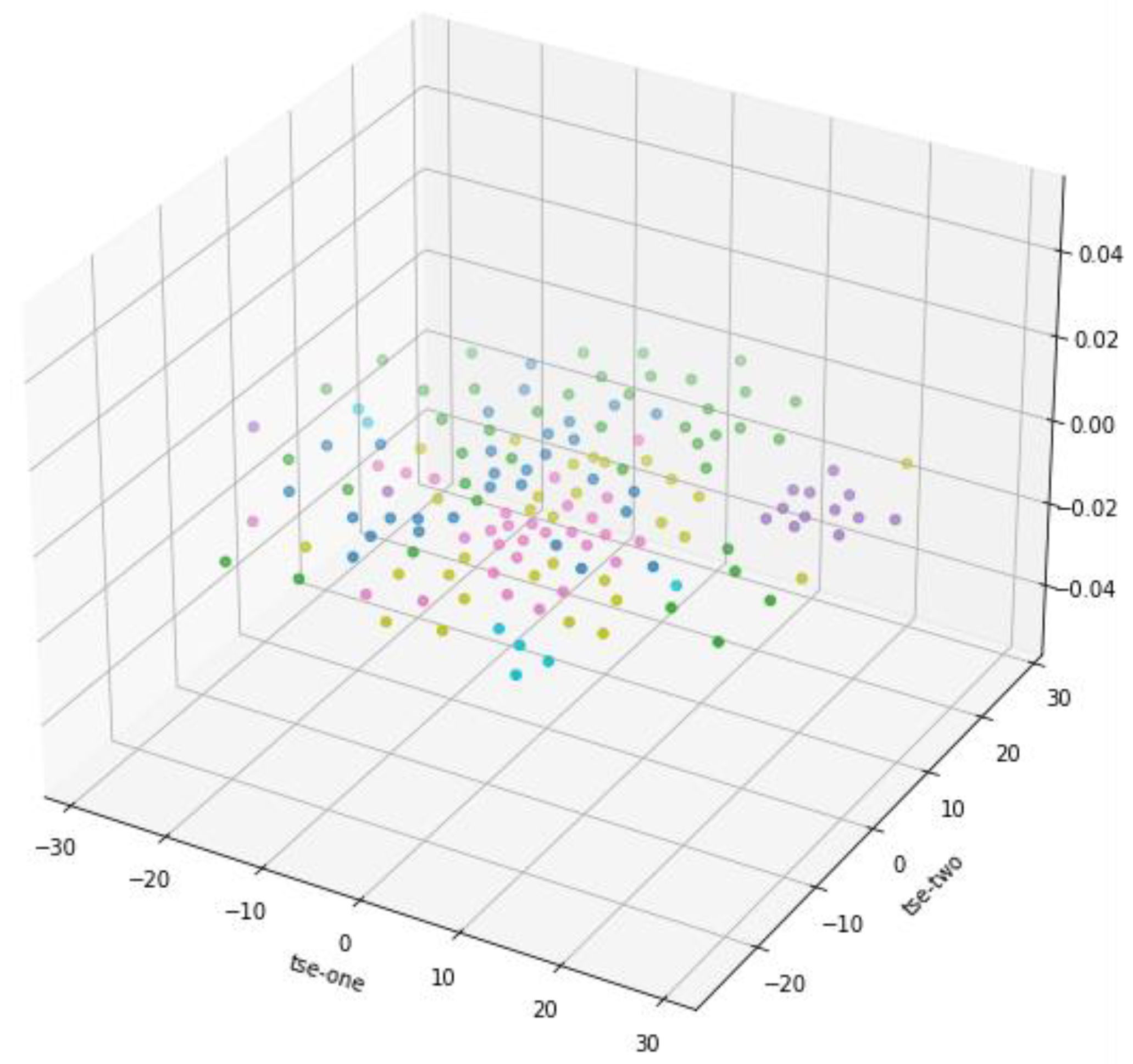

Table 1.
Multi-classification results of XGBoost model.
| Precision | Recall | f1-Score | Support | |
|---|---|---|---|---|
| 0 | 0.50 | 0.67 | 0.57 | 9 |
| 1 | 0.93 | 0.78 | 0.85 | 18 |
| 2 | 0.83 | 1.00 | 0.91 | 5 |
| 3 | 1.00 | 0.77 | 0.87 | 13 |
| 4 | 1.00 | 0.50 | 0.67 | 14 |
| 5 | 1.00 | 1.00 | 1.00 | 2 |
| micro avg | 0.85 | 0.72 | 0.78 | 61 |
| macro avg | 0.88 | 0.79 | 0.81 | 61 |
| weighted avg | 0.89 | 0.72 | 0.78 | 61 |
| samples avg | 0.69 | 0.72 | 0.70 | 61 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



