
Submitted:
24 October 2024
Posted:
25 October 2024
You are already at the latest version
Abstract
Neurological disorders such as Autism Spectrum Disorder (ASD), Schizophrenia (SCH), Bipolar Disorder (BD), and Major Depressive Disorder (MDD) affect millions of people worldwide, yet their molecular mechanisms remain poorly understood. This study applied the Comparative Analysis of Shapley values (CASh) to transcriptomic data from nine datasets associated with these complex disorders, demonstrating its effectiveness in identifying differentially expressed genes (DEGs). CASh, which integrates Game Theory with Bootstrap resampling, offers a robust alternative to traditional statistical methods by evaluating the contribution of each gene within the broader context of the entire dataset. Unlike conventional approaches, CASh is highly effective at detecting subtle but meaningful molecular patterns that are often missed. These findings highlight the potential of CASh to enhance the precision of transcriptomic analysis, providing deeper insights into the molecular signatures of these disorders and establishing a solid basis for improving diagnostic techniques and developing more targeted therapeutic interventions.
Keywords:
autism
; bipolar disorder
; major depressive disorder
; microarrays
; schizophrenia
; systems biology
; transcriptomics
1. Introduction
Complex disorders affecting neurological processes are responsible of great health, social and economic costs worldwide. Despite the heterogeneity of these complex disorders, they all pose a significant global burden, since the misunderstanding of their causes and associated factors that intensify the importance of these phenotypes is the main cause of the insufficiency of diagnosis, and also the lack of effectiveness in medical treatment for patients negatively impacts the well-being of those affected.
Autistic Spectrum Disorder (ASD) (Hirota & King, 2023; Lord et al., 2018) is a phenotype that contains from the most severe autism, when social and communicative functions are very limited, until Asperger syndrome, characterized by mild symptoms. Anyway, all diagnostic features show a rigid behavior, a pathological selection for some issues, and the capacity for attention and communication is affected (Sharma et al., 2018). Some body systems are also affected, as digestive (Martin et al., 2018; Srikantha & Mohajeri, 2019), immune (Heidari et al., 2021; Meltzer & Van de Water, 2017; Ormstad et al., 2018; Robinson-Agramonte et al., 2022), circulatory (Kealy et al., 2020; Mouridsen et al., 2016) and nervous (Doroszkiewicz et al., 2021; Matta et al., 2019; Sharon et al., 2016). Microbiota (Kang et al., 2017) and genetic causes (Qiu et al., 2022) have been proposed in the early development of this disorder, and studies support that risk of suffering ASD rises when relatives are affected (Arberas & Ruggieri, 2019). These symptoms are harmful for patients’ autonomy and the welfare of caregivers (Dissanayake et al., 2019; Ruzzo et al., 2019). The World Health Organization communicated in 2023 that one out of one hundred children are suffering this disorder and prevalence is rising in the previous few years. Due to that, and given the fact that origins and development of this condition are not agreed by specialists and researchers in this field of medicine (Silverman et al., 2010; Tseng et al., 2022), plenty of research teams are thinking of strategies in order to discover the etiology and main factors for understanding this disorder.
Schizophrenia is a neurological disorder that is characterized by positive (hallucinations, lack of social skills and cognitive distortions) and negative (general apathy, in social and job issues) (Khan et al., 2013) symptoms. This disease is linked to increased vulnerability to cardiovascular (Barnett et al., 2007; Hagi et al., 2021), metabolic (Trubetskoy et al., 2022) and infectious (Fuglewicz et al., 2017) diseases, which rise the risk of an early death. Furthermore, it has a direct link with suicide index growth (Balhara & Verma, 2012; Carlborg et al., 2010; Sher & Kahn, 2019), and also caregivers and nearly people are negatively affected in social terms, since patients suffer from diminished autonomy (Gulayín, 2022; Ribé et al., 2018). Its prevalence worldwide is 24 million people (McGrath et al., 2008; Saha et al., 2005), with a percentage of 0.32%, 0.45% in adults (World Health Organization, 2022), and it tends to appear in teenagers at an advanced age, probably with bonds to neural pruning (Germann et al., 2021). Up to date, the origin of this disorder remains unknown (Pino et al., 2014). In this line, there is some consensus in the relevance of some gene factors implied in its onset (Trubetskoy et al., 2022), but that are not determinant to its origin (Vilain et al., 2013), considering that other factors as social environment, drug abuse (including alcohol) (Häfner & an der Heiden, 1997; Janoutová et al., 2016; Stilo & Murray, 2019), and neural pruning, usual in adolescence, can influence in a decisive way (Germann et al., 2021). Myelin sheaths (Valdés-Tovar et al., 2022) and central nervous system architecture (Bobilev et al., 2020; Cheng et al., 2022; Heckers & Konradi, 2002) are also bonded with this disorder. The most extended belief nowadays is that schizophrenia is a multifactorial disorder (Kahn & Sommer, 2015; Morera-Fumero & Abreu-Gonzalez, 2013). In order to clarify the disease causes, omics techniques as transcriptomics have been applied (Gandal et al., 2018). Nevertheless, this is a pathological situation that harms in a severe way the life quality of patients, what generates a medical and social interest that concerns to pharma industry, which intends to alleviate this suffering with drugs that minimize the secondary effects associated to available treatments (Krause et al., 2018), usually adverse for daily life of patients (Perlick et al., 2010). Thus, efficient research is crucial to solve the social and economic problems attached to this disease (Evans-Lacko et al., 2014).
Bipolar disorder (BD) (Smith et al., 2012; Tondo et al., 2017) is a neurological condition characterized by the alternance of maniac episodes (euphoria, excessive joy, uncontrolled enthusiasm, etc.) with depressive ones (anhedonia, sadness, lack of interest for living, etc.) (Fagiolini et al., 2015). Genetic causes have been studied (Gandal et al., 2018), and some environmental factors as alcoholism and other types of drug abuse has been proposed as disease cause (Aldinger & Schulze, 2017). The development of genomics and transcriptomics may help to understand the disorder and treat it efficiently. Its prevalence was 40 millions of people in 2019 (Institute of Health Metrics and Evaluation, 2022), and the suffering rises suicide index for these patients (Beyer & Weisler, 2016; Clemente et al., 2015; Miller & Black, 2020). There is still not much understanding about this disorder, but some drugs, including lithium, have been reported to alleviate its symptoms (Katz et al., 2022; Malhi et al., 2017).
Major depressive disorder (MDD) (Filatova et al., 2021) is a neurologic disease of unknown origin (Gómez Maquet et al., 2020), with more severe symptoms that common depression (Kennedy, 2008). Among these are anhedonia, sadness and lack of desire to live (Bauer et al., 2019). Genetic causes are considered, which has led to the development of transcriptomics and epigenetic studies (Kendall et al., 2021), but also a physiological and hormonal origin are reported, as well as environmental factors like stress, psychological and social aspects (Suda & Matsuda, 2022). Due to the fact that its origin remains unknown, it is classified as a complex disorder (Harder et al., 2022), which causes a great social and economic burden for the community environment of the affected people (Greenberg et al., 2021; Keshavarz et al., 2022). World prevalence is about 350 million of people (Gutiérrez-Rojas et al., 2020; Smith et al., 2019), but there is not much consensus. In fact, this prevalence differs between regions (3% in Japan and 16.3% in USA) (Gutiérrez-Rojas et al., 2020; Smith et al., 2019). Every year even 850,000 suicides have been registered because of major depressive disorder (Li et al., 2022; Serra et al., 2022). Different techniques, such as omics ones and neuroimage, and several biomarkers, as certain fatty acids and miRNA have been used, but there is no consensus (Figueroa-Hall et al., 2020; Gadad et al., 2018; Zhou et al., 2019). Nowadays, there are lots of medicines that treat this disease, taking advantage of the limited knowledge we have about encephalon.
Despite high prevalence worldwide, the origins of these disorders are still unknown. Because of that, it is necessary to apply techniques able to detect key factors for prevention and treatment, pointing towards its main causes and improving as much as possible the health and quality of life of these patients.
Advances in omics technologies, particularly microarray analysis, have revolutionized the comprehensive exploration of gene expression patterns associated with neurological conditions (Bettencourt et al., 2023; Legati et al., 2021; Xu et al., 2022). Microarray technology enables the simultaneous measurement of thousands of genes, providing deep insights into the dysregulated molecular pathways implicated in the pathogenesis of various diseases (Bryant et al., 2004; Copland et al., 2003; Krokidis & Vlamos, 2018; Rai et al., 2016; Ward, 2006). A critical aspect of microarray data analysis is the identification of differentially expressed genes (DEGs), which serve as key indicators in understanding disease mechanisms. Traditionally, these analyses have relied on ranking genes based on individual p-values; however, this approach does not always correlate with biological significance. In some cases, very small p-values, indicative of high statistical significance, may not correspond to biologically relevant signals, while larger p-values, often disregarded, could be linked to genes crucial for specific biological processes (Esteban & Wall, 2011). Classical microarray analysis methods typically utilize Welch’s t-test and linear models such as Empirical Bayes to identify DEGs by comparing gene expression levels between experimental groups or conditions (Jeffery et al., 2006; Selvaraj & Natarajan, 2011). However, these traditional approaches may miss significant gene expression changes, particularly in complex diseases like those affecting the brain, which are characterized by heterogeneous molecular profiles (Ganapathy et al., 2019; Villani & Marzetti, 2023).
To address the limitations of p-value-based approaches, which often result in the excessive suppression of biologically relevant signals due to multiple testing correction methods, more robust methodologies have been developed (Breitling & Herzyk, 2005; Cordero et al., 2007; Esteban & Wall, 2011). Notably, one such method integrates Game Theory, utilizing a computational index known as the Shapley value (Esteban & Wall, 2011). This approach provides a more nuanced assessment of gene significance by evaluating the cumulative contribution of each gene within the context of the entire gene set analyzed. The Shapley value quantifies the importance of each gene by considering its contribution in conjunction with the contributions of all other genes in the same experiment (Moretti & Patrone, 2008). By combining Game Theory with traditional statistical analyses, this methodology offers a powerful tool for enhancing the detection and interpretation of meaningful gene expression differences (Esteban & Wall, 2011).
We applied the microarray games methodology in this study, specifically harnessing Shapley values, to analyze gene expression data related to various neurological pathologies. This approach integrates Game Theory to improve the detection and functional analysis of genes involved in complex biological conditions, such as ASD, schizophrenia, bipolar disorder, and major depressive disorder (Esteban & Wall, 2011). By evaluating the average marginal contribution of each gene across all possible coalitions, this technique reveals critical insights into the genetic underpinnings of these diseases, potentially leading to innovative diagnostic and therapeutic strategies. The game-theoretic approach not only enhances the identification of key genetic players but also enriches our understanding of their biological roles within complex, multi-genic pathologies.
To achieve a comprehensive understanding of gene expression profiles associated with four prevalent neurological pathologies, we employed two distinct methods for microarray data analysis: (i) a conventional approach utilizing Welch’s t-test and Empirical Bayes methods, and (ii) a complementary analysis based on the Comparative Analysis of Shapley value (CASh) method, derived from Game Theory. Previous research (Castro-Martínez et al., 2024; Esteban & Wall, 2011) has demonstrated that the CASh method significantly increases the power to detect differentially expressed genes (DEGs), providing a more robust framework for analyzing complex biological data.
2. Materials and Methods
2.1. Microarray Expression Data Acquisition, Processing and Exploratory Analysis
Microarray data were sourced from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/). For dataset selection, raw data from Affymetrix commercial microarrays—specifically, the Affymetrix Human Genome U133A Array (HG-U133A), Affymetrix Human Genome U133 Plus 2.0 Array (HG-U133_Plus_2), and Affymetrix Human Gene 1.1 ST Array [transcript (gene) version] (HuGene-1_1-st)—were prioritized whenever available.
CEL files from three autism datasets—GSE6575, GSE18123, and GSE25507 (Alter et al., 2011; Gregg et al., 2008; Kong et al., 2012); two schizophrenia datasets—GSE17612 and GSE62333 (Cattane et al., 2015; Maycox et al., 2009); two bipolar disorder datasets—GSE5389 and GSE7036 (Matigian et al., 2007; Ryan et al., 2006); and two datasets encompassing schizophrenia, bipolar disorder, and major depressive disorder samples—GSE12654 and GSE53987 (Iwamoto et al., 2004; Lanz et al., 2019) were retrieved from the GEO repository. Raw data were downloaded for each dataset, followed by preprocessing, quality control, and normalization using relative log expression (RLE), normalized unscaled standard error (NUSE), and Robust Multi-Array Average expression measure (RMA) methods. These processes were carried out using the ‘affy’ (version 1.82.0) and ‘affyPLM’ (version 1.80.0) packages in RStudio (version 2021.09.20) (Bolstad et al., 2003; Bolstad et al., 2005; Irizarry et al., 2003). Finally, expression matrices were generated, and samples were classified into experimental and control groups for further analysis (Supplementary Table 1).
Each dataset was processed independently to identify differentially expressed genes (DEGs). Two distinct approaches were employed for differential expression analysis between patients and controls: (i) a conventional approach utilizing Welch’s t-test and Empirical Bayes methods, and (ii) an alternative method based on the Comparative Analysis of Shapley value (CASh) technique.
To gain a comprehensive understanding of gene expression patterns, we applied various exploratory techniques commonly used in microarray data analysis. Principal Component Analysis (PCA), heatmaps, and volcano plots were generated to evaluate the distribution of gene expression patterns. PCA was used to illustrate gene expression distribution at two levels: the entire gene set in each dataset versus the differentially expressed genes identified by CASh analysis (p-value < 0.01). Heatmaps were created to visualize DEGs detected through both Empirical Bayes (raw p-value < 0.05) and CASh (p-value < 0.01) analyses, highlighting the clustering of samples according to disease status. Additionally, volcano plots were employed to compare the p-values obtained from Empirical Bayes and CASh analyses, providing a visual representation of the statistical relationships between the methods used for detecting DEGs (see Supplementary Figures 1, 2, and 3 for further details).
2.1.1. Classical Approaches
Conventional analyses for detecting DEGs were conducted using Welch’s t-test, implemented through the ‘multtest’ (2.60.0) package in RStudio (version 2021.09.0) (Pollard et al., 2005). In microarray experiments, the small number of replicates and the large number of genes typically analyzed pose significant challenges, leading to the issue of low statistical power with ordinary t-tests. This limitation makes t-tests less effective for filtering out regulated genes (Åstrand et al., 2008; Gottardo et al., 2003). Moreover, most multiple testing adjustments tend to be quite conservative, particularly when the number of replicates is small (Gottardo et al., 2003). To address this issue, we employed Bayesian-based methods, specifically the Empirical Bayes approach, as implemented in the Bioconductor ‘limma’ R package (bioconductor.org/packages/release/bioc/html/limma.html).
Significant DEGs were identified following multiple testing correction using the Benjamini & Hochberg method to control for False Discovery Rates (FDR) (Benjamini & Hochberg, 1995).
2.1.2. Comparative Analysis of Shapley Value (CASh) Approach
We utilized the Comparative Analysis of Shapley value (CASh) method to identify DEGs by assessing their cooperative contribution to overall gene expression changes (Moretti et al., 2008). The Shapley value, a concept rooted in Game Theory, quantifies the marginal contribution of each gene to the collective expression changes observed within the dataset (Moretti, 2010). CASh combines the Microarray Game algorithm, which is applied to transcriptomic data from microarray experiments, with the Bootstrap technique that involves random resampling of certain values to mitigate the impact of potential outliers in the data matrix (Cesari et al., 2018; Moretti, 2010; Moretti et al., 2008, 2010). In this approach, gene expression is treated as a cooperative game, where each gene collaboratively contributes to the observed expression changes, providing a more nuanced understanding of gene interactions within the dataset.
In this context, a cooperative game is defined by a set of players (genes) and a characteristic function that assigns a value to each subset of genes, representing their combined contribution. The Shapley value for a gene is calculated as its average marginal contribution across all possible subsets of genes, providing a robust measure of each gene's importance in the study. In the following lines of this section, we reproduce the mathematical formulation as described elsewhere (Castro-Martínez et al., 2024; Esteban & Wall, 2011).
Formally, given a coalitional game for each player the Shapley value is defined by:
where is a permutation of players, is the set of players that precede player in the permutation and is the cardinality of .
We refer to a Boolean matrix (see below) , where is the number of arrays, and the Boolean values 0–1 represent two complementary expression properties, for example, normal expression (coded by 0) and over-expression (coded by 1). Let be the -th column of , we define the support of , denoted by , as the set The microarray game corresponding to is defined as the coalitional game , where is such that is the rate of occurrences of coalition as a winning coalition, i.e., as a superset of the supports in the Boolean matrix ; in formula, , for each , is defined as the value
where is the cardinality of the set with the set of arrays and Since it is computationally too expensive to calculate the Shapley value of game according to relation , Moretti et al. (2008) introduced an easy way to calculate for whatever microarray game We have adapted the scripts (available under request) from these authors run under R (r-project.org/).
In our study, the CASh method was applied to detect DEGs using two levels of stringency by setting cutoff p-values at 0.01 (more restrictive) and 0.05 (less restrictive). These genes were then analyzed to distinguish between over-regulated and under-regulated expression levels, based on standard deviations from the control group. Boolean matrices were constructed to represent these expression states, which were subsequently used to define microarray games and calculate the corresponding Shapley values.
A final matrix was generated from the original data, incorporating the expression levels of a selected number of genes and samples. This matrix included genes with raw p-values below 0.01 or 0.05 and categorized the samples into distinct groups (e.g., patients with specific conditions and healthy controls). To identify over-regulated gene expression levels relative to controls, each continuous value in the gene expression vector was coded as 1 if it was equal to or greater than the mean plus the standard deviation of the control group expressions, and as 0 otherwise. This process resulted in a Boolean matrix with values {0, 1} reflecting these conditions.
A similar approach was employed to identify under-regulated gene expressions, where each value less than the mean minus the standard deviation of control expressions was coded as 1, with all other values coded as 0. This produced another Boolean matrix with rows corresponding to genes and columns to samples. These Boolean matrices were then divided according to sample group distinctions, forming separate matrices for each group. From these matrices, microarray games were defined for each condition, and Shapley values were calculated to assess the significance of each gene’s contribution to the conditions under study.
To mitigate the influence of random high Shapley values, a resampling procedure was applied, similar to that described by Moretti et al. (2008). Bootstrap resampling with 1,000 iterations was performed for each analysis. This method, known as Comparative Analysis of Shapley value (CASh), refines the selection of genes significantly associated with the conditions under investigation.
To further reduce the likelihood of false positives, corrections for multiple testing were applied, and Shapley values were compared against statistically significant thresholds. Additionally, Fold Changes (FC) were evaluated, with genes exhibiting p-values below 0.01 and 0.05 and |FC| > 2 considered statistically significant.
2.2. Gene Set Enrichment Analysis and Functional Annotation
In this study, we utilized the g:Profiler functional profiling tool (version e111_eg58_p18_30541362), specifically the g:GOSt module (https://biit.cs.ut.ee/gprofiler/gost), to perform an extensive analysis of the biological processes and pathways influenced by differentially expressed genes (DEGs). This tool leverages Gene Ontology (GO) terms to create a comprehensive, annotated landscape of gene functions and interactions (Kolberg et al., 2023; Raudvere et al., 2019). Gene Ontology provides a structured vocabulary that classifies and integrates biological data across species into three main categories: biological processes, cellular components, and molecular functions. By inputting the list of DEGs into g:GOSt, the tool maps these genes to known GO terms, allowing us to identify which biological pathways and processes are enriched in these genes. This enrichment analysis is instrumental in understanding the roles these DEGs may play in the specific conditions under study. The g:GOSt module performs its analysis by comparing the input gene list against databases of known gene and protein functions, identifying statistically significant over-representations of specific functions or pathways. This is achieved using various statistical methods, including Fisher’s Exact Test, to calculate enrichment p-values, which help determine which processes or pathways are more involved with the DEGs than expected by chance. The results from g:GOSt not only highlight the predominant biological themes associated with the DEGs but also provide insights into the potential molecular mechanisms driving the disease or condition. For example, if a significant number of DEGs are involved in inflammatory response pathways, it may indicate that inflammation plays a crucial role in the pathology of the condition being studied. Moreover, the outcomes from such analyses can guide experimental design by identifying key pathways that could be targeted for further experimental validation or therapeutic intervention. Tools like g:Profiler are indispensable in the genomic era, enabling researchers to translate large datasets of gene expression information into actionable biological insights (Kolberg et al., 2023; Raudvere et al., 2019).
During the analysis of gene expression data, it is crucial to ensure that transcript identifiers (IDs) are accurately annotated and standardized to official gene symbols. This step is fundamental for consolidating data from different sources and facilitating meaningful biological interpretation. To achieve this, we used the g:Convert tool available on the g:Profiler web server (https://biit.cs.ut.ee/gprofiler/convert). This tool converts various biological identifiers into recognized gene symbols, enhancing the consistency and reliability of genomic data analysis. The g:Convert module supports a wide range of biological identifiers, including Ensembl IDs, UniProt IDs, RefSeq, and others, allowing researchers to input data from various experimental outputs and databases. Upon inputting transcript IDs into g:Convert, the tool maps these IDs to the official gene symbols based on the most up-to-date and comprehensive databases. This ensures that subsequent analyses, such as gene expression profiling or functional enrichment, are conducted on verified and universally accepted nomenclature, thereby reducing the risk of errors and inconsistencies. In cases where transcript names are ambiguous, which can occur due to multiple identifiers for a single gene or updates in genomic databases, we prioritized IDs with the most extensive Gene Ontology (GO) annotations. This approach is based on the rationale that identifiers with more comprehensive annotations are likely more researched and documented, offering a higher degree of reliability. By selecting IDs with the most GO annotations, we aimed to enhance the robustness of our dataset, ensuring that the functional analysis reflects well-supported gene functions and interactions. The use of the g:Convert tool in this manner not only streamlines the process of gene annotation but also significantly enhances the quality of the data being analyzed. Accurate annotation is critical, as it directly impacts the interpretation of biological data and the conclusions drawn from research studies. As gene databases are continually updated and refined, tools like g:Convert are invaluable for maintaining the accuracy and relevance of genomic research, providing researchers with confidence in their analytical outputs (Kolberg et al., 2023; Raudvere et al., 2019).
In addition, we conducted an in-depth analysis of Gene Ontology (GO) categories, which categorize gene products based on their involvement in biological processes (BP), cellular components (CC), and molecular functions (MF). To determine the significance of these GO categories, we applied a rigorous statistical criterion, the Benjamini-Hochberg False Discovery Rate (FDR), requiring that GO terms exhibit an FDR value below 0.05 to be considered significantly enriched. This stringent threshold ensures that only the most robust associations are identified, minimizing the likelihood of false positives. We then identified the top ten significantly enriched GO terms within each category for further investigation. To visually represent the findings, the top ten significantly enriched GO terms in each category were plotted for CASh 0.05 comparisons using the ‘ggplot2’ (version 3.5.1) package in RStudio (Wilkinson, 2011).
3. Results
3.1. Datasets and Samples Analyzed
Gene expression data from nine datasets, encompassing a total of 506 samples, were accessed for this study. Table 1 provides an overview of the main characteristics of these datasets.
To detect differentially expressed genes (DEGs), we employed two distinct strategies. First, conventional methods based on Welch’s t-test and Empirical Bayes were applied. Following this, we conducted an alternative analysis using the CASh method. The conventional approach, utilizing Welch’s t-test and Empirical Bayes, failed to identify any DEGs. In contrast, the CASh method successfully revealed several transcripts when using both 0.01 and 0.05 cutoff p-values for the preselection of DEGs (Table 2). Complete lists of DEGs detected for each dataset after these comparisons are provided in Supplementary Table 2. Our analyses demonstrate that the application of the CASh method significantly improves the detection of differentially expressed genes across the nine datasets analyzed.
3.2. Functional Enrichment Analysis of the Differentially Expressed Genes
Due to the restrictive criteria applied when using the CASh 0.01 method and the FDR correction of p-values, the number of DEGs detected was insufficient to identify significantly enriched pathways associated with some gene sets (data not shown). However, functional enrichment analysis using the DEGs identified with the CASh 0.05 method revealed several significantly enriched processes in the analyzed datasets.
In the Autism Spectrum Disorder datasets (GSE6575, GSE18123, and GSE25507), DEGs were primarily associated with biological processes (BP) such as structure development and cardiac muscle cell development, while cellular components (CC) were mainly related to intracellular organelles (Figure 1).
For the schizophrenia datasets (GSE17612, GSE62333, GSE12654, and GSE53987), the most significantly enriched BPs were related to the regulation of programmed cell death, regulation of primary metabolic processes, and multicellular organism development. The CC results highlighted cytoplasm, nucleoplasm, and extracellular space, while molecular function (MF) analysis identified activities mainly associated with glycine-tRNA ligase activity (Figure 2).
In the bipolar disorder datasets (GSE5389, GSE7036, GSE12654, and GSE53987), gene set enrichment analysis revealed telomeric and metabolic processes as a significantly enriched BP. Additionally, synapse, phosphatidylcholine-sterol O-acyltransferase activator activity and chromosome, and protein binding were identified as significantly enriched CC and MF, respectively (Figure 3).
Finally, in the major depressive disorder datasets (GSE12654 and GSE53987), gene set enrichment analysis highlighted epiboly and wound healing spreading of cells as significantly enriched BPs, while nucleoplasm, site of polarized growth and growth cone were identified as significantly enriched CCs and protein binding as significantly enriched MF(Figure 4).
4. Discussion
Neurological pathologies inflict significant suffering and pose substantial burdens on millions of people worldwide. In recent years, the advent of omics technologies has enabled a comprehensive exploration of the molecular patterns associated with many of the most common neurological conditions. Microarray technology, which emerged nearly three decades ago with the goal of studying whole gene expression profiles, has since provided unprecedented insights into the dysregulated molecular pathways involved in disease pathogenesis (Ducray et al., 2007; Shai, 2006). In the present study, we analyzed data from nine datasets generated using Affymetrix microarray devices, including three datasets from Autism Spectrum Disorder, two from Schizophrenia, two from Bipolar Disorder, and two datasets encompassing samples from Schizophrenia, Bipolar Disorder, and Major Depressive Disorder.
Raw data were downloaded from the GEO public repository, and gene expression files were pre-processed, quality controlled, and normalized. To detect differentially expressed genes (DEGs), we employed two strategies: (i) a traditional approach using classical statistical t-tests and (ii) an alternative approach utilizing the CASh method (Moretti et al., 2008). The traditional t-test approach identified few DEGs, whereas the CASh method revealed a significant number of statistically relevant genes across the nine datasets analyzed. The t-test identifies genes based on their differential expression between two conditions, considering a gene significant when its p-value falls below a pre-established threshold (0.05 adjusted p-value in our study). In contrast, the CASh method not only considers the expression of each gene under two conditions but also evaluates the contribution of each gene across all possible permutations using the Shapley value as a measure. This holistic approach mitigates the impact of confounding variables by considering the overall gene network rather than isolated gene expressions. However, a current limitation of the CASh method is that it does not explicitly account for potential confounding effects, which should be addressed in future applications (Cesari et al., 2018; Moretti et al., 2008, 2010; Sun et al., 2020). In summary, CASh offers a more nuanced understanding of gene interactions and their collective impact on disease pathophysiology.
Interestingly, the functional enrichment analysis of the DEGs detected using the CASh method, confirmed previous findings on the molecular bases of the neurological pathologies studied. For instance, processes related to cardiac muscle cell development in ASD samples are directly linked to vascular abnormalities observed in patients with this phenotype (Yao et al., 2006). In the case of Schizophrenia, the regulation of primary metabolic processes and glycine-tRNA ligase activity emerged as significant processes, which are particularly relevant given the metabolic issues associated with Schizophrenia (Von Hausswolff-Juhlin et al., 2009). Similarly, Bipolar Disorder was linked to several key findings in our study, including the positive regulation of lipoprotein lipase activity and synapse and phosphatidylcholine-sterol O-acyltransferase activator activity, which align with the known association of this disorder with altered fatty acids (Saunders et al., 2016). For Major Depressive Disorder, characterized by inflammation and neurological damage, we identified processes such as "wound healing spreading of cells" and "growth cone" as significant in the context of differential gene expression.
5. Conclusions and Limitations
This study highlights the power of Comparative Analysis of Shapley values (CASh) in revealing complex genetic insights into neurological disorders such as Autism Spectrum Disorder (ASD), Schizophrenia, Bipolar Disorder, and Major Depressive Disorder. CASh has been proven as highly effective in identifying differentially expressed genes, many of which are missed by traditional statistical methods, offering a more nuanced understanding of the molecular mechanisms underlying these conditions. These findings open new opportunities for developing innovative diagnostic and therapeutic strategies that may shed light in the etiology of these complex conditions.
However, several limitations should be considered. The inherent complexity of microarray data—such as noise, batch effects, and variability in sample quality—can introduce biases that affect the accuracy of gene expression analysis, despite the rigorous preprocessing and normalization applied. Additionally, the reliance on public datasets may bring biases related to differences in data collection methods, patient selection, and experimental design, potentially limiting the generalizability of our results. To mitigate these issues, future studies should be conducted to validate the findings by using more diverse cohorts of patients.
Looking ahead, integrating CASh with complementary omics technologies, such as proteomics and metabolomics, promises a more comprehensive view of the pathophysiological processes in brain diseases. This combined approach could significantly improve the development of multi-marker panels, enhancing diagnostic accuracy. Longitudinal studies using CASh could also track disease progression and treatment responses, providing insights into how gene expression evolves over time in relation to disease states.
A further challenge is the computational intensity of CASh, particularly with large datasets. The method requires substantial computational resources, and interpreting Shapley values can be complex. Simplifying the approach—through algorithm optimization or data reduction—would make CASh more accessible for routine clinical and research applications. Additionally, CASh does not account for post-transcriptional modifications or protein-level interactions, which are critical for a complete understanding of disease mechanisms. Future work could address this by integrating CASh with proteomic and metabolomic data to offer deeper insights at the protein level.
Another key limitation is the lack of experimental validation of the identified differentially expressed genes. To confirm the biological relevance of these findings, future studies should incorporate in vitro functional assays, such as gene knockdown or overexpression experiments. Moreover, in vivo studies in animal models would help to further elucidate the roles of these genes in disease mechanisms and assess their potential as therapeutic targets.
Achieving the full potential of CASh will require strong interdisciplinary collaboration. Geneticists, neurologists, oncologists, and bioinformaticians must work together to conduct large-scale studies that validate and refine the gene signatures identified, translating these discoveries into practical clinical applications. By advancing our understanding of the genetic basis of neurological disorders, this research contributes to precision medicine approaches, ultimately improving patient outcomes and reducing the global burden of these conditions.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Exploratory analysis results: Principal Component Analysis (PCA) showing the distribution of gene expression patterns across all the datasets. HPC: hippocampus, PFC: pre-frontal cortex, STR: striatum; Figure S2: Exploratory analysis results: side-by-side volcano plots showing the comparison between the different tests statistics applied to each dataset. HPC: hippocampus, PFC: pre-frontal cortex, STR: striatum; Figure S3: Exploratory analysis results: heatmap showing the distribution of the differentially expressed genes identified by different methods across all the datasets. HPC: hippocampus, PFC: pre-frontal cortex, STR: striatum; Table S1: Technical description of the datasets analyzed in the present study. HPC: hippocampus, PFC: pre-frontal cortex, STR: striatum, BD: bipolar disorder, SCH: schizophrenia, MDD: major depressive disorder; Table S2: Differentially expressed genes obtained for each dataset after statistical analyses. HPC: hippocampus, PFC: pre-frontal cortex, STR: striatum; Table S1: Technical description of the datasets analyzed in the present study. HPC: hippocampus, PFC: pre-frontal cortex, STR: striatum, BD: bipolar disorder, SCH: schizophrenia, MDD: major depressive disorder; Table S3: Functional enrichment analysis of the differentially expressed genes obtained in each dataset through the application of Comparative Analysis of Shapley values with raw p-values 0.01 and 0.05. HPC: hippocampus, PFC: pre-frontal cortex, STR: striatum, BD: bipolar disorder, SCH: schizophrenia, MDD: major depressive disorder.
Author Contributions
Conceptualization, F.J.E. and J.A.C.M.; methodology, F.J.E., E.V. and J.A.C.M.; software, F.J.E. and J.A.C.M.; validation, L.D.B. and F.J.E.; formal analysis, J.A.C.M.; investigation, J.A.C.M., E.V., L.D.B. and F.J.E.; resources, F.J.E.; data curation, E.V., L.D.B. and F.J.E.; writing—original draft preparation, J.A.C.M. and E.V.; writing—review and editing, J.A.C.M., E.V., L.D.B. and F.J.E.; visualization, J.A.C.M.; supervision, E.V., L.D.B., F.J.E.; project administration, F.J.E.; funding acquisition, F.J.E. All authors have read and agreed to the published version of the manuscript.
Funding
The research group receives funding for research from the University of Jaén (PAIUJA-EI_CTS02_2023) and from the Junta de Andalucía (BIO-302). F.J.E. is partially financed by the Ministry of Science and Innovation, the State Research Agency (AEI), and the European Regional Development Fund (ERDF - Ref: PID2021-122991NB-C21).
Data Availability Statement
Microarray data were obtained from Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo) as stated above. The custom scripts used for data analysis are deposited in the public repository Zenodo and are available through https://zenodo.org/records/11222132.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Aldinger, F.; Schulze, T. G. Environmental factors, life events, and trauma in the course of bipolar disorder. Psychiatry Clin. Neurosci. 2017, 71, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Alter, M. D.; Kharkar, R.; Ramsey, K. E.; Craig, D. W.; Melmed, R. D.; Grebe, T. A.; Bay, R. C.; Ober-Reynolds, S.; Kirwan, J.; Jones, J. J.; Turner, J. B.; Hen, R.; Stephan, D. A. Autism and increased paternal age related changes in global levels of gene expression regulation. PloS One 2011, 6, e16715. [Google Scholar] [CrossRef]
- Arberas, C.; Ruggieri, V. Autismo: aspectos genéticos y biológicos. Medicina (B Aires).
- Åstrand, M.; Mostad, P.; Rudemo, M. Empirical Bayes models for multiple probe type microarrays at the probe level. BMC Bioinformatics 2008, 9, 156. [Google Scholar] [CrossRef] [PubMed]
- Balhara, Y. P.; Verma, R. Schizophrenia and suicide. East Asian Arch. Psychiatry 2012, 22, 126–133. [Google Scholar]
- Barnett, A. H.; Mackin, P.; Chaudhry, I.; Farooqi, A.; Gadsby, R.; Heald, A.; Hill, J.; Millar, H.; Peveler, R.; Rees, A.; Singh, V.; Taylor, D.; Vora, J.; Jones, P. B. Minimising metabolic and cardiovascular risk in schizophrenia: Diabetes, obesity and dyslipidaemia. J. Psychopharmacol. 2007, 21, 357–373. [Google Scholar] [CrossRef]
- Bauer, M.; Rush, A. J.; Ricken, R.; Pilhatsch, M.; Adli, M. Algorithms For Treatment of Major Depressive Disorder: Efficacy and Cost-Effectiveness. Pharmacopsychiatry 2019, 52, 117–125. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling The False Discovery Rate—A Practical And Powerful Approach To Multiple Testing. J. R. Stat. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Bettencourt, C.; Skene, N.; Bandres-Ciga, S.; Anderson, E.; Winchester, L. M.; Foote, I. F.; Schwartzentruber, J.; Botia, J. A.; Nalls, M.; Singleton, A.; Schilder, B. M.; Humphrey, J.; Marzi, S. J.; Toomey, C. E.; Kleifat, A. A.; Harshfield, E. L.; Garfield, V.; Sandor, C.; Keat, S.; Tamburin, S.; Frigerio, C. S.; Lourida, I.; the Deep Dementia Phenotyping (DEMON) Network; Ranson, J. M.; Llewellyn, D. J. Artificial intelligence for dementia genetics and omics. Alzheimers Dement. 2023, 19, 5905–5921. [Google Scholar] [CrossRef]
- Beyer, J. L.; Weisler, R. H. Suicide Behaviors in Bipolar Disorder: A Review and Update for the Clinician. Psychiat. Clin. North Am. 2016, 39, 111–123. [Google Scholar] [CrossRef]
- Bobilev, A. M.; Perez, J. M.; Tamminga, C. A. Molecular alterations in the medial temporal lobe in schizophrenia. Schizophr. Res. 2020, 217, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Bolstad, B.; Collin, F.; Brettschneider, J.; Simpson, K.; Cope, L.; Irizarry, R.; Speed, T. P. Quality Assessment of Affymetrix GeneChip Data. En Bioinformatics and computational biology solutions using R and bioconductor 2005, 33–47. [Google Scholar] [CrossRef]
- Bolstad, B. M.; Irizarry, R. A.; Åstrand, M.; Speed, T. P. A Comparison of Normalization Methods for High Density Oligonucleotide Array Data Based on Variance and Bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef]
- Breitling, R.; Herzyk, P. Rank-based methods as a non-parametric alternative of the T-statistic for the analysis of biological microarray data. J. Bioinform. Comput. Biol. 2005, 3, 1171–1189. [Google Scholar] [CrossRef] [PubMed]
- Bryant, P. A.; Venter, D.; Robins-Browne, R.; Curtis, N. Chips with everything: DNA microarrays in infectious diseases. Lancet Infect. Dis. 2004, 4, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Carlborg, A.; Winnerbäck, K.; Jönsson, E. G.; Jokinen, J.; Nordström, P. Suicide in schizophrenia. Expert Review of Neurotherapeutics 2010, 10, 1153–1164. [Google Scholar] [CrossRef] [PubMed]
- Castro-Martínez, J.A.; Vargas, E.; Díaz-Beltrán, L.; Esteban, F.J. Comparative Analysis of Shapley values enhances transcriptomics insights across some common uterine pathologies. Genes 2024, 15, 723. [Google Scholar] [CrossRef] [PubMed]
- Cattane, N.; Minelli, A.; Milanesi, E.; Maj, C.; Bignotti, S.; Bortolomasi, M.; Chiavetto, L. B.; Gennarelli, M. Altered Gene Expression in Schizophrenia: Findings from Transcriptional Signatures in Fibroblasts and Blood. PLoS ONE 2015, 10, e0116686. [Google Scholar] [CrossRef]
- Cesari, G.; Algaba, E.; Moretti, S.; Nepomuceno, J. A. An application of the Shapley value to the analysis of co-expression networks. Appl. Netw. Sci. 2018, 3, 1. [Google Scholar] [CrossRef]
- Cheng, W.; van der Meer, D.; Parker, N.; Hindley, G.; O’Connell, K. S.; Wang, Y.; Shadrin, A. A.; Alnæs, D.; Bahrami, S.; Lin, A.; Karadag, N.; Holen, B.; Fernandez-Cabello, S.; Fan, C. C.; Dale, A. M.; Djurovic, S.; Westlye, L. T.; Frei, O.; Smeland, O. B.; Andreassen, O. A. Shared genetic architecture between schizophrenia and subcortical brain volumes implicates early neurodevelopmental processes and brain development in childhood. Mol. Psychiatry 2022, 27, 5167–5176. [Google Scholar] [CrossRef]
- Clemente, A. S.; Diniz, B. S.; Nicolato, R.; Kapczinski, F. P.; Soares, J. C.; Firmo, J. O.; Castro-Costa, É. Bipolar disorder prevalence: A systematic review and meta-analysis of the literature. Rev. Bras. Psiquiatr. 2015, 37, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Copland, J. A.; Davies, P. J.; Shipley, G. L.; Wood, C. G.; Luxon, B. A.; Urban, R. J. The use of DNA microarrays to assess clinical samples: The transition from bedside to bench to bedside. Recent Prog. Horm. Res. 2003, 58, 25–53. [Google Scholar] [CrossRef] [PubMed]
- Cordero, F.; Botta, M.; Calogero, R. A. Microarray data analysis and mining approaches. Brief. Funct. Genomic. Proteomic. 2007, 6, 265–281. [Google Scholar] [CrossRef]
- Dissanayake, C.; Searles, J.; Barbaro, J.; Sadka, N.; Lawson, L. P. Cognitive and behavioral differences in toddlers with autism spectrum disorder from multiplex and simplex families. Autism Res. 2019, 12, 682–693. [Google Scholar] [CrossRef]
- Doroszkiewicz, J.; Groblewska, M.; Mroczko, B. The Role of Gut Microbiota and Gut–Brain Interplay in Selected Diseases of the Central Nervous System. Int. J. Mol. Sci. 2021, 22, 10028. [Google Scholar] [CrossRef] [PubMed]
- Ducray, F.; Honnorat, J.; Lachuer, J. DNA microarray technology: Principles and applications to the study of neurological disorders. Revue Neurologique 2007, 163, 409–420. [Google Scholar] [CrossRef]
- Esteban, F. J.; Wall, D. P. Using game theory to detect genes involved in Autism Spectrum Disorder. TOP 2011, 19, 121–129. [Google Scholar] [CrossRef]
- Evans-Lacko, S.; Courtin, E.; Fiorillo, A.; Knapp, M.; Luciano, M.; Park, A.L.; Brunn, M.; Byford, S.; Chevreul, K.; Forsman, A. K.; Gulacsi, L.; Haro, J. M.; Kennelly, B.; Knappe, S.; Lai, T.; Lasalvia, A.; Miret, M.; O’Sullivan, C.; Obradors-Tarragó, C.; Rüsch, N.; Sartorius, N.; Švab, V.; van Weeghel, J.; Van Audenhove, C.; Wahlbeck, K.; Zlati, A.; McDaid, D.; Thornicroft, G.; ROAMER Consortium. The state of the art in European research on reducing social exclusion and stigma related to mental health: A systematic mapping of the literature. Eur. Psychiatr. 2014, 29, 381–389. [Google Scholar] [CrossRef]
- Fagiolini, A.; Coluccia, A.; Maina, G.; Forgione, R. N.; Goracci, A.; Cuomo, A.; Young, A. H. Diagnosis, Epidemiology and Management of Mixed States in Bipolar Disorder. CNS Drugs 2015, 29, 725–740. [Google Scholar] [CrossRef]
- Figueroa-Hall, L. K.; Paulus, M. P.; Savitz, J. Toll-Like Receptor Signaling in Depression. Psychoneuroendocrinology 2020, 121, 104843. [Google Scholar] [CrossRef]
- Filatova, E. V.; Shadrina, M. I.; Slominsky, P. A. Major Depression: One Brain, One Disease, One Set of Intertwined Processes. Cells 2021, 10, 1283. [Google Scholar] [CrossRef]
- Fuglewicz, A. J.; Piotrowski, P.; Stodolak, A. Relationship between toxoplasmosis and schizophrenia: A review. Adv. Clin. Exp. Med. 2017, 26, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Gadad, B. S.; Jha, M. K.; Czysz, A.; Furman, J. L.; Mayes, T. L.; Emslie, M. P.; Trivedi, M. H. Peripheral Biomarkers of Major Depression and Antidepressant Treatment Response: Current Knowledge and Future Outlooks. J. Affect. Disord. 2018, 233, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, A.; Mishra, A.; Soni, M. R.; Kumar, P.; Sadagopan, M.; Kanthi, A. V.; Patric, I. R. P.; George, S.; Sridharan, A.; Thyagarajan, T. C.; Aswathy, S. L.; Vidya, H. K.; Chinnappa, S. M.; Nayanala, S.; Prakash, M. B.; Raghavendrachar, V. G.; Parulekar, M.; Gowda, V. K.; Nampoothiri, S.; Menon, R. M.; Pachat, D.; Udani, V.; Naik, N.; Kamate, M.; Devi, A. R. R.; Kunju, P. A. M.; Nair, M.; Hedge, A. U.; Kumar, M. P.; Sundaram, S.; Tilak, P.; Puri, R. D.; Shah, K.; Sheth, J.; Hasan, Q.; Sheth, F.; Agrawal, P.; Katragadda, S.; Veeramachaneni, V.; Chandru, V.; Hariharam, R.; Mannan, A. U. Multi-gene testing in neurological disorders showed an improved diagnostic yield: Data from over 1000 Indian patients. J. Neurol. 2019, 266, 1919–1926. [Google Scholar] [CrossRef] [PubMed]
- Gandal, M. J.; Zhang, P.; Hadjimichael, E.; Walker, R. L.; Chen, C.; Liu, S.; Won, H.; van Bakel, H.; Varghese, M.; Wang, Y.; Shieh, A. W.; Haney, J.; Parhami, S.; Belmont, J.; Kim, M.; Moran Losada, P.; Khan, Z.; Mleczko, J.; Xia, Y.; Dai, R.; Wang, D.; Yang, Y. T.; Xu, M.; Fish, K.; Hof, P. R.; Warrell, J.; Fiztgerald, D.; White, K.; Jaffe, A. E.; Psychencode Consortium; Peters, M. A.; Gerstein, M.; Liu, C.; Iakoucheva, L. M.; Pinto, D.; Geschwind, D. H. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 2018, 362, eaat8127. [Google Scholar] [CrossRef]
- Germann, M.; Brederoo, S. G. , Sommer, I. E. C. Abnormal synaptic pruning during adolescence underlying the development of psychotic disorders. Curr. Opin. Psychiatry 2021, 34, 222–227. [Google Scholar] [CrossRef]
- Gómez Maquet, Y. , Ángel, J. D., Cañizares, C., Lattig, M. C., Agudelo, D. M., Arenas, Á., Ferro, E. The role of stressful life events appraisal in major depressive disorder. Rev. Colomb. Psiquiatr. (Engl. Ed.), 2020, 49, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Gottardo, R. , Pannucci, J. A., Kuske, C. R.; Brettin, T. Statistical analysis of microarray data: A Bayesian approach. Biostatistics 2003, 4, 597–620. [Google Scholar] [CrossRef]
- Greenberg, P. E.; Fournier, A.A.; Sisitsky, T.; Simes, M.; Berman, R.; Koenigsberg, S. H.; Kessler, R. C. The Economic Burden of Adults with Major Depressive Disorder in the United States (2010 and 2018). PharmacoEconomics 2021, 39, 653–665. [Google Scholar] [CrossRef]
- Gregg, J. P.; Lit, L.; Baron, C. A.; Hertz-Picciotto, I.; Walker, W.; Davis, R. A.; Croen, L. A.; Ozonoff, S.; Hansen, R.; Pessah, I. N.; Sharp, F. R. Gene expression changes in children with autism. Genomics 2008, 91, 22–29. [Google Scholar] [CrossRef]
- Gulayín, M. E. Burden in family caregivers of people with schizophrenia: A literature review. Vertex Rev. Argent. Psiquiatr. 2022, XXXIII, 50–65. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Rojas, L.; Porras-Segovia, A.; Dunne, H.; Andrade-González, N.; Cervilla, J. A. Prevalence and correlates of major depressive disorder: A systematic review. Rev. Bras. Psiquiatr. 2020, 42, 657–672. [Google Scholar] [CrossRef] [PubMed]
- Häfner, H.; an der Heiden, W. Epidemiology of schizophrenia. Can. J. Psychiatry 1997, 42, 139–151. [Google Scholar] [CrossRef]
- Hagi, K.; Nosaka, T.; Dickinson, D.; Lindenmayer, J. P.; Lee, J.; Friedman, J.; Boyer, L.; Han, M.; Abdul-Rashid, N. A.; Correll, C. U. Association Between Cardiovascular Risk Factors and Cognitive Impairment in People With Schizophrenia: A Systematic Review and Meta-analysis. JAMA Psychiatry 2021, 78, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Harder, A.; Nguyen, T.D. , Pasman, J. A., Mosing, M. A., Hägg, S., Lu, Y. Genetics of age-at-onset in major depression. Transl. Psychiatry 2022, 12, 124. [Google Scholar] [CrossRef]
- Heckers, S. , Konradi, C. Hippocampal neurons in schizophrenia. J. Neural Transm. (Vienna) 2002, 109, 891–905. [Google Scholar] [CrossRef]
- Heidari, A. , Rostam-Abadi, Y., Rezaei, N. The immune system and autism spectrum disorder: Association and therapeutic challenges. Acta Neurobiol. Exp. 2021, 81, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Hirota, T.; King, B. H. Autism Spectrum Disorder: A Review. JAMA 2023, 329, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Institute of Health Metrics and Evaluation. Global Health Data Exchange (GHDx) 2022.
- Irizarry, R. A.; Bolstad, B. M.; Collin, F.; Cope, L. M.; Hobbs, B.; Speed, T. P. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003, 31, e15. [Google Scholar] [CrossRef]
- Iwamoto, K.; Kakiuchi, C.; Bundo, M.; Ikeda, K.; Kato, T. Molecular characterization of bipolar disorder by comparing gene expression profiles of postmortem brains of major mental disorders. Mol. Psychiatry 2004, 9, 406–416. [Google Scholar] [CrossRef]
- Janoutová, J.; Janácková, P.; Serý, O.; Zeman, T.; Ambroz, P.; Kovalová, M.; Varechová, K.; Hosák, L.; Jirík, V.; Janout, V. Epidemiology and risk factors of schizophrenia. Neuro Endocrinol. Lett. 2016, 37, 1–8. [Google Scholar]
- Jeffery, I. B.; Higgins, D. G.; Culhane, A. C. Comparison and evaluation of methods for generating differentially expressed gene lists from microarray data. BMC Bioinformatics 2006, 7, 359. [Google Scholar] [CrossRef] [PubMed]
- Kahn, R. S.; Sommer, I. E. The neurobiology and treatment of first-episode schizophrenia. Mol. Psychiatry 2015, 20, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.W.; Adams, J. B.; Gregory, A. C.; Borody, T.; Chittick, L.; Fasano, A.; Khoruts, A.; Geis, E.; Maldonado, J.; McDonough-Means, S.; Pollard, E. L.; Roux, S.; Sadowsky, M. J.; Lipson, K. S.; Sullivan, M. B.; Caporaso, J. G.; Krajmalnik-Brown, R. Microbiota Transfer Therapy alters gut ecosystem and improves gastrointestinal and autism symptoms: An open-label study. Microbiome 2017, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Katz, I. R.; Rogers, M. P.; Lew, R.; Thwin, S. S.; Doros, G.; Ahearn, E.; Ostacher, M. J.; DeLisi, L. E.; Smith, E. G.; Ringer, R. J.; Ferguson, R.; Hoffman, B.; Kaufman, J. S.; Paik, J. M.; Conrad, C. H.; Holmberg, E. F.; Boney, T. Y.; Huang, G. D.; Liang, M. H.; Li+ plus Investigators. Lithium Treatment in the Prevention of Repeat Suicide-Related Outcomes in Veterans With Major Depression or Bipolar Disorder: A Randomized Clinical Trial. JAMA Psychiatry 2022, 79, 24–32. [Google Scholar] [CrossRef]
- Kealy, J.; Greene, C.; Campbell, M. Blood-brain barrier regulation in psychiatric disorders. Neurosci. Lett. 2020, 726, 133664. [Google Scholar] [CrossRef] [PubMed]
- Kendall, K. M.; Van Assche, E.; Andlauer, T. F. M.; Choi, K. W.; Luykx, J. J.; Schulte, E. C.; Lu, Y. The genetic basis of major depression. Psychol. Med. 2021, 51, 2217–2230. [Google Scholar] [CrossRef]
- Kennedy, S. H. Core symptoms of major depressive disorder: Relevance to diagnosis and treatment. Dialogues Clin. Neurosci. 2008, 10, 271–277. [Google Scholar] [CrossRef]
- Keshavarz, K.; Hedayati, A.; Rezaei, M.; Goudarzi, Z.; Moghimi, E.; Rezaee, M.; Lotfi, F. Economic burden of major depressive disorder: A case study in Southern Iran. BMC Psychiatry 2022, 22, 577. [Google Scholar] [CrossRef]
- Khan, Z. U.; Martin-Montañez, E.; Muly, E. C. Schizophrenia: Causes and treatments. Curr. Pharm. Des. 2013, 19, 6451–6461. [Google Scholar] [CrossRef]
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Adler, P.; Vilo, J.; Peterson, H. G:Profiler-interoperable web service for functional enrichment analysis and gene identifier mapping (2023 update). Nucleic Acids Res. 2023, 51, W207–W212. [Google Scholar] [CrossRef]
- Kong, S. W.; Collins, C. D.; Shimizu-Motohashi, Y.; Holm, I. A.; Campbell, M. G.; Lee, I.H.; Brewster, S. J.; Hanson, E.; Harris, H. K.; Lowe, K. R.; Saada, A.; Mora, A.; Madison, K.; Hundley, R.; Egan, J.; McCarthy, J.; Eran, A.; Galdzicki, M.; Rappaport, L.; Kunkel, L. M.; Kohane, I. S. Characteristics and predictive value of blood transcriptome signature in males with autism spectrum disorders. PloS One 2012, 7, e49475. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.; Zhu, Y.; Huhn, M.; Schneider-Thoma, J.; Bighelli, I. , Chaimani, A., Leucht, S. Efficacy, acceptability, and tolerability of antipsychotics in children and adolescents with schizophrenia: A network meta-analysis. Eur. Neuropsychopharmacol. 2018, 28, 659–674. [Google Scholar] [CrossRef] [PubMed]
- Krokidis, M. G. , Vlamos, P. Transcriptomics in amyotrophic lateral sclerosis. Front. Biosci. (Elite Ed.) 2018, 10, 103–121. [Google Scholar] [CrossRef]
- Lanz, T. A. , Reinhart, V., Sheehan, M. J., Rizzo, S. J. S., Bove, S. E., James, L. C., Volfson, D., Lewis, D. A., Kleiman, R. J. Postmortem transcriptional profiling reveals widespread increase in inflammation in schizophrenia: A comparison of prefrontal cortex, striatum, and hippocampus among matched tetrads of controls with subjects diagnosed with schizophrenia, bipolar or major depressive disorder. Transl. Psychiatry 2019, 9, 151. [Google Scholar] [CrossRef]
- Legati, A. , Giacopuzzi, E., Spinazzi, M., Lek, M. Editorial: Application of Omics Approaches to the Diagnosis of Genetic Neurological Disorders. Front. Neurol. 2021, 12, 712010. [Google Scholar] [CrossRef]
- Li, X. , Mu, F., Liu, D., Zhu, J., Yue, S., Liu, M., Liu, Y., Wang, J. Predictors of suicidal ideation, suicide attempt and suicide death among people with major depressive disorder: A systematic review and meta-analysis of cohort studies. J. Affect. Disord. 2022, 302, 332–351. [Google Scholar] [CrossRef] [PubMed]
- Lord, C. , Elsabbagh, M., Baird, G., Veenstra-Vanderweele, J. Autism spectrum disorder. Lancet 2018, 392, 508–520. [Google Scholar] [CrossRef] [PubMed]
- Malhi, G. S. , Gessler, D., Outhred, T. The use of lithium for the treatment of bipolar disorder: Recommendations from clinical practice guidelines. J. Affect. Disord. 2017, 217, 266–280. [Google Scholar] [CrossRef] [PubMed]
- Martin, C. R. , Osadchiy, V., Kalani, A., Mayer, E. A. The Brain-Gut-Microbiome Axis. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Matigian, N. , Windus, L., Smith, H., Filippich, C., Pantelis, C., McGrath, J., Mowry, B., Hayward, N. Expression profiling in monozygotic twins discordant for bipolar disorder reveals dysregulation of the WNT signalling pathway. Mol. Psychiatry 2007, 12, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Matta, S. M. , Hill-Yardin, E. L., Crack, P. J. The influence of neuroinflammation in Autism Spectrum Disorder. Brain Behav. Immun. 2019, 79, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Maycox, P. R. , Kelly, F., Taylor, A., Bates, S., Reid, J., Logendra, R., Barnes, M. R., Larminie, C., Jones, N., Lennon, M., Davies, C., Hagan, J. J., Scorer, C. A., Angelinetta, C., Akbar, M. T., Hirsch, S., Mortimer, A. M., Barnes, T. R. E., de Belleroche, J. Analysis of gene expression in two large schizophrenia cohorts identifies multiple changes associated with nerve terminal function. Mol. Psychiatry 2009, 14, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J. , Saha, S., Chant, D., Welham, J. Schizophrenia: A concise overview of incidence, prevalence, and mortality. Epidemiol. Rev. 2008, 30, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, A. , Van de Water, J. The Role of the Immune System in Autism Spectrum Disorder. Neuropsychopharmacology 2017, 42, 284–298. [Google Scholar] [CrossRef]
- Miller, J. N. , Black, D. W. Bipolar Disorder and Suicide: A Review. Curr. Psychiatry Rep. 2020, 22, 6. [Google Scholar] [CrossRef] [PubMed]
- Morera-Fumero, A. L. , Abreu-Gonzalez, P. Role of Melatonin in Schizophrenia. Int. J. Mol. Sci. 2013, 14, 9037–9050. [Google Scholar] [CrossRef]
- Moretti, S. Statistical analysis of the Shapley value for microarray games. Comput. Oper. Res. 2010, 37, 1413–1418. [Google Scholar] [CrossRef]
- Moretti, S. , Fragnelli, V., Patrone, F., Bonassi, S. Using coalitional games on biological networks to measure centrality and power of genes. Bioinformatics 2010, 26, 2721–2730. [Google Scholar] [CrossRef]
- Moretti, S. , Patrone, F. Transversality of the Shapley value. TOP 2008, 16, 1–41. [Google Scholar] [CrossRef] [PubMed]
- Moretti, S. , van Leeuwen, D., Gmuender, H., Bonassi, S., van Delft, J., Kleinjans, J., Patrone, F., Merlo, D. F. Combining Shapley value and statistics to the analysis of gene expression data in children exposed to air pollution. BMC Bioinformatics 2008, 9, 361. [Google Scholar] [CrossRef] [PubMed]
- Mouridsen, S. E. , Rich, B., Isager, T. Diseases of the circulatory system among adult people diagnosed with infantile autism as children: A longitudinal case control study. Res. Dev. Disabil. 2016, 57, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Ormstad, H. , Bryn, V., Saugstad, O. D., Skjeldal, O., Maes, M. Role of the Immune System in Autism Spectrum Disorders (ASD). CNS Neurol. Disord. Drug Targets 2018, 17, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Perlick, D. A. , Rosenheck, R. A., Kaczynski, R., Swartz, M. S., Canive, J. M., Lieberman, J. A. Impact of antipsychotic medication on family burden in schizophrenia: Longitudinal results of CATIE trial. Schizophr. Res. 2010, 116, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Pino, O. , Guilera, G., Gómez-Benito, J., Najas-García, A., Rufián, S., Rojo, E. Neurodevelopment or neurodegeneration: Review of theories of schizophrenia. Actas Esp. Psiquiatri. 2014, 42, 185–195. [Google Scholar]
- Pollard, K. S. , Dudoit, S., van der Laan, M. J. Multiple Testing Procedures: The multtest Package and Applications to Genomics. En R. Gentleman, V. J. Carey, W. Huber, R. A. Irizarry, S. Dudoit (Eds.), Bioinformatics and Computational Biology Solutions Using R and Bioconductor 2005, 249-271. Springer. [CrossRef]
- Qiu, S. , Qiu, Y., Li, Y., Cong, X. Genetics of autism spectrum disorder: An umbrella review of systematic reviews and meta-analyses. Transl. Psychiatry 2022, 12, 249. [Google Scholar] [CrossRef]
- Rai, G. , Rai, R., Saeidian, A. H., Rai, M. Microarray to deep sequencing: Transcriptome and miRNA profiling to elucidate molecular pathways in systemic lupus erythematosus. Immunol. Res. 2016, 64, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Raudvere, U. , Kolberg, L., Kuzmin, I., Arak, T., Adler, P., Peterson, H., Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef]
- Ribé, J. M. , Salamero, M., Pérez-Testor, C., Mercadal, J., Aguilera, C., Cleris, M. Quality of life in family caregivers of schizophrenia patients in Spain: Caregiver characteristics, caregiving burden, family functioning, and social and professional support. Int. J. Psychiatry Clin. Pract. 2018, 22, 25–33. [Google Scholar] [CrossRef]
- Robinson-Agramonte, M. de L. A., Noris García, E., Fraga Guerra, J., Vega Hurtado, Y., Antonucci, N., Semprún-Hernández, N., Schultz, S., Siniscalco, D. Immune Dysregulation in Autism Spectrum Disorder: What Do We Know about It? Int. J. Mol. Sci. 2022, 23, 3033. [Google Scholar] [CrossRef] [PubMed]
- Ruzzo, E. K. , Pérez-Cano, L., Jung, J. Y., Wang, L. K., Kashef-Haghighi, D., Hartl, C., Singh, C., Xu, J., Hoekstra, J. N., Leventhal, O., Leppä, V. M., Gandal, M. J., Paskov, K., Stockham, N., Polioudakis, D., Lowe, J. K., Prober, D. A., Geschwind, D. H., Wall, D. P. Inherited and De Novo Genetic Risk for Autism Impacts Shared Networks. Cell 2019, 178, 850–866. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M. M. , Lockstone, H. E., Huffaker, S. J., Wayland, M. T., Webster, M. J., Bahn, S. Gene expression analysis of bipolar disorder reveals downregulation of the ubiquitin cycle and alterations in synaptic genes. Mol. Psychiatry 2006, 11, 965–978. [Google Scholar] [CrossRef]
- Saha, S. , Chant, D., Welham, J., McGrath, J. A systematic review of the prevalence of schizophrenia. PLoS Med. 2005, 2, e141. [Google Scholar] [CrossRef] [PubMed]
- Saunders, E. F. H. , Ramsden, C. E., Sherazy, M. S., Gelenberg, A. J., Davis, J. M., Rapoport, S. I. Omega-3 and Omega-6 Polyunsaturated Fatty Acids in Bipolar Disorder. J. Clin. Psychiatry 2016, 77, e1301–e1308. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, S. , Natarajan, J. Microarray Data Analysis and Mining Tools. Bioinformation 2011, 6, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Serra, G. , De Crescenzo, F., Maisto, F., Galante, J. R., Iannoni, M. E., Trasolini, M., Maglio, G., Tondo, L., Baldessarini, R. J., Vicari, S. Suicidal behavior in juvenile bipolar disorder and major depressive disorder patients: Systematic review and meta-analysis. J. Affect. Disord. 2022, 311, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Shai, R. M. Microarray tools for deciphering complex diseases. Front. Biosci. 2006, 11, 1414–1424. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S. R. , Gonda, X., Tarazi, F. I. Autism Spectrum Disorder: Classification, diagnosis and therapy. Pharmacol. Therapeut. 2018, 190, 91–104. [Google Scholar] [CrossRef]
- Sharon, G. , Sampson, T. R., Geschwind, D. H., Mazmanian, S. K. The Central Nervous System and the Gut Microbiome. Cell 2016, 167, 915–932. [Google Scholar] [CrossRef] [PubMed]
- Sher, L. , Kahn, R. S. Suicide in Schizophrenia: An Educational Overview. Medicina (Kaunas) 2019, 55, 361. [Google Scholar] [CrossRef]
- Silverman, J. L. , Yang, M., Lord, C., Crawley, J. N. Behavioural phenotyping assays for mouse models of autism. Nat. Rev. Neurosci. 2010, 11, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Smith, D. J. , Whitham, E. A., Ghaemi, S. N. Bipolar disorder. Handb. Clin. Neurol. 2012, 106, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Smith, R. C. , Osborn, G. G., Dwamena, F. C., D’Mello, D., Freilich, L., Laird-Fick, H. S. Major Depression and Related Disorders. En Essentials of Psychiatry in Primary Care: Behavioral Health in the Medical Setting 2019, McGraw-Hill Education. accessmedicine.mhmedical.com/content.aspx? 1163. [Google Scholar]
- Srikantha, P. , Mohajeri, M. H. The Possible Role of the Microbiota-Gut-Brain-Axis in Autism Spectrum Disorder. International Journal of Molecular Sciences 2019, 20, 2115. [Google Scholar] [CrossRef] [PubMed]
- Stilo, S. A. , Murray, R. M. Non-Genetic Factors in Schizophrenia. Curr. Psychiatry Rep. 2019, 21, 100. [Google Scholar] [CrossRef] [PubMed]
- Suda, K. , Matsuda, K. How Microbes Affect Depression: Underlying Mechanisms via the Gut–Brain Axis and the Modulating Role of Probiotics. Int. J. Mol. Sci. 2022, 23, 1172. [Google Scholar] [CrossRef] [PubMed]
- Sun, M. W. , Moretti, S., Paskov, K. M., Stockham, N. T., Varma, M., Chrisman, B. S., Washington, P. Y., Jung, J. Y., Wall, D. P. Game theoretic centrality: A novel approach to prioritize disease candidate genes by combining biological networks with the Shapley value. BMC Bioinformatics 2020, 21, 356. [Google Scholar] [CrossRef] [PubMed]
- Tondo, L. , Vázquez, G. H., Baldessarini, R. J. Depression and Mania in Bipolar Disorder. Current Neuropharmacology 2017, 15, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Trubetskoy, V. , Pardiñas, A. F., Qi, T., Panagiotaropoulou, G., Awasthi, S., Bigdeli, T. B., Bryois, J., Chen, C.Y., Dennison, C. A., Hall, L. S., Lam, M., Watanabe, K., Frei, O., Ge, T., Harwood, J. C., Koopmans, F., Magnusson, S., Richards, A. L., Sidorenko, J., … Schizophrenia Working Group of the Psychiatric Genomics Consortium. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 2022, 604, 502–508. [Google Scholar] [CrossRef]
- Tseng, C.E. J. , McDougle, C. J., Hooker, J. M., Zürcher, N. R. Epigenetics of Autism Spectrum Disorder: Histone Deacetylases. Biol. Psychiatry 2022, 91, 922–933. [Google Scholar] [CrossRef]
- Valdés-Tovar, M. , Rodríguez-Ramírez, A. M., Rodríguez-Cárdenas, L., Sotelo-Ramírez, C. E., Camarena, B., Sanabrais-Jiménez, M. A., Solís-Chagoyán, H., Argueta, J., López-Riquelme, G. O. Insights into myelin dysfunction in schizophrenia and bipolar disorder. World J. Psychiatry 2022, 12, 264–285. [Google Scholar] [CrossRef]
- Vilain, J. , Galliot, A.M., Durand-Roger, J., Leboyer, M., Llorca, P.M., Schürhoff, F., Szöke, A. Environmental risk factors for schizophrenia: A review. L’Encephale 2013, 39, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Villani, E. R. , Marzetti, E. Molecular Signals and Genetic Regulations of Neurological Disorders. Int. J. Mol. Sci. 2023, 24, 5902. [Google Scholar] [CrossRef] [PubMed]
- Von Hausswolff-Juhlin, Y. , Bjartveit, M., Lindström, E., Jones, P. Schizophrenia and physical health problems. Acta Psychiatr. Scand. 2009, 119, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Wang Q, Wei J, Pan Y, Xu S. An efficient empirical Bayes method for genomewide association studies. J. Anim. Breed. Genet. 2016, 133, 253–63. [Google Scholar] [CrossRef] [PubMed]
- Ward, K. Microarray technology in obstetrics and gynecology: A guide for clinicians. Am. J. Obstet. Gynecol. 2006, 195, 364–372. [Google Scholar] [CrossRef]
- Wilkinson, L. ggplot2: Elegant Graphics for Data Analysis by H. WICKHAM. Biometrics 2011, 67, 678–679. [Google Scholar] [CrossRef]
- World Health Organization. Schizophrenia. 2022.
- Xu, J. , Mao, C., Hou, Y., Luo, Y., Binder, J. L., Zhou, Y., Bekris, L. M., Shin, J., Hu, M., Wang, F., Eng, C., Oprea, T. I., Flanagan, M. E., Pieper, A. A., Cummings, J., Leverenz, J. B., Cheng, F. Interpretable deep learning translation of GWAS and multi-omics findings to identify pathobiology and drug repurposing in Alzheimer’s disease. Cell Rep. 2022, 41, 111717. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y. , Walsh, W. J., McGinnis, W. R., Praticò, D. Altered vascular phenotype in autism: Correlation with oxidative stress. Arch. Neurol. 2006, 63, 1161–1164. [Google Scholar] [CrossRef]
- Zhou, X. , Liu, L., Lan, X., Cohen, D., Zhang, Y., Ravindran, A. V., Yuan, S., Zheng, P., Coghill, D., Yang, L., Hetrick, S. E., Jiang, X., Benoliel, J.J., Cipriani, A., Xie, P. Polyunsaturated fatty acids metabolism, purine metabolism and inosine as potential independent diagnostic biomarkers for major depressive disorder in children and adolescents. Mol. Psychiatry 2019, 24, 1478–1488. [Google Scholar] [CrossRef]
Figure 1.
Gene Set Enrichment Analysis results showing the significantly enriched Gene Ontology (GO) terms of the differentially expressed genes in Autism Spectrum Disorder datasets: (a) GSE6575 dataset; (b) GSE18123 dataset; (c) GSE25507 dataset. For each dataset, significantly enriched molecular functions (GO:MF), biological processes (GO:BP) and cellular components (GO:CC) are shown in green, blue and orange, respectively.
Figure 1.
Gene Set Enrichment Analysis results showing the significantly enriched Gene Ontology (GO) terms of the differentially expressed genes in Autism Spectrum Disorder datasets: (a) GSE6575 dataset; (b) GSE18123 dataset; (c) GSE25507 dataset. For each dataset, significantly enriched molecular functions (GO:MF), biological processes (GO:BP) and cellular components (GO:CC) are shown in green, blue and orange, respectively.
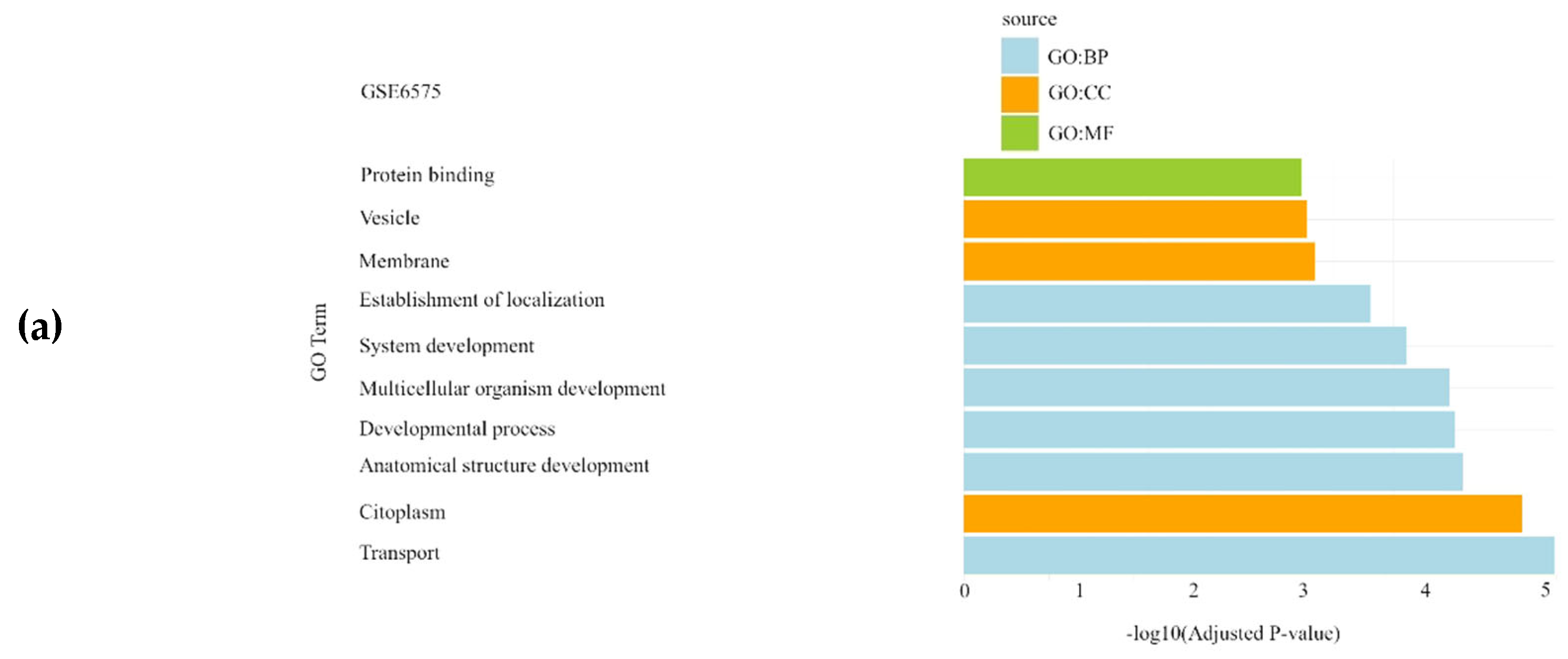
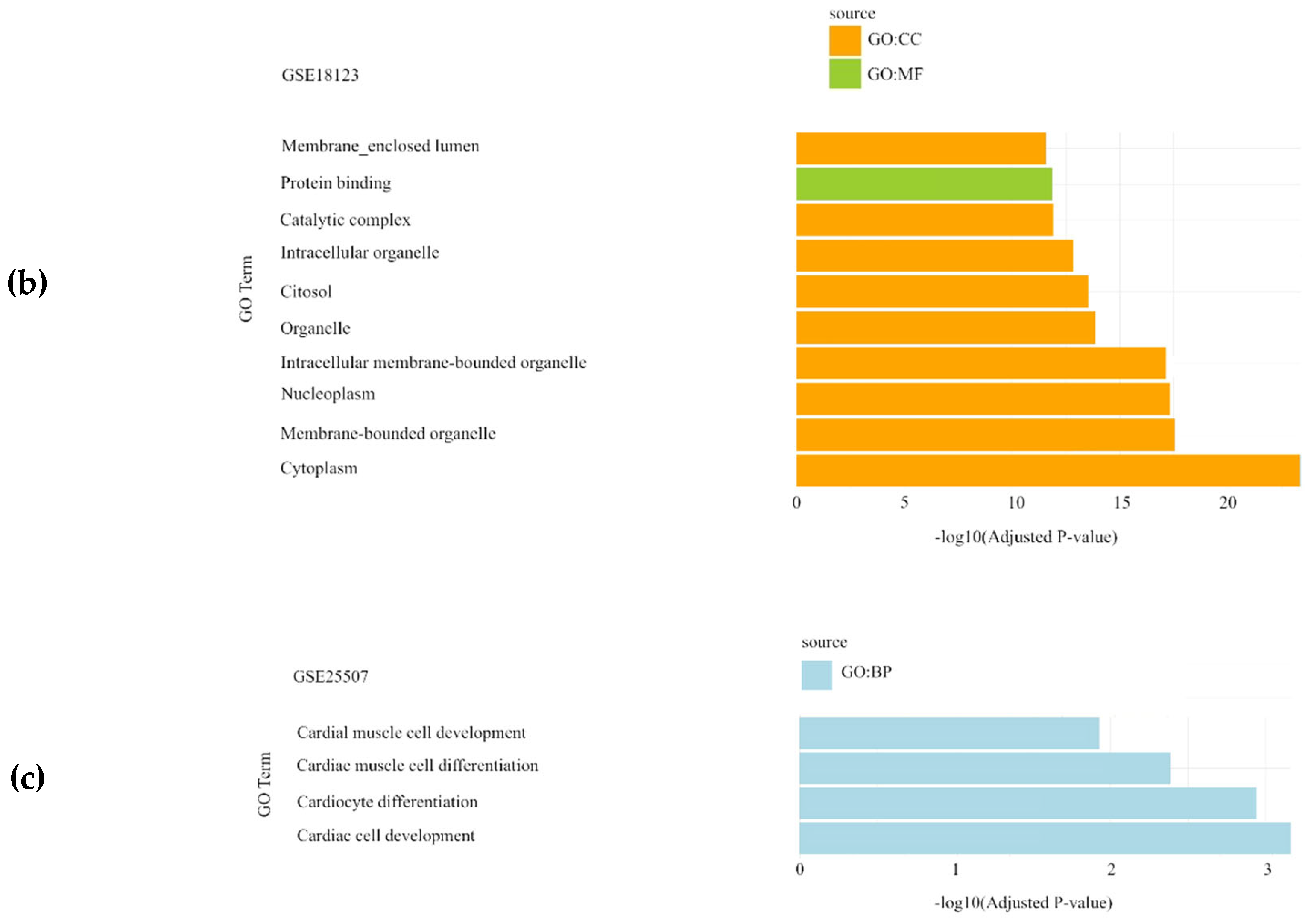
Figure 2.
Gene Set Enrichment Analysis results showing the significantly enriched Gene Ontology (GO) terms of the differentially expressed genes in schizophrenia datasets: (a) GSE62333 dataset; (b) GSE12654_SCH dataset; (c) GSE53987_HPC dataset; (d) GSE53987_PFC dataset; (e) GSE53987_STR dataset. For each dataset, significantly enriched molecular functions (GO:MF), biological processes (GO:BP) and cellular components (GO:CC) are shown in green, blue and orange, respectively. GSE17612 has no results in GO. SCH: schizophrenia; HPC: hippocampus; PFC: pre-frontal cortex; STR: striatum.
Figure 2.
Gene Set Enrichment Analysis results showing the significantly enriched Gene Ontology (GO) terms of the differentially expressed genes in schizophrenia datasets: (a) GSE62333 dataset; (b) GSE12654_SCH dataset; (c) GSE53987_HPC dataset; (d) GSE53987_PFC dataset; (e) GSE53987_STR dataset. For each dataset, significantly enriched molecular functions (GO:MF), biological processes (GO:BP) and cellular components (GO:CC) are shown in green, blue and orange, respectively. GSE17612 has no results in GO. SCH: schizophrenia; HPC: hippocampus; PFC: pre-frontal cortex; STR: striatum.
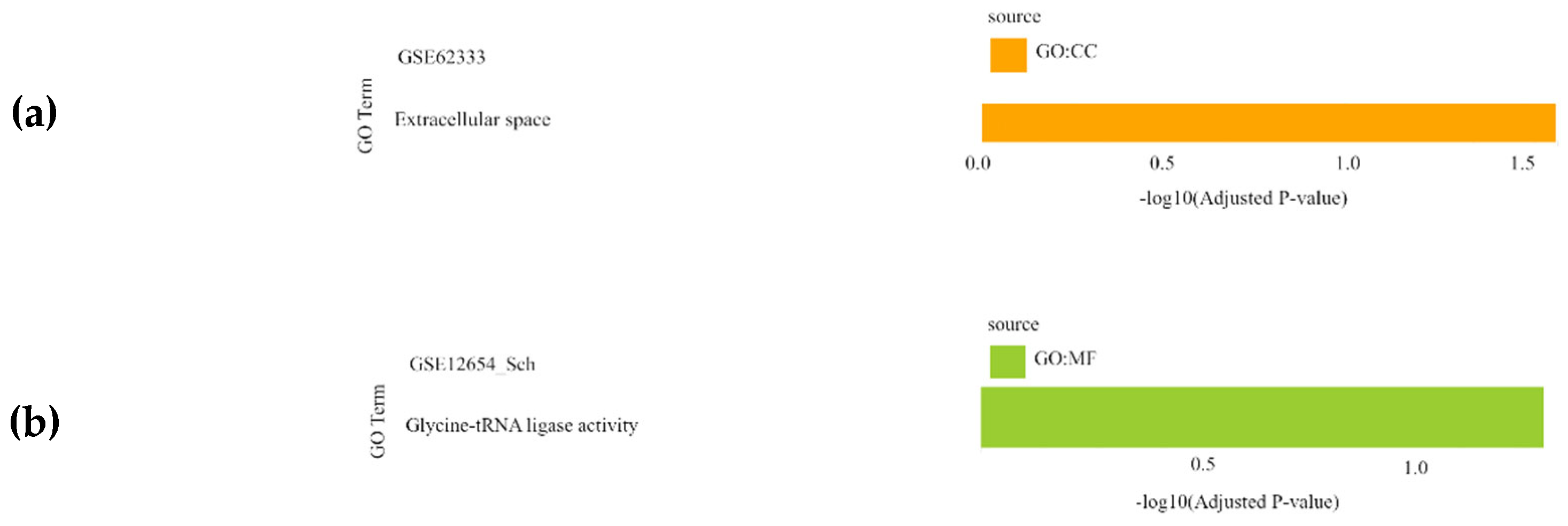
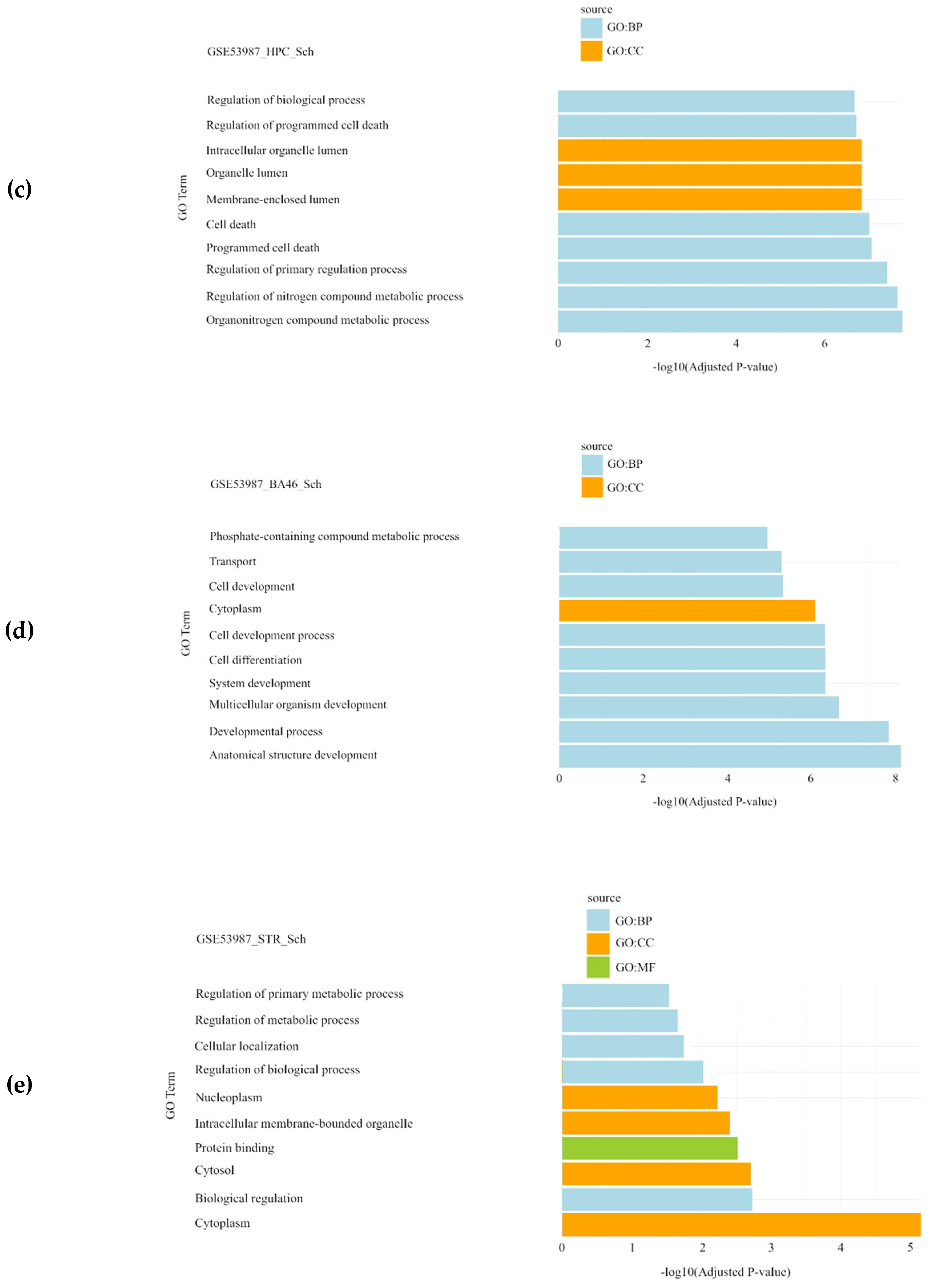
Figure 3.
Gene Set Enrichment Analysis results showing the significantly enriched Gene Ontology (GO) terms of the differentially expressed genes in bipolar disorder datasets: (a) GSE5389 dataset; (b) GSE7036 dataset; (c) GSE53987_PFC dataset. For each dataset, significantly enriched molecular functions (GO:MF), biological processes (GO:BP) and cellular components (GO:CC) are shown in green, blue and orange, respectively. GSE12654, GSE53987_HPC and GSE53987_STR have no results in GO. BD: bipolar disorder; HPC: hippocampus; PFC: pre-frontal cortex; STR: striatum.
Figure 3.
Gene Set Enrichment Analysis results showing the significantly enriched Gene Ontology (GO) terms of the differentially expressed genes in bipolar disorder datasets: (a) GSE5389 dataset; (b) GSE7036 dataset; (c) GSE53987_PFC dataset. For each dataset, significantly enriched molecular functions (GO:MF), biological processes (GO:BP) and cellular components (GO:CC) are shown in green, blue and orange, respectively. GSE12654, GSE53987_HPC and GSE53987_STR have no results in GO. BD: bipolar disorder; HPC: hippocampus; PFC: pre-frontal cortex; STR: striatum.

Figure 4.
Gene Set Enrichment Analysis results showing the significantly enriched Gene Ontology (GO) terms of the differentially expressed genes in major depression datasets: (a) GSE53987_HPC dataset; (b) GSE53987_PFC dataset. For each dataset, significantly enriched molecular functions (GO:MF), biological processes (GO:BP) and cellular components (GO:CC) are shown in green, blue and orange, respectively. GSE12654 and GSE53987_STR have no results in GO. MDD: major depressive disorder; HPC: hippocampus; PFC: pre-frontal cortex; STR: striatum.
Figure 4.
Gene Set Enrichment Analysis results showing the significantly enriched Gene Ontology (GO) terms of the differentially expressed genes in major depression datasets: (a) GSE53987_HPC dataset; (b) GSE53987_PFC dataset. For each dataset, significantly enriched molecular functions (GO:MF), biological processes (GO:BP) and cellular components (GO:CC) are shown in green, blue and orange, respectively. GSE12654 and GSE53987_STR have no results in GO. MDD: major depressive disorder; HPC: hippocampus; PFC: pre-frontal cortex; STR: striatum.
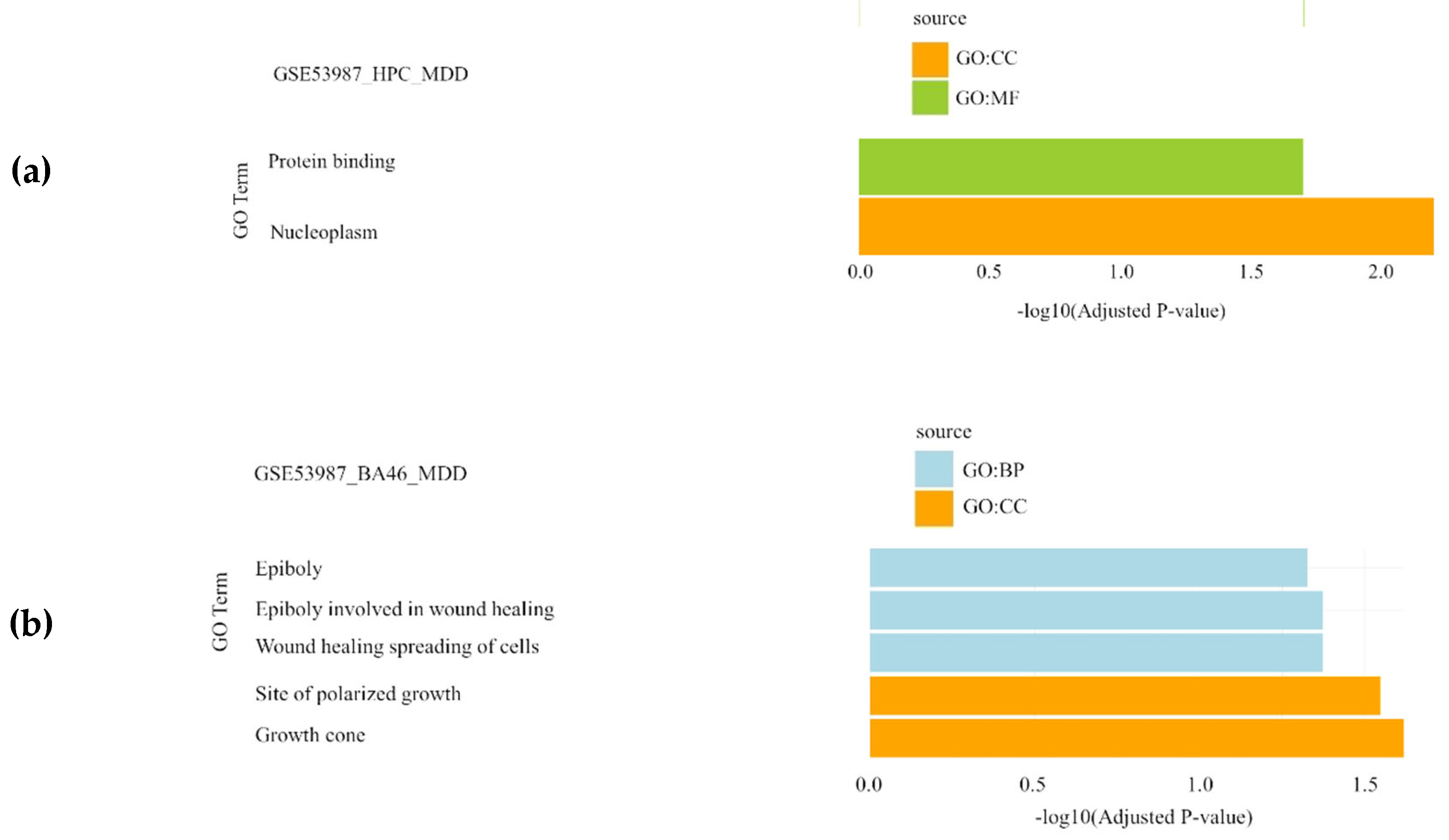

Table 1.
Summary of Gene Expression Omnibus (GEO) datasets analyzed in our study. For each study, number and description of samples are shown. SCH: Schizophrenia; BD: Bipolar Disorder; MDD: Major Depressive Disorder.
Table 1.
Summary of Gene Expression Omnibus (GEO) datasets analyzed in our study. For each study, number and description of samples are shown. SCH: Schizophrenia; BD: Bipolar Disorder; MDD: Major Depressive Disorder.
| Phenotype group | Dataset ID | No. of samples | Description of samples |
|---|---|---|---|
| GSE6575 | 25 | Whole blood autism (n=14) vs. controls (n=11) | |
| Autism | GSE18123 | 23 | Whole blood autism (n=13) vs. controls (n=10) |
| GSE25507 | 26 | Peripheral blood lymphocytes (n=12) vs. controls (n=14) | |
| Schizophrenia | GSE17612 | 30 | Brain tissue (n=17) vs. controls (n=13) |
| GSE62333 | 25 | Skin fibroblasts (n=11) vs. controls (n=14) | |
| Bipolar disorder | GSE5389 | 17 | Brain tissue (n=7) vs. controls (n=10) |
| GSE7036 | 6 | Lymphoblastoid cell lines (n=3) vs. controls (n=3) | |
| Miscellanea (SCH, BD, MDD) | GSE12654 | 38 | Brain tissue (n=24) vs. controls (n=14) |
| GSE53987 | 186 | Brain tissue (n=135) vs. controls (n=51) |
Table 2.
Number of differentially expressed genes (DEGs) detected after the analysis using conventional techniques based on Welch's t-test and Empirical Bayes (EBayes) (Wang et al., 2016), and alternative approaches rooted on Comparative Analysis of Shapley value (CASh) method with cutoff raw p-values of 0.01 or 0.05, respectively. FDR corrected p-values are included where indicated. SCH: Schizophrenia; BD: Bipolar Disorder; MDD: Major Depressive Disorder; HPC: hippocampus; PFC: pre-frontal cortex; STR: striatum.
Table 2.
Number of differentially expressed genes (DEGs) detected after the analysis using conventional techniques based on Welch's t-test and Empirical Bayes (EBayes) (Wang et al., 2016), and alternative approaches rooted on Comparative Analysis of Shapley value (CASh) method with cutoff raw p-values of 0.01 or 0.05, respectively. FDR corrected p-values are included where indicated. SCH: Schizophrenia; BD: Bipolar Disorder; MDD: Major Depressive Disorder; HPC: hippocampus; PFC: pre-frontal cortex; STR: striatum.
| Dataset ID | Welch’s t-test |
EBayes FDR<0.01 | EBayes FDR<0.05 | CASh 0.05 FDR<0.05 |
CASh 0.01 | CASh 0.05 | |
|---|---|---|---|---|---|---|---|
| GSE6575 GSE18123 GSE25507 |
0 | 0 | 0 | 0 | 204 (87 ↑, 117 ↓) | 930 (324 ↑, 606 ↓) | |
| 947 | 205 | 2973 | 45 (12 ↑, 33 ↓) | 879 (467 ↑, 412 ↓) | 1862 (1027 ↑, 835 ↓) | ||
| 0 | 0 | 0 | 0 | 28 (10 ↑, 18 ↓) | 141 (41 ↑, 100 ↓) | ||
| GSE17612 GSE62333 |
0 | 0 | 0 | 0 | 1 (1 ↑, 0 ↓) | 11 (8 ↑, 3 ↓) | |
| 5 | 0 | 5 | 0 | 68 (33 ↑, 35 ↓) | 164 (95 ↑, 69 ↓) | ||
| GSE5389 | 1 | 0 | 2 | 0 | 40 (24 ↑, 16 ↓) | 162 (103 ↑, 59 ↓) | |
| GSE7036 | 0 | 0 | 0 | 0 | 8 (4 ↑, 4 ↓) | 35 (12 ↑, 23 ↓) | |
| GSE12654_SCH | 0 | 0 | 0 | 0 | 2 (2 ↑, 0 ↓) | 8 (4 ↑, 4 ↓) | |
| GSE12654_BD | 0 | 0 | 0 | 0 | 0 | 8 (6 ↑, 2 ↓) | |
| GSE12654_MDD | 0 | 0 | 0 | 0 | 0 | 0 | |
| GSE53987_HPC_SCH | 283 | 2 | 1393 | 4 (0 ↑, 4 ↓) | 794 (595 ↑, 199 ↓) | 655 (357 ↑, 298 ↓) | |
| GSE53987_HPC_BD | 0 | 0 | 0 | 0 | 41 (14 ↑, 27 ↓) | 152 (48 ↑, 104 ↓) | |
| GSE53987_HPC_MDD | 0 | 0 | 0 | 0 | 47 (14 ↑, 33 ↓) | 163 (41 ↑, 122 ↓) | |
| GSE53987_PFC_SCH | 0 | 0 | 32 | 0 | 182 (106 ↑, 76 ↓) | 354 (179 ↑, 175 ↓) | |
| GSE53987_PFC_BD | 0 | 0 | 0 | 0 | 157 (54 ↑, 103 ↓) | 394 (141 ↑, 253 ↓) | |
| GSE53987_PFC_MDD | 0 | 0 | 0 | 0 | 61 (11 ↑, 50 ↓) | 175 (34 ↑, 141 ↓) | |
| GSE53987_STR_SCH | 0 | 0 | 1 | 0 | 81 (36 ↑, 45 ↓) | 258 (139 ↑, 119 ↓) | |
| GSE53987_STR_BD | 0 | 0 | 0 | 0 | 42 (10 ↑, 32 ↓) | 77 (33 ↑, 44 ↓) | |
| GSE53987_STR_MDD | 0 | 0 | 0 | 0 | 19 (8 ↑, 11 ↓) | 32 (12 ↑, 30 ↓) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



