
Submitted:
24 October 2024
Posted:
24 October 2024
You are already at the latest version
Abstract
Adopting an immune response and tissue-based nomenclature in food allergies brings the field closer to precision and personalized medicine. An updated classification of non-IgE-mediated food hypersensitivity highlights mechanisms through a complex pathophysiological perspective and endotype-driven framework. This approach aligns with the essential goals of discovering new diagnostic and therapeutic biomarkers, enhancing the understanding of clinical presentations, and refining immunotherapy and targeted treatments, such as monoclonal antibodies and small molecule drugs, to improve patient management and quality of life. Endotype-focused and molecular-based decision-making also endows translational and clinical research, fostering the ongoing scientific quest for more innovative solutions.
Keywords:
non-IgE-mediated hypersensitivity
; food allergy
; classification
; mechanisms
1. Introduction
The rising prevalence of food allergies worldwide is an important public health concern, significantly impacting the quality of life for patients and their families. Due to current challenges related to both underdiagnosis and overdiagnosis, there is a constant need for accurate diagnosis and personalized management of food allergies. In response to this, the latest EAACI position papers on food allergy should be discussed from the perspective of the new terminology of allergy and hypersensitivity reactions, to provide valuable information for allergists and other medical professionals [1,2,3].
According to the new EAACI guidelines, food allergy is an abnormal or exaggerated reaction to food-related stimuli which involves various types of hypersensitivity reactions engaging antibody-mediated, immune cell-mediated, tissue-driven mechanisms and/or direct response to chemicals, resulting in the development of respiratory, skin, eye, gastrointestinal and other symptoms, including anaphylaxis. A hypersensitivity reaction to food is defined as an adverse reaction to food referring to an undesirable, uncomfortable or damaging response that arises from an immune system overreaction and/or tissue cell dysfunction, causing objectively reproducible symptoms or signs initiated by exposure to a food-related defined stimulus at a dose tolerated by normal individuals [1,4,5].
The term atopy has limited use nowadays as it is based mainly on the symptomatic definition of diseases and does not represent the current understanding of the pathophysiology. In the past, atopy was defined as the personal or familial tendency to produce IgE antibodies in response to low doses of allergens, usually proteins, and to develop typical symptoms such as asthma, rhinoconjunctivitis, or eczema/dermatitis. More recently, it was proposed that atopy may be considered the familial tendency to develop Th2 responses against common environmental antigens, thus keeping both IgE-associated, extrinsic and non-IgE-associated, intrinsic subtypes of atopic dermatitis within the definition of atopy [6,7].
The conventional approach to food allergies involving non-IgE mediated mechanisms according to the latest EAACI guidelines is presented in Table 1 [3].
The need for an additional approach to food allergy classification derives from the new epithelial barrier impairment theory and recent advances in pathophysiological mechanisms, potential novel molecular diagnostic biomarkers related to pathogenic processes or responses to therapeutic interventions, and molecularly targeted pharmacotherapeutic strategies, particularly biologics. An up-to-date classification should incorporate this information using the latest terminology based on the mechanisms of hypersensitivity reactions. The practical implementation of this classification into daily practice needs to be underlined, particularly for non-IgE-mediated food allergies, which are frequently inadequately understood [1,8,9,10,11].
Our expanded classification of food-induced hypersensitivity adverse reactions goes beyond the framework of the traditional approach to food allergies to provide a broader perspective of all the complex and multiple mechanisms that can be engaged in hypersensitivity reactions induced by exposure to foods or food products through oral, inhalation and cutaneous routes. We have focused on non-IgE-mediated hypersensitivity reactions to foods whose pathogenic mechanisms are less known or approached in everyday clinical practice, and we have included them in the new EAACI nomenclature structure, as schematically presented in Figure 1. In addition, the knowledge of foods involved in each type of reaction is necessary for an optimal approach to collecting a patient’s medical history. Understanding the pathophysiology of rare inborn errors of immunity and other genetic disorders presenting with food hypersensitivity reactions and how these monogenic alterations involving disruptions in specific biological processes lead to disease can provide insights into similar pathways involved in common food allergies, highlighting the essential contribution of some molecular pathways in disease pathogenesis. Updated disease endotypes and pathophysiological mechanisms are identified and presented in detail for these food allergy phenotypes.
2. Classification of Hypersensitivity Reactions to Foods Focused on Non-IgE Mediated Mechanisms
2.1. Type I Hypersensitivity Reactions to Foods Involving Associated non-IgE-Mediated Mechanisms
Typical IgE-dependent mast cell-mediated reactions are immediate hypersensitivity reactions induced by IgE specific against allergens. Systemic reactions are triggered when allergens enter the circulation and activate IgE-sensitized extravascular mast cells, causing immediate discharge of pre-stored proinflammatory mediators. Although perivascular macrophages are capable of transendothelial sampling slowly after exposure to blood-borne antigens, these are rapidly adsorbed by the lamellipodia of CD301b+ perivascular dendritic cells that protrude through the endothelial wall into the vessels. Such cells continuously sample blood and spontaneously shed antigen-bound microvesicles (generated by vacuolar protein VPS4) to neighbouring IgE-bearing mast cells in the perivascular region, which vigorously degranulate. Allergen-specific IgE is bound to tissue-resident mast cells and circulating basophils through its high-affinity receptor named FcεRI. The allergen crosslinks surface-bound IgE upon re-exposure, triggering basophil and mast cell anaphylactic degranulation. The spleen tyrosine kinase and Bruton’s tyrosine kinase are essential cytoplasmic non-receptor kinases for signalling through the high-affinity receptor FcεRI on these cells. Activation of intracellular signalling cascades results in rapid degranulation with immediate release of preformed mediators, such as histamine and tryptase, carboxypeptidase A, and proteoglycans, generation of arachidonic acid metabolites (lipid mediators), and later production of inflammatory cytokines and chemokines, responsible for inducing signs and symptoms in systemic allergic reactions to ingested foods [12,13,14].
Regarding such type I immediate hypersensitivity reactions to foods, class 1 food allergy is the primary route of sensitization in the gastrointestinal tract and transcutaneous (with disrupted skin barrier) and manifestations with food ingestion always appear. It is common in children but can also occur in adults, and symptoms with food inhalation are occasional. In class 2 food allergy, IgE sensitization is against cross-reactive allergens present in airborne sources; it is common in adults but can also occur in children/adolescents. Class 3 food allergy has the sensitization route respiratory and transcutaneous (with disrupted skin barrier); it is common in adults at the workplace but can also occur in the domestic setting with food preparation; symptoms with food ingestion are rare and with food inhalation are always present [15,16].
More than 170 foods have been associated with IgE-mediated class 1 food allergies. According to the recent EAACI guidelines, the most commonly involved foods are those traditionally considered the "eight big food allergens": cow's milk, hen's eggs, shellfish, fish, peanuts, tree nuts, wheat, soya, along with other plant allergens, such as legumes, sesame, vegetables and fruits. In Europe, it is mandatory to label food products with the 14 main allergens of plant origins, namely cereals containing gluten, peanuts, soya, tree nuts, lupin, celery, mustard, and sesame, of animal origins, namely cow's milk, eggs, crustaceans, molluscs, fish, and sulfites [3,17,18,19].
Typical manifestations of IgE-mediated food allergy include urticaria, angioedema, oral/pharyngeal pruritus/swelling and other gastrointestinal manifestations, rhinitis and asthma symptoms, and anaphylaxis. Food allergy as an exacerbating factor for asthma is uncommon and occurs primarily in young children. Confirmed food allergy is a risk factor for asthma-related mortality. Food chemicals, such as sulfites, may also trigger asthma symptoms, mainly when asthma is poorly controlled, involving type VII hypersensitivity reactions. Moreover, there is little evidence to support any general involvement of other dietary substances, including benzoate, tartrazine, and monosodium glutamate, in worsening asthma [3,20].
Food allergy dependent on cofactors or augmentation factors involves associated non-IgE-mediated mechanisms. Food- and cofactor/risk factor-induced allergic reactions represent distinctive phenotypes of IgE-mediated food allergy, which appear to be preferentially associated with specific allergens. Their most common and severe phenotype is food anaphylaxis dependent on augmentation factors or food-dependent exercise-induced anaphylaxis (FDEIA). Such food allergic reactions appear within six hours of consumption of the offending food allergen, typically within two to four hours, in the context of a combination of food intake with cofactors, especially exercise, acetylsalicylic acid/non-steroidal anti-inflammatory drugs (NSAIDs) and alcohol consumption. Food-dependent exercise-induced allergic reactions (FDEIAR) produce a limited amount of specific IgE, indicating an intermediate level between asymptomatic (tolerant) sensitized subjects and patients with classic food allergies. Moreover, it was recently suggested that FDEIAR and classic food allergies can coexist in sensitized patients. While FDEIAR can be associated with various foods in different geographic regions, specific sources are more commonly implicated. Wheat is a top food allergen, but fruits and vegetables are also commonly involved [21,22,23,24].
Cofactor-induced anaphylaxis is increasingly associated with wheat allergy and lipid transfer protein allergy. Wheat-dependent exercise-induced anaphylaxis (WDEIA) is the best-described phenotype of FDEIA, more accurately defined as wheat allergy dependent on augmentation factors (WALDA). It is common among young adults and characterized by IgE sensitization to omega-5 gliadin Tri a 19 from hexaploid wheat (Triticum aestivum) as the most specific biomarker. Other wheat allergens involved include non-specific lipid transfer protein (LTP) Tri a 14. LTPs are also significant allergens responsible for other FDEIAR in Italy and other Mediterranean countries [23,24,25]. Although raised levels of serum histamine and tryptase during FDEIA indicate IgE-dependent mast cell degranulation, the pathophysiological mechanisms may include exercise- and NSAIDs-induced increase in intestinal permeability and facilitation of allergen absorption into the circulation, higher core temperature and exercise-induced blood flow redistribution with allergen transport to mast cell-containing tissues, increases in endogenous norepinephrine levels and plasma osmolality. In WDEIA, physical effort after eating produces IL-6 in contracting skeletal muscles, enhancing the expression of tissue transglutaminase enzymes localized beneath the gut epithelium, resulting in food allergen peptides aggregation and amplification of IgE cross-linking. Alcohol has been related to an increase of histamine levels by inhibition of diamino oxidase and increasing extracellular adenosine by inhibition of its uptake. Many of these associated non-IgE-mediated mechanisms have limited evidence, and further studies are needed [23,26,27,28].
Concerning the role of IgG-mediated food allergic reactions, in the particular LTP case, anaphylaxis may be elicited via IgE or both IgE and IgG, with the involvement of neutrophils and not only of mast cells and basophils, although other allergens may act likewise. LTPs are abundant allergens in fruits, vegetables, and nuts, and about 40% of food-related anaphylaxis induced by LTPs requires nonsteroidal anti-inflammatory drugs (NSAIDs) as a cofactor. LTP-specific IgG1 and particularly IgG3 levels are found in patients with LTP anaphylaxis, with notable increases in the expression of markers of activation and trafficking of neutrophils and, to a much lesser degree, in patients with LTP anaphylaxis having NSAID as cofactor. Although patients with NSAID-related LTP-induced anaphylaxis are defined by a baseline repression of IFN–gamma–regulated genes and IFN-gamma levels, patients with NSAID-independent LTP-induced anaphylaxis have an inflammatory-like syndrome with apparent neutrophilic involvement and increased FcγRI expression. Levels of LTP-specific IgG1 and IgG3, ligands for FcγRI, are also increased in patients with NSAID-independent LTP-induced anaphylaxis, which might be considered a potential diagnostic tool [29].
Poppy seeds used for baking and in other food products can be the cause of severe IgE-mediated allergic reactions. However, they can also induce nonimmunological hypersensitivity, and physical exercise may be a cofactor in such reactions. Unwashed or unprocessed poppy seeds can be contaminated with opium alkaloids, such as morphine and codeine, from the latex of the poppy plant. Opioid-induced degranulation of mast cells provokes the release of histamine and other preformed mediators due to mast cell Mas-related G protein-coupled receptor X2 (MRGPRX2) activation, representing a pharmacological-type type VII reaction [1,30,31,32,33].gamma
Cross-reactivity between aeroallergens and foods in patients with respiratory allergy may induce class 2 IgE-mediated food allergy, with manifestations ranging from oral allergy syndrome to severe anaphylaxis. Clinical entities due to IgE sensitization to cross-reactive aeroallergen and food allergen components are described for many sources of plant origin (pollen-food syndromes and associations, such as birch-apple, birch-soy, cypress-peach and celery-mugwort-spice syndromes, and mugwort-peach, mugwort-mustard, ragweed-melon-banana associations) and of invertebrate, mammalian or avian origin (mite-shrimp, cat-pork, and bird-egg syndromes). Clinical cases of IgE-mediated allergic reactions to ingestion of food products containing pollen grains of cross-reactive entomophilous plants in patients with respiratory allergy to anemophilous Asteraceae weed pollen are also mentioned for herbal medicine products, artisanal honey and bee pollen supplements, along with IgE-mediated allergic reactions to foods contaminated with mites or fungi in patients with respiratory allergy to these aeroallergens. Associated non-IgE-mediated mechanisms have also been reported in some of these IgE-mediated syndromes, such as in mite ingestion-associated exercise-induced anaphylaxis. Patients with oral mite anaphylaxis also present an increased prevalence of NSAID hypersensitivity. Even though no salicylates were detected in mite-contaminated wheat flour, the opisthosomal gland secretion from pyroglyphid mites contains 2-formyl-3-hydrobenzyl formate salicylaldehyde analogue [16,23,34,35,36].
Galactose α-1,3-galactose (α-Gal) syndrome is a phenotype of food allergy based on the production of specific IgE antibodies to the α-Gal oligosaccharide. It is characterised by delayed type I allergy to red meat manifested as late-onset anaphylaxis, urticaria or angioedema after ingestion of pig, beef or lamb meat, and systemic allergic reactions with immediate onset at parenteral exposure to drugs significantly containing α-Gal, such as chimeric monoclonal antibodies (e.g. cetuximab), antivenom products, colloidal plasma volume substitutes, vaccines and other drugs containing gelatin and/or other proteins of porcine/bovine origin. Repeated tick bites represent the primary cause of the specific IgE responses to this epitope, the saliva of Ixodidae ticks and their gastrointestinal tract containing the nonprimate mammalian blood group α-Gal carbohydrate. After ingesting α-Gal-containing red meat, the type I hypersensitivity reactions have a late onset, the oligosaccharide having a prolonged chylomicron transport from the human intestine through mesenteric lymph nodes in circulation. Early onset of symptoms has also been reported after red meat consumption with concomitant cofactors such as exercise, acetylsalicylic acid/NSAIDs and alcohol consumption involving associated non-IgE-mediated mechanisms [37,38]. Similarly, subjects with repeated jellyfish stings, such as surfers, are at risk for the development of food allergy to Japanese fermented soybeans (natto). The suspected allergen is the protein poly-γ-glutamic acid (PGA), present in natto bacteria Bacillus subtilis and cnidarian nematocyte capsules. Both delayed and immediate reactions were reported. PGA is a water-soluble, slowly biodegradable, high molecular weight biopolymer. Moreover, the tyramine content in natto might trigger symptoms via a non-immunologic mechanism after natto ingestion [39,40].
Occupational rhinoconjunctivitis/asthma due to inhalation exposure to aerosolized food proteins is a class 3 IgE-mediated food allergy. High-molecular-weight agents, such as wheat flour allergens and some low-molecular-weight agents, such as epigallocatechin gallate from green tea, induce IgE-mediated reactions. Other low-molecular-weight agents used as antioxidants or preservatives are involved through pharmacological-type or not well-characterized mechanisms, such as metabisulfite. Moreover, inhaling vegetable dust from grains and seeds containing endotoxins can induce inflammatory responses, resulting in organic dust toxic syndrome, which commonly presents with asthma-like symptoms and should be differentiated from occupational asthma. While the mechanism for seafood proteins is commonly IgE-mediated, digestive enzymes such as trypsin from pilchard, salmon, and king crab may activate protease-activated receptor-2 (PAR-2) on airway epithelial cells, inducing airway inflammation via IL-8 expression [15,41,42,43,44].
Contact urticaria syndrome (CUS) encompasses several forms of immediate contact skin reactions (ICSR) triggered by foods. It can manifest as immunologic contact urticaria (ICU) and/or protein contact dermatitis (PCD). A wide variety of food compounds are responsible for both occupational and non-occupational exposures. Common causes include high molecular weight food proteins such as animal proteins from fish, shellfish, and meat products and plant-derived proteins from fruits, vegetables, spices, and grains, such as wheat. Smaller molecular weight food processing enzymes like α-amylase can also be implicated. Occupations involving food handling, such as food preparation workers, cooks and chefs, bakers, butchers and meat cutters, are at a higher risk. Lipophilic substances in certain foods can penetrate the skin, especially through the hair follicles. Furthermore, various factors contributing to reduced stratum corneum barrier integrity facilitate the penetration of high molecular weight proteins into the skin, including pre-existing dermatitis such as atopic dermatitis, irritant contact dermatitis, excessive hand washing, chemical damage from detergents, and physical damage from microtraumas, wounds, and burns. Coexisting allergic disorders, such as allergic rhinitis and asthma or eczema, are risk factors for ICU, while atopic dermatitis was reported in up to half of the PCD cases [45,46,47,48]. While ICU is a type I hypersensitivity reaction that occurs in patients with IgE antibodies against specific foods, the pathogenesis of PCD comprises a combination of type I, immediate, IgE-mediated hypersensitivity reaction, type IV, delayed, cell-mediated hypersensitivity reaction, and/or a delayed hypersensitivity reaction due to IgE-bearing Langerhans' cells [47,48,49,50].
2.2. Type II Hypersensitivity Reactions via Antibody-Mediated Cytotoxicity
In these antibody-mediated reactions, antibodies (IgG or IgM) are directed against cellular antigens, resulting in cell destruction without inflammation (e.g., autoimmune hemolytic anaemia, immune thrombocytopenia) or antibody-mediated cellular and tissue damage with inflammation (e.g., gluten ataxia, pemphigus vulgaris) [1,51,52,53].
IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X-linked) syndrome, a rare inborn error of immunity due to loss-of-function mutations in the gene encoding the FOXP3 transcription factor with the lack of and/or impaired function of CD4+CD25+FOXP3+ regulatory T cells (Tregs), is characterized by early-onset dermatitis with food allergies associated with multiorgan autoimmunity. Severe food allergy as a variant of IPEX syndrome may be caused by a deletion in a noncoding region of the FOXP3 gene. Extremely elevated IgE levels accompanied by intense peripheral eosinophilia and evidence of overt Th2 skewing are revealed in IPEX syndrome, along with high rates of autoantibodies typical for autoimmune enteropathy (anti-enterocyte, harmonin and villin autoantibodies), type 1 diabetes mellitus (antibodies against pancreatic islet cells, insulin, or anti-glutamate decarboxylase) and thyroiditis (anti-microsome peroxidase antibodies and anti-thyroglobulin). Many children with IPEX syndrome present food allergies and associated type II hypersensitivity reactions characteristic of autoimmune cytopenias (anti-platelets and anti-neutrophils antibodies, positive Coombs test) [54,55,56,57,58].
Interestingly, the association between celiac disease (CD) and immune thrombocytopenia (ITP) has been reported. Screening for CD is considered in children with ITP, and some patients recovered from ITP after starting a gluten-free diet. Moreover, adult patients with chronic ITP tend to present more frequently CD-related autoantibodies, including anti-gliadin IgG and anti-endomysium IgG [59,60,61].
Gluten ataxia is a sporadic ataxia triggered by the ingestion of gluten, with positive serum antigliadin antibodies, with or without gluten-sensitive enteropathy, usually presenting with pure cerebellar ataxia, responding to a strict gluten-free diet. While developing anti-tissue transglutaminase 2 IgA (anti-TG2) antibodies is linked with the gastrointestinal disease, anti-TG6 IgG and IgA antibodies are prevalent in gluten ataxia, independent of intestinal involvement. There is evidence for cross-reactivity due to molecular mimicry of antigliadin antibodies with the Purkinje cells, a unique type of neurons located in the cerebellar cortex, of cerebellar IgA deposits that contain TG-6, and inflammation as a prominent feature in the pathogenesis of this condition [62,63,64,65,66].
Pemphigus is characterized by acantholysis (loss of keratinocyte to keratinocyte adhesion) induced by the binding of circulating IgG autoantibodies to intercellular adhesion molecules. IgG autoantibodies against desmoglein 3 are characteristic of mucosal pemphigus vulgaris; those against desmoglein 1 are linked to pemphigus foliaceus, while those against desmoglein 1 and desmoglein 3 are linked to mucocutaneous pemphigus vulgaris. Dietary factors have been implicated as inducers of pemphigus vulgaris and pemphigus foliaceus based on case reports. However, the current evidence does not support a robust link between diet and pemphigus. Diet-related pemphigus linked to thiol allyl compounds from garlic, leek and onion may be explained by antibody-mediated immunologic acantholysis related to the inhibition of enzymes that aggregate keratinocytes and disturbance to cell adhesion by the formation of thiol-cysteine bonds with the release of sequestered antigens or stimulation of neoantigen formation. Both thiol allyl compounds and tannins, such as those from red wine, tea and red chilli pepper, may be incorporated into the Malpighian epithelia, leading to nonimmunologic biochemical acantholysis [67,68,69,70,71].
2.3. Type III Hypersensitivity Reactions via Immune Complexes
Type III hypersensitivity reactions are abnormal immune responses mediated by the formation of immune complexes acting as circulating antigen-antibody aggregates, which migrate out of plasma. The immune complex deposition in tissues activates the classical pathway, leading to the release of C3a and C5a, which then recruit monocytes and neutrophils, inflammatory cells that discharge lysosomal enzymes and free radicals at the site of immune complexes, driving inflammatory tissue damage [72].
Hypersensitivity pneumonitis (HP), also known as extrinsic allergic alveolitis, may be induced by food-related sources of exposure, such as foodstuff and food processing. Plant or animal proteins and especially food-associated fungi may cause occupational HP, including nut dust (tiger nut alveolitis), fish dust (fish meal workers' lung), Penicillium glabrum (salami factory workers' lung), P. camemberti, P. roqueforti, P. verrucosum, P. caseifulvum and Aspergillus clavatus (cheese-worker's lung, cheese washer's lung), Botrytis cinerea (wine workers' lung), and Rhizopus/Mucor stolonifer (paprika slicers' lung) [73,74,75,76,77]. HP's acute phase represents a type III allergic reaction, while its chronic phase mainly involves a type IVa reaction. Acute HP is mediated by immune complexes, genetically predisposed individuals exposed to an environmental culprit antigen became sensitized with detected serum specific antibodies, usually IgG, and with subsequent exposures develop episodic lung inflammation with immune complex formation and an influx of neutrophils. Subacute and chronic HP result from CD4+ Th1 lymphocyte-mediated delayed hypersensitivity, causing bronchiolocentric granulomatous lymphocytic alveolitis. This requires, among others, the expression of Th1 cytokines, including TNF-α, IL-12, and IFN-γ, as well as a toll-like receptor TLR9-mediated dendritic cell response, which is believed to promote Th1 responses and prevent Th2 skewing during the expansion of the adaptive immune response. Complex cell recruitment and homing processes that involve the upregulation of multiple chemokines, such as CCL5, CCL4, CXCL9 and CXCL10, contribute to interstitial and intra-alveolar immune cell infiltration. IFNγ and TNF promote the accumulation, activation and aggregation of macrophages, resulting in the development of granulomatous inflammation. During chronic HP, switching from a Th1 to a Th2 environment may contribute to the profibrotic response. Furthermore, increased Th17 cells promote lung inflammation and may contribute to progressive lung fibrosis due to HP in specific individuals [1,75,77].
In Heiner syndrome, a rare cow's milk-induced pulmonary hypersensitivity syndrome that primarily affects infants, characterized by food-induced pulmonary hemosiderosis (hemosiderin-laden macrophages in alveoli), digestive bleeding and poor growth, the formation of immune complexes (type III reaction) is strongly suspected along with cell-mediated (type IV) reactions and may contribute to the development of the disease. It was suggested that cow's milk antigens may trigger immune complex reactions resulting in multiorgan abnormalities at pulmonary, gastrointestinal and renal levels. Deposits of immunoglobulins, complement, fibrin and milk protein antigens diffusely scattered in the lung tissue and granular immuno-deposits along the glomerular basement membrane were described. Although high values of serum specific IgG against bovine milk proteins were detected, it is not sufficiently explicable why only some children develop such precipitating antibodies and if these precipitins play a significant causative role [78,79,80].
In the case of systemic contact dermatitis to foods, the hematogenous spread of the allergens is supposed to cause cutaneous reactions through mechanisms including type IV and type III responses and unspecific cytokine release. However, the evidence favouring a type-III hypersensitivity reaction is considered insufficient [46,48,81].
2.4. Type IV Hypersensitivity Reactions via Cell-Mediated Mechanisms
2.4.1. Type IVa via T1 Immune Responses
Allergic contact dermatitis (ACD) is classically considered a type IVa delayed hypersensitivity reaction. Although ACD to foods is considered relatively rare, a wide variety of allergenic haptens from vegetables, fruits and many spices have been identified, such as diallyl disulfide from garlic (Amaryllidaceae/Alliaceae family), allyl isothiocyanate from cabbage, cauliflower, broccoli, radish (Brassicaceae/Cruciferae family), sesquiterpene lactones such as lactucin and lactucopicrin from lettuce, endive (Asteraceae family) and costunolide from bay laurel leaf, cinnamic aldehyde (cinnamal) from cinnamon (Lauraceae family), vanillin from vanilla (Orchidaceae family), eugenol from cloves (Myrtaceae family), limonene, citral and geraniol from peel of orange, bergamot, lemon, lime (Rutaceae family), linalool from coriander and falcarinol from carrot (Apiaceae/Umbelliferae family), urushiol from mango fruit peel and cardol from cashew nut shell oil (Anacardiaceae family). Food additives may also induce ACD, particularly antioxidants, such as sodium metabisulfite/sulfites, butylated hydroxyanisole/hydroxytoluene, gallates, tocopherol, and preservatives such as parabens, benzoic acid/benzoates, sorbic acid/sorbates, and emulsifier propylene glycol. Bleaching agents such as benzoyl peroxide and persulfates are other potential triggers but are no longer used in flour in many countries. Domestic and especially occupational exposures with higher risks involve food handling. Because some agents used as food additives or naturally present in spices are also possibly present in cosmetics and perfumes, other exposures are encountered that can result in ACD [46,82,83,84].
Classically, type IVa hypersensitivity in ACD is considered a T1 response mediated by memory Th1 and Tc1 cells. ACD is a complex immunological allergic disease that supposes two temporally dissociated phases involving complex and interlinked innate and adaptive immune responses. The initial sensitization phase (afferent or induction phase) includes the events from the first skin contact with the allergenic hapten to the development of effector T cells. It lasts 10–15 days and usually has no clinical consequences with no initial hapten-specific skin inflammation. In the advanced elicitation phase (effector or challenge phase), when re-exposed to the sensitizing agent, sensitised subjects present clinical manifestations of ACD, usually within 24-72 h [85,86,87,88].
The initiating molecular event for skin sensitization is the binding of a xenobiotic hapten to a skin protein. Low molecular weight (LMW) reactive chemicals (< 1000 Da) are too small to be recognized by the immune system and must first react with a carrier protein. The hapten penetrates the stratum corneum, conjugates with a self-protein or amino acids (a process called haptenization) via covalent binding (nonmetal haptens), and forms a hapten-protein complex (haptenated protein) to serve as an allergen that stimulates the immune system and induces sensitization. Food-related haptens are either electrophilic (such as α-methylene-γ-butyrolactones) or nonelectrophilic haptens (thiol haptenation via disulfide formation, such as diallyl disulfide). Some other are considered prehaptens (activated by nonenzymatic air oxidation, such as linalool and limonene) or prohaptens (skin enzymatic activation, such as urushiol oxidized to electrophilic orthoquinone), while others are prehaptens and prohaptens (such as geraniol) [83,89,90,91,92]. Another key event is the hapten activation of epidermal innate immune responses by complex mechanisms involving damage-associated molecular patterns (DAMPs), the inflammasome, and inflammatory cytokines. Keratinocyte activation results in upregulation of inflammatory cytokines, such as IL-1α, TNF-α, and IL-18 [87,93,94].
The next key step is the initiation of activation of adaptive responses with the mobilization and contribution of skin antigen-presenting cells (APCs). LCs are critically important epidermal APC characterized by the expression of Langerin (CD207, a C-type lectin receptor required for hapten/antigen recognition), CD1a (structurally related to MHC-I), and Birbeck granules (internalized langerin with hapten/antigen) which reside in the supra-basal keratinocyte layer (stratum spinosum). Other skin APCs are inflammatory dendritic epidermal cells (IDECs) residing in the basal keratinocyte layer (stratum basale) and dermal dendritic cells (dDCs). Haptens penetrating the skin either directly activate LCs or form a hapten–carrier protein complex that is taken up by skin-resident LCs. The hapten-loaded LCs are activated with upregulation of the surface maturation marker CD83 and costimulatory molecule CD86 and migrate from the skin to local draining lymph nodes (LNs). Activated LCs present upregulated CXCR4 which ligand is CXCL12 detected in lymphatic vessels, and CCR7 which interacts with its only two ligands, CCL19 and CCL21, expressed mainly in the high endothelial venules and lymph node parenchyma. Accordingly, lymphatic migration is CXCR4- and CCR7-dependent [85,88,95,96].
The central event in immune sensitization is the presentation of the allergen by matured LCs to antigen-responsive T lymphocytes in the draining LNs resulting in the formation of primed effector and memory specific T lymphocytes. A large diversity of T cell polarization was reported, with effector lymphocytes mainly producing typically type 1 (IFN-γ and TNF-α), but also type 17 and type 22 cytokines. Therefore, although historically considered a Th1-dominated or a mixed Th1/Th2 response, ACD is increasingly recognized as involving Th17 and Th22 polarization. Moreover, certain chemicals, such as fragrance components, demonstrated a particularly strong Th2 polarization with some Th22 and smaller Th1/Th17 contributions [88,95,97].
The second elicitation phase in ACD is dominated by the recruitment of effector T cells toward the site of the allergen re-exposure to the same (or cross-reactive) sensitizing agent, their activation being followed by T cell-mediated tissuar damage with keratinocyte-directed cytotoxicity mediated by CD8+ T-cells and inflammatory cell infiltration mainly comprising of lymphocytes and neutrophils. T cells represent, together with the directly interacting APCs, the most important effector cells in this phase of allergic contact dermatitis. CD8+ cytotoxic memory Tc1 cells are considered the key hapten-specific effector T cells recruited early after exposure both in the epidermis and dermis, and mainly resonsible for epidermal-dermal interface dermatitis with spongiosis. The skin damage is associated with the accumulation of Tc1/Th1 lymphocytes. CD4+ Th1 cells, producing high amounts of IFN-γ and TNF-α, display cytotoxic activity against keratinocytes and may cooperate with CD8+ T cells in amplifying the inflammatory response. Other cell types than Langerhans cells also function as APCs in the elicitation phase, including keratinocytes, mast cells and infiltrating macrophages. Mast cells also contribute to the rapid neutrophil infiltration in response to haptens by enhancing neutrophil extravasation from the bloodstream to the skin lesion via degranulation of TNF-α into the blood vessels. The memory immune response in type IVa reactions is amplified by innate immune cells ILC1 and classically-activated, Th1 skewed, M1 macrophages, among others. Activated macrophages release various inflammatory mediators, such as ROS, proteases and pro-inflammatory cytokines, contributing to tissue damage [1,87,98].
Photoallergic contact dermatitis (PACD) to foods is considered rare. It is a T cell-mediated immune response similar to ACD, but the allergen is previously photoactivated by sunlight or artificial UVA light. Several reported cases of PACD to garlic without occupational exposure have been reported, the allergen being diallyl disulfide [46,48,99,100].
Fixed food eruption (FFE) is an uncommon condition when ingestion of food causes a skin reaction in a particular site/sites, similar to a fixed drug eruption. The pathomechanisms may be similar to fixed drug eruption and different from systemic contact dermatitis. It involves activating the cutaneous immune system by ingesting the allergen and memory CD8+ T cells specific to food that reside and persist in the skin. They are reactivated on re-exposure and mediate keratinocyte apoptosis. Reported causes of FFE include asparagus, strawberries, kiwi fruits, cashew nuts, almonds, hazelnuts, walnuts, pistachio nuts, peanuts, lentils, fish, molluscs and crustaceans, tonic water (quinine) and synthetic food colouring (tartrazine) [101,102,103,104,105].
Celiac disease (CD) is considered a Th1 disease involving mechanisms of type IVa hypersensitivity mediated by gliadin-specific Th1 cells, which induce chronic intestinal inflammation. CD is characterized by chronic immune-mediated enteropathy triggered by ingesting dietary gluten and related prolamines in genetically predisposed individuals (HLA-DQ2 and HLA-DQ8 haplotypes). The 33-mer peptide, a specific fragment derived from the α-gliadin protein found in the wheat storage protein gluten, is highly resistant to digestion, its p31-43 and p57-68 sequences constituting significant shorter fragments. Gliadin peptides containing T-cell epitopes resist gastrointestinal degradation and enter the lamina propria via transcytosis or paracellular routes. They are deamidated by tissue transglutaminase (TG2/tTG) present in the gut, which is the main autoantigen in CD. TG2 modifies glutamine to glutamic acid within gliadin, a critical step that causes gliadin peptides to have a stronger affinity for HLA-DQ2/DQ8 molecules on antigen presenting cells. Subsequent presentation to CD4+ T-cells results in inflammation and mucosal epithelial cell damage. Inflammatory cytokines such as IFN-γ, TNF-a, and IL-21 secreted by activated gluten-specific CD4+ T cells perpetuate the Th1 response and induce an inflammatory condition of the small intestine, with consequential intraepithelial lymphocytosis and an alteration of the duodenal mucosa architecture with villous atrophy and crypt hyperplasia. Furthermore, T cell-dependent B cells produce of autoantibodies against tTG and antibodies against deamidated gliadin peptides, which brands CD as a mixed allergic-autoimmune disease. The undigested gliadin p57-68 peptide induces an adaptive Th1 pro-inflammatory response, while the p31-43 peptide induces a stress/innate immune response involving increased expression of IL-15 via alterations in vesicular trafficking and CFTR (cystic fibrosis transmembrane conductance regulator) inhibition in intestinal epithelial cells. IL-15 is a cytokine crucial in generating epithelial damage in active CD. Moreover, IL-15 also stimulates the generation of ILC1, NK, and memory CD8 T cells. It was revealed that it stimulates the expansion of intestinal intraepithelial lymphocytes (IELs) with cytotoxic activity, inducing enterocyte killing and enhancing the production of IL-21 by IELs. IEL proliferation and perforin/granzyme-dependent cytotoxicity is further promoted by IL-21, which also amplifies the Th1 response in CD. Epithelial damage results in the release of nuclear alarmins such as TSLP, IL-33, and HMGB1. TSLP can increase in the initial stages of CD and appear not to be influenced by the gluten-free diet, indicating the persistence of an underlying inflammation more marked in patients with refractory disease. ILC1s and these alarmins are essential in generating and maintaining inflammation in CD [106,107,108,109,110].
The number of intraepithelial lymphocytes bearing the infrequent γ-δ T-cell receptor (TCRγδ+) is increased in patients with active CD. However, their pathogenetic role, compared with the lamina propria lymphocytes, is debatable. Moreover, the relative pathogenic importance of humoral versus the established role of cellular immunity in CD is also uncertain. Besides the lamina propria effector CD4+ T cells, activated by the gluten-derived peptides, which represent an effector T cell subset producing proinflammatory cytokines, additional subsets of T cells have regulatory functions (Treg). These subsets include CD4+ type 1 regulatory T cells (Tr1) and CD4+CD25+ T cells expressing the master transcription factor forkhead box P3 (Foxp3) also with important roles in CD pathogenesis. The major autoantigen in CD is considered tTG-2, and autoantibodies against it are able to block intestinal epithelial differentiation. The serological determination of CD-associated antibodies has a crucial diagnosis role, mentioning anti-tTG IgA and IgG antibodies, anti-endomysial IgA antibodies (targeting tTG-2), along with IgA and IgG antibodies against deamidated gliadin peptide (DGP). Combing detection of IgA and IgG antibodies against recombinant human tTG and IgG antibodies to deaminated gliadin-analogue fusion peptide (GAF-3X) ensures a great diagnostic performance [111,112,113,114,115]. Autoantibodies targeting the TG2 epitopes are produced during gluten intake, and their production stops on a gluten-free diet [116].
In the chronic phase of atopic dermatitis (AD), the Th1 immune responses play a pathogenesis role. Particularly in patients with the intrinsic phenotype, where normal serum levels of IgE are detected, Th1 responses are more robust than in patients with the extrinsic phenotype. Based on IFNG gene expression and the release of IFN-γ by lesional T cells, high and low IFNG DA endotypes correspond to intrinsic and extrinsic AD phenotypes, respectivelly. Th1 cells mainly produce IFN-γ, TNF-α, and GM-CSF, which are detected in the chronic phase of the disease. The Th1 cell infiltration may be a compensatory reaction to attenuate excessive type 2 deviation in AD. The role of Th1 cytokines in AD skin barrier remains ambiguous, variable effects of TNF-α and IFN-γ on epidermal lipid metabolism and their levels being reported, the Th1 cytokine IFN-γ instead induces apoptosis in keratinocytes and further promotes the alteration of the stratum corneum barrier. The IFN-γ receptor protein complex is the heterodimer of two chains, IFNGR1 and IFNGR2. Intracellular signalling occurs via activating the JAK1-2/STAT1 pathway [117,118,119,120,121].
In patients with moderate-to-severe AD, expression of Th1-related (IFNG, IL12/23p40, STAT1, and CXCL9) biomarkers increase with age. The immune system responses in patients with intrinsic AD reveal increased Th1 signalling (IFN-γ, CXCL9, CXCL10) and African American AD patients exhibit a reduction in the Th1 responses compared to European Americans. Another mechanism possibly intricated in Th1 and Th17/Th22 cell infiltration in chronic AD involves endothelin 1 produced by keratinocytes participating in the complex chronic immune responses in inflammatory skin conditions. IL-25 up-regulates the production of endothelin 1, while this peptide also up-regulates the production of IL-25 in keratinocytes [117,122,123,124,125].
In patients with delayed-type food allergy and chronic AD, Th1 cells mainly drive pathogenesis. Moreover, activated eosinophils produce matrix metalloproteinase-9 (MMP-9), leading to the independent release of IL-1 β by the inflammasome/caspase-1. Eosinophils may be responsible for switching Th2-dominated immune responses to Th1 ones in chronic lesions and delayed-type food allergy via IL-12 production. Moreover, it is presumed that conditions mediated by an independent Th1 pathway, such as delayed-type food allergy, will reveal a lower expression of Th17- and Th2-related cytokines. CXCL9, a chemokine related to Th1 responses, is a potential AD biomarker in this framework, and a CXCL9-high AD endotype was revealed [126,127,128,129,130]. Moreover, endothelin-1, a peptide involved in dendritic cell-mediated Th1 responses, related to chronicity of AD and decreasing ocular surface mucin from conjunctival goblet cells, has been recently reported with an endotype-phenotype association [127,130].
2.4.2. Type IVb via T2 Immune Responses
The most characteristic expressions of type IVb hypersensitivity reactions are revealed in eosinophilic oesophagitis (EoE), atopic dermatitis (T2 endotype), and protein-contact dermatitis. Type IVb reactions are mediated by Th2 cells, which get their phenotype upon exposure to IL-4, basophils or NK-T cells. Th2 cells produce high amounts of IL-4, IL-5, IL-9, IL-13, IL-31 and eotaxins 1-3. IL-4 and IL-13 are the key cytokines of type IVb hypersensitivity and induce a class switch to IgE in B cells. IL-13 is responsible for the tissue remodelling accompanying chronicity in type IVb hypersensitivity, while IL-5 mediates the bone marrow expansion of eosinophils, circulating eosinophilia and recruitment of eosinophils to the sites of inflammation and their survival in the tissues. Eosinophils degranulate releasing their endogenous proteases into the microenvironment causing further tissue injury, chronic tissue damage and barrier disruption. IL-31 mainly produced by Th2 cells causes neurogenic inflammation, and IL-9 produced by Th9 cells enhances IL-4-mediated synthesis of IgE by B cells and is an important growth factor for mast cells. Most of the identified pathogenic drivers of this type 2 inflammation are mainly related to these cells. Epithelial barrier defects and dysbiotic microbiomes are also involved in chronic type 2 inflammatory conditions [1,132].
Eosinophilic gastrointestinal diseases (EGID) are chronic, immune-mediated disorders characterized by a pathologic increase in eosinophil-predominant inflammation in specific regions of the gastrointestinal tract in the absence of secondary causes of eosinophilia. Besides eosinophilic esophagitis (EoE) with esophageal involvement alone, any other location of involvement is termed non-EoE EGID, including eosinophilic gastritis (EoG), eosinophilic enteritis (EoN), and eosinophilic colitis (EoC). Normally, eosinophils are resident cells in the lamina propria of the gastrointestinal tract, with their numbers increasing toward the distal segments, peaking in the cecum and appendix, but are absent in the esophagus. Therefore, in contrast to the esophagus, establishing histologic thresholds indicative of pathology is more challenging [133,134,135].
Eosinophilic esophagitis (EoE) is a chronic and progressive immune-mediated disease of the esophagus defined clinically by symptoms of esophageal dysfunction and histologically by an eosinophil-predominant infiltration of the esophageal squamous epithelium. It is characterized by allergen-driven type IVb hypersensitivity responses with eosinophilic Th2-predominant inflammation and epithelial barrier defects, along with tissue remodelling. Esophageal inflammation in EoE involves more frequently the entire esophagus, followed by isolated distal and proximal involvement. EoE may progress from an eosinophil-rich inflammatory stage to a fibrostenotic one, with subepithelial collagen deposition, smooth muscle hypertrophy, and angiogenesis [136,137,138].
EoE may be provoked by either ingested food allergens or by local contact with aeroallergens. The six most common foods identified to trigger EoE are milk, wheat, egg, soy, tree nuts/peanuts, and fish/shellfish. Aeroallergens possibly incriminated are either inhaled and locally deposited or, in some cases, swallowed (oral or sublingual immunotherapy), the interplay between respiratory and food allergens being complex since they may also cross-react. Although EoE has a genetic predisposition, environmental factors appear to have a more significant role. The impairment of esophageal epithelium integrity, potentially facilitated by gastric acid exposure and/or carriage of genetic variants altering epithelial barrier function, permits entrance of food or aeroallergens with initiation of immune responses [139,140,141,142,143].
Exogenous antigens trigger the production of epithelial-derived alarmin cytokines TSLP and IL-33, the latest via activating the intracellular allergen sensor RIPK1-caspase-8 ripoptosome. The esophageal mucosal barrier dysfunction due to dysregulated endogenous proteases and an abnormal epithelium allows the translocation of allergens to the dendritic cells, which process and present them to CD4+ T cells. TSLP and IL-33 influence the dendritic cells to mature Th2-biased effector T cells and stimulate group 2 innate lymphocytes (ILC2s). Both secrete type 2 cytokines, IL-4, IL-5, and IL-13, which recruit and activate eosinophils, basophils and mast cells. Mast cells in the oesophagus are resident cells, as opposed to eosinophils, mainly involved in the earliest phases of EoE pathogenesis. TSLP is a member of the IL-2 cytokine family functioning as a regulator of the T2 response by driving the production of IL-4, IL-5, IL-9, and IL-13, as well as by having pro-inflammatory effects. TSLP-induced STAT5 transcription factor phosphorylation in circulating CD4+ T cells is important, the number of circulating and esophageal CD4+ T cells responsive to TSLP being correlated with the number of esophageal eosinophils. EoE presents increased baseline expression of markers for eosinophilic inflammation (eosinophil cationic protein), genes associated with a mast cell signature (carboxypeptidase CPA3) or those involved in Th2 associated allergic inflammation (IL5 and IL13) including the hallmark EoE genes for eosinophil chemotaxis (CCL26), T cell activation (TSLP), tissue remodeling periostin (POSTN) and barrier function desmoglein (DSG1) [141,142,144,145].
Esophageal eosinophilia in EoE is mainly driven by STAT6-dependent local expression of CCL26 (eotaxin-3). STAT6 is the key transcription factor activated by IL-4 and IL-13 which has a role in the development of Th2 cells, promotes type 2 immune responses, and induces calpain 14 (CAPN14) expression involved in epithelial repair. GATA-3, a master transcription factor of Type 2 immune responses, was also found to have increased expression in EoE. IL-4 mediated induction of eotaxin-3 secretion is sensitive to Ca2+ signaling and the non-gastric P2-type H+/K+ ATPase ATP12A, expressed in esophageal epithelium and upregulated in active EoE, may be an essential factor in this pathway. Moreover, IL-13 stimulated secretion of eotaxin-3 by esophageal epithelial cells is dependent on the aryl hydrocarbon receptor (AHR) functioning as an environmental sensor [138,142,144,146,147].
IL-13, produced in the esophagus by infiltrating immune cells, including Th2 cells, eosinophils and mast cells, has a central role, its up-regulation in the esophageal tissue driving esophageal eosinophilia, epithelial hyperplasia, and esophageal remodelling (fibrosis, angiogenesis). Additionally, IL-13 induces the expression of CCL26, which promotes eosinophil chemotaxis and recruitment into the tissue. Mast cells and eosinophils propagate allergic inflammation through cytokine and inflammatory mediator production (e.g., PGD2, leukotrienes, granule enzymes), further contributing to immune cell activation and epithelial changes impairing barrier function. Immune activation is characterised by an increase in type 2 cells, allergic effector cells (eosinophils, mast cells, basophils) and elevated soluble Th2 mediators (IL-13, IL-4, IL-5, CCL26). A feed-forward cycle progresses, causing chronic inflammation that stimulates tissue remodelling/fibrosis via cytokine TGF-β, epithelial-mesenchymal transition, and pro- and anti-fibrotic mediator modulation. Enhanced infiltration of amphiregulin-producing Th2 cells is detected in the fibrotic areas; therefore, it is considered that amphiregulin further contributes to esophageal fibrosis in EoE. TGF-β produced by infiltrating eosinophils and mast cells is increased in EoE esophageal tissue and promotes esophageal remodelling by inducing fibroblast activation and secretion of extracellular matrix (ECM) proteins (e.g., collagen, fibronectin). Moreover, the decreased expression of the membrane protein tetraspanin 12 (TSPAN12) in endothelial cells induces profibrotic mediators in fibroblast, and thrombospondin-1 (TSP-1) expression is capable of inducing fibroblast collagen I production, suggesting a profibrotic role in EoE. Although IgE and IgG4 antibodies derived from B-cells are increased in EoE, they do not have a prominent role in the development of EoE [138,139,142,148,149].
Despite being a phenotypic feature of EoE and contributing to its pathogenesis, eosinophils are not the main pathophysiological driver of the disease and are secondarily attracted to the oesophagus. Furthermore, EoE-like variants devoid of eosinophils have been recently identified, suggesting that the spectrum of inflammatory conditions targeting the esophagus is complex. The role of eosinophils in EoE might be less critical than type 2 inflammatory players residing in the esophageal mucosa, such as Treg cells and mast cells [136,141,142,146].
In eosinophilic gastritis (EoG)/eosinophilic enteritis (EoN), eosinophilic infiltration of the gastrointestinal tract is a fundamental histopathological characteristic and is driven by several Th2-dependent cytokines and induced by ingested food allergens similar to those involved in EoE. The Th2-mediated inflammatory cytokines IL-4, IL-5, and IL-13 play essential roles in eosinophilic release, migration, and degranulation. In the bone marrow compartment, IL-3, IL-5, and GM-CSF stimulate the maturation of eosinophils. Further, IL-5 regulates the release of eosinophils from the bone marrow, while eotaxin-1 promotes chemotaxis and migration toward tissue. IL-5 and GM-CSF additionally regulate eosinophil activation and survival. It was suggested that eosinophils accumulate in tissues when eotaxin-1 levels surpass IL-5 levels, whereas higher IL-5 levels promote their presence in peripheral blood. Eotaxin-1 and α4β7 integrin regulate eosinophilic migrating into the gastrointestinal mucosa. Enhanced IL-4, IL-5, and IFN-γ expression in peripheral T cells is revealed in patients with EoG/EoN. The Th2 cytokines IL-4, IL-5, IL-13 and the eosinophil-related chemokine eotaxin-3 CCL26 are upregulated in gastric biopsy specimens, while IL-3, IL-5, and GM-CSF are detected in duodenal and colonic tissue in the vast majority of patients with EoG/EoN. Upon reaching the target tissue, eosinophils are activated and degranulate, releasing toxic proteins like major basic protein (MBP), eosinophil peroxidase (EPO), eosinophil-derived neurotoxin (EDN), and eosinophil cationic protein (ECP), which are cytotoxic to the gastrointestinal epithelium and secrete cytokines that enhance the inflammatory responses. Epithelial cells secrete the long isoform of TSLP with pro-inflammatory actions. Activated B cells produce IgE, which binds to the FcεRI receptor on mast cells, inducing mast cell degranulation. The chemoattractant receptor CRTH2 is located on the surface of eosinophils, mast cells, and basophils and mediates chemotaxis. Sialic acid-binding immunoglobulin-like lectin Siglec-8, highly specific for eosinophils and mast cells, is involved in eosinophil apoptosis [134,150,151,152,153].
The eosinophilic colitis (EoC) pathophysiology is multifactorial and involves Th2 reactions. In addition to eosinophil-predominant inflammation, the accumulation of mast cells in the interstitium of the colon wall suggests their pathogenic role. In adults, the pathogenesis is believed to be mainly CD4+ mediated, while in children, it is more linked to food allergies, such as cow’s milk allergy. It's important to note that the most common manifestation of EoC is food protein-induced allergic proctocolitis, primarly involving important type V reaction [152,154].
Protein contact dermatitis (PCD) may also reveal characteristics of a type IVb hypersensitivity reaction. Th2 cells are the leading players in this T2 immune response. Repeated epicutaneous exposure to papain, but not bovine serum albumin, induces serum allergen-specific antibody production and Th2 differentiation in skin-draining lymph nodes. Papain is used in meat tenderizer powders, marinades and spice mixes. Such an allergenic protein with protease activity may differentiate Th17 and induce Th2 and Th17/Th22 recall responses at epicutaneous challenge sites. Moreover, in the case of occupational repeated epicutaneous exposure to fish parasite Anisakis proteins, IL-4 drives systemic sensitization, while IL-13 plays a central role in the case of PCD. Protein contact dermatitis (PCD) may also reveal characteristics of a type IVb hypersensitivity reaction. Th2 cells are the leading players in this T2 immune response. Repeated epicutaneous exposure to papain, but not bovine serum albumin, induces serum allergen-specific antibody production and Th2 differentiation in skin-draining lymph nodes. Papain is used in meat tenderizer powders, marinades and spice mixes. Such an allergenic protein with protease activity may differentiate Th17 and induce Th2 and Th17/Th22 recall responses at epicutaneous challenge sites. Moreover, in repeated occupational epicutaneous exposure to fish parasite Anisakis proteins, IL-4 drives systemic sensitization, while IL-13 plays a central role in the case of PCD [1,155,156,157].
Systemic contact dermatitis (SCD) or systemically reactivated allergic contact dermatitis represents a type of hypersensitivity reaction caused by systemic exposure to a specific or chemically related hapten in a patient with previous T cell-mediated contact sensitization by cutaneous route. SCD induced by ingested food is uncommon, and its pathogenesis involves mainly specific type IV T cell responses. Th1 and Th2 cells, cytotoxic T lymphocytes, and NK cells play a crucial role by secreting many proinflammatory cytokines. Immediate type I reactions and components of the complement system are also suspected to be involved [48,158,159]. Some foods, spices and drinks containing hapten ingredients to which subjects become sensitized topically and have the potential to cause SCD. Allergenic triggers present in unheated garlic (diallyl disulfide), lettuce (lactucin and lactucopicrin), bay laurel leaf (costunolide), raw cashew nuts contaminated with cashew nut shell oil (cardol, anacardic acid), tonic water (quinine), and food additives (emulsifier propylene glycol, sweetener aspartame) are also known to cause food-related SCD [81,160,161,162,163].
Diverse naturally occurring food fragrances/flavourings acting as allergenic triggers in SCD are commonly found in spices such as cinnamon (eugenol, cinnamates, benzoates), cloves (eugenol, vanillin), nutmeg (isoeugenol), vanilla (vanillin, benzoic acid), curry (eugenol, cinnamates, vanillin, ferulic acid), but also in citrus peels (cinnamates and ferulic acid), tomatoes (eugenol, cinnamates, ferulic acid, coniferyl alcohol). Other similar compounds are found in flavoured confections with artificial vanilla (vanillin, added eugenol), chocolate (eugenol, cinnamates, vanillin), cola drinks (eugenol, cinnamates, vanillin, ferulic acid) and flavoured alcoholic beverages, such as vermouth and liqueurs. All these favouring haptens are Balsam of Peru/Myroxylon pereirae resin ingredients and fragrances, usually representing the contact sensitizers in patients with SCD to the mentioned foods. They may be classified as cinnamate (cinnamal/cinnamic aldehyde, cinnamyl alcohol, benzyl cinnamate), eugenol, vanillin, benzoate, ferulic acid or coniferin groups. Patients with contact dermatitis and sensitization to Myroxylon pereirae resin and/or fragrances may develop SCD from the above mentioned spices, flavourings, and foods. Possible explanations are cross-reactivity (structurally related fragrances), metabolization or oxidation of one fragrance molecule into another, such as cinnamyl alcohol into cinnamal, concomitant sensitizations from coupled exposures or independent sensitizations over time [46,48,164,165,166]. Myroxylon pereirae resin ingredients may provoke a Th2 response in certain individuals. The role of Th2 pathways is also supported by studies demonstrating the beneficial effects of disrupting the Th2 pathway for the treatment of SCD. Moreover, SCD also requires antigen-specific cutaneous leukocyte antigen CLA-positive T lymphocytes and skin-resident memory lymphocytes [167,168,169].
SCD may be caused by metals such as nickel, cobalt, chromium in sensitized patients following oral ingestion. Oral exposure to nickel is not known to sensitise, but high nickel content in the diet of some nickel-sensitive subjects can provoke/aggravate eczematous lesions, condition known as haematogenous contact eczema or SCD, and various cutaneous and extra-cutaneous manifestations defined as systemic nickel allergy syndrome (SNAS). The peripheral CLA+CD45+RO+CD8+ cells are reduced after oral challenge with nickel, these being presumably skin homing effector cells in SCD. In SNAS, a nickel-related mucosa pro-inflammatory process alters the intestinal barrier functions [170,171,172]. Foods associated with a high nickel intake include cocoa, chocolate, coffee, crustaceans and molluscs, and seeds of leguminous plants. In addition, canned meat and acidic foods cooked using stainless steel cookware and utensils are important sources of nickel and chromium. Chromium may also be present as picolinate, polynicotinate, or chloride in food supplements [81,173,174,175,176].
Inborn errors of immunity with atopic phenotypes are suspected when red flags represented by high levels of Th2 biomarkers, such as increased total serum IgE and eosinophilia, are detected. Among hyper-IgE syndromes (HIES), the prototypic one caused by mutations in the transcription factor STAT3 gene and identified as autosomal dominant HIES (AD-HIES or STAT3-HIES), formerly known as Job syndrome, tends to present with lower lifetime frequency and severity of food allergy than atopic dermatitis patients, despite extremely high total serum IgE levels, this being at least partially explained by the essential role of STAT3 signalling in mast cell degranulation. Instead, the dominant-negative loss-of-function pathogenic variants of the caspase activation and recruitment domain CARD11 gene, encoding a scaffold protein required for lymphocyte antigen receptor signalling, result in a distinctive HIES known as CARD11-associated atopy with dominant interference of NF-κB signalling (CADINS) disease. Patients with CADINS present Th2 skewed immune response with increased IgE level and eosinophilia, severe atopic dermatitis with food allergy, chronic urticaria, recurrent infections and autoimmunity [177,178,179].
A combined immunodeficiency generally less profound than severe combined immune deficiency (SCID), known as autosomal recessive DOCK8 deficiency, due to a genetic defect in the dedicator of cytokinesis DOCK8 gene, impairing T-cell receptor signalling and cytoskeletal remodelling, is characterized by severe dermatitis with food allergies, elevated serum IgE levels and eosinophilia, associated with recurrent and severe infections with human papilloma virus, herpes viruses, molluscum contagiosum and other bacterial and fungal infections, risk of malignancies and autoimmunity. The mechanisms involved in the increased prevalence and severity of food allergy in DOCK8 deficiency include inadequate ability to activate STAT3 during TLR9 stimulation for inhibition of IgE isotype switching, defective immune synapse formation diminishing TCR signal strength, thereby favouring Th2 polarization, augmented TFh13 cells promoting high-affinity allergen-specific IgE for mast cell degranulation and anaphylaxis, cytothripsis of migrating mononuclear phagocytes with IL-1β secretion inducing GM-CSF production by T cells, which in turn boosts overall T cell production of IL-4, IL-5, and IL-13, and increased ILC2 cells in the gastrointestinal tract via the DOCK8’s ability to promote Cdc42 activity [177,179,180].
Wiskott–Aldrich syndrome (WAS) is an X-linked recessive disease due to mutations in the WAS protein (WASp), which plays a critical role along with DOCK8 in the initiation of TCR-driven actin polymerization. WAS is characterized by atopic dermatitis with an increased prevalence of food allergy, thrombocytopenia with small platelets, and combined immune deficiency with recurrent bacterial/viral infections. A combined immunodeficiency associated with susceptibility to EBV lymphoproliferative conditions named RLTPR (RGD, leucine-rich repeat, tropomodulin and proline-rich-containing protein) deficiency due to mutations in capping protein regulator and myosin linker CARMIL2 affecting the CD28-responsive pathway in T cells and the BCR-responsive pathway in B cells, presents high IgE levels, severe atopic dermatitis with food allergies, allergic asthma and cold urticaria, as well as a predisposition to a variety of other severe infectious diseases. Among SCID, atopic dermatitis with food allergy and eosinophilia are frequently reported in Adenosine deaminase (ADA)-SCID. Other severe primary immune deficiencies with elevated IgE and increased Th2 cytokine production include IPEX and Omenn syndromes [177,179].
Comèl-Netherton syndrome (CNS) is rare autosomal recessive HIES characterized by the diagnostic triad of chronic inflammatory skin lesions (congenital ichthyosiform erythroderma and ichthyosis linearis circumflexa), specific bamboo-like hair-shaft defect (trichorrhexis invaginata), and high serum IgE levels and a high prevalence of allergic rhinitis, asthma and food allergies. CNS is caused by loss-of-function mutations in the SPINK5 gene encoding LEKTI, which is expressed in the stratified epithelium of the skin and sinonasal mucosa. The consequence of LEKTI deficiency is a loss of inhibition of serine proteinases, leading to unopposed activity of kallikrein-related peptidase 5 (KLK5), which activates KLK7, KLK14, and elastase 2 (ELA2), leading to a profound skin barrier defect with an early and accelerated percutaneous allergen sensitization. KLK5 also activates protease-activated receptor PAR-2 expressed on the surface of keratinocytes with increased production of thymic stromal lymphopoietin (TSLP), enhancing the allergic predisposition. Moreover, PAR-2 leads to increased expression of TNF-α, IL-8, and ICAM-1, thus augmenting the inflammatory process. IL-4 and IL-13 are key cytokines of the Th2 inflammation present in CNS. Allergic reactions are predominantly associated with such Th2 responses in patients with ichthyosis linearis circumflexa CNS phenotype, while Th9 responses are prominent in those with the scaly erythroderma CNS phenotype. Notably, the IL-17/IL-36 pathways predominate in both clinical subtypes. IgE-mediated food allergies in CNS are common, often multiple and with a high risk of anaphylaxis. Specific IgE antibodies to allergens triggering clinical symptoms are against common food allergens, such as cow’s milk, egg, wheat, and nuts. Limited data revealed that all patients with CNS have serum IgE antibodies against multiple potentially anaphylactic stable allergens, such as LTPs, storage proteins and tropomyosins, and against at least one cross-reactive allergen such as PR-10 protein or profilin panallergens. The severe skin barrier defect in CNS with increased allergen penetration contributes to rapid and vast allergic IgE sensitization. Because LEKTI is not expressed in the gut epithelium, further studies are needed to elucidate whether the early and broad sensitization to LTP allergens, which are more ingestion than inhalation-induced, could be explained in CNS by a functional barrier defect of the intestinal mucosa and whether the intestinal microbiome may contribute to this effect. Moreover, EoE was reported in Netherton syndrome and both LEKTI deficiency in the esophagus and high level of IgE-mediated food reactivity were suspected to be involved [181,182,183,184,185].
Genetic connective tissue disorders with hypermobility syndromes, such as Loeys-Dietz syndrome (an autosomal dominant HIES due to mutations in TGF-β receptors 1 and 2, with enhanced TGF-β signalling), ERBIN deficiency (an autosomal dominant HIES due to ERBB2-interacting protein deficiency with increased TGF-β pathway activation via STAT-3), and hypermobile Ehlers-Danlos syndrome (with increased activity of TGF-β due to altered binding by extracellular matrix), are associated with Th2 responses and high prevalence of EoE. These inherited diseases link the TGF-β pathway defects with Th2 responses in humans [186,187,188].
Atopic dermatitis (AD), a common chronic systemic inflammatory disease with skin manifestations affecting children and adults, has a complex pathophysiology involving dysregulated T cell-mediated immune response mainly involving type 2 cytokines (IL-4, IL-13, IL-31) and JAK/STAT signalling pathways, but also other T cell responses, neuroinflammation mediators and epidermal barrier dysfunction. The dysregulated activity of specific types of immune cells as the driving force involved in non-IgE-associated intrinsic AD phenotype (the “inside-out hypothesis”), while the epithelial barrier dysfunction is critical in the IgE-associated extrinsic AD phenotype (the “outside-in hypothesis”) [7,189,190,191]. According to the dual allergen exposure hypothesis, sensitization to food allergens may occur in genetically susceptible individuals with inflamed eczematous skin and impaired skin barrier enableling enhanced penetration of food antigens, when they are cutaneously exposed to low-dose of such allergens in early life, prior to obtaining tolerance by oral exposure, causing T cell deviation towards a Th2 allergenic type and subsequent food allergy; whereas oral tolerance to food antigens is generally promoted through early higher dose oral exposure causing T cell deviation towards tolerogenic Th1 and Treg subtypes [192,193,194,195].
The hallmark T2 cytokines, IL-4, IL-13, and IL-31, and their inflammatory pathways synergistically contribute to AD's pathogenesis regarding immune and barrier abnormalities and critical symptoms, such as pruritus [196]. Acute lesions in AD are primarily triggered through IL-22, with lesser contributions from IL-17 and IFN-γ, significantly accompanied by Th2 cytokines including IL-4/IL-13 and IL-31, while the expression of Th2 and Th22 cytokines is further increased in chronic lesions, with a higher activation of Th1 and Th17 responses [122.197].
IL-4 and IL-13 are the critical cytokines driving the initiation and chronicity of type 2 (T2) inflammation, a dominant inflammatory pathway in AD. The most common endotypes for AD are type-2 (T2) and non-T2. Other recently distinguished are type-17 and type-22 subtypes and mixed types such as T2/type 1 and T2/type 17, revealing the significant involvement of effector Th2, Th22, Th1, and Th17 cells and associated pro-inflammatory cytokine expression [198,199,200].
The alarmins TSLP, IL-25, and IL-33 released from keratinocytes stimulate a type 2 response through ILC2, Th2 cells, and Tfh2 cells, generated after activation of Langerhans cells and inflammatory dendritic epidermal cells. IL-4, IL-13, and IL-5 are released from such lymphocytes. IL-4 and IL-13 activate type I (IL-4Rα/CD132) and type II (IL-4Rα/IL-13Rα1) receptors on B cells, keratinocytes, and sensory neurons, activating JAK-STAT pathways. They are critical drivers of T2 inflammation in AD, aggravating the genetically inherited skin barrier dysfunction, inducing itch and contributing to microbiome dysbiosis. Neuronal itch is induced by type 2 cytokines IL-4, IL-13, IL-31, and alarmins IL-33 and TSLP, involved in the itch-scratch cycle. Furthermore, IL-4 and IL-13 upregulate the production of TSLP, IL-25, and IL-33 in keratinocytes. Antimicrobial peptides, such as human β-defensin 3, are decreased in keratinocytes in response to IL-4 and IL-13. T2 cytokines also exacerbate the imbalance of skin microbiota, while IL-5 is recruiting eosinophils, thus perpetuating a vicious circle. IL-13 upregulates dipeptidyl peptidase-4 (DPP-4) and periostin expression, which are indicators of IL-13 activity. Recently, IL-13-high AD, DPP-4-high AD, periostin-high AD and eosinophil-high AD endotype-phenotype associations were revealed [117,128,201,202]. Malassezia cells and their allergens, recognized by TLR2 on keratinocytes and dendritic cells, cause the release of pro-inflammatory cytokines, elicit the production of fungal specific IgE antibodies through dendritic cells and T cell-mediated activation of B cells, and via autoreactive T cells which can crossreact between fungal and human manganese-dependent superoxide dismutase play a role in the pathogenesis of AD and associated head and neck dermatitis as another endotype-phenotype association [203,204].
When dealing with food allergies in patients with AD, it's crucial to differentiate between children and adults, immediate and delayed reactions, and IgE- and non-IgE-mediated reactions. Additionally, both genetic and dietary factors play a role in the ethnic variations in food allergies related to AD [205,206].
AD and food allergy represent a unique phenotype distinguishable from AD without food allergy. A complex combination of epidermal structural genetic mutations and cytokine activation causes alterations in nonlesional skin surface with reduced content of the structural protein filaggrin, its breakdown products, urocanic acid and pyroglutamic acid, functioning as natural moisturizing factor ingredients, and ultralong-chain lipids, such as omega-esterified fatty acid sphingosine ceramide, which control skin water retention and prevent allergen penetration. Children with AD and food allergy also exhibit a high dendritic cell and immune activation (including type 2) signature in their nonlesional skin, comparable to lesional skin, involving type 2 cytokines, such as IL-4/IL-13, IL-31, TSLP, and IL-33 or inflammatory cytokines such as TNF-α which can cause a substantial reduction in filaggrin expression. Moreover, increased expression of cytokeratins such as KRT16, a marker of keratinocyte proliferation, may be considered an indicator of enhanced type 2 responses in the nonlesional, clinically normal-appearing skin of patients with AD and food allergy, which can also explain the association of pruritus and food allergy [207]. Children with AD and multiple allergen sensitization to foods may undergo the so-called atopic march, which is currently considered a not-so-frequent evolution trajectory due to the persistence of fetal T2 signalling. IL-33 and IL-9, often associated with early sensitization, are upregulated in AD infants [208].
The skin-gut mechanisms by which inflamed skin communicates to the intestine to modulate the intestinal epithelial and immune environment, to promote pathologic type 2-skewed immune responses and the development of food allergy, involve the systemic circulation of inflammatory mediators and cells. Elevated circulating IgE may help to shape the intestinal immune environment by promoting mast cell maturation and survival through the engagement of FcεRI. IL-33, an alarmin released from damaged keratinocytes elevated in lesional skin and in the serum of patients with AD, is thought to act on intestinal ILC2s to promote production of IL-13. The IL-4/IL-13 signalling is also involved in the enrolment of intestinal mast cells related to food allergy. The influence of IL-31 in the intestine is not well understood but may also contribute to a pro-inflammatory state and promote Th2 cytokine responses [209,210].
Patterns of clinical reactivity to foods in children with AD include, besides IgE-mediated immediate-type noneczematous reactions, non-IgE-mediated AD delayed exacerbation reactions, and IgE- and non-IgE-mediated mixed noneczematous and eczematous reactions. The pattern of delayed exacerbation of AD is not clearly defined in adult-onset AD [211,212,213,214,215]. More than 50% of children with AD that can be exacerbated with foods will react with a worsening of skin eczema alone or in addition to immediate symptoms. The most common foods in children are cow’s milk, hen’s egg or peanuts, and soy, wheat, rye, oat, or corn. Older patients with AD also react to foods, although reactions to food allergens such as hen’s eggs or cow’s milk are not as common as in children. Subgroups of both age groups with AD can also react to pollen-associated foods. Excess feeding with foods containing nickel, such as chocolate, possibly exacerbates skin lesions in some patients with non-IgE-associated AD. IgE-associated and IgE-independent T-cell-mediated responses appear relevant in clinical eczematous mixed reactions. Ingestion of food allergens triggering dermatitis in sensitized patients requires a Th2-skewed immune response with CLA+T cells homing to sites where resident memory T lymphocytes reside. Food-triggered AD can, to some extent, be regarded as a form of systemic contact dermatitis [101,215,216,217,218].
Late eczematous reactions to foods in AD are typically non-IgE-mediated and associated with antigen-specific T cells. IL-22 is considered a critical cytokine for the pathogenicity of skin-activated allergen-specific T cells that can migrate and induce such reactions [217,219,220]. In addition, late-onset reactions to oral food challenges may be linked to low serum IL-10 concentrations, while IL-17- and IL-32-producing regulatory B cells, which are CD5+CD19low cells producing IL-10 and TGF-β and expressing the Foxp3 transcription factor, are related to tolerance to food allergens in terms of late eczematous reactions in AD [221,222,223]. Skin-homing CLA+ T cells migrate into the skin via molecular interactions of CLA, LFA-1, and VLA-4 expressed on T cells, with E-selectin, ICAM-1 and VCAM-1 present on cutaneous endothelial cells. Supplementary interactions between CCR4 and CCR10 expressed on CLA+ T cells, and their ligands CCL17 and CCL27 produced by keratinocytes, are needed during this process. Local activation of CLA+ T cells is involved in cutaneous immune responses. The migration of activated T cells to other target organs of inflammation has been demonstrated in food allergen-specific and skin-homing T cells sensitized in the gut and can migrate into the skin, causing AD lesions [224,225]. Most skin-infiltrating lymphocytes in AD patients express CCR10, indicating that CCL27/CTACK-CCR10 interactions are critical in T cell-mediated skin inflammation. Besides this cutaneous T cell-attracting chemokine expressed by keratinocytes, which mediates the migration of lymphocytes into the skin, additional emerging potential AD biomarkers include other Th2-related chemokines, such as CCL18/pulmonary and activation-regulated chemokine, CCL17/thymus and activation-regulated chemokine CCL22/macrophage-derived chemokine and CCL26/eotaxin-3 [128,129,226,227]. CCL17/TARC, CCL18/PARC, and CCL22/MDC, all associated with Th2 pathogenesis, are upregulated in AD with food allergen sensitization profiles. Although blood biomarkers can correlate with AD disease activity, their interpretation may be limited by the potential contribution within the circulation from other allergic comorbidities such as asthma and allergic rhinitis [207,228]. CLA+ T cells are memory cells that recognize epitopes in food allergens such as milk and peanuts. T-cell activation in AD also suggests that ongoing skin inflammation is driven by cutaneous T-cell reservoirs and lymph node-homing populations, explaining disease exacerbations across different skin regions. Moreover, IL-13, together with IL-4 and Th2-related chemokines CCL17 and CCL22, are preferentially produced by circulating memory CLA+ T cells upon activation [194,229,230,231,232]. When comparing T cells from AD and psoriasis patients, differential expression of the CCL22 gene in CD8+ T cells was detected, and increased expression of IL-19 and IL-24 has been linked with severe AD [233].
2.4.3. Type IVc via T3 Immune Responses
In type IVc hypersensitivity, Th17 cells play a crucial role by producing IL-17 family cytokines that recruit and activate neutrophils and enhance Th2 cytokine production, leading to inflammation. This mechanism is particularly relevant to conditions like atopic dermatitis with normal serum IgE levels and greater susceptibility to irritant contact dermatitis. IL-17 is a pleiotropic cytokine belonging to the IL-17 family, including IL-17A (commonly known as IL-17), IL-17E (also known as IL-25), and IL-17F. IL-17A and IL-17F are the closest members, with 50% homology [1,234].
In food allergy, inflammatory responses involving IL-17 may be involved. Few studies found increased serum IL-17 levels in adults and children with food IgE-mediated hypersensitivity. In adult patients with an elevated concentration of allergen-specific IgE, serum IL-17A concentrations are higher than in healthy subjects. Furthermore, increased food allergen-specific IgE levels are positively correlated to IL-17E serum concentrations in children [235,236,237].
Food protein-induced enterocolitis syndrome (FPIES) is a non-IgE-mediated food allergy in which the mechanisms responsible for immune recognition of food antigens are unclear. An increase in circulating neutrophils post-challenge is part of the criteria for defining positive FPIES oral food challenges, and acute FPIES reactions are associated with significant IL-17 inflammatory signatures. It was recently revealed that pediatric FPIES reactions are associated with a significant Th17 cytokine response with systemic release of IL-17A, IL-17C, and IL-22 during challenges. Such reactions are associated with activating CD3+CD4-CD8- CD161+ and +T lymphocytes; however, the potential sources of IL-17 include Th17 cells. Similarly, in adult FPIES patients an elevation in serum IL-17 following a positive oral food challenge has been reported [238,239,240].
Celiac disease (CD) is classically considered a Th1 disease, but Th17, IL-17A, and IL-21 also have complex roles in its pathogenesis. Both Th1 and Th17 cells are present in duodenal biopsies from CD patients. The mucosal Th17 cells participate in the onset and development of CD. Th17 cells from CD patients but not from control subjects proliferate in response to the gliadin challenge. Gluten peptides, as well as CD-associated bacteria, induce IL-17A responses. In active CD, IL-17A is produced by both duodenal gliadin-reactive Th17 and Tc17. Levels of IL-17A are significantly higher in biopsies from patients with active CD compared to controls. The IL-17A-producing cells from CD patients also express two cytokines with dual, opposite effects, IL-22 and TGFβ, both having anti-inflammatory and pro-inflammatory actions. It is essential to mention that there is an overproduction of IL-21 in the intestinal mucosa of active CD, and the blockade of IL-21 activity reduces Th1-associated transcription factor T-bet and IFNγ secretion of Th1 cells infiltrating the upper bowel mucosa in CD [108,241,242,243,244]. An increased mucosal IL-17A response has been suggested to correlate with villous atrophy. Mucosal upregulation of Th17 immunity occurs at the late stage of disease and is downregulated with gluten-free diet [245,246,247].
Dermatitis herpetiformis (DH) is a gluten-induced skin disorder characterized by subepidermal granular IgA deposits and a variable degree of enteropathy identical to CD. The increase in small bowel mucosal intraepithelial TCRγδ lymphocytes, an almost pathognomonic finding in CD, is present in nearly 95% of DH patients with villous atrophy and about 70% of those with non-atrophic damage. However, many patients have no gastrointestinal symptoms. The epidermal transglutaminase (TG3/eTG) found in the skin is the main autoantigen in DH, as opposed to TG2 in CD. DH starts as a TG2 immune response in the gut, which evolves into a TG3 response in the papillary dermis. By epitope spreading, circulating IgA class autoantibodies also form against TG3. Both patients with CD and DH produce anti-TG3 antibodies. Still, in DH, these have high affinity and thus form immune complexes with TG3 produced by keratinocytes and deposit within dermal papillae, triggering a local inflammatory response that is predominantly neutrophilic. The histopathological hallmarks of DH are neutrophilic microabscesses in the dermal papillae. The serum levels of cytokine IL-17A, anti-tTG and anti-DGP are significantly higher in patients with DH than in healthy controls. DH patients reveal an increased frequency of skin-derived T cells producing TNF-α compared to those with CD. Moreover, a cross-reactive T-cell response toward the two transglutaminases isoforms, TG2 and TG3, is detected. Serum anti-TG2 IgA is a specific marker for gluten-induced enteropathy in both CD and DH, but most DH patients also have circulating anti-TG3 IgA even if anti-TG2 ones are absent [248,249,250,251,252].
In several atopic dermatitis (AD) phenotypes, including pediatric AD, AD in patients of Asian ancestry, and non-IgE-associated AD, the role of Th17 pathway has been documented, therefore it is considered that the Th17 pathway plays a role in chronic AD, with differences depending on ethnicity and age [122,253,254,255]. It was revealed that peripheral blood IL-17+ CD4+ T-cells are increased in number in AD patients and correlated with the disease severity. IL-17 produced by Th17 lymphocytes is thought to coordinate local tissue inflammation by increasing the synthesis of proinflammatory and neutrophil chemotactic cytokines, such as IL-6, GM-CSF, TNF-α or IL-1β [255,256,257,258]. In patients with moderate-to-severe AD, expression of Th17-related (IL17A and IL20) biomarkers increase with age. Moreover, a high activation of Th17 responses is observed in chronic AD lesions [122,125,259]. The immune system responses in patients with intrinsic AD reveal increased Th17/Th22 activation (IL-17A, CCL20, Elafin, and IL-22). This even suggests some immunological overlap with psoriasis. The Asian AD phenotype is characterized by greater expression of Th17-related markers (IL-17A, IL-19, CCL20) along with upregulation of IL-22 and the IL-17/IL-22–induced S100A12 in comparison to European-American patients with AD, but not tothe levels found in psoriasis. Additionally, African American AD patients exhibit a reduction in the Th17 compared to European Americans [122,123,124,129]. During the chronic phase of AD, Th17 and Th22 cytokines cooperatively modulate local inflammation by upregulating proinflammatory cytokines that stimulate epidermal hyperplasia. IL-17A is produced mainly by Th17 cells, while the key source for IL-17C is epithelial cells. IL-17C appears to trigger a stimulation autocrine loop in keratinocytes. In these cells, IL-17A has two receptor complexes, IL-17RA/IL-17RC and IL-17RA/IL-17RD. Intracellular signalling of both receptors occurs via downstream adaptor protein ACT1, engaging TNF receptor-associated factor TRAF6, thus activating the p38 MAPK and NF-κB pathways. Thus, IL-17A promotes keratinocyte proliferation directly, but it also indirectly stimulates it by inducing the production of IL-19 from them. IL-17A is unlikely to trigger JAK-STAT pathways directly but may activate STAT3 via IL-19 signalling. IL-17A can enhance the expression of involucrin and filaggrin-2, and may not directly induce skin barrier dysfunction. Moreover, IL-17-associated AD epidermal lesions and skin lipid abnormalities attributed to IL-17 direct effect or via activation of Th2 responses are not clearly identified [117,120,261]. A potential AD biomarker in this context is CXCL2, a chemokine associated with Th17 responses. Moreover, IL-17 and IL-32-producing Foxp3-expressing regulatory B cells are related to tolerance to food allergens regarding late eczematous reactions in AD [126,221,222].
A category of type IV reactions in AD involves Th22 cells, playing a tissue-protective role in the early stages and involved in tissue remodelling in the chronic phase. The activation of Th22 cells predominates mainly in the acute phase of AD, accompanied by a shift in balance towards Th2 responses. These cells appeared to downregulate genes involved in terminal differentiation of epidermal keratinocytes and tight junction products such as claudins, contributing to skin barrier impairment. Nevertheless, in the chronic phase of AD, Th2 and Th22 responses are also enhanced despite predominant Th1 responses. Patients with Asian ancestry have higher Th22 activation than Europeans, while African Americans have this activation reduced [1,257]. Th22 is an important source of IL-22, documented as one of the pathogenic cytokines in AD. Th17 cells also produce IL-22, along with IL-26, which may play a significant role in combining Th17 and Th2 responses, leading to the development of AD. IL-22, mainly produced by these cells, is a crucial mediator of epidermal hyperplasia. It binds IL-22R1/IL-10R2 complex, activates the JAK1/TYK2-STAT3 pathway and inhibits the expression of filaggrin, loricrin, and involucrin. IL-22 induces keratinocyte proliferation and epidermal thickening in AD. At the same time, it inhibits differentiation and maturation and downregulates the expression of barrier function-related factors, contributing to lichenification [118,119,256,257,258]. In addition, IL-22 is a critical cytokine for the pathogenicity of skin-activated allergen-specific T cells able to migrate and induce late eczematous reactions to food allergens in AD. Recently, IL-22-high AD and CXCL2-high AD endotypes were revealed [128,219].
Comèl-Netherton syndrome (CNS), an inherited HIES due to biallelic mutations in the serine protease inhibitor Kazal-type 5 (SPINK5) gene and characterised by a high prevalence of food allergies, also displays a prevailing Th17 immune response. The role of Th17 responses in its pathogenesis has recently been demonstrated for inflammatory skin lesions [181,263,264,265].
2.5. Type V Tissue-Driven Hypersensitivity Reactions via Epithelial Barrier Impairment
Food allergies particularly linked to epithelial barrier dysfunction include food anaphylaxis dependent on augmentation factors increasing intestinal permeability, food protein-induced enterocolitis syndrome, food protein-induced enteropathy, food protein-induced allergic proctocolitis, eosinophilic esophagitis and celiac disease. Although FPIAP manifestations are isolated to the gastrointestinal tract, the other disorders have symptoms and signs beyond the gastrointestinal tract [79].
Regarding food anaphylaxis dependent on cofactors/augmentation factors increasing intestinal permeability, extensive research has explored the role of exercise and other cofactors in food allergic reactions. The most prevalent cofactor altering intestinal permeability is exercise, along with alcohol and NSAIDs ingestion. Exercise may enhance allergen bioavailability by increasing intestinal permeability and allergen absorption, but this seems specific to patients with food allergies and does not occur in control subjects. Exercise may also induce increased blood circulation in skeletal muscles, heart, skin and lungs, resulting in relative hypoperfusion of the gastrointestinal tract with increased permeability. In the case of exercise and NSAID cofactors, it was suggested that an increase in gastrointestinal permeability occurs independently of mast cell activation due to disrupted tight junctions caused by the inhibition of PGE2 production in enterocytes mediated by ClC-2 chloride channels [24,28,266,267,268]. Alcohol increases intestinal permeability mainly through oxidative stress with damaging the intestinal mucosa and increasing the risk of developing anaphylaxis [28,269].
Non-IgE-mediated food allergies, typically presenting in breastfed infants, which involve type V tissue-driven hypersensitivity reactions, include food protein-induced enterocolitis syndrome involving the entire gastrointestinal tract, food protein-induced enteropathy affecting the small bowel, and food protein-induced allergic proctitis and proctocolitis involving the rectum and colon [1,270].
In food protein-induced enterocolitis syndrome (FPIES), food allergens, such as cow's milk, soya, rice, oats, wheat, avocado and fish, may activate T cells in the intestinal epithelial lining, causing local inflammation and increased intestinal permeability with fluid shifts and augmentation in antigen influx. The amount of food required to provoke symptoms varies widely, reflecting the individual degree of hypersensitivity. Key manifestations in FPIES are repetitive protracted vomiting after food ingestion in its acute form and intermittent progressive vomiting and diarrhoea in its chronic form [8,271,272].
FPIES reactions with increased intestinal permeability are associated with broad systemic innate immune activation (neutrophils, monocytes, eosinophils, natural killer cells, lymphocytes), marked Th17 cytokine response and mucosal barrier damage. This innate immune activation in peripheral blood is noticed in the absence of a detectable food antigen-specific antibody response in the FPIES acute form and a lack of direct evidence for a pathogenic antigen-specific T cell response. Symptomatic pediatric FPIES challenges are associated with significant elevations of innate inflammatory markers, such as IL-8, LIF, TNFa, IL-10, and IL-6, along with increased levels of IL-2 (T cell activation marker), cytokines of the IL-17 family, macrophage inflammatory protein CCL20 (ligand of CCR6 on Th17 cells) and mucosal damage marker REG1A. These biomarkers are not increased in asymptomatic challenges or IgE-mediated allergy. Moreover, activating the purinergic pathway during FPIES reactions reveals a mechanism connecting inflammation to neuronal pathways mediating vomiting. The purine metabolite adenosine induces serotonin release from the enterochromaffin cells of the upper gastrointestinal epithelium, which activates vagal afferent nerves that terminate in the vomiting center [273,274]. Circulating and intestinal mucosal tissue eosinophils are activated in acute FPIES reactions, which are associated with the systemic immune events. A significant increase in CD69 expression on eosinophils after an acute reaction was detected, with no significant change in peripheral eosinophil numbers, but with a post-challenge increase in levels of fecal eosinophil-derived neurotoxin (EDN), considered a granule-derived alarmin [275,276].
In food protein-induced enteropathy (FPE), cell-mediated hypersensitivity to food antigens, such as cow's milk, soya, egg, and wheat, may occur due to disruption in the mucosal intestinal barrier. Food-specific T cell infiltration, cytotoxic CD8+ T cells and increased density of intraepithelial γδ-TCR+ cells were reported. The structural damage to the jejunal mucosa is associated with malabsorption, the cardinal manifestations of FPE being chronic diarrhoea and failure to thrive. Typical celiac disease antibodies are absent in FPE [270,272,277].
In food protein-induced allergic proctocolitis (FPIAP), it is suggested that the delayed development of the gastrointestinal immune system with impaired communication between local and systemic immunity is linked to increased intestinal permeability, leading to hypersensitivity to specific foods such as cow's milk, hen's egg, and soya. The main clinical manifestation is rectal bleeding with mucus-bloody stools in an otherwise healthy-looking infant. Interestingly, it was recently discovered that the serum levels of the macrophage inflammatory protein CCL20 are reduced in FPIAP. A localized rectosigmoid inflammatory response may be explained by the fact that immunoglobulins in breast milk bind to dietary proteins, which are only released after being enzymatically cleaved in the distal colon [270,272,277,278,279]. Eosinophils are the dominant inflammatory cells present in rectosigmoid biopsies of patients with FPIAP. When activated, they secrete eosinophil-derived neurotoxin (EDN), its elevated fecal/rectal swab levels been correlated with the eosinophil activation and degranulation [275,280,281].
Other non-IgE-mediated food allergies or with mixed mechanisms involving type V tissue-driven hypersensitivity reactions are celiac disease and eosinophilic esophagitis, respectively.
In celiac disease (CD) pathogenesis, disassembling the epithelial tight junction (TJ) and augmented paracellular permeability is an obvious hallmark. Gliadin specific peptides or bacteria cause a chemokine receptor CXCR-3-mediated, MyD88-dependent luminal zonulin release from intestinal epithelial cells (IECs). Zonulin binds epidermal growth factor receptor (EGFR) in IECs directly binding and/or through protease activated receptor 2 (PAR2) transactivation, leading to PCK-α dependent tight junction disassembly. Increased intestinal permeability leads to paracellular flux of antigens into the lamina propria where they are able to interact with the immune system. The immune reaction leads to TNFβ and IFNγ production, which alter the MLCK activity leading to higher TJs disruption and increased permeability. At the same time, activated B cells differentiate into IgA plasma cells, including secretory IgA antibodies (sIgA) that are secreted to the luminal area. The disruption of the epithelial junctional complex, especially tight junctions (TJs), increases intestinal permeability and triggers an enhanced immune response to gluten, leading to the development of CD. In the active disease, anomalies in desmosomes, dilatation of intercellular space, and shorter microvilli are reported, along with broader adherence junctions. The inflammatory environment in CD favours damage to the epithelial cells and alterations of the barrier with villous atrophy. TSLP levels can increase in the initial stages of the disease and then lower in the more advanced stages of inflammation. Moreover, serum TSLP appears not to be influenced by the gluten-free diet [108,282,283,284].
In active CD, the secretory IgA antibodies (sIgA), which in homeostasis represent the luminal immunoglobulin that protects intestinal epithelia from antigens, create immune complexes with gliadin and allow intestinal epithelial transcellular transport of intact gliadin peptides into the lamina propria via binding to the transferrin receptor CD71 at the apical surface, promoting an enhanced loop activation of the immune system. This receptor is confined in homeostasis to the basolateral membrane but shows an ectopic expression at the apical membrane of enterocytes in CD. Thus, the retrotransport of IgA-gliadin immune complexes in active CD triggers exacerbated adaptive and innate immune responses and promotes an enhanced loop immune activation that results in mucosal lesions [282,284,285].
Non-celiac wheat sensitivity (less accurately named non-celiac “gluten sensitivity”) is a not well-defined condition more common than CD, characterized by both intestinal and extra-intestinal manifestations related to the consumption of gluten-containing foods in the absence of a diagnosis of celiac disease or wheat allergy (autoimmunity and IgE-mediated hypersensitivity are not pathogenetic mechanisms). An increase in the Claudin-4 component of tight junctions, responsible for paracellular permeability, has been observed. This suggests an impaired intestinal barrier. Additionally, it has been noted that serum zonulin, a protein involved in regulating intestinal barrier permeability, decreases with a gluten-free diet. Furthermore, it is believed that intestinal dysbiosis may contribute to the symptoms of non-celiac wheat sensitivity (NCWS), possibly due to increased fermentation. Fermentable oligosaccharides, disaccharides, monosaccharides, and polyols (FODMAPs) are also implicated in this condition [23,286,287,288,289].
Eosinophilic esophagitis (EoE) pathogenesis is dominated by impaired epithelial barrier function and dysregulated immune cell responses influenced by genetic polymorphisms, generating a feed-forward cycle leading to loss of immunologic tolerance to exogenous allergens and chronic eosinophilic inflammation. Esophageal barrier dysfunction in EoE is mainly driven by the type 2 cytokines IL-13 and IL-4 through effects on epithelial differentiation and causing loss of barrier proteins, such as the desmosome desmoglein-1 (DSG1) and epithelial differentiation proteins filaggrin (FLG) and involucrin (IVL). Likewise, a dysregulated protease/antiprotease response has been revealed in the esophageal epithelium in active EoE. In addition to inflammatory mediators, genetic predisposition and environmental factors contribute to the establishment and maintenance of esophageal barrier dysfunction [138,139,290]. In EoE, the abnormal epithelial barrier is associated with reduced intercellular junction protein expression. Thus, antigens can easily penetrate the epithelium and initiate inflammatory responses and systemic sensitization. An increased expression of thymic stromal lymphopoietin (TSLP), cathelicidin and proteases, such as the kallikrein-5 and -7, was reported along with an abnormal expression of epithelial cell proteins especially those implicated in adhesion and integrity, such as a reduced expression of lymphoepithelial Kazal-type-related inhibitor (LEKTI), filaggrin, E-cadherin, claudin, occludin, demoglein-1, independent of corticosteroid therapy. Furthermore, lower cathelicidin expression and numbers of CD1a+ cells were noted in corticosteroid-treated EoE. Expression of epithelial-derived nuclear alarmins TSLP, IL-25, and IL-33 is elevated in esophageal biopsies, TSLP being correlated with eosinophil extracellular traps (EETs) formation due to infiltrating eosinophils. TSLP high expression is also associated with exaggerated basophil responses. Therefore, the TSLP-basophil axis is essential in the pathogenesis of EoE. In addition, IL-33 may play a pivotal role in the etiology of EoE by activating the IL-13 pathway. Tissue eosinophils express the IL-33 receptor ST2 and type 2 cytokines in EoE, and those that lack antigen-specific T cell receptors maintain their ability to respond to IL-33, thus explaining the continued inflammation after removal of dietary antigens [275,291,292,293,294].
Mast cells accumulate in the epithelium of patients with EoE and disrupt the function of the esophageal epithelial barrier. IgE-activated mast cells induce this dysfunction via the downregulation of barrier proteins and antiprotease expression, which may in part be mediated by OSM, a member of the IL-6 cytokine family, among other proinflammatory mediators. OSM expression is increased in active EoE and OSM receptor-expressing esophageal epithelial cells are detected within the esophageal tissue in EoE. Stimulation of such esophageal epithelial cells with oncostatin M (OSM) decreases the expression of filaggrin and desmoglein-1 and increases the protease calpain-14 (CAPN14) involved in epithelial homeostasis. CAPN14, a calcium-dependent cysteine protease specific to the esophagus, is also induced by IL-13 and, being overexpressed by the esophageal epithelia in EoE. IL-13 epithelial stimulation results overexpression of CAPN14 with diminished barrier function and architectural changes indicative of barrier impairment, such as epidermal clefting and loss of the normal expression of filaggrin and DSG1. Evidence highlighting the essential contribution of this pathway in EoE is the fact that severe dermatitis-multiple allergies-metabolic wasting (SAM) syndrome also known as congenital erythroderma-hypotrichosis-recurrent infections-multiple food allergies syndrome, a rare genodermatitis caused by recessive homozygous or compound heterozygous loss-of-function mutations in DSG1 gene, or dominant heterozygous mutations in desmoplakin (DSP) gene, is associated with disrupted epithelial barrier and EoE [187,290,295,296,297].
The SAM syndrome is classified among the inherited skin disorders with food allergies having the cutaneous barrier dysfunction as important pathogenetic mechanism, together with Comèl-Netherton syndrome (mutations in the SPINK5 gene), ichthyosis vulgaris (mutations in filaggrin FLG gene), and the inflammatory peeling skin syndrome (mutations in corneodesmosin CDSN gene) [177,179,298].
The autosomal dominant CARD14 deficiency is associated with atopic dermatitis (AD) and recurrent infections, the mechanism involving skin barrier dysfunction. Loss-of-function mutations in the caspase recruitment domain-containing protein CARD14 gene, a major regulator of nuclear factor kB (NF-kB), are associated severe AD and food allergy, while dominant gain-of-function mutations in the same gene are associated with psoriasis. AD-associated mutations in CARD14 impair NF-kB activation and CARD14 loss of function impairs epidermal secretion of antimicrobial peptides hBD1, hBD2, and hCCL20, involved in regulation of cutaneous inflammatory circuits and protecting the skin against infections [177,179,259].
Atopic dermatitis (AD) is a common chronic inflammatory disorder marked by skin barrier dysfunction. High levels of IgE and dysfunctional/low levels of filaggrin may predispose patients with AD to food allergy. Although only some patients with AD and food allergies have filaggrin gene mutations, this strong association indicates that cutaneous barrier impairment contributes to the development of both conditions [129,207,299,300]. The downregulation of epidermal differentiation complex molecules, such as filaggrin, involucrin, and loricrin, is the fundamental characteristic of the lesional skin of AD and is associated with skin barrier dysfunction. In AD, the disturbed epidermal barrier leads to increased permeation of allergens, which encounter Langerhans and dendritic cells, activating Th2 lymphocytes to produce IL-4 and IL-13. Th2 and Th22 cytokines (IL-4/IL-13 and IL-22) can inhibit terminal differentiation and contribute to barrier defects [117,261,301]. IL-4 and especially IL-13, the two central T2 cytokines associated with AD pathogenesis produced by ILC2s and Th2 cells, via binding to IL-4Rα/IL-13Rα1 on keratinocytes and activating downstream JAK1/TYK2/JAK2 and subsequently STAT6/STAT3, down-regulate the aryl hydrocarbon receptor-mediated transcription of filaggrin, loricrin and involucrin, inhibit the nuclear translocation of the Ovo-like transcriptional repressor OVOL1, thus disrupting the skin barrier function. Pathogenic abnormality in the barrier and water-holding functions of the stratum corneum in AD is mainly attributable to decreased levels of total ceramides. Th2-associated cytokines downregulate fatty acid elongase and β-glucocerebrosidase involved in lipid chain elongation and ceramide synthesis. The decrease in ceramide is also due to abnormal expression of sphingomyelin/glucosylceramide deacylase, the β-subunit of acid ceramidase. Moreover, IL-4 and IL-13 also stimulate the expression of 3-β-hydroxysteroid dehydrogenase-1, resulting in decreased levels of triglyceride concentration in keratinocytes, further damaging the skin barrier, and concomitantly increase collagen disposition, resulting in skin remodelling and lichenification. The barrier dysfunction triggers the colonization of Staphylococcus aureus and transepidermal entry of allergens. IL-18, a proinflammatory cytokine-stimulated by epithelial damage and staphylococcal colonization, promotes T2-mediated inflammation by activating basophils and mast cells [117,120,190,302]. IL-4/IL-13-mediated STAT6 activation upregulates periostin and then enhances IL-24 production. IL-24 binds to IL-20Rβ/IL-22Rα or IL-20Rβ/IL-20Rα, activates the JAK1/TYK2-STAT3 pathway, and inhibits the expression of filaggrin. Moreover, IL-22 binds the IL-22R1/IL-10R2 complex, activates the JAK1/TYK2-STAT3 pathway, and inhibits the expression of filaggrin, loricrin, and involucrin [117,161].
2.6. Type VI Tissue-Driven Hypersensitivity Reactions via Metabolic-Induced Immune Dysregulation
Such hypersensitivity reactions should be primary approached in the framework of adipose tissue inflammation and metabolic dysfunction in obesity and high-fat diets (HFDs). Obesity is characterized by chronic low-grade inflammation due to the release of adipokines and proinflammatory cytokines from adipose tissue, such as TNF-α, IL-6 and leptin. The persistent inflammatory state disrupts normal immune regulation and alters the function of immune cells, including T-lymphocytes and macrophages [303,304,305].
Many metabolic changes impair the function of immune cells and affect their ability to manage dietary antigens properly, leading to an overactive immune response to food allergens. In addition, obesity and HFDs also lead to intestinal inflammation with an increased number of T-lymphocytes, mature dendritic cells and macrophages. Proinflammatory cytokines, including TNF-α, IFN-γ and IL-1β, contribute to the development of inflammation in the epithelial compartments and intestinal lamina propria, which generates a deterioration of the intestinal barrier function. Moreover, obesity and HFDs decrease the expression of anti-inflammatory receptors PPARγ and change the composition of the intestinal microbiome, leading to a further alteration of intestinal wall permeability [306,307]. The disruption of immune tolerance and the increased permeability of the intestinal barrier, driven by altered metabolism and inflammation in obesity, facilitate the development of food allergies. Type VI hypersensitivity reactions associated with inflammation and tissue damage favour food allergy manifestations ranging from urticaria and gastrointestinal distress to severe anaphylaxis. Therefore, obesity and HFDs are associated with the development and course of food allergy. The interplay between metabolic disorders and immune dysfunction highlights the complex pathogenesis of food allergies in obese individuals, emphasizing the need for combined targeted interventions [306,309].
PGM3 deficiency, a rare congenital glycosylation disorder classified as an autosomal recessive HIES caused by mutations of the phosphoglucomutase PGM3 gene, links glycosylation defects to severe dermatitis with elevated serum IgE and food allergies, autoimmunity, immune deficiency, and neurocognitive impairment. PGM3 is a crucial enzyme in the synthesis of uridine diphosphate N-acetylglucosamine, critical for multiple glycosylation pathways. Altered protein glycosylation can lead to multisystem abnormalities, such as impaired immune functions, PGM3-deficient T cells producing excessive Th17 and Th2 cytokines which contribute to inflammatory and autoimmune manifestations, and abnormal neuronal development with probable brain hypomyelination [177,297,310,311].
Another important issue to be discussed at this point is the approach of non-immune adverse reactions to foods represented by “intolerances” due to metabolic (e.g. lactose intolerance) or disturbed transport mechanisms (e.g. fructose malabsorption). Such food intolerances can present some symptoms similar to food allergy but does not involve the immune system [5,312].
Food intolerances due to metabolic causes are not immunologically mediated and, therefore, they are not exactly type VI hypersensitivity reactions, but are worth discussing briefly. Examples of such nonallergic adverse food reactions due to enzyme deficiencies include lactose intolerance (due to primary lactase deficiency or lactase non-persistence, secondary, congenital or developmental lactase deficiency), hereditary fructose intolerance (aldolase B deficiency) and non-hereditary fructose malabsorption, congenital saccharose intolerance and acquired forms of sucrase-isomaltase deficiency. Hereditary galactosemia is due to defects in galactokinase, galactose-1-phosphate uridylyltransferase and UDP-galactose 4-epimerase. Other metabolic conditions are aldehyde dehydrogenase deficiency with flushing after alcohol, and glucose-6-phosphate dehydrogenase deficiency with hemolysis after ingestion of vicine and covicine-containing fava beans. In intolerance to FODMAPs (fermentable oligosaccharides, disaccharides, monosaccharides and polyols), short-chain carbohydrates that include lactose, fructose when in excess of glucose, sugar polyols such as sorbitol and mannitol, fructans, and galactooligosaccharides such as stachyose and raffinose, are involved, being poorly absorbed and fermented by intestinal bacteria. The consumption of high amounts of FODMAPs leads to the excessive production of short-chain fatty acids and a large amount of gas, including carbon dioxide, hydrogen, and methane, which are responsible for luminal distention and water retention [3,289,313,314,315].
2.7. Type VII Hypersensitivity Reactions via Direct Cellular and Inflammatory Responses to Chemical Substances
Hypersensitivity related to direct cellular responses to chemicals is involved in the pathophysiological mechanisms of food-dependent NSAID-induced anaphylaxis, non-immunological contact urticaria, phototoxic contact dermatitis to foods, as well as other often not well understood so-called “idiosyncratic” or “pseudo-allergic reactions”.
The role of nonsteroidal anti-inflammatory drugs is well documented as triggering cofactor in food-dependent NSAID-induced anaphylaxis (FDNIA) related to food lipid transfer proteins (LTPs), in circa 35% of cases, but also to wheat, peanuts, sunflower seeds and shellfish. In the complex pathophysiological mechanisms of FDNIA, nonselective NSAIDs inhibit COX-1, leading to the inhibition of mucosal protective prostaglandin synthesis, thus exposing the gastrointestinal tissues to the harmful effects of gastric acid and bile. NSAIDs also uncouple mitochondrial oxidative phosphorylation and induce mitochondrial damage that leads to epithelial dysfunction with increased intestinal permeability and allergen absorption. Moreover, NSAIDs directly impact mast cells and basophils IgE activation, amplifying their activation and degranulation. An increased expression of the adenosine receptors ADORA3 and enzymes involved in nucleotide metabolism was revealed in patients with FDNIA related to food LTPs, thus the amplified expression of ATP ectonucleotidases and robust activation of ADORA3 potentiate FcεRI-induced degranulation of mast cells in FDNIA. Interestingly, in cases of FDNIA related to wheat, augmentation by acetylsalicylic/acid aspirin in a dose-dependent manner but also urticaria/anaphylaxis to food additives or foods with natural salicylates such as pickles and curry was reported [28,316,317,318,319].
Non-immunological contact urticaria (NICU) to food represents a transient wheal-and-flare reaction occurring in individuals without previous sensitization, restricted to the skin area in contact with the eliciting food. Common contact low molecular weight agents in foods causing NICU are usually natural organic compounds commonly used as a food preservatives, such as sorbic acid and benzoic acid, or flavoring agents, such as benzaldehyde with almond-like aroma and cinnamic acid and cinnamaldehyde with cinnamon-like aroma [46,47,48,320].
Phototoxic contact dermatitis (PTCD) to food or phytophotodermatitis is typically due to topical skin exposure to certain plant-derived substances that cause a phototoxic reaction. It usually occurs by co-exposure to edible plants with parts contaning furocoumarins and solar radiation. The most common inducers of PTCD are stems and leaves of Apiaceae (Umbelliferae) vegetables, especially celery, parsley, parsnip, and peel and pulp of Rutaceae fruits such as lemon, lime, bergamot orange, grapefruit. These contain methoxypsoralen furocoumarins, which are inactive but, following exposure to UVA radiation (320–400 nm), became toxic for keratinocytes and cause dermatitis with unusual configurations and long lasting residual hyperpigmentation. Bartenders who prepare mixed drinks by squeezing citrus fruits outdoors, photo-exposed gardeners, and food industry workers who handle vegetables such as celery are at greater risk. Generalized phototoxic reactions may occur when ingesting large amounts of celery following PUVA therapy or tanning salon sessions [46,47,48,320].
In non-celiac wheat sensitivity (NCWS), amylase/trypsin inhibitors in wheat, proteins with parasitic enzyme-inhibiting properties, activate the toll-like receptor TLR4-MD2-CD14 complex on intestinal innate immune cells, triggering the secretion of proinflammatory chemokines and cytokines. This process promotes intestinal and extra-intestinal inflammation, contributing to the symptoms of NCWS. In patients classified as NCGS an increased number of duodenal intraepithelial lymphocytes and the presence of eosinophils at the lamina propria level were reported [23,286,289,321,322].
Finally, another issue to be mentioned is the approach of non-immune (not immunologically mediated) adverse reactions to food chemicals represented by the often not well-understood so-called other idiosyncratic or pseudo-allergic reactions. Neither serum specific IgE nor IgG against food allergens is involved. Food-specific IgG4 does not indicate food intolerances but rather a physiological response of the immune system after exposure to food components [5,312,323].
Natural food chemicals have been attributed to a wide range of adverse food reactions. Biogenic amines in foods are produced by bacteria during fermentation, storage or decay, and may be involved in pseudoallergic reactions when ingested. Pathophysiological discussions are frequently focussed on high histamine-containing foods, even though other vasoactive amines such as phenylethylamine, serotonin, tyramine, tryptamine, cadaverine and/or biogenic diamine putrescine and its polyamine metabolites spermine and spermidine, also cause adverse reactions or may affect histamine metabolism [324,325,326]. For some individuals, ingesting foods with high levels of natural biogenic amines can cause symptoms that may mimic IgE-mediated food reactions. High histamine-containing foods (e.g. aged cheese, fermented meat/fish, sauerkraut), histamine-releasing foods (e.g. strawberry, papaya, kiwi, pineapple, wine), phenylethylamine (chocolate), serotonin (banana, tomato), tyramine (pickled fish, aged cheese) and tryptamine (tomato, plum) may induce pseudoallergic reactions by pharmacological-type mechanisms [3,79,313]. The amount of these biogenic amines in food ingested tends to be more directly related to the severity of manifestations than immunologic reactions to food. Moreover, some additives affect biogenic amine content; for example, strawberries treated with ascorbic acid show a noticeable decrease in histamine concentration [313,327].
Regarding so-called histamine intolerance, ingesting histamine-rich foods is often suspected of causing (non-specific) symptoms, even though supporting scientific data are limited. No pathophysiologic mechanism, such as deficiency in one of the enzymes that degrade histamine, has been demonstrated in prospective, controlled studies. Histamine is degraded in the intestine by diamine oxidase and histamine N-methyl-transferase. There are still no objective biomarkers for the adverse reactions to ingested histamine. Several medications, such as metamizole, metronidazole, metoclopramide, and verapamil, have been supposed to have a negative effect on histamine-degrading enzymes, primarily diamine oxidase, but the relevance of these interactions needs to be further validated [313,324].
Salicylate intolerance has been defined as a nonspecific pseudoallergic reaction to salicylic acid, its derivatives or other related organic or inorganic acids of similar chemical structure. Acetyl salicylate is a potent inhibitor of COX-1, an isoform of the enzyme cyclooxygenase (COX), which prevents the conversion of arachidonic acid to cyclic prostanoids, whereas salicylic acid inhibits COX-2 gene expression. Spices containing high levels of natural salicylates have a high bioavailability and inhibit both COX-1 and COX-2, which may indicate additional dietary factors involved [326,328,329].
Food additive intolerance occurs less often than is supposed by patients and the media. Despite the high number of food additives, only several chemicals have been suspected in pseudoallergic adverse reactions, mediated through non-immunologic mechanisms. Although case reports have pointed to the potential for IgE-mediated anaphylaxis to natural dyes carmine (E120) from Dactylopius coccus, annatto (E160b) from Bixa orellana, and natural emulsifiers/thickeners such as plant-derived cellulose gum/carboxymethyl cellulose (E466), guar gum (E412) from Cyamopsis tetragonolobus, gum arabic (E414) from Acacia spp., fruit-derived pectin (E440), seaweed-derived carrageenan (E417), and animal collagen-derived gelatin (E441), the mechanisms behind sodium benzoate (E211) preservative and monosodium glutamate (E621) flavor enhancer food-associated reactions are not clearly elucidated, rare case reports suggesting involvement of non-IgE mediated basophil activation [326,330,331,332,333].
Chronic urticaria, recurrent angioedema and nonallergic asthma have been associated with pseudoallergic reactions to food additives. This association is controversial for atopic dermatitis and gastrointestinal diseases. Food additives that may be used in selected cases for in vivo and/or ex vivo provocations include synthetic food colours, such as azo dyes tartrazine (E102, now considered well-tolerated in patients with aspirin-exacerbated respiratory disease), sunset yellow (E110), azorubine (E122), amaranth (E123), cochineal red (E124), brilliant black (E151), and quinoline yellow (E104), xanthene erythrosine (E127), triarylmethane Patent blue V (E131), indigo carmine (E132) dyes; preservatives sorbic acid (E200), sodium benzoate (E211), propylparaben (E216), potassium metabisulfite (E224); flavour enhancer monosodium glutamate (E621), and artificial non-saccharide sweetener aspartame (E951). Monosodium glutamate (MSG), used primarily in Asian cuisine, may induce a nonallergic MSG symptom complex (formerly named the Chinese restaurant syndrome) when ingested in quantities more significant than those encountered in a regular European diet, the mechanism proposed involving an exaggerated sensitivity to this compound, which is metabolized after ingestion to glutamate, a major excitatory amino acid neurotransmitter [330,332,333,334,335].
Sulphur dioxides and sulphites (E220-E228) can cause potentially severe exacerbations in as many as 5% of patients with asthma, whereas individuals without asthma are rarely affected. Sulfite-sensitive patients more often have severe asthma. They may also cause urticaria and angioedema, and, less commonly, anaphylaxis. Both the European Union and the US Food and Drug Administration have regulated the use of these preservatives, requiring foods containing more than 10 ppm (10 mg/kg) to be labelled, but there is significant variation between levels for the same foods. Sulphites, readily metabolized to sulphate by sulphite oxidase, mainly occur in the human diet due to the addition of sodium/potassium sulphite/metabisulphite to foods to prevent enzymatic and non-enzymatic browning, control oxidation and prevent bacterial growth. The highest levels of sulfites are found in commercially prepared dried fruits (such as apricots) and processed potatoes, cider, beer and wines, wine vinegar and sauerkraut juice and some seafood such as shrimps. Sulphite preservatives are banned in fresh fruits and vegetables. The probability of a reaction is dependent on the nature of the food, the level and form of residual sulfite, the sensitivity of the patient, and the mechanism of the sulfite-induced reaction. Increased histamine and other mediators release from blood basophils was postulated in anaphylaxis, with no specific IgE detected, and monoclonal mast cell activation syndrome was reported as possibly associated. Other mechanisms are more probably involved in most reactions, with inhalation of sulphur dioxide, generated in the stomach from ingested sulphites causing bronchospasm by stimulation of irritant receptors and cholinergic pathways, or deficiency of mitochondrial sulphite oxidase in urticaria [20,326,331,333].
Gustatory nonimmunologic reactions induced especially by spicy foods are due to abnormal gustatory reflexes involving parasympathetic innervation are present in gustatory rhinitis, Frey syndrome or gustatory sweating, ‘crocodile tear syndrome’ or gustatory lacrimation, and gustatory otorrhoea. Transient receptor potential cation channel agonists present in foods are involved in the pathophysiology of gustatory rhinitis, a non-allergic, non-inflammatory condition characterized by rhinorrhea triggered by ingesting hot and spicy foods. TRPV1 agonist capsaicin, a pungent agent in chili peppers, red cayenne, tabasco sauce, horseradish, red and black pepper, and TRPA1 agonists found in garlic, such as organosulfur compounds allicin, diallyl disulfide and allyl isothiocyanate, activate primary sensory neurons via these thermosensitive chemical sensors. Stimulation of C fibres leads to the local release of several neuropeptides, including substance P and calcitonin gene-related peptide, with increased vasodilation and vascular permeability. Moreover, enhanced chemosensory sensitivity was detected in idiopathic rhinitis [336,337,338,339]. Spicy and sour foods may also induce gustatory sweating and/or unilateral flushing in the cutaneous distribution of the auriculotemporal nerve as a consequence of aberrant regenerated parasympathetic nerve fibers after parotid surgery in adults or facial trauma in childhood, condition known as auriculotemporal (Frey) syndrome [340,341,342].
Toxic nonimmunologic reactions can occur due to direct noxious effects of contaminated foods. Bacterial food poisoning is a main example that can produce gastrointestinal and neurologic manifestations [79].
Noninfectious food poisoning provoked by biotoxins are usually represented by amatoxin-containing wild mushroom poisoning, shellfish and pufferfish poisoning syndromes, and scombroid poisoning. The α-amanitin and other amatoxins are produced primarily by Amanita, Lepiota, and Galerina species of mushrooms. Marine dinoflagellate-produced saxitoxin, brevetoxin and okadaic acid from bivalve molluscs are involved in paralytic, neurotoxic, and diarrheic shellfish poisoning, respectively, while tetrodotoxin in pufferfish poisoning, and ciguatoxins in ciguatera tropical fish poisoning. Histamine intoxication most commonly occurs following the consumption of spoiled fish, particularly from the family Scombridae. Scombroid poisoning is related to fish with high levels of free histidine in their muscle tissues are refrigerated inadequately at sea or improperly stored at any time before consumption, and bacterial overgrowth sustains the conversion of histidine into histamine by bacterial enzyme histidine decarboxylase. Dark-fleshed fish from the Scombridae family, such as mackerel and tuna, and nonscombroid representatives, such as amberjack, have also been implicated; thus, this condition is better termed histamine fish poisoning. Toxic levels of histamine and other toxins are not destroyed by cooking or subsequent refrigeration. Although direct cellular responses to toxins are incriminated in such food poisonings and their clinical manifestations may overlap with food allergies, they must not be considered hypersensitivity reactions, as they can affect anyone who ingests contaminated food [343,344,345,346,347].
3. Conclusion
The significant advantage of the immune response and tissue-based new nomenclature approach in food allergies is helping to move the field towards precision and personalized medicine. An up-to-date classification of non-IgE-mediated hypersensitivity reactions to foods focused on mechanisms with a detailed pathophysiological approach and endotype-driven thinking in food allergies supports the fundamental aim of clinical immunology to lead to new diagnostic and therapy-related biomarkers, better understanding of clinical manifestations, improved immunotherapy and targeted therapeutic strategies, including monoclonal antibodies and small molecule drugs, and thus to refine disease management and improve the patients’ quality of life. The rationale for the use and the potential development of molecular biomarkers are kept in mind, such as disease risk, diagnostic, disease staging, and disease prognostic biomarkers, along with proof-of-mechanism, drug activity, pharmacodynamic and therapeutic response predictive biomarkers. Moreover, molecularly targeted therapies such as biological therapeutics, also called biologics, are constantly considered in precision medicine judgment. Endotype thinking and molecular decision-making can also support future translational and clinical research, encouraging professionals to continue their scientific work to explore more innovative strategies.
Author Contributions
All authors have contributed to the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Jutel, M.; Agache, I.; Zemelka-Wiacek, M.; Akdis, M.; Chivato, T.; Del Giacco, S.; Gajdanowicz, P.; Gracia, I.E.; Klimek, L.; Lauerma, A. , et al. Nomenclature of allergic diseases and hypersensitivity reactions: Adapted to modern needs: An EAACI position paper. Allergy 2023, 78, 2851–2874. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.N.; Xiang, L. Interpretation of 2023 EAACI guidelines on the diagnosis of IgE-mediated food allergy. Zhonghua Yu Fang Yi Xue Za Zhi 2024, 58, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.F.; Riggioni, C.; Agache, I.; Akdis, C.A.; Akdis, M.; Alvarez-Perea, A.; Alvaro-Lozano, M.; Ballmer-Weber, B.; Barni, S.; Beyer, K. , et al. EAACI guidelines on the diagnosis of IgE-mediated food allergy. Allergy 2023, 78, 3057–3076. [Google Scholar] [CrossRef] [PubMed]
- Johansson, S.G.; Hourihane, J.O.; Bousquet, J.; Bruijnzeel-Koomen, C.; Dreborg, S.; Haahtela, T.; Kowalski, M.L.; Mygind, N.; Ring, J.; van Cauwenberge, P. , et al. A revised nomenclature for allergy. An EAACI position statement from the EAACI nomenclature task force. Allergy 2001, 56, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Ring, J.; Jutel, M.; Papadopoulos, N.; Pfaar, O.; Akdis, C. Provocative proposal for a revised nomenclature for allergy and other hypersensitivity diseases. Allergy 2018, 73, 1939–1940. [Google Scholar] [CrossRef]
- Schmid-Grendelmeier, P.; Simon, D.; Simon, H.U.; Akdis, C.A.; Wüthrich, B. Epidemiology, clinical features, and immunology of the "intrinsic" (non-IgE-mediated) type of atopic dermatitis (constitutional dermatitis). Allergy 2001, 56, 841–849. [Google Scholar] [CrossRef]
- Wollenberg, A.; Christen-Zäch, S.; Taieb, A.; Paul, C.; Thyssen, J.P.; de Bruin-Weller, M.; Vestergaard, C.; Seneschal, J.; Werfel, T.; Cork, M.J. , et al. ETFAD/EADV Eczema task force 2020 position paper on diagnosis and treatment of atopic dermatitis in adults and children. J Eur Acad Dermatol Venereol 2020, 34, 2717–2744. [Google Scholar] [CrossRef]
- Meyer, R.; Chebar Lozinsky, A.; Fleischer, D.M.; Vieira, M.C.; Du Toit, G.; Vandenplas, Y.; Dupont, C.; Knibb, R.; Uysal, P.; Cavkaytar, O.; Nowak-Wegrzyn, A.; Shah, N.; Venter, C. Diagnosis and management of Non-IgE gastrointestinal allergies in breastfed infants—An EAACI Position Paper. Allergy 2020, 75, 14–32. [Google Scholar] [CrossRef]
- Pat, Y.; Yazici, D.; D'Avino, P.; Li, M.; Ardicli, S.; Ardicli, O.; Mitamura, Y.; Akdis, M.; Dhir, R.; Nadeau, K.; et al. Recent advances in the epithelial barrier theory. Int Immunol 2024, 36, 211–222. [Google Scholar] [CrossRef]
- Popescu, F.D. Molecular biomarkers for grass pollen immunotherapy. World J Methodol 2014, 4, 26–45. [Google Scholar] [CrossRef]
- Riggioni, C.; Oton, T.; Carmona, L.; Du Toit, G.; Skypala, I.; Santos, A.F. Immunotherapy and biologics in the management of IgE-mediated food allergy: Systematic review and meta-analyses of efficacy and safety. Allergy 2024, 79, 2097–2127. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.W.; Suwanpradid, J.; Kim, I.H.; Staats, H.F.; Haniffa, M.; MacLeod, A.S.; Abraham, S.N. Perivascular dendritic cells elicit anaphylaxis by relaying allergens to mast cells via microvesicles. Science 2018, 362, eaao0666. [Google Scholar] [CrossRef]
- Lin, E.V.; Suresh, R.V.; Dispenza, M.C. Bruton's tyrosine kinase inhibition for the treatment of allergic disorders. Ann Allergy Asthma Immunol 2024, 133, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Peavy, R.D.; Metcalfe, D.D. Understanding the mechanisms of anaphylaxis. Curr Opin Allergy Clin Immunol 2008, 8, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Jeebhay, M.F.; Moscato, G.; Bang, B.E.; Folletti, I.; Lipińska-Ojrzanowska, A.; Lopata, A.L.; Pala, G.; Quirce, S.; Raulf, M.; Sastre, J.; et al. Food processing and occupational respiratory allergy - An EAACI position paper. Allergy 2019, 74, 1852–1871. [Google Scholar] [CrossRef] [PubMed]
- Popescu, F.D. Cross-reactivity between aeroallergens and food allergens. World J Methodol 2015, 5, 31–50. [Google Scholar] [CrossRef] [PubMed]
- Benedé, S.; Garrido-Arandia, M.; Martín-Pedraza, L.; Bueno, C.; Díaz-Perales, A.; Villalba, M. Multifactorial modulation of food-induced anaphylaxis. Front Immunol 2017, 8, 552. [Google Scholar] [CrossRef]
- Mortz, C.G.; Eller, E.; Garvik, O.S.; Kjaer, H.F.; Zuberbier, T.; Bindslev-Jensen, C. Challenge-verified thresholds for allergens mandatory for labeling: How little is too much for the most sensitive patient? Allergy 2024, 79, 1306–1316. [Google Scholar] [CrossRef]
- Spolidoro, G.C.I.; Ali, M.M.; Amera, Y.T.; Nyassi, S.; Lisik, D.; Ioannidou, A.; Rovner, G.; Khaleva, E.; Venter, C.; van Ree, R.; et al. Prevalence estimates of eight big food allergies in Europe: Updated systematic review and meta-analysis. Allergy 2023, 78, 2361–2417. [Google Scholar] [CrossRef]
- Global Initiative for Asthma. Global Strategy for Asthma Management and Prevention, 2024. Updated May 2024. Available from: https://ginasthma.org/wp-content/uploads/2024/05/GINA-2024-Strategy-Report-24_05_22_WMS.pdf (Accessed 8 September 2024).
- Chiang, V.; Mak, H.W.F.; Yeung, M.H.Y.; Kan, A.K.C.; Au, E.Y.L.; Li, P.H. Epidemiology, outcomes, and disproportionate burden of food-dependent exercise-induced anaphylaxis from the Hong Kong Multidisciplinary Anaphylaxis Management Initiative (HK-MAMI). J Allergy Clin Immunol Glob 2023, 2, 100127. [Google Scholar] [CrossRef]
- Kulthanan, K.; Ungprasert, P.; Jirapongsananuruk, O.; Rujitharanawong, C.; Munprom, K.; Trakanwittayarak, S.; Pochanapan, O.; Panjapakkul, W.; Maurer, M. Food-Dependent Exercise-Induced Wheals/Angioedema, Anaphylaxis, or Both: A Systematic Review of Phenotypes. J Allergy Clin Immunol Pract 2023, 11, 1926–1933. [Google Scholar] [CrossRef] [PubMed]
- Preda, M.; Popescu, F.D.; Vassilopoulou, E.; Smolinska, S. Allergenic Biomarkers in the Molecular Diagnosis of IgE-Mediated Wheat Allergy. Int J Mol Sci 2024, 25, 8210. [Google Scholar] [CrossRef] [PubMed]
- Scala, E.; Villella, V.; Asero, R. Food-dependent exercise-induced allergic reactions in Lipid Transfer Protein (LTP) hypersensitive subjects: new data and a critical reappraisal. Eur Ann Allergy Clin Immunol 2024. [Google Scholar] [CrossRef]
- Chong, T.; Olivieri, B.; Skypala, I.J. Food-triggered anaphylaxis in adults. Curr Opin Allergy Clin Immunol 2024, 24, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Jeong, K. Food-dependent exercise-induced anaphylaxis: The need for better understanding and management of the disease. Allergy Asthma Immunol Res 2022, 14, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Cano, R.; San Bartolome, C.; Casas-Saucedo, R.; Araujo, G.; Gelis, S.; Ruano-Zaragoza, M.; Roca-Ferrer, J.; Palomares, F.; Martin, M.; Bartra, J. , et al. Immune-mediated mechanisms in cofactor-dependent food allergy and anaphylaxis: Effect of cofactors in basophils and mast cells. Front Immunol 2021, 11, 623071. [Google Scholar] [CrossRef]
- Scherf, K.A.; Lindenau, A.C.; Valentini, L.; Collado, M.C.; García-Mantrana, I.; Christensen, M.; Tomsitz, D.; Kugler, C.; Biedermann, T.; Brockow, K. Cofactors of wheat-dependent exercise-induced anaphylaxis do not increase highly individual gliadin absorption in healthy volunteers. Clin Transl Allergy 2019, 9, 19. [Google Scholar] [CrossRef]
- Muñoz-Cano, R.; Pascal, M.; Bartra, J.; Picado, C.; Valero, A.; Kim, D.K.; Brooks, S.; Ombrello, M.; Metcalfe, D.D.; Rivera, J.; Olivera, A. Distinct transcriptome profiles differentiate nonsteroidal anti-inflammatory drug-dependent from nonsteroidal anti-inflammatory drug-independent food-induced anaphylaxis. J Allergy Clin Immunol 2016, 137, 137–146. [Google Scholar] [CrossRef]
- Baldo, B.A.; Pham, N.H. Opioid toxicity: histamine, hypersensitivity, and MRGPRX2. Arch Toxicol 2023, 97, 359–375. [Google Scholar] [CrossRef]
- Kauppila, G.R.; Eagen, K.V. Opioid Use Disorder from Poppy Seed Tea Use: A Case Report. Am J Case Rep 2023, 24, e938675. [Google Scholar] [CrossRef]
- Kowalczyk, A.; Kuczyńska, R.; Żbikowska-Gotz, M.; Bartuzi, Z.; Krogulska, A. Anaphylaxis in an 8-Year-Old Boy Following the Consumption of Poppy Seed. J Investig Allergol Clin Immunol 2020, 30, 288–289. [Google Scholar] [CrossRef] [PubMed]
- Podzhilkova, A.; Nagl, C.; Hummel, K.; Bindslev-Jensen, C.; Eller, E.; Mortz, C.G.; Bublin, M.; Hoffmann-Sommergruber, K. Poppy Seed Allergy: Molecular Diagnosis and Cross-Reactivity With Tree Nuts. J Allergy Clin Immunol Pract 2024, 12, 2144–2154.e11. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Borges, M.; Fernández-Caldas, E.; Capriles-Hulett, A.; Caballero-Fonseca, F. Mite-induced inflammation: More than allergy. Allergy Rhinol (Providence) 2012, 3, e25–e29. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Borges, M.; Suárez Chacón, R.; Capriles-Hulett, A.; Caballero-Fonseca, F.; Fernández-Caldas, E. Anaphylaxis from ingestion of mites: pancake anaphylaxis. J Allergy Clin Immunol 2013, 131, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Werfel, T.; Asero, R.; Ballmer-Weber, B.K.; Beyer, K.; Enrique, E.; Knulst, A.C.; Mari, A.; Muraro, A.; Ollert, M.; Poulsen, L.K. , et al. Position paper of the EAACI: food allergy due to immunological cross-reactions with common inhalant allergens. Allergy 2015, 70, 1079–1090. [Google Scholar] [CrossRef]
- Darsow, U.; Gelincik, A.; Jappe, U.; Platts-Mills, T.A.; Ünal, D.; Biedermann, T. Algorithms in allergy: An algorithm for alpha-Gal syndrome diagnosis and treatment, 2024 update. Allergy 2024. [Google Scholar] [CrossRef]
- Popescu, F.D.; Cristea, O.M.; Ionica, F.E.; Vieru, M. Drug allergies due to IgE sensitization to α-Gal. Farmacia 2019, 67, 43–49. [Google Scholar] [CrossRef]
- Ikemoto, C.; Tamagawa-Mineoka, R.; Masuda, K.; Iida, S.; Inomata, N.; Katoh, N. Immediate-onset anaphylaxis of Bacillus subtilis-fermented soybeans (natto). J Dermatol 2014, 41, 857–858. [Google Scholar] [CrossRef]
- Inomata, N.; Chin, K.; Aihara, M. Anaphylaxis caused by ingesting jellyfish in a subject with fermented soybean allergy: possibility of epicutaneous sensitization to poly-gamma-glutamic acid by jellyfish stings. J Dermatol 2014, 41, 752–753. [Google Scholar] [CrossRef]
- Lopata AL, Jeebhay MF. Airborne seafood allergens as a cause of occupational allergy and asthma. Curr Allergy Asthma Rep. 2013, 13, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.K.; Seternes, O.M.; Larsen, M.; Kishimur, H.; Rudenskaya, G.N.; Bang, B. Purified sardine and king crab trypsin display individual differences in PAR-2-, NF-κB-, and IL-8 signaling. Toxicol. Environ. Chem. 2011, 93, 1991–2011. [Google Scholar] [CrossRef]
- Pougnet, R.; Loddé, B.; Lucas, D.; Jégaden, D.; Bell, S.; Dewitte, J.D. A case of occupational asthma from metabisulphite in a fisherman. Int. Marit. Health 2010, 62, 180–184. [Google Scholar] [PubMed]
- Smit, L.A.; Wouters, I.M.; Hobo, M.M.; Eduard, W.; Doekes, G.; Heederik, D. Agricultural seed dust as a potential cause of organic dust toxic syndrome. Occup. Environ. Med. 2006, 63, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Amaro, C.; Goossens, A. Immunological occupational contact urticaria and contact dermatitis from proteins: a review. Contact Dermatitis 2008, 58, 67–75. [Google Scholar] [CrossRef]
- Brancaccio, R.R.; Alvarez, M.S. Contact allergy to food. Dermatol. Ther. 2004, 17, 302–313. [Google Scholar] [CrossRef]
- Giménez-Arnau, A.M.; Pesqué, D.; Maibach, H.I. Contact Urticaria Syndrome: a comprehensive review. Curr. Dermatol. Rep. 2022, 11, 194–201. [Google Scholar] [CrossRef]
- Walter, A.; Seegräber, M.; Wollenberg, A. Food-related contact dermatitis, contact urticaria, and atopy patch test with food. Clin. Rev. Allergy Immunol. 2019, 56, 19–31. [Google Scholar] [CrossRef]
- Ashbaugh, A.G.; Abel, M.K.; Murase, J.E. Protein causes of urticaria and dermatitis. Immunol. Allergy Clin. North Am. 2021, 41, 481–491. [Google Scholar] [CrossRef]
- Vethachalam, S. Persaud, Y. Contact Urticaria. [Updated 2023 Jul 31]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: www.ncbi.nlm.nih.gov/books/NBK549890/.
- Bajwa, S.F.; Mohammed, R.H. Type II hypersensitivity reaction. [Updated 2023 Jul 4]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan–. Available from: www.ncbi.nlm.nih.gov/books/NBK563264.
- Dispenza, M.C. Classification of hypersensitivity reactions. Allergy Asthma Proc. 2019, 40, 470–473. [Google Scholar] [CrossRef]
- Justiz Vaillant, A.A.; Vashisht, R.; Zito, P.M. Immediate hypersensitivity reactions (Archived). 2023 May 29. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan–. [PubMed]
- Husebye, E.S.; Anderson, M.S.; Kämpe, O. Autoimmune polyendocrine syndromes. N. Engl. J. Med. 2018, 378, 1132–1141. [Google Scholar] [CrossRef]
- Lampasona, V.; Passerini, L.; Barzaghi, F.; Lombardoni, C.; Bazzigaluppi, E.; Brigatti, C.; Bacchetta, R.; Bosi, E. Autoantibodies to harmonin and villin are diagnostic markers in children with IPEX syndrome. PLoS One 2013, 8, e78664. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Lee, K.H.; Jeon, B.; Ochs, H.D.; Lee, J.S.; Gee, H.Y.; Seo, S.; Geum, D.; Piccirillo, C.A.; Eisenhut, M.; van der Vliet, H.J.; Lee, J.M.; Kronbichler, A.; Ko, Y.; Shin, J.I. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome: a systematic review. Autoimmun. Rev. 2020, 19, 102526. [Google Scholar] [CrossRef] [PubMed]
- Torgerson, T.R.; Linane, A.; Moes, N.; Anover, S.; Mateo, V.; Rieux-Laucat, F.; Hermine, O.; Vijay, S.; Gambineri, E.; Cerf-Bensussan, N.; Fischer, A.; et al. Severe food allergy as a variant of IPEX syndrome caused by a deletion in a noncoding region of the FOXP3 gene. Gastroenterology 2007, 132, 1705–1717. [Google Scholar] [CrossRef]
- Verbsky, J.W.; Chatila, T.A. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) and IPEX-related disorders: an evolving web of heritable autoimmune diseases. Curr. Opin. Pediatr. 2013, 25, 708–714. [Google Scholar] [CrossRef]
- Altintas, A.; Pasa, S.; Cil, T.; Bayan, K.; Gokalp, D.; Ayyildiz, O. Thyroid and celiac diseases autoantibodies in patients with adult chronic idiopathic thrombocytopenic purpura. Platelets 2008, 19, 252–257. [Google Scholar] [CrossRef]
- Fisgin, T.; Yarali, N.; Duru, F.; Usta, B.; Kara, A. Hematologic manifestation of childhood celiac disease. Acta Haematol. 2004, 111, 211–214. [Google Scholar] [CrossRef]
- Guarina, A.; Marinoni, M.; Lassandro, G.; Saracco, P.; Perrotta, S.; Facchini, E.; Notarangelo, L.D.; Russo, G.; Giordano, P.; Romano, F.; et al. Association of immune thrombocytopenia and celiac disease in children: a retrospective case control study. Turk. J. Haematol. 2021, 38, 175–180. [Google Scholar] [CrossRef]
- Hadjivassiliou, M.; Aeschlimann, P.; Sanders, D.S.; Mäki, M.; Kaukinen, K.; Grünewald, R.A.; Bandmann, O.; Woodroofe, N.; Haddock, G.; Aeschlimann, D.P. Transglutaminase 6 antibodies in the diagnosis of gluten ataxia. Neurology 2013, 80, 1740–1745. [Google Scholar] [CrossRef]
- Hadjivassiliou, M.; Davies-Jones, G.A.; Sanders, D.S.; Grünewald, R.A. Dietary treatment of gluten ataxia. J Neurol Neurosurg Psychiatry 2003, 74, 1221–1224. [Google Scholar] [CrossRef]
- Lauret, E.; Rodrigo, L. Celiac disease and autoimmune-associated conditions. Biomed Res Int. 2013, 2013, 127589. [Google Scholar] [CrossRef]
- Sapone, A.; Bai, J.C.; Ciacci, C.; Dolinsek, J.; Green, P.H.; Hadjivassiliou, M.; Kaukinen, K.; Rostami, K.; Sanders, D.S.; Schumann, M.; et al. Spectrum of gluten-related disorders: consensus on new nomenclature and classification. BMC Med. 2012, 10, 13. [Google Scholar] [CrossRef] [PubMed]
- Todd, P.K.; Shakkottai, V.G. Overview of cerebellar ataxia in adults. In: Hurtig, H.I., ed. UpToDate. Wolters Kluwer. Updated August 5, 2024. Available online: www.uptodate.com/contents/overview-of-cerebellar-ataxia-in-adults (accessed on 8 September 2024).
- Brenner, S.; Goldberg, I. Drug-induced pemphigus. Clin Dermatol. 2011, 29, 455–457. [Google Scholar] [CrossRef] [PubMed]
- Brenner, S.; Ruocco, V.; Ruocco, E.; Russo, A.; Tur, E.; Luongo, V.; Lombardi, M.L. In vitro tannin acantholysis. Int J Dermatol. 2000, 39, 738–742. [Google Scholar] [CrossRef] [PubMed]
- Halil Yavuz, I.; Ozaydın Yavuz, G. The role of pentraxin 3 in pemphigus vulgaris. Postepy Dermatol Alergol. 2020, 37, 503–507. [Google Scholar] [CrossRef]
- Hertl, M.; Sitaru, C. Pathogenesis, clinical manifestations, and diagnosis of pemphigus. In: Zone, J.J., ed. UpToDate. Wolters Kluwer. Updated January 25, 2024. Available online: www.uptodate.com/contents/pathogenesis-clinical-manifestations-and-diagnosis-of-pemphigus (accessed on 8 September 2024).
- Ruocco, V.; Brenner, S.; Ruocco, E. Pemphigus and diet: does a link exist? Int J Dermatol. 2001, 40, 161–163. [Google Scholar] [CrossRef]
- Usman, N.; Annamaraju, P. Type III Hypersensitivity Reaction. [Updated 2023 May 22]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available online: www.ncbi.nlm.nih.gov/books/NBK559122/.
- Costabel, U.; Miyazaki, Y.; Pardo, A.; Koschel, D.; Bonella, F.; Spagnolo, P.; Guzman, J.; Ryerson, C.J.; Selman, M. Hypersensitivity pneumonitis. Nat Rev Dis Primers 2020, 6, 65. [Google Scholar] [CrossRef]
- King, T.E. Hypersensitivity pneumonitis (extrinsic allergic alveolitis): Epidemiology, causes, and pathogenesis. In: Flaherty, K.R., Ed. UpToDate. Wolters Kluwer. Updated August 01, 2023. Available online: www.uptodate.com/contents/hypersensitivity-pneumonitis-extrinsic-allergic-alveolitis-epidemiology-causes-and-pathogenesis (Accessed 8 September 2024).
- Kongsupon, N.; Walters, G.I.; Sadhra, S.S. Occupational causes of hypersensitivity pneumonitis: a systematic review and compendium. Occup Med 2021, 71, 255–259. [Google Scholar] [CrossRef]
- Quirce, S.; Vandenplas, O.; Campo, P.; Cruz, M.J.; de Blay, F.; Koschel, D.; Moscato, G.; Pala, G.; Raulf, M.; Sastre, J.; Siracusa, A.; Tarlo, S.M.; Walusiak-Skorupa, J.; Cormier, Y. Occupational hypersensitivity pneumonitis: an EAACI position paper. Allergy 2016, 71, 765–779. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A.; King, T.E., Jr. Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med 2012, 186, 314–324. [Google Scholar] [CrossRef]
- Arasi, S.; Mastrorilli, C.; Pecoraro, L.; Giovannini, M.; Mori, F.; Barni, S.; Caminiti, L.; Castagnoli, R.; Liotti, L.; Saretta, F. , et al. Heiner Syndrome and Milk Hypersensitivity: An Updated Overview on the Current Evidence. Nutrients 2021, 13, 1710. [Google Scholar] [CrossRef]
- Cox, A.L.; Sicherer, S.H. Classification of adverse food reactions. J Food Allergy 2020, 2, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Park, M.; Jung, J.H.; Kim, S.Y.; Kim, Y.H.; Hahn, S.M.; Kim, S.; Lee, M.J.; Shim, H.S.; Sohn, M.H.; Kim, K.W.; Kim, M.J. Children with Heiner Syndrome: A Single-Center Experience. Children 2021, 8, 1110. [Google Scholar] [CrossRef] [PubMed]
- Aquino, M.; Rosner, G. Systemic Contact Dermatitis. Clin Rev Allergy Immunol 2019, 56, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Killig, C.; Werfel, T. Contact reactions to food. Curr Allergy Asthma Rep 2008, 8, 209–214. [Google Scholar] [CrossRef]
- Rozas-Muñoz, E.; Lepoittevin, J.P.; Pujol, R.M.; Giménez-Arnau, A. Allergic contact dermatitis to plants: understanding the chemistry will help our diagnostic approach. Actas Dermosifiliogr 2012, 103, 456–477. [Google Scholar] [CrossRef]
- Vergara, C.; Mauro, M.; Belloni Fortina, A.; Giulioni, E.; Larese Filon, F. Occupational Contact Dermatitis in Food Handlers: A 26-Year Retrospective Multicentre Study in North-East Italy (Triveneto Group on Research on Contact Dermatitis). Dermatitis 2024. [Google Scholar] [CrossRef]
- Yamaguchi, H.L.; Yamaguchi, Y.; Peeva, E. Role of Innate Immunity in Allergic Contact Dermatitis: An Update. Int J Mol Sci. 2023, 24, 12975. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Leonard, A.; Guttman-Yassky, E. The Unique Molecular Signatures of Contact Dermatitis and Implications for Treatment. Clin Rev Allergy Immunol. 2019, 56, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Brites, G.S.; Ferreira, I.; Sebastião, A.I.; Silva, A.; Carrascal, M.; Neves, B.M.; Cruz, M.T. Allergic contact dermatitis: From pathophysiology to development of new preventive strategies. Pharmacol Res. 2020, 162, 105282. [Google Scholar] [CrossRef]
- Scheinman, P.L.; Vocanson, M.; Thyssen, J.P.; Johansen, J.D.; Nixon, R.L.; Dear, K.; Botto, N.C.; Morot, J.; Goldminz, A.M. Contact dermatitis. Nat Rev Dis Primers. 2021, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Chipinda, I.; Hettick, J.M.; Siegel, P.D. Haptenation: chemical reactivity and protein binding. J Allergy (Cairo) 2011, 2011, 839682. [Google Scholar] [CrossRef] [PubMed]
- Lepoittevin, J.P.; Berl, V.; Giménez-Arnau, E. Alpha-methylene-gamma-butyrolactones: versatile skin bioactive natural products. Chem Rec 2009, 9, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.W.; Benezra, C. Quantitative structure-activity relationships for skin sensitization potential of urushiol analogues. Contact Dermatitis 1993, 29, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Sukakul, T.; Bruze, M.; Svedman, C. Fragrance Contact Allergy—A Review Focusing on Patch Testing. Acta Derm Venereol 2024, 104, adv40332. [Google Scholar] [CrossRef]
- Azeem, M.; Kader, H.; Kerstan, A.; Hetta, H.F.; Serfling, E.; Goebeler, M.; Muhammad, K. Intricate Relationship Between Adaptive and Innate Immune System in Allergic Contact Dermatitis. Yale J Biol Med 2020, 93, 699–709. [Google Scholar]
- Sakamoto, E.; Katahira, Y.; Mizoguchi, I.; Watanabe, A.; Furusaka, Y.; Sekine, A.; Yamagishi, M.; Sonoda, J.; Miyakawa, S.; Inoue, S. , et al. Chemical- and Drug-Induced Allergic, Inflammatory, and Autoimmune Diseases Via Haptenation. Biology 2023, 12, 123. [Google Scholar] [CrossRef]
- Aleksic, M.; Rajagopal, R.; de-Ávila, R.; Spriggs, S.; Gilmour, N. The skin sensitization adverse outcome pathway: exploring the role of mechanistic understanding for higher tier risk assessment. Crit Rev Toxicol 2024, 54, 69–91. [Google Scholar] [CrossRef]
- Rodrigues Neves, C.; Gibbs, S. Progress on Reconstructed Human Skin Models for Allergy Research and Identifying Contact Sensitizers. Curr Top Microbiol Immunol 2021, 430, 103–129. [Google Scholar] [CrossRef]
- Dhingra, N.; Shemer, A.; Correa da Rosa, J.; Rozenblit, M.; Fuentes-Duculan, J.; Gittler, J.K.; Finney, R.; Czarnowicki, T.; Zheng, X.; Xu, H.; et al. Molecular profiling of contact dermatitis skin identifies allergen-dependent differences in immune response. J Allergy Clin Immunol 2014, 134, 362–372. [Google Scholar] [CrossRef]
- Alcain, J.; Infante Cruz, A.D.P.; Barrientos, G.; Vanzulli, S.; Salamone, G.; Vermeulen, M. Mechanisms of unconventional CD8 Tc2 lymphocyte induction in allergic contact dermatitis: Role of H3/H4 histamine receptors. Front Immunol 2022, 13, 999852. [Google Scholar] [CrossRef]
- Afvari, S.; Zippin, J.H. Type I hypersensitivity in photoallergic contact dermatitis. JAAD Case Rep 2023, 44, 47–49. [Google Scholar] [CrossRef] [PubMed]
- Scheman, A.; Gupta, S. Photoallergic contact dermatitis from diallyl disulfide. Contact Dermatitis 2001, 45, 179. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, K.; Issa, C.J.; Nedorost, S.T.; Lio, P.A. Is food-triggered atopic dermatitis a form of systemic contact dermatitis? Clin Rev Allergy Immunol 2024, 66, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gil-Pallares, P.; Alvarez-Garcia, O.; González-Moure, C.; Castro-Murga, M.; Monteagudo-Sánchez, B. Fixed food eruption caused by Maja squinado (European spider crab) in a child. Contact Dermatitis 2020, 83, 510–512. [Google Scholar] [CrossRef] [PubMed]
- McWilliam, V.; Koplin, J.; Lodge, C.; Tang, M.; Dharmage, S.; Allen, K. The prevalence of tree nut allergy: A systematic review. Curr Allergy Asthma Rep 2015, 15, 54. [Google Scholar] [CrossRef]
- Sharma, L.; Agarwal, R.; Chopra, A.; Mitra, B. A cross-sectional observational study of clinical spectrum and prevalence of fixed food eruption in a tertiary care hospital. Indian Dermatol Online J 2020, 11, 361–366. [Google Scholar] [CrossRef]
- Volz, T.; Berner, D.; Weigert, C.; Röcken, M.; Biedermann, T. Fixed food eruption caused by asparagus. J Allergy Clin Immunol 2005, 116, 1390–1392. [Google Scholar] [CrossRef]
- Lania, G.; Nanayakkara, M.; Maglio, M.; Auricchio, R.; Porpora, M.; Conte, M.; De Matteis, M.A.; Rizzo, R.; Luini, A.; Discepolo, V.; et al. Constitutive alterations in vesicular trafficking increase the sensitivity of cells from celiac disease patients to gliadin. Commun Biol 2019, 2, 190. [Google Scholar] [CrossRef]
- Maiuri, L.; Villella, V.R.; Raia, V.; Kroemer, G. The gliadin-CFTR connection: New perspectives for the treatment of celiac disease. Ital J Pediatr 2019, 45, 40. [Google Scholar] [CrossRef]
- Rizzi, A.; Di Gioacchino, M.; Gammeri, L.; Inchingolo, R.; Chini, R.; Santilli, F.; Nucera, E.; Gangemi, S. The emerging role of innate lymphoid cells (ILCs) and alarmins in celiac disease: An update on pathophysiological insights, potential use as disease biomarkers, and therapeutic implications. Cells 2023, 12, 1910. [Google Scholar] [CrossRef]
- Tye-Din, J.A.; Galipeau, H.J.; Agardh, D. Celiac disease: A review of current concepts in pathogenesis, prevention, and novel therapies. Front Pediatr 2018, 6, 350. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Vargas, J.; Green, P.H.R.; Bhagat, G. Innate lymphoid cells and celiac disease: Current perspective. Cell Mol Gastroenterol Hepatol 2021, 11, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Bijelić, B.; Matić, I.Z.; Besu, I.; Janković, L.; Juranić, Z.; Marušić, S.; Andrejević, S. Celiac disease-specific and inflammatory bowel disease-related antibodies in patients with recurrent aphthous stomatitis. Immunobiology 2019, 224, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Camarca, A.; Rotondi Aufiero, V.; Mazzarella, G. Role of regulatory T cells and their potential therapeutic applications in celiac disease. Int J Mol Sci 2023, 24, 14434. [Google Scholar] [CrossRef]
- Cellier, C.; Patey, N.; Mauvieux, L.; Jabri, B.; Delabesse, E.; Cervoni, J.P.; Burtin, M.L.; Guy-Grand, D.; Bouhnik, Y.; Modigliani, R.; Barbier, J.P.; Macintyre, E.; Brousse, N.; Cerf-Bensussan, N. Abnormal intestinal intraepithelial lymphocytes in refractory sprue. Gastroenterology 1998, 114, 471–481. [Google Scholar] [CrossRef]
- Halttunen, T.; Mäki, M. Serum immunoglobulin A from patients with celiac disease inhibits human T84 intestinal crypt epithelial cell differentiation. Gastroenterology 1999, 116, 566–572. [Google Scholar] [CrossRef]
- Husby, S.; Murray, J.A.; Katzka, D.A. AGA clinical practice update on diagnosis and monitoring of celiac disease—changing utility of serology and histologic measures: Expert review. Gastroenterology 2019, 156, 885–889. [Google Scholar] [CrossRef]
- Görög, A.; Antiga, E.; Caproni, M.; Cianchini, G.; De, D.; Dmochowski, M.; Dolinsek, J.; Drenovska, K.; Feliciani, C.; Hervonen, K.; et al. S2k guidelines (consensus statement) for diagnosis and therapy of dermatitis herpetiformis initiated by the European Academy of Dermatology and Venereology (EADV). J Eur Acad Dermatol Venereol 2021, 35, 1251–1277. [Google Scholar] [CrossRef]
- Furue, M. Regulation of Skin Barrier Function via Competition between AHR Axis versus IL-13/IL-4‒JAK‒STAT6/STAT3 Axis: Pathogenic and Therapeutic Implications in Atopic Dermatitis. J Clin Med 2020, 9, 3741. [Google Scholar] [CrossRef]
- Huang, I.H.; Chung, W.H.; Wu, P.C.; Chen, C.B. JAK-STAT signaling pathway in the pathogenesis of atopic dermatitis: An updated review. Front Immunol 2022, 13, 1068260. [Google Scholar] [CrossRef]
- Kogame, T.; Egawa, G.; Kabashima, K. Exploring the role of Janus kinase (JAK) in atopic dermatitis: a review of molecular mechanisms and therapeutic strategies. Immunol Med 2023, 46, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, P.R.; Seminario-Vidal, L.; Abe, B.; Ghobadi, C.; Sims, J.T. Cytokines and Epidermal Lipid Abnormalities in Atopic Dermatitis: A Systematic Review. Cells 2023, 12, 2793. [Google Scholar] [CrossRef] [PubMed]
- Wasserer, S.; Jargosch, M.; Mayer, K.E.; Eigemann, J.; Raunegger, T.; Aydin, G.; Eyerich, S.; Biedermann, T.; Eyerich, K.; Lauffer, F. Characterization of High and Low IFNG-Expressing Subgroups in Atopic Dermatitis. Int J Mol Sci 2024, 25, 6158. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, L.; D'Onghia, M.; Lazzeri, L.; Rubegni, G.; Cinotti, E. Blocking the IL-4/IL-13 Axis versus the JAK/STAT Pathway in Atopic Dermatitis: How Can We Choose? J Pers Med 2024, 14, 775. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Ahn, K. Atopic dermatitis endotypes: knowledge for personalized medicine. Curr Opin Allergy Clin Immunol 2022, 22, 153–159. [Google Scholar] [CrossRef]
- Sanyal, R.D.; Pavel, A.B.; Glickman, J.; Chan, T.C.; Zheng, X.; Zhang, N.; Cueto, I.; Peng, X.; Estrada, Y.; Fuentes-Duculan, J.; et al. Atopic dermatitis in African American patients is TH2/TH22-skewed with TH1/TH17 attenuation. Ann Allergy Asthma Immunol 2019, 122, 99–110.e6. [Google Scholar] [CrossRef]
- Zhou, L.; Leonard, A.; Pavel, A.B.; Malik, K.; Raja, A.; Glickman, J.; Estrada, Y.D.; Peng, X.; Del Duca, E.; Sanz-Cabanillas, J.; et al. Age-specific changes in the molecular phenotype of patients with moderate-to-severe atopic dermatitis. J Allergy Clin Immunol 2019, 144, 144–156. [Google Scholar] [CrossRef]
- Esnault, S.; Kelly, E.A.; Johnson, S.H.; DeLain, L.P.; Haedt, M.J.; Noll, A.L.; Sandbo, N.; Jarjour, N.N. Matrix Metalloproteinase-9-Dependent Release of IL-1β by Human Eosinophils. Mediators Inflamm. 2019, 7479107. [Google Scholar] [CrossRef]
- Packi, K.; Matysiak, J.; Klimczak, S.; Matuszewska, E.; Bręborowicz, A.; Pietkiewicz, D.; Matysiak, J. Analysis of the Serum Profile of Cytokines Involved in the T-Helper Cell Type 17 Immune Response Pathway in Atopic Children with Food Allergy. Int. J. Environ. Res. Public Health 2022, 19, 7877. [Google Scholar] [CrossRef]
- Park, C.O.; Kim, S.M.; Lee, K.H.; Bieber, T. Biomarkers for Phenotype-Endotype Relationship in Atopic Dermatitis: A Critical Review. EBioMedicine 2024, 103, 105121. [Google Scholar] [CrossRef]
- Renert-Yuval, Y.; Thyssen, J.P.; Bissonnette, R.; Bieber, T.; Kabashima, K.; Hijnen, D.; Guttman-Yassky, E. Biomarkers in Atopic Dermatitis—A Review on Behalf of the International Eczema Council. J. Allergy Clin. Immunol. 2021, 147, 1174–1190.e1. [Google Scholar] [CrossRef] [PubMed]
- Sutton, C.E.; Lalor, S.J.; Sweeney, C.M.; Brereton, C.F.; Lavelle, E.C.; Mills, K.H. Interleukin-1 and IL-23 Induce Innate IL-17 Production from γδ T Cells, Amplifying Th17 Responses and Autoimmunity. Immunity 2009, 31, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Kido-Nakahara, M.; Onozuka, D.; Izuhara, K.; Saeki, H.; Nunomura, S.; Takenaka, M.; Matsumoto, M.; Kataoka, Y.; Fujimoto, R.; Kaneko, S.; et al. Exploring Patient Background and Biomarkers Associated with the Development of Dupilumab-Associated Conjunctivitis and Blepharitis. Allergol. Int. 2024, 73, 332–334. [Google Scholar] [CrossRef] [PubMed]
- Kolkhir, P.; Akdis, C.A.; Akdis, M.; Bachert, C.; Bieber, T.; Canonica, G.W.; Guttman-Yassky, E.; Metz, M.; Mullol, J.; Palomares, O.; Renz, H.; Ständer, S.; Zuberbier, T.; Maurer, M. Type 2 Chronic Inflammatory Diseases: Targets, Therapies and Unmet Needs. Nat Rev Drug Discov 2023, 22, 743–767. [Google Scholar] [CrossRef] [PubMed]
- Dellon, E.S.; Gonsalves, N.; Abonia, J.P.; Alexander, J.A.; Arva, N.C.; Atkins, D.; Attwood, S.E.; Auth, M.K.H.; Bailey, D.D.; Biederman, L.; et al. International Consensus Recommendations for Eosinophilic Gastrointestinal Disease Nomenclature. Clin Gastroenterol Hepatol 2022, 20, 2474–2484.e3. [Google Scholar] [CrossRef]
- Li, K.; Ruan, G.; Liu, S.; Xu, T.; Guan, K.; Li, J.; Li, J. Eosinophilic Gastroenteritis: Pathogenesis, Diagnosis, and Treatment. Chin Med J (Engl) 2023, 136, 899–909. [Google Scholar] [CrossRef]
- Neely, J.L.; Reimers, A.; Taylor, S.; Garg, S.; Masuda, M.Y.; Schroeder, S.; Wright, B.L.; Doyle, A.D. GATA-3 and T-bet as Diagnostic Markers of Non-Esophageal Eosinophilic Gastrointestinal Disease. Allergy 2022, 77, 1042–1044. [Google Scholar] [CrossRef]
- Greuter, T.; Straumann, A.; Fernandez-Marrero, Y.; Germic, N.; Hosseini, A.; Chanwangpong, A.; Yousefi, S.; Simon, D.; Collins, M.H.; Bussmann, C.; et al. A Multicenter Long-Term Cohort Study of Eosinophilic Esophagitis Variants and Their Progression to Eosinophilic Esophagitis Over Time. Clin Transl Gastroenterol 2024, 15, e00664. [Google Scholar] [CrossRef]
- Sorge, A.; Aldinio, G.; Marinoni, B.; Visaggi, P.; Penagini, R.; Maniero, D.; Ghisa, M.; Marabotto, E.; de Bortoli, N.; Pasta, A.; Dipace, V.; Calabrese, F.; Vecchi, M.; Savarino, E.V.; Coletta, M. Distribution of Esophageal Inflammation in Patients with Eosinophilic Esophagitis and Its Impact on Diagnosis and Outcome. Dig Liver Dis 2024, 56, S1590–S865800967. [Google Scholar] [CrossRef]
- Underwood, B.; Troutman, T.D.; Schwartz, J.T. Breaking Down the Complex Pathophysiology of Eosinophilic Esophagitis. Ann Allergy Asthma Immunol 2023, 130, 28–39. [Google Scholar] [CrossRef]
- Alsohaibani, F.I.; Peedikayil, M.C.; Alzahrani, M.A.; Azzam, N.A.; Almadi, M.A.; Dellon, E.S.; Al-Hussaini, A.A. Eosinophilic Esophagitis: Current Concepts in Diagnosis and Management. Saudi J Gastroenterol 2024, 30, 210–227. [Google Scholar] [CrossRef] [PubMed]
- Cianferoni, A.; Shuker, M.; Brown-Whitehorn, T.; Hunter, H.; Venter, C.; Spergel, J.M. Food Avoidance Strategies in Eosinophilic Oesophagitis. Clin Exp Allergy 2019, 49, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Ridolo, E.; Barone, A.; Ottoni, M.; Peveri, S.; Montagni, M.; Nicoletta, F. The New Therapeutic Frontiers in the Treatment of Eosinophilic Esophagitis: Biological Drugs. Int J Mol Sci 2024, 25, 1702. [Google Scholar] [CrossRef]
- Rossi, C.M.; Santacroce, G.; Lenti, M.V.; di Sabatino, A. Eosinophilic Esophagitis in the Era of Biologics. Expert Rev Gastroenterol Hepatol 2024, 18, 271–281. [Google Scholar] [CrossRef]
- Wilson, B.E.; Sacta, M.A.; Wright, B.L.; Spergel, J.; Wolfset, N. The Relationship Between Eosinophilic Esophagitis and Immunotherapy. Immunol Allergy Clin North Am 2024, 44, 281–291. [Google Scholar] [CrossRef]
- Franciosi, J.P.; Mougey, E.B.; Dellon, E.S.; Gutierrez-Junquera, C.; Fernandez-Fernandez, S.; Venkatesh, R.D.; Gupta, S.K. Proton Pump Inhibitor Therapy for Eosinophilic Esophagitis: History, Mechanisms, Efficacy, and Future Directions. J Asthma Allergy 2022, 15, 281–302. [Google Scholar] [CrossRef]
- Rochman, Y.; Kotliar, M.; Ben-Baruch Morgenstern, N.; Barski, A.; Wen, T.; Rothenberg, M.E. TSLP Shapes the Pathogenic Responses of Memory CD4+ T Cells in Eosinophilic Esophagitis. Sci Signal 2023, 16, eadg6360. [Google Scholar] [CrossRef]
- Greuter, T.; Straumann, A.; Fernandez-Marrero, Y.; Germic, N.; Hosseini, A.; Yousefi, S.; Simon, D.; Collins, M.H.; Bussmann, C.; Chehade, M.; et al. Characterization of Eosinophilic Esophagitis Variants by Clinical, Histological, and Molecular Analyses: A Cross-Sectional Multi-Center Study. Allergy 2022, 77, 2520–2533. [Google Scholar] [CrossRef]
- Odiase, E.; Zhang, X.; Chang, Y.; Nelson, M.; Balaji, U.; Gu, J.; Zhang, Q.; Pan, Z.; Spechler, S.J.; Souza, R.F. In Esophageal Squamous Cells From Eosinophilic Esophagitis Patients, Th2 Cytokines Increase Eotaxin-3 Secretion Through Effects on Intracellular Calcium and a Non-Gastric Proton Pump. Gastroenterology 2021, 160, 2072–2088.e6. [Google Scholar] [CrossRef]
- Kaneko, T.; Iwamura, C.; Kiuchi, M.; Kurosugi, A.; Onoue, M.; Matsumura, T.; Chiba, T.; Nakayama, T.; Kato, N.; Hirahara, K. Amphiregulin-Producing TH2 Cells Facilitate Esophageal Fibrosis of Eosinophilic Esophagitis. J Allergy Clin Immunol Glob 2024, 3, 100287. [Google Scholar] [CrossRef]
- Shoda, T.; Wen, T.; Caldwell, J.M.; Ben-Baruch Morgenstern, N.; Osswald, G.A.; Rochman, M.; Mack, L.E.; Felton, J.M.; Abonia, J.P.; et al. Loss of Endothelial TSPAN12 Promotes Fibrostenotic Eosinophilic Esophagitis via Endothelial Cell-Fibroblast Crosstalk. Gastroenterology 2022, 162, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, J.M.; Collins, M.H.; Stucke, E.M.; Putnam, P.E.; Franciosi, J.P.; Kushner, J.P.; Abonia, J.P.; Rothenberg, M.E. Histologic Eosinophilic Gastritis Is a Systemic Disorder Associated with Blood and Extragastric Eosinophilia, TH2 Immunity, and a Unique Gastric Transcriptome. J Allergy Clin Immunol 2014, 134, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, S.; Shoda, T.; Ishimura, N.; Ohta, S.; Ono, J.; Azuma, Y.; Okimoto, E.; Izuhara, K.; Nomura, I.; Matsumoto, K.; Kinoshita, Y. Serum Biomarkers for the Diagnosis of Eosinophilic Esophagitis and Eosinophilic Gastroenteritis. Intern Med 2017, 56, 2819–2825. [Google Scholar] [CrossRef]
- Marasco, G.; Visaggi, P.; Vassallo, M.; Fiocca, M.; Cremon, C.; Barbaro, M.R.; De Bortoli, N.; Bellini, M.; Stanghellini, V.; Savarino, E.V.; Barbara, G. Current and Novel Therapies for Eosinophilic Gastrointestinal Diseases. Int J Mol Sci 2023, 24, 15165. [Google Scholar] [CrossRef]
- Sunkara, T.; Rawla, P.; Yarlagadda, K.S.; Gaduputi, V. Eosinophilic Gastroenteritis: Diagnosis and Clinical Perspectives. Clin Exp Gastroenterol 2019, 12, 239–253. [Google Scholar] [CrossRef]
- Troncone, R.; Discepolo, V. Colon in Food Allergy. J Pediatr Gastroenterol Nutr 2009, 48 Suppl 2, S89–91. [Google Scholar] [CrossRef]
- Nieuwenhuizen, N.; Herbert, D.R.; Brombacher, F.; Lopata, A.L. Differential Requirements for Interleukin (IL)-4 and IL-13 in Protein Contact Dermatitis Induced by Anisakis. Allergy 2009, 64, 1309–1318. [Google Scholar] [CrossRef]
- Ogasawara, A.; Yuki, T.; Takai, T.; Yokozeki, K.; Katagiri, A.; Takahashi, Y.; Yokozeki, H.; Basketter, D.; Sakaguchi, H. Epicutaneous Challenge with Protease Allergen Requires Its Protease Activity to Recall TH2 and TH17/TH22 Responses in Mice Pre-sensitized via Distant Skin. J Immunotoxicol 2021, 18, 118–126. [Google Scholar] [CrossRef]
- Ogasawara, A.; Yuki, T.; Katagiri, A.; Lai, Y.T.; Takahashi, Y.; Basketter, D.; Sakaguchi, H. Proteolytic Activity Accelerates the TH17/TH22 Recall Response to an Epicutaneous Protein Allergen-Induced TH2 Response. J Immunotoxicol 2022, 19, 27–33. [Google Scholar] [CrossRef]
- Nowak, D.; Gomułka, K.; Dziemieszonek, P.; Panaszek, B. Systemowe Kontaktowe Zapalenie Skóry [Systemic Contact Dermatitis]. Postepy Hig Med Dosw 2016, 70, 124–134. [Google Scholar] [CrossRef]
- Scott, J.F.; Hammond, M.I.; Nedorost, S.T. Food Avoidance Diets for Dermatitis. Curr Allergy Asthma Rep 2015, 15, 60. [Google Scholar] [CrossRef] [PubMed]
- Burden, A.D.; Wilkinson, S.M.; Beck, M.H.; Chalmers, R.J. Garlic-Induced Systemic Contact Dermatitis. Contact Dermatitis 1994, 30, 299–300. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, T.K.; Zug, K.A. Systemic Contact Dermatitis to Raw Cashew Nuts in a Pesto Sauce. Am J Contact Dermat 1998, 9, 51–54. [Google Scholar] [PubMed]
- Kothari, R.; Kishore, K.; Sandhu, S.; Bhatnagar, A.; Pal, R.; Chand, S. Systemic Contact Dermatitis to Spices: Report of a Rare Case. Contact Dermatitis 2022, 86, 323–325. [Google Scholar] [CrossRef]
- Paulsen, E. Systemic Allergic Dermatitis Caused by Sesquiterpene Lactones. Contact Dermatitis 2017, 76, 1–10. [Google Scholar] [CrossRef]
- de Groot, A.C. Myroxylon Pereirae Resin (Balsam of Peru) - A Critical Review of the Literature and Assessment of the Significance of Positive Patch Test Reactions and the Usefulness of Restrictive Diets. Contact Dermatitis 2019, 80, 335–353. [Google Scholar] [CrossRef]
- Dooms-Goossens, A.; Dubelloy, R.; Degreef, H. Contact and Systemic Contact-Type Dermatitis to Spices. Dermatol Clin 1990, 8, 89–93. [Google Scholar] [CrossRef]
- Scheman, A.; Rakowski, E.M.; Chou, V.; Chhatriwala, A.; Ross, J.; Jacob, S.E. Balsam of Peru: Past and Future. Dermatitis 2013, 24, 153–160. [Google Scholar] [CrossRef]
- Ahuja, K.; Issa, C.J.; Nedorost, S.T.; Lio, P.A. Is Food-Triggered Atopic Dermatitis a Form of Systemic Contact Dermatitis? Clin Rev Allergy Immunol 2024, 66, 1–13. [Google Scholar] [CrossRef]
- Bhatia, J.; Sarin, A.; Wollina, U.; Lotti, T.; Navarini, A.A.; Mueller, S.M.; Grabbe, S.; Saloga, J.; Rokni, G.R.; Goldust, M. Review of Biologics in Allergic Contact Dermatitis. Contact Dermatitis 2020, 83, 179–181. [Google Scholar] [CrossRef]
- Gimenez-Rivera, V.A.; Patel, H.; Dupuy, F.P.; Allakhverdi, Z.; Bouchard, C.; Madrenas, J.; Bissonnette, R.; Piccirillo, C.A.; Jack, C. NOD2 Agonism Counter-Regulates Human Type 2 T Cell Functions in Peripheral Blood Mononuclear Cell Cultures: Implications for Atopic Dermatitis. Biomolecules 2023, 13, 369. [Google Scholar] [CrossRef] [PubMed]
- Mikajiri, R.; Fukunaga, A.; Miyoshi, M.; Maeshige, N.; Washio, K.; Masaki, T.; Nishigori, C.; Yamamoto, I.; Toda, A.; Takahashi, M.; Asahara, S.I.; Kido, Y.; Usami, M. Dietary Intervention for Control of Clinical Symptom in Patients with Systemic Metal Allergy: A Single Center Randomized Controlled Clinical Study. Kobe J Med Sci 2024, 69, E129–E143. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, L.; Arena, A.; Arena, E.; Zambito, M.; Ingrassia, A.; Valenti, G.; Loschiavo, G.; D'Angelo, A.; Saitta, S. Systemic Nickel Allergy Syndrome: Epidemiological Data from Four Italian Allergy Units. Int J Immunopathol Pharmacol 2014, 27, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Veien, N.K. Systemic Contact Dermatitis. Int J Dermatol 2011, 50, 1445–1456. [Google Scholar] [CrossRef]
- EFSA Panel on Contaminants in the Food Chain (CONTAM); Schrenk, D. ; Bignami, M.; Bodin, L.; Chipman, J.K.; Del Mazo, J.; Grasl-Kraupp, B.; Hogstrand, C.; Hoogenboom, L.R.; Leblanc, J.C.; Nebbia, C.S.; et al. Update of the Risk Assessment of Nickel in Food and Drinking Water. EFSA J 2020, 18, e06268. [Google Scholar] [CrossRef]
- Roach, K.; Roberts, J. A Comprehensive Summary of Disease Variants Implicated in Metal Allergy. J Toxicol Environ Health B Crit Rev 2022, 25, 279–341. [Google Scholar] [CrossRef]
- Sharma, A.D. Low Chromate Diet in Dermatology. Indian J Dermatol 2009, 54, 293–295. [Google Scholar] [CrossRef]
- Tramontana, M.; Bianchi, L.; Hansel, K.; Agostinelli, D.; Stingeni, L. Nickel Allergy: Epidemiology, Pathomechanism, Clinical Patterns, Treatment and Prevention Programs. Endocr Metab Immune Disord Drug Targets 2020, 20, 992–1002. [Google Scholar] [CrossRef]
- Castagnoli, R.; Lougaris, V.; Giardino, G.; Volpi, S.; Leonardi, L.; La Torre, F.; Federici, S.; Corrente, S.; Cinicola, B.L.; Soresina, A.; et al. Immunology Task Force of the Italian Society of Pediatric Allergy and Immunology (SIAIP). Inborn Errors of Immunity with Atopic Phenotypes: A Practical Guide for Allergists. World Allergy Organ J 2021, 14, 100513. [Google Scholar] [CrossRef]
- Diaz-Cabrera, N.M.; Bauman, B.M.; Iro, M.A.; Dabbah-Krancher, G.; Molho-Pessach, V.; Zlotogorski, A.; Shamriz, O.; Dinur-Schejter, Y.; Sharon, T.D.; Stepensky, P.; et al. Management of Atopy with Dupilumab and Omalizumab in CADINS Disease. J Clin Immunol 2024, 44, 48. [Google Scholar] [CrossRef]
- Vaseghi-Shanjani, M.; Smith, K.L.; Sara, R.J.; Modi, B.P.; Branch, A.; Sharma, M.; Lu, H.Y.; James, E.L.; Hildebrand, K.J.; Biggs, C.M.; Turvey, S.E. Inborn Errors of Immunity Manifesting as Atopic Disorders. J Allergy Clin Immunol 2021, 148, 1130–1139. [Google Scholar] [CrossRef] [PubMed]
- Su, H.C. Insights into the Pathogenesis of Allergic Disease from Dedicator of Cytokinesis 8 Deficiency. Curr Opin Immunol 2023, 80, 102277. [Google Scholar] [CrossRef] [PubMed]
- Barbieux, C.; Bonnet des Claustres, M.; de la Brassinne, M.; Bricteux, G.; Bagot, M.; Bourrat, E.; Hovnanian, A. Duality of Netherton Syndrome Manifestations and Response to Ixekizumab. J Am Acad Dermatol 2021, 84, 1476–1480. [Google Scholar] [CrossRef] [PubMed]
- Hannula-Jouppi, K.; Laasanen, S.L.; Heikkilä, H.; Tuomiranta, M.; Tuomi, M.L.; Hilvo, S.; Kluger, N.; Kivirikko, S.; Hovnanian, A.; Mäkinen-Kiljunen, S.; et al. IgE Allergen Component-Based Profiling and Atopic Manifestations in Patients with Netherton Syndrome. J Allergy Clin Immunol 2014, 134, 985–988. [Google Scholar] [CrossRef]
- Paluel-Marmont, C.; Bellon, N.; Barbet, P.; Leclerc-Mercier, S.; Hadj-Rabia, S.; Dupont, C.; Bodemer, C. Eosinophilic Esophagitis and Colonic Mucosal Eosinophilia in Netherton Syndrome. J Allergy Clin Immunol 2017, 139, 2003–2005.e1. [Google Scholar] [CrossRef]
- Prodinger, C.; Yerlett, N.; MacDonald, C.; Chottianchaiwat, S.; Goh, L.; Du Toit, G.; Mellerio, J.E.; Petrof, G.; Martinez, A.E. Prevalence of and Risk Factors for Nutritional Deficiency and Food Allergy in a Cohort of 21 Patients with Netherton Syndrome. Pediatr Allergy Immunol 2023, 34, e13914. [Google Scholar] [CrossRef]
- Stuvel, K.; Heeringa, J.J.; Dalm, V.A.S.H.; Meijers, R.W.J.; van Hoffen, E.; Gerritsen, S.A.M.; van Zelm, M.C.; Pasmans, S.G.M.A. Comel-Netherton Syndrome: A Local Skin Barrier Defect in the Absence of an Underlying Systemic Immunodeficiency. Allergy 2020, 75, 1710–1720. [Google Scholar] [CrossRef]
- Abonia, J.P.; Wen, T.; Stucke, E.M.; Grotjan, T.; Griffith, M.S.; Kemme, K.A.; Collins, M.H.; Putnam, P.E.; Franciosi, J.P.; von Tiehl, K.F.; Tinkle, B.T.; Marsolo, K.A.; Martin, L.J.; Ware, S.M.; Rothenberg, M.E. High Prevalence of Eosinophilic Esophagitis in Patients with Inherited Connective Tissue Disorders. J Allergy Clin Immunol 2013, 132, 378–386. [Google Scholar] [CrossRef]
- O'Shea, K.M.; Aceves, S.S.; Dellon, E.S.; Gupta, S.K.; Spergel, J.M.; Furuta, G.T.; Rothenberg, M.E. Pathophysiology of Eosinophilic Esophagitis. Gastroenterology 2018, 154, 333–345. [Google Scholar] [CrossRef]
- Lyons, J.J.; Liu, Y.; Ma, C.A.; Yu, X.; O'Connell, M.P.; Lawrence, M.G.; Zhang, Y.; Karpe, K.; Zhao, M.; Siegel, A.M.; et al. ERBIN Deficiency Links STAT3 and TGF-β Pathway Defects with Atopy in Humans. J Exp Med 2017, 214, 669–680. [Google Scholar] [CrossRef]
- Chaudhary, F.; Agrawal, D.K. Ethnic and Racial Disparities in Clinical Manifestations of Atopic Dermatitis. Arch Intern Med Res 2024, 7, 114–133. [Google Scholar] [CrossRef] [PubMed]
- Savva, M.; Papadopoulos, N.G.; Gregoriou, S.; Katsarou, S.; Papapostolou, N.; Makris, M.; Xepapadaki, P. Recent Advancements in the Atopic Dermatitis Mechanism. Front Biosci 2024, 29, 84. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.L.; Guttman-Yassky, E.; Pawlikowski, J.; Ghorayeb, E.G.; Ota, T.; Lebwohl, M.G. Interleukin-1α Inhibitor Bermekimab in Patients with Atopic Dermatitis: Randomized and Nonrandomized Studies. Arch Dermatol Res 2024, 316, 589. [Google Scholar] [CrossRef] [PubMed]
- Brough, H.A.; Nadeau, K.C.; Sindher, S.B.; Alkotob, S.S.; Chan, S.; Bahnson, H.T.; Leung, D.Y.M.; Lack, G. Epicutaneous Sensitization in the Development of Food Allergy: What is the Evidence and How Can This Be Prevented? Allergy 2020, 75, 2185–2205. [Google Scholar] [CrossRef]
- Graham, F. , Eigenmann PA. Atopic dermatitis and its relation to food allergy. Curr Opin Allergy Clin Immunol. 2020, 20, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Hervé, P.L.; Dioszeghy, V.; Matthews, K.; Bee, K.J.; Campbell, D.E.; Sampson, H.A. Recent Advances in Epicutaneous Immunotherapy and Potential Applications in Food Allergy. Front Allergy 2023, 4, 1290003. [Google Scholar] [CrossRef]
- Singh, A.M.; Anvari, S.; Hauk, P.; Lio, P.; Nanda, A.; Sidbury, R.; Schneider, L. Atopic Dermatitis and Food Allergy: Best Practices and Knowledge Gaps—A Work Group Report from the AAAAI Allergic Skin Diseases Committee and Leadership Institute Project. J Allergy Clin Immunol Pract 2022, 10, 697–706. [Google Scholar] [CrossRef]
- Dubin, C.; Del Duca, E.; Guttman-Yassky, E. The IL-4, IL-13 and IL-31 Pathways in Atopic Dermatitis. Expert Rev Clin Immunol 2021, 17, 835–852. [Google Scholar] [CrossRef]
- Huang IH, Chung WH, Wu PC, Chen CB. JAK-STAT signaling pathway in the pathogenesis of atopic dermatitis: An updated review. Front Immunol. 2022, 13, 1068260. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Agache, I.; Song, Y.; Posso, M.; Alonso-Coello, P.; Rocha, C.; Solà, I.; Beltran, J.; Akdis, C.A.; Akdis, M.; Brockow, K.; et al. Efficacy and safety of dupilumab for moderate-to-severe atopic dermatitis: A systematic review for the EAACI biologicals guidelines. Allergy 2021, 76, 45–58. [Google Scholar] [CrossRef]
- Agache, I.; Akdis, C.A.; Akdis, M.; Brockow, K.; Chivato, T.; Del Giacco, S.; Eiwegger, T.; Eyerich, K.; Giménez-Arnau, A.; Gutermuth, J.; et al. EAACI Biologicals Guidelines—dupilumab for children and adults with moderate-to-severe atopic dermatitis. Allergy 2021, 76, 988–1009. [Google Scholar] [CrossRef] [PubMed]
- Czarnowicki, T.; He, H.; Krueger, J.G.; Guttman-Yassky, E. Atopic dermatitis endotypes and implications for targeted therapeutics. J Allergy Clin Immunol 2019, 143, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Maintz, L.; Welchowski, T.; Herrmann, N.; Brauer, J.; Traidl-Hoffmann, C.; Havenith, R.; Müller, S.; Rhyner, C.; Dreher, A.; Schmid, M.; Bieber, T. ; CK-CARE study group. IL-13, periostin and dipeptidyl-peptidase-4 reveal endotype-phenotype associations in atopic dermatitis. Allergy 2023, 17 January. [CrossRef]
- Song, A.; Lee, S.E.; Kim, J.H. Immunopathology and immunotherapy of inflammatory skin diseases. Immune Netw 2022, 22, e7. [Google Scholar] [CrossRef] [PubMed]
- de Wijs, L.E.M.; Nguyen, N.T.; Kunkeler, A.C.M.; Nijsten, T.; Damman, J.; Hijnen, D.J. Clinical and histopathological characterization of paradoxical head and neck erythema in patients with atopic dermatitis treated with dupilumab: A case series. Br J Dermatol 2020, 183, 745–749. [Google Scholar] [CrossRef]
- Glatz, M.; Bossard, P.P.; Hoetzenecker, W.; Schmid-Grendelmeier, P. The role of Malassezia spp. in atopic dermatitis. J Clin Med 2015, 4, 1217–1228. [Google Scholar] [CrossRef]
- Tokura, Y.; Hayano, S. Subtypes of atopic dermatitis: From phenotype to endotype. Allergol Int 2022, 71, 14–24. [Google Scholar] [CrossRef]
- Zuberbier, T.; Abdul Latiff, A.; Aggelidis, X.; Augustin, M.; Balan, R.G.; Bangert, C.; Beck, L.; Bieber, T.; Bernstein, J.A.; Bertolin Colilla, M.; et al. A concept for integrated care pathways for atopic dermatitis—A GA2LEN ADCARE initiative. Clin Transl Allergy 2023, 13, e12299. [Google Scholar] [CrossRef]
- Leung, D.Y.M.; Calatroni, A.; Zaramela, L.S.; LeBeau, P.K.; Dyjack, N.; Brar, K.; David, G.; Johnson, K.; Leung, S.; Ramirez-Gama, M.; et al. The nonlesional skin surface distinguishes atopic dermatitis with food allergy as a unique endotype. Sci Transl Med 2019, 11, eaav2685. [Google Scholar] [CrossRef]
- Bousquet, J.; Melén, E.; Haahtela, T.; Koppelman, G.H.; Togias, A.; Valenta, R.; Akdis, C.A.; Czarlewski, W.; Rothenberg, M.; Valiulis, A.; et al. Rhinitis associated with asthma is distinct from rhinitis alone: The ARIA-MeDALL hypothesis. Allergy 2023, 78, 1169–1203. [Google Scholar] [CrossRef]
- Davis, K.L.; Claudio-Etienne, E.; Frischmeyer-Guerrerio, P.A. Atopic dermatitis and food allergy: More than sensitization. Mucosal Immunol, 2024; S1933-021900059-X. [Google Scholar] [CrossRef]
- Sernicola, A.; Amore, E.; Rizzuto, G.; Rallo, A.; Greco, M.E.; Battilotti, C.; Svara, F.; Azzella, G.; Nisticò, S.P.; Dattola, A.; Chello, C.; Pellacani, G.; Grieco, T. Dupilumab as Therapeutic Option in Polysensitized Atopic Dermatitis Patients Suffering from Food Allergy. Nutrients 2024, 16, 2797. [Google Scholar] [CrossRef]
- Bergmann, M.M.; Caubet, J.C.; Boguniewicz, M.; Eigenmann, P.A. Evaluation of food allergy in patients with atopic dermatitis. J Allergy Clin Immunol Pract 2013, 1, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Breuer, K.; Heratizadeh, A.; Wulf, A.; Baumann, U.; Constien, A.; Tetau, D.; Kapp, A.; Werfel, T. Late eczematous reactions to food in children with atopic dermatitis. Clin Exp Allergy 2004, 34, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Manam, S.; Tsakok, T.; Till, S.; Flohr, C. The association between atopic dermatitis and food allergy in adults. Curr Opin Allergy Clin Immunol 2014, 14, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Papapostolou, N.; Xepapadaki, P.; Gregoriou, S.; Makris, M. Atopic Dermatitis and Food Allergy: A Complex Interplay What We Know and What We Would Like to Learn. J Clin Med 2022, 11, 4232. [Google Scholar] [CrossRef] [PubMed]
- Werfel, T.; Breuer, K. Role of food allergy in atopic dermatitis. Curr Opin Allergy Clin Immunol 2004, 4, 379–385. [Google Scholar] [CrossRef]
- Dębińska, A.; Sozańska, B. Epicutaneous sensitization and food allergy: preventive strategies targeting skin barrier repair—facts and challenges. Nutrients 2023, 15, 1070. [Google Scholar] [CrossRef]
- Werfel, T.; Ballmer-Weber, B.; Eigenmann, P.A.; Niggemann, B.; Rancé, F.; Turjanmaa, K.; Worm, M. Eczematous reactions to food in atopic eczema: position paper of the EAACI and GA2LEN. Allergy 2007, 62, 723–728. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Hirasawa, N.; Asakawa, S.; Okita, K.; Tokura, Y. Intrinsic atopic dermatitis shows high serum nickel concentration. Allergol Int 2015, 64, 282–284. [Google Scholar] [CrossRef]
- Glocova, I.; Brück, J.; Geisel, J.; Müller-Hermelink, E.; Widmaier, K.; Yazdi, A.S.; Röcken, M.; Ghoreschi, K. Induction of skin-pathogenic Th22 cells by epicutaneous allergen exposure. J Dermatol Sci 2017, 87, 268–277. [Google Scholar] [CrossRef]
- Tsuge, I.; Kondo, Y.; Tokuda, R.; Kakami, M.; Kawamura, M.; Nakajima, Y.; Komatsubara, R.; Yamada, K.; Urisu, A. Allergen-specific helper T cell response in patients with cow's milk allergy: Simultaneous analysis of proliferation and cytokine production by carboxyfluorescein succinimidyl ester dilution assay. Clin Exp Allergy 2006, 36, 1538–1545. [Google Scholar] [CrossRef]
- Noh, J.; Noh, G. Allergen-specific responses of CD19(high) and CD19(low) B Cells in Non-IgE-mediated food allergy of late eczematous reactions in atopic dermatitis: presence of IL-17- and IL-32-producing regulatory B cells (Br17 & Br32). Inflamm Allergy Drug Targets 2012, 11, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Noh, J.; Noh, G.; Kim, H.S.; Kim, A.R.; Choi, W.S. Allergen-specific responses of CD19(+)CD5(+)Foxp3(+) regulatory B cells (Bregs) and CD4(+)Foxp3(+) regulatory T cell (Tregs) in immune tolerance of cow milk allergy of late eczematous reactions. Cell Immunol 2012, 274, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Sütas, Y.; Kekki, O.M.; Isolauri, E. Late onset reactions to oral food challenge are linked to low serum interleukin-10 concentrations in patients with atopic dermatitis and food allergy. Clin Exp Allergy 2000, 30, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Abernathy-Carver, K.J.; Sampson, H.A.; Picker, L.J.; Leung, D.Y. Milk-induced eczema is associated with the expansion of T cells expressing cutaneous lymphocyte antigen. J Clin Invest 1995, 95, 913–918. [Google Scholar] [CrossRef]
- Nicolàs, L.S.S.; Czarnowicki, T.; Akdis, M.; Pujol, R.M.; Lozano-Ojalvo, D.; Leung, D.Y.M.; Guttman-Yassky, E.; Santamaria-Babí, L.F. CLA+ memory T cells in atopic dermatitis. Allergy 2024, 79, 15–25. [Google Scholar] [CrossRef]
- Glickman, J.W.; Han, J.; Garcet, S.; Krueger, J.G.; Pavel, A.B.; Guttman-Yassky, E. Improving evaluation of drugs in atopic dermatitis by combining clinical and molecular measures. J Allergy Clin Immunol Pract 2020, 8, 3622–3625.e19. [Google Scholar] [CrossRef]
- Homey, B.; Alenius, H.; Müller, A.; Soto, H.; Bowman, E.P.; Yuan, W.; McEvoy, L.; Lauerma, A.I.; Assmann, T.; Bünemann, E.; et al. CCL27-CCR10 interactions regulate T cell-mediated skin inflammation. Nat Med 2002, 8, 157–165. [Google Scholar] [CrossRef]
- Leonard, A.; Wang, J.; Yu, L.; Liu, H.; Estrada, Y.; Greenlees, L.; McPhee, R.; Ruzin, A.; Guttman-Yassky, E.; Howell, M.D. Atopic Dermatitis Endotypes Based on Allergen Sensitization, Reactivity to Staphylococcus aureus Antigens, and Underlying Systemic Inflammation. J Allergy Clin Immunol Pract 2020, 8, 236–247.e3. [Google Scholar] [CrossRef]
- Acevedo, N.; Benfeitas, R.; Katayama, S.; Bruhn, S.; Andersson, A.; Wikberg, G.; Lundeberg, L.; Lindvall, J.M.; Greco, D.; Kere, J.; et al. Epigenetic alterations in skin homing CD4+CLA+ T cells of atopic dermatitis patients. Sci Rep 2020, 10, 18020. [Google Scholar] [CrossRef]
- Czarnowicki, T.; Malajian, D.; Shemer, A.; Fuentes-Duculan, J.; Gonzalez, J.; Suárez-Fariñas, M.; Krueger, J.G.; Guttman-Yassky, E. Skin-homing and systemic T-cell subsets show higher activation in atopic dermatitis versus psoriasis. J Allergy Clin Immunol 2015, 136, 208–211. [Google Scholar] [CrossRef]
- Langan, S.M.; Irvine, A.D.; Weidinger, S. Atopic dermatitis. Lancet 2020, 396, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Sans-De San Nicolàs, L.; Figueras-Nart, I.; Bonfill-Ortí, M.; De Jesús-Gil, C.; García-Jiménez, I.; Guilabert, A.; Curto-Barredo, L.; Bertolín-Colilla, M.; Ferran, M.; Serra-Baldrich, E.; et al. SEB-induced IL-13 production in CLA+ memory T cells defines Th2 high and Th2 low responders in atopic dermatitis. Allergy 2022, 77, 3448–3451. [Google Scholar] [CrossRef] [PubMed]
- Soul, J.; Carlsson, E.; Hofmann, S.R.; Russ, S.; Hawkes, J.; Schulze, F.; Sergon, M.; Pablik, J.; Abraham, S.; Hedrich, C.M. Tissue gene expression profiles and communication networks inform candidate blood biomarker identification in psoriasis and atopic dermatitis. Clin Immunol 2024, 265, 110283. [Google Scholar] [CrossRef]
- Chang, S.H.; Dong, C. Signaling of interleukin-17 family cytokines in immunity and inflammation. Cell Signal 2011, 23, 1069–1075. [Google Scholar] [CrossRef]
- Herberth, G.; Daegelmann, C.; Röder, S.; Behrendt, H.; Krämer, U.; Borte, M.; Heinrich, J.; Herbarth, O.; Lehmann, I.; LISAplus study group. IL-17E but not IL-17A is associated with allergic sensitization: results from the LISA study. Pediatr Allergy Immunol 2010, 21, 1086–1090. [Google Scholar] [CrossRef]
- Hofmann, M.A.; Fluhr, J.W.; Ruwwe-Glösenkamp, C.; Stevanovic, K.; Bergmann, K.C.; Zuberbier, T. Role of IL-17 in atopy—A systematic review. Clin Transl Allergy 2021, 11, e12047. [Google Scholar] [CrossRef]
- Żbikowska-Gotz, M.; Pałgan, K.; Gawrońska-Ukleja, E.; Kuźmiński, A.; Przybyszewski, M.; Socha, E.; Bartuzi, Z. Expression of IL-17A concentration and effector functions of peripheral blood neutrophils in food allergy hypersensitivity patients. Int J Immunopathol Pharmacol 2016, 29, 90–98. [Google Scholar] [CrossRef]
- Berin, M.C.; Lozano-Ojalvo, D.; Agashe, C.; Baker, M.G.; Bird, J.A.; Nowak-Wegrzyn, A. Acute FPIES reactions are associated with an IL-17 inflammatory signature. J Allergy Clin Immunol. 2021, 148, 895–901.e6. [Google Scholar] [CrossRef]
- Lozano-Ojalvo, D.; Chen, X.; Dunkin, D.; Agashe, C.; Baker, M.G.; Bird, J.A.; Molina, E.; Nowak-Wegrzyn, A.; Berin, M.C. Untargeted serum metabolomic analysis reveals a role for purinergic signaling in FPIES. J Allergy Clin Immunol. 2023, 151, 797–802. [Google Scholar] [CrossRef]
- Gonzalez-Delgado, P.; Anvari, S.; Barrachina, J.; Portillo, A.L.J.; Jimenez, T.; Marco de la Calle, F.M.; Fernandez, J. Egg-induced adult food protein-induced enterocolitis syndrome: Clinical phenotypes, natural history and immunological characteristics. J Allergy Clin Immunol Pract. 2024, 12, 1657–1659. [Google Scholar] [CrossRef]
- Fina, D.; Sarra, M.; Caruso, R.; Del Vecchio Blanco, G.; Pallone, F.; MacDonald, T.T.; Monteleone, G. Interleukin 21 contributes to the mucosal T helper cell type 1 response in coeliac disease. Gut 2008, 57, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Omidian, Z.; Ahmed, R.; Giwa, A.; Donner, T.; Hamad, A.R.A. IL-17 and limits of success. Cell Immunol 2019, 339, 33–40. [Google Scholar] [CrossRef]
- Ortega, C.; Fernández, S.; Estévez, O.A.; Aguado, R.; Molina, I.J.; Santamaría, M. IL-17 producing T cells in celiac disease: angels or devils? Int Rev Immunol 2013, 32, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Sjöberg, V.; Sandström, O.; Hedberg, M.; Hammarström, S.; Hernell, O.; Hammarström, M.L. Intestinal T-cell responses in celiac disease - impact of celiac disease associated bacteria. PLoS One 2013, 8, e53414. [Google Scholar] [CrossRef]
- Cicerone, C.; Nenna, R.; Pontone, S. Th17, intestinal microbiota and the abnormal immune response in the pathogenesis of celiac disease. Gastroenterol Hepatol Bed Bench 2015, 8, 117–122. [Google Scholar] [PubMed]
- Lahdenperä, A.I.; Fälth-Magnusson, K.; Högberg, L.; Ludvigsson, J.; Vaarala, O. Expression pattern of T-helper 17 cell signaling pathway and mucosal inflammation in celiac disease. Scand J Gastroenterol 2014, 49, 145–156. [Google Scholar] [CrossRef]
- Madi, M.; Abdelsalam, M.; Elakel, A.; Zakaria, O.; AlGhamdi, M.; Alqahtani, M.; AlMuhaish, L.; Farooqi, F.; Alamri, T.A.; Alhafid, I.A.; Alzahrani, I.M.; Alam, A.H.; Alhashmi, M.T.; Alasseri, I.A.; AlQuorain, A.A. Salivary interleukin-17A and interleukin-18 levels in patients with celiac disease and periodontitis. PeerJ 2024, 12, e17374. [Google Scholar] [CrossRef]
- Caproni, M.; Capone, M.; Rossi, M.C.; Santarlasci, V.; Maggi, L.; Mazzoni, A.; Rossettini, B.; Renzi, D.; Quintarelli, L.; Bianchi, B.; et al. T Cell Response Toward Tissue-and Epidermal-Transglutaminases in Coeliac Disease Patients Developing Dermatitis Herpetiformis. Front Immunol 2021, 12, 645143. [Google Scholar] [CrossRef]
- Collin, P.; Salmi, T.T.; Hervonen, K.; Kaukinen, K.; Reunala, T. Dermatitis herpetiformis: a cutaneous manifestation of coeliac disease. Ann Med 2017, 49, 23–31. [Google Scholar] [CrossRef]
- Fernández-Bañares, F.; Crespo, L.; Planella, M.; Farrais, S.; Izquierdo, S.; López-Palacios, N.; Roy, G.; Vidal, J.; Núñez, C. Improving the Diagnosis of Dermatitis Herpetiformis Using the Intraepithelial Lymphogram. Nutrients 2024, 16, 232. [Google Scholar] [CrossRef]
- Nguyen, C.N.; Kim, S.J. Dermatitis Herpetiformis: An Update on Diagnosis, Disease Monitoring, and Management. Medicina (Kaunas) 2021, 57, 843. [Google Scholar] [CrossRef] [PubMed]
- Velikova, T.; Shahid, M.; Ivanova-Todorova, E.; Drenovska, K.; Tumangelova-Yuzeir, K.; Altankova, I.; Vassileva, S. Celiac-Related Autoantibodies and IL-17A in Bulgarian Patients with Dermatitis Herpetiformis: A Cross-Sectional Study. Medicina 2019, 55, 136. [Google Scholar] [CrossRef] [PubMed]
- Bakker, D.S.; Nierkens, S.; Knol, E.F.; Giovannone, B.; Delemarre, E.M.; van der Schaft, J.; van Wijk, F.; de Bruin-Weller, M.S.; Drylewicz, J.; Thijs, J.L. Confirmation of multiple endotypes in atopic dermatitis based on serum biomarkers. J Allergy Clin Immunol 2021, 147, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Peled, A.; Sarig, O.; Sun, G.; Samuelov, L.; Ma, C.A.; Zhang, Y.; Dimaggio, T.; Nelson, C.G.; Stone, K.D.; Freeman, A.F.; et al. Loss-of-function mutations in caspase recruitment domain-containing protein 14 (CARD14) are associated with a severe variant of atopic dermatitis. J Allergy Clin Immunol 2019, 143, 173–181.e10. [Google Scholar] [CrossRef]
- Tsai, Y.C.; Tsai, T.F. Overlapping Features of Psoriasis and Atopic Dermatitis: From Genetics to Immunopathogenesis to Phenotypes. Int J Mol Sci 2022, 23, 5518. [Google Scholar] [CrossRef]
- Goh, M.S.; Yun, J.S.; Su, J.C. Management of atopic dermatitis: a narrative review. Med J Aust 2022, 216, 587–593. [Google Scholar] [CrossRef]
- Laska, J.; Tota, M.; Łacwik, J.; Sędek, Ł.; Gomułka, K. IL-22 in Atopic Dermatitis. Cells 2024, 13, 1398. [Google Scholar] [CrossRef]
- Sugaya, M. The Role of Th17-Related Cytokines in Atopic Dermatitis. Int J Mol Sci 2020, 21, 1314. [Google Scholar] [CrossRef]
- Tsoi, L.C.; Rodriguez, E.; Stölzl, D.; Wehkamp, U.; Sun, J.; Gerdes, S.; Sarkar, M.K.; Hübenthal, M.; Zeng, C.; Uppala, R.; et al. Progression of acute-to-chronic atopic dermatitis is associated with quantitative rather than qualitative changes in cytokine responses. J Allergy Clin Immunol 2020, 145, 1406–1415. [Google Scholar] [CrossRef]
- Weidinger, S.; Novak, N. Atopic dermatitis. Lancet 2016, 387, 1109–1122. [Google Scholar] [CrossRef]
- Furue, M. Regulation of Filaggrin, Loricrin, and Involucrin by IL-4, IL-13, IL-17A, IL-22, AHR, and NRF2: Pathogenic Implications in Atopic Dermatitis. Int J Mol Sci 2020, 21, 5382. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Honda, T.; Kabashima, K. Multipolarity of cytokine axes in the pathogenesis of atopic dermatitis in terms of age, race, species, disease stage and biomarkers. Int Immunol 2018, 30, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Giancotta, C.; Colantoni, N.; Pacillo, L.; Santilli, V.; Amodio, D.; Manno, E.C.; Cotugno, N.; Rotulo, G.A.; Rivalta, B.; Finocchi, A.; Cancrini, C.; Diociaiuti, A.; El Hachem, M.; Zangari, P. Tailored treatments in inborn errors of immunity associated with atopy (IEIs-A) with skin involvement. Front Pediatr 2023, 11, 1129249. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, I.; Knöpfel, N.; Theiler, M.; Bonnet des Claustres, M.; Barbieux, C.; Schwieger-Briel, A.; Brunner, C.; Donghi, D.; Buettcher, M.; Meier-Schiesser, B.; Hovnanian, A.; Weibel, L. Secukinumab Therapy for Netherton Syndrome. JAMA Dermatol 2020, 156, 907–911. [Google Scholar] [CrossRef]
- Pontone, M.; Giovannini, M.; Filippeschi, C.; Oranges, T.; Pedaci, F.A.; Mori, F.; Barni, S.; Barbati, F.; Consonni, F.; Indolfi, G.; Lodi, L.; Azzari, C.; Ricci, S.; Hovnanian, A. Biological treatments for pediatric Netherton syndrome. Front Pediatr 2022, 10, 1074243. [Google Scholar] [CrossRef]
- Christensen, M.J.; Eller, E.; Kjaer, H.F.; Broesby-Olsen, S.; Mortz, C.G.; Bindslev-Jensen, C. Exercise-induced anaphylaxis: causes, consequences, and management recommendations. Expert Rev Clin Immunol 2019, 15, 265–273. [Google Scholar] [CrossRef]
- Morita, E.; Chinuki, Y.; Kohno, K.; Matsuo, H. Cofactors of wheat-dependent exercise-induced anaphylaxis increase gastrointestinal gliadin absorption by an inhibition of prostaglandin production. Clin Exp Allergy 2023, 53, 359–361. [Google Scholar] [CrossRef]
- Shin, M. Food allergies and food-induced anaphylaxis: role of cofactors. Clin Exp Pediatr 2021, 64, 393–399. [Google Scholar] [CrossRef]
- Ferrier, L.; Bérard, F.; Debrauwer, L.; Chabo, C.; Langella, P.; Buéno, L.; Fioramonti, J. Impairment of the intestinal barrier by ethanol involves enteric microflora and mast cell activation in rodents. Am J Pathol 2006, 168, 1148–1154. [Google Scholar] [CrossRef]
- Feuille, E.; Nowak-Węgrzyn, A. Food Protein-Induced Enterocolitis Syndrome, Allergic Proctocolitis, and Enteropathy. Curr Allergy Asthma Rep 2015, 15, 50. [Google Scholar] [CrossRef]
- Banerjee, A.; Nobleza, K.; Haddad, C.; Eubanks, J.; Rana, R.; Rider, N.L.; Pompeii, L.; Nguyen, D.; Anvari, S. Applying Market Basket Analysis to Determine Complex Coassociations Among Food Allergens in Children With Food Protein-Induced Enterocolitis Syndrome (FPIES). Health Serv Res Manag Epidemiol 2024, 11, 23333928241264020. [Google Scholar] [CrossRef] [PubMed]
- Labrosse, R.; Graham, F.; Caubet, J.C. Non-IgE-Mediated Gastrointestinal Food Allergies in Children: An Update. Nutrients 2020, 12, 2086. [Google Scholar] [CrossRef] [PubMed]
- Berin, M.C.; Lozano-Ojalvo, D.; Agashe, C.; Baker, M.G.; Bird, J.A.; Nowak-Wegrzyn, A. Acute FPIES reactions are associated with an IL-17 inflammatory signature. J Allergy Clin Immunol 2021, 148, 895–901.e6. [Google Scholar] [CrossRef] [PubMed]
- Lozano-Ojalvo, D.; Chen, X.; Dunkin, D.; Agashe, C.; Baker, M.G.; Bird, J.A.; Molina, E.; Nowak-Wegrzyn, A.; Berin, M.C. Untargeted serum metabolomic analysis reveals a role for purinergic signaling in FPIES. J Allergy Clin Immunol 2023, 151, 797–802. [Google Scholar] [CrossRef]
- Rizzi, A.; Lo Presti, E.; Chini, R.; Gammeri, L.; Inchingolo, R.; Lohmeyer, F.M.; Nucera, E.; Gangemi, S. Emerging Role of Alarmins in Food Allergy: An Update on Pathophysiological Insights, Potential Use as Disease Biomarkers, and Therapeutic Implications. J Clin Med 2023, 12, 2699. [Google Scholar] [CrossRef]
- Wada, T.; Matsuda, Y.; Toma, T.; Koizumi, E.; Okamoto, H.; Yachie, A. Increased CD69 Expression on Peripheral Eosinophils from Patients with Food Protein-Induced Enterocolitis Syndrome. Int Arch Allergy Immunol 2016, 170, 201–205. [Google Scholar] [CrossRef]
- Zubeldia-Varela, E.; Barker-Tejeda, T.C.; Blanco-Pérez, F.; Infante, S.; Zubeldia, J.M.; Pérez-Gordo, M. Non-IgE-Mediated Gastrointestinal Food Protein-Induced Allergic Disorders: Clinical Perspectives and Analytical Approaches. Foods 2021, 10, 2662. [Google Scholar] [CrossRef]
- Koksal, B.T.; Barıs, Z.; Sencelikel, T.; Ozcay, F.; Ozbek, O.Y. Food Protein-Induced Allergic Proctocolitis in Infants Is Associated with Low Serum Levels of Macrophage Inflammatory Protein-3a. J Pediatr Gastroenterol Nutr 2024, 78, 211–216. [Google Scholar] [CrossRef]
- Tsabouri, S.; Nicolaou, N.; Douros, K.; Papadopoulou, A.; Priftis, K.N. Food Protein Induced Proctocolitis: A Benign Condition with an Obscure Immunologic Mechanism. Endocr Metab Immune Disord Drug Targets 2017, 17, 32–37. [Google Scholar] [CrossRef]
- de Boer, J.; Deb, C.; Bornstein, J.; Horvath, K.; Mehta, D.; Smadi, Y. Using Eosinophil Biomarkers from Rectal Epithelial Samples to Diagnose Food Protein-Induced Proctocolitis: A Pilot Study. J Pediatr Gastroenterol Nutr 2020, 71, e109–e112. [Google Scholar] [CrossRef]
- Rycyk, A.; Cudowska, B.; Lebensztejn, D.M. Eosinophil-Derived Neurotoxin, Tumor Necrosis Factor Alpha, and Calprotectin as Non-Invasive Biomarkers of Food Protein-Induced Allergic Proctocolitis in Infants. J Clin Med 2020, 9, 3147. [Google Scholar] [CrossRef] [PubMed]
- Jauregi-Miguel, A. The Tight Junction and the Epithelial Barrier in Coeliac Disease. Int Rev Cell Mol Biol 2021, 358, 105–132. [Google Scholar] [CrossRef] [PubMed]
- Sowińska, A.; Morsy, Y.; Czarnowska, E.; Oralewska, B.; Konopka, E.; Woynarowski, M.; Szymańska, S.; Ejmont, M.; Scharl, M.; Bierła, J.B.; Wawrzyniak, M.; Cukrowska, B. Transcriptional and Ultrastructural Analyses Suggest Novel Insights into Epithelial Barrier Impairment in Celiac Disease. Cells 2020, 9, 516. [Google Scholar] [CrossRef] [PubMed]
- Taraz, T.; Mahmoudi-Ghehsareh, M.; Asri, N.; Nazemalhosseini-Mojarad, E.; Rezaei-Tavirani, M.; Jahani-Sherafat, S.; Naseh, A.; Rostami-Nejad, M. Overview of the Compromised Mucosal Integrity in Celiac Disease. J Mol Histol 2024, 55, 15–24. [Google Scholar] [CrossRef]
- Matysiak-Budnik, T.; Moura, I.C.; Arcos-Fajardo, M.; Lebreton, C.; Ménard, S.; Candalh, C.; Ben-Khalifa, K.; Dugave, C.; Tamouza, H.; van Niel, G.; et al. Secretory IgA Mediates Retrotranscytosis of Intact Gliadin Peptides via the Transferrin Receptor in Celiac Disease. J Exp Med 2008, 205, 143–154. [Google Scholar] [CrossRef]
- Barbaro, M.R.; Cremon, C.; Morselli-Labate, A.M.; Di Sabatino, A.; Giuffrida, P.; Corazza, G.R.; Di Stefano, M.; Caio, G.; Latella, G.; Ciacci, C.; et al. Serum Zonulin and Its Diagnostic Performance in Non-Coeliac Gluten Sensitivity. Gut 2020, 69, 1966–1974. [Google Scholar] [CrossRef]
- Rotondi Aufiero, V.; Fasano, A.; Mazzarella, G. Non-Celiac Gluten Sensitivity: How Its Gut Immune Activation and Potential Dietary Management Differ from Celiac Disease. Mol Nutr Food Res 2018, 62, e1700854. [Google Scholar] [CrossRef]
- Sapone, A.; Lammers, K.M.; Mazzarella, G.; Mikhailenko, I.; Cartenì, M.; Casolaro, V.; Fasano, A. Differential Mucosal IL-17 Expression in Two Gliadin-Induced Disorders: Gluten Sensitivity and the Autoimmune Enteropathy Celiac Disease. Int Arch Allergy Immunol 2010, 152, 75–80. [Google Scholar] [CrossRef]
- Zingone, F.; Bertin, L.; Maniero, D.; Palo, M.; Lorenzon, G.; Barberio, B.; Ciacci, C.; Savarino, E.V. Myths and Facts about Food Intolerance: A Narrative Review. Nutrients 2023, 15, 4969. [Google Scholar] [CrossRef]
- Kleuskens, M.T.A.; Bek, M.K.; Al Halabi, Y.; Blokhuis, B.R.J.; Diks, M.A.P.; Haasnoot, M.L.; Garssen, J.; Bredenoord, A.J.; van Esch, B.C.A.M.; Redegeld, F.A. Mast Cells Disrupt the Function of the Esophageal Epithelial Barrier. Mucosal Immunol 2023, 16, 567–577. [Google Scholar] [CrossRef]
- Pyon, G.C.; Masuda, M.Y.; Putikova, A.; Luo, H.; Gibson, J.B.; Dao, A.D.; Ortiz, D.R.; Heiligenstein, P.L.; Bonellos, J.J.; LeSuer, W.E.; Pai, R.K.; Garg, S.; Rank, M.A.; Nakagawa, H.; Kita, H.; Wright, B.L.; Doyle, A.D. Tissue-Specific Inducible IL-33 Expression Elicits Features of Eosinophilic Esophagitis. J Allergy Clin Immunol 2024, S0091-6749, 00910–00912. [CrossRef]
- Simon, D.; Page, B.; Vogel, M.; Bussmann, C.; Blanchard, C.; Straumann, A.; Simon, H.U. Evidence of an Abnormal Epithelial Barrier in Active, Untreated and Corticosteroid-Treated Eosinophilic Esophagitis. Allergy 2018, 73, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Smolinska, S.; Antolín-Amérigo, D.; Popescu, F.D.; Jutel, M. Thymic Stromal Lymphopoietin (TSLP), Its Isoforms and the Interplay with the Epithelium in Allergy and Asthma. Int J Mol Sci 2023, 24, 12725. [Google Scholar] [CrossRef] [PubMed]
- Uchida, A.M.; Lenehan, P.J.; Vimalathas, P.; Miller, K.C.; Valencia-Yang, M.; Qiang, L.; Canha, L.A.; Ali, L.R.; Dougan, M.; Garber, J.J.; Dougan, S.K. Tissue Eosinophils Express the IL-33 Receptor ST2 and Type 2 Cytokines in Patients with Eosinophilic Esophagitis. Allergy 2022, 77, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Litosh, V.A.; Rochman, M.; Rymer, J.K.; Porollo, A.; Kottyan, L.C.; Rothenberg, M.E. Calpain-14 and Its Association with Eosinophilic Esophagitis. J Allergy Clin Immunol 2017, 139, 1762–1771.e7. [Google Scholar] [CrossRef]
- McAleer, M.A.; Pohler, E.; Smith, F.J.; Wilson, N.J.; Cole, C.; MacGowan, S.; Koetsier, J.L.; Godsel, L.M.; Harmon, R.M.; Gruber, R.; et al. Severe Dermatitis, Multiple Allergies, and Metabolic Wasting Syndrome Caused by a Novel Mutation in the N-terminal Plakin Domain of Desmoplakin. J Allergy Clin Immunol 2015, 136, 1268–1276. [Google Scholar] [CrossRef]
- Pan, C.; Zhao, A.; Li, M. Atopic Dermatitis-like Genodermatosis: Disease Diagnosis and Management. Diagnostics (Basel) 2022, 12, 2177. [Google Scholar] [CrossRef]
- Ogrodowczyk, A.; Markiewicz, L.; Wróblewska, B. Mutations in the Filaggrin Gene and Food Allergy. Prz Gastroenterol 2014, 9, 200–207. [Google Scholar] [CrossRef]
- Irvine, A.D.; McLean, W.H.; Leung, D.Y. Filaggrin mutations associated with skin and allergic diseases. N Engl J Med 2011, 365, 1315–1327. [Google Scholar] [CrossRef]
- Tham, E.H.; Leung, D.Y. Mechanisms by Which Atopic Dermatitis Predisposes to Food Allergy and the Atopic March. Allergy Asthma Immunol Res 2019, 11, 4–15. [Google Scholar] [CrossRef]
- Leonard, A.; Guttman-Yassky, E. The Unique Molecular Signatures of Contact Dermatitis and Implications for Treatment. Clin Rev Allergy Immunol 2019, 56, 1–8. [Google Scholar] [CrossRef]
- Sashikawa-Kimura, M.; Takada, M.; Hossain, M.R.; Tsuda, H.; Xie, X.; Komine, M.; Ohtsuki, M.; Imokawa, G. Overexpression of the β-Subunit of Acid Ceramidase in the Epidermis of Mice Provokes Atopic Dermatitis-like Skin Symptoms. Int J Mol Sci 2024, 25, 8737. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Autieri, M.V.; Scalia, R. Adipose Tissue Inflammation and Metabolic Dysfunction in Obesity. Am J Physiol Cell Physiol 2021, 320, C375–C391. [Google Scholar] [CrossRef]
- Khanna, D.; Khanna, S.; Khanna, P.; Kahar, P.; Patel, B.M. Obesity: A Chronic Low-Grade Inflammation and Its Markers. Cureus 2022, 14, e22711. [Google Scholar] [CrossRef] [PubMed]
- López-Ortega, O.; Moreno-Corona, N.C.; Cruz-Holguin, V.J.; Garcia-Gonzalez, L.D.; Helguera-Repetto, A.C.; Romero-Valdovinos, M.; Arevalo-Romero, H.; Cedillo-Barron, L.; León-Juárez, M. The Immune Response in Adipocytes and Their Susceptibility to Infection: A Possible Relationship with Infectobesity. Int J Mol Sci 2022, 23, 6154. [Google Scholar] [CrossRef] [PubMed]
- Morąg, B.; Kozubek, P.; Gomułka, K. Obesity and Selected Allergic and Immunological Diseases—Etiopathogenesis, Course and Management. Nutrients 2023, 15, 3813. [Google Scholar] [CrossRef]
- Zhang, P. The Role of Diet and Nutrition in Allergic Diseases. Nutrients 2023, 15, 3683. [Google Scholar] [CrossRef]
- Gargano, D.; Appanna, R.; Santonicola, A.; De Bartolomeis, F.; Stellato, C.; Cianferoni, A.; Casolaro, V.; Iovino, P. Food Allergy and Intolerance: A Narrative Review on Nutritional Concerns. Nutrients 2021, 13, 1638. [Google Scholar] [CrossRef]
- Poto, R.; Fusco, W.; Rinninella, E.; Cintoni, M.; Kaitsas, F.; Raoul, P.; Caruso, C.; Mele, M.C.; Varricchi, G.; Gasbarrini, A. The Role of Gut Microbiota and Leaky Gut in the Pathogenesis of Food Allergy. Nutrients 2023, 16, 92. [Google Scholar] [CrossRef]
- Tangye, S.G.; Al-Herz, W.; Bousfiha, A.; Cunningham-Rundles, C.; Franco, J.L.; Holland, S.M.; Klein, C.; Morio, T.; Oksenhendler, E.; Picard, C.; Puel, A.; et al. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol 2022, 42, 1473–1507. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, X.; Ichikawa, M.; Lyons, J.J.; Datta, S.; Lamborn, I.T.; Jing, H.; Kim, E.S.; Biancalana, M.; Wolfe, L.A.; et al. Autosomal Recessive Phosphoglucomutase 3 (PGM3) Mutations Link Glycosylation Defects to Atopy, Immune Deficiency, Autoimmunity, and Neurocognitive Impairment. J Allergy Clin Immunol 2014, 133, 1400–1409. [Google Scholar] [CrossRef]
- Stukus, D.R.; Mikhail, I. Pearls and Pitfalls in Diagnosing IgE-Mediated Food Allergy. Curr Allergy Asthma Rep 2016, 16, 34. [Google Scholar] [CrossRef] [PubMed]
- Commins, S.P. Food Intolerance and Food Allergy in Adults: An Overview. In: Sicherer, S.H., ed. UpToDate. Wolters Kluwer. Updated May 30, 2024. Available online: www.uptodate.com/contents/food-intolerance-and-food-allergy-in-adults-an-overview (accessed on 8 September 2024).
- Demirbas, D.; Coelho, A.I.; Rubio-Gozalbo, M.E.; Berry, G.T. Hereditary Galactosemia. Metabolism 2018, 83, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Pasta, A.; Formisano, E.; Calabrese, F.; Plaz Torres, M.C.; Bodini, G.; Marabotto, E.; Pisciotta, L.; Giannini, E.G.; Furnari, M. Food Intolerances, Food Allergies and IBS: Lights and Shadows. Nutrients 2024, 16, 265. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.T.; Li, A.; Banerjee, J.; Gao, Z.G.; Kambayashi, T.; Jacobson, K.A.; Civan, M.M. The Role of Activated Adenosine Receptors in Degranulation of Human LAD2 Mast Cells. Purinergic Signal 2014, 10, 465–475. [Google Scholar] [CrossRef]
- Matsukura, S.; Aihara, M.; Sugawara, M.; Kunimi, Y.; Matsuki, M.; Inoue, Y.; Kambara, T.; Ikezawa, Z. Two Cases of Wheat-Dependent Anaphylaxis Induced by Aspirin Administration but Not by Exercise. Clin Exp Dermatol 2010, 35, 233–237. [Google Scholar] [CrossRef]
- Muñoz-Cano, R.; Pascal, M.; Araujo, G.; Goikoetxea, M.J.; Valero, A.L.; Picado, C.; Bartra, J. Mechanisms, Cofactors, and Augmenting Factors Involved in Anaphylaxis. Front Immunol 2017, 8, 1193. [Google Scholar] [CrossRef]
- Pascal, M.; Muñoz-Cano, R.; Milà, J.; Sanz, M.L.; Diaz-Perales, A.; Sánchez-López, J.; García-Moral, A.; Juan, M.; Valero, A.; Yagüe, J.; Picado, C.; Bartra, J. Nonsteroidal Anti-Inflammatory Drugs Enhance IgE-Mediated Activation of Human Basophils in Patients with Food Anaphylaxis Dependent on and Independent of Nonsteroidal Anti-Inflammatory Drugs. Clin Exp Allergy 2016, 46, 1111–1119. [Google Scholar] [CrossRef]
- Süß, H.; Dölle-Bierke, S.; Geier, J.; Kreft, B.; Oppel, E.; Pföhler, C.; Skudlik, C.; Worm, M.; Mahler, V. Contact Urticaria: Frequency, Elicitors and Cofactors in Three Cohorts (Information Network of Departments of Dermatology; Network of Anaphylaxis; and Department of Dermatology, University Hospital Erlangen, Germany). Contact Dermatitis 2019, 81, 341–353. [Google Scholar] [CrossRef]
- Geisslitz, S.; Weegels, P.; Shewry, P.; Zevallos, V.; Masci, S.; Sorrells, M.; Gregorini, A.; Colomba, M.; Jonkers, D.; Huang, X. , et al. Wheat Amylase/Trypsin Inhibitors (ATIs): Occurrence, Function and Health Aspects. Eur J Nutr 2022, 61, 2873–2880. [Google Scholar] [CrossRef]
- Junker, Y.; Zeissig, S.; Kim, S.J.; Barisani, D.; Wieser, H.; Leffler, D.A.; Zevallos, V.; Libermann, T.A.; Dillon, S.; Freitag, T.L. , et al. Wheat Amylase Trypsin Inhibitors Drive Intestinal Inflammation via Activation of Toll-Like Receptor 4. J Exp Med 2012, 209, 2395–2408. [Google Scholar] [CrossRef]
- Stapel, S.O.; Asero, R.; Ballmer-Weber, B.K.; Knol, E.F.; Strobel, S.; Vieths, S.; Kleine-Tebbe, J.; EAACI Task Force. Testing for IgG4 Against Foods is Not Recommended as a Diagnostic Tool: EAACI Task Force Report. Allergy 2008, 63, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Reese, I.; Ballmer-Weber, B.; Beyer, K.; Fuchs, T.; Kleine-Tebbe, J.; Klimek, L.; Lepp, U.; Niggemann, B.; Saloga, J.; Schäfer, C.; et al. German Guideline for the Management of Adverse Reactions to Ingested Histamine: Guideline of the German Society for Allergology and Clinical Immunology (DGAKI), the German Society for Pediatric Allergology and Environmental Medicine (GPA), the German Association of Allergologists (AeDA), and the Swiss Society for Allergology and Immunology (SGAI). Allergo J Int 2017, 26, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Reese, I.; Zuberbier, T.; Bunselmeyer, B.; Erdmann, S.; Henzgen, M.; Fuchs, T.; Jäger, L.; Kleine-Tebbe, J.; Lepp, U.; Niggemann, B.; Raithel, M.; Saloga, J.; Vieths, S.; Werfel, T. Diagnostic Approach for Suspected Pseudoallergic Reaction to Food Ingredients. J Dtsch Dermatol Ges 2009, 7, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Skypala, I.J.; Williams, M.; Reeves, L.; Meyer, R.; Venter, C. Sensitivity to Food Additives, Vaso-Active Amines and Salicylates: A Review of the Evidence. Clin Transl Allergy 2015, 5, 34. [Google Scholar] [CrossRef]
- Mohamed, G.G.; El-Hameed, A.K.; El-Din, A.M.; El-Din, L.A. High Performance Liquid Chromatography, Thin Layer Chromatography and Spectrophotometric Studies on the Removal of Biogenic Amines from Some Egyptian Foods Using Organic, Inorganic and Natural Compounds. J Toxicol Sci 2010, 35, 175–187. [Google Scholar] [CrossRef]
- Hare, L.G.; Woodside, J.V.; Young, I.S. Dietary Salicylates. J. Clin. Pathol. 2003, 56, 649–650. [Google Scholar] [CrossRef]
- Paterson, J.R.; Srivastava, R.; Baxter, G.J.; Graham, A.B.; Lawrence, J.R. Salicylic Acid Content of Spices and Its Implications. J. Agric. Food Chem. 2006, 54, 2891–2896. [Google Scholar] [CrossRef]
- Kang, M.G.; Song, W.J.; Park, H.K.; Lim, K.H.; Kim, S.J.; Lee, S.Y.; Kim, S.H.; Cho, S.H.; Min, K.U.; Chang, Y.S. Basophil Activation Test with Food Additives in Chronic Urticaria Patients. Clin. Nutr. Res. 2014, 3, 9–16. [Google Scholar] [CrossRef]
- Ortolani, C.; Bruijnzeel-Koomen, C.; Bengtsson, U.; Bindslev-Jensen, C.; Björkstén, B.; Høst, A.; Ispano, M.; Jarish, R.; Madsen, C.; Nekam, K. , et al. Controversial Aspects of Adverse Reactions to Food. European Academy of Allergology and Clinical Immunology (EAACI) Reactions to Food Subcommittee. Allergy 1999, 54, 27–45. [Google Scholar] [CrossRef]
- Reese, I.; Zuberbier, T.; Bunselmeyer, B.; Erdmann, S.; Henzgen, M.; Fuchs, T.; Jäger, L.; Kleine-Tebbe, J.; Lepp, U.; Niggemann, B. , et al. Diagnostic Approach for Suspected Pseudoallergic Reaction to Food Ingredients. J. Dtsch. Dermatol. Ges. 2009, 7, 70–77. [Google Scholar] [CrossRef]
- Simon, R.A. Allergic and Asthmatic Reactions to Food Additives. In Sicherer, S.H., Ed.; UpToDate. Wolters Kluwer. Updated August 2024. Available online: www.uptodate.com/contents/allergic-and-asthmatic-reactions-to-food-additives (Accessed 8 September 2024).
- Treudler, R.; Simon, J.C. Anaphylaxis to Food Additives. Allergo J. Int. 2022, 31, 141–144. [Google Scholar] [CrossRef]
- Zanfirescu, A.; Ungurianu, A.; Tsatsakis, A.M.; Nițulescu, G.M.; Kouretas, D.; Veskoukis, A.; Tsoukalas, D.; Engin, A.B.; Aschner, M.; Margină, D. A Review of the Alleged Health Hazards of Monosodium Glutamate. Compr. Rev. Food Sci. Food Saf. 2019, 18, 1111–1134. [Google Scholar] [CrossRef] [PubMed]
- Georgalas, C.; Jovancevic, L. Gustatory Rhinitis. Curr. Opin. Otolaryngol. Head Neck Surg. 2012, 20, 9–14. [Google Scholar] [CrossRef]
- Liva, G.A.; Karatzanis, A.D.; Prokopakis, E.P. Review of Rhinitis: Classification, Types, Pathophysiology. J. Clin. Med. 2021, 10, 3183. [Google Scholar] [CrossRef]
- Van Gerven, L.; Alpizar, Y.A.; Steelant, B.; Callebaut, I.; Kortekaas Krohn, I.; Wouters, M.; Vermeulen, F.; Boeckxstaens, G.; Talavera, K.; Hellings, P.W. Enhanced Chemosensory Sensitivity in Patients with Idiopathic Rhinitis and Its Reversal by Nasal Capsaicin Treatment. J. Allergy Clin. Immunol. 2017, 140, 437–446.e2. [Google Scholar] [CrossRef]
- Watanabe, T.; Terada, Y. Food Compounds Activating Thermosensitive TRP Channels in Asian Herbal and Medicinal Foods. J. Nutr. Sci. Vitaminol. 2015, 61, S86–88. [Google Scholar] [CrossRef]
- de Bree, R.; van der Waal, I.; Leemans, C.R. Management of Frey Syndrome. Head Neck 2007, 29, 773–778. [Google Scholar] [CrossRef]
- Izikson, L.; English, J.C. III.; Zirwas, M.J. The Flushing Patient: Differential Diagnosis, Workup, and Treatment. J. Am. Acad. Dermatol. 2006, 55, 193–208. [Google Scholar] [CrossRef]
- Young, A.; Okuyemi, O.T. Frey Syndrome. [Updated 2023 Jan 12]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: www.ncbi.nlm.nih.gov/books/NBK562247.
- Diaz, J.H. Amatoxin-Containing Mushroom Poisonings: Species, Toxidromes, Treatments, and Outcomes. Wilderness Environ Med 2018, 29, 111–118. [Google Scholar] [CrossRef]
- Predy, G.; Honish, L.; Hohn, W.; Jones, S. Was it Something She Ate? Case Report and Discussion of Scombroid Poisoning. CMAJ 2003, 168, 587–588. [Google Scholar]
- Smolinska, S.; Jutel, M.; Crameri, R.; O'Mahony, L. Histamine and Gut Mucosal Immune Regulation. Allergy 2014, 69, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Vilariño, N.; Louzao, M.C.; Abal, P.; Cagide, E.; Carrera, C.; Vieytes, M.R.; Botana, L.M. Human Poisoning from Marine Toxins: Unknowns for Optimal Consumer Protection. Toxins 2018, 10, 324. [Google Scholar] [CrossRef] [PubMed]
- Smolinska, S.; Winiarska, E.; Globinska, A.; Jutel, M. Histamine: A Mediator of Intestinal Disorders—A Review. Metabolites 2022, 12, 895. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic updated classification of the hypersensitivity reactions to foods [adapted after 1].
Figure 1.
Schematic updated classification of the hypersensitivity reactions to foods [adapted after 1].
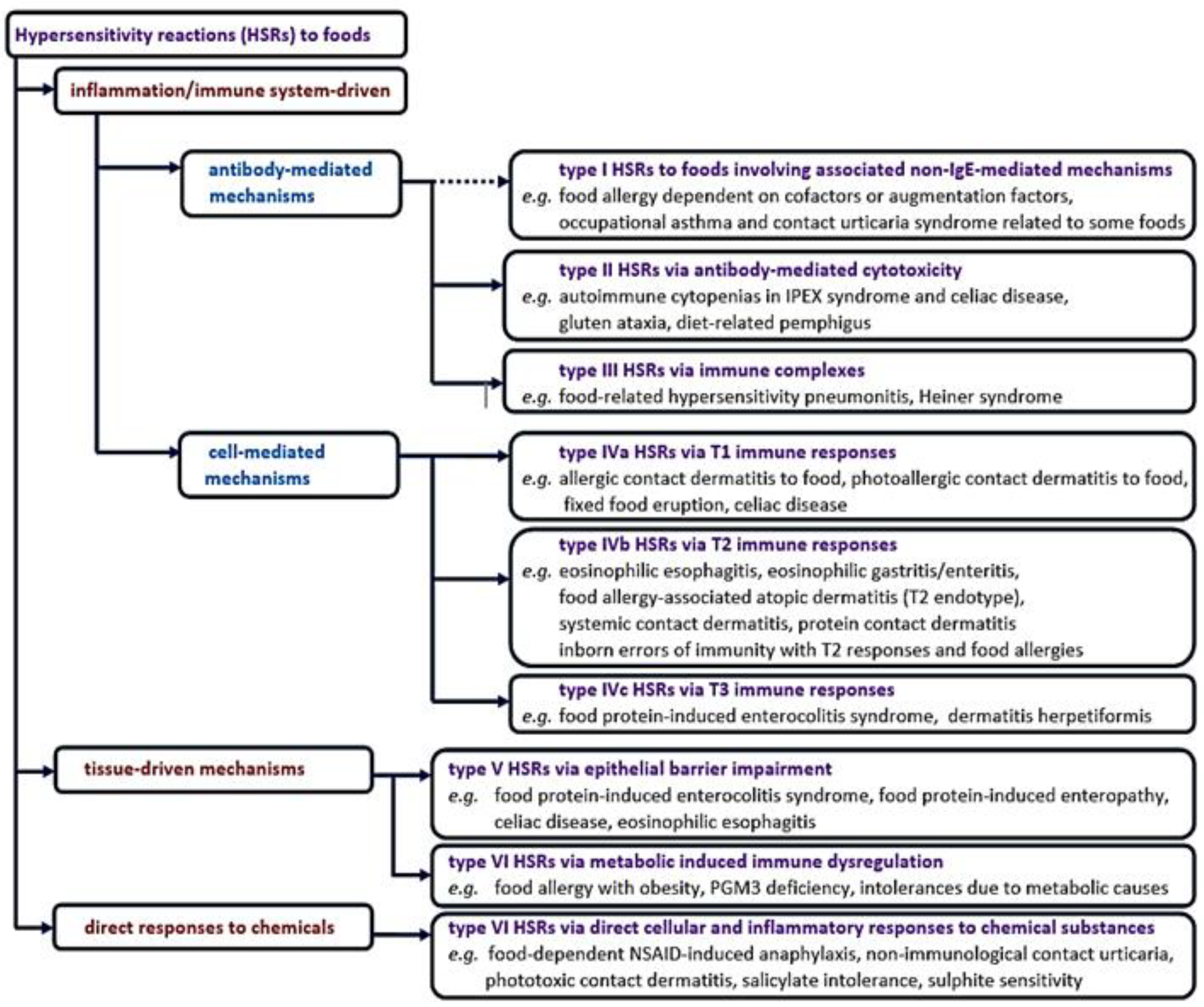

Table 1.
Conventional classification of non-IgE-mediated hypersensitivity reactions to foods [3].
Table 1.
Conventional classification of non-IgE-mediated hypersensitivity reactions to foods [3].
| Subtypes of non-IgE mediated food hypersensitivity reactions | Examples |
| non-IgE mediated food allergies | contact dermatitis, food protein-induced enterocolitis syndrome, food protein-induced allergic proctitis/proctocolitis, food protein-induced enteropathy, Heiner syndrome, celiac disease, dermatitis herpetiformis |
| mixed IgE and non-IgE mediated food allergies | |
| eosinophilic oesophagitis, eosinophilic gastritis/enteritis, food-exacerbation of atopic eczema, food-exacerbation of asthma |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



