
Submitted:
24 October 2024
Posted:
25 October 2024
Read the latest preprint version here
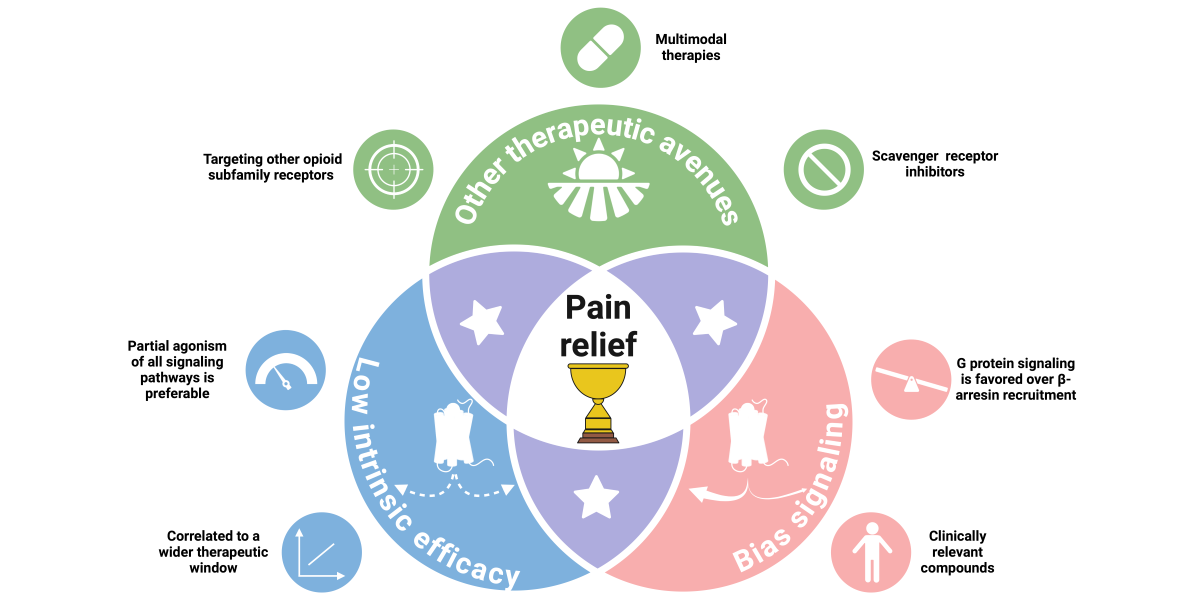
Abstract
Opioid analgesics have been used for more than 5,000 years and remain the principal pain medications prescribed today. Although morphine is considered the gold standard for pain relief, this µ-opioid receptor (MOP)-selective agonist provides only moderate relief for many chronic pain conditions and promotes several unwanted effects that can adversely affect patient quality-of-life, prevent adherence to treatment or generate addiction. In addition to the lack of progress in the development of better analgesics, there have been no significant breakthroughs to date that have addressed side effects mentioned above. Fortunately, a better understanding of opioid pharmacology has restored hope for the development of better and safer pain medications. In this review, we describe how clinically approved opioids were initially characterized as biased ligands and what impact this approach could have on clinical practice. We also looked at the preclinical and clinical development of MOP biased agonists, with an emphasis on the oliceridine story, as the first specifically designed biased painkiller. Moreover, we explore the discrepancies between ligands with low intrinsic efficacy and those with biased properties. Finally, we examine the rationale behind biased ligand development during the opioid crisis era.
Keywords:
1. Introduction
2. Shaping Analgesic Drug Design by Understanding Opioid Pharmacology
3. GPCRs: From Simple Switches to Integrated Signaling Processors
4. Old Drugs, New Tricks: MOP-Biased Agonists in Clinically Approved Opioids
5. Oliceridine, the Prototype MOP-Biased Agonist
6. MOP-Biased Ligands in Preclinical Development
7. MOP-Biased Ligands Versus Low Intrinsic Efficacy Ligands at the µ-Opioid Receptor
8. Novel MOP Ligands in the Current Opioid Crisis Era
9. Alternatives to Biased Signaling at the MOP
10. The Remaining Unknown Ingredients for an Ideal Analgesic
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cohen, S.P.; Vase, L.; Hooten, W.M. Chronic pain: An update on burden, best practices, and new advances. Lancet 2021, 397, 2082–2097. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Blanco, C. The changing opioid crisis: Development, challenges and opportunities. Mol. Psychiatry 2020, 26, 218–233. [Google Scholar] [CrossRef] [PubMed]
- Duarte, D.F. Opium and opioids: A brief history. 2005, 55, 135–146.
- Santoro, D.; Savica, V. Development of the concept of pain in history. J. Nephrol. 2011, 24, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Abdel Shaheed, C.; Maher, C.G.; Williams, K.A.; Day, R.; McLachlan, A.J. Efficacy, Tolerability, and Dose-Dependent Effects of Opioid Analgesics for Low Back Pain: A Systematic Review and Meta-analysis. JAMA Intern Med. 2016, 176, 958–968. [Google Scholar] [CrossRef] [PubMed]
- de Leon-Casasola, OA. Opioids for chronic pain: New evidence, new strategies, safe prescribing. Am J Med. 2013, 126, S3–11. [Google Scholar] [CrossRef]
- Kissin, I. The Development of New Analgesics Over the Past 50 Years: A Lack of Real Breakthrough Drugs. Anesthesia Analg. 2010, 110, 780–789. [Google Scholar] [CrossRef]
- WHO Expert Committee. The selection and use of essential medicines: Report of the WHO Expert Committee, 2017 (including the 20th WHO Model List of Essential Medicines and the 6th WHO Model List of essential Medicines for Children). WHO technical report series ; no 1006. Geneva: World Health Organization; 2017. p. 604.
- Shipton, E.A.; Shipton, E.E.; Shipton, A.J. A Review of the Opioid Epidemic: What Do We Do About It? Pain Ther. 2018, 7, 23–36. [Google Scholar] [CrossRef]
- Manchikanti L, Helm S, 2nd, Fellows B, Janata JW, Pampati V, Grider JS; et al. Opioid epidemic in the United States. Pain Physician. 2012, 15, ES9–38.
- Webster, L.; Schmidt, W.K. Dilemma of Addiction and Respiratory Depression in the Treatment of Pain: A Prototypical Endomorphin as a New Approach. Pain Med. 2019, 21, 992–1004. [Google Scholar] [CrossRef]
- Wang, Y.; Zhuang, Y.; DiBerto, J.F.; Zhou, X.E.; Schmitz, G.P.; Yuan, Q.; Jain, M.K.; Liu, W.; Melcher, K.; Jiang, Y.; et al. Structures of the entire human opioid receptor family. Cell 2023, 186, 413–427. [Google Scholar] [CrossRef]
- Matthes HW, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I; et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature. 1996, 383, 819–823.
- A Hsia, J.; Moss, J.; Hewlett, E.L.; Vaughan, M. ADP-ribosylation of adenylate cyclase by pertussis toxin. Effects on inhibitory agonist binding. J. Biol. Chem. 1984, 259, 1086–1090. [Google Scholar] [CrossRef] [PubMed]
- Taussig, R.; Iniguez-Lluhi, J.A.; Gilman, A.G. Inhibition of adenylyl cyclase by Gi alpha. Science. 1993, 261, 218–221. [Google Scholar] [CrossRef]
- Ippolito DL, Temkin PA, Rogalski SL, Chavkin C. N-terminal tyrosine residues within the potassium channel Kir3 modulate GTPase activity of Galphai. J Biol Chem. 2002, 277, 32692–32696.
- Sadja, R.; Alagem, N.; Reuveny, E. Gating of GIRK Channels. Neuron 2003, 39, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Díaz, A.; Flórez, J.; Pazos, A.; Hurlé, M.A. Opioid tolerance and supersensitivity induce regional changes in the autoradiographic density of dihydropyridine-sensitive calcium channels in the rat central nervous system. Pain 2000, 86, 227–235. [Google Scholar] [CrossRef]
- Díaz, A.; Ruíz, F.; Flórez, J.; Pazos, A.; A Hurlé, M. Regulation of dihydropyridine-sensitive Ca++ channels during opioid tolerance and supersensitivity in rats. . 1995, 274, 1538–1544. [Google Scholar]
- Wickman, K.; Clapham, D.E. Ion channel regulation by G proteins. Physiol. Rev. 1995, 75, 865–885. [Google Scholar] [CrossRef]
- Dang, V.C.; Christie, M.J. Mechanisms of rapid opioid receptor desensitization, resensitization and tolerance in brain neurons. Br. J. Pharmacol. 2012, 165, 1704–1716. [Google Scholar] [CrossRef]
- Groer, C.E.; Schmid, C.L.; Jaeger, A.M.; Bohn, L.M. Agonist-directed interactions with specific beta-arrestins determine mu-opioid receptor trafficking, ubiquitination, and dephosphorylation. J Biol Chem. 2011, 286, 31731–31741. [Google Scholar] [CrossRef]
- Melief, E.J.; Miyatake, M.; Bruchas, M.R.; Chavkin, C. Ligand-directed c-Jun N-terminal kinase activation disrupts opioid receptor signaling. Proc. Natl. Acad. Sci. 2010, 107, 11608–11613. [Google Scholar] [CrossRef] [PubMed]
- Whistler, J.L.; Chuang, H.-H.; Chu, P.; Jan, L.Y.; von Zastrow, M. Functional Dissociation of μ Opioid Receptor Signaling and Endocytosis. Neuron 1999, 23, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Gondin AB, Halls ML, Canals M, Briddon SJ. GRK Mediates mu-Opioid Receptor Plasma Membrane Reorganization. Front Mol Neurosci. 2019;12:104.
- Raffa, R.B.; Martinez, R.P.; Connelly, C.D. G-protein antisense oligodeoxyribonucleotides and μ-opioid supraspinal antinociception. Eur. J. Pharmacol. 1994, 258, R5–R7. [Google Scholar] [CrossRef] [PubMed]
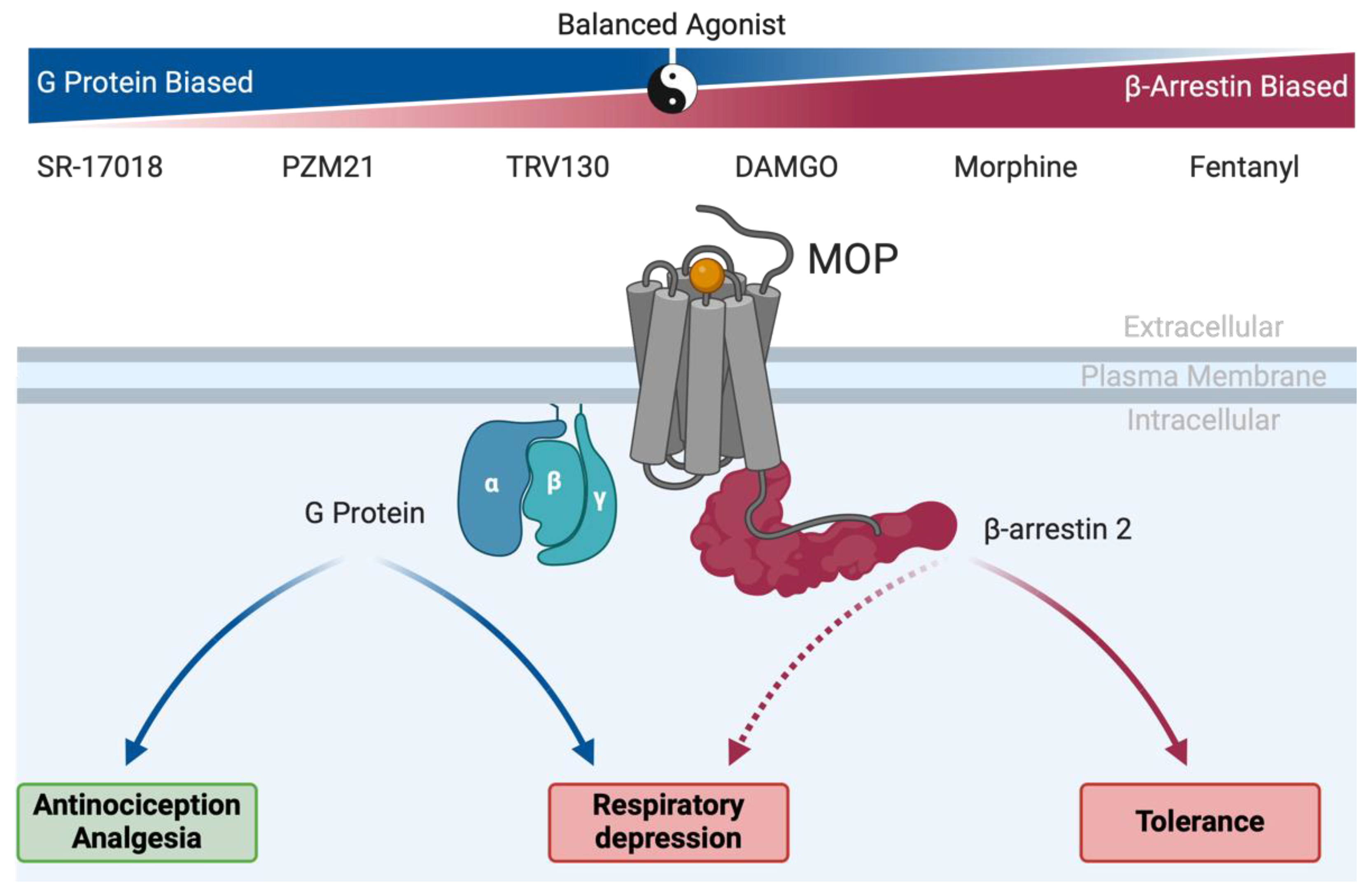
- Bohn, L.M.; Lefkowitz, R.J.; Gainetdinov, R.R.; Peppel, K.; Caron, M.G.; Lin, F.T. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science. 1999, 286, 2495–2498. [Google Scholar] [CrossRef] [PubMed]
- Raehal, K.M.; Walker, J.K.L.; Bohn, L.M. Morphine Side Effects in β-Arrestin 2 Knockout Mice. J. Pharmacol. Exp. Ther. 2005, 314, 1195–1201. [Google Scholar] [CrossRef]
- Bohn, L.M.; Gainetdinov, R.R.; Lin, F.T.; Lefkowitz, R.J.; Caron, M.G. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000, 408, 720–723. [Google Scholar] [CrossRef]
- Gavériaux-Ruff, C.; Kieffer, B. Opioid receptor genes inactivated in mice: The highlights. Neuropeptides 2002, 36, 62–71. [Google Scholar] [CrossRef]
- Yang, C.H.; Huang, H.W.; Chen, K.H.; Chen, Y.S.; Sheen-Chen, S.M.; Lin, C.R. Antinociceptive potentiation and attenuation of tolerance by intrathecal beta-arrestin 2 small interfering RNA in rats. Br J Anaesth. 2011, 107, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Kliewer A, Schmiedel F, Sianati S, Bailey A, Bateman JT, Levitt ES; et al. Phosphorylation-deficient G-protein-biased mu-opioid receptors improve analgesia and diminish tolerance but worsen opioid side effects. Nat Commun. 2019, 10, 367.
- Bull, F.A.; Baptista-Hon, D.T.; Sneddon, C.; Wright, L.; Walwyn, W.; Hales, T.G. Src Kinase Inhibition Attenuates Morphine Tolerance without Affecting Reinforcement or Psychomotor Stimulation. Anesthesiology 2017, 127, 878–889. [Google Scholar] [CrossRef]
- Kliewer A, Gillis A, Hill R, Schmiedel F, Bailey C, Kelly E; et al. Morphine-induced respiratory depression is independent of beta-arrestin2 signalling. Br J Pharmacol. 2020, 177, 2923–2931.
- Chen YJ, Oldfield S, Butcher AJ, Tobin AB, Saxena K, Gurevich VV; et al. Identification of phosphorylation sites in the COOH-terminal tail of the mu-opioid receptor. J Neurochem. 2013, 124, 189–199.
- Miess E, Gondin AB, Yousuf A, Steinborn R, Mosslein N, Yang Y; et al. Multisite phosphorylation is required for sustained interaction with GRKs and arrestins during rapid mu-opioid receptor desensitization. Sci Signal. 2018, 11, 539.
- Gregory, K.J. Commentary on “Morphine-induced respiratory depression is independent of beta-arrestin2 signalling”. Br J Pharmacol. 2020, 177, 2904–2905. [Google Scholar] [CrossRef] [PubMed]
- Lam H, Maga M, Pradhan A, Evans CJ, Maidment NT, Hales TG; et al. Analgesic tone conferred by constitutively active mu opioid receptors in mice lacking beta-arrestin 2. Mol Pain. 2011, 7, 24.
- Stone, L.S.; Molliver, D.C. In search of analgesia: Emerging roles of GPCRs in pain. Mol Interv. 2009, 9, 234–251. [Google Scholar] [CrossRef]
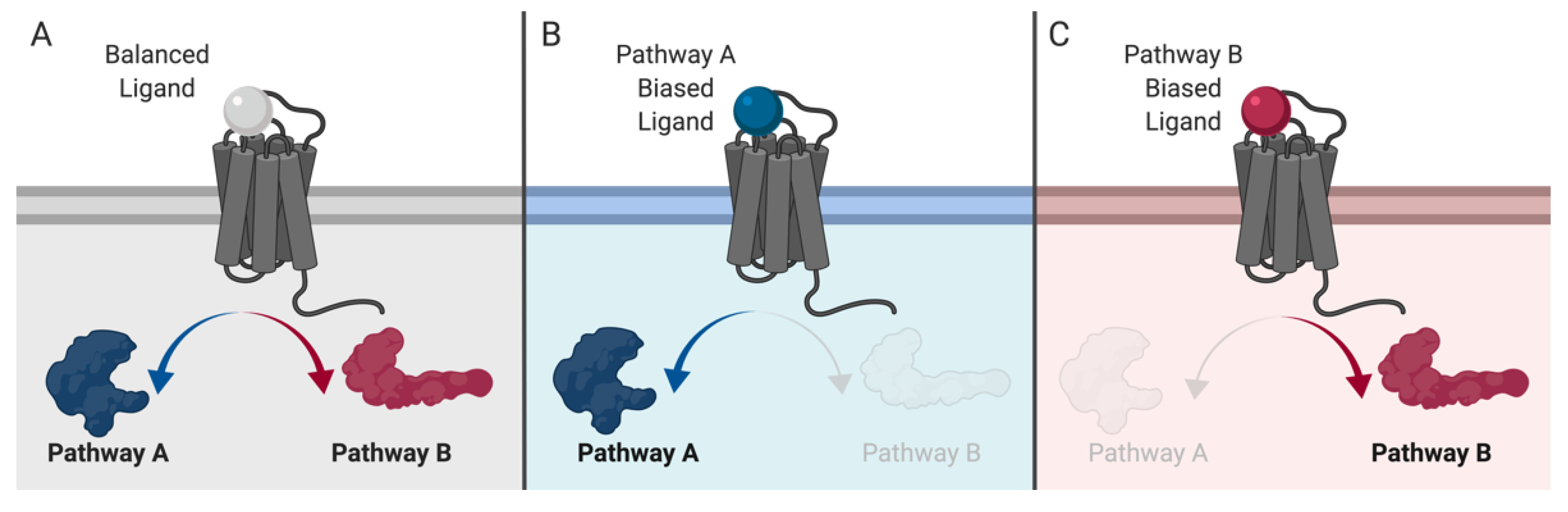
- Rajagopal, S.; Rajagopal, K.; Lefkowitz, R.J. Teaching old receptors new tricks: Biasing seven-transmembrane receptors. Nat. Rev. Drug Discov. 2010, 9, 373–386. [Google Scholar] [CrossRef]
- Audet, M.; Bouvier, M. Restructuring G-Protein- Coupled Receptor Activation. Cell 2012, 151, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Kenakin, T. Functional Selectivity and Biased Receptor Signaling. J. Pharmacol. Exp. Ther. 2010, 336, 296–302. [Google Scholar] [CrossRef]
- Wootten, D.; Christopoulos, A.; Marti-Solano, M.; Babu, M.M.; Sexton, P.M. Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 2018, 19, 638–653. [Google Scholar] [CrossRef]
- van der Westhuizen ET, Breton B, Christopoulos A, Bouvier M. Quantification of ligand bias for clinically relevant beta2-adrenergic receptor ligands: Implications for drug taxonomy. Mol Pharmacol. 2014, 85, 492–509.
- Kenakin, T.; Christopoulos, A. Signalling bias in new drug discovery: Detection, quantification and therapeutic impact. Nat. Rev. Drug Discov. 2012, 12, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Besserer-Offroy, E.; Sarret, P. Sending out Biased Signals: An Appropriate Proposition for Pain? Douleur et Analgésie. 2019, 32, 108–110. [Google Scholar] [CrossRef]
- Michel, M.C.; Charlton, S.J. Biased Agonism in Drug Discovery-Is It Too Soon to Choose a Path? Mol Pharmacol. 2018, 93, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Urs NM, Gee SM, Pack TF, McCorvy JD, Evron T, Snyder JC; et al. Distinct cortical and striatal actions of a beta-arrestin-biased dopamine D2 receptor ligand reveal unique antipsychotic-like properties. Proc Natl Acad Sci USA. 2016, 113, E8178–E86.
- Schattauer, S.S.; Kuhar, J.R.; Song, A.; Chavkin, C. Nalfurafine is a G-protein biased agonist having significantly greater bias at the human than rodent form of the kappa opioid receptor. Cell Signal. 2017, 32, 59–65. [Google Scholar] [CrossRef]
- Kenakin, T. Bias translation: The final frontier? Br J Pharmacol. 2024, 181, 1345–1360. [Google Scholar] [CrossRef]
- Kenakin, T. Know your molecule: Pharmacological characterization of drug candidates to enhance efficacy and reduce late-stage attrition. Nat. Rev. Drug Discov. 2024, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Keith DE, Murray SR, Zaki PA, Chu PC, Lissin DV, Kang L; et al. Morphine activates opioid receptors without causing their rapid internalization. J Biol Chem. 1996, 271, 19021–19024.
- Ehrlich, A.T.; Kieffer, B.L.; Darcq, E. Current strategies toward safer mu opioid receptor drugs for pain management. Expert Opin. Ther. Targets 2019, 23, 315–326. [Google Scholar] [CrossRef]
- McPherson J, Rivero G, Baptist M, Llorente J, Al-Sabah S, Krasel C; et al. mu-opioid receptors: Correlation of agonist efficacy for signalling with ability to activate internalization. Mol Pharmacol. 2010, 78, 756–766.
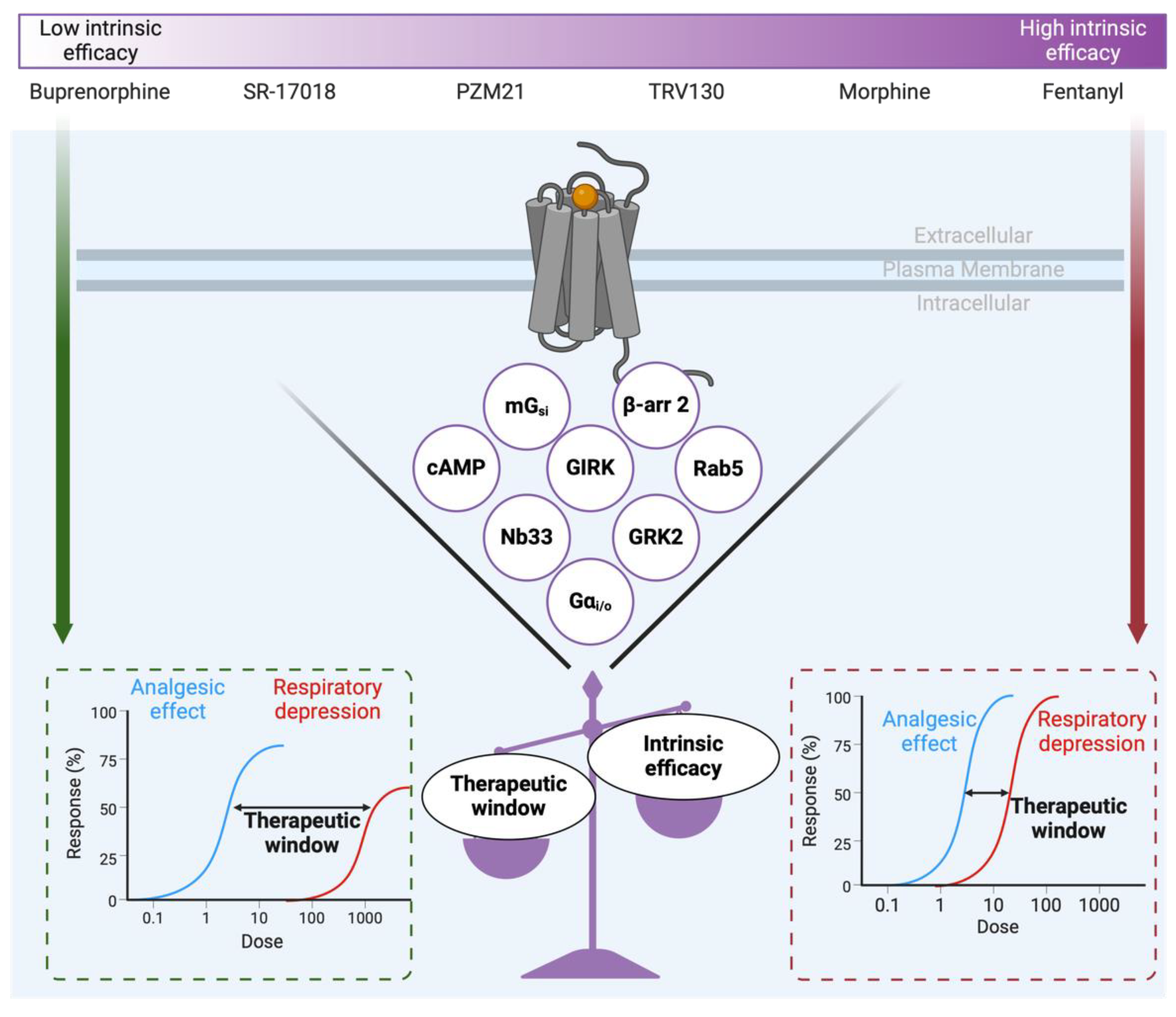
- Schmid, C.L.; Kennedy, N.M.; Ross, N.C.; Lovell, K.M.; Yue, Z.; Morgenweck, J.; Cameron, M.D.; Bannister, T.D.; Bohn, L.M. Bias Factor and Therapeutic Window Correlate to Predict Safer Opioid Analgesics. Cell 2017, 171, 1165–1175. [Google Scholar] [CrossRef] [PubMed]
- Butler, S. Buprenorphine—Clinically useful but often misunderstood. Scand. J. Pain 2013, 4, 148–152. [Google Scholar] [CrossRef]
- Lutfy, K.; Cowan, A. Buprenorphine: A Unique Drug with Complex Pharmacology. Curr. Neuropharmacol. 2004, 2, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.P.; Pasternak, G.; Behm, B. Treating Chronic Pain: An Overview of Clinical Studies Centered on the Buprenorphine Option. Drugs 2018, 78, 1211–1228. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, A.T.; Darcq, E. Recommending buprenorphine for pain management. Pain Manag. 2019, 9, 13–16. [Google Scholar] [CrossRef]
- Vlok, R.; An, G.H.; Binks, M.; Melhuish, T.; White, L. Sublingual buprenorphine versus intravenous or intramuscular morphine in acute pain: A systematic review and meta-analysis of randomized control trials. Am J Emerg Med. 2019, 37, 381–386. [Google Scholar] [CrossRef]
- Gudin, J.; Fudin, J.; Nalamachu, S. Levorphanol use: Past, present and future. Postgrad. Med. 2015, 128, 46–53. [Google Scholar] [CrossRef]
- Prommer, E. Levorphanol: The forgotten opioid. Support. Care Cancer 2006, 15, 259–264. [Google Scholar] [CrossRef]
- Prommer, E. Levorphanol: Revisiting an Underutilized Analgesic. Palliat. Care: Res. Treat. 2014, 8, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Le Rouzic, V.; Narayan, A.; Hunkle, A.; Marrone, G.F.; Lu, Z.; Majumdar, S.; Xu, J.; Pan, Y.-X.; Pasternak, G.W. Pharmacological Characterization of Levorphanol, a G-Protein Biased Opioid Analgesic. Anesthesia Analg. 2019, 128, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.S.; Upputuri, O.; Mantha, S.S.P.; Rayani, B.K. Levorphanol: Rewinding an Old, Bygone Multimodal Opioid Analgesic! Indian J Palliat Care. 2019, 25, 483–484. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.C.; Fudin, J.; Raffa, R.B. Is levorphanol a better option than methadone? Pain Med. 2015, 16, 1673–1679. [Google Scholar] [CrossRef] [PubMed]
- Chen X-T, Pitis P, Liu G, Yuan C, Gotchev D, Cowan CL; et al. Structure–Activity Relationships and Discovery of a G Protein Biased μ Opioid Receptor Ligand, [(3-Methoxythiophen-2-yl)methyl]({2-[(9R)-9-(pyridin-2-yl)-6-oxaspiro-[4.5]decan-9-yl]ethyl})amine (TRV130), for the Treatment of Acute Severe Pain. Journal of Medicinal Chemistry. 2013, 56, 8019–8031.
- DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM; et al. A G protein-biased ligand at the mu-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther. 2013, 344, 708–717.
- Trevena Inc. Trevena Announces Submission of New Drug Application to U.S. FDA for Oliceridine Injection. 2017 [cited 2020-03-24]; Available from: https://www.trevena.com/investors/press-releases/detail/127/trevena-announces-submission-of-new-drug-application-to.
- Trevena Inc. Trevena Announces FDA Acceptance for Review of New Drug Application for Oliceridine Injection. 2018 [cited 2020-03-24]; Available from: https://www.trevena.com/investors/press-releases/detail/129/trevena-announces-fda-acceptance-for-review-of-new-drug.
- Trevena Inc. Trevena Announces Oliceridine FDA Advisory Committee Meeting Outcome. 2018 [cited 2020-03-24]; Available from: https://www.trevena.com/investors/press-releases/detail/154/trevena-announces-oliceridine-fda-advisory-committee.
- FDAAADPAC Transcript for the , 2018 Meeting of the Anesthetic and Analgesic Drug Products Advisory Committee (AADPAC). 2018 [cited 2020-03-24]; Available from: https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/AnestheticAndAnalgesicDrugProductsAdvisoryCommittee/UCM629602.pdf. 11 October.
- Trevena Inc. Trevena Announces Receipt of Type A Meeting Minutes and Provides Regulatory Update for Oliceridine. 2019 [cited 2020-03-14]; Available from: https://www.trevena.com/investors/press-releases/detail/160/trevena-announces-receipt-of-type-a-meeting-minutes-and.
- Trevena Inc. Trevena Announces Initiation of Healthy Volunteer Study for Oliceridine. 2019 [cited 2020-05-12]; Available from: https://www.trevena.com/investors/press-releases/detail/5/trevena-announces-initiation-of-healthy-volunteer-study-for.
- Nafziger, A.N.; Arscott, K.A.; Cochrane, K.; Skobieranda, F.; Burt, D.A.; Fossler, M.J. The Influence of Renal or Hepatic Impairment on the Pharmacokinetics, Safety, and Tolerability of Oliceridine. Clin. Pharmacol. Drug Dev. 2019, 9, 639–650. [Google Scholar] [CrossRef]
- Trevena Inc. Trevena Resubmits New Drug Application for Oliceridine. 2020 [cited 2020-06-16]; Available from: https://www.trevena.com/investors/press-releases/detail/224/trevena-resubmits-new-drug-application-for-oliceridine.
- Gan, T.; Wase, L. Oliceridine, a G protein-selective ligand at the mu-opioid receptor, for the management of moderate to severe acute pain. Drugs Today 2020, 56, 269–286. [Google Scholar] [CrossRef]
- Trevena Inc. Trevena Announces FDA Approval of OLINVYK™ (oliceridine) injection. 2020 [cited 2024-08-18]; Available from: https://www.trevena.com/investors/press-releases/detail/234/trevena-announces-fda-approval-of-olinvyk-oliceridine-injection.
- US Food & Drug Administration. FDA Approves New Opioid for Intravenous Use in Hospitals, Other Controlled Clinical Settings. 2020 [cited; Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-new-opioid-intravenous-use-hospitals-other-controlled-clinical-settings.
- Shah, A.; Shah, R.; Fahim, G.; A Brust-Sisti, L. A Dive Into Oliceridine and Its Novel Mechanism of Action. Cureus 2021, 13. [Google Scholar] [CrossRef]
- Azzam, A.A.H.; Lambert, D.G. Preclinical discovery and development of oliceridine (Olinvyk(R)) for the treatment of post-operative pain. Expert Opin Drug Discov. 2022 Mar;17(3):215-23.
- Trevena Inc. Trevena, Inc. Announces DEA Scheduling of OLINVYK™ (oliceridine) injection. 2020 [cited; Available from: https://www.trevena.com/investors/press-releases/detail/244/trevena-inc-announces-dea-scheduling-of-olinvyk-oliceridine-injection.
- US Food & Drug Administration. Second Cycle Review And Summary Basis for Approval—Oliceridine injection (NDA 210-730). 2020 2020-08-07 [cited 210730Orig1s000 ]; Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/210730Orig1s000MultidisciplineR.pdf.
- Trevena Inc. Trevena Announces Approval of OLINVYK in China. 2023 [cited 2024-08-18]; Available from: https://www.trevena.com/investors/press-releases/detail/320/trevena-announces-approval-of-olinvyk-in-china.
- Ayad, S.; Demitrack, M.A.; Burt, D.A.; Michalsky, C.; Wase, L.; Fossler, M.J.; Khanna, A.K. Evaluating the Incidence of Opioid-Induced Respiratory Depression Associated with Oliceridine and Morphine as Measured by the Frequency and Average Cumulative Duration of Dosing Interruption in Patients Treated for Acute Postoperative Pain. Clin. Drug Investig. 2020, 40, 755–764. [Google Scholar] [CrossRef]
- Brzezinski, M.; Hammer, G.B.; Candiotti, K.A.; Bergese, S.D.; Pan, P.H.; Bourne, M.H.; Michalsky, C.; Wase, L.; Demitrack, M.A.; Habib, A.S. Low Incidence of Opioid-Induced Respiratory Depression Observed with Oliceridine Regardless of Age or Body Mass Index: Exploratory Analysis from a Phase 3 Open-Label Trial in Postsurgical Pain. Pain Ther. 2021, 10, 457–473. [Google Scholar] [CrossRef] [PubMed]
- Hammer, G.B.; Khanna, A.K.; Michalsky, C.; Wase, L.; Demitrack, M.A.; Little, R.; Fossler, M.J.; Ayad, S. Oliceridine Exhibits Improved Tolerability Compared to Morphine at Equianalgesic Conditions: Exploratory Analysis from Two Phase 3 Randomized Placebo and Active Controlled Trials. Pain Ther. 2021, 10, 1343–1353. [Google Scholar] [CrossRef] [PubMed]
- Beard, T.L.; Michalsky, C.; Candiotti, K.A.; Rider, P.; Wase, L.; Habib, A.S.; Demitrack, M.A.; Fossler, M.J.; Viscusi, E.R. Oliceridine is Associated with Reduced Risk of Vomiting and Need for Rescue Antiemetics Compared to Morphine: Exploratory Analysis from Two Phase 3 Randomized Placebo and Active Controlled Trials. Pain Ther. 2021, 10, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.M.; DeBoer, E. State and Future Science of Opioids and Potential of Biased-ligand Technology in the Management of Acute Pain After Burn Injury. J. Burn. Care Res. 2023, 44, 524–534. [Google Scholar] [CrossRef]
- Manglik, A.; Lin, H.; Aryal, D.K.; McCorvy, J.D.; Dengler, D.; Corder, G.; Levit, A.; Kling, R.C.; Bernat, V.; Hübner, H.; et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature 2016, 537, 185–190. [Google Scholar] [CrossRef]
- Emery, M.A.; Eitan, S. Members of the same pharmacological family are not alike: Different opioids, different consequences, hope for the opioid crisis? Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2019;92:428-49.
- Kudla L, Bugno R, Skupio U, Wiktorowska L, Solecki W, Wojtas A; et al. Functional characterization of a novel opioid, PZM21, and its influence on behavioural responses to morphine. Br J Pharmacol. 2019 Jul 26.
- Hill R, Disney A, Conibear A, Sutcliffe K, Dewey W, Husbands S; et al. The novel mu-opioid receptor agonist PZM21 depresses respiration and induces tolerance to antinociception. Br J Pharmacol. 2018, 175, 2653–2661.
- Grim, T.W.; Acevedo-Canabal, A.; Bohn, L.M. Toward Directing Opioid Receptor Signaling to Refine Opioid Therapeutics. Biol. Psychiatry 2019, 87, 15–21. [Google Scholar] [CrossRef]
- de Waal PW, Shi J, You E, Wang X, Melcher K, Jiang Y; et al. Molecular mechanisms of fentanyl mediated beta-arrestin biased signaling. PLoS Comput Biol. 2020, 16, e1007394.
- Ramos-Gonzalez N, Groom S, Sutcliffe KJ, Bancroft S, Bailey CP, Sessions RB; et al. Carfentanil is a beta-arrestin-biased agonist at the mu opioid receptor. Br J Pharmacol. 2023, 180, 2341–2360.
- Huang W, Manglik A, Venkatakrishnan AJ, Laeremans T, Feinberg EN, Sanborn AL; et al. Structural insights into micro-opioid receptor activation. Nature. 2015, 524, 315–321.
- Zhuang Y, Wang Y, He B, He X, Zhou XE, Guo S; et al. Molecular recognition of morphine and fentanyl by the human mu-opioid receptor. Cell. 2022, 185, 4361–4375.
- Kelly, E.; Conibear, A.; Henderson, G. Biased Agonism: Lessons from Studies of Opioid Receptor Agonists. Annu. Rev. Pharmacol. Toxicol. 2023, 63, 491–515. [Google Scholar] [CrossRef] [PubMed]
- Kise, R.; Inoue, A. GPCR signaling bias: An emerging framework for opioid drug development. J. Biochem. 2024, 175, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Gillis, A.; Gondin, A.B.; Kliewer, A.; Sanchez, J.; Lim, H.D.; Alamein, C.; Manandhar, P.; Santiago, M.; Fritzwanker, S.; Schmiedel, F.; et al. Low intrinsic efficacy for G protein activation can explain the improved side effect profiles of new opioid agonists. Sci. Signal. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Stoeber, M.; Jullié, D.; Lobingier, B.T.; Laeremans, T.; Steyaert, J.; Schiller, P.W.; Manglik, A.; von Zastrow, M. A Genetically Encoded Biosensor Reveals Location Bias of Opioid Drug Action. Neuron 2018, 98, 963–976. [Google Scholar] [CrossRef]
- Stahl, E.L.; Bohn, L.M. Low Intrinsic Efficacy Alone Cannot Explain the Improved Side Effect Profiles of New Opioid Agonists. Biochemistry 2021, 61, 1923–1935. [Google Scholar] [CrossRef]
- Clark, D.J.; Schumacher, M.A. America’s Opioid Epidemic: Supply and Demand Considerations. Anesthesia Analg. 2017, 125, 1667–1674. [Google Scholar] [CrossRef]
- Killeen, N.; Lakes, R.; Webster, M.; Killoran, S.; McNamara, S.; Kavanagh, P.; Eagleton, M.; McCormack, S.; Micheau, E.; Moughty, A.; et al. The emergence of nitazenes on the Irish heroin market and national preparation for possible future outbreaks. Addiction 2024, 119, 1657–1658. [Google Scholar] [CrossRef]
- Kozell LB, Eshleman AJ, Wolfrum KM, Swanson TL, Bloom SH, Benware S; et al. Pharmacologic Characterization of Substituted Nitazenes at mu, kappa, and Delta Opioid Receptors Suggests High Potential for Toxicity. J Pharmacol Exp Ther. 2024, 389, 219–228.
- Bendjilali-Sabiani, J.-J.; Eiden, C.; Lestienne, M.; Cherki, S.; Gautre, D.; Broek, T.V.D.; Mathieu, O.; Peyrière, H. Isotonitazene, a synthetic opioid from an emerging family: The nitazenes. Therapies 2024. [Google Scholar] [CrossRef]
- Amaducci, A.; Aldy, K.; Campleman, S.L.; Li, S.; Meyn, A.; Abston, S.; Culbreth, R.E.; Krotulski, A.; Logan, B.; Wax, P.; et al. Naloxone Use in Novel Potent Opioid and Fentanyl Overdoses in Emergency Department Patients. JAMA Netw. Open 2023, 6, e2331264–e2331264. [Google Scholar] [CrossRef] [PubMed]
- Häuser, W.; Schug, S.; Furlan, A.D. The opioid epidemic and national guidelines for opioid therapy for chronic noncancer pain: A perspective from different continents. PAIN Rep. 2017, 2, e599. [Google Scholar] [CrossRef]
- Rudd, R.A.; Seth, P.; David, F.; Scholl, L. Increases in Drug and Opioid-Involved Overdose Deaths—United States, 2010-2015. MMWR Morb Mortal Wkly Rep. 2016, 65, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Busse, J.W.; Craigie, S.; Juurlink, D.N.; Buckley, D.N.; Wang, L.; Couban, R.J.; Agoritsas, T.; Akl, E.A.; Carrasco-Labra, A.; Cooper, L.; et al. Guideline for opioid therapy and chronic noncancer pain. Can. Med Assoc. J. 2017, 189, E659–E666. [Google Scholar] [CrossRef]
- Bohn, L.M.; Gainetdinov, R.R.; Sotnikova, T.D.; Medvedev, I.O.; Lefkowitz, R.J.; Dykstra, L.A.; Caron, M.G. Enhanced Rewarding Properties of Morphine, but not Cocaine, in βarrestin-2 Knock-Out Mice. J. Neurosci. 2003, 23, 10265–10273. [Google Scholar] [CrossRef]
- Zamarripa, C.A.; Edwards, S.R.; Qureshi, H.N.; Yi, J.N.; Blough, B.E.; Freeman, K.B. The G-protein biased mu-opioid agonist, TRV130, produces reinforcing and antinociceptive effects that are comparable to oxycodone in rats. Drug Alcohol Depend. 2018, 192, 158–162. [Google Scholar] [CrossRef] [PubMed]
- A Altarifi, A.; David, B.; Muchhala, K.H.; E Blough, B.; Akbarali, H.; Negus, S.S. Effects of acute and repeated treatment with the biased mu opioid receptor agonist TRV130 (oliceridine) on measures of antinociception, gastrointestinal function, and abuse liability in rodents. J. Psychopharmacol. 2017, 31, 730–739. [Google Scholar] [CrossRef]
- Liang, D.Y.; Li, W.W.; Nwaneshiudu, C.; Irvine, K.A.; Clark, J.D. Pharmacological Characters of Oliceridine, a mu-Opioid Receptor G-Protein[FIGURE DASH]Biased Ligand in Mice. Anesth Analg. 2018 Jul 21.
- Soergel DG, Subach RA, Burnham N, Lark MW, James IE, Sadler BM; et al. Biased agonism of the mu-opioid receptor by TRV130 increases analgesia and reduces on-target adverse effects versus morphine: A randomized, double-blind, placebo-controlled, crossover study in healthy volunteers. Pain. 2014, 155, 1829–1835.
- Grim TW, Schmid CL, Stahl EL, Pantouli F, Ho JH, Acevedo-Canabal A; et al. A G protein signaling-biased agonist at the mu-opioid receptor reverses morphine tolerance while preventing morphine withdrawal. Neuropsychopharmacology. 2020, 45, 416–425.
- Quirion, B.; Bergeron, F.; Blais, V.; Gendron, L. The Delta-Opioid Receptor; a Target for the Treatment of Pain. Front. Mol. Neurosci. 2020, 13, 52. [Google Scholar] [CrossRef]
- Blaine, A.T.; van Rijn, R.M. Receptor expression and signaling properties in the brain, and structural ligand motifs that contribute to delta opioid receptor agonist-induced seizures. Neuropharmacology. 2023, 232, 109526. [Google Scholar] [CrossRef]
- Conibear AE, Asghar J, Hill R, Henderson G, Borbely E, Tekus V; et al. A Novel G Protein-Biased Agonist at the delta Opioid Receptor with Analgesic Efficacy in Models of Chronic Pain. J Pharmacol Exp Ther. 2020, 372, 224–236.
- PharmNovo, A. PharmNovo announces positive Phase I results for neuropathic pain management drug PN6047. 2024 [cited 2024-08-18]; Available from: https://www.mediconvillage.se/pharmnovo-announces-positive-phase-i-results-for-neuropathic-pain-management-drug-pn6047/.
- Cayir, S.; Zhornitsky, S.; Barzegary, A.; Sotomayor-Carreño, E.; Sarfo-Ansah, W.; Funaro, M.C.; Matuskey, D.; Angarita, G. A review of the kappa opioid receptor system in opioid use. Neurosci. Biobehav. Rev. 2024, 162, 105713. [Google Scholar] [CrossRef] [PubMed]
- Santino, F.; Gentilucci, L. Design of kappa-Opioid Receptor Agonists for the Development of Potential Treatments of Pain with Reduced Side Effects. Molecules. 2023, 28. [Google Scholar]
- Dalefield, M.L.; Scouller, B.; Bibi, R.; Kivell, B.M. The Kappa Opioid Receptor: A Promising Therapeutic Target for Multiple Pathologies. Front. Pharmacol. 2022, 13, 837671. [Google Scholar] [CrossRef] [PubMed]
- Mores, K.L.; Cummins, B.R.; Cassell, R.J.; van Rijn, R.M. A Review of the Therapeutic Potential of Recently Developed G Protein-Biased Kappa Agonists. Front. Pharmacol. 2019, 10, 407. [Google Scholar] [CrossRef] [PubMed]
- Valentino, R.J.; Volkow, N.D. Untangling the complexity of opioid receptor function. Neuropsychopharmacology 2018, 43, 2514–2520. [Google Scholar] [CrossRef] [PubMed]
- Meyrath M, Szpakowska M, Zeiner J, Massotte L, Merz MP, Benkel T; et al. The atypical chemokine receptor ACKR3/CXCR7 is a broad-spectrum scavenger for opioid peptides. Nat Commun. 2020, 11, 3033.
- Gach-Janczak, K.; Biernat, M.; Kuczer, M.; Adamska-Bartłomiejczyk, A.; Kluczyk, A. Analgesic Peptides: From Natural Diversity to Rational Design. Molecules 2024, 29, 1544. [Google Scholar] [CrossRef]
- Palmer, C.B.; Meyrath, M.; Canals, M.; Kostenis, E.; Chevigné, A.; Szpakowska, M. Atypical opioid receptors: Unconventional biology and therapeutic opportunities. Pharmacol. Ther. 2021, 233, 108014. [Google Scholar] [CrossRef]
- O’Neill, A.; Lirk, P. Multimodal Analgesia. Anesthesiol Clin. 2022 Sep;40(3):455-68.
- Young, A.; Buvanendran, A. Recent Advances in Multimodal Analgesia. Anesthesiol. Clin. 2012, 30, 91–100. [Google Scholar] [CrossRef]
- Giummarra, M.J.; Gibson, S.J.; Allen, A.R.; Pichler, A.S.; Arnold, C.A. Polypharmacy and Chronic Pain: Harm Exposure Is Not All about the Opioids. Pain Med. 2015, 16, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, R.S.; Arumugam, S.; Lau, C.S. Use of preoperative gabapentin significantly reduces postoperative opioid consumption: A meta-analysis. J. Pain Res. 2016, ume 9, 631–640. [Google Scholar] [CrossRef]
- Pergolizzi, J.; de Laar, v.; Langford, R.; Mellinghoff, H.-U.; Merchante, M.; Nalamachu, S.; O’Brien, J.; Perrot, S.; Raffa, R.B. Tramadol/paracetamol fixed-dose combination in the treatment of moderate to severe pain. J. Pain Res. 2012, 5, 327–346. [Google Scholar] [CrossRef] [PubMed]
- Pergolizzi, J.; Varrassi, G.; LeQuang, J.A.K.; Breve, F.; Magnusson, P. Fixed Dose Versus Loose Dose: Analgesic Combinations. Cureus 2023, 15, e33320. [Google Scholar] [CrossRef]
- Frantz, S. The trouble with making combination drugs. Nat Rev Drug Discov. 2006, 5, 881–882. [Google Scholar] [PubMed]
- Gascon, N.; Almansa, C.; Merlos, M.; Vela, J.M.; Encina, G.; Morte, A.; Smith, K.; Plata-Salamán, C. Co-crystal of tramadol-celecoxib: Preclinical and clinical evaluation of a novel analgesic. Expert Opin. Investig. Drugs 2019, 28, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Caputi, F.F.; Nicora, M.; Simeone, R.; Candeletti, S.; Romualdi, P. Tapentadol: An analgesic that differs from classic opioids due to its noradrenergic mechanism of action. Minerva Medica 2019, 110, 62–78. [Google Scholar] [CrossRef] [PubMed]
- Barakat, A. Revisiting Tramadol: A Multi-Modal Agent for Pain Management. CNS Drugs 2019, 33, 481–501. [Google Scholar] [CrossRef]
- Cannella, N.; Lunerti, V.; Shen, Q.; Li, H.; Benvenuti, F.; Soverchia, L.; Narendran, R.; Weiss, F.; Ciccocioppo, R. Cebranopadol, a novel long-acting opioid agonist with low abuse liability, to treat opioid use disorder: Preclinical evidence of efficacy. Neuropharmacology 2024, 257, 110048. [Google Scholar] [CrossRef]
- Kiguchi, N.; Ding, H.; Ko, M.-C. Author response for “Therapeutic potentials of NOP and MOP receptor coactivation for the treatment of pain and opioid abuse”. 2020. [CrossRef]
- Sanam, M.; Ashraf, S.; Saeed, M.; Khalid, A.; Abdalla, A.N.; Qureshi, U.; Ul-Haq, Z. Cebranopadol: An Assessment for Its Biased Activation Potential at the Mu Opioid Receptor by DFT, Molecular Docking and Molecular Dynamic Simulation Studies. ChemistrySelect 2023, 8. [Google Scholar] [CrossRef]
- Kleczkowska, P.; Lipkowski, A.W.; Tourwe, D.; Ballet, S. Hybrid Opioid/Non-Opioid Ligands in Pain Research. Curr. Pharm. Des. 2014, 19, 7435–7450. [Google Scholar] [CrossRef] [PubMed]
- Kenakin, T. Biased Receptor Signaling in Drug Discovery. Pharmacol. Rev. 2019, 71, 267–315. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Maudsley, S.; Gesty-Palmer, D. Translating in vitro ligand bias into in vivo efficacy. Cell. Signal. 2018, 41, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.S.; Sundrum, T.; Hay, D.L. PACAP receptor pharmacology and agonist bias: Analysis in primary neurons and glia from the trigeminal ganglia and transfected cells. Br. J. Pharmacol. 2014, 171, 1521–1533. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, A.T.; Semache, M.; Gross, F.; Da Fonte, D.F.; Runtz, L.; Colley, C.; Mezni, A.; Le Gouill, C.; Lukasheva, V.; Hogue, M.; et al. Biased Signaling of the Mu Opioid Receptor Revealed in Native Neurons. iScience 2019, 14, 47–57. [Google Scholar] [CrossRef]
- Rivero G, Llorente J, McPherson J, Cooke A, Mundell SJ, McArdle CA; et al. Endomorphin-2: A biased agonist at the mu-opioid receptor. Mol Pharmacol. 2012, 82, 178–188.
- Kelly, E. Efficacy and ligand bias at the mu-opioid receptor. Br J Pharmacol. 2013, 169, 1430–1446. [Google Scholar] [CrossRef]
- Pedersen MF, Wrobel TM, Marcher-Rorsted E, Pedersen DS, Moller TC, Gabriele F; et al. Biased agonism of clinically approved mu-opioid receptor agonists and TRV130 is not controlled by binding and signaling kinetics. Neuropharmacology. 2019, 107718.


| Compound | Structure | Biased toward1 | Contradictory literature |
|---|---|---|---|
| Morphine | 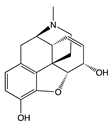 |
β-arrestins [55,68,147] |
No bias [101,148,149] |
| Fentanyl | 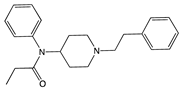 |
β-arrestins [55,95] |
No bias [96] |
| Buprenorphine | 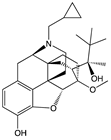 |
G protein [101,117,147,150] |
|
| Levorphanol | 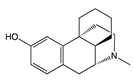 |
G protein [64] |
|
| Oliceridine | 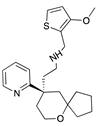 |
G protein [67,68] |
Tendency towards G protein [101] |
| PZM21 | 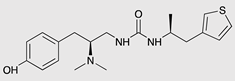 |
G protein [90,93] |
Tendency towards G protein [101] |
| SR-17018 | 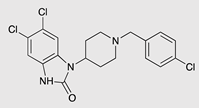 |
G protein [55] |
Tendency towards G protein [101] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



