1. Introduction
After several decades of intensive research, antisense oligonucleotides (ASOs) have emerged as a next-generation therapeutic strategy for a range of diseases, including neurodegenerative disorders, cancer, and rare genetic conditions. ASOs are short, synthetic molecules, typically 15-30 nucleotides in length, designed to modulate gene expression [
1]. They function by hybridizing to a target mRNA strand through Watson-Crick base pairing, leading to one of two primary mechanisms of action: RNase H-mediated degradation or splice switching. In the first mechanism, the ASO-mRNA duplex is recognized by the enzyme RNase H1, which directs the mRNA for degradation, thereby reducing the production of a disease-causing protein [
2]. Alternatively, splice-switching oligonucleotides (SSOs) are designed to bind to splicing regulatory regions in pre-mRNA, creating a steric block of splicing factors that influences exon inclusion or exclusion [
3]. This mechanism can restore the reading frame of a protein which has been disrupted by a mutation. Currently five SSOs have gained FDA approval, including Nusinersen for the treatment of spinal muscular atrophy, as well as Eteplirsen, Golodirsen, Viltolarsen, and Casimersen for duchenne muscular dystrophy [
4]. A major advantage of ASOs lies in their ability to target traditionally "undruggable" proteins. While only around 2% of the human proteome is accessible to small molecule drugs, ASOs can be designed to target virtually any gene, requiring only the mRNA target sequence [
5]. This versatility has fueled significant investment in ASO research and commercialization.
Like any therapeutic innovation, ASOs face certain limitations. Oral administration is inefficient, necessitating alternative delivery methods such as subcutaneous injection or intravenous infusion [
6]. After injection, up to 40% of ASOs tend to accumulate in the liver, while another significant portion are rapidly filtered by the kidneys and excreted in the urine [
7,
8]. Due to their nucleic acid-like structure, ASOs are also susceptible to degradation by nuclease enzymes, both in the circulation and following cellular entry [
9]. Direct delivery to target tissues, such as through intravitreal or intrathecal injections, can mitigate some of these challenges, though these routes often carry additional risks and potential adverse effects. To address these challenges, extensive research has focused on modifying the ASO backbone, 2’ sugar, and nucleobases, as well as exploring alternative oligonucleotide chemistries [
10]. These structural modifications have significantly enhanced ASOs' resistance to nuclease degradation, improved their solubility, and increased their overall therapeutic efficacy.
Despite these advances, due to the mechanism of ASO uptake, several key limitations persist. ASOs are large, negatively charged molecules, which prevents them from passively diffusing across the cell membrane. Although the precise mechanisms of ASO internalization are not fully understood, endocytosis is known to play a major role. After binding to cell surface proteins, ASOs are typically internalized through clathrin- or caveolin-mediated endocytosis [
11]. These pathways are commonly referenced in ASO research but are by no means exclusive. Studies using clathrin and caveolin inhibitors have demonstrated minimal impact on ASO internalization, suggesting that other mechanisms are also involved [
12]. Additional pathways, such as macropinocytosis and the CLIC/GEEC pathway, have been reported to contribute to ASO uptake.
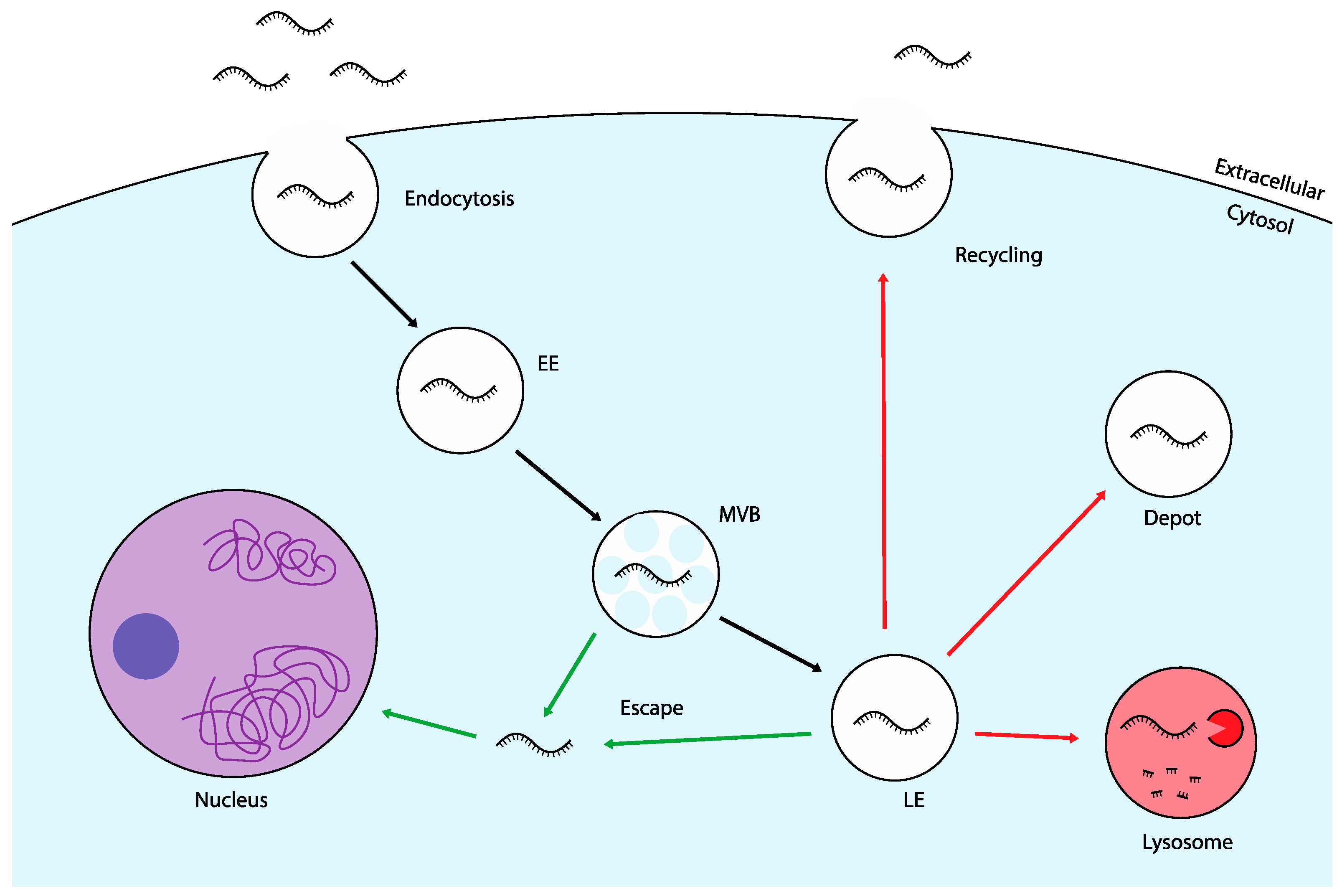
Once internalized, ASOs are encapsulated in membrane-bound structures called endosomes. These early endosomes (EEs) undergo a maturation process into multivesicular bodies (MVBs) and late endosomes (LEs), from which several intracellular trafficking routes can occur. One common pathway involves the fusion of LEs with lysosomes, which serve as the cell's primary degradation centers [
13]. Other pathways include transport to autophagosomes or the Golgi apparatus, both of which have been implicated in ASO trafficking [
14,
15]. Alternatively, ASOs can be directed to recycling endosomes, which return to the cell surface, or retained in stable structures known as "depot" endosomes, leading to long-term cellular storage [
16,
17] (
Figure 1). The major challenge across all trafficking routes is the retention of ASOs within membrane-bound vesicles. Whether degraded in lysosomes, trapped in endosomes, or recycled back out of the cell, ASOs are largely prevented from reaching their target mRNA and exerting a therapeutic effect [
18]. This is known as the "endosomal escape problem," a major barrier in ASO delivery. Among various parameters affecting ASO efficacy, endosomal escape has been shown to correlate most strongly with transfection efficiency, underscoring its critical role in successful delivery [
19]. In fact, studies suggest that only 1-2% of exogenously administered oligonucleotides manage to escape endosomal entrapment and reach the cytosol [
20]. Endosomal escape, while a significant bottleneck, is not the final hurdle in ASO delivery. After escaping endosomal entrapment, ASOs freely diffuse through the cytoplasm with only a portion successfully reaching the nucleus for internalization [
21]. Particularly for SSOs, which modulate pre-mRNA splicing, nuclear accumulation is critical for therapeutic efficacy.
Although these delivery barriers have long been recognized, overcoming them remains a significant challenge. Given the clinical success of approved ASOs, even small improvements in endosomal escape or nuclear localization could lead to significant gains in therapeutic efficacy. This review aims to provide an overview of the current understanding of these processes, alongside innovative approaches that could pave the way for improved ASO delivery systems. Continued exploration in these areas holds the promise of overcoming existing limitations, ultimately advancing the development of ASO-based therapies and expanding their applicability in treating a broader range of conditions.
2. Endosomal Escape and Nuclear Localization of ASOs
It is evident that some natural mechanisms of endosomal escape must occur, as freely delivered ASOs can reach their intracellular targets exerting a therapeutic effect. However, the precise processes by which ASOs naturally escape from endosomes and localize to the nucleus remain poorly understood. One theory suggests that biological membranes, including endosomal membranes, undergo spontaneous fluctuations [
22]. These fluctuations may create temporary openings in the membrane, allowing ASOs to escape. The random and transient nature of these events, with rapid resealing of the membrane, could explain why only a small fraction of ASOs successfully escape endosomal entrapment.
Another proposed mechanism for endosomal escape involves membrane fusion events. During endosomal maturation MVBs form, consisting of numerous intraluminal vesicles (ILVs) [
23]. The process of ILV formation requires the endosomal membrane to pinch off, creating an opportunity for the intraluminal contents, including ASOs, to escape. Additionally, these ILVs can undergo a process known as retrofusion, in which they re-fuse with the limiting membrane of the MVB [
24]. These fusion events that occur during the formation and maturation of MVBs may significantly contribute to the release of ASOs. This hypothesis is supported by data indicating that the majority of endocytosed RNA therapeutics are released from intermediate endosomal compartments like MVBs [
20,
25]. There is a general consensus that early escape in the trafficking process is crucial for effective cytosolic release, while ASOs that are retained are likely to be degraded in lysosomes. However, visualizing such rapid escape events at the nanometer scale is technically challenging, resulting in limited empirical evidence to support these hypotheses. Consequently, the exact location and mechanisms of ASO escape remain unresolved questions.
Recent evidence highlights the importance of endosome-Golgi transport pathways in facilitating endosomal escape. For instance, studies have shown that following the administration of ASOs, COPII-coated vesicles, which are typically involved in the anterograde transport of proteins from the endoplasmic reticulum (ER) to the Golgi apparatus, exhibit a significant association with ASO-containing LEs [
26]. Moreover, the knockdown of COPII tethering proteins has been shown to impair endosomal escape, supporting the hypothesis that the binding of these vesicles to endosomes could enhance the release of ASOs. Further investigations have identified Golgi-derived vesicles containing the mannose-6-phosphate receptor (M6PR) as crucial players in this process [
27]. M6PR vesicles are primarily responsible for transporting hydrolase enzymes to the endo-lysosomal system; it is posited that fusion events between these vesicles and the endosomal membrane may facilitate ASO escape.
The few ASOs that successfully escape endosomal entrapment disperse throughout the cell before encountering another barrier: the nuclear membrane. Some oligonucleotides have been observed to freely diffuse into the nucleus, likely leveraging their small size to pass through the nuclear pore complex [
28,
29]. However, nuclear import is a highly regulated process influenced by factors such as ASO size, charge, and secondary structure. For instance, decreasing temperature has been shown to inhibit the nuclear transport of phosphorothioate (PS)-modified ASOs more significantly than their unmodified counterparts [
30]. This finding suggests that the modified ASOs may rely on active transport mechanisms to traverse the nuclear pore complex. Other exogenous RNA molecules have been reported to enter the nucleus via interactions with nuclear shuttle proteins, such as importins, and a similar active transport mechanism may be utilized by ASOs [
31].
While the potential for ASOs to effectively reach their intracellular targets is evident, the precise mechanisms underlying their natural escape from endosomes and subsequent localization to the nucleus remain poorly understood. In reality, a combination of different mechanisms may be at play, depending on the specific cell type and cargo involved. Given the complexity and variability of these processes, continued research is essential to elucidate the multifaceted mechanisms that govern ASO trafficking and localization.
3. Traditional Endosomal Escape Strategies
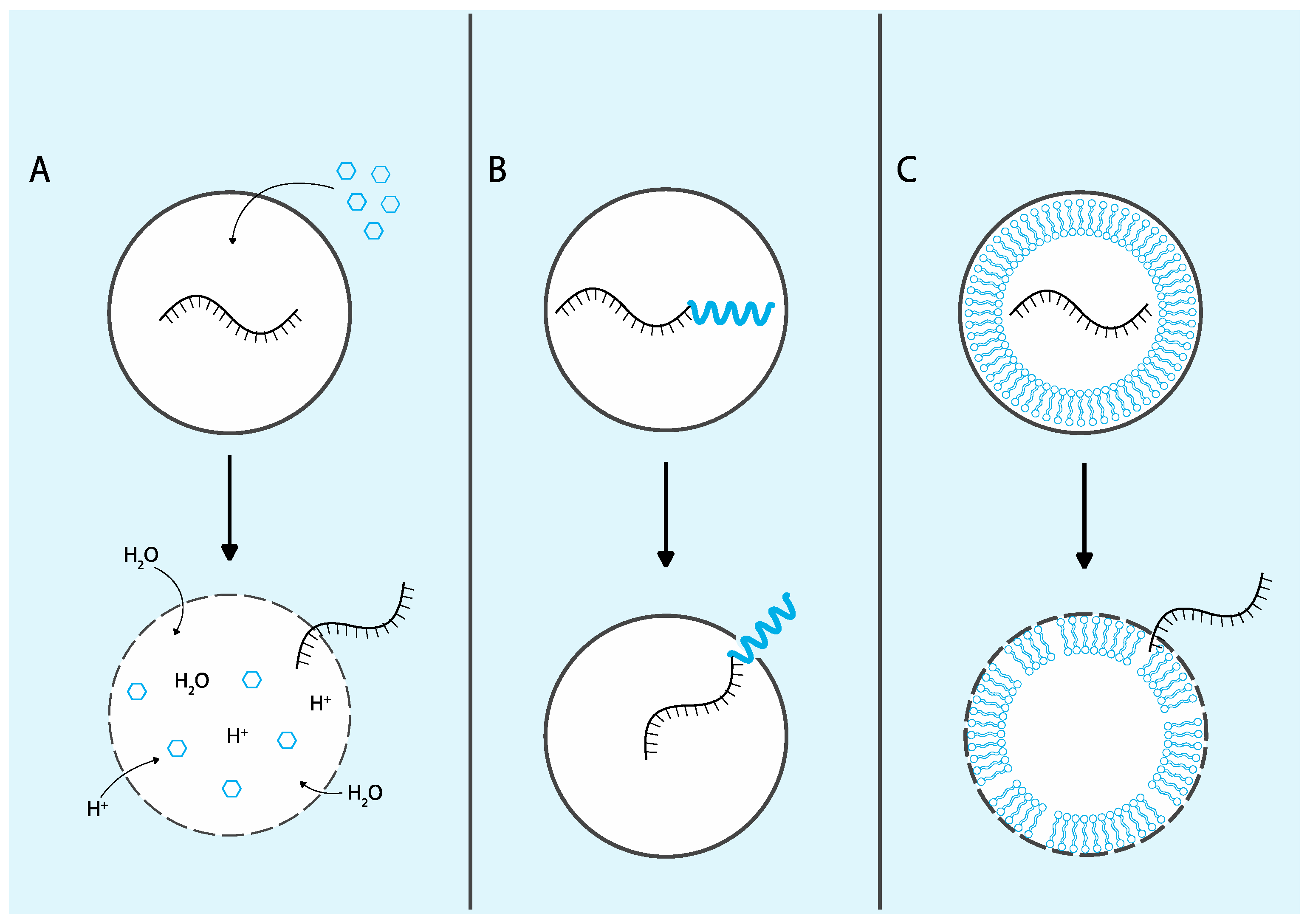
Given the significant challenge that endosomal escape presents for the delivery of ASOs and other biologic therapeutics, extensive research has focused on strategies to enhance this critical process. Many delivery approaches exploit materials that respond to endogenous or exogenous stimuli to trigger endosomal escape events. Endosomal compartments experience a stepwise reduction in pH, with values decreasing from approximately 6.5 in early endosomes (EEs) to around 4.5 in lysosomes [
32]. Many strategies leverage this characteristic by utilizing pH-responsive materials that remain inert in the extracellular environment (pH 7.0) but activate their escape mechanisms specifically within endosomal compartments. In addition to pH sensitivity, endosomal escape can also be facilitated by exogenous stimuli such as light, magnetic fields, or ultrasound, which can induce membrane destabilization [
33]. This section will review the mechanisms and advancements of these traditional endosomal escape strategies, including endosomal-disrupting small molecules, proteins and peptides, and non-viral delivery vehicles (
Figure 2).
3.1. Endosomal Disrupting Small Molecules
Small molecules offer significant advantages over other biologic delivery enhancers in terms of solubility, stability, and the potential for oral administration. As a result, there has been considerable interest in identifying small molecule endosomal escape enhancers. One notable example is the antimalarial drug chloroquine. At cytosolic pH levels, chloroquine readily enters cells, but it becomes protonated in the acidic environment of endosomes due to its weakly basic properties [
34]. This protonation increases osmotic pressure and destabilizes the endosomal membrane, a phenomenon known as the proton sponge effect, also referred to as the pH buffering effect, osmotic swelling, or osmotic lysis. The underlying mechanism of the proton sponge effect is linked to the activity of the V-ATPase, a membrane proton pump in endo/lysosomes which maintains the intraluminal pH at a specific level [
35]. When pH buffering molecules enter, the V-ATPase compensates by pumping more protons into the vesicle. This influx of protons is accompanied by the entry of chloride ions and water, significantly increasing osmotic pressure and creating openings in the membrane for escape [
36]. These characteristics have made chloroquine a prototypical small molecule endosomal escape enhancer. Other cationic amphiphilic drugs (CADs), such as siramesine and amitriptyline, have also been shown to enhance the delivery of RNA therapeutics, presumably through a similar mechanism [
37]. However, a significant drawback of these CADs is that the resulting endosomal membrane rupture can induce unacceptable levels of cellular stress.
Toxicity limitations have not deterred further exploration of potential small molecule endosomal escape enhancers. Researchers at the University of North Carolina conducted a screening of small molecules from their compound library of UNC molecules for the ability to enhance ASO delivery [
38]. Notably, one compound, UNC7938, was identified as significantly increasing ASO activity by promoting the release of ASOs from endosomal compartments. Follow-up studies confirmed that UNC7938 also enhances the in vivo efficacy of an exon-skipping ASO in the Mdx mouse model [
39]. The mechanism of action for UNC7938 involves destabilizing the endosomal membrane, differentiating it from CADs like chloroquine, which primarily rely on increasing osmotic pressure [
40]. While UNC7938 has demonstrated promise, safety assessments have yielded mixed results; some studies reported a 90% cell death rate in vitro, whereas no evidence of liver or kidney toxicity was observed in vivo [
41,
39]. In contrast, the less potent small molecule UNC4267 has also been shown to enhance endosomal escape with fewer toxic effects [
42]. However, this reduced toxicity correlates with decreased membrane disruption and, consequently, less effective endosomal escape. It appears that enhancing endosomal escape with such drugs necessitates a certain degree of toxicity, largely relegating their application to in vitro transfection enhancers.
3.2. Proteins and Peptides
Early research into protein-mediated endosomal escape primarily focused on endolytic peptides that induce membrane destabilization in response to the low pH of endosomes, facilitating escape [
43]. However, these treatments encounter similar toxicity limitations as those seen with endolytic small molecules. To mitigate toxicity concerns, alternative peptide-based strategies have been developed. This section will review two promising peptide platforms: biomimetics and cell-penetrating peptides (CPPs), which can effectively target endosomal membranes and promote the release of therapeutic payloads into the cytosol.
3.2.1. Biomimetics
To enhance endosomal escape, some of the most efficient models are found in bacterial toxins and viruses, which enter cells via endocytosis but require escape from endosomes to exert their effects. Given the few billion years that bacteria and viruses have had to evolve such mechanisms, their high efficiency is not surprising. These proteins serve as the gold standard for designing endosomal escape peptides.
One notable mechanism utilized by bacterial toxins is the action of pore-forming proteins. In the acidic environment of endosomes, bacterial toxins like Listeriolysin O (LLO) and Perfringolysin O (PFO) can create pores that facilitate escape into the cytosol [
44]. Researchers have explored models based on these proteins, demonstrating that mutant forms of LLO significantly enhance the endosomal escape of gold nanoparticles [
45]. While these proteins are effective at perforating endosomes, they can also form pores in the plasma membrane, potentially causing significant cellular damage. To mitigate such negative effects, innovative strategies like co-administration with toxin-neutralizing antibodies have been investigated [
46]. This approach employed a PFO-antibody conjugate to prevent toxin activity in the cytoplasm, allowing PFO to be released only after endocytosis, increasing the therapeutic window five-fold.
Other toxin-mimetics, such as modified diphtheria, shiga, and botulinum toxins, have been engineered for intracellular delivery with various strategies to reduce toxicity [47-49]. Toxin proteins are often composed of functional domains, each with specific roles such as toxicity or membrane fusion. By selectively targeting certain domains, researchers can achieve their desired effects. For example, in an attenuated diphtheria toxin designed for siRNA delivery, only the active domain responsible for toxicity was modified [
47]. This approach preserved effective uptake and endosomal escape by retaining the translocation and receptor binding domains while significantly reducing toxicity. Alternatively, toxic domains can be completely removed. Park et al. focused solely on the endosomal escape domain of botulinum neurotoxin and conjugated it to their protein delivery system [
49]. This strategy resulted in a 100-fold increase in cytosolic delivery, attributed to enhanced endosomal escape.
Enveloped viruses, such as influenza A, utilize a membrane fusion mechanism for endosomal escape. This process is mediated by the hemagglutinin (HA) glycoprotein on the viral surface [
50]. The acidic pH of endosomes triggers a conformational change in HA, initiating fusion with the endosomal membrane [
51]. In contrast to the approximately 1% escape rate observed for ASO therapeutics, HA has been estimated to achieve an escape efficiency in the range of 30-70% [
17]. Given the impressive escape capabilities of such proteins, researchers have begun exploring biomimetic versions of HA. Dr. Steven Dowdy’s lab is currently developing a synthetic HA-like delivery system aimed at creating a universal endosomal escape domain (uEED) [
52]. This system utilizes a hydrophilic outer domain to mask the internal membrane fusion domain. Upon endosomal entry, the low pH environment prompts the shedding of the masking domain, allowing for endosomal escape. A significant advantage of this approach is that synthetic proteins do not elicit the same immunogenic response as natural viral proteins. Moreover, promoting fusion with the endosomal membrane reduces the risk of membrane rupture, thus limiting toxicity.
Overall, proteins derived from bacterial toxins and viruses demonstrate remarkable efficiency in endosomal escape. However, the toxicity associated with pore formation and potential adverse immune responses remain significant challenges for clinical drug delivery applications. Ongoing research continues to develop strategies aimed at minimizing these negative side effects.
3.2.2. Cell Penetrating Peptides
Cell-penetrating peptides (CPPs) are short peptide sequences that, as their name suggests, can be readily internalized by cells. Most CPPs are rich in positively charged amino acids, such as arginine and lysine, which enhance their interaction with the negatively charged plasma membrane [
53]. While the positive charge of CPPs facilitates cellular uptake, it can complicate ASO delivery due to the negative charge of ASOs, leading to the formation of CPP-ASO aggregates. Therefore, this strategy is more commonly used for neutrally charged ASOs, such as phosphorodiamidate morpholino oligonucleotides (PMOs). Indeed, CPP-PMO conjugates have been shown to significantly enhance ASO therapeutic efficacy both in vitro and in vivo [
54].
Despite their ability to improve the delivery of therapeutics, the cellular uptake process of CPPs still predominantly relies on endocytosis, resulting in endosomal entrapment. Therefore, additional strategies are necessary to promote endosomal escape for these peptides. A key feature in designing CPPs for endosomal escape involves leveraging the natural pH gradient found within the endosomal pathway. Such peptides can exist as random coils at physiological pH but adopt an alpha-helical structure in the acidic environment of endosomes, facilitating membrane penetration [
55]. Other factors, including the histidine-arginine ratio, hydrophobicity, and helicity, are also crucial determinants of a peptide’s escape ability [56-58]. One of the most well studied CPPs is TAT, a positively charged protein derived from the HIV virus [
59]. Notably, the protein transduction domain (PTD) of TAT is recognized for its strong ability to facilitate cell entry. A dimerized form of TAT (dfTAT), linked by a disulfide bond, is believed to significantly enhance endosomal escape by disrupting the endosomal membrane [
60].
Non-canonical cyclic CPPs have also emerged as promising endosomal escape agents. For instance, cFΦR4, a cyclic variant of an arginine-rich CPP, has been shown to significantly enhance cytosolic delivery compared to its linear counterparts [
61]. Importantly, these cyclic peptides were found to escape from early endosomal compartments, limiting potential degradation of their cargo by enzymes. Other studies on cyclic CPPs confirmed their capacity for high cytosolic delivery, although there was no evidence of endosomal rupture, suggesting that an alternative mechanism to membrane disruption is at play [
62]. Using model membrane systems and confocal microscopy, researchers visualized the formation of small, distinct budding vesicles. This led to a proposed mechanism whereby protein-membrane interactions induce membrane curvature and the creation of miniature vesicles following endocytosis. These small vesicles, once formed, are unstable; upon release, a loss of their pH gradient causes them to collapse and release their contents into the cytosol. Subsequent studies corroborated the vesicle budding and collapse hypothesis in other CPPs and the diphtheria toxin [
63,
64]. Such peptides improved cytosolic delivery by up to 120% compared to the mere 2% observed with traditional CPPs like TAT. While cyclic CPPs have been previously studied for protein delivery, they have recently been validated with oligonucleotides, enhancing the efficacy of SSOs administered to mouse models of two neuromuscular disorders: duchenne muscular dystrophy and facioscapulohumeral muscular dystrophy [
65].
While CPPs certainly increase ASO efficacy, whether this enhancement arises primarily from improved endosomal escape or through other mechanisms remains a complex question. In addition to endocytosis, CPPs may also enter cells via direct translocation through the plasma membrane [
66]. Reports indicate that CPPs, traditionally thought to enhance endosomal escape, show negligible cytosolic delivery when this direct translocation pathway is inhibited by membrane depolarization [
67]. This finding suggests that endocytosis followed by endosomal escape may not be the primary mechanism for ASO delivery via CPPs. Further studies employing a Split Luciferase Endosomal Escape Quantification (SLEEQ) assay have indicated that endosomal escape is not significantly enhanced using CPPs [
68]. One hypothesis for these findings is that CPPs may merely facilitate the endocytosis of ASOs without enhancing their escape from endosomes, meaning the delivery benefits of CPPs could stem simply from increasing cellular uptake. Additionally, a portion of peptides capable of directly translocating the plasma membrane could be responsible for the increased therapeutic efficacy. Ultimately the evidence is quite mixed on the specific uptake pathways of CPPs and how they improve delivery. The precise mechanisms of delivery may vary based on the specific CPP in question and the cargo it is delivering.
3.3. Non-viral Delivery Vehicles
An increasingly popular strategy to enhance endosomal escape involves the use of non-viral delivery vehicles. Unlike viral delivery systems, such as AAV9, which are frequently employed for transgenic applications, non-viral delivery vehicles offer a promising alternative. Viruses naturally possess endosomal escape mechanisms that facilitate the delivery of genetic material to the cytosol, making them a logical choice for therapeutic delivery. However, concerns regarding toxicity and immunogenicity associated with viral vectors have limited their practical applications [
69]. Considering these limitations, research has shifted significantly toward non-viral methods. Non-viral delivery vehicles can improve the stability and cellular uptake of ASOs while also being engineered for enhanced endosomal escape [
70]. This section will explore several innovative non-viral delivery formulations specifically engineered to enhance endosomal escape, focusing on lipid-based systems and exogenous stimuli-responsive vehicles.
3.3.1. Lipid Delivery Vehicles
Lipid nanoparticles (LNPs) are widely used for delivery of nucleic acids such as in mRNA vaccines, but they still face the challenge of poor cytosolic delivery, like freely delivered ASOs [
20]. To address this limitation, researchers have focused on modifying LNP systems to improve endosomal escape. A key approach involves the use of cationic lipids, which enhances cellular uptake and facilitates interactions with the negatively charged endosomal membrane [
71]. This interaction triggers a membrane fusion mechanism in which the lipids transition from a lamellar to an inverted hexagonal phase, promoting fusion with the endosomal membrane and enabling cargo release into the cytosol [
72]. However, cationic lipids also pose toxicity risks, potentially initiating apoptotic and inflammatory pathways [
73].
As a result, much of the research has shifted toward using ionizable LNPs. These contain lipid head groups that remain neutral at physiological pH but become protonated in the acidic environment of the endosome. This property allows them to stay inert during delivery and cellular uptake, but actively associate with the endosomal membrane to promote escape once inside the endosome. Nanoparticles composed of ionizable lipids such as DLin-MC3-DMA and ALC-0315 have demonstrated increased transfection efficacy, likely due to enhanced endosomal escape [
74]. This type of system can reduce the cytotoxicity seen with traditional cationic lipids while retaining high delivery efficiency.
Alternatively, anionic LNPs offer a lower toxicity option, however these formulations face challenges in encapsulating negatively charged ASOs due to electrostatic repulsion. To improve encapsulation, cations such as calcium can be incorporated during liposome formation, resulting in the generation of calcium phosphate nanoparticles that reduce nucleic acid repulsion [
75]. Calcium-phosphate anionic liposomes loaded with siRNA have demonstrated more effective endosomal escape than the cationic transfection reagent lipofectamine 6000 [
76]. Perhaps more importantly, toxicity was significantly reduced with this anionic delivery formulation compared with the cationic lipofectamine during longer transfection durations.
Lipid-based delivery vehicles remain a promising approach for enhancing the delivery of RNA therapeutics by improving stability, uptake, and endosomal escape. In addition to cationic, anionic, or ionizable lipids, helper lipids such as cholesterol, phospholipids, and PEGylated lipids play an important role in improving escape efficiency [
77]. These helpers influence the positioning of nanoparticle lipids, promoting more effective interactions with cellular membranes and facilitating endosomal escape [
78]. Ongoing research into optimizing LNP composition holds potential to further enhance delivery efficacy while minimizing associated toxicity.
3.3.3. Exogenous Stimuli Responsive Delivery Vehicles
In addition to endogenous factors like pH, endosomal escape can be triggered by external stimuli such as light, magnetism, and ultrasound, offering a more controlled approach to enhance the delivery of therapeutics. One of the most well-studied methods, photochemical internalization (PCI), uses photosensitive molecules that produce reactive oxygen species (ROS) upon exposure to light [
79]. Following endocytosis, ROS formation induces perforation of the endosomal membrane allowing for therapeutic release. Specifically, the formation of a highly reactive singlet oxygen which readily oxidizes biological materials, has been shown to be essential for this PCI mediated endosomal escape [
80].
A second light-activated method involves a photothermal mechanism, which utilizes photothermally responsive materials, such as gold nanoparticles (AuNPs), to achieve endosomal escape. When irradiated, AuNPs absorb light and generate localized heat, causing the formation of vapor nanobubbles in the surrounding fluid [
81]. These nanobubbles physically disrupt the endosomal membrane, facilitating the release of therapeutic cargo. Other photothermal materials such as melanin-poly-l-lysine polymers have also been employed to enhance the delivery of siRNA via this mechanism [
82].
Magnetic fields offer another strategy for inducing endosomal escape. Magnetic nanoparticles, typically composed of iron oxide, respond to an alternating magnetic field by generating heat or physically rotating [
83,
84]. This heating effect, similar to the photothermal mechanism, destabilizes the membrane and promotes the release of cargo. Alternatively, the mechanical disruption caused by nanoparticle rotation can also facilitate escape. This approach has been successfully applied in the delivery of siRNA using iron oxide nanocages [
85].
Ultrasound is another promising technique for enhancing endosomal escape, primarily through cavitation, a process in which ultrasound-responsive materials such as gas-filled microbubbles expand and collapse in response to focused ultrasound waves [
86]. The mechanical forces generated by this cavitation can disrupt the endosomal membrane, allowing for the release of therapeutic cargo. Co-delivery of DNA complexes with microbubbles has demonstrated increased transfection efficacy following ultrasound exposure [
87]. Other studies suggest that ultrasound alone, without the need for microbubbles, may be sufficient to permeabilize membranes and improve delivery efficiency, though the exact mechanisms remain under investigation [
88].
The use of exogenous stimuli, including light, magnetic fields, and ultrasound, provides a method for controlled, localized, and on-demand induction of endosomal escape for ASO delivery. Magnetic and ultrasonic approaches offer the advantage of deeper tissue penetration compared to light-based methods, which are limited to a few millimeters in biological tissues [
89]. Despite their potential, these strategies rely on the physical perturbation of the endosomal membrane, which can pose challenges related to tissue damage or off-target effects. Optimizing stimulation parameters is a key focus for making these non-invasive methods safer and more efficient for therapeutic delivery.
4. Alternative Strategies to the Endosomal Escape Problem
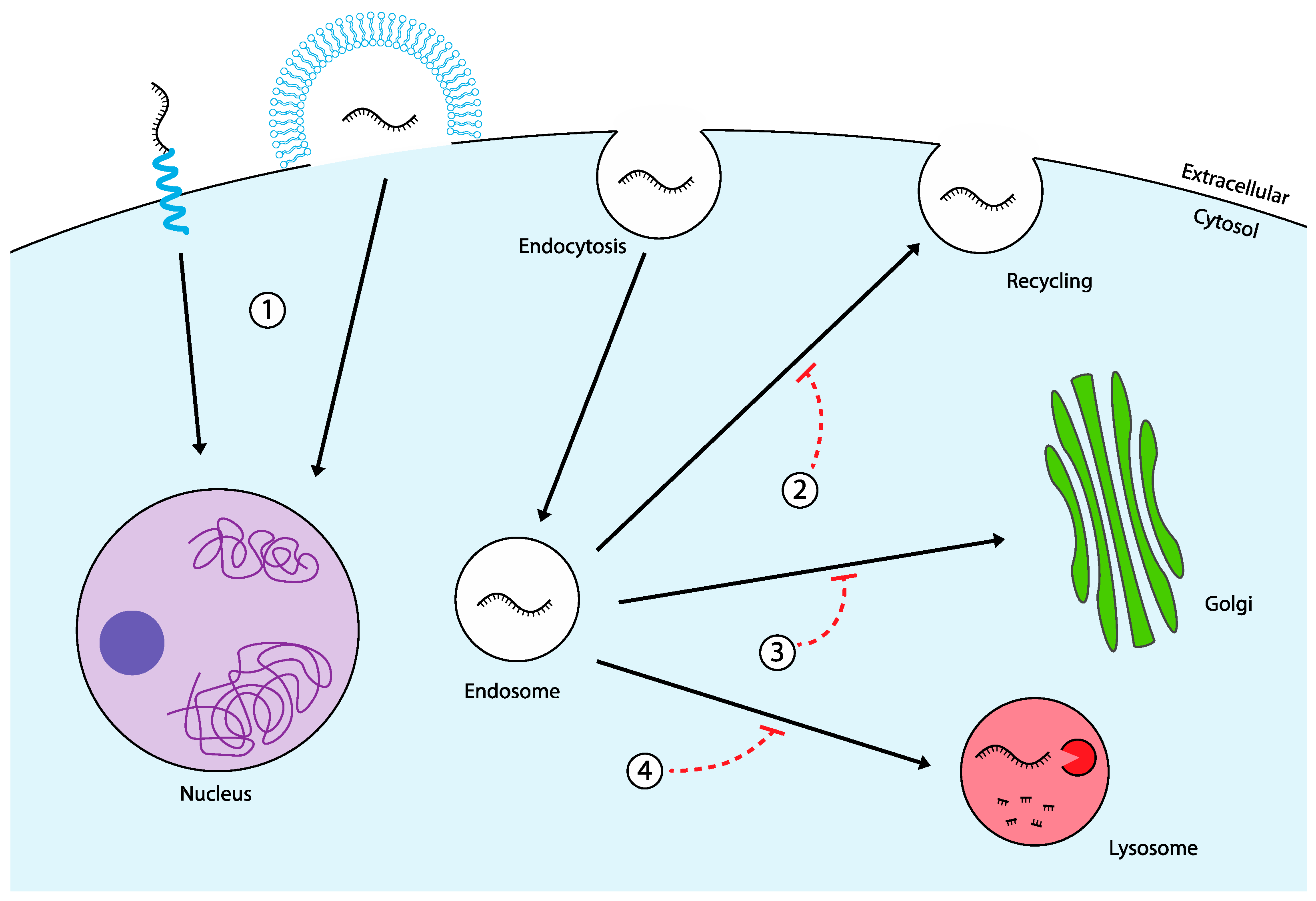
Extensive research has been dedicated to developing strategies for enhancing endosomal escape in ASO therapeutics. However, despite various advancements, no single approach has yet proven to be both highly effective and safe for widespread clinical application. This underscores the need for novel, innovative solutions to overcome the challenges of endosomal entrapment. In this section, we explore two promising alternative strategies: bypassing the endosomal pathway altogether via direct cytosolic delivery, and the targeted manipulation of intracellular trafficking pathways to reroute ASOs for more efficient delivery (
Figure 3). These approaches hold potential to sidestep the limitations of current endosomal escape methods while improving therapeutic efficacy and safety.
4.1. Direct Cytosolic Entry
A logical approach to preventing endosomal entrapment is to bypass the endo-lysosomal system entirely by delivering ASOs directly into the cytosol. While endocytosis is the dominant pathway for ASO cellular uptake, it is not the only route. One method to differentiate between endocytosis and membrane fusion is by conducting transfection experiments at varying temperatures. Since endocytosis is energy-dependent, lowering the temperature should inhibit this process. However, some lipid nanoparticles (LNPs) have been shown to enter cells even at 4°C, suggesting that these nanoparticles can fuse directly with the plasma membrane, releasing their cargo into the cytosol through an energy-independent mechanism [
90]. Similar results have been observed with siRNA delivered via lipoplexes, where approximately 5% of uptake occurs through endocytosis-independent pathways [
91]. Moreover, this study found inhibiting endocytosis does not significantly reduce siRNA activity, indicating that membrane translocation could be the primary route of productive delivery. Building on this insight, recent delivery formulations have been designed specifically to leverage plasma membrane fusion for enhanced cytosolic entry.
One approach to enhancing direct cytosolic delivery involves modifying LNPs with coiled-coil peptides that mimic the function of SNARE proteins. SNAREs are endogenous proteins responsible for promoting vesicle fusion with plasma membranes in various biological processes [
92]. Upon the binding of complementary SNARE proteins, spontaneous energy-independent membrane fusion occurs. This mechanism was mimicked to deliver mRNA using LNPs modified with the coiled-coil lipopeptide CPE4 [
93]. The CPE4 platform demonstrated enhanced gene delivery even in the presence of endocytosis inhibitors, suggesting direct fusion with the plasma membrane.
In addition to fusogenic proteins, peptides rich in arginine residues have been found to directly penetrate the plasma membrane. This capability is attributed to the presence of guanidinium groups, with guanidinium-modified CPPs having delivered ASOs into cells via direct translocation [
94]. The role of arginine in promoting cytosolic delivery has also been extended to various nanoparticle formulations. For example, quantum dot nanoparticles terminated with arginine have been demonstrated to cross the plasma membrane via direct translocation [
95]. Additionally, arginine-containing polymers can form membrane pores to deliver plasmid DNA (pDNA) directly into the cytosol [
96]. While some designs effectively leverage this mechanism for delivery, not all arginine-containing nanoparticles easily enter cells. Key factors such as nanoparticle size and hardness play critical roles in determining the specific uptake pathway [
97,
98].
4.2. Manipulation of Intracellular Trafficking
To explore potential enhancers of oligonucleotide delivery, numerous small molecule screenings have been conducted to assess their impact on ASO activity. While some small molecules improve ASO effectiveness by disrupting the endosomal membrane as previously discussed, other studies have identified small molecules that enhance ASO activity through different mechanisms, particularly by modifying intracellular trafficking pathways [
38,
98]. This suggests that targeting intracellular trafficking may offer an alternative approach to overcoming the challenges of endosomal escape.
Following endocytic uptake, one potential trafficking pathway involves the recycling of endosomes back to the plasma membrane, where their contents are expelled via exocytosis. One study found that up to 70% of siRNA delivered by LNPs is expelled through this recycling process, suggesting that inhibiting endosomal recycling might enhance delivery efficiency [
16]. Initial evidence for this approach focused on the protein Niemann-Pick type C1 (NPC-1), a key regulator of the endosomal recycling pathway. NPC-1 knockout cells have been shown to retain LNPs in endosomes for a longer duration [
16]. Moreover, inhibiting NPC-1 with the small molecule NP3.47 has been shown to significantly increase siRNA accumulation in endosomes, resulting in enhanced gene silencing [
100]. Although this strategy does not directly improve endosomal escape, retaining cargo inside the cell longer could increase the probability of escape events.
Recently, the synthetic sphingolipid analog SH-BC-893 was shown to increase the cytosolic delivery of ASOs from 3% to 10% when co-administered [
41]. SH-BC-893 inhibits the small GTPase ARF-6, which in its active form promotes the movement of endosomes to the plasma membrane, further supporting the idea that limiting endosomal recycling can improve delivery [
101]. Interestingly, SH-BC-893’s effect is not limited to recycling inhibition; it also prevents lysosomal fusion. This dual action, blocking both recycling and degradation pathways, provides a greater opportunity for ASOs to escape endosomes. Other studies have reported similar findings, with molecules like NH
4+ inhibiting lysosomal fusion and significantly boosting ASO activity [
102].
Small molecules like Retro-1, an inhibitor of endo-Golgi retrograde transport, have also been shown to significantly enhance the endosomal escape and activity of ASOs [
103]. Retro-1 was originally identified in a screen for compounds affecting the trafficking of bacterial toxins, many of which use the retrograde transport pathway to move from endosomes to the Golgi and eventually into the cytosol [
104]. Blocking this pathway was effective at inhibiting toxin effects, but how this inhibition enhances ASO escape remains unclear. If ASOs were transported through the Golgi-ER pathway on their way to the nucleus, one would expect Retro-1 to reduce their activity, not increase it. One hypothesis is that preventing retrograde transport retains ASOs in late endosomes, which may be more amenable to cytosolic escape. Another theory suggests that Retro-1 may directly act on late endosomes to promote ASO release, although no evidence of endosomal rupture or pH change has been observed, distinguishing it from small molecule endolytic agents [
103]. Interestingly, ASO-Retro-1 conjugates have shown much lower efficacy compared to ASOs administered with Retro-1 separately, suggesting that Retro-1 needs cytosolic access to reach its target effector [
105]. Adding to the complexity, different Retro-1 analogs have been found to selectively affect either ASOs or toxins, with only Retro-1 impacting both [
106]. This implies that, while similar, ASO and toxin trafficking mechanisms differ in key aspects highlighting the specificity of its action in ASO trafficking.
While these findings emphasize the significant role of intracellular trafficking in ASO delivery efficiency, targeting key peptide mediators in these pathways poses challenges. Many such proteins are not easily druggable, limiting the ability to specifically manipulate them [
107]. Furthermore, vesicle and protein interactions serve essential physiological functions that could be disrupted by chemical interventions. For instance, NPC-1 inhibition, although beneficial for prolonging ASO retention in endosomes, is not ideal therapeutically due to its association with lysosomal lipid storage disorders [
108]. Similarly, impaired lysosomal degradation is linked to numerous diseases, as the inability to clear cellular waste products can lead to harmful accumulations [
109]. It remains unclear whether temporary inhibition of these pathways via pharmacological agents would cause long-term adverse effects, but this remains an important concern. Examples of direct cytosolic delivery and intracellular trafficking manipulation have demonstrated promising results for ASO delivery. While these alternative strategies may not entirely solve the endosomal escape problem, when used in conjunction with traditional endosomal escape platforms, they could offer greater therapeutic potential.
5. Nuclear Localization of ASOs
While endosomal escape is often regarded as the major bottleneck in ASO delivery, it is not the final hurdle. For SSOs, nuclear localization is crucial to modulate splicing as pre-mRNA is located exclusively in the nucleus. Additionally, ASOs that function via RNase H1-mediated degradation are active in both the cytoplasm and nucleus, but achieving high nuclear levels is key for optimal gene silencing [
110]. Studies using subcellular fractionation show a direct correlation between the nuclear concentration of ASOs and the extent of mRNA knockdown. However, most ASOs remain in the cytosol after transfection [
111]. The cytoplasmic retention of ASOs exposes them to degradation by intracellular nucleases before interaction with target mRNA [
9]. This highlights an additional challenge in ASO delivery: promoting efficient nuclear localization following endosomal escape.
Numerous factors influence the nuclear localization of ASOs, particularly through ASO-protein interactions. One of the earliest structural modifications made to ASOs, the phosphorothioate (PS) backbone, significantly enhances their activity, potentially due to an increased binding affinity for cellular proteins that regulate various ASO functions [
112]. A key aspect of nuclear localization is the formation of ASO aggregates known as PS-bodies. These small, membraneless structures form in the nucleus after ASO delivery and are induced by interactions between ASOs and proteins, which act as nucleation sites for aggregation [
113]. Although the exact role of PS-bodies remains unclear, they are believed to serve as storage sites for ASOs, RNA, and associated proteins. Some studies suggest that PS-bodies do not significantly impact ASO activity, while others indicate they may play a crucial role in nuclear localization. For instance, Liang et al. demonstrated that the β subunit of chaperonin T-complex 1 (TCP-1β) associates with ASOs inside PS-bodies and enhances their activity [
114]. Additionally, the nuclear localization of ASOs and PS-bodies appears to depend on the function of Ran GTPase, indicating an active transport mechanism. Ultimately, while the significance of PS-body formation in the nuclear localization and activity of ASOs remains an open question, it could represent an important area for further research aimed at optimizing ASO therapeutic strategies.
Intracellular trafficking pathways also significantly influence the nuclear localization of ASOs. Research indicates that ASO activity is more pronounced in cell types with efficient trafficking to the nuclear region [
115]. In this context, the perinuclear localization of endosomes during transport has been closely associated with ASO efficacy. The migration of endosomes toward the nucleus, facilitated by transport along microtubules, is influenced by various endoplasmic reticulum (ER) tethering proteins, including RNF26, SQSTM1, and UBE2J1 [
116]. These interactions suggest that the connectivity between endosomes and other organelles may mediate effective nuclear transport. The localization of ASO-containing endosomes to perinuclear regions allows escaped ASOs to enter the nucleus more rapidly, thereby enhancing their functional capacity. Conversely, the accumulation of lysosomes in perinuclear regions has been linked to increased ASO degradation, which ultimately reduces their activity [
117]. This highlights the importance of not only the trafficking pathways themselves but also the spatial organization of organelles in regulating the nuclear localization of ASOs.
5.1. Nuclear Localization Signals
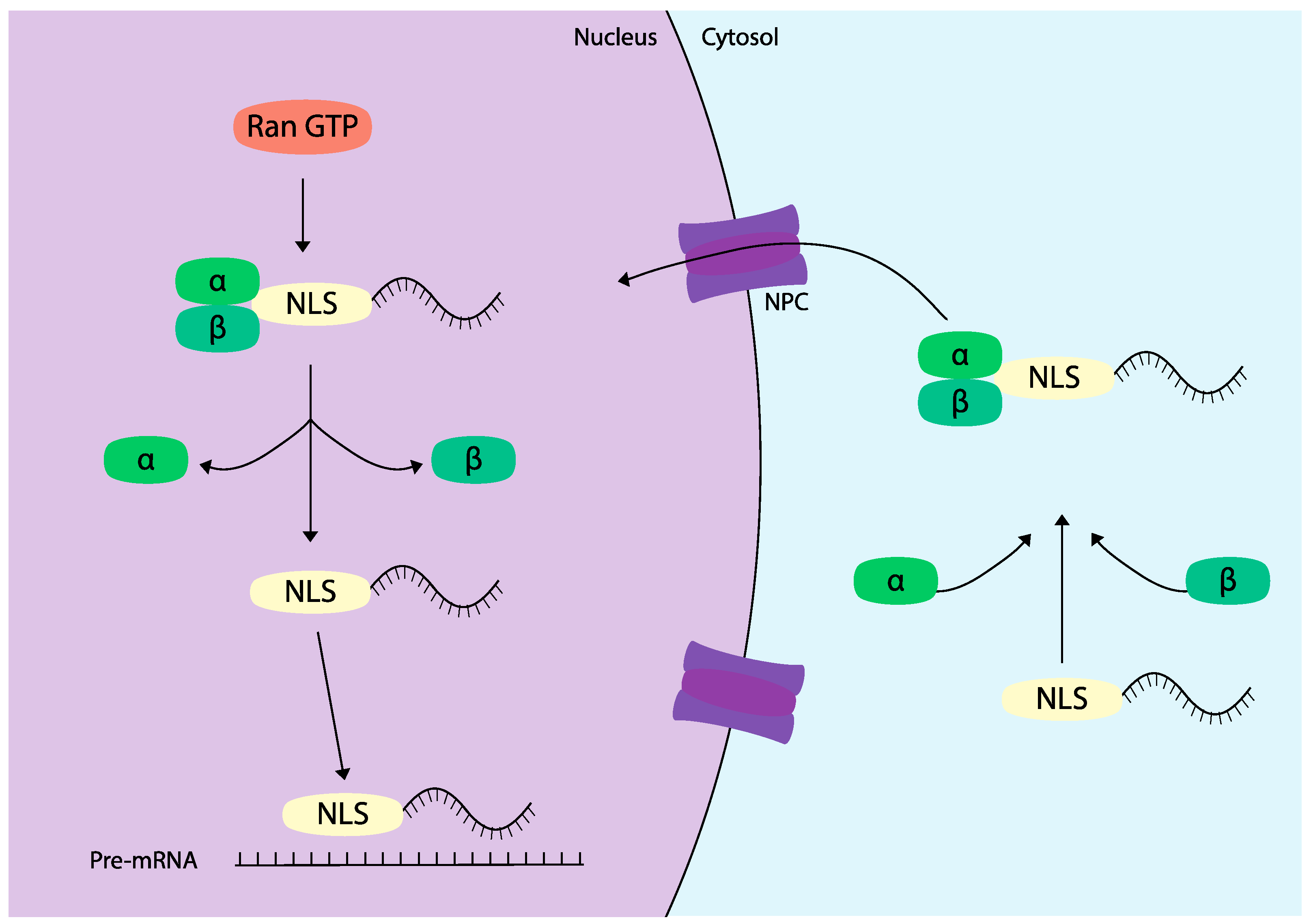
Compared to endosomal escape, far fewer strategies have been explored to enhance nuclear localization of ASOs. One primary method involves the use of nuclear localization signals (NLS), which are short, predominantly cationic peptide sequences that facilitate the transport of proteins into the nucleus [
118]. NLS sequences are found in various viral and endogenous proteins that play crucial roles in physiological processes such as transcription regulation and cell cycle control. This molecular mechanism of nuclear transport heavily relies on a family of transport proteins known as importins. Upon binding to an NLS, importins shuttle proteins into the nucleus through the nuclear pore complex via an active transport process mediated by Ran GTPase [
119] (
Figure 4).
The application of NLS in drug delivery has primarily focused on larger molecules like proteins or plasmid DNA (pDNA), due to their inability to passively cross the nuclear membrane, compared with smaller ASO therapeutics which can more readily traverse this barrier [
120]. However, the penetration of ASOs into the nucleus can still be restricted by factors such as their size, secondary structure, and the potential need for active transport [
30]. Moreover, the permeability of the nuclear pore complex can vary based on cell type and environmental conditions, further influencing ASO entry [
121].
The ability of NLS to promote nuclear transport has made them a promising focus for enhancing ASO delivery and overcoming barriers to effective gene therapy. Similar to the previously discussed CPP-ASO conjugates, ASOs can also be conjugated to NLS peptides to improve their nuclear uptake. Early studies in this area explored the conjugation of ASOs to NLS derived from various sources, including the SV40 T-antigen, influenza virus nucleoprotein, and HIV-1 TAT protein [
21]. A key finding from Kubo et al. demonstrated that unmodified ASOs were evenly distributed throughout the cytoplasm and nucleus, with similar localization patterns observed for ASOs conjugated to a control peptide. In contrast, NLS-ASO conjugates exhibited a marked preference for nuclear localization. This enhanced nuclear accumulation significantly increased antisense activity, with the most effective NLS from the SV40 T-antigen boosting treatment efficacy from 87.6% to 99.6% within 24 hours. The SV40 T-antigen was the first NLS identified, with its corresponding peptide sequence PKKKRKV now recognized as a prototypical NLS [
118]. Beyond enhancing nuclear localization, the cationic nature of NLS peptides may also improve cellular uptake through mechanisms similar to those of cationic CPPs, potentially facilitating better overall delivery of ASOs.
Additionally, combining NLS with other delivery strategies, such as nanoparticle encapsulation or lipid-based systems, has the potential to yield synergistic effects that maximize the therapeutic efficacy of ASOs. For example, Tarvirdipour et al. designed a peptide-based nanoparticle functionalized with the NLS sequence KRKR specifically to deliver ASOs targeting the Bcl-2 gene [
122]. This innovative delivery system demonstrated a notable increase in the association of nanoparticles with the nuclear membrane, as well as a higher proportion of nanoparticles entering the nucleus compared to those lacking the NLS. Importantly, this enhanced nuclear accumulation corresponded with a significantly greater knockdown of the target gene. In a separate study, the incorporation of an NLS into poly-lactic-co-glycolic acid (PLGA) polymers not only enhanced nuclear localization but also improved nanoparticle loading, suggesting that NLS can confer additional stability benefits to the nanoparticles themselves [
123]. These findings underscore the multifaceted advantages of NLS in the context of nanoparticle design, as they not only facilitate nuclear transport but may also enhance the overall stability and loading capacity of the delivery systems. Recent advances in peptide engineering have led to the development of modified NLS sequences that could further enhance specificity and efficiency in targeting.
Despite the potential benefits of incorporating nuclear localization signals (NLS) into ASO delivery systems, a recent study has revealed some contrasting findings regarding their effectiveness. Among seven different NLS tested, only one sequence, SKKKKTKVC derived from the transcription factor TFIIE-β, successfully enhanced the nuclear localization of ASOs [
124]. However, this increased localization did not translate into enhanced antisense activity. The researchers attributed this discrepancy to endosomal entrapment in the perinuclear region, noting that treatment with the endolytic small molecule chloroquine restored robust splice switching.
These findings underscore that while nuclear localization is crucial, endosomal escape remains a significant barrier to effective ASO delivery. Combinatorial approaches, such as pairing an NLS with fusogenic peptides, may provide a dual mechanism to enhance ASO efficacy by promoting both nuclear entry and endosomal escape [
125]. By targeting both pathways, such strategies could lead to a more comprehensive enhancement of ASO activity. Furthermore, a deeper understanding of the molecular mechanisms underlying nuclear localization and endosomal trafficking is essential for optimizing ASO therapies. This knowledge could inform the design of multifunctional delivery systems that not only improve nuclear access but also facilitate endosomal escape, ultimately enhancing the therapeutic potential of ASOs. Continued research in this area may lead to innovative strategies that overcome existing limitations and improve the overall effectiveness of ASO-based therapies.
6. Conclusions and Future Perspectives
ASOs hold immense potential to transform the treatment landscape for various conditions by directly targeting disease-causing genes. However, their therapeutic efficacy is significantly hindered by challenges such as endosomal entrapment and inadequate nuclear localization. Despite the numerous strategies proposed to mitigate these issues, no single platform has emerged as a comprehensive solution or achieved clinical applicability. To address these limitations, we advocate for focused research in several key areas:
Cell Type and Cargo-Specific Understanding of ASO Intracellular Trafficking: A critical aspect of enhancing endosomal escape lies in thoroughly understanding how ASOs are trafficked within cells and their natural mechanisms for escaping endosomal entrapment. Although recent studies have improved our comprehension of these processes, emerging evidence indicates that trafficking mechanisms can vary significantly based on cell type and the specific cargo involved. For example, while the small molecule retro-1 enhances ASO efficacy, it has no effect on siRNA delivery [
103]. Additionally, different endosomal escape enhancers have demonstrated varying effectiveness when ASOs are delivered via lipid nanoparticles compared to cholesterol conjugates [
98]. Therefore, further research tailored to the endosomal escape mechanisms of specific cargos (such as ASOs, siRNAs, mRNAs, and pDNAs), delivery systems (including free oligonucleotides, nanoparticle-encapsulated ASOs, and peptide-conjugated formats), and the relevant cell types is essential for the practical application of these approaches.
Focus on Non-Toxic Endosomal Escape Strategies: One of the most significant challenges regarding endosomal escape is achieving a balance between escape efficiency and cytotoxicity. The endo-lysosomal system plays a crucial role in intracellular trafficking and the clearance of cellular waste products. Disrupting this system can lead to cellular stress and detrimental effects. For instance, the formation of breaks in the endosomal membrane can trigger a membrane damage response mediated by galectins, initiating downstream inflammatory pathways that can cause cytotoxicity [
126]. Consequently, strategies that avoid physically damaging the endosomal membrane such as promoting membrane fusion or facilitating vesicle budding/collapse, are the most promising for minimizing potential toxicity warranting further investigation.
Alternative and Combinatorial Strategies: Current delivery approaches alone are insufficient to overcome the rate-limiting barriers associated with ASO delivery. We propose expanding research efforts to explore combinatorial strategies that integrate multiple techniques to optimize ASO delivery. For instance, the synergistic use of nanoparticles and peptides in conjunction with small molecule modulators of intracellular trafficking could significantly enhance delivery efficiency. While improving endosomal escape is crucial, the significance of efficient nuclear localization must also be emphasized. Nuclear accumulation is essential for ASO functionality, and exploring innovative strategies to facilitate this process could dramatically improve therapeutic outcomes. Incorporating NLS peptides into existing delivery platforms presents a promising avenue to tackle these challenges.
In summary, addressing the barriers to effective ASO delivery necessitates a multifaceted approach that prioritizes understanding intracellular trafficking mechanisms, developing non-toxic escape strategies, and exploring innovative combinatorial methods. Continued research in these areas could pave the way for more effective ASO-based therapies, especially for diseases where precise gene expression regulation is critical. By overcoming these challenges, ASOs can realize their full therapeutic potential, ultimately benefiting a wide range of patients.
Author Contributions
Original draft preparation, R.A.; Review and editing, R.A. and T.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
We thank the University of Alberta Faculty of Medicine and Dentistry, the University of Alberta Faculty of Graduate and Postdoctoral Studies, the Friends of Garrett Cumming Research Fund, Muscular Dystrophy Canada, the Canadian Institute of Health Research (CIHR), the Women and Children’s Health Research Institute (WCHRI), the HM Toupin Neurological Science Research Fund, Alberta Innovates: Health Solutions (AIHS), Jesse’s Journey, the U.S. Department of Defense, and the Heart and Stroke Foundation Canada for their support.
Conflicts of Interest
T.Y. is a cofounder and shareholder of OligomicsTx Inc., which aims to commercialize antisense technology. The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Rinaldi, C.; Wood, M.J.A. Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 2018, 1, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Lima, W.F.; Zhang, H.; Fan, A.; Sun, H.; Crooke, S.T. Determination of the role of the human RNase H1 in the pharmacology of DNA-like antisense drugs. J. Biol. Chem. 2004, 279, 17181–17189. [Google Scholar] [CrossRef]
- Havens, M.A.; Hastings, M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef] [PubMed]
- Ersöz, E.; Demir-Dora, D. Unveiling the potential of antisense oligonucleotides: Mechanisms, therapies, and safety insights. Drug Dev. Res. 2024, 85. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, J.; Gao, Y.; Li, Y.; Li, Y. Strategies for targeting undruggable targets. Expert. Opin. Drug Discov. 2022, 17, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Tillman, L.G.; Geary, R.S.; Hardee, G.E. Oral delivery of antisense oligonucleotides in man. J. Pharm. Sci. 2008, 97, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Bijsterbosch, M.K.; Manoharan, M.; Rump, E.T.; De Vrueh, R.L.; van Veghel, R.; Tivel, K.L.; Biessen, E.A.; Bennett, C.F.; Cook, P.D.; van Berkel, T.J. In vivo fate of phosphorothioate antisense oligodeoxynucleotides: predominant uptake by scavenger receptors on endothelial liver cells. Nucleic Acids Res. 1997, 25, 3290–3296. [Google Scholar] [CrossRef]
- Iversen, F.; Yang, C.; Dagnæs-Hansen, F.; Schaffert, D.H.; Kjems, J.; Gao, S. Optimized siRNA-PEG conjugates for extended blood circulation and reduced urine excretion in mice. Theranostics 2013, 3, 201–209. [Google Scholar] [CrossRef]
- Fisher, T.L.; Terhorst, T.; Cao, X.; Wagner, R.W. Intracellular disposition and metabolism of fluorescently-labeled unmodified and modified oligonucleotides microinjected into mammalian cells. Nucleic Acids Res. 1993, 21, 3857–3865. [Google Scholar] [CrossRef]
- Dhuri, K.; Bechtold, C.; Quijano, E.; Pham, H.; Gupta, A.; Vikram, A.; Bahal, R. Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. J. Clin. Med. 2020, 9, 2004. [Google Scholar] [CrossRef]
- Juliano, R.L.; Carver, K.; Cao, C.; Ming, X. Receptors, endocytosis, and trafficking: the biological basis of targeted delivery of antisense and siRNA oligonucleotides. J. Drug Target. 2013, 21, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Koller, E.; Vincent, T.M.; Chappell, A.; De, S.; Manoharan, M.; Bennett, C.F. Mechanisms of single-stranded phosphorothioate modified antisense oligonucleotide accumulation in hepatocytes. Nucleic Acids Res. 2011, 39, 4795–4807. [Google Scholar] [CrossRef] [PubMed]
- Racoosin, E.L.; Swanson, J.A. Macropinosome maturation and fusion with tubular lysosomes in macrophages. J. Cell Biol. 1993, 121, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Ochaba, J.; Powers, A.F.; Tremble, K.A.; Greenlee, S.; Post, N.M.; Matson, J.E.; MacLeod, A.R.; Guo, S.; Aghajan, M. A novel and translational role for autophagy in antisense oligonucleotide trafficking and activity. Nucleic Acids Res. 2019, 47, 11284–11303. [Google Scholar] [CrossRef]
- Fichter, K.M.; Ingle, N.P.; McLendon, P.M.; Reineke, T.M. Polymeric nucleic acid vehicles exploit active interorganelle trafficking mechanisms. ACS Nano 2013, 7, 347–364. [Google Scholar] [CrossRef]
- Sahay, G.; Querbes, W.; Alabi, C.; Eltoukhy, A.; Sarkar, S.; Zurenko, C.; Karagiannis, E.; Love, K.; Chen, D.; Zoncu, R.; et al. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat. Biotechnol. 2013, 31, 653–658. [Google Scholar] [CrossRef]
- Dowdy, S.F. Endosomal escape of RNA therapeutics: How do we solve this rate-limiting problem? RNA 2023, 29, 396–401. [Google Scholar] [CrossRef]
- Crooke, S.T.; Wang, S.; Vickers, T.A.; Shen, W.; Liang, X.H. Cellular uptake and trafficking of antisense oligonucleotides. Nat Biotechnol. 2017, 35, 230–237. [Google Scholar] [CrossRef]
- Jiang, Y.; Lu, Q.; Wang, Y.; Xu, E.; Ho, A.; Singh, P.; Wang, Y.; Jiang, Z.; Yang, F.; Tietjen, G.T.; et al. Quantitating Endosomal Escape of a Library of Polymers for mRNA Delivery. Nano Lett. 2020, 20, 117–1123. [Google Scholar] [CrossRef]
- Gilleron, J.; Querbes, W.; Zeigerer, A.; Borodovsky, A.; Marsico, G.; Schubert, U.; Manygoats, K.; Seifert, S.; Andree, C.; Stöter, M.; et al. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat. Biotechnol. 2013, 31, 638–346. [Google Scholar] [CrossRef]
- Kubo, T.; Zhelev, Z.; Bakalova, R.; Ohba, H.; Doi, K.; Fujii, M. Controlled intracellular localization and enhanced antisense effect of oligonucleotides by chemical conjugation. Org. Biomol. Chem. 2005, 3, 3257–3259. [Google Scholar] [CrossRef] [PubMed]
- Monzel, C.; Sengupta, K. Measuring shape fluctuations in biological membranes. J. Phys. D Appl. Phys. 2016, 49, 243002. [Google Scholar] [CrossRef]
- Woodman, P.G.; Futter, C.E. ; Multivesicular bodies: co-ordinated progression to maturity. Curr. Opin. Cell Biol. 2008, 20, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Perrin, P.; Janssen, L.; Janssen, H.; van den Broek, B.; Voortman, L.M.; van Elsland, D.; Berlin, I.; Neefjes, J. Retrofusion of intralumenal MVB membranes parallels viral infection and coexists with exosome release. Curr. Biol. 2021, 31, 3884–3893. [Google Scholar] [CrossRef]
- Paramasivam, P.; Franke, C.; Stöter, M.; Höijer, A.; Bartesaghi, S.; Sabirsh, A.; Lindfors, L.; Arteta, M.Y.; Dahlén, A.; Bak, A.; et al. Endosomal escape of delivered mRNA from endosomal recycling tubules visualized at the nanoscale. J. Cell Biol. 2022, 221, e202110137. [Google Scholar] [CrossRef]
- Liang, X.H.; Sun, H.; Nichols, J.G.; Allen, N.; Wang, S.; Vickers, T.A.; Shen, W.; Hsu, C.W.; Crooke, S.T. COPII vesicles can affect the activity of antisense oligonucleotides by facilitating the release of oligonucleotides from endocytic pathways. Nucleic Acids Res. 2018, 46, 10225–10245. [Google Scholar] [CrossRef]
- Liang, X.H.; Sun, H.; Hsu, C.W.; Nichols, J.G.; Vickers, T.A.; De Hoyos, C.L.; Crooke, S.T. Golgi-endosome transport mediated by M6PR facilitates release of antisense oligonucleotides from endosomes. Nucleic Acids Res. 2020, 48, 1372–1391. [Google Scholar] [CrossRef] [PubMed]
- Chin, D.J.; Green, G.A.; Zon, G.; Szoka Jr, F.C.; Straubinger, R.M. Rapid nuclear accumulation of injected oligodeoxyribonucleotides. New Biol. 1990, 2, 1091–1100. [Google Scholar]
- Leonetti, J.P.; Mechti, N.; Degols, G.; Gagnor, C.; Lebleu, B. Intracellular distribution of microinjected antisense oligonucleotides. Proc. Natl. Acad. Sci. USA 1991, 88, 2702–2706. [Google Scholar] [CrossRef]
- Lorenz, P.; Misteli, T.; Baker, B.F.; Bennett, C.F.; Spector, D.L. ; Nucleocytoplasmic shuttling: a novel in vivo property of antisense phosphorothioate oligodeoxynucleotides. Nucleic Acids Res. 2000, 28, 582–592. [Google Scholar] [CrossRef]
- Ma, J.; Dissanayaka Mudiyanselage, S.D.; Park, W.J.; Wang, M.; Takeda, R.; Liu, B.; Wang, Y. A nuclear import pathway exploited by pathogenic noncoding RNAs. Plant Cell 2022, 34, 3543–3556. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.B.; Dammer, E.B.; Ren, R.J.; Wang, G. The endosomal-lysosomal system: from acidification and cargo sorting to neurodegeneration. Transl. Neurodegener. 2015, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Cabral, H. Enhancing Targeted Drug Delivery through Cell-Specific Endosomal Escape. ChemMedChem 2024, 19, e202400274. [Google Scholar] [CrossRef] [PubMed]
- Hajimolaali, M.; Mohammadian, H.; Torabi, A.; Shirini, A.; Khalife Shal, M.; Barazandeh Nezhad, H.; Iranpour, S.; Baradaran Eftekhari, R.; Dorkoosh, F. Application of chloroquine as an endosomal escape enhancing agent: new frontiers for an old drug. Expert. Opin. Drug Deliv. 2021, 18, 877–889. [Google Scholar] [CrossRef]
- Scott, C.C.; Gruenberg, J. Ion flux and the function of endosomes and lysosomes: pH is just the start: the flux of ions across endosomal membranes influences endosome function not only through regulation of the luminal pH. Bioessays 2011, 33, 103–110. [Google Scholar] [CrossRef]
- Behr, J.P. The Proton Sponge: a Trick to Enter Cells the Viruses Did Not Exploit. CHIMIA 1997, 51, 34–36. [Google Scholar] [CrossRef]
- Du Rietz, H.; Hedlund, H.; Wilhelmson, S.; Nordenfelt, P.; Wittrup, A. Imaging small molecule-induced endosomal escape of siRNA. Nat. Commun. 2020, 11, 1809. [Google Scholar] [CrossRef]
- Yang, B.; Ming, X.; Cao, C.; Laing, B.; Yuan, A.; Porter, MA.; Hull-Ryde, E.A.; Maddry, J.; Suto, M.; Janzen, W.P.; Juliano, R.L. High-throughput screening identifies small molecules that enhance the pharmacological effects of oligonucleotides. Nucleic Acids Res. 2015, 43, 1987–1996. [Google Scholar] [CrossRef]
- Bizot, F.; Fayssoil, A.; Gastaldi, C.; Irawan, T.; Phongsavanh, X.; Mansart, A.; Tensorer, T.; Brisebard, E.; Garcia, L.; Juliano, R.L.; et al. Oligonucleotide Enhancing Compound Increases Tricyclo-DNA Mediated Exon-Skipping Efficacy in the Mdx Mouse Model. Cells 2023, 12, 702. [Google Scholar] [CrossRef]
- Juliano, R.L.; Wang, L.; Tavares, F.; Brown, E.G.; James, L.; Ariyarathna, Y.; Ming, X.; Mao, C.; Suto, M. Structure-activity relationships and cellular mechanism of action of small molecules that enhance the delivery of oligonucleotides. Nucleic Acids Res. 2018, 46, 1601–1613. [Google Scholar] [CrossRef]
- Finicle, B.T.; Eckenstein, K.H.; Revenko, A.S.; Anderson, B.A.; Wan, W.B.; McCracken, A.N.; Gil, D.; Fruman, D.A.; Hanessian, S.; Seth, P.P.; et al. Simultaneous inhibition of endocytic recycling and lysosomal fusion sensitizes cells and tissues to oligonucleotide therapeutics. Nucleic Acids Res. 2023, 51, 1583–1599. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ariyarathna, Y.; Ming, X.; Yang, B.; James, L.I.; Kreda, S.M.; Porter, M.; Janzen, W.; Juliano, R.L. A Novel Family of Small Molecules that Enhance the Intracellular Delivery and Pharmacological Effectiveness of Antisense and Splice Switching Oligonucleotides. ACS Chem. Biol. 2017, 12, 1999–2007. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Khan, J.M. pH-sensitive endosomolytic peptides in gene and drug delivery: Endosomal escape and current challenges. J. Drug Deliv. Sci. Technol. 2022, 76, 103786. [Google Scholar] [CrossRef]
- Panjwani, A.; Strauss, M.; Gold, S.; Wenham, H.; Jackson, T.; Chou, J.J.; Rowlands, D.J.; Stonehouse, N.J.; Hogle, J.M.; Tuthill, T.J. Capsid protein VP4 of human rhinovirus induces membrane permeability by the formation of a size-selective multimeric pore. PLoS Pathog. 2014, 10, e1004294. [Google Scholar] [CrossRef] [PubMed]
- Plaza-Ga, I.; Manzaneda-González, V.; Kisovec, M.; Almendro-Vedia, V.; Muñoz-Úbeda, M.; Anderluh, G.; Guerrero-Martínez, A.; Natale, P.; López Montero, I. pH-triggered endosomal escape of pore-forming Listeriolysin O toxin-coated gold nanoparticles. J. Nanobiotechnology 2019, 17, 108. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.J.; Liu, D.V.; Sklaviadis, D.; Gui, D.Y.; Vander Heiden, M.G.; Wittrup, K.D. Antibody-mediated neutralization of perfringolysin o for intracellular protein delivery. Mol. Pharm, 2015, 12, 1992–2000. [Google Scholar] [CrossRef]
- Arnold, A.E.; Smith, L.J.; Beilhartz, G.L.; Bahlmann, L.C.; Jameson, E.; Melnyk, R.A.; Shoichet, M.S. Attenuated diphtheria toxin mediates siRNA delivery. Sci. Adv. 2020, 6, eaaz4848. [Google Scholar] [CrossRef]
- Hadjerci, J.; Billet, A.; Kessler, P.; Mourier, G.; Ghazarian, M.; Gonzalez, A.; Wunder, C.; Mabrouk, N.; Tartour, E.; Servent, D.; et al. Engineered Synthetic STxB for Enhanced Cytosolic Delivery. Cells 2023, 12, 1291. [Google Scholar] [CrossRef]
- Park, S.G.; Lee, H.B.; Kang, S. Development of plug-and-deliverable intracellular protein delivery platforms based on botulinum neurotoxin. Int. J. Biol. Macromol. 2024, 261 Pt 1, 129622. [Google Scholar] [CrossRef]
- Dawre, S.; Maru, S. Human respiratory viral infections: Current status and future prospects of nanotechnology-based approaches for prophylaxis and treatment. Life Sci. 2021, 278, 119561. [Google Scholar] [CrossRef]
- Lagache, T.; Sieben, C.; Meyer, T.; Herrmann, A.; Holcman, D. Stochastic Model of Acidification, Activation of Hemagglutinin and Escape of Influenza Viruses from an Endosome. Front. Phys. 2017, 5, 25. [Google Scholar] [CrossRef]
- Jadhav, S.G.; Setten, R.L.; Medina, C.; Cui, X.S.; Dowdy, S.F. Design, Synthesis, and Biochemical Analysis of a Molecule Designed to Enhance Endosomal Escape. AAPS J. 2023, 26, 10. [Google Scholar] [CrossRef] [PubMed]
- Derakhshankhah, H.; Jafari, S. ; Cell penetrating peptides: A concise review with emphasis on biomedical applications. Biomed. Pharmacother. 2018, 108, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Wu, L.C.L.; Wood, J.A.; Yao, M.; Treleaven, C.M.; Estrella, N.L.; Wentworth, B.M.; Hanson, G.J.; Passini, M.A. A cell-penetrating peptide enhances delivery and efficacy of phosphorodiamidate morpholino oligomers in mdx mice. Mol. Ther. Nucleic Acids 2022, 30, 17–27. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, H.O.; McCaffrey, J.; McCrudden, C.M.; Zholobenko, A.; Ali, A.A.; McBride, J.W.; Massey, A.S.; Pentlavalli, S.; Chen, K.H.; Cole, G.; et al. Development and characterization of self-assembling nanoparticles using a bio-inspired amphipathic peptide for gene delivery. J. Control Release 2014, 10, 141–149. [Google Scholar] [CrossRef]
- Liu, Y.; Wan, H.H.; Tian, D.M.; Xu, X.J.; Bi, C.L.; Zhan, X.Y.; Huang, B.H.; Xu, Y.S.; Yan, L.P. Development and Characterization of High Efficacy Cell-Penetrating Peptide via Modulation of the Histidine and Arginine Ratio for Gene Therapy. Materials 2021, 14, 4674. [Google Scholar] [CrossRef]
- Allen, J.; Pellois, J.P. ; Hydrophobicity is a key determinant in the activity of arginine-rich cell penetrating peptides. Sci. Rep. 2022, 12, 15981. [Google Scholar] [CrossRef]
- Takada, H.; Tsuchiya, K.; Demizu, Y. Helix-Stabilized Cell-Penetrating Peptides for Delivery of Antisense Morpholino Oligomers: Relationships among Helicity, Cellular Uptake, and Antisense Activity. Bioconjug Chem. 2022, 33, 1311–1318. [Google Scholar] [CrossRef]
- Clark, E.; Nava, B.; Caputi, M. Tat is a multifunctional viral protein that modulates cellular gene expression and functions. Oncotarget 2017, 8, 27569–27581. [Google Scholar] [CrossRef]
- Erazo-Oliveras, A.; Najjar, K.; Dayani, L.; Wang, T.Y.; Johnson, G.A.; Pellois, J.P. Protein delivery into live cells by incubation with an endosomolytic agent. Nat. Methods 2014, 11, 861–867. [Google Scholar] [CrossRef]
- Qian, Z.; LaRochelle, J.R.; Jiang, B.; Lian, W.; Hard, R.L.; Selner, N.G.; Luechapanichkul, R.; Barrios, A.M.; Pei, D. Early endosomal escape of a cyclic cell-penetrating peptide allows effective cytosolic cargo delivery. Biochemistry 2014, 53, 4034–4046. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Martyna, A.; Hard, R.L.; Wang, J.; Appiah-Kubi, G.; Coss, C.; Phelps, M.A.; Rossman, J.S.; Pei, D. Discovery and Mechanism of Highly Efficient Cyclic Cell-Penetrating Peptides. Biochemistry 2016, 55, 2601–2612. [Google Scholar] [CrossRef] [PubMed]
- Sahni, A.; Qian, Z.; Pei, D. Cell-Penetrating Peptides Escape the Endosome by Inducing Vesicle Budding and Collapse. ACS Chem. Biol. 2020, 15, 2485–2492. [Google Scholar] [CrossRef] [PubMed]
- Sahni, A.; Pei, D. Bacterial Toxins Escape the Endosome by Inducing Vesicle Budding and Collapse. ACS Chem. Biol. 2021, 16, 2415–2422. [Google Scholar] [CrossRef]
- Li, X.; Kheirabadi, M.; Dougherty, P.G.; Kamer, K.J.; Shen, X.; Estrella, N.L.; Peddigari, S.; Pathak, A.; Blake, S.L.; Sizensky, E.; et al. The endosomal escape vehicle platform enhances delivery of oligonucleotides in preclinical models of neuromuscular disorders. Mol. Ther. Nucleic Acids 2023, 33, 273–285. [Google Scholar] [CrossRef]
- Liu, B.R.; Chiou, S.H.; Huang, Y.W.; Lee, H.J. Bio-Membrane Internalization Mechanisms of Arginine-Rich Cell-Penetrating Peptides in Various Species. Membranes 2022, 12, 88. [Google Scholar] [CrossRef]
- Serulla, M.; Anees, P.; Hallaj, A.; Trofimenko, E.; Kalia, T.; Krishnan, Y.; Widmann, C. Plasma membrane depolarization reveals endosomal escape incapacity of cell-penetrating peptides. Eur. J. Pharm. Biopharm. 2023, 184, 116–124. [Google Scholar] [CrossRef]
- Teo, S.L.Y.; Rennick, J.J.; Yuen, D.; Al-Wassiti, H.; Johnston, A.P.R.; Pouton, C.W. Unravelling cytosolic delivery of cell penetrating peptides with a quantitative endosomal escape assay. Nat. Commun. 2021, 12, 3721. [Google Scholar] [CrossRef]
- Ertl, H.C.J. Immunogenicity and toxicity of AAV gene therapy. Front. Immunol. 2022, 13, 975803. [Google Scholar] [CrossRef]
- Huang, S.; Hao, X.Y.; Li, Y.J.; Wu, J.Y.; Xiang, D.X.; Luo, S. Nonviral delivery systems for antisense oligonucleotide therapeutics. Biomater. Res. 2022, 26, 49. [Google Scholar] [CrossRef]
- Sun, D.; Lu, ZR. Structure and Function of Cationic and Ionizable Lipids for Nucleic Acid Delivery. Pharm. Res. 2023, 40, 27–46. [Google Scholar] [CrossRef] [PubMed]
- Koltover, I.; Salditt, T.; Rädler, J.O.; Safinya, C.R. An inverted hexagonal phase of cationic liposome-DNA complexes related to DNA release and delivery. Science 1998, 281, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Lonez, C.; Vandenbranden, M.; Ruysschaert, J.M. Cationic lipids activate intracellular signaling pathways. Adv. Drug Deliv. Rev. 2012, 64, 1749–1758. [Google Scholar] [CrossRef] [PubMed]
- Ferraresso, F.; Strilchuk, A.W.; Juang, L.J.; Poole, L.G.; Luyendyk, J.P.; Kastrup, C.J. Comparison of DLin-MC3-DMA and ALC-0315 for siRNA Delivery to Hepatocytes and Hepatic Stellate Cells. Mol. Pharm. 2022, 19, 2175–2182. [Google Scholar] [CrossRef]
- Kovtun, A.; Heumann, R.; Epple, M. ; Calcium phosphate nanoparticles for the transfection of cells. Biomed. Mater. Eng. 2009, 19, 241–247. [Google Scholar] [CrossRef]
- Han, X.; Lu, Y.; Xu, Z.; Chu, Y.; Ma, X.; Wu, H.; Zou, B.; Zhou, G. Anionic liposomes prepared without organic solvents for effective siRNA delivery. IET Nanobiotechnol. 2023, 17, 269–280. [Google Scholar] [CrossRef]
- Hald Albertsen, C.; Kulkarni, J.A.; Witzigmann, D.; Lind, M.; Petersson, K.; Simonsen, J.B. The role of lipid components in lipid nanoparticles for vaccines and gene therapy. Adv. Drug Deliv. Rev. 2022, 188, 114416. [Google Scholar] [CrossRef]
- Ermilova, I.; Swenson, J. DOPC versus DOPE as a helper lipid for gene-therapies: molecular dynamics simulations with DLin-MC3-DMA. Phys. Chem. Chem. Phys. 2020, 22, 28256–28268. [Google Scholar] [CrossRef]
- Jerjes, W.; Theodossiou, T.A.; Hirschberg, H.; Høgset, A.; Weyergang, A.; Selbo, P.K.; Hamdoon, Z.; Hopper, C.; Berg, K. Photochemical Internalization for Intracellular Drug Delivery. From Basic Mechanisms to Clinical Research. J. Clin. Med. 2020, 9, 528. [Google Scholar] [CrossRef]
- Ohtsuki, T.; Miki, S.; Kobayashi, S.; Haraguchi, T.; Nakata, E.; Hirakawa, K.; Sumita, K.; Watanabe, K.; Okazaki, S. The molecular mechanism of photochemical internalization of cell penetrating peptide-cargo-photosensitizer conjugates. Sci. Rep. 2015, 5, 18577. [Google Scholar] [CrossRef]
- Fraire, J.C.; Houthaeve, G.; Liu, J.; Raes, L.; Vermeulen, L.; Stremersch, S.; Brans, T.; García-Díaz Barriga, G.; De Keulenaer, S.; Van Nieuwerburgh, F.; et al. Vapor nanobubble is the more reliable photothermal mechanism for inducing endosomal escape of siRNA without disturbing cell homeostasis. J. Control Release 2020, 319, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Fan, B.; Gao, W.; Li, L.; Li, T.; Sun, J.; Peng, X.; Li, X.; Wang, Z.; Wang, B.; et al. Enhanced endosomal escape by photothermal activation for improved small interfering RNA delivery and antitumor effect. Int. J. Nanomed. 2018, 13, 4333–4344. [Google Scholar] [CrossRef] [PubMed]
- Domenech, M.; Marrero-Berrios, I.; Torres-Lugo, M.; Rinaldi, C. Lysosomal membrane permeabilization by targeted magnetic nanoparticles in alternating magnetic fields. ACS Nano 2013, 7, 5091–5101. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.A.; Fang, J.; Paragodaarachchi, A.; Kodama, K.; Yakobashvili, D.; Ichiyanagi, Y.; Matsui, H. Magnetically Induced Brownian Motion of Iron Oxide Nanocages in Alternating Magnetic Fields and Their Application for Efficient siRNA Delivery. Nano Lett. 2022, 22, 8852–8859. [Google Scholar] [CrossRef]
- Kang, M.A.; Rao, P.P.; Matsui, H.; Mahajan, S.S. Delivery of mGluR5 siRNAs by Iron Oxide Nanocages by Alternating Magnetic Fields for Blocking Proliferation of Metastatic Osteosarcoma Cells. Int. J. Mol. Sci. 2022, 23, 7944. [Google Scholar] [CrossRef]
- Zhao, Y.Z.; Du, L.N.; Lu, C.T.; Jin, Y.G.; Ge, S.P. Potential and problems in ultrasound-responsive drug delivery systems. Int. J. Nanomed. 2013, 8, 1621–1633. [Google Scholar] [CrossRef]
- Qiu, Y.; Luo, Y.; Zhang, Y.; Cui, W.; Zhang, D.; Wu, J.; Zhang, J.; Tu, J. The correlation between acoustic cavitation and sonoporation involved in ultrasound-mediated DNA transfection with polyethylenimine (PEI) in vitro. J. Control Release 2010, 145, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Haag, P.; Frauscher, F.; Gradl, J.; Seitz, A.; Schäfer, G.; Lindner, J.R.; Klibanov, A.L.; Bartsch, G.; Klocker, H.; Eder, I.E. Microbubble-enhanced ultrasound to deliver an antisense oligodeoxynucleotide targeting the human androgen receptor into prostate tumours. J. Steroid Biochem. Mol. Biol. 2006, 102, 103–113. [Google Scholar] [CrossRef]
- Li, B.; Zhao, M.; Lai, W.; Zhang, X.; Yang, B.; Chen, X.; Ni, Q. Activatable NIR-II Photothermal Lipid Nanoparticles for Improved Messenger RNA Delivery. Angew. Chem. Int. Ed. Engl. 2023, 62, e202302676. [Google Scholar] [CrossRef]
- Yap, S.L.; Yu, H.; Li, S.; Drummond, C.J.; Conn, C.E.; Tran, N. Cell interactions with lipid nanoparticles possessing different internal nanostructures: Liposomes, bicontinuous cubosomes, hexosomes, and discontinuous micellar cubosomes. J. Colloid. Interface Sci. 2024, 656, 409–423. [Google Scholar] [CrossRef]
- Lu, J.J.; Langer, R.; Chen, J. A novel mechanism is involved in cationic lipid-mediated functional siRNA delivery. Mol. Pharm. 2009, 6, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Jahn, R.; Cafiso, D.C.; Tamm, L.K. Mechanisms of SNARE proteins in membrane fusion. Nat. Rev. Mol. Cell Biol. 2024, 25, 101–118. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Shen, M.; Pattipeiluhu, R.; Zhou, X.; Zhang, Y.; Bakkum, T.; Sharp, T.H.; Boyle, A.L.; Kros, A. Efficient mRNA delivery using lipid nanoparticles modified with fusogenic coiled-coil peptides. Nanoscale 2023, 15, 15206–15218. [Google Scholar] [CrossRef]
- Gupta, A.; Gupta, S.; Das, U.; Sinha, S. Guanidinium-Functionalized Flexible Azaproline Transporter for Efficient Intracellular Delivery of Proapoptotic Peptide and PDL1 Antisense Morpholino Oligo in Human Carcinoma Cells In Vitro. Bioconjug Chem. 2022, 33, 907–917. [Google Scholar] [CrossRef]
- Ghosh, S.; Panja, P.; Dalal, C.; Jana, N.R. Arginine-Terminated, Chemically Designed Nanoparticle for Direct Cell Translocation. ACS Appl. Bio Mater. 2019, 2, 339–348. [Google Scholar] [CrossRef]
- Kang, Z.; Liu, Q.; Zhang, Z.; Zheng, Y.; Wang, C.; Pan, Z.; Li, Q.; Liu, Y.; Shi, L. Arginine-Rich Polymers with Pore-Forming Capability Enable Efficient Intracellular Delivery via Direct Translocation Across Cell Membrane. Adv. Healthc. Mater. 2022, 11, e2200371. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.K.; Debnath, K.; Arora, H.; Seth, P.; Jana, N.R.; Jana, N.R. Direct Cellular Delivery of Exogenous Genetic Material and Protein via Colloidal Nano-Assemblies with Biopolymer. ACS Appl. Mater. Interfaces 2022, 14, 3199–3206. [Google Scholar] [CrossRef] [PubMed]
- Panja, P.; Jana, N.R. Lipid-Raft-Mediated Direct Cytosolic Delivery of Polymer-Coated Soft Nanoparticles. J. Phys. Chem. B 2020, 124, 5323–5333. [Google Scholar] [CrossRef]
- Gilleron, J.; Paramasivam, P.; Zeigerer, A.; Querbes, W.; Marsico, G.; Andree, C.; Seifert, S.; Amaya, P.; Stöter, M.; Koteliansky, V.; et al. Identification of siRNA delivery enhancers by a chemical library screen. Nucleic Acids Res. 2015, 43, 7984–8001. [Google Scholar] [CrossRef]
- Wang, H.; Tam, Y.Y.; Chen, S.; Zaifman, J.; van der Meel, R.; Ciufolini, M.A.; Cullis, P.R. The Niemann-Pick C1 Inhibitor NP3.47 Enhances Gene Silencing Potency of Lipid Nanoparticles Containing siRNA. Mol. Ther. 2016, 24, 2100–2108. [Google Scholar] [CrossRef]
- Schweitzer, J.K.; Sedgwick, A.E.; D'Souza-Schorey, C. ARF6-mediated endocytic recycling impacts cell movement, cell division and lipid homeostasis. Semin. Cell Dev. Biol. 2011, 22, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Castanotto, D.; Liu, X.; Shemi, A.; Stein, C.A. Ammonium and arsenic trioxide are potent facilitators of oligonucleotide function when delivered by gymnosis. Nucleic Acids Res. 2018, 46, 3612–3624. [Google Scholar] [CrossRef] [PubMed]
- Ming, X.; Carver, K.; Fisher, M.; Noel, R.; Cintrat, J.C.; Gillet, D.; Barbier, J.; Cao, C.; Bauman, J.; Juliano, R.L. The small molecule Retro-1 enhances the pharmacological actions of antisense and splice switching oligonucleotides. Nucleic Acids Res. 2013, 41, 3673–3687. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Selyunin, A.; Mukhopadhyay, S. Targeting the Early Endosome-to-Golgi Transport of Shiga Toxins as a Therapeutic Strategy. Toxins 2020, 12, 342. [Google Scholar] [CrossRef] [PubMed]
- Agramunt, J.; Pedroso, E.; Kreda, S.M.; Juliano, R.L.; Grandas, A. Retro-1-Oligonucleotide Conjugates. Synthesis and Biological Evaluation. Molecules 2019, 24, 579. [Google Scholar] [CrossRef]
- Yang, B.; Ming, X.; Abdelkafi, H.; Pons, V.; Michau, A.; Gillet, D.; Cintrat, J.C.; Barbier, J.; Juliano, R. Retro-1 Analogues Differentially Affect Oligonucleotide Delivery and Toxin Trafficking. ChemMedChem 2016, 11, 2506–2510. [Google Scholar] [CrossRef]
- Wagenaar, T.R.; Tolstykh, T.; Shi, C.; Jiang, L.; Zhang, J.; Li, Z.; Yu, Q.; Qu, H.; Sun, F.; Cao, H.; et al. Identification of the endosomal sorting complex required for transport-I (ESCRT-I) as an important modulator of anti-miR uptake by cancer cells. Nucleic Acids Res. 2015, 43, 1204–1215. [Google Scholar] [CrossRef]
- Vanier, M.T. Niemann-Pick diseases. Handb. Clin. Neurol. 2013, 113, 1717–1721. [Google Scholar] [CrossRef]
- Platt, F.M.; d'Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Primers 2018, 4, 27. [Google Scholar] [CrossRef]
- Liang, X.H.; Sun, H.; Nichols, J.G.; Crooke, S.T. ; RNase H1-Dependent Antisense Oligonucleotides Are Robustly Active in Directing RNA Cleavage in Both the Cytoplasm and the Nucleus. Mol. Ther. 2017, 25, 2075–2092. [Google Scholar] [CrossRef]
- Pendergraff, H.; Schmidt, S.; Vikeså, J.; Weile, C.; Øverup, C.W.; Lindholm, M.; Koch, T. Nuclear and Cytoplasmatic Quantification of Unconjugated, Label-Free Locked Nucleic Acid Oligonucleotides. Nucleic Acid. Ther. 2020, 30, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Vickers, T.A.; Liang, X.H. Phosphorothioate modified oligonucleotide-protein interactions. Nucleic Acids Res. 2020, 48, 5235–5253. [Google Scholar] [CrossRef]
- Lorenz, P.; Baker, B.F.; Bennett, C.F.; Spector, D.L. Phosphorothioate antisense oligonucleotides induce the formation of nuclear bodies. Mol. Biol. Cell 1998, 9, 1007–1023. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Shen, W.; Sun, H.; Prakash, TP.; Crooke, S.T. TCP1 complex proteins interact with phosphorothioate oligonucleotides and can co-localize in oligonucleotide-induced nuclear bodies in mammalian cells. Nucleic Acids Res. 2014, 42, 7819–7832. [Google Scholar] [CrossRef]
- Linnane, E.; Davey, P.; Zhang, P.; Puri, S.; Edbrooke, M.; Chiarparin, E.; Revenko, A.S.; Macleod, A.R.; Norman, J.C.; Ross, S.J. Differential uptake, kinetics and mechanisms of intracellular trafficking of next-generation antisense oligonucleotides across human cancer cell lines. Nucleic Acids Res. 2019, 47, 4375–4392. [Google Scholar] [CrossRef]
- Liang, X.H.; Nichols, J.G.; Tejera, D.; Crooke, S.T. Perinuclear positioning of endosomes can affect PS-ASO activities. Nucleic Acids Res. 2021, 49, 12970–12985. [Google Scholar] [CrossRef]
- Zhao, J.C.; Saleh, A.; Crooke, S.T. SIDT2 Inhibits Phosphorothioate Antisense Oligonucleotide Activity by Regulating Cellular Localization of Lysosomes. Nucleic Acid. Ther. 2023, 33, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Wu, T.; Zhang, B.; Liu, S.; Song, W.; Qiao, J.; Ruan, H. Types of nuclear localization signals and mechanisms of protein import into the nucleus. Cell Commun. Signal 2021, 19, 60. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Yamada, K.; Yoneda, Y. Importin α: a key molecule in nuclear transport and non-transport functions. J. Biochem. 2016, 160, 69–75. [Google Scholar] [CrossRef]
- Ciolina, C.; Byk, G.; Blanche, F.; Thuillier, V.; Scherman, D.; Wils, P. Coupling of nuclear localization signals to plasmid DNA and specific interaction of the conjugates with importin alpha. Bioconjug Chem. 1999, 10, 49–55. [Google Scholar] [CrossRef]
- Raices, M.; D'Angelo, M.A. Analysis of Nuclear Pore Complex Permeability in Mammalian Cells and Isolated Nuclei Using Fluorescent Dextrans. Methods Mol. Biol. 2022, 2502, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Tarvirdipour, S.; Skowicki, M.; Schoenenberger, C.A.; Kapinos, L.E.; Lim, R.Y.H.; Yaakov Benenson, Y.; Palivan, C.G. A self-assembling peptidic platform to boost the cellular uptake and nuclear delivery of oligonucleotides. Biomater. Sci. 2022, 10, 4309–4323. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.; Bahal, R. Investigation of PLGA nanoparticles in conjunction with nuclear localization sequence for enhanced delivery of antimiR phosphorothioates in cancer cells in vitro. J. Nanobiotechnology 2019, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.C.; Becker, J.P.; Slominski, D.; Halloy, F.; Søndergaard, C.; Ravn, J.; Hall, J. Peptide Conjugates of a 2'-O-Methoxyethyl Phosphorothioate Splice-Switching Oligonucleotide Show Increased Entrapment in Endosomes. ACS Omega 2023, 8, 40463–40481. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Vidal, P.; Chaloin, L.; Heitz, F.; Divita, G. A new peptide vector for efficient delivery of oligonucleotides into mammalian cells. Nucleic Acids Res. 1997, 25, 2730–2736. [Google Scholar] [CrossRef]
- Omo-Lamai, S.; Wang, Y.; Patel, M.N.; Essien, E.O.; Shen, M.; Majumdar, A.; Espy, C.; Wu, J.; Channer, B.; Tobin, M.; et al. Lipid Nanoparticle-Associated Inflammation is Triggered by Sensing of Endosomal Damage: Engineering Endosomal Escape Without Side Effects. bioRxiv 2024. [Google Scholar] [CrossRef]
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).