Submitted:
27 October 2024
Posted:
28 October 2024
You are already at the latest version
Abstract
A comprehensive understanding of the mechanisms underlying how Lentinula edodes handles heat stress (HS) is crucial for the development of superior HS-resistant strains. However, the mechanism by which the two sexually compatible haploid nuclei respond to heat stress remains largely unknown. Here, we examined the nuclei-specific (SP3 and SP30) HS-resistant mechanisms using integrated metabolomic and transcriptomic analysis. Results showed HS elicited the boost of ROS and hampered the mycelium growth for both monokaryons. Metabolome and transcriptome data demonstrated that the two sexually compatible monokaryons responded differently to HS. For SP3, the differentially expressed genes were significantly enriched in MAPK signaling, cell cycle and sugar metabolism, whereas those for SP30 were enriched in glyoxylate and dicarboxylate metabolism, and protein processing. The differentially accumulated metabolites of both strains were enriched in the glycerophospholipid metabolism, alpha-linolenic acid metabolism, biosynthesis of cofactors, etc, but with different regulatory modes. The enriched KEGG pathways for SP3 mostly tend to be down-regulated, whereas those in SP30 showed a contrary trend. The genes in MAPK signaling pathway might be associated with the glycerophospholipid metabolism in SP3, but not in SP30. Omics-integration analysis depicted distinguishable regulatory networks and discovered totally different hub genes of the two strains. Our findings revealed for the first time that the two sexually compatible nuclei responded to HS with different mechanisms and provided candidate metabolites, responsive genes and regulatory pathways for further experimental validation.
Keywords:
Lentinula edodes
; Heat stress
; metabolomics
; transcriptomics
; multi-omics integration
1. Introduction
Lentinula edodes, known as xianggu in China and shiitake in Japan, is the top 2 widely cultivated and consumed mushroom in the world. It represents 26% of the world’s total mushroom production [1]. Shiitake is recognized for its distinguishable taste, flavor, and its nutritional and medical values. It contains many biologically active compounds, including polysaccharides, lentinan, LEM and KS–2, ergosterol, nucleic acid derivatives, water–soluble lignin, eritadenine, etc., conferring it promising pharmacology effects comprising antitumor, immunomodulatory, hypocholesterolemic, antibacterial, antifungal, anti–inflammatory and antioxidant, etc. [2,3]. In China, Shiitake has been used in Chinese medicine for more than 2000 years [4].
Shiitake is broadly distributed in the subtropical to temperate regions of the northern hemisphere, and its cultivation happens worldwide, whereas most of its production is from east Aisa such as China, Japan and South Korea [5,6]. The optimum temperature for the growth of L. edodes mycelia and fruiting is 24–27 °C and 8–20 °C , respectively [7]. For shiitake production, heat stress acted as one of the major environmental stressors limiting its productivity. Temperature higher than 25°C resulted in a highly reduced commercial value of the shiitake fruiting body (small fruiting bodies, thin cap, and less open umbrellas) [8]. Temperature higher than 30°C severely inhibits mycelia growth and hinder the fruiting body formation process [9]. Consequently, heat stress can cause significant yield loss and quality deterioration of shiitake. Unfortunately, extreme heat events are expected to intensify due to the ongoing global climate change [10].
There are plenty of literatures that have documented impacts of heat stress on mycelium development of shiitake, ranging from the morphological changes to gene expression, and metabolic reprogramming. Morphologically, heat stress markedly hampers mycelium growth, and weakens the regenerate mycelium colony when heat stress is removed [4,11]. Using the microscope and SEM (scanning electron microscope), damages on the cell surface and loss of cytoplasmic inclusions could be observed upon exposure of heat [12]. Shiitake mycelium also responds to heat stress on physiological level by activates the antioxidative system to defend the damage on mycelium cell [13]. In general, heat stress can induce the elevation of reactive oxygen species (ROS) level, which subsequently triggers lipid peroxidation in cell membrane. As a results, the enzymatic (superoxide dismutase, catalase, peroxidase, etc.) and non-enzymatic (reduced glutathione, trehalose, indoleacetic acid, linoleic acid, etc.) antioxidants are synthesized to scavenge the excessive ROS to relieve cell injuries [4,8,11,12,14]. The omics-based approaches offer powerful strategies to gain a global landscape of mechanisms by which shiitake copes with heat stress. Using a combined proteome and transcriptome approach, wang et al. [12] found that the heat shock proteins (HSPs) and IAA were integral to the heat resistance of shiitake mycelium. Using transcriptomics techniques, xu et al. [15] investigated the mechanism by which 2,4-Dichlorophenoxyacetic acid (2,4-D) improving the L. edodes thermotolerance.
Probably because the dikaryotic stage represents the longest period in the life circle of shiitake, all literatures addressing its heat-resistance mechanisms has focused on the dikaryotic mycelium. Undoubtedly, the dikaryon-based heat-resistance research is of crucial importance to understand how L. edodes against thermostress. Nonetheless, with the aim of breeding for the superior heat-resistant cultivars, the importance of the dikaryon-based documents could be compromised, because current shiitake breeding practices are basically using monokaryons from the parents as parents. However, so far, no studies have investigated the mechanism by which monokaryotic mycelium confront high temperature stress. Recent genome sequencing studies on the two sexually compatible monokaryons from one L. edodes strains showed that their genomic structures harbored pronounced differences [16,17], implying that the two haploid nuclei might handle high temperature scenario differently. More recently, Yoo et al. [18] demonstrated that the monokaryons from two L. edodes strains, which were characterized by thermo-tolerance and thermo-sensitivity, might have different thermo-tolerance by a comparative genomic investigation. Unfortunately, in this study physiological and molecular evidence are lacking.
To fill the gap in understanding the monokaryons-specific mechanisms in response to heat stress, we investigated the morphological, physiological, metabolic and transcriptomic variations of two sexually compatible monokaryons from a widely cultivated L. edodes strain under normal and high temperature conditions. Our results, for the first time, demonstrate the mechanisms by which the two sexually compatible monokaryons cope with thermostress, deepening the understanding of the heat-resistance mechanisms of L. edodes.
2. Results
2.1. Thermostress Triggered ROS Production and Inhibited Mycelium Growth of the Two Monokaryons
We first examined the heat stress (HS) responses of the two sexually compatible monokaryons on the morphological and physiological levels (Figure 1). Results demonstrated that HS markedly inhibited the growth rate of mycelium for both monokaryons. When HS was removed, the regenerated mycelium appeared weak compared to the old mycelium formed prior to HS exposure (Figure 1a,b). Based on the average growth rate of the two monokaryons before and after HS, we found that the growth inhibition rate of SP3 strains was significantly higher than that of SP30 strains (Figure 1c). It suggested that the growth of SP3 might be inhibited more severely than SP30 subjected to HS. DCFH-DA and NBT staining showed that ROS contents were comparable for the two strains under room temperature, whereas were significantly increased when challenged with HS (Figure 1d,f). We also measured the H2O2 content by enzymatic assay, which showed consistent results with the ROS staining assay. The MDA production, a marker of the cell membrane lipid peroxidation, was induced significantly under HS (Figure 1e).
2.2. The Two Monokaryons Showed Distinct Metabolic Profiles under Normal and HS Conditions
Non-targeted metabolomics was applied to examine the metabolic variations of mycelium of the two monokaryons. In total, 1122 metabolites were detected (Supplementary Table S1). Based on the HMDB superclass, 33.59% of those compounds were lipids and lipid-like molecules, and 23.33% of those were organic acids and derivatives (Supplementary Figure S1A). KEGG functional annotation analysis demonstrated that most of those compounds were related to the category of metabolism, followed by the environmental information processing and genetic information processing (Supplementary Figure S1B).
The upset plot showed that there were 285 and 382 differential accumulated metabolites (DAMs) for SP3 and SP30 subjected to HS, respectively. The number of the up-regulated metabolites in SP30 was 1.8 times higher than that in SP3. There were 58 and 141 DAMs were specifically up-regulated for SP3 and SP30 under HS, respectively (Figure 2A). Principal component analysis demonstrated that the metabolic profiles of the two strains exhibited distinct differences and were also distinguishable to those subjected to HS. Interestingly, the first component (PC1), which explained 33.0% of the total variation, could explain the differences resulted from the two strains; where the second component (PC2), representing 17.1% of the total variation, could explain the variation derived from HS treatment (Figure 2B). The loading plot depicted the top 20 compounds contributed to the PCA model (Figure 2C). Figure 2D demonstrated the top 20 metabolites contributed to PC1 and PC2, respectively. With the heatmap analysis we could observe that those metabolites could classify into 3 clusters (a, b, c). The metabolites in cluster “a” were clearly related to the HS condition, whereas the cluster “b” and “c” were associated with monokaryon SP30 and SP3, respectively (Figure 2E).
2.3. The Two Monokaryons Showed Distinct Transcriptomic Profiles under Normal and HS Conditions
To identify genes and corresponding functional pathways associated with HS for the two monokaryons, we performed a comparative transcriptomics analysis. A total of 129.13 Gb clean data was obtained with a high score of quality (Q30 rate 93.58%) (Supplementary Table S2). The average mapping rate was 89.96%, and more than 84% of genes were coding sequencing for all samples (Supplementary Tables S3 and S4). The functional annotations could be found in Supplementary Table S5.
In consistent with the metabolome data, the transcriptomic profiles of the two monokaryons showed distinct variations under normal and HS conditions. The first component (PC1), which explained 65.4% of the total variation, could explain the differences resulting from the two strains; where the second component (PC1), representing 13.44% of the total variation, could explain the variation derived from the HS treatment (Figure 3A). There were 1484 and 1899 DEGs were identified from SP3 and SP30 in response to HS, respectively. The number of both the up and down-regulated genes in SP30 was higher than that in SP3 in response to HS, particularly for the up-regulated genes in SP30, which were 1.6 times higher than the numbers in SP3 (Figure 3B,C). Using the KEGG enrichment analysis, we observed that the DEGs in SP3 subjected to HS were significantly enriched in environmental information processing (MAPK signaling pathways), cell cycle, and metabolism (biosynthesis of nucleotide sugar, and amino sugar and nucleotide sugar metabolism), whereas those in SP30 were glyoxylate and dicarboxylate metabolism, and genetic information processing (protein processing in endoplasmic reticulum) (Figure 3D,E).
2.4. Identification of the Key Metabolic Pathway of the Two Monokaryons in Response to HS
With the volcano analysis, we found that 418 metabolites were differentially accumulated between monokaryon SP3 and SP30 (Figure 4A). Those DAMs were mainly enriched in the pathways of the “glycerophospholipid metabolism”, “alpha-linolenic acid metabolism”, “nucleotide metabolism”, etc. (Figure 4B). There were 124 metabolites that were up-regulated, and 181 metabolites were down-regulated when SP3 were treated with heat (Figure 4C). Those DAMs in SP3 were mainly enriched in KEGG pathways of “glycerophospholipid metabolism”, “alpha-linolenic acid metabolism”, “biosynthesis of cofactors”, etc. (Figure 4D). As for SP30, 222 metabolites were up-regulated and 159 metabolites were down-regulated under HS (Figure 4E). Those DAMs in SP30 were largely enriched in the KEGG pathways of the “alpha-linolenic acid metabolism”, “linolenic acid metabolism”, “glycerophospholipid metabolism” as showed in Figure 4F. Interestingly, for SP3 most of the top 20 altered metabolic pathways tend to be down-regulated, whereas those in SP30 presented an opposite trend. Of the top 20 altered metabolic pathways, 11 pathways were the same, while the trends were different. It worth to note that the top changed pathway “glycerophospholipid metabolism” was significantly (p0.001) down-regulated in SP3, while was significantly up-regulated (p0.01), highlighting the strain-specific differences of the metabolic profile in response to HS.
2.5. Glycerophospholipid Metabolism Pathway Analysis
Glycerophospholipid metabolism has been shown to be involved in heat stress responses in both plant [19,20,21] and edible fungi [11,22,23]. The metabolomic data showed that 16 and 13 DAMs were detected in SP3 and SP30 under HS in the glycerophospholipid metabolism pathway, respectively (supplementary Tables S6 and S7), and the transcriptomics showed that only 3 and 10 DEGs were found in the pathway in SP3 and SP30 under HS, respectively (supplementary Table S8). According to the KEGG pathway, we mapped those genes in the glycerophospholipid metabolism (Figure 5a,b). It showed that the glycerophospholipid metabolism was modulated differently in the two monokaryons in response to HS. Upon exposure to HS, only one gene was involved in the conversion of phosphatidyl-L-serine to phosphatidylethanolamine in SP3, while for SP30 four genes were involved. The phosphatidylethanolamine was then catalyzed by different enzymes by which different downstream metabolites were synthesized. To discover more DEGs which are putatively associated with regulation of glycerophospholipid metabolism, we conducted association analysis using the DEMs detected in glycerophospholipid metabolism pathway and all DEGs. The Arrow plot demonstrated a high agreement between the DEMs and DEGs data sets and the discriminative ability of each data set (Supplementary Figure S2). Using the sPLS model, we were able to detect two highly associated communities (correlation coefficients ) in the two monokaryons, respectively (Figure 5). The communities of SP3 consisted of 11 genes and 3 metabolites, where 2 metabolites were correlated with 7 genes in subcommunity I, and 1 metabolite was correlated with 4 genes in subcommunity II. As for SP30, 2 metabolites were correlated with 4 genes in subcommunity I, and 2 metabolites were correlated with 3 genes in subcommunity II (Figure 5c–f). For SP3, it was worth noticing that most of the highly associated genes were annotated as being involved in MAPK signaling pathways, which were completely different from what was observed in SP30 (Figure 5e,f).
2.6. Integrative Analysis of Upregulated Metabolites with Transcriptome
To unearth the gene regulatory network of the upregulated metabolites elicited by HS, a integrative analysis was performed using the upregulated metabolites with the DEGs of the two monokaryons. First, we examined the agreement between the upregulated metabolites and DEGs data sets using the sPLS regression model. The resulting arrow plots demonstracted a good association between the two dataset for both monokaryons (Figure 6a,b). For strain SP3, the integration analysis revealed two distinct regulatory subnetwork as a consequence of HS. The big subnetwork (subnetwork I) comprised 33 metabolites and 8 putative regulatory genes, and the subnetwork II comprised 4 metabolites and 3 putative regulatory genes. Interestingly, in subnetwork I the metabolites were all negatively correlated with genes, whereas in subnetwork II those were all positively correlated. The metabolites in subnetwork II were negatively correlated with genes in subnetwork I (Figure 6c). The top 5 hub genes included pb011955, pb003418, gb002771, pb007688, gb006174, and 3 of those genes were annotated to heat shock protein (Table 1). For SP30, 2 seperated subnetwork were found, of which the smaller subnetwork were partitioned to 2 subcommunites (subnetwork II and subnetwork III). The subnetwork I included negatively correlated metabolites and DEGs, whereas the subnetwork II and III consisted of postively correlated metabolites and DEGs (Figure 6d). The top 5 hub genes were pb006533, pb003154, gb005258, gb007318, pb008404 in SP30, which were completely different with what discovered in SP3 (Table 1).
3. Discussion
Unlike the sexual reproduction of plants and animals, of which the sperm nuclei and egg nuclei are fused to form a diploid zygote (2n), the haploid nuclei with different mating type, however, stay apart in fungal cells (n+n) [24]. The constituent nuclei can therefore be recovered from the heterokaryons as haploid monokaryons, and the intact and original genome combination can be maintained forever. Consequently, in mushroom breeding sexually compatible haploid monokaryons are important breeding materials. Recent studies showed that the two separated nuclei functioning differently during development and interactions with the surrounding environmental factors [16,25,26]. Therefore, it is crucial to uncover how the two genetically different nuclei handle environmental stimulus.
For L. edodes, heat resistance is one of the most important goals for its breeding, as high temperature events have frequently occurred in all production areas and severely reduced its productivity and fruiting body quality. The prerequisite for the improvement of its heat-resistance genetically is the knowledge regarding how shiitake addresses the high temperature condition on molecular levels. Though plenty of work has been done to elucidate the heat-resistant mechanism of shiitake, it is still far from being completely clear. Particularly, how the two physically separated monokaryons in the heterokaryotic cell function when confront high temperature is still in their infancy. To the best of our knowledge, our work is the first pioneer study attempt to uncover the mechanism by which the two sexually compatible haploid nucleus handle heat stress.
Using the combination of the omics-based strategy and phenotypic parameters, we depicted the overall bare-bones how the two haploid nucleus tickled HS. Consistent with the findings in diploid mycelium, ROS production was also elicited by high temperatures, which might contribute to the decreased growth rate of the mycelium. The extent of growth inhibition by HS suggested that the two monokaryons might have different capacity to resist HS. Further, the metabolome and transcriptome data supported the finding that the two nucleus SP3 and SP30 responded differently when challenged with high temperatures. We also found that SP30 has a greater number of DAMs and DEGs in comparison with SP3 under HS. The KEGG enrichment analysis showed that the DAMs for SP3 and SP30 enriched in many different pathways. More interestingly, the enriched KEGG pathways for SP3 mostly tend to be down-regulated, whereas those in SP30 showed a contrary trend. The glycerophospholipid metabolism pathways, which has been shown to associated with heat-resistant in edible fungi [11,22,23], were enriched in both strains in our study but with different regulatory mechanisms. This result can be a good supplement to our previous findings, which described that the glycerophospholipid metabolism was markedly enriched in the heterokaryotic L. edodes mycelium in response to HS [11]. As only a limited number of DEGs were found directly in the glycerophospholipid metabolism pathway, we performed an integrative analysis using the detected glycerophospholipid metabolites with all DEGs. We discovered that the genes in MAPK signaling pathway might be highly associated with the glycerophospholipid metabolism in SP3, which was not found in SP30. Taking together, our results support the previous findings that the glycerophospholipid metabolism pathway is related to HS responses in fungi.
Thanks to the strong correlation between our metabolome and transcriptome dataset, we were able to construct a regulatory network of the DAMs and DEGs to delve deeper on the mechanisms by which the two monokaryons cope with HS. Our omics-integration analysis revealed distinguishable regulatory networks structures between the two strains. The two monokaryons’ network possessed totally different hub genes, further supporting our idea that the two monokaryons handle HS in different strategies. In strain SP3, we were able to discover some highly correlated heat shock proteins (HSPs) which were not found in SP30. These results agreed with the importance of the role of HSPs in L. edodes heat-resistance found in dikaryotic mycelium [12,14,27], but highlight the different regulatory mechanisms for the nucleus side by side in a cell.
4. Materials and Methods
4.1. Fungal Strains and Cultivation
The two sexually compatible monokaryotic strains SP3 and SP30 were obtained from the dikaryotic L. edodes strain JZB2102217, which is widely cultivated in northern China, using protoplasting method as described by Gao et al. [17]. Mycelia from original cultures were punched out using a cork borer (1 cm diameter), and then inoculate in petri dishes (10 cm diameter) containing 35 mL of potato dextrose agar (PDA) medium as described previously [11,17]. Prior to inoculation, a sterilized cellophane membrane was placed on the surface of the PDA medium for efficient collection of the mycelium samples. The two strains were cultivated in a growth incubator at 25 °C and in permanent darkness. After 6 days of growth at 25 °C, the fungal cultures were divided into the control and treatment groups, where the latter were subjected to heat exposure at 37 °C for 12 h.
4.2. Measurement of Growth Rate
The colony diameter was recorded every two days. The inhibition rate was calculated as follows: growth inhibition rate=(G1−G2/G2)×100%, where G1 and G2 denoted the growth rate of the fungal colony after and before heat treatment. Growth rate was calculated according to the diameter change during the period of growth before and after heat treatment (mm/d).
4.3. Detection of Reactive Oxygen Species and Malondialdehyde Content
Reactive oxygen species (ROS) production was measured according to the previously described method [11,28] with modifications. For 2′,7′-dichlorodihydrofluorescein diacetate (DCHF-DA) staining, sterile glass coverslips were obliquely inserted into the petri dishes just after the inoculation of fungal blocks, which were allowed the fungal mycelium to grow on later. When the hyphae grew on the coverslips, the coverslips with hyphae were incubated in 10 μmol/L of DCHF-DA (Solarbio, Beijing, China) phosphate-buffered saline (PBS) solution for 25 min under dark conditions for ROS visualization. After staining, ROS production was visualized using a fluorescence microscope (Olympus, IX71, Tokyo, Japan). For nitroblue tetrazolium (NBT, Beyotime Biotechnology, Beijing, China) staining, mycelia pellets were incubated in 0.5 μg/ml NBT aqueous solution for 2 h, and the intensity was monitored under a light microscope. The hydrogen peroxide (H2O2) content was measured using assay kit following the manufacturer’s instructions (Solarbio Life Sciences, Beijing, China). For MDA content measurement, 0.1g of fresh mycelium samples were used for MDA extraction following the manufacture’s instruction (Solarbio, Beijing, China).
4.4. RNA Isolation, Sequencing, Differential Expression Analysis and Functional Annotation
Total RNA was extracted from mycelia of L. edodes (five replicates) using TRIzol® Reagent according to the manufacturer’s instruction (Invitrogen, Carlsbad, CA, USA). The RNA quantity determination, libraries construction, and Illumina (Illumina, San Diego, CA, USA) sequencing were performed according to the protocols described by Guo et al. [11]. The raw paired end reads were trimmed and quality controlled by SeqPrep (https://github.com/jstjohn/SeqPrep) and Sickle (https://github.com/najoshi/sickle) with default parameters. Then clean reads were separately aligned to reference genome (GCA_015476405.1) with orientation mode using HISAT2 (http://ccb.jhu.edu/software/hisat2/index.shtml) software [29]. The mapped reads of each sample were assembled by StringTie in a reference-based approach [30].
Gene expression levels (fragments per kilobases per million reads) were calculated using the RESM software (http://deweylab.biostat.wisc.edu/rsem/) [31]. Differentially expressed genes (DEGs) were identified using the DESeq2 [32] with the following general criteria:|log2FC| > 1 and padjust ≤ 0.05. The padjust was calculated using the BH (FDR correction with Benjamini/Hochberg) methods [33]. Transcripts were annotated using the functional database including Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), NCBI non-redundant protein sequences (NR), Swiss-Prot, Protein family (Pfam), and Evolutionary Genealogy of Genes: Non-supervised Orthologous Groups (EggNOG) [34].
4.5. Non-Target Metabolomics Analysis
Fifty mg of mycelium was used for metabolites measurement using an ultra-high-performance liquid-chromatography (UHPLC) system (Thermo Electron Corporation, San Jose, CA, USA), coupled with Thermo UHPLC-Q Exactive Mass Spectrometer (MS/MS) (Thermo Electron Corporation, San Jose, CA, USA). The mycelium sample preparation, metabolites extraction, chromatographical measurement, and raw chromatographical data analysis were performed following the previously described protocols [11]. Metabolic features detected at least 80% in any set of samples were retained. After filtering, minimum metabolite values were imputed for specific samples in which the metabolite levels fell below the lower limit of quantitation and each metabolic features were normalized by sum. The internal standard was used for data QC (reproducibility). Metabolic features which the relative standard deviation (RSD) of QC > 30% were discarded. Compounds were identified by comparison of accurate mass, MS/MS fragments spectra and isotope ratio difference against biochemical databases Human metabolome database (HMDB) (http://www.hmdb.ca/) and Metlin database (https://metlin.scripps.edu/). The mass tolerance between the measured m/z values and the exact mass of the components of interest was ± 10 ppm. Mass features without MS/MS spectra were tentatively annotated on MS1 level using 5.0 mDa tolerance. The metabolite abundances were quantified according to their peak areas.
Differentially expressed metabolites (VIP in OPLSDA model ≥ 1, false discovery rate (FDR)≤0.05, Fold change1.) between groups were mapped into biochemical pathways through metabolic enrichment and pathway analysis based on database search (KEGG, http://www. genome.jp/kegg/). To identify statistically significantly enriched pathway, scipy.stats (Python packages) (https://docs.scipy.org/doc/scipy/) was used with Fisher’s exact test.
4.6. Statistical Analysis
Principal Component Analysis (PCA) and orthogonal partial least square regression discriminant analysis (OPLS-DA) were performed to examine variations and reduce dimensions of the metabolomics and transcriptomic data. PCA was conducted using the “FactoMineR” [35] and “factoextra” [36] package in R (version 4.4.1). OPLS-DA was performed using the SIMCA 14.1 (Umetrics, Umeå, Sweden). The robustness of OPLS-DA models was assessed by seven-fold cross-validation. The reliability of the predictive models was assessed by analysis of variance testing of cross-validated predictive residuals (CV-ANOVA), R2 and Q2 which provide information for interpretability and predictability, respectively. The p value was estimated with paired student’s t-test on single dimensional statistical analysis. To test the overall associations between DEGs and DEMs, we performed Procrustes analysis using the R package “vegan” (version 2.7-0) [37]. Data were logarithmically (log10) transformed, centered, and pareto scaled prior to multivariate analyses [41]. For integration and visualization of transcriptome × metabolome associations, the sparse partial least squares (sPLS) regression method [38] was implemented using the R package “mixOmics” [39].
5. Conclusions
Breeding of heat-resistant strains is critical for L. edodes production, since its cultivation is frequently threatened by high temperature stress. However, unlike plants and animals, the two sexually different nucleus stay side by side in cells and harbor different genetical structures, and contribute differently during development. However, the mechanism by which the two nuclei handle HS remains unclear. Here, we found for the first time that the two sexually compatible monokaryons regulated HS using different molecular machineries and provided informative findings from the aspect of metabolomics and transcriptomics. These results offer a preliminary outline for the nuclei-specific strategy to cope with HS, and candidate metabolites, responsive genes and regulatory pathways for further experimental validation.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: The chemical diversity and summary of the KEGG functional annotation of all detected metabolites; Figure S2: The Arrow plot demonstrated agreement between DEMs detected in glycerophospholipid metabolism pathway and all DEGs. Table S1: The clean metabolome data set obtained from the raw LC-MS measurement; Table S2: Data size and quality of the transcriptome data from all samples; Table S3: The mapping quality of the reads to the reference genome; Table S4: The positions of the reads on the reference genome; Table S5: Statistics of the annotated genes in different databases. Table S6: The differentially accumulated metabolites detected in the glycerophospholipid metabolism pathways in SP3 under heat stress; Table S7: The differentially accumulated metabolites detected in the glycerophospholipid metabolism pathways in SP30 under heat stress; Table S8: The differentially expressed genes detected in the glycerophospholipid metabolism for SP3 and SP30 under heat stress.
Author Contributions
Conceptualization, Y.G. and S.W.; methodology, Y.G.; software, W.J.; validation, Y.G., Y.Z. and M.T.; formal analysis, Y.G. W.J. and Q.G.; investigation, Y.G., W.J. Y.Z. and M.T.; resources, Y.L. and Q.G.; data curation, Y.G. and W.J.; writing—original draft preparation, Y.G.; writing—review and editing, Q.G., Y.L. and S.W.; visualization, Y.G. and W.J.; supervision, Y.L. and S.W.; project administration, Y.G. and S.W.; funding acquisition, Y.G. and S.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Beijing Academy of Agriculture and Forestry Sciences (BAAFS) Program (grant number: KJCX20230419), the China Agriculture Research System (grant number: CARS-20), and the Beijing Innovation Consortium of Agriculture Research System (grant number: BAIC03).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original data presented in the study are openly available in the National Genomics Data Center, China National Center for Bioinformation, under project of PRJCA031400.
Acknowledgments
The authors thank Ms. ZJ song for technical assistance on the recovery of strains.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Singh, M.; Kamal, S.; Sharma, V.P. Status and trends in world mushroom production-III-world production of different mushroom species in 21st century. Mushroom Res. 2020, 29, 75–111. [Google Scholar] [CrossRef]
- Ahmad, I.; Arif, M.; Xu, M.; Zhang, J.; Ding, Y.; Lyu, F. Therapeutic values and nutraceutical properties of shiitake mushroom (Lentinula edodes): A review. Trends Food Sci. Technol. 2023, 134, 123–135. [Google Scholar] [CrossRef]
- Xu, X.; Yu, C.; Liu, Z.; Cui, X.; Guo, X.; Wang, H. Chemical composition, antioxidant and anti-inflammatory activity of shiitake mushrooms (lentinus edodes). J. Fungi 2024, 10, 552. [Google Scholar] [CrossRef]
- Zhang, Q.; Feng, R.; Miao, R.; Lin, J.; Cao, L.; Ni, Y.; Li, W.; Zhao, X. Combined transcriptomics and metabolomics analysis reveals the molecular mechanism of heat tolerance of Le023M, a mutant in Lentinula edodes. Heliyon 2023, 9. [Google Scholar] [CrossRef]
- Sierra-Patev, S.; Min, B.; Naranjo-Ortiz, M.; Looney, B.; Konkel, Z.; Slot, J.C.; Sakamoto, Y.; Steenwyk, J.L.; Rokas, A.; Carro, J. A global phylogenomic analysis of the shiitake genus Lentinula. Proc. Natl. Acad. Sci. U. S. A 2023, 120, e2214076120. [Google Scholar] [CrossRef]
- Menolli Jr, N.; Sanchez-Ramirez, S.; Sanchez-Garcia, M.; Wang, C.; Patev, S.; Ishikawa, N.K.; Mata, J.L.; Lenz, A.R.; Vargas-Isla, R.; Liderman, L. Global phylogeny of the shiitake mushroom and related Lentinula species uncovers novel diversity and suggests an origin in the Neotropics. Mol. Phylogenet. and Evo. 2022, 173, 107494. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Chen, M.; Zhao, Y.; Zha, L.; Yang, H.; Wu, Y. GC–MS-based nontargeted and targeted metabolic profiling identifies changes in the Lentinula edodes mycelial metabolome under high-temperature stress. Int. J. Mol. Sci. 2019, 20, 2330. [Google Scholar] [CrossRef]
- Yang, H.; Jiang, J.; Chen, M.; Song, X.; Yu, C.; Chen, H.; Zhao, Y. Homologous delta-12 fatty acid desaturase (fad2) genes affect gene expression and linoleic acid levels in Lentinula edodes under heat stress. J. Fungi 2024, 10, 496. [Google Scholar] [CrossRef]
- Shen, Y.; Cai, W.; Zhou, S.; Jin, Q.; Feng, W.; Fan, L.; Song, T.; Li, L. Phenotype analysis of mycelium growth regeneration after heat stress in a Lentinula edodes F2 population. J. Hortic. Sci. Biotechnol 2017, 92, 397–403. [Google Scholar] [CrossRef]
- González-García, M.P.; Conesa, C.M.; Lozano-Enguita, A.; Baca-González, V.; Simancas, B.; Navarro-Neila, S.; Sánchez-Bermúdez, M.; Salas-González, I.; Caro, E.; Castrillo, G. Temperature changes in the root ecosystem affect plant functionality. Plant Commun. 2023, 4. [Google Scholar] [CrossRef]
- Guo, Y.; Gao, Q.; Fan, Y.; Song, S.; Yan, D.; Zhao, J.; Chen, Y.; Liu, Y.; Wang, S. Two strains of Lentinula edodes differ in their transcriptional and metabolic patterns and respond differently to thermostress. J. Fungi 2023, 9, 179. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.-Z.; Ma, C.-J.; Luo, Y.; Zhou, S.-S.; Zhou, Y.; Ma, X.-L.; Cai, Y.-L.; Yu, J.-J.; Bian, Y.-B.; Gong, Y.-H. Proteome and transcriptome reveal involvement of heat shock proteins and indoleacetic acid metabolism process in Lentinula edodes thermotolerance. Cell. Physiol. Biochem. 2018, 50, 1617–1637. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Zhang, S.; Wu, J.; Sun, X.; Ma, A. Heat stress in macrofungi: effects and response mechanisms. Appl. Microbiol. Biotechnol. 2021, 105, 7567–7576. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Luo, Y.; Wang, C.; Zhou, Y.; Mou, C.; Kang, H.; Xiao, Y.; Bian, Y.; Gong, Y.H. Hsp40 protein LeDnaJ07 enhances the thermotolerance of Lentinula edodes and regulates IAA biosynthesis by interacting LetrpE. Front. Microbiol. 2020, 11, 707. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Zhou, S.; Song, J.; Zhong, H.; Zhu, T.; Gong, Y.; Zhou, Y.; Bian, Y. Comparative transcriptome analysis provides insights into the mechanism by which 2,4-dichlorophenoxyacetic acid improves thermotolerance in Lentinula edodes. Front. Microbiol. 2022, 13, 910255. [Google Scholar] [CrossRef] [PubMed]
- Shen, N.; Xie, H.; Liu, K.; Li, X.; Wang, L.; Deng, Y.; Chen, L.; Bian, Y.; Xiao, Y. Near-gapless genome and transcriptome analyses provide insights into fruiting body development in Lentinula edodes. Int. J. Biol. Macromol. 2024, 263, 130610. [Google Scholar] [CrossRef]
- Gao, Q.; Yan, D.; Song, S.; Fan, Y.; Wang, S.; Liu, Y.; Huang, Y.; Rong, C.; Guo, Y.; Zhao, S. Haplotype-resolved genome analyses reveal genetically distinct nuclei within a commercial cultivar of Lentinula edodes. J. Fungi 2022, 8, 167. [Google Scholar] [CrossRef]
- Yoo, S.-i.; Moon, S.; Hong, C.P.; Park, S.-G.; Shim, D.; Ryu, H. Genome sequencing of Lentinula edodes revealed a genomic variant block associated with a thermo-tolerant trait in fruit body formation. J. Fungi 2024, 10, 628. [Google Scholar] [CrossRef]
- Feng, B.; Xiong, J.; Tao, L. How rice plants response to abiotic stresses. Int. J. Mol. Sci. 2023, 24, 12806. [Google Scholar] [CrossRef]
- Zhou, J.; Tang, X.; Li, J.; Dang, S.; Ma, H.; Zhang, Y. Comparative transcriptomic and metabolomic analyses provide insights into the responses to high temperature stress in alfalfa (Medicago sativa L.). BMC Plant Bio. 2024, 24, 776. [Google Scholar] [CrossRef]
- Sun, M.; Huang, D.; Zhang, A.; Khan, I.; Yan, H.; Wang, X.; Zhang, X.; Zhang, J.; Huang, L. Transcriptome analysis of heat stress and drought stress in pearl millet based on Pacbio full-length transcriptome sequencing. BMC Plant Bio. 2020, 20, 323. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Ge, J.; He, B.; Zhang, Z.; Hu, Z.; Li, Y.; Zeng, B. Transcriptomic analysis reveals Aspergillus oryzae responds to temperature stress by regulating sugar metabolism and lipid metabolism. Plos one 2022, 17, e0274394. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Zhao, M.; Wu, X.; Zhang, J. Metabolic response of Pleurotus ostreatus to continuous heat stress. Front. Microbiol 2020, 10, 3148. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuis, B.P.; James, T.Y. The frequency of sex in fungi. Philos. T. R. Soc. B. 2016, 371, 20150540. [Google Scholar] [CrossRef] [PubMed]
- Sperschneider, J.; Yildirir, G.; Rizzi, Y.S.; Malar C, M.; Mayrand Nicol, A.; Sorwar, E.; Villeneuve-Laroche, M.; Chen, E.C.H.; Iwasaki, W.; Brauer, E.K.; et al. Arbuscular mycorrhizal fungi heterokaryons have two nuclear populations with distinct roles in host–plant interactions. Nat. Microbiol. 2023, 8, 2142–2153. [Google Scholar] [CrossRef] [PubMed]
- Gehrmann, T.; Pelkmans, J.F.; Ohm, R.A.; Vos, A.M.; Sonnenberg, A.S.M.; Baars, J.J.P.; Wösten, H.A.B.; Reinders, M.J.T.; Abeel, T. Nucleus-specific expression in the multinuclear mushroom-forming fungus <i>Agaricus bisporus</i> reveals different nuclear regulatory programs. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 4429–4434. [Google Scholar] [PubMed]
- Ling, Y.-Y.; Ling, Z.-L.; Zhao, R.-L. Construction of a heat-resistant strain of Lentinus edodes by fungal Hsp20 protein overexpression and genetic transformation. Front. Microbiol. 2022, 13, 1009885. [Google Scholar] [CrossRef] [PubMed]
- Ren, A.; Liu, R.; Miao, Z.G.; Zhang, X.; Cao, P.F.; Chen, T.X.; Li, C.Y.; Shi, L.; Jiang, A.L.; Zhao, M.W. Hydrogen-rich water regulates effects of ROS balance on morphology, growth and secondary metabolism via glutathione peroxidase in Ganoderma lucidum. Environ. Microbiol. 2017, 19, 566–583. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinf. 2011, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biol. 2014, 15, 1–21. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc., Ser. B, Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Zhang, J.; Ye, L.; Chen, Q.; Wang, F. Response analysis of Pinus sibirica to pine wood nematode infection through transcriptomics and metabolomics study. Front. Plant Sci. 2024, 15, 1383018. [Google Scholar] [CrossRef]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: an R package for multivariate analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef]
- Kassambara, A.; Mundt, F. Package ‘factoextra’. Extract and visualize the results of multivariate data analyses. Available online: https://cran.r-project.org/web/packages/factoextra/index.html (accessed on 25.10.2024.
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Lê Cao, K.-A.; Rossouw, D.; Robert-Granié, C.; Besse, P. A sparse PLS for variable selection when integrating omics data. Stat. Appl. Genet. Mol. Biol. 2008, 7. [Google Scholar] [CrossRef]
- Rohart, F.; Gautier, B.; Singh, A.; Lê Cao, K.-A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef]
Figure 1.
The morphological and physiological responses of SP3 and SP30 strains under control (25 °C) and heat stress (HS, 37 °C); The fungal hyphae of two strains were able to recovery growth at 2 days after HS; (a) The morphology of SP3 and SP30 cultures at 1 and 15 days after inoculation (7th days after HS); (b) The fungal colony diameters of different strains. HS was conducted at 6th day after inoculation; (c) The growth rate and growth inhibition rate of two strains in response to HS; (d) The ROS staining (bar = 50 μm) and mean fluorescence intensity. Different letters indicate significant differences between groups (ANOVA, p < 0.05,). Data represents as mean ± se (n = 5); (e) The H2O2 and MDA contents of the two strains with and without HS. The values above the boxes showed the p value of Kruskal-Wallis test; (f) NBT staining of the two strains with and without HS.
Figure 1.
The morphological and physiological responses of SP3 and SP30 strains under control (25 °C) and heat stress (HS, 37 °C); The fungal hyphae of two strains were able to recovery growth at 2 days after HS; (a) The morphology of SP3 and SP30 cultures at 1 and 15 days after inoculation (7th days after HS); (b) The fungal colony diameters of different strains. HS was conducted at 6th day after inoculation; (c) The growth rate and growth inhibition rate of two strains in response to HS; (d) The ROS staining (bar = 50 μm) and mean fluorescence intensity. Different letters indicate significant differences between groups (ANOVA, p < 0.05,). Data represents as mean ± se (n = 5); (e) The H2O2 and MDA contents of the two strains with and without HS. The values above the boxes showed the p value of Kruskal-Wallis test; (f) NBT staining of the two strains with and without HS.
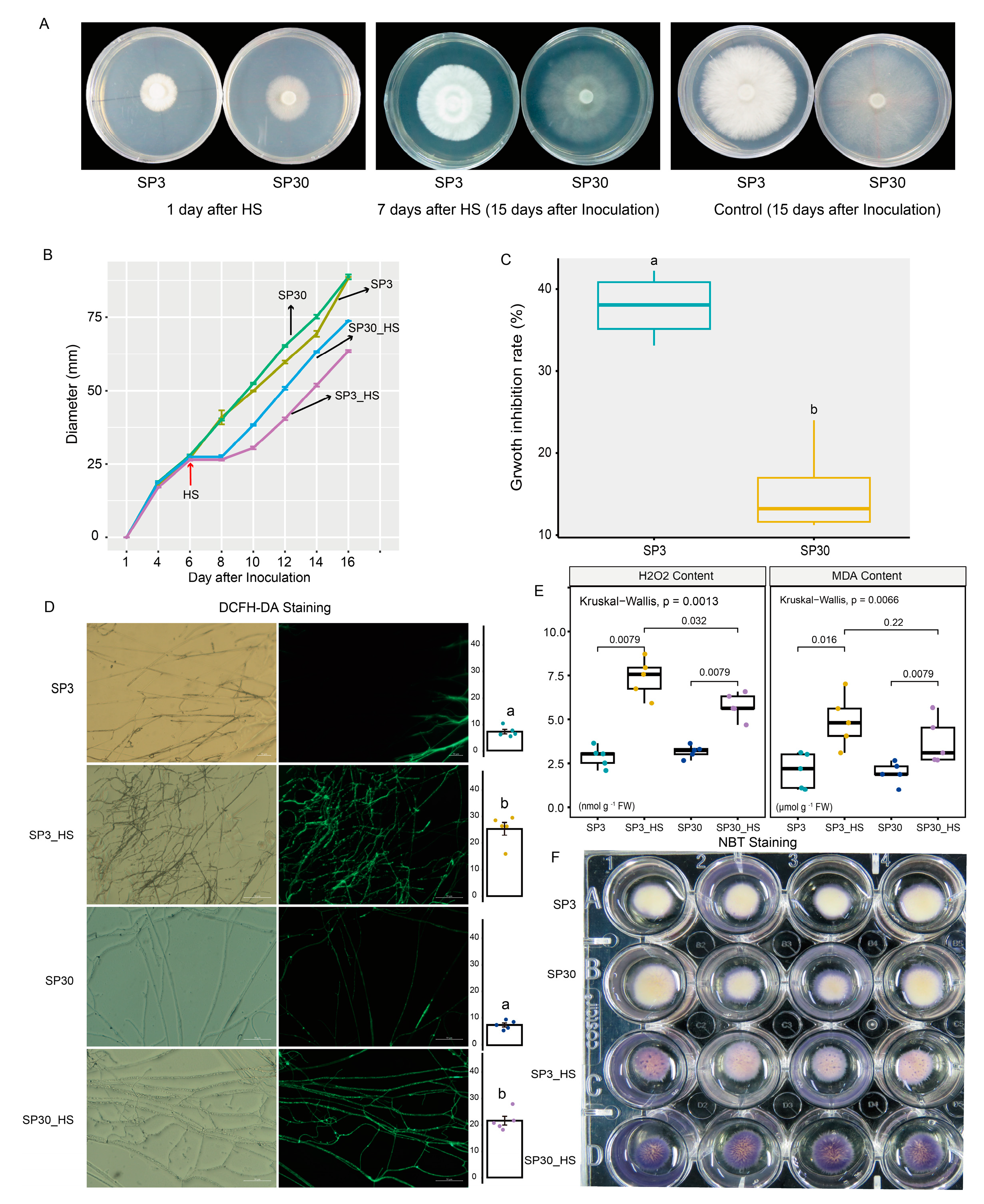
Figure 2.
Metabolites detected from SP3 and SP30 strains under control (25 °C) and heat stress (HS, 37 °C). (a) The Venn analysis for the differential accumulated metabolites (DEMs). The DEMs were identified following the criteria: false discovery rate (FDR)0.05, Variable influence on projection (VIP)1 in OPLS-DA model, Fold change1. (b) PCA score plot generated using all the metabolites. (c) PCA loading plot showed the top 20 metabolites contributed to the PCA model. (d) Contributions of the top 20 metabolites for PC1 and PC2. (e) Heatmap analysis of the top 20 metabolites for PC1 and PC2. The dendrograms showed the hierarchical cluster analysis for the two strains and metabolites. .
Figure 2.
Metabolites detected from SP3 and SP30 strains under control (25 °C) and heat stress (HS, 37 °C). (a) The Venn analysis for the differential accumulated metabolites (DEMs). The DEMs were identified following the criteria: false discovery rate (FDR)0.05, Variable influence on projection (VIP)1 in OPLS-DA model, Fold change1. (b) PCA score plot generated using all the metabolites. (c) PCA loading plot showed the top 20 metabolites contributed to the PCA model. (d) Contributions of the top 20 metabolites for PC1 and PC2. (e) Heatmap analysis of the top 20 metabolites for PC1 and PC2. The dendrograms showed the hierarchical cluster analysis for the two strains and metabolites. .
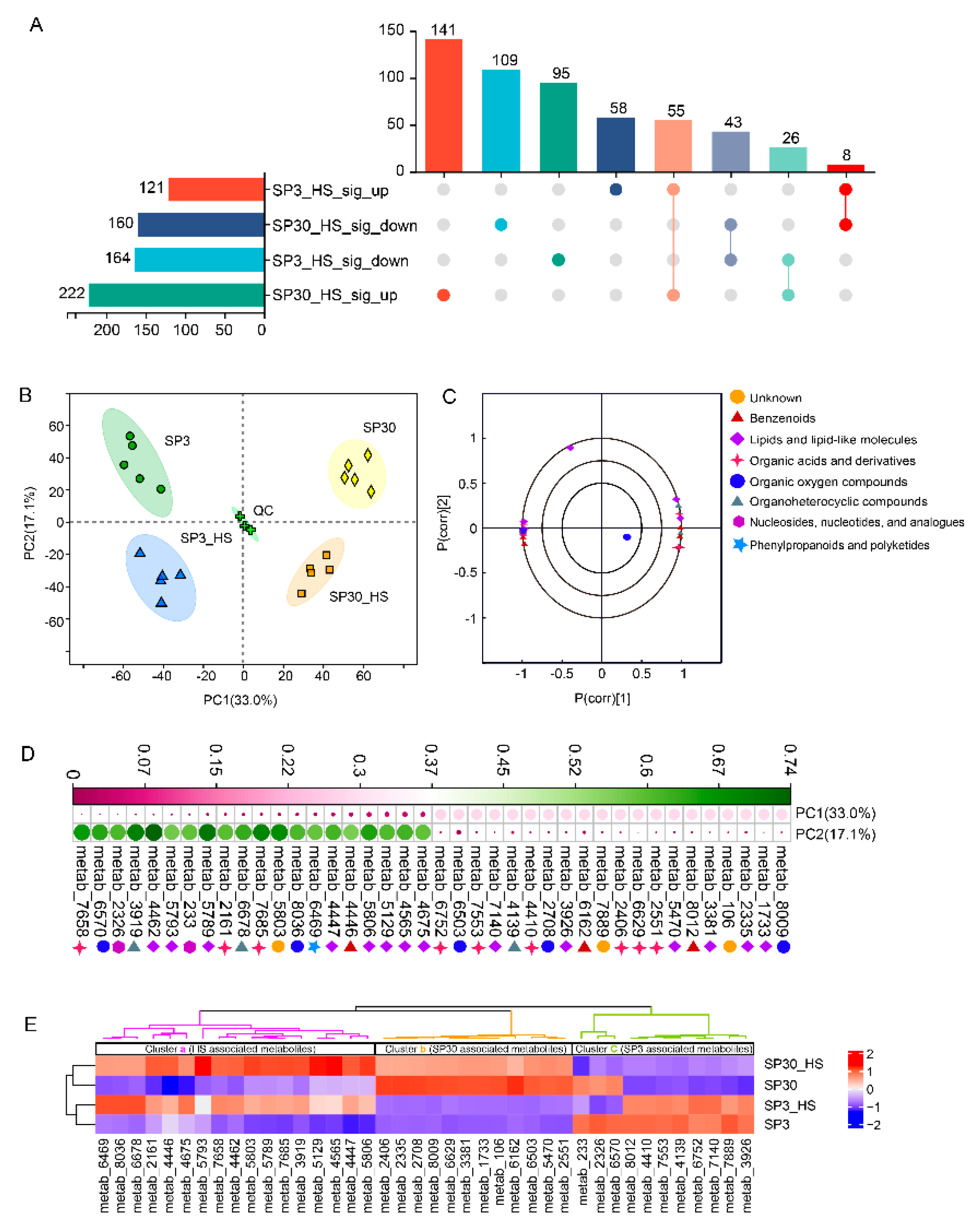
Figure 3.
Transcriptomic analysis of SP3 and SP30 in response to heat stress. (a) Score plot of principal component analysis. (b), (c) Scatter plots of all expressed genes for SP30 and SP3 under HS, respectively. X-axis and Y-axis present log10 value of gene expression. Blue points represent down-regulation genes, red points denote upregulation genes and grey points denote genes with no significant differences. (d), (e) KEGG enrichment analysis of the deferentially expressed genes for SP3 and SP30 under HS, respectively.
Figure 3.
Transcriptomic analysis of SP3 and SP30 in response to heat stress. (a) Score plot of principal component analysis. (b), (c) Scatter plots of all expressed genes for SP30 and SP3 under HS, respectively. X-axis and Y-axis present log10 value of gene expression. Blue points represent down-regulation genes, red points denote upregulation genes and grey points denote genes with no significant differences. (d), (e) KEGG enrichment analysis of the deferentially expressed genes for SP3 and SP30 under HS, respectively.
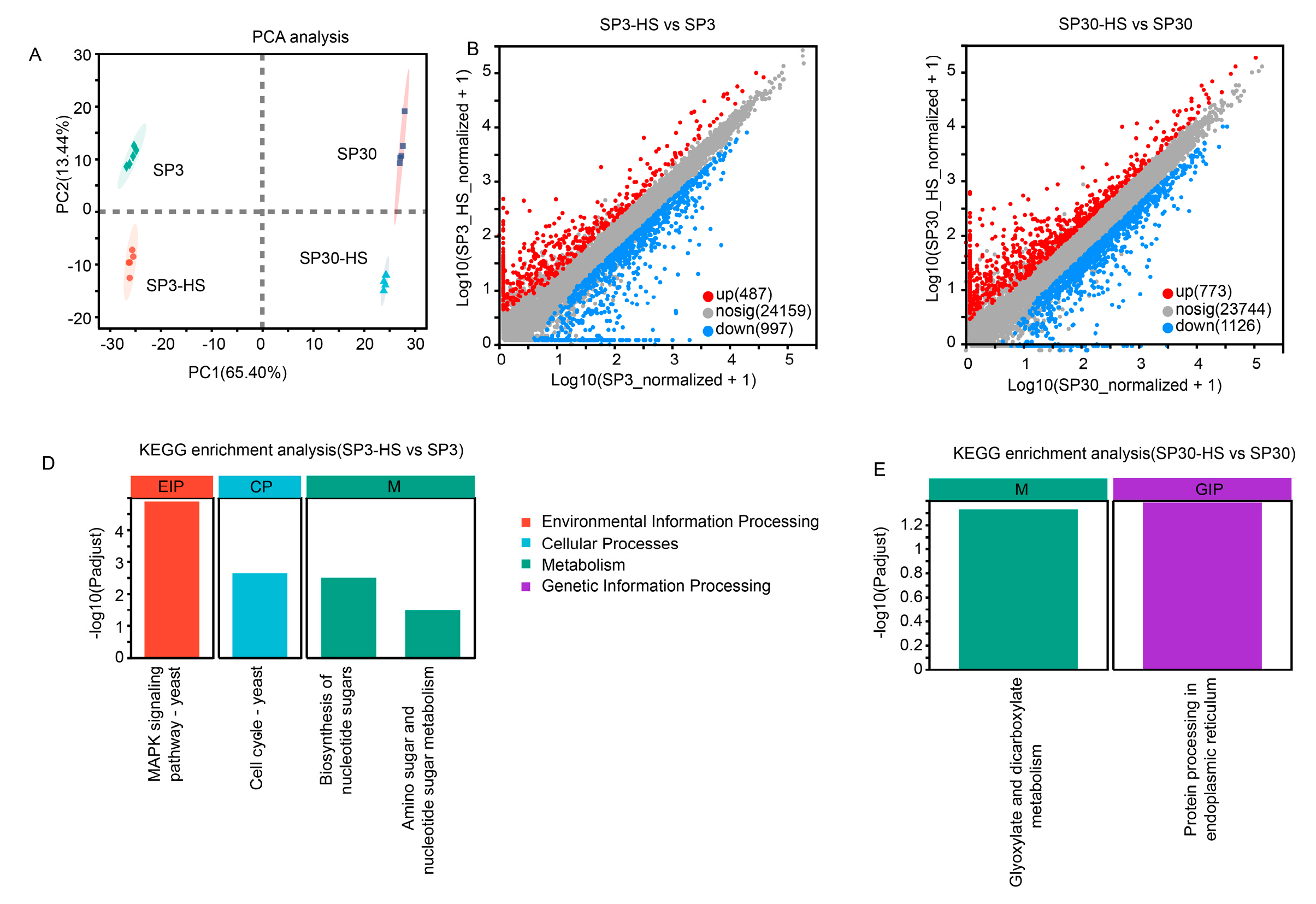
Figure 4.
Differentially accumulated metabolites (DAMs) analysis of SP3 and SP30 under normal and HS condition. (a), (b) Volcano plot depicts the DAMs, and their KEGG enrichment analysis between the two strains under normal condition, respectively. (c), (d) Volcano plot depicts the DAMs, and their KEGG enrichment analysis of SP3 under HS condition, respectively. (e), (f) Volcano plot depicts the DAMs, and their KEGG enrichment analysis of SP30 under HS condition, respectively. For volcano plot, the size of points indicates the VIP value in the corresponding OPLS-DA model. For the differential abundance score analysis, the size of the point denotes the number of metabolites in the pathways. The length of the line represents the absolute value of DA score. Positive and negative DA scores represent the pathway tend to be up- and down-regulated, respectively.
Figure 4.
Differentially accumulated metabolites (DAMs) analysis of SP3 and SP30 under normal and HS condition. (a), (b) Volcano plot depicts the DAMs, and their KEGG enrichment analysis between the two strains under normal condition, respectively. (c), (d) Volcano plot depicts the DAMs, and their KEGG enrichment analysis of SP3 under HS condition, respectively. (e), (f) Volcano plot depicts the DAMs, and their KEGG enrichment analysis of SP30 under HS condition, respectively. For volcano plot, the size of points indicates the VIP value in the corresponding OPLS-DA model. For the differential abundance score analysis, the size of the point denotes the number of metabolites in the pathways. The length of the line represents the absolute value of DA score. Positive and negative DA scores represent the pathway tend to be up- and down-regulated, respectively.
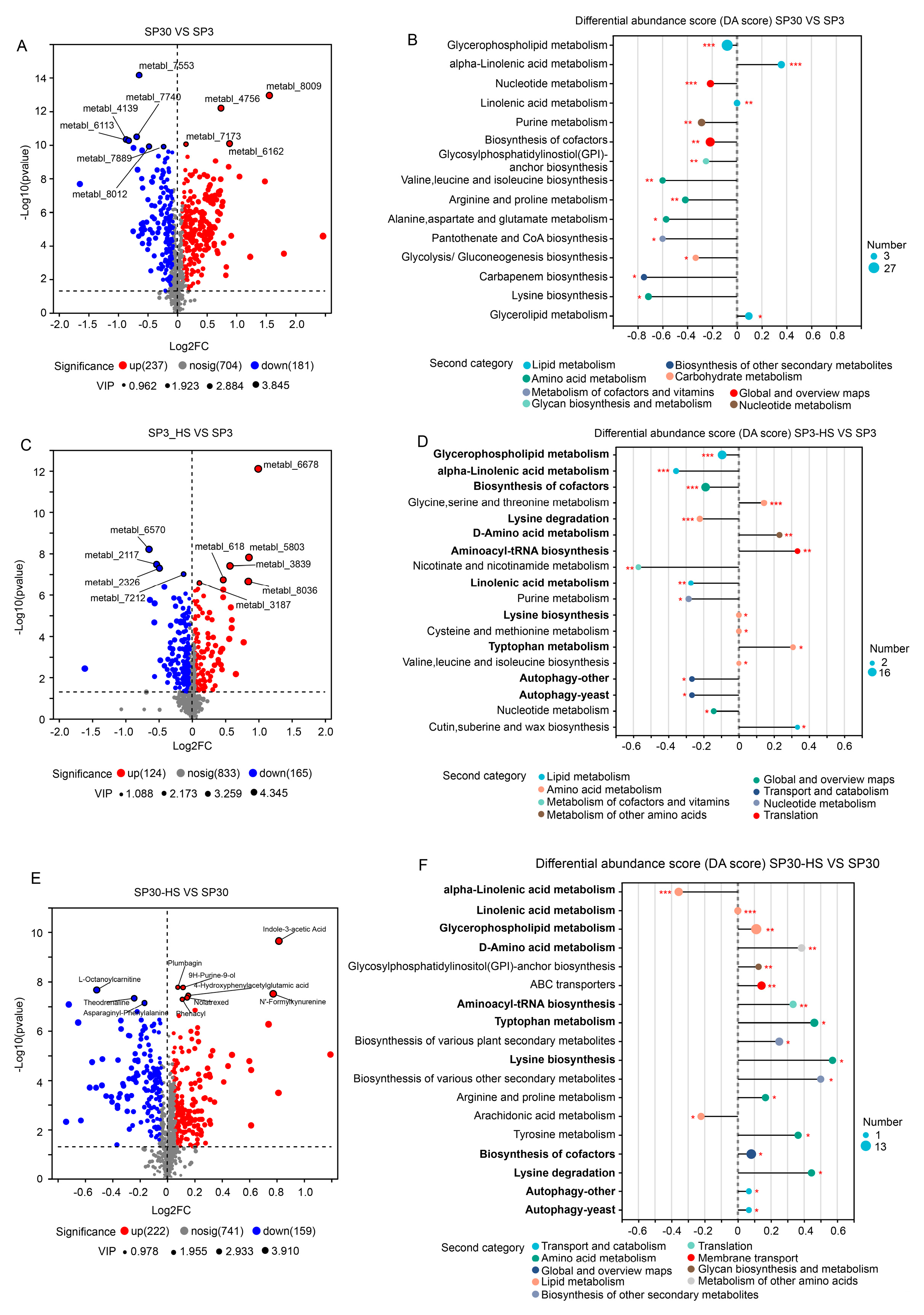
Figure 5.
Putative modulation of glycerophospholipid metabolism pathway in SP3 and SP30 under heat stress. (a), (b) Simplified representation of DEGs and DEMs in glycerophospholipid metabolism pathway. (c), (d), (e), (f) Integration analysis of DEMs in glycerophospholipid metabolism pathway and DEGs using sPLS method (cut off threhold 0.9). .
Figure 5.
Putative modulation of glycerophospholipid metabolism pathway in SP3 and SP30 under heat stress. (a), (b) Simplified representation of DEGs and DEMs in glycerophospholipid metabolism pathway. (c), (d), (e), (f) Integration analysis of DEMs in glycerophospholipid metabolism pathway and DEGs using sPLS method (cut off threhold 0.9). .
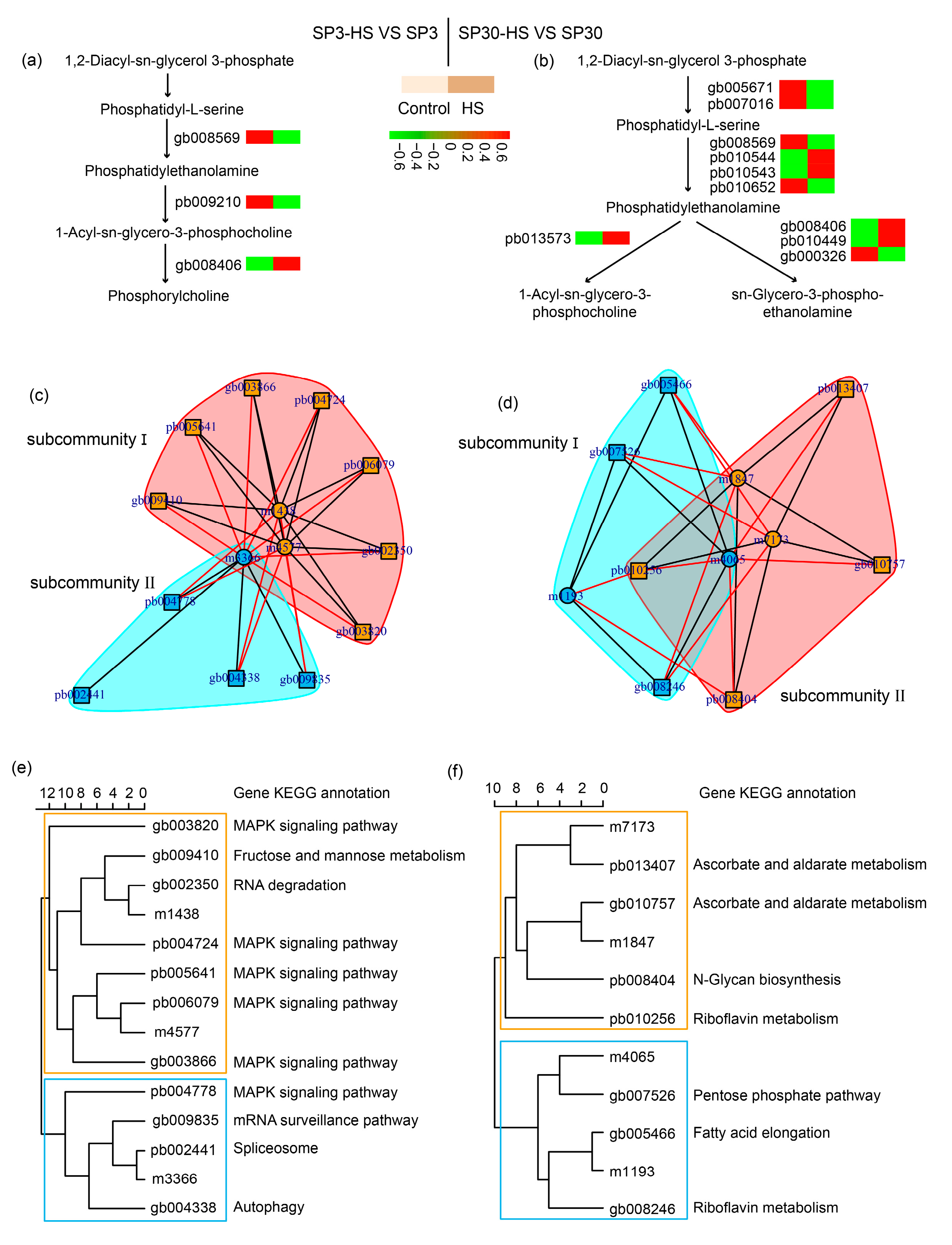
Figure 6.
Transcriptome–metabolome wide association analysis of SP3 and SP30 under HS. (a), (b) The sparse partial least squares (sPLS) association analysis between the upregulated metabolites and all DEGs. (c), (d) The integration network of highly associated genes and metabolites at threshold of 0.9. Positive correlation were depicted with red edges, and negative correlations were represented using blue and light blue edges. The wideth of edges denoted the correlation coefficients between nodes.
Figure 6.
Transcriptome–metabolome wide association analysis of SP3 and SP30 under HS. (a), (b) The sparse partial least squares (sPLS) association analysis between the upregulated metabolites and all DEGs. (c), (d) The integration network of highly associated genes and metabolites at threshold of 0.9. Positive correlation were depicted with red edges, and negative correlations were represented using blue and light blue edges. The wideth of edges denoted the correlation coefficients between nodes.
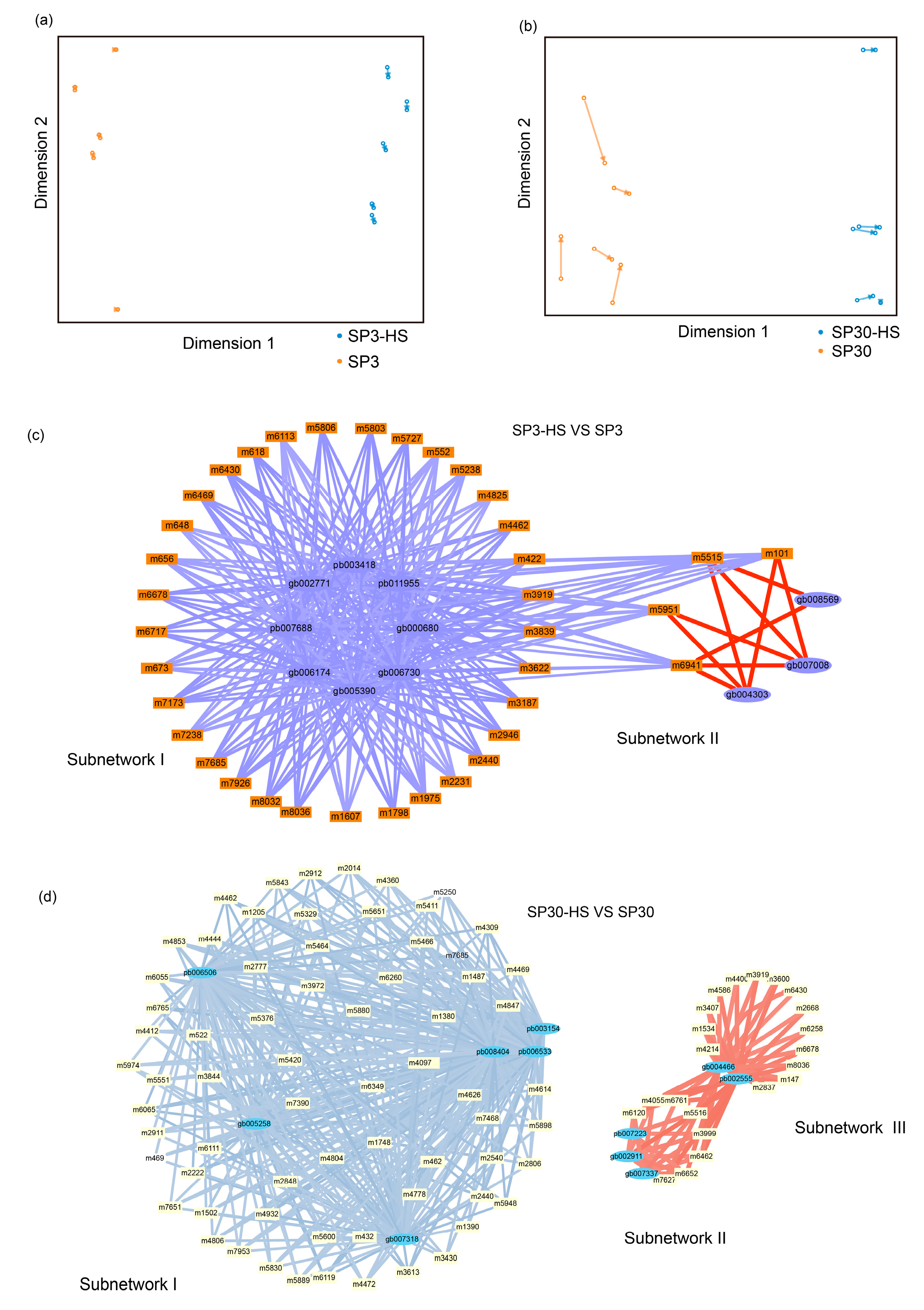

Table 1.
The top 5 hub genes for strain SP3 and SP30 under heat stress. Hub genes were identified by the number of edges adjacent to the node i.e. node degree in the network. .
Table 1.
The top 5 hub genes for strain SP3 and SP30 under heat stress. Hub genes were identified by the number of edges adjacent to the node i.e. node degree in the network. .
| HSSP3 VS SP3 | ||||
| Gene ID | Log2FC | Padjust | KEGG | Swiss-Prot description |
| pb011955 | 3.015065 | 8.11E-205 | Protein processing in endoplasmic reticulum | Heat shock protein 16 |
| pb003418 | 2.197605 | 9.04E-178 | Longevity regulating pathway | Heat shock protein 104 |
| gb002771 | 3.061065 | 2.23E-101 | Longevity regulating pathway | Heat shock protein 104 |
| pb007688 | 1.614501 | 1.09E-134 | Biotin metabolism | Biotin--protein ligase |
| gb006174 | 1.614501 | 1.09E-134 | Biotin metabolism | Biotin--protein ligase |
| HSSP30VS SP30 | ||||
| pb006533 | -5.35584 | 2.07E-241 | - | |
| pb003154 | -1.10127 | 1.19E-67 | - | Uncharacterized secreted protein ARB_0804 |
| gb005258 | -6.18295 | 0 | - | - |
| gb007318 | -1.38438 | 1.03E-70 | - | gmc oxidoreductase |
| pb008404 | -1.24555 | 9.01E-170 | Protein processing in endoplasmic reticulum | Probable mannosyl-oligosaccharide alpha-1,2-mannosidase 1B |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



