
Submitted:
25 October 2024
Posted:
29 October 2024
You are already at the latest version
Abstract
Cosmopolitan character, and proven high and differentiated efficiency of Cd hyperaccumultor Solanum nigrum L. suggest possibility of optimizing its Cd phytoremediation capacity and applicability through searching among remote ecotypes/genotypes. However, the extensive studies on this hyperaccumulator have been limited to Far East (Asian) regions. Pioneer pot experiments on Middle European ecotype of S. nigrum with the use of concentration range 0-50 mg kg-1 Cd in soil, revealed its comparable to Asian ecotypes Cd phytoremediation capacity, but fundamentally different Cd tolerance threshold. It manifested itself in sharp biomass decline at Csoil ≈ 10 mg kg-1 Cd in Asian ecotypes versus gradual mild biomass decrease within the whole Csoil ≈ 0 -50 mg kg-1 Cd range with no crisis symptoms in Middle European ecotype. In its A50 variety adapted by using seeds originated from the first-generation plants grown in soil with Csoil ≈ 50 mg kg-1 Cd, cadmium tolerance, accumulation performance and all physiological parameters (chlorophyll, carotenoids, RuBisCO, and first- and second line defense antioxidant activity) were significantly enhanced while cell damage by ROS was considerably lesser. This makes Middle European ecotype and its adapted variety A50 particularly useful to sustainable decontamination of heavily polluted “hot spots” in the degraded post-industrial areas.
Keywords:
Cadmium phytoremediation
; Solanum nigrum L. Middle European vs. Asian ecotypes
; Cd hyperaccumulative properties
; hyperaccumulation mechanism differentiation
; adapted variety efficiency
; physiological parameter response
; ecotype stress tolerance
; heavily polluted soil decontamination
1. Introduction
Cadmium (Cd) is one of the most widespread problematic non-essential priority pollutants of soils due to multitude of historic and contemporary, geogenic and anthropogenic diffuse pollution sources (emissions to air and waste from smelting/mining industries and fossil fuel- or biomass-fired power plants, fertilizers, agrochemicals and manure, sewage sludge and waste-based soil amendments applied to soil within circular economy implementation, and often contaminated irrigation water) [1]. High mobility in the environment and long range transmission of pollutants (LRTP) with air fluxes caused occurrence of high diffuse soil pollution with Cd far away from emission sources [2]. High susceptibility to accumulation in tissues of some basic agricultural products (e.g. rice, root and leafy vegetables) may increase Cd daily intake by consumers to the level exceeding daily tolerable limits, thus particularly strongly endangering human health. Widespread Cd pollution of soils has induced searching for non-invasive, efficient and cost-effective methods of its reduction to the level assuring food safety and ecological sustainability (in the farmlands, dictated mostly by cultivated crops and their ability to accumulate Cd in the edible parts). Development in the last decades of phytoremediation methods with the use of plants hiperaccumulating pollutants create promising prospects of non-invasive, low-cost, operationally simple and environmentally acceptable reduction of soil pollution. However, due to non-essentiality and strong toxicity of Cd to plants, only scarce species display tolerance to Cd and hyperaccumultion properties. In hyperaccumulator database 2017, only 7 species were registered as Cd hyperaccumulators in 2017 [3], and not many species, mostly endemic or exotic, were added to this list in the following years. Among them, entirely Solanum nigrum L. discovered as efficient Cd hyperaccumulator recently [4], occurs as a common wild plant worldwide. Moreover, it appeared that different ecotypes of S. nigrum grown far away from each other, display significantly different Cd tolerance and hyperaccumulation capacity, thus creating possibility of selecting the most efficient hyperaccumulator just by comparing different ecotypes [5]. Surprisingly enough, despite high interest to phytoremediation of soils polluted with Cd, and hundreds of studies on S. nigrum, they are concentrated mostly in Far East region, while the reports originating from other parts of the world are scarce and often thematically marginal [6,7,8,9,10,11,12,13,14]. Simultaneously, since the beginning of the industrial revolution at the end of 18th century, Cd remains to be one of the most problematic pollutants in Northern Hemisphere, and specifically in Europe due to historically the longest strong impact of agrochemicals and emissions from heavy industry and power production, and long-rang transport with LRTP, Poland and Germany being currently the biggest Cd emitters and suppliers to land of many other European countries [15]. In surface soils of mining areas, heavy industrial regions, urban areas and farmlands with extensive agriculture in Europe, USA and Canada, Cd occurs in concentrations at the level of 10 mg kg-1 up to roughly 100-300 mg kg-1, occasionally reaching even bigger values, which leads to excessive levels of Cd in edible plants dangerous to human health [1]. Such areas, mostly thickly populated (e.g. in Upper Silesia industrial region in Poland or in Ruhr in Germany) must be efficiently remediated. Phytoremediation appears to be the best non-invasive, cost-effective and sustainable option, and S. nigrum as common cosmopolitan weed plant of well confirmed Cd hyperaccumulative properties seems to be a perfect candidate for this purpose.
Solanum nigrum L. ssp. nigrum is an annual herb belonging to large Solanaceae family, the section Solanum identified as Cd hyperaccumulator appeared to have naturally high Cd hyperaccumulation capacity independent on soil properties [4,16,17,18]. Simultaneously, the recent study [5] revealed that the ecotypes of the same species Solanum nigrum L. ssp. nigrum grown in different remote sites isolated from each other may show distinctly differentiated Cd hyperaccumulation capacities that was reflected also by different tolerance of studied ecotypes to Cd stress. Notwithstanding this, all different ecotypes of S. nigrum show diverse but strong tolerance to Cd stress and could effectively protect themselves from Cd toxicity.
The aim of this pioneer study was to assess Cd hyperaccumulating capacity of a common Middle-European ecotype of S. nigrum that has never been studied before, and its adaptability to Cd stress. It was hypothesized that its natural tolerance to Cd stress may be enhanced in the next generation of plants growing in soil with high concentrations of Cd due to inherited adaptation ability. This would allow to use the adapted native plants for effective phytoremediation of highly Cd-polluted areas occurring in thickly populated industrial and post-industrial regions and contribute to revival of their environmental status and sustainability.
2. Materials and Methods
2.1. Soil Characteristics and Experimental Design
In the experiment, seeds of a Middle-European ecotype of Solanum nigrum L. ssp. nigrum originated from the City Botanical Garden in Zabrze (Upper Silesia, Poland) (N50°17'45.81" E18°45'52.37") where the plants were grown freely as weeds, were used as a primary experimental material. The experiment was carried out at the Institute of Environmental Engineering, Polish Academy of Sciences in Zabrze. The topsoil (0-20 cm) was collected from the area of seeds origin and analyzed according to standard methods of soil analysis [19]. The soil was slightly acidic (pH 6.57), total organic carbon TOC 16.73 g kg-1, total N 0.73 g kg-1, available P 15.33 mg kg-1, available K 189.05 mg kg-1, Cd 0.22 mg kg-1, Pb 17,15 mg kg-1, and Zn 37.88 mg kg-1. Cadmium (Cd) and other PTE - Potentially Toxic Elements (Pb, Zn) represented low natural level. The test soil was brown soil according to the World Reference Base for Soil Resources WRB (IUSS Working Group WRB 2022) [20] classification, of loamy texture.
After being sieved through a 1mm sieve and homogenized, naturally dried at the exposure to the open air soil was divided into 10 kg DW portions, next spiked with CdCl2 2.5 H2O (added in solution as superior pure reagent) and equilibrated for three months. In total, an untreated control sample (T0, 0.22 mg kg-1 Cd) and four Cd treatments (nominal concentrations 10, 20, 30, 50 mg kg-1Cd) were applied. Measured actual Cd concentrations in homogenized and equilibrated soil are given in Table 1. The research was conducted as pot experiment, in three independent biological replicates.
Homogenized and equilibrated soil of each Cd treatment was put into pots φ=25 cm and H=20 cm. (2.5 kg per pot, with 3 g (NH4)2SO4 added as fertilizer). For the experiment, S. nigrum seeds collected from plants grown in the natural unpolluted soil (N0) and in soil treated with 50 mg kg-1 Cd in the previous year (A50), were sterilized with 0.1% HgCl2 for 10 min, soaked in tap water for 2 days, and sown into each pot, 20-30 seeds per pot. After sowing and moisturizing the soil to 50% of water capacity, the pots were placed into the foliar garden tunnel. During the experiment, the pots were randomly replaced several times. The soil moisture in the pots was maintained at approximately 50-60% of the water capacity using tap water. After reaching approximately 5 cm in height, plants in each pot were thinned to 10 uniform seedlings. The experiment was ended when about 70% of the plants were in flowering stage. During the plant growth, humidity and temperature in the tunnel were continuously automatically detected (for 90 days, from June until the end of August). The S. nigrum plants were harvested after 90 days at the mature stage and directed to analysis.
2.2. Experimental Methods and Sample Analysis
2.2.1. Cd uptake, Accumulation and Translocation in S. nigrum
The methods of experimental procedure and sample analysis were, in general, similar to those applied in another study [5]. The harvested S. nigrum plants were washed three times with tap water, next with deionized water, and separated into root (underground) and shoot (aboveground part consisting of stem, leaf and flowers). The plant material was then oven dried at 105 ℃ for 30 min, followed by drying at 75 ℃ to constant weight (with 0.0001 g accuracy). The biomass (Bmn) was measured as dry weight (DW) of underground and aboveground plant parts, where n-index means root (R) and shoot (S).
The Cd concentrations in plant material (root and shoot) were determined after grinding into fine powder and open digestion of about 0.5 g of a powdered mateial with the mixture of concentrated nitric and perchloric acids in proportion 15:5, until the mineralization the next day, followed by quantitative fitration and Cd determination with ICP-OES (OPTIMA 2000, Perkin-Elmer). For quality assurance/quality control (QA/QC), the standard reference material (NIST SRM 1547, peach leaves) was used.
Soil samples were analyzed for Cd content following similar procedure with the use. of microwave-assisted digestion (Anton Paar Multiwave 3000 SOL). The digestion was carried out at 1400W, IR = 240°C and pressure values p = 60bar. After mineralization and quantitative filtration,Cd contents in soil wee determined with the use of the same ICP-OES (OPTIMA 2000, Perkin-Elmer).
On the basis of Cd concentrations (Cn) and biomass (Bmn) determined in the specific parts n of the plants, where n-index means root (R) and shoot (S), the Cd enrichment factors of the related parts of the plants EFn = Cn/Csoil, translocation factors TF = CS/CR, accumulated Cd loads Ln =Cn Bmn, and load translocation factors LTF = LS/LR – from root to shoot were calculated according to Dai et al. [5,21]. In these equations, Cn – Cd concentrations in root and shoot, respectively (mg kg-1), Ln – accumulated loads of Cd in root and shoot, respectively (µg pot-1), Bmn – biomass of root and shoot, respectively (g pot-1).
2.2.2. Impact of Cd on the Physiological Parameters of S. nigrum
To assess the mechanisms of plant defence and adaptability to Cd stress, the physiological parameters were determined in fresh leaves of all S.nigrum samples from each pot, i.e. as the results of experiment conducted in three independent biological replicates. The analysis was performed at the Institute of Biology, University of Szczecin (Poland). In this research, a considerably broader scope of physiological parameters was investigted than presented in other research on Solanum nigrum L as Cd hyperaccumulator [5,21,22,23]. Except photosynthetic pigments, applied methods of other physiological parameter assessment differed in some significant details (mostly in units used) with respect to these presented in the relevant publications. This did not allow a direct comparability of results, however clearly presented trends. To avoid misinterpretation, the methods used for the physological parameter assessment are presented here in more detail.
Photosynthetic pigments concentration was determined using Lichtenthaler and Buschmann protocol [24]. To estimate chlorophyll a, chlorophyll b and carotenoids, leaves were homogenized and extracted in 80 % (v/v) acetone (FW : acetone, 1:10, w/v). After rotation and centrifugation, supernatant was used for measurements of absorbance values at 470, 645, and 665 nm by UV-Vis spectrophotometer (ThermoFisher Scientific, Madison, USA) and subsequent calculations of chlorophyll a (Chl a), chloropjyll b (Chl b) and caratenoid. The results were expressed as mg g−1 FW.
Superoxide anion and hydrogen peroxide (ROS) content was assessed according to the procedure described by Cembrowska-Lech [25] using dihydroethdium (DHE) (ThermoFisher) for O2•-, and CDCDHFDA-AM (6-carboxy-2',7'-dichlorodihydrofluorescein diacetate) (ThermoFisher) for H2O2 analysis. The labelled cells were analyzed using flow cytometer (Partec) with an air-cooled 20 mV argon-ion laser. The relative O2•- and H2O2 level was expressed as the mean fluorescence intensity (percentage of the control).
Enzyme extraction and western blot analysis were performed after fine grinding plant samples in liquid N2 with the use of a Retsch MM200 (Haan, Germany) laboratory ball mill, homogenization in the lysis buffer, and subsequent boiling and centrifugation. Samples containing 50 µg protein were loaded per line and separated on 12% SDS-PAGE gel [26]. After electrophoresis, the gels were electroblotted onto PVDF membranes (Millipore). Following triple washing in TBST, the blotting membranes were incubated in a blocking solution and probed with the polyclonal antibody: Cu/ZnSOD (AS10 652, Agrisera, Vännäs, Sweden), MnSOD (AS09 524, Agrisera, Vännäs, Sweden), FeSOD (AS06 125, Agrisera, Vännäs ,Sweden), CAT (AS09 501, Agrisera, Vännäs, Sweden), APX (AS08 368, Agrisera, Vännäs, Sweden), GR (AS06 181, Agrisera, Vännäs, Sweden), GPX (AS06 183, Agrisera, Vännäs, Sweden), RbcL (AS03 037, Agrisera, Vännäs, Sweden), and RbcS (AS07 259, Agrisera, Vännäs, Sweden). The membranes were then washed three times in TBST and probed with peroxidase conjugated secondary antibody (AS09 602 or AS09 603, Agrisera, Vännäs, Sweden). The immunoblots were incubated with a detection solution containing acetate buffer, diaminobenzidine and H2O2. The data are as immunoblot band visualization and the band intensities were determined using the Fiji ImageJ software v2.9.0 [27].
Superoxide dismutase (EC 1.15.1.1) activity was tested according to Giannopolitis and Ries [28] by the inhibition of NBT chloride photoreduction. The assay was carried out using the following reaction mixture: 0.1 M potassium phosphate buffer (pH 7.8), 1.3 µM riboflavine, 13 mM methionine, 63 µM NBT, 0.1 mM EDTA and 100 µL of the enzymatic extract. The reaction mixture was illuminated (50 µmol m−2 s−1) at 25 °C for 10 min and the absorbance measured at 560 nm. One unit of SOD activity was defined as the amount of the enzyme required to inhibit the reduction of NBT by 50% under the specified conditions. SOD activity of the extracts was expressed as U mg−1 protein.
Catalase (EC 1.11.1.6) activity was measured according to Rao et al. [29]. The enzyme activity was monitored spectrophotometrically at 240 nm for 60 s using the following mixture: 50 mM potassium phosphate buffer (pH 7.0), 14.3 mM H2O2 and 100 µL of enzymatic extract. Purified CAT was used as a calibration standard. CAT activity was expressed as U mg−1 protein. Data for both enzyme activities were expressed as means of independent biological replicates ± SD.
Glutathione reductase (EC 1.8.1.7) activity was analyzed as described by Esterbauer and Grill [30] by following the rate of NADPH oxidation at 340 nm for 3 min. The assay mixture contained: 0.1 mM potassium phosphate buffer (pH 7.8), 0.5 mM NADPH, 10 mM oxidized glutathione (GSSG), 10 mM EDTA and 100 µl of enzyme extract. The GR activity was expressed as nmol NADPH min-1 mg-1 protein.
Glutathione peroxidase (EC 1.11.1.9) activity was assessed as described by Nagalakshmi and Prasad [31] by following the rate of NADPH oxidation at 340 nm for 5 min. The reaction mixture contained: 0.5 M potassium phosphate buffer (pH 8.2), 10 mM EDTA, 1.14 M NaCl, 10 mM GSH, 2 mM NADPH, and 2.5 mM H2O2, and 100 µl of enzyme extract. The reaction was started by adding 2 U of GR. The GS activity was expressed as nmol NADPH min-1 mg-1 protein.
The protein content in the enzymatic extracts was assayed by Bradford’s method [32], using bovine serum albumin (BSA) as a standard. Glutathione in the reduced (GSH) and oxidized (GSSG) form were assayed following procedure described by Smith [33], which comprised grinding plant samples in liquid N2, extraction in sulphosalicylic acid (1 : 10, w/v), centrifugation, and measuring absorbance values in the neutralized supernatant for total glutathione (GSH + GSSG) and in GSSG alone after GSH masking, in double-extracted and suitably incubated samples at 412 nm and 25°C with the use of UV-Vis spectrophotometer(Thermo Fisher Scientific, Madison, USA. The results were expressed as molar concentrations of GSH (in nmol GSH g−1 FW) and GSSG (in nmol GSSG g−1 FW).
The ascorbic acid (AsA) contents were determined as described by Kampfenkel et al. [34] in supernatants of adequately prepared extracts from plant samples, by measuring absorbance values at 525 nm on UV-Vis spectrophotometer and subsequent calculations of molar concentrations of AsA (mmol AsA g−1 FW).
Lipid peroxidation was estimated by determination of malondialdehyde (MDA) contents according to Bailly et al. [35], by measuring absorbance values at 532 and 600 nm with the use of extinction coefficient 155 mM–1 cm–1. Results were expressed as μmol g–1.
2.3. Data Processing and Statistical Analysis
The experiments were conducted in three biological replicates and the results were expressed as mean ± sd. Data processing, averages, standard deviation values (sd), accunulated Cd loads Ln, enrichment factor (EF) and translocation factor (TF, LTF) calculations were performed using Microsoft Excel 365. The means were analyzed for significance using one–way analysis of variance, ANOVA (Statistica for Windows v. 13.0, Stat–Soft Inc., Tulsa, OK, USA). Data were checked for normality and homogeneity of variance. When the data did not conform to the normal distribution or failed to pass the variance homogeneity test, non-parametric Kruskal-Wallis Test was used to analyze the differences between groups. Tukey HSD (Honestly Significant Difference) multiple comparisons test was used to check the significance of differences (p <0.05).
3. Results and Discussion
3.1. Effect of Adaptation to Cd Stress on Biomass of Middle -European ecotype of S. nigrum
There were no distinct morphological differences either between root or shoot of Middle-European ecotype of S. nigrum L. ssp. nigrum, both naturally non-adapted (N0) and adapted in the first-generation to high concentration of Cd in soil (A50). Both N0 and A50 varieties retained similar growth and healthy appearance within the whole range of Cd treatments of soil (from 0 to 50 mg kg-1DW) that corresponded to the actual measured mean concentrations of Cd in soil from 0.22 mg kg-1 to 50.49 mg kg-1 (Table 1). However, more intense green color of leaves and analytical data clearly show the trends related to adaptation of S nigrum to high Cd concentrations in soil
Non-adapted variety N0 displayed mild downward trend of root biomass with the increase of Cd contents in soil, scarcely by 36% at T50 compared to T0. Shoot biomass showed even more gentle decline (by 26%), at several-fold bigger biomass of shoot than that of root (S/R biomass ratio ranged from 5.7 to 7.3 (mean 6.5) within all soil treatment range, i.e. from T0 to T50 (Csoil = 0.22 - 50.49 mg kg-1).
In turn, the declining trends of biomass of adapted variety A50 with increase of Cd stress from T0 to T50 appeared to be distinctly weaker, reaching 30% for roots and only 5% for shoots, at S/R biomass ratio varying from 6.0 to 8.4 (mean 7.2). Increase of S/R ratio at T50 was mostly due to the lesser relative impact of Cd stress on shoot biomass and clear signs of resistance mechanism, particularly visible in A50.
The mean root biomass of adapted varietyA50 in T0—T20 range (Csoil = 0.22-20.44 mg kg-1 Cd, DW) was roughly from 3 to 10 % lower than that of non-adapted one N0, while the mean shoot biomass of A50 was more uniformly, from 8 to 9% lower, respectively. However, at higher Cd concentrations in soil ((T30-T50, Csoil = 30.09 - 50.49 mg kg-1 Cd, DW), the biomass both of root and shoot of adapted (A50) variety surpassed that of non-adapted (N0) one, while an excess biomass showed clearly increasing trend (from 3% to 29%, and from10% to 18 % in root and shoot, respectively). This evidently resulted from the generally high resistance of Middle European S. nigrum ecotype to Cd, but considerably weaker impact of high concentrations of Cd in soil on the adapted plant growth.
There are no data on other ecotypes of S. nigrum grown under the same as this soil/climate conditions. However, the identical comparative pot experiments performed on cinnamon soil type (Xantic Ali-Udic Cambisols) [20], with the use of remote Asian ecotypes, two Chinese (Shenyang SY and Hanzhong HZ) and one Japanese ones (Kyoto KT) [5], showed very close root and shoot biomass values of Middle European and Asian ecotypes at T0 treatment, but considerably lesser resistance of Asian ecotypes to growing Cd content in soil. Already at Csoil >10 mg kg-1 Cd, fast decrease of biomass (DW) of Asian ecotypes was observed. The reported biomass decline was up to 2.5-4-fold (by 62-76%) of root, and up to over 3-fold (by 69-72%) of shoot at Csoil ≈ 50 mg kg-1 Cd, while Middle European ecotype showed scarcely up to 36% and 26% of root biomass, and by 26% and 5% of shoot biomass decline, respectively in N0 and A50 varieties. Much lesser S/R ratio (3.4-3.8) of Asian ecotypes than Middle European N0 and A50 varieties (S/R = 7.3 and 8.1, respectively) at Csoil ≈ 50 mg kg-1 Cd indicated considerably stronger adverse impact of high Cd stress on Asian ecotype growth.
Pot experiments on Korean ecotype S.nigrum L. from Daegu (35088’N, 128059’E) cultivated in growth chamber at 25 0C, 14 h photoperiod and 60% relative humidity in sand soaked with half-strength Hoagland nutrient solution also indicated decrease of biomass (DW) of leaves, stems and roots by 79, 75.6, and 75%, respectively at exposure of plants to 50-80 mg kg-1Cd [36]. Korean ecotype, at comparable to Chinese and Japanese ecotypes shoot biomass, and about two-fold bigger root biomass of S.nigrum grown in untreated medium, displayed its very sharp decline at the Cd range from 10 to 50 mg kg-1 Cd, but no significant changes at further increase of Cd concentrations to 80 mg kg-1. This indicates the launch of a new defence mechanism under the deep stress conditions. The sensitivity.of plants to Cd in this experiment might be additionally affected with the growth medium close to hydroponic conditions that usually results in bigger Cd accumultion in plants, but in adequately higher stress than at Cd uptake by plants from soil.
One more comparative pot experiment conducted at a glasshouse on the Australian {Melbourne) and Chinese (SY) ecotypes grown in a burozem soil from La Trobe University campus (Melbourne) in a glasshouse at 20±5 0C and natural light showed about 30% lower biomass of Ausrealian ecotype, and similar resistance to Cd content in soil [16]. Concentration of 20 mg-1 kg Cd in soil was a treshhold content assuring plant resilience, while sharp decline of biomass by 85-90% at 40 mg-1 kg Cd in soil and no further significant changes up to 80 mg-1 kg Cd in soil indicated similar reaction of studied Chinese (SY) and Australian S. nigrum ecotypes to stress as it was oberved with respect to Korean ecotype. It appears that under the very high stress, new defence mechanisms start to act, however after a very serious damage to plant physiology.
In contrast, such breakdown of defence mechanisms has not been observed in the Middle European ecotype of S. nigrum, in both non-adapted N0 and adapted A50 varieties, where decline of biomass occurred gradually and smoothly, and at a retively small scale, in particular compared to Asian/Australian ecotypes. There is no other evidence of diffeent reaction of European ecotypes to Cd stress, however shoot biomass (DW) reduction by 64 % only in Provence (France) ecotype of S. nigrum in pot experiment at 100 mg-1 kg Cd in a clayey loam soil [12] confirms considerably higher natural resistance of European ecotypes.
3.2. Cd Accumulation, Enrichment and Translocation in Natural (N0) and Adapted (A50) Plants Exposed to the Growing Cd Stress
Increasing contents of Cd in soil resulted in growing concentrations of Cd in roots and shoots of the Middle-European ecotype of S. nigrum, both of non-adapted (N0) and adapted to high concentrations of Cd (A50). However, Cd uptake by both varieties differed substantially. Non-adapted variety appeared to accumulate bigger contents of Cd in a root system within all studied rank of Cd concentrations in soil, evidently due to its lesser transport with xylem sap to shoots (Table 2). This manifested itself in a noticeably bigger Cd concentrations, and even more visible accumulation of Cd loads in shoots. of adapted variety A50 within all range of elevated Cd concentrations in soil (T10 -T50).
The values of enrichment and translocation factors justified high Cd-hyperaccumulative properties of non-adapted Middle-European ecotype of S. nigrum (N0). Enrichment factor EF for roots ranged roughly from 2.6.to 6.4, and for shoots from 3.4 to 7.7, at translocation factor TF exceeding or close to 1, up to T20 (Csoil= 20.44 mg kg-1Cd). At higher Cd contents in soil, TF was <1, but still high, while Cd concentrations in shoot were well above 100 mg kg-1Cd, thus meeting the hyperaccumulator requirements.
Adapted variety (A50) under the same growing conditions displayed somewhat lesser range of EF for roots (from1.1 to 5.5) and bigger for shoots (from 2.2 to 8.6), at TF>1 up to T30 (Csoil= 30.09 mg kg-1Cd). All Cd concentrations in shoot of A50 at Csoil >10 mg kg-1Cd were above 100 mg kg-1Cd, and bigger than in N0, thus confirming somewhat stronger Cd hyperaccumulative properties measured by its accumulation in shoot.
Actual accumulated Cd loads within the investigated concentration range in soil highlighted lesser Cd accumulation in A50 root system than in N0 due to the bigger Cd load translocation from root to shoot, showing regular declining trend with the increase of Cd concentrations in soil (LTF = 8.8 to 4.0 for N0 vs. LTF = 12.3 to 5.2 for A50). Remarkable, total Cd loads accumulated in roots and shoots of both N0 and A50 at increasing concentrations of Cd in soil appeared to be similar to each other (Table 2). This means that the total amounts of Cd accumulated in adapted A50 and non-adapted N0 varieties are basically similar, while long-range xylem sap transport from roots to leave vacuoles appeared to be more intense in adapted variety A50. The pattern of Cd accumulation in root and shoot of both varieties of S. nigrum as a hyperaccumulator clearly illustrates the difference of Cd uptake and translocation between Cd excluders and hyperaccumulators. Namely, in Cd excluders, such as e.g. S. torvum, reduced concentrations of Cd in roots of a plant growing in Cd-contaminated soil is mainly due to Cd -induced drought stress in root system. The transcriptional regulation of dehydration-related genes inhibiting water transport in aquaporins thus causing a drought effect, constitutes defensive mechanism against Cd penetration into plant tissues [21,37]. It results also in reduced Cd flow from roots through stem to leaves. In contrast, in A50, reduced Cd content in root compared to N0, was caused entirely by increased xylem sap long range flow rate at the lack of barriers in Cd uptake from soil. Compared to Asian ecotypes, both Middle European varieties showed similar Cd accumulation capacity within tolerable to Asian ecotypes Cdsoil range not causing biomass reduction (e.g. CR = 57.8 -81.4 mg kg-1, CS = 78.7-102 mg kg-1, and LT= 856-1348 µg pot -1 for SY, HZ and KT [5] vs. CR = 50.8 -55.5 mg kg-1, CS = 81.1-90.7 mg kg-1, and LT= 1067-1085 µg pot -1 at Csoil = 10 mg kg-1Cd for N0 and A50 (Table 2). However, sensitivity to Cd stress at Csoil >10 mg kg-1Cd results in declining biomass and total Cd accumulation capacity, at deteriorating physiological parameters but increased Cd concentrations in tissues of Asian ecotypes. Opposite, Middle Europan varieties showed high resistance to Cd stress in the whole range of studied soil concentrations, up to Csoil = 50 mg kg-1Cd, displaying lesser Cd concentrations in plant tissues, but higher total accumulated loads of Cd and their overall good physiological status (e.g. CR = 301 - 342 mg kg-1, CS = 318-387 mg kg-1, and LT= 1083-1781 µg pot -1 for SY, HZ and KT [5] vs. CR = 324 -280 mg kg-1, CS = 175-165 mg kg-1, and LT= 2106-2269 µg pot -1 at Csoil = 50 mg kg-1Cd for N0 and A50 – Table 2).Within this study, no limit of Cd tolerance of Middle European ecotype of S. nigrum, either non-adapted, or adapted to Cd stress was reached. Although, among very scarce European studies on S. nigrum, one study on the exposure to Csoil = 100 mg kg-1Cd, i.e. to twice as big concentration as studied here, of the local South-West European ecotype revealed still high Cd accumulation capacity and declining tolerance [12]. Concentrations of Cd in root and shoot (DW) of this ecotype appeared to be comparable to that assessed for Middle European ecotype and reached CR = 377 mg kg-1, CS=107 mg kg-1 at Csoil= 100 mg kg-1Cd vs. CR=324=280 mg kg-1, and CS = 175-165 mg kg-1 at Csoil= 50 mg kg-1Cd assessed for Middle-European ecotype at two times lesser Cd concentration in soil. This indicates further gradual decrease by about 39% of Cd accumulation in shoot provided that these European ecotypes are indeed react similarly to Cd stress.
These data require conducting further comparative experiments of different remote ecotypes under the same soil/climate conditions. Comparing the results obtained in this study and available so far, besides different tolerance to Cd stress of various remote Asian/Far East and European ecotypes of S. nigrum, two diverse defense mechanisms were indicated. One, observed in Asian/Far East ecotypes is more conservative and displays almost unrestricted translocation of Cd with xylem sap from root to shoot until deep breakdown of physiological functions of a plant under critical level of Cd stress and further partial adaptation of a plant to functioning under excessive stress. This manifests itself in continuously high accumulation of Cd in shoot at poor condition of a plant and deep biomass reduction [5,16].
Another mechanism displayed by the Middle-European ecotype of S. nigrum in this study in N0 and A50 varieties, but in particular in the adapted variety A50, is directed to the protection of physiological functions of a plant by active molecular mechanisms in roots controlling Cd loading capacity into the xylem for long-distance transport from root to shoot and storage in leaf vacuoles [5,38].
This resulted in the lack of the breakdown caused by Cd stress in the Middle European ecotype, but also in the lower concentrations of Cd accumulated both in root and shoot tissues at the critical contents of this metal in soil (Table 2). The Cd accumulation in root was somewhat lower, and in shoot somewhat higher in adapted A50 than in non-adapted N0 variety practically in all Cd-fortified soils, however within non-significant level (p<0.05). Consequently, compared to N0, A50 displayed lower enrichment factors EF in root, but higher in shoot, and higher translocation factor TF that at T0 –T30 range was >1, while non-adapted N0 variety complied with this requirement only within T0-T10 range
Therefore, the overall effect of Middle-European S. nigrum ecotype adaptation to the high Cd content in soil manifested itself in higher tolerance of A50 variety to Cd stress that resulted both in bigger shoot biomass and either higher Cd concentrations in xylem sap at the loading stage from roots, or greater long-range xylem sap flow rate from roots to leave vacuoles. Much lesser Cd accumulation in stem than in leaves observed in the South-West European [12] and also in Asian (SY) [5] ecotypes suggests rather this last mechanism. Moreover, despite considerably higher concentrations of Cd in tissues of Asian S. nigrum ecotypes SY, NZ and KT [5], their evidently weaker protection mechanisms against stress and strong reduction of biomass preceded by a physiological breakdown caused that the total Cd accumulation capacity of Middle-European ecotype at critical Cd contents in soil appeared to be visibly higher (by 49%, 26% and 15% in N0, and by 52%, 32% and 22%, respectively to A50.
3.3. Effect of Adaptation to High Cd Contents in Soil on Physiological Properties of Middle-European ecotype of Solanum nigrum
3.3.1. Photosynthetic Pigments
The trends in photosynthetic pigment contents in leaves of non-adapted N0 variety of the studied Middle-European ecootype of S.nigrum L, despite lack of visual changes in the plant/leaves status, showed general similarity to these observed in Asian ecotypes [5]. Chlorphyll a, chlorophyl b and caratenoid contents displayed no significant changes only at T10, while at higher Cd concentrtions in soil, a significant decline of all pigment contents occurred (Figure 1), However, at T30 the declining trend ceased and no further significant (p<0.05) chanes were observed in chlorphyll a and carotene content,while in chlorophyll b a mild decline occurred alreadyat T≥20. In contrast, A50 variety showed considerably higher than N0 all photosynthetic pigment contents at all treasments (by 24%, 23% and 14% at T0 and by 59%, 57% and 18% at T50, respectively), and mild decline at bigger concentrations of Cd in soil (by 34%, 28% and 45% for chlorphyll a, chlorophyl b and caratenoids vs. 64% for chlorophyll a and b and 43% for caratenoids in N0 at T50).
The adapted A50 variety of Middle-European S. nigrum ecotype appeared to be more enriched in photosynthetic pigments and more resistant to Cd stress than non-adapted N0 with respect to their content in leaves. These data showed that both varieties are tolerant to Cd, in particular A50 that showed mild declineand retained relatively high photosynthetic parameters at the whole range of Cd treatments. The photosynthetic performance of leaves of non-adapted N0 varieties of the studied Middle-European ecootype of S. nigrum L, despite lack of visual changes in the plant/leaves status showed higher sensitivity to Cd stress
3.3.2. RuBisCO Activities
Activities of RuBisCO proteins representing both large RbcL and small RbcS subunits (Figure 2a, b) responsible for converting atmosphric CO2 into organic carbon forms, shoved different response to oxidative stress in non-adapted N0 and adapted A50 varieties. At T0, opposite to photosyntetic pigments, no difference in the activity of both subunits were observed. Increase of abiotic oxidative stress caused by the growing content of Cd in soil resulted in the regular decline of RbcLand RbcS in N0, more pronouced with respect to BbcS, where complete disapearance of this subunit at T50 occurred (Figure 2b). It is considered that exprssion of RbcS regulates the size of the RuBisCO pool and therefore affects the overall catalytic efficiency of the RuBisCO complex mostly located in the RbcL This is well correlates with regularly declining RbcL activity in N0 variety, upto 78% at T50 compared to T0 (Figure 2a).
3.3.3. Reacrtive Oxygen Species (ROS)
Generated in excess reactive oxygen species (ROS), the superoxide anion (O2•‒) and hydrogen peroxide (H2O2) involved in physiological stress and pathological processes in the apoplast of plant cells, displayed distint increase in leaves of both non-adapted N0 and adapted A50 varieties in parallel with increase of Cd content in soil, indicating growing oxidative sress (Figure 3). However, no signs of crisis situation and deep cell damage with respect to superoxide anion content was observed: the increase had a regular character, by 105% and 69%, respectively in the range between T0 and T50 for N0 and A50. The O2• level was nonetheless considerably (by 31%) bigger in leaves of N0 than in A50 (Figure 3a) indicating lesser oxidative stress of adapted variety.
Oxidative stress caused by hydrogen peroxide (H2O2) also appeared to be more pronounced for N0 variety, where substantially higher level of this ROS was detected in its leaves at T50 compared to T30 treatment (by 74%), while increase of H2O2 in the range T0 –T30 treatments was weaker (by 58%). In contrast, adapted A50 variety did not show much growth of H2O2 level in the whole Cd concentration range (scarcely by 29% compared to T0). This indicated its high rsistance to Cd stress practically up to the highest T50 treatment, at distinctly lower ROS level in the leaves of adapted A50 varety, while sharp up-regulaton of H2O2 at T30-T50 range in N0 showed the symptoms of a greater stress at T50.
3.3.4. First Line Defence Antioxidant Activities
Among antioxidant defence systems in plants, key atioxdant enzymes catalaze (CAT), superoxide dismutazes (SOD) and glutathione peroxdase (GPX) are considered the most important and efficient antioxdants that prevent or supprers the formation of free radicals and ROS in plant cells. Specifically, CAT degrades H2O2 into H2O and O2. SOD catalyzes the dismutation of two superoxide (O2●-) anions and water into H2O2 and O2 [39,40,41].
The activities of these first line antioxidant enzymes in leaves of non-adapted N0 and adapted A50 varieries of Middle-European ecotype of S.nigrum showed Cd stress-related changes following specific patterns. In N0 leaves, up-regulation of all key enzyes (CAT, SOD, GPX) indcating growing defence mechanism was observed up to T10 (with respect to SOD up to T20), followed by down-regulation due to weakening of he defence system (Figure 4). The strongest relative up-regulation trends were observed in SOD contents (up to 32% at T20) and the weakest in GPX activties (up to 12% at T10). The strongest relative down-regulation occurred in CAT activities (by 60% betwen T10 and T50 treatments).
In contrst, in adapted vaiety A50, the first line antioxidant activities, after reaching the highest level at T10 (by 24% for CAT, 20 % for SOD and 15% for GPX) , practically held it, showing only slight decrease above the initial level at T0 (CAT, GPX), while SOD activity kept sligt growing trend up to T50 (Figure 4).
Worth mentioning, that SOD activities were always bigger in N0 than in A50 at all Cd treatments , except that at T50, when decline of SOD in N0 reversed relations. Of three SOD isozymes disingiushed by covalently linked metal ions and cellular locations (Cu/ZnSOD, MnSOD and FeSOD) [39], Cu/ZnSOD isozyme showed the same N0/A50 relations as SOD enzyme indicating its predominance in both varieties (Figure 5a). In the adapted A50 variety, activities of all three isozymes showed increase at T10 (by ~19%, 13% and 21%, respectively) and stable level (p<0.05) in the Cd range T10-T50, for MnSOD and FeSOD considerably higher than in N0 (Figure 5a, b, c). Opposite, in non-adapted variety N0 activities of MnSOD isozyme (Figure 5b) showed slight increase at T10 (by 7% compared to T0), followed by the decrease (by 32%) in the range T10-T30 and stabilization at T30-T50 (p< 0.05). Activities of FeSOD in N0 remained at the same lower level compared to T0 within all Cd treatment range (by19%, p<0.05). Differential expresson of antioxidant isozymes in leaves of S. nigrum exposed to Cd stress indicated substantal differences in production superoxide anions in specific celular locations. Since Cu/ZnSOD isozyme that shows the biggest affinity to overall SOD pattern and expression dynamics is found in cytoplasm and in chloroplasts, it indcates that superoxide anions requiring deactivation are generated mostly in these cellular locatons. Lesser activity changes showed MnSOD located in mitochondria, while the most stable activity displayed FeSOD found in chloroplsts.
3.3.5. Second Line Defence Antioxidant Activities
In reducing Cd stress , also the group of scavenging antioxidants takes important part. This second line defence antioxidants [41] scavenge and neutralize free radicals by donating electron to them, thus converting themselves into free radicals, but of lesser damaging effect. They, in turn, are easily neutralized by other antioxidants of this group. The prominent representatives of this group are ascorbate and glutathione enzymatic (APX, AsA, GR) and non-enzymatic cmpounds (GSH, GSSG). Besides direct scavenging ROS, they perform other functions that enhance antioxidant capacity and reduce oxidative stress [42]. Of these antioxidants, a very strong reaction to increasing Cd stress in S. nigrum showed ascorbate antioxidant compounds: ascorbate peroxidaze APX and ascorbate AsA (Figure 6). APX occurs in cytosol and organelles (chloroplast, mitocondria and peroxisome) and ascorbate AsA present also in nucleus and vacuoles of leave cells [40].
With respect to APX in the leaves of non-adapted N0 variety of S. nigrum, typical for this ecotype increase of its acivity (by 14% in T10 related to T0) occurred, indicating activation of its defence functions. However, it was followed by the breakdown of defence capacity and fast decline of APX activity up to its thorough extinction within the T10 – T50 interval of Cd treatments. An enzymatic co-factor ascorbate (AsA) was affcted even more, showing sharp decrease (by 33% compared to T0) up to total extinction at T30.
Ascorbate compounds enrichment in the leaves of adapted variety A50 appeared to be considerably bigger and showed much higher defence capacity. After increase of APX at T10 by 28% compared to T0, its defence activity continuously exceeded control up to T30, while at T50, APX activity declined scarcely by 37% related to T0 (Figure 6a). Also AsA contents in adapted A50 variety in the range T0-T30 appeared to be much less responsive to Cd stress than in N0 , and was extinct only at T50 (Figure 6b).
Simultaneously, the levels of second line glutathione antioxidant compounds present in plant cells (GR, GSH) and the level of disulfide GSSG generation in both S. nigrum varieties suggest much weaker response of these compounds to increasing Cd stress (Figure 6 c–e). The level of glutathione reductase (GR) appeared to be similar in both varieties and did not show significant (p<0.05) changes within the whole T0 – T50 range of Cd treatments (Figure 6c). In turn, the contents of non-enzymatic antioxidant glutathione (GSH) was also stable in both varieties. The differences in AsA (Figure 6b) and GSH (Figure 6d) trends in response to Cd stress suggest also other mecanisms besides regulation of Ascorbate-Glutathione (AsA-GSH) pathway [42] in detoxifying ROS in this Middle-European ecotype of S. nigrum. Whle AsA showed fast decline, GSH contents were basically constant, in A50 about 30 % bigger than in N0. GSH displayed very slight general increasing trend at T10 followed by the same slight (above T0 level) decrease in the T10-T50 range in N0 variety and no significant changes in adapted A50 variety (Figure 6d). Opposite, the levels of oxidized GSH (GSSG) were in non-adapted variety N0 somewhat (7-10%) higher than in adapted A50, at the similar slight decreasing trend (Figure 6 e). This resulted in the basically constant ratio GSH/GSSG at T10-T50, higher than at T0 in both vareties (1.0 > 0.9 in N0 and 1.40 > 1.29 in A50), and significantly lower in N0 than in A50 (1.0 vs. 1.40), which indicated increased defence capacity in both varieties at Cd inctrease in soil and lesser oxidative stress in the A50 variety, at generally weak reaction of second line glutathione antioxidant compounds to oxidative stress. Along with moderate reduction of the first line defence antioxidant GPX in N0 at the highest Cd treatment T50 in soil, and higher its activity in N50 at T50 than T0, this points to the strong glutathione-based ROS -resistance mechanism on the one hand, and lesser involvement of glutathione compounds into Cd-induced ROS detoxification in comparison with CAT, SOD and ascorbate on the other hand.
High resistance to Cd stress of the Middle European ecotype of S.nigrum was confirmed by the trends in the generation of an indicator of plant cell damage malondialdehyde MDA in leaves of studied non-adapted N0 and adapted A50 varieties (Figure 6f). In the Cd range from T0 to T20 treatments, MDA contents in N0 increased reflecing growing oxidative damade, however in the range T20-T30, MDA remaied at the constant level. Formation of MDA in A50 was significantly smaller (by21-46%), stabilizing at T30-T50 level of Cd treatment. Such trends in MDA generaton indicate high tolerance of both varieties of the Middle-European ecotype of S.nigrunm to abiotic stress caused by the growing contents of Cd in soil, and significanly lesser oxidative damage in A50 variety. Remarkable, that both varieties of Middle European ecotype, non-adapted N0 and adapted A50,opposite to Asian ecotypes [5] showed strong resistance to the similar high-level of Cd stress (T30-T50), and differ mostly by the level of oxidative damage (by 46 -22% lesser in A50).
4. Conclusions
The study in its entirety confirmed the assumption that different ecotypes of cosmopolitan Cd hyperaccumulator S. nigrum L ssp. nigrum, which orignate from remote geographical locations, may significantly differ with respect to Cd hyperaccumuation capacity, tolerance and regulation mechanisms. It appeared that the studied East European ecotype, compared to Asian ecotypes [5], displayed much bigger tolerance to abiotic stress, in addition significantly enhanced by adaptation through using seeds trom the first-generation of plants grown in soil with high content of Cd (50 mg Cd kg-1.). This maniested itself in the mild root and shoot biomass reducion at T50 and much higher shoot to root biomass ratio (S/R),both in non-adapted N0, but in particular in A50 (adapted variety).compared to the. Asian ecotypes exposed to the same contents of Cd in soil (in the range from 0 to 50 mg kg-1Cd).and showing fast reducton of bomass already at Csoil=10 mg Cd kg-1.
There appeared to be also substantial differences between Asian and Middle European ecotypes, and N0 and A50 varieties with respect to Cd enrichment and translocation in plant tissues. While Asian ecotypes continued to accumulate growing contents of Cd under the critical stress conditions and fast declining biomass, Midle European ecotype retained good health status and strong resistance to Cd stress at the whole range of the studied concentrations in soil, up to Csoil=50 mg Cd kg-1,at lesser Cd concentrations in plant tissues, but similar or higher accumulated Cd loads, both in shoots and total, in shoots and roots, compared to Asian ecotypes.
Adaptation to Cd stress in A50 variety resulted in enhancing all physiological paraeters of plant crucial for its development, growth and tolerance to abiotic stress (chlorophyll a and b, carotenoids, RuBisCO, first line defence (neutralizing) antioxidants (CAT, SOD and its isozymes, GPX), and second line defense (scavenging) antioxidants (APX, AsA, GR, GSH). This was admittedly confirmed by clearly lower Cd stress indicated by lesser MDA generation and higher GSH/GSSG ratio for adapted variety A50 compred to non-adapted N0.
Overall, selected Asian ecotypes of S. nigrum ssp nigrum appear to show somewhat better Cd phytoremediation performance than Middle European ectype within Cd tolerance range of Asian ecotypes, mostly at the moderate soil contamination level up to 10 mg Cd kg-1.. On the other hand, Middle European ecotype stands out in much higher treshhold of tolerance to Cd stress, reaching beyond the highest tested Cd content in soil (50 mg Cd kg-1), probably close to 100 mg Cd kg-1.. Moreover, adapted variety A50 of the Middle European ecotype shows further considerable enhancement of the plant resistance to Cd stress and the highest Cd accumulation capacity and healthy state of the plant at the biggest tested Cd concentratons in soil. This makes Middle European ecotype and its adapted variety A50 particularly useful to sustainable decontamination of heavily polluted “hot spots” in the degraded post-industrial areas.
It should be added, that significant difference between remote ecotypes of Cd hyperaccumultor Solanum nigrum L. ssp. nigrum in Cd accumulation capacity and tolerance to Cd stress suggests possible consierable differentiation of molecular mechanisms resposible for these properties in the multitude of ecotypes and genotypes . growing worldwide. Some recent rsearch indicate occurrence of differential responsive mechanisms to low and high Cd exposure in the same plant [43], thus even bigger differentiation at the molecular level can be expected in the remote ecotypes/ genotypes. Elucidation of these mechnisms may optimize phytoremediation of contaminted lands without using any supportive chemical or other environmentally problematic or expensive means.
Funding
This research was conducted within the statute activity of the Institute of Environmental Engineering, Polish Academy of Sciences in Zabrze, Poland (grant number. 1a-133/2022). The research received no other external funding.
Author Contributions
Conceptualization, I. Twardowska; methodology, I. Twardowska, L. Skuza and D. Cembrowska-Lech; software, E. Miszczak and S. Stefaniak; validation, I Twardowska, and L. Skuza ; formal analysis, E. Miszczak and S. Stefaniak; investigation, E. Miszczak and D. Cembrowska-Lech, ; resources, I. Twardowska, E. Miszczak, L. Skuza; data curation, E. Miszczak and S. Stefaniak.; writing - original draft, I. Twardowska and D. Cembrowska-Lech; writing and editing, I. Twardowska.; visualization, E. Miszczak; supervision, I. Twardowska; project administration, I. Twardowska.; funding acquisition, I. Twardowska. All authors have read and agreed to the published version of the manuscript.”.
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable.
Data Availability Statement
Data available on request.
Acknowledgments
The physiological research were performed in the framework of collaboration with the Institute of Biology, University of Szczecin, Poland. The invaluable assistance of Dr. Agnieszka Zawisza-Raszka, Director of the City Botanical Garden in Zabrze and of its staff in acquiring local ecotype of Solanum nigrum L. ssp. nigrum and soil is highly appreciated.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Kabata-Pendias, A. Trace Elements in Soils and Plants, 4th ed.; CRC Press Boca Raton, FL, 2011; pp. 266.
- Miszczak, E.; Stefaniak, S.; Michczyński, A.; Steinnes, E.; Twardowska, I. A novel approach to peatlands as archives of total cumulative spatial pollution loads from atmospheric deposition of airborne elements complementary to EMEP data: priority pollutants (Pb, Cd, Hg). Sci. Total. Environ. 2020, 705, 135776. [Google Scholar] [CrossRef] [PubMed]
- Reeves, R.D.; Baker, A.J.M.; Jaffr´e, T.; Erskine, P.D.; Echevarria, G.; van der Ent, A. A global database for plants that hyperaccumulate metal and metalloid trace elements. New Phytol. 2018, 218, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.H.; Zhou, Q.X.; Wang, X.; Zhang, K.S.; Guo, G.L.; Ma, L.Q. A newly-discovered Cd-hyperaccumulator Solanum nigrum L. Chin. Sci. Bull. 2005, 50, 33–38. [Google Scholar] [CrossRef]
- Dai, H.; Wei, S.; Twardowska, I.; Hou, N.; Zhang, Q. Cosmopolitan cadmium hyperaccumulator Solanum nigrum: Exploring cadmium uptake, transport and physiological mechanisms of accumulation in different ecotypes as a way of enhancing its hyperaccumulative capacity. Journal of Environmental Management, 2022, 320, 115878. [Google Scholar] [CrossRef]
- Marques, A.P.G.C.; Rangel, A.O.S.S.; Castro, P.M.L. Effect of arsenic, lead and zinc on seed germination and plant growth in black nightshade (Solanum nigrum L. ) vs. clover (Trifolium incarnatum L.). Fresenius Environmental Bulletin, 2007, 16, 896–903. [Google Scholar]
- Teixeira, J.; de Sousa, A.; Azenha, M.; Moreira, J.T.; Fidalgo, F.; Silva, A.F.; Faria, J.L. Solanum nigrum L. weed plants as a remediation tool for metalaxyl-polluted effluents and soils. Chemosphere 2011, 85, 744–750. [Google Scholar] [CrossRef]
- Teixeira, J.; Ferraz, P.; Gouveia, C.; Azevedo, F.; Neves, S.; Fidalgo, F.; Silva, A.M. Targeting key metabolic points for an enhanced phytoremediation of wastewaters pre-treated by the photo-Fenton process using Solanum nigrum L. Ecotoxicol. Environ. Saf. 2015, 120, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Ferraz, P.; Fidalgo, F.; Almeida, A.; Teixeira, J. Phytostabilization of nickel by the zinc and cadmium hyperaccumulator Solanum nigrum L. Are metallothioneins involved? Plant Physiol. Biochem. 2012, 57, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.Z.U.; Rizwan, M.; Ali, S.; Ok, Y.S.; Ishaque, W.; Nawaz, M.F.; Akmal, F.; Waqar, M. Remediation of heavy metal contaminated soils by using Solanum nigrum: A review. Ecotoxicol. Environ. Saf. 2017, 143, 236–248. [Google Scholar] [CrossRef]
- Sousa, B.; Soares, C.; Oliveira, F.; Martins, M.; Branco-Neves, S.; Barbosa, B.; Ataíde, I.; Teixeira, J.; Azenha, M.; Azevedo, R.A.; et al. Foliar application of 24-epibrassinolide improves Solanum nigrum L. tolerance to high levels of Zn without affecting its remediation potential. Chemosphere 2019, 244, 125579. [Google Scholar] [CrossRef]
- Pons, M.-L.; Collin, B.; Doelsch, E.; Chaurand, P.; Fehlauer, T.; Levard, C.; Keller, C.; Rose, J. X-ray absorption spectroscopy evidence of sulfur-bound cadmium in the Cd-hyperaccumulator Solanum nigrum and the non-accumulator Solanum melongena. Environ. Pollut. 2021, 279, 116897. [Google Scholar] [CrossRef] [PubMed]
- Al-Huqail, A.A. Stimulating the efficiency of Cd-phytoremediation from contaminated soils by Solanum nigrum L.: Effect of foliar and soil application of yeast extract. South Afr. J. Bot. 2023, 161, 512–518. [Google Scholar] [CrossRef]
- Sahito, Z.A.; Zehra, A. , Song Yu S. ; Chen S., Arif M.A.R.; Raza S.T., Lahori A.H., Mai Ali Mwaheb M.A.M.A.; He Z.; Yang X. Folic acid supplementation improves seed germination, seedling growth and cadmium uptake in a mining ecotype of Solanum nigrum L. Environmental Technology and Innovationvol 2024, 34, 103600. [Google Scholar]
- EMEP, 2022. EMEP Status Report 2/2022. Assessment of heavy metal and POP pollution on global, regional and national scales. Joint MSC-E, CCC, CEIP, IMR, CEIMAT, INERIS, ENEA and FMI Report. http://www.msceast.org/reports/2_2022.pdf (assessed 27.09. 2024.
- Wei, S.; Clark, G.; Dornila, A.I.; Monsant, A.C. Cd hyperacumulative characteristics of Australia ecotype Solanum nigrum L. and its implication in screening hyperaccumulator. Int. J. Phytoremediation 2013, 15, 199–205. [Google Scholar]
- Dou, X.; Dai, H.; Skuza, L.; Wei, S. Strong accumulation capacity of hyperaccumulator Solanum nigrum L. for low or insoluble Cd compounds in soil and its implication for phytoremediation. Chemosphere 2020, 260, 127564. [Google Scholar]
- Yang, X.; Qin, J.; Li, J.; Lai, Z.; Li, H. Upland rice intercropping with Solanum nigrum inoculated with arbuscular mycorrhizal fungi reduces grain Cd while promoting phytoremediation of Cd-contaminated soil. Journal of Hazardous Materials, 2021, 406, 124325. [Google Scholar] [CrossRef] [PubMed]
- Ditzler, C. , Scheffe, K., and Monger, H.C. (eds.). Soil survey manual. Soil Science Division Staff USDA Handbook 18. Government Printing Office, Washington, D.C; 2017.
- IUSS Working Group WRB. World Reference Base for Soil Resources. In International Soil Classification System for Naming Soils and Creating Legends for Soil Maps, 4th ed.; International Union of Soil Sciences: Vienna, Austria, 2022. [Google Scholar]
- Dai, H.; Wei, S.; Twardowska, I.; Zhang, Q. In search of the exclusion/lowaccumulation mechanisms: cadmium uptake and accumulation from soil by cultivated (Solanum melongena L. ) and wild eggplants (Solanum torvum L.). J. Clean. Prod. 2021, 323, 12941. [Google Scholar]
- Dai, H.; Wei, S.; Noori, A. The mechanism of chelator improved the tolerance and accumulation of poplar to Cd explored through differential expression protein based on iTRAQ. J. Hazard. Mater. 2020, 393, 122370. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Wei, S.; Pogrzeba, M.; Krzyżak, J.; Rusinowski, S.; Zhang, Q. The cadmium accumulation differences of two Bidens pilosa L. ecotypes from clean farmlands and the changes of some physiology and biochemistry indices. Ecotoxicol. Environ. Saf. 2020, 209, 111847. [Google Scholar] [CrossRef]
- Lichtenthaler, H.K.; Buschmann, C. Extraction of Phtosynthetic Tissues:Chlorophylls and Carotenoids. Curr. Protoc. Food Anal. Chem. 2001, 1, F4–2. [Google Scholar] [CrossRef]
- Cembrowska-Lech, D. Tissue Printing and Dual Excitation Flow Cytometry for Oxidative Stress—New Tools for Reactive Oxygen Species Research in Seed Biology. Int. J. Mol. Sci. 2020, 21, 8656. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head phase of bacteriophage T4. Nature 1970, 227, 680–5. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Giannopolitis, C.N.; Ries, S.K. Superoxide dismutases: I. Occurrence in higher plants. Plant Physiol. 1977, 59, 309–314. [Google Scholar] [PubMed]
- Rao, M.V.; Paliyath, G.; Ormrod, D.P. Ultraviolet-B- and Ozone-Induced Biochemical Changes in Antioxidant Enzymes of Arabidopsis thaliana. Plant Physiol. 1996, 110, 125–136. [Google Scholar] [CrossRef]
- Esterbauer, H.; Grill, D. Seasonal Variation of Glutathione and Glutathione Reductase in Needles of Picea abies. Plant Physiol. 1978, 61, 119–121. [Google Scholar] [CrossRef]
- Nagalakshmi, N.; Prasad, M. Responses of glutathione cycle enzymes and glutathione metabolism to copper stress in Scenedesmus bijugatus. Plant Sci. 2001, 160, 291–299. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities utilizing the principle of protein dye binding. Anal Biochem 1976, 72, 248–54. [Google Scholar] [CrossRef]
- Smith, I.K. Stimulation of Glutathione Synthesis in Photorespiring Plants by Catalase Inhibitors. Plant Physiol. 1985, 79, 1044–1047. [Google Scholar] [CrossRef]
- Kampfenkel, K.; Van Montagu, M. ; Inze,` D. Extraction and determination of ascorbate and dehydroascorbate from plant tissue. Anal Biochem 1995, 225, 165–167. [Google Scholar]
- Bailly, C. , Benamar A., Corbineau F., Côme D. 1996. Changes in malondialdehyde content and in superoxide dismutase, catalase and glutathione reductase activities in sunflower seeds as related to deterioration during accelerated aging. Physiol. Plant. 97:104-110.
- Khan, A.R.; Ullah, I.; Khan, A.L.; Hong, S.-J.; Waqas, M.; Park, G.-S.; Kwak, Y.; Choi, J.; Jung, B.-K.; Park, M.; et al. Phytostabilization and Physicochemical Responses of Korean Ecotype Solanum nigrum L. to Cadmium Contamination. Water, Air, Soil Pollut. 2014, 225, 2147. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Fukuoka, H.; Arao, T.; Ohyama, A.; Nunome, T.; Miyatake, K.; Negoro, S. Gene expression analysis in cadmium-stressed roots of a low cadmium-accumulating solanaceous plant, Solanum torvum. J. Exp. Bot. 2009, 61, 423–437. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Sun, J.; Du, L.; Liu, X. Comparative transcriptome analysis of cadmium responses in Solanum nigrum and Solanum torvum. New Phytol. 2012, 196, 110–124. [Google Scholar] [CrossRef]
- Baek, K.-H.; Skinner, D.Z. Production of reactive oxygen species by freezing stress and the protective roles of antioxidant enzymes in plants. J. Agric. Chem. Environ. 2012, 01, 34–40. [Google Scholar] [CrossRef]
- Racchi, M.L. Antioxidant Defenses in Plants with Attention to Prunus and Citrus spp. Antioxidants 2013, 2, 340–369. [Google Scholar] [CrossRef]
- Ighodaro, O.M.; Akinloye, O.A. First line defense antioxidants – superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): their fundamental role in the entire antioxidant defense grid. Alexandria J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef]
- Hasanuzzaman, M.; Bhuyan, M.H.M.B.; Anee, T.I.; Parvin, K.; Nahar, K.; Mahmud, J.A.; Fujita, M. Regulation of Ascorbate-Glutathione Pathway in Mitigating Oxidative Damage in Plants under Abiotic Stress. Antioxidants 2019, 8, 384. [Google Scholar] [CrossRef]
- Song, L.-Y.; Liu, X.; Zhang, L.-D.; Hu, W.-J.; Xu, C.-Q.; Li, J.; Song, S.-W.; Guo, Z.-J.; Sun, C.-Y.; Tang, H.-C.; et al. Proteomic analysis reveals differential responsive mechanisms in Solanum nigrum exposed to low and high dose of cadmium. J. Hazard. Mater. 2023, 448, 130880. [Google Scholar] [CrossRef]
Figure 1.
Effects of different Cd treatments on (a) chlorophyll a, (b) chlorophyll b content in non-adapted N0 and adapted A50 varieties of S. nigrum L. 1) 0 to 50 means control and soil treatments with Cd (mg kg-1); 2) Data over bars for the same treatments marked by the same capital letters are not significantly different at p<0.05. Data for different treatments marked by the same lowercase letters are not significantly different at p<0.05.
Figure 1.
Effects of different Cd treatments on (a) chlorophyll a, (b) chlorophyll b content in non-adapted N0 and adapted A50 varieties of S. nigrum L. 1) 0 to 50 means control and soil treatments with Cd (mg kg-1); 2) Data over bars for the same treatments marked by the same capital letters are not significantly different at p<0.05. Data for different treatments marked by the same lowercase letters are not significantly different at p<0.05.
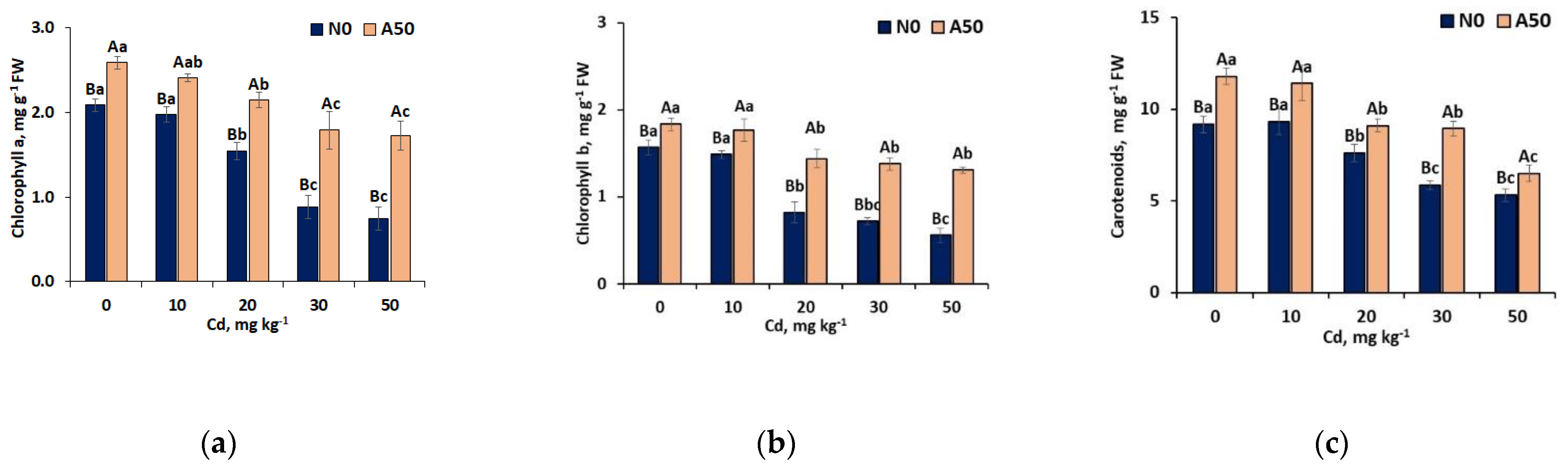
Figure 2.
Effects of different Cd treatments on (a) RbcL and (b) RbcS content in non-adapted N0 and adapted A50 varieties of S. nigrum L. 1) 0 to 50 means control and soil treatments with Cd (mg kg-1); 2) Data over bars for the same treatments marked by the same capital letters are not significantly different at p<0.05. Data for different treatments marked by the same lowercase letters are not significantly different at p<0.05.
Figure 2.
Effects of different Cd treatments on (a) RbcL and (b) RbcS content in non-adapted N0 and adapted A50 varieties of S. nigrum L. 1) 0 to 50 means control and soil treatments with Cd (mg kg-1); 2) Data over bars for the same treatments marked by the same capital letters are not significantly different at p<0.05. Data for different treatments marked by the same lowercase letters are not significantly different at p<0.05.
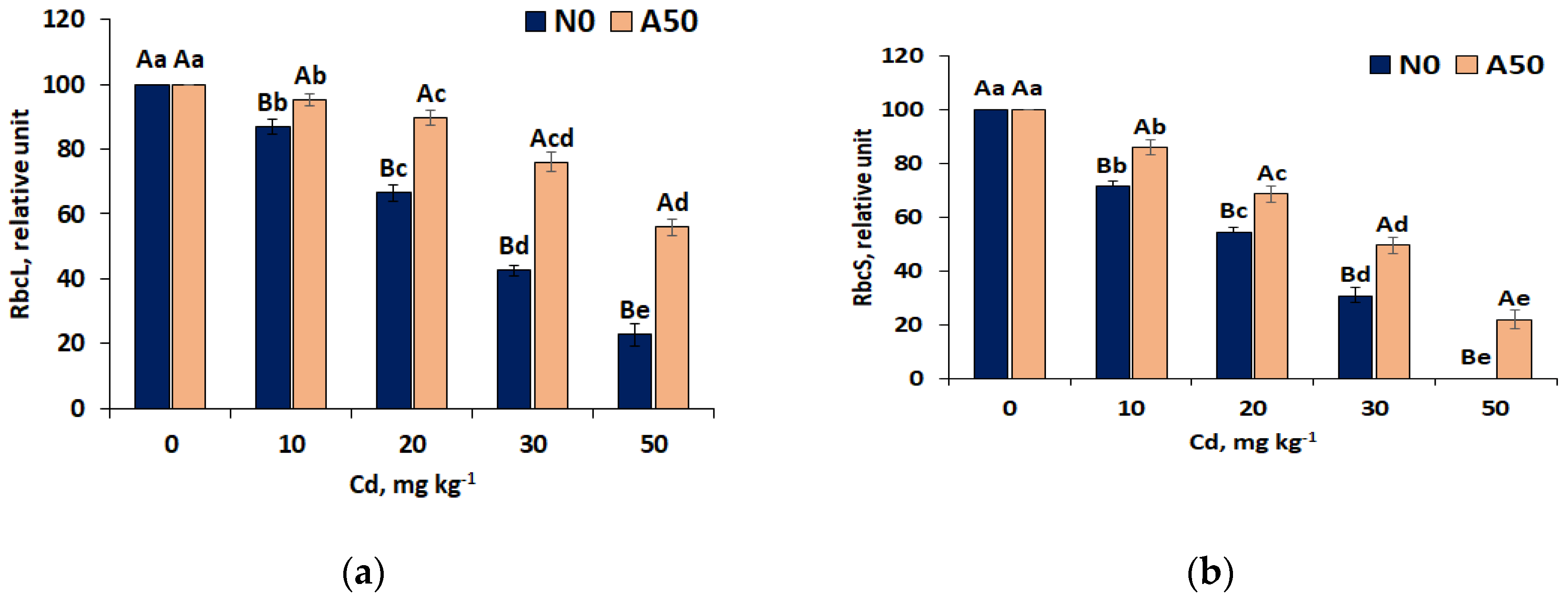
Figure 3.
Figure 3. Effects of different Cd treatments on ROS contents: (a) superoxide anion and (b) hydrogen peroxide in in non-adapted N0 and adapted A50 varieties of S. nigrum L. 1) 0 to 50 means control and soil treatments with Cd (mg kg-1); 2) Data over bars for the same treatments marked by the same capital letters are not significantly different at p<0.05. Data for different treatments marked by the same lowercase letters are not significantly different at p<0.05.
Figure 3.
Figure 3. Effects of different Cd treatments on ROS contents: (a) superoxide anion and (b) hydrogen peroxide in in non-adapted N0 and adapted A50 varieties of S. nigrum L. 1) 0 to 50 means control and soil treatments with Cd (mg kg-1); 2) Data over bars for the same treatments marked by the same capital letters are not significantly different at p<0.05. Data for different treatments marked by the same lowercase letters are not significantly different at p<0.05.
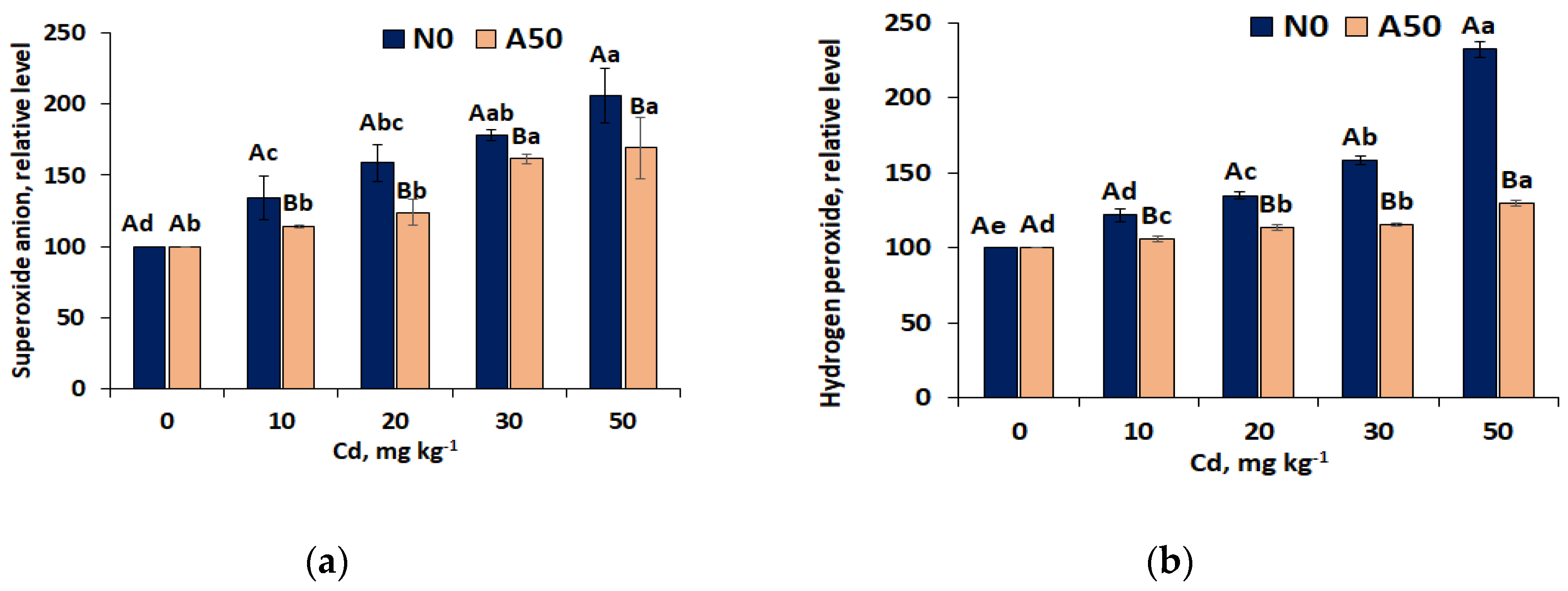
Figure 4.
Response of the first-line defence antioxidant (a)CAT, (b) SOD and (c) GPX) activity in leaves of the non-adapted N0 and adapted A50 varieties of S. nigrum to the growing Cd stress. 1) 0 to 50 means control and soil treatments with Cd (mg kg-1); 2) Data over bars for the same treatments marked by the same capital letters are not significantly different at p<0.05. Data for different treatments marked by the same lowercase letters are not significantly different at p<0.05.
Figure 4.
Response of the first-line defence antioxidant (a)CAT, (b) SOD and (c) GPX) activity in leaves of the non-adapted N0 and adapted A50 varieties of S. nigrum to the growing Cd stress. 1) 0 to 50 means control and soil treatments with Cd (mg kg-1); 2) Data over bars for the same treatments marked by the same capital letters are not significantly different at p<0.05. Data for different treatments marked by the same lowercase letters are not significantly different at p<0.05.
Figure 5.
Response of SOD isozyme (a) Cu/ZnSOD, (b) MnSOD (c) FeSOD activities in leaves of the non-adapted N0 and adapted A50 varieties of S. nigrum to the growing Cd stress. 1) 0 to 50 means control and soil treatments with Cd (mg kg-1); 2) Data over bars for the same treatments marked by the same capital letters are not significantly different at p<0.05. Data for different treatments marked by the same lowercase letters are not significantly different at p<0.05.
Figure 5.
Response of SOD isozyme (a) Cu/ZnSOD, (b) MnSOD (c) FeSOD activities in leaves of the non-adapted N0 and adapted A50 varieties of S. nigrum to the growing Cd stress. 1) 0 to 50 means control and soil treatments with Cd (mg kg-1); 2) Data over bars for the same treatments marked by the same capital letters are not significantly different at p<0.05. Data for different treatments marked by the same lowercase letters are not significantly different at p<0.05.
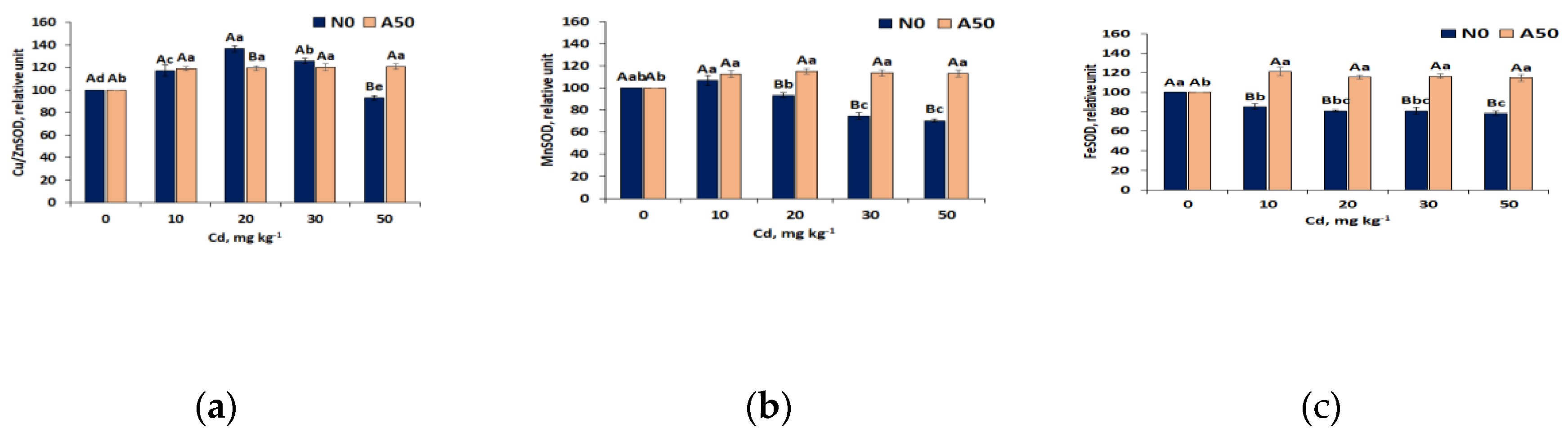
Figure 6.
Response of the second-line defense antioxidants ascorbate: (a) APX, (b) AsA and glutathione compounds: (c) GR, (d) GSH, (e) GSSG activity and (f) MDA content in leaves of non-adapted N0 and adapted A50 varieties of S. nigrum L.to the growing Cd stress. 1) 0 to 50 means control and soil treatments with Cd (mg kg-1); 2) Data over bars for the same treatments marked by the same capital letters are not significantly different at p<0.05. Data for different treatments marked by the same lowercase letters are not significantly different at p<0.05.
Figure 6.
Response of the second-line defense antioxidants ascorbate: (a) APX, (b) AsA and glutathione compounds: (c) GR, (d) GSH, (e) GSSG activity and (f) MDA content in leaves of non-adapted N0 and adapted A50 varieties of S. nigrum L.to the growing Cd stress. 1) 0 to 50 means control and soil treatments with Cd (mg kg-1); 2) Data over bars for the same treatments marked by the same capital letters are not significantly different at p<0.05. Data for different treatments marked by the same lowercase letters are not significantly different at p<0.05.
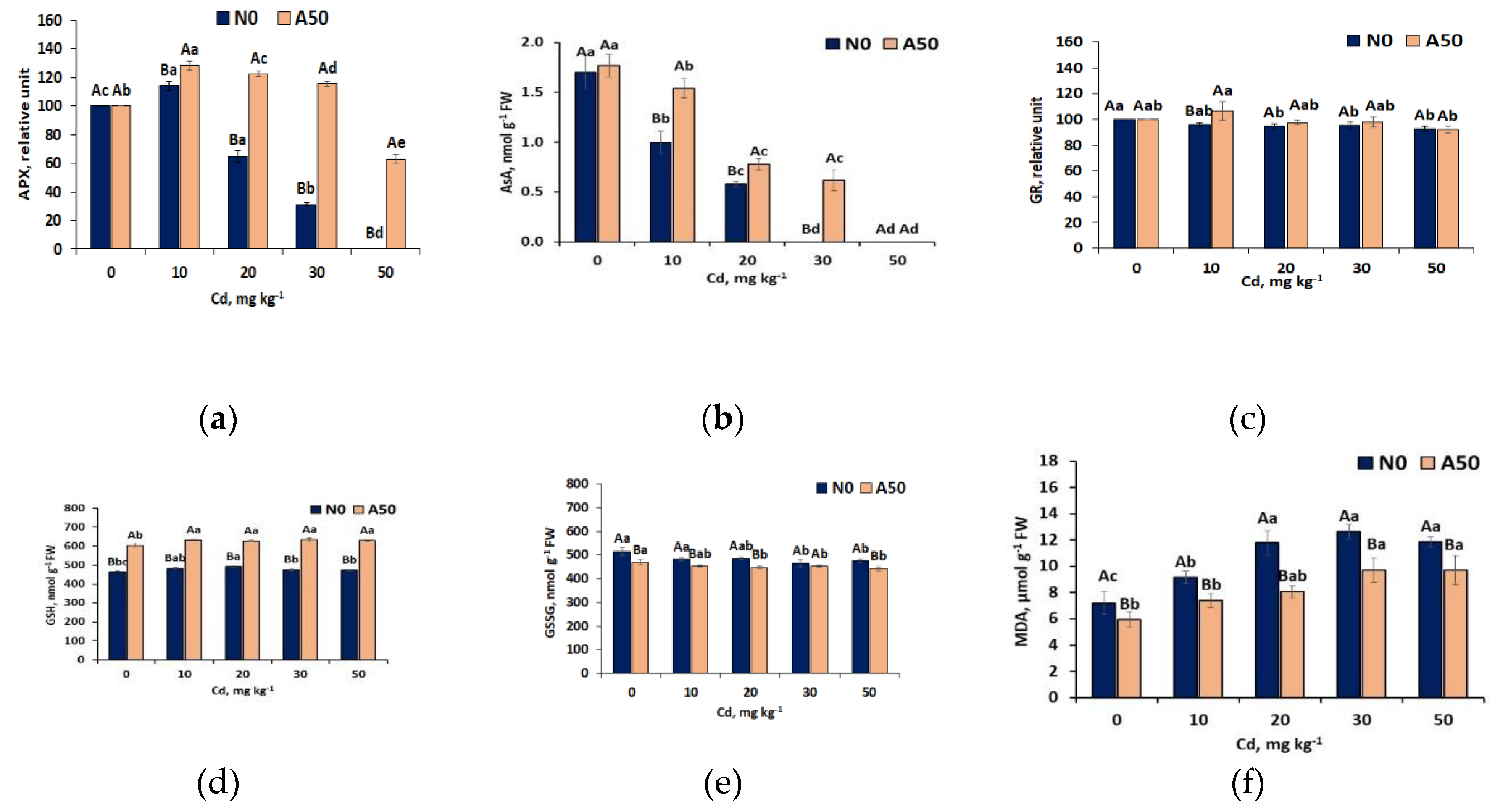

Table 2.
Effects of different Cd treatments on the biomass of Middle-European ecotype of S. nigrum L. non-adapted (N0) and adapted to Cd stress (A50).
Table 2.
Effects of different Cd treatments on the biomass of Middle-European ecotype of S. nigrum L. non-adapted (N0) and adapted to Cd stress (A50).
| T-Treatments mg Cd kg-1 | 0 | 10 | 20 | 30 | 50 |
|---|---|---|---|---|---|
| Cdsoil mg Cd kg-1 |
0.22±0.08 | 10.52±0.87 | 20.44±1.96 | 30.09±1.52 | 50.49±2.29 |
| BmR - Biomass of root (g pot -1 DW) | |||||
| N0 | 2.06±0.41Aa | 2.02±0.62Aa | 1.59±0.48Aa | 1.34±0.85Aa | 1.31±0.15Aa |
| A50 | 2.00±0.28Aa | 1.89±0.08Aa | 1.43±0.37Aa | 1.38±0.70Aa | 1.41±0.33Aa |
| BmS -Biomass of shoot (g pot -1 DW) | |||||
| N0 | 13.00±3.51Aa | 11.90±2.11Aa | 11.00±1.29Aa | 8.26±3.96Aa | 9.65±0.68Aa |
| A50 | 12.01±1.80Aa | 10.86±3.69Aa | 9.98±1.51Aa | 9.08±4.40Aa | 11.38±1.01Aa |
Note: 1)T (0=50) mean soil treatments with Cd (0, 10, 20, 30, 40, 50 mg Cd kg-1, respectively; 2)Data in each line marked with the same capital letters letters are not significantly different at p < 0.05; Data in each column/ section marked with the same lowercase are not significantly different at p < 0.05.
Table 2.
Cd accumulation, enrichment and translocation of Cd in non-adapted (N0) and adapted (A50) Middle-European ecotype of.S. nigrum L. at different Cd concentrations in soil.
Table 2.
Cd accumulation, enrichment and translocation of Cd in non-adapted (N0) and adapted (A50) Middle-European ecotype of.S. nigrum L. at different Cd concentrations in soil.
| T | Csoil mg kg-1Symbol | Cn mg kg-1 | EFn | TF | Ln µg pot -1 | LTF | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| N0 | A50 | N0 | A50 | N0 | A50 | N0 | A50 | N0 | A50 | ||
| Root | |||||||||||
| 0 | 0.22±0.08 | 0.57±0.14dA | 0.24±0.02cB | 2.59±0.63bA | 1.09±0.08bB | 1.20±0.49cA | 0.48±0.06bA | ||||
| 10 | 10.52±0.87 | 55.49±7.96cA | 50.83±11.66bcA | 5.27±0.76aA | 4.83±1.11aA | 108.6±21.9bcA | 95.90±21.58bA | ||||
| 20 | 20.44±1.96 | 127.20±11.95bA | 104.21±21.42bA | 6.22±0.56aA | 5.10±1.04aA | 201.8±68.6bA | 150.1±58.2bA | ||||
| 30 | 30.09±1.52 | 166.74±28.40bA | 110.16±25.99bB | 5.54±0.94aA | 3.66±0.86aB | 210.4±124.5bA | 157.6±92.6bA | ||||
| 50 | 50.49±2.29 | 324.07±31.20aA | 279.91±49.01aA | 6.42±0.62aA | 5.54±0.97aA | 420.8±17.5aA | 401.7±142.1aA | ||||
| Shoot | |||||||||||
| 0 | 0.22±0.08 | 0.82±0.34dA | 0.49±0.08cA | 3.72±1.53bA | 2.23±0.34cA | 1.40±0.23bB | 2.06±0.27aA | 10.9±5.92cA | 5.85±0.81cA | 8.77±1.63aB | 12.26±0.86aA |
| 10 | 10.52±0.87 | 81.05±7.81cA | 90.67±7.61bA | 7.70±0.74aA | 8.62±0.72aA | 1.48±0.27aA | 1.83±0.28aA | 957.9±127.2bA | 988.8±377.7bA | 8.91±0.71aA | 10.51±3.71abA |
| 20 | 20.44±1.96 | 111.05±2.15bA | 129.27±34.06aA | 5.43±0.10bA | 6.32±1.67bA | 0.88±0.10cB | 1.23±0.11bA | 1222.0±157.7abA | 1269.1±289.9abA | 6.44±1.82abA | 8.79±1.46abA |
| 30 | 30.09±1.52 | 103.03±7.81bA | 117.01±16.04abA | 3.42±0.26bcA | 3.89±0.53cA | 0.62±0.06cB | 1.11±0.33bcA | 835.69±377.0bA | 1015.2±403.3bA | 4.41±1.11bA | 7.43±2.55abA |
| 50 | 50.49±2.29 | 174.65±9.92aA | 164.55±13.52aA | 3.46±0.20bcA | 3.26±0.27cA | 0.54±0.04cA | 0.60±0.15cA | 1685.51±154.6aA | 1867.6±151.6aA | 4.00±0.20bA | 5.11±1.96bA |
| Total | |||||||||||
| 0 | 0.22±0.08 | 12.15±6.40cA | 6.33±0.86cA | ||||||||
| 10 | 10.52±0.87 | 1066.6±148.9bA | 1084.7±386.1bA | ||||||||
| 20 | 20.44±1.96 | 1423.8±183.9bA | 1419.2±344.7abA | ||||||||
| 30 | 30.09±1.52 | 1046.1±500.7bA | 1172.8±490.5bA | ||||||||
| 50 | 50.49±2.29 | 2106.3±85.9aA | 2269.3±240.6aA | ||||||||
1 Csoil (mg kg-1) - measured actual Cd concentrations in soil at T (treatments): 0, 10, 20, 30, 50 mg Cd kg-1, respectively; 2 N0 – S. nigrum not adapted and A50 – S.nighrum adapted to high cncentrations of Cd (50 mg Cd kg-1 ) in soil; 3 Enrichment factors EFn = Cn/Csoil, Translocation factors TF = Cshoot/Croot where Cn –concentrations in root and shoot, respectively; Accumulated loads Ln (µg pot1) = Cn Bmn, where Cn – concentrations in root and shoot, respectively (µg g-1); Bmn – biomass of root and shoot, respectively (g pot-1); Ltotal = Lroot+Lshoot – total accumulated load in root and shoot; Accumulated load translocation factor LTF – load translocation factor from root to shoot; 4 Data in each section/column marked by the same lowercase letters are not significantly different at p < 0.05; Data in each section/line marked by the same capital letters are not significantly different at p < 0.05.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



