
Submitted:
28 October 2024
Posted:
30 October 2024
You are already at the latest version
Abstract
Hearing loss (HL) is the most common disorder in newborns with a highly heterogeneous genetic background. Despite significant progress in screening and identifying genes related with congential hearing loss, there are still candidate genes implicated in HL that remain undiscovered. We investigated HL in 43 China families that segregate bilateral sensorineural via whole-exome sequencing(WES) and Sanger sequencing. Variants were found in 10 known non-syndromic hearing loss (NSHL) genes, 5 known syndromic hearing loss(SHL) genes and a candidate HL genes ATP7B. RNA sequencing reveals ATP7B mRNA expression of in developing and adult cochlea. Immunohistochemistry of the adult mouse cochlear tissue revealed prominent expression of ATP7B in the organ of Corti and the spiral ganglion neuron. Overall, we propose a new candidate genes ATP7B for congential hearing loss and novel variants in known HL genes, which expand our understanding of the etiology of HL.
Keywords:
congential hearing loss
; congential deafness
; etiology
; gene diagnosis
1. Introduction
Hearing loss(HL) is the most common disorder that has an incidence of 2–3 out of 1000 live births [1]. Genetics accounted for at least 50% of congential hearing loss [2]. Regional and ethnic differences of genetics between populations exist [3]. To date, more than 120 non-syndromic hearing loss(NSHL) genes have been identified [4,5]. GJB2 is the most common gene associated with autosomal recessive (AR) NSHL among East Asian population [6,7].
Identifying the genetic etiology of HL is essential for customizing personalized treatment plans, evaluating hearing recovery after cochlear implant, and genetic counseling [8,9]. Patients carrying different pathogenic variants exhibit a wide spectrum of phenotypes and vastly differing outcomes after cochlear implantation, thus a precise genetic diagnosis for HL patients may predict the potential benefits of cochlear implants [10]. Variants limited to presynaptic areas such as cochlear hair cells, supporting cells, or stria vascularis regions may result in better hearing recovery effects after cochlear implantation, while variants limited to the postsynaptic to auditory central region may result in uneven or poor hearing recovery after cochlear implantation [10,11,12]. Genetic testing combined with genetic counseling for congenital hearing loss may also reduced the incidence of congenital hearing loss in newborns.
Despite significant progress in screening and identifying genes related with congential hearing loss, there are still candidate genes implicated in HL that remain undiscovered [13,14,15,16]. Whole-exome sequencing (WES) have recently enhanced the discovery of rare or novel variants in known and candidate genes related hearing loss. In this study, we investigated HL in 43 China families that segregate bilateral sensorineural HL via WES and Sanger sequencing, and identified rare and novel variants in known and candidate genes, which are valuable for a deeper understanding of the relationship between genotype and phenotype of HL.
2. Materials and Methods
Sample Collection and Clinical Evaluation
Participants were enrolled from our outpatient department. Approval from the Institutional Review Boards of Tongji Medical College, Huazhong University of Science and Technology (2022 S041). Before participation, the study was clearly explained to each participant or their parents. All participant or their parents and they have signed written initial informed consent per the Declaration of Helsinki. All of the probands underwent evaluation include family and clinical history, available medical records, physical examination, tympanometry and audiological examinations, such as pure tone audiometry (PTA), audiometry Distortion Product Otoacoustic Emission (DPOAE), and auditory steady-state–evoked responses (ASSR) or auditory brainstem response (ABR). 3-5ml of peripheral blood samples from the probands and related family members was collected and subjected to whole-exome sequencing. Further clinical evaluation is used to determine whether the patient and family members have the syndrome. Temporal bone magnetic resonance imaging(MRI) or computed tomography(CT) is used to identify any possible developmental malformation.
Whole-Exome Sequencing and Data Analyses
Genomic DNA (gDNA) samples from 43 probands from 43 families underwent WES. The method has been described in detail in our previous articles. gDNA was extracted from the samples using the QIAamp DNA Blood Maxi Kit. Fragmentation of the genomic DNA was performed to generate the exome library. The libraries were sequenced on the BGISEQ-500 platform. Sequencing data were compared with the human genome reference (GRCh37/hg19) to detect target regions, single-nucleotide variants (SNVs), and INDEL calling. Identified SNVs and indels were compared with the information available in multiple databases (National Center for Biotechnology Information GenBank database, the Database of Single Nucleotide Polymorphisms, the 1000 Genomes Database). Pedigree mapping of the suspected variants was amplified via Sanger sequencing or WES.
RNA Expression Profiling of ATP7B in Mice Cochlear Tissues
Publicly available datasets in Gene Expression Omnibus (GEO) database and SHIELD (Shared Harvard Inner-Ear Laboratory Database) were utilized for statistical analysis and evaluation of ATP7B RNA expression levels in the mice cochlea. RNA sequencing data for hair cells and surrounding cells from the cochlea and utricle at different developmental stages (E16, P0, P4, and P7) originated from the dataset (GSE60019). We also analyzed the expression of the ATP7B genes in adult CBA/J mice cochlear RNA sequencing data from dataset (GSE111347) that includes the transcriptome of cochlear pillar and Deiters’ cells of adult CBA/J mice (28–35 days after birth), and dataset (GSE56866) that also includes the transcriptome of cochlear inner hair cells (IHC) and outer hair cells (OHC) of either sex adult CBA/J mice (25–35 days after birth). Cell-specific expression of the ATP7B gene in two datasets containing adult mouse cochlear RNA sequencing was comprehensively analyzed using Transcripts Per Kilobase Million (TPM).
Immunofluorescence of ATP7B Expression in Cochlear Tissue
To observe the regions where ATP7B is expressed in the inner ears of auditory mature mice, we prepared cochlear samples and conducted immunofluorescence staining. Cochlear tissues were dissected from C57BL/6 mice at postnatal day 18(P18). After the mice were euthanized, the cochlear tissue were carefully dissected from the temporal bones and rapidly moved into 4% paraformaldehyde at 4°C overnight and then treat the cochlear tissues with 10% EDTA-Na2 at 4°C overnight to soften the tissues. The cochlear tissues were embedded in optimal cutting temperature compound (Servicebio, G6059) and sectioned on a microtome (Leica,CM1950) to 10 μm sections at -25°C. The prepared tissue sections are stored at -80°C. The cochlear tissue specimens were incubated in 10% donkey serum for 1 h at room temperature, and then placed in ATP7B primary antibody(ABclonal, A5676, 1:200 diluted) and incubated overnight at 4°C. Then the cochlear tissue specimens were washed with 0.1% Tween20 in PBS for four times and incubated with Alexa Fluor®488 Donkey anti Rabbit secondary antibodies (AntGene, ANT024s) for 2 hour at room temperature. After incubation with a secondary antibodies, specimens werewashed with 0.1% Tween20 in PBS for three times. DAPI (AntGene, ANT165) and phalloidin (Yeasen, 40736ES75) were used for nuclear and F-actin staining, respectively. Images were obtained with a Nikon laser scanning confocal microscope.
3. Results
The Pedigree Analysis of the Families in this Cohort
This cohort consisted of 43 unrelated Chinese families. The participants in this study were recruited across the country with the majority from Hubei province. 79.1% (34/43) families could be explained by carring genomic variants leaving 20.9% (9/43) families without genetic causes identified through WES (Figure 1. A). The number of NIHL family and SHL family was 36 (84.8%) and 7 (15.2%) , respectively (Figure 1. B). 76.5% (26/34 genetic causes) families exhibited a likely AR mode of inheritance. 20.6% (7/34 genetic causes) families showed a pattern of an autosomal dominant (AD) mode of inheritance. 2.9% family (1/34 genetic causes) had a possible X-linked dominant mode of inheritance(Figure 1. C). 79.1% (34/43) families have members with prelingual HL (Figure 1. D).
Bioinformatic and Molecular Analysis
Identified variants in this study include missense variants (27), insertion/deletion (23) , nonsense (6), synonymous (1), and non-coding variants (6) (Figure 1. E). Ten known NSHL genes were identified in 27 of the 43 families (Table 1). These genes included GJB2, SLC26A4, MYO7A, DIAPH3, PTPRQ, LOXHD1, CDH23, MPZL2, EYA4, and OTOA. Five known SHL genes were identified in 6 of the 43 families (Table 2). These genes included TCOF1, EYA1, FDXR, AIFM1, and SOX10. ATP7B was segregate in SHL family as a candidate SHL gene (Table 3). Variants in GJB2 (14 families, 32.6%) and SLC26A4 (5 families, 11.6%) accounted for the majority (44.2%). EYA1 variants were found in two families, and variants in 13 other genes were only observed in one family. Twenty of the variants have been reported to be associated with the occurrence of hearing loss, while thirteen of the novel variants were found to segregate in 11/43 HL families, inlcude DIAPH3 c.2256_2257insT, PTPRQ c.6293T>C, CDH23 c.4859T>A, MPZL2 c.393_436+21del, TCOF1 c.3997_4007del, LOXHD1 c.2438T>A and c.1759C>T, EYA1 c.1350_1353delTAATinsCAGACA, FDXR c.1069G>T and c.364C>T, ATP7B c.4014T>A, SOX10 c.133del, AIFM1 c.1771-14T>A.
Non-syndromic hearing loss gene variants. Fourteen families segregated variants in GJB2 (OMIM: 121011), while five families segregated variants in SLC26A4 (OMIM: 605646), which are the most common HL genes in East Asia [17,18]. All the variants (N=12) identified in these families were classified as pathogenic and likely pathogenic based on the ACMG-AMP classification guidelines for HL. MYO7A (OMIM: 276903) variants c.1183C>T (p.Arg395Cys) and c.3696_3706del (p.Arg1232SerTer72) were segregated from NSHL phenotype of proband in Fam 20. It has been Fam 21 was found to have a DIAPH3 (OMIM: 603550) c.2256_2257insT (p.Ser752SerfsTer12) variant segregating with the HL phenotype. In Fam 21, the mother of the proband was confirmed by Sanger sequencing to carry DIAPH3 c.2256_2257insT (Figure 2.A). The mother's three brothers also affected HL, although they did not undergo genetic testing. PTPRQ (OMIM: 603317) is associated with ADHL (OMIM: 617663) and ARNSHL (OMIM: 613391). Novel heterozygous variants PTPRQ c.6293T>C (p.Leu2098Ser) were segregating in Fam 22 (Figure 2.B). Proband showed normal vestibular function until now. LOXHD1 (OMIM: 613072) is associated with ARNSHL (OMIM: 613079). Novel compound heterozygous variants LOXHD1 c.2438T>A (p.Leu813Ter) and c.1759C>T (p.Arg587Trp) were segregating in Fam 23 (Figure 2.C). CDH23 (OMIM: 605516) is involved in Usher syndrome 1D (OMIM: 601067) and ARNSHL (OMIM: 601386). Fam 24 was found to have Homozygous CDH23 c.4859T>A (p.Val1620Glu) segregating with the profound HL phenotype (Figure 2.D). The proband was 13 years old at the time of testing and had not yet exhibited vestibular dysfunction or pigmentary retinopathy. Fam 25 segregates compound heterozygous variants MPZL2 (OMIM: 604873) c.393_436+21del and c.220C>T (p.Gln74Ter) (Figure 2.E). MPZL2 has been associated with ARNSHL(OMIM: 618145). The feature of Fam26 are an HL father and two HL boys, which both carring EYA4 (OMIM: 603550) c.1759C>T ( p.Arg587Ter) variant (Figure 3.F). EYA4 has been associated with ARNSHL(OMIM: 601316). No clinical signs were observed after the genetics tests that would indicate that affected family members have disorder affecting other organs relate to syndromic HL. We also identified a novel compound heterozygous variants c.2359G>T(p.Glu787Ter) and c.2353A>C(p.Thr785Pro) in OTOA (Figure 2.G). OTOA (OMIM: 607038) has been associated with ARNSHL(607039).
Syndromic hearing loss gene variants. Novel heterozygous variants TCOF1 (OMIM: 606847) c.3997_4007del (p.Ser1333GlnfsTer16) were identified to be likely cause of SHL in Fam 28 (Figure 3.A). The proband in Fam28 has been clinically diagnosed with Treacher Collins syndrome (OMIM:154500). It was reported that clinical features of include downward-slanting palpebral fissures, malar hypoplasia, conductive deafness, mandibular hypoplasia, atresia of external ear canal, microtia, coloboma of the lower eyelid, asymmetry, projection of scalp hair onto the lateral cheek, cleft palate, choanal stenosis/atresia, cardiac malformation. Two families (Fam29 and Fam30) segregate variants EYA1 (OMIM: 601653) c.1225C>T (p.Asn451ArgfsTer18) and c.1350_1353delTAATinsCAGACA (p.Pro458Ala) (Figure 3.B,C). EYA1 is associated with Branchiootic syndrome 1 (OMIM: 602588). Both probands were observed to have clinical features of branchial cervical fistulae, preauricular pits and cup shaped Pinnae. Novel compound heterozygous variants FDXR c.1069G>T (p.Val357Leu) and c.364C>T (p.Arg122Cys) were identified to be likely cause of SHL in Fam 31 (Figure 3.D). FDXR (OMIM: 103270) variants FDXR c.364C>T (p.Arg122Cys) and c.1069G>T (p.Val357Leu) could cause Auditory neuropathy and optic atrophy (OMIM: 617717) or Multiple mitochondrial dysfunctions syndrome 9B (OMIM: 620887). Proband with severe HL in Fam 31 have bilateral optic nerve atrophy and retinitis pigmentosa, while the brother of the probands in Fam 31 have Down syndrome(OMIM: 190685). Sanger sequencing confirms that the mother of the proband carries a heterozygous variant c.364C>T (p.Arg122Cys) in FDXR, and the father of the proband does not carry FDXR variant. In Fam 33, novel heterozygous variants SOX10 (OMIM: 602229) C.133del (p.Gly38AlafsTer71) and heterozygous variants SLC26A4 (OMIM: 605646) c.1975G>C were both identified in proband (Figure 3.E). SOX10 variants has been associated with Waardenburg syndrome 2E (OMIM:611584) and Waardenburg syndrome 4C (OMIM:613266). The proband have blue eyes and profound HL. The mother of the proband in Fam 33, also have blue eyes and HL, and was identfied carring heterozygous variant C.133del in SOX10 by WES sequencing. The father of the proband in Fam 33, with the HL phenotype, and was identfied carring compound heterozygous variants c.1975G>C and c.1919G>A (p.Trp640Ter) in SLC26A4. In addition, the younger brother of the proband's father in Fam 33 also suffers from HL and was identified to carry a compound heterozygous variant c.1975G>C and c.1919G>A in SLC26A4. Additionally, the proband in Fam 34 was identified to carry novel variants AIFM1 c.1771-14T>A (Figure 3.F). AIFM1 (OMIM: 300169) have been associated with X-linked deafness-5 with peripheral neuropathy (DFNX5, OMIM: 300614), Cowchock syndrome (OMIM: 300169), and Combined oxidative phosphorylation deficiency 6 (OMIM: 300816). The proband in Fam 34 showed delayed development at 6 months after birth. Then the proband underwent a series of clinical examinations and was diagnosed with profound sensorineural hearing loss. MRI shows bilateral widening ventricular. The proband has multiple epileptic seizures. By age 5 years, the proband could not stand or walk alone, has poor eye contact, and is almost unresponsive to external sound stimuli. At present, the proband underwent cochlear implantation at the age of 6 and received continuous speech and motor rehabilitation training. However, the parents of the proband do not believe that cochlear implantation and speech rehabilitation training increase the proband's response to external sound stimuli. The parents of the proband do not carry the variant in AIFM1. The AIFM1 variant in the proband may be spontaneous mutation.
Hearing loss candidate genes variants. ATP7B (OMIM: 606882) is with autosomal recessive disorder Wilson disease (OMIM: 277900). In Fam 32, the proband was identified to carry compound heterozygous variant of ATP7B c.4014T>A (p.Ile1338Ile) and c.3446G>A (p.Gly1149Glu) (Figure 4. A). Variant ATP7B c.3446G>A was predicted to be likely pathogenic [19], while the clinical features of ATP7B c.4014T>A carrier could be conflicting interpretat according to gnomAD. It has been reported that synonymous mutation c.4014T>A(p.Ile1338Ile) could cause exon skipping in the ATP7B mRNA transcript [20].
Expression of HL Candidate Genes ATP7B in the Mouse Inner Ear
We used various publicly available RNA sequencing and microarray data sets to investigated expression of HL candidate gene ATP7B in the developing and adult mouse cochlear. ATP7B shows a low mRNA expression in the developing cochlear epithelium include cochlear hair cells(HC) and cochlear surrounding cells(SC) throughout E14, P0, P4 and P7 (Figure 4. B). The expressions of ATP7B mRNA were detected in pillar cells (PC) at adult CBA/J mice cochlear, while less ATP7B mRNA were detected in inner hair cells (IHC), outter hair cells (OHC), and Deiters’ cells (DC) (Figure 4. C).
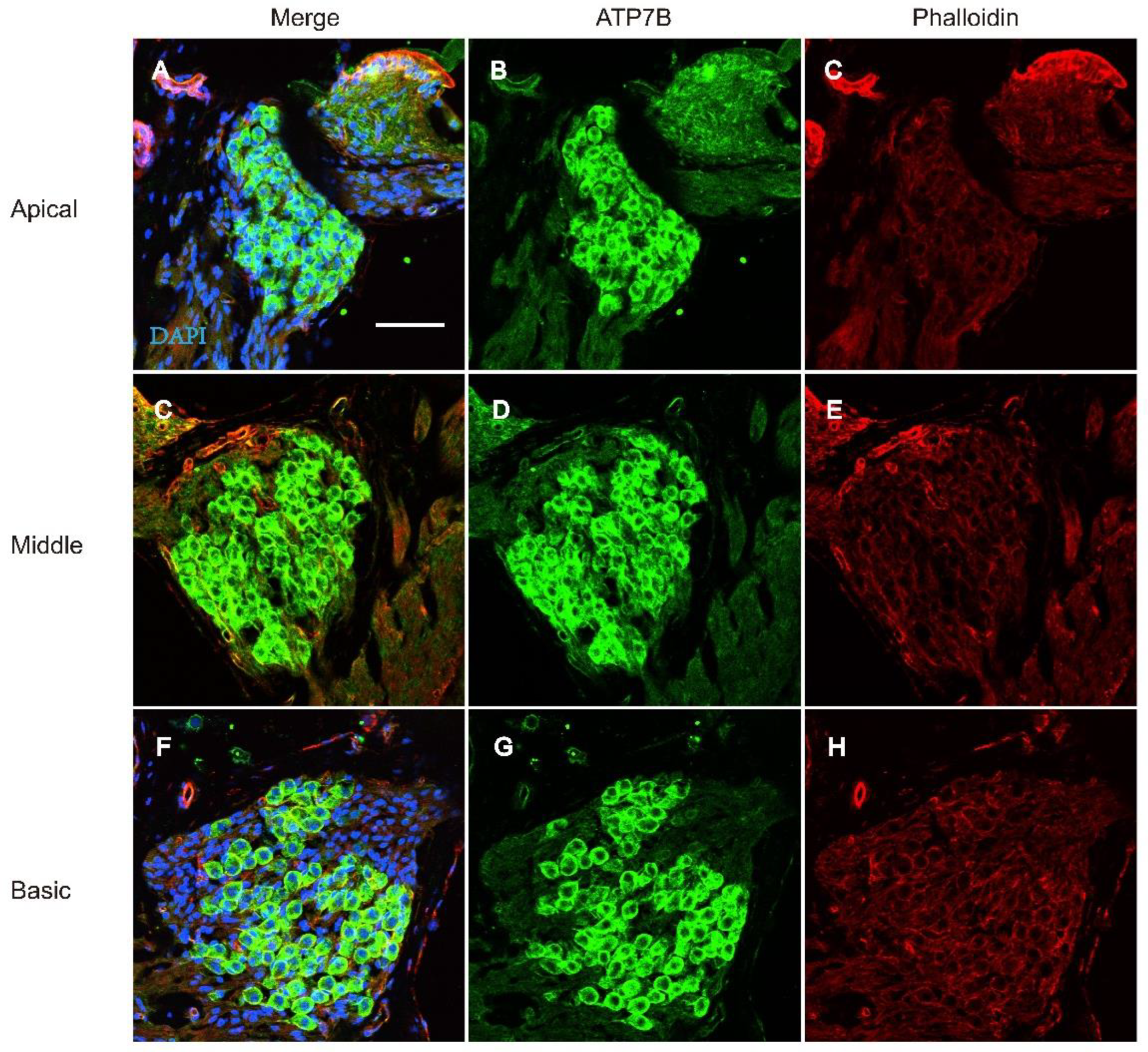
The onset of mice hearing arround P12 and the hearing threshold of mice remains stable around P18. To detect the expression of ATP7B protein in the adult mouse cochlea, we performed immunofluorescence staining on cochlear tissues of C57BL/6 mice at P25. ATP7B were detect to mainly expressed in the spiral ganglions (SGN) and organ of Corti (OC), include IHC, OHC, PC, DC, and Claudius' cells (CC) (Figure 5. A-L). Immunofluorescence results also demonstrates that ATP7B were expressed in the cytoplasmic region of the cochlear outer hair cells (Figure 5. M-O) and spiral ganglions cell (Figure 6. A-H).
3.2. Figures, Tables and Schemes
Figure 1.
Principal component analysis and overview of gene variants in this cohort. A: Frequency of the known HI and candidate genes in chinese families included in this cohort. B: Proportions of non-syndromic hearing loss and syndromic hearing loss families in this cohort. C: Proportions of hereditary mode of variants in known and novel candidate HL genes were identified. D: Proportions of pre-lingual and post-lingual hearing loss of the probands in this cohort. E: Proportions of variant types in known and novel candidate HL genes were identified.
Figure 1.
Principal component analysis and overview of gene variants in this cohort. A: Frequency of the known HI and candidate genes in chinese families included in this cohort. B: Proportions of non-syndromic hearing loss and syndromic hearing loss families in this cohort. C: Proportions of hereditary mode of variants in known and novel candidate HL genes were identified. D: Proportions of pre-lingual and post-lingual hearing loss of the probands in this cohort. E: Proportions of variant types in known and novel candidate HL genes were identified.
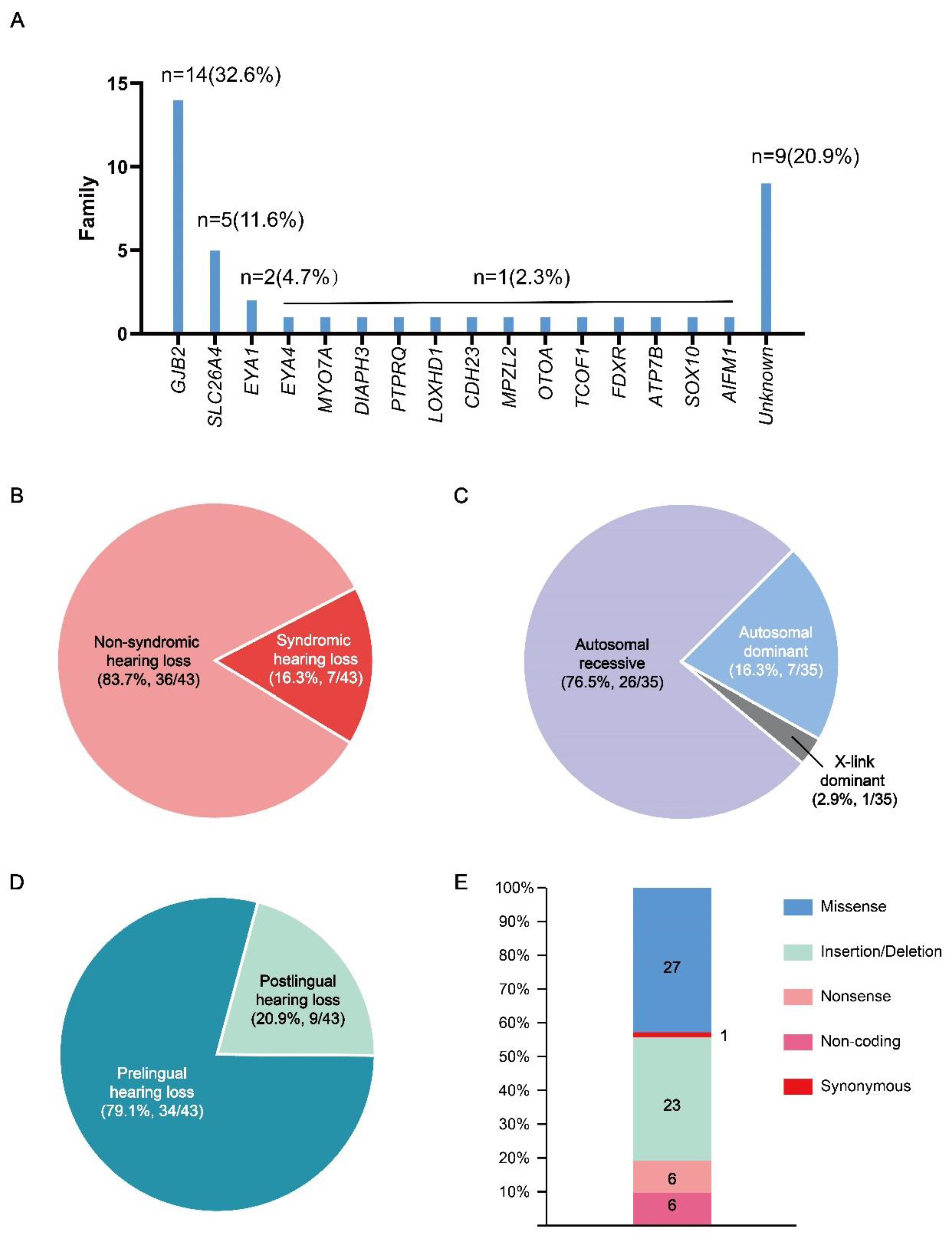
Figure 2.
Pedigrees of families segregation novel non-sydromic hearing loss genes variants. The segregation of non-sydromic hearing loss genes was shown in the respective families. A: Fam21, B: Fam22, C: Fam23, D: Fam24, E: Fam25, F: Fam26, G: Fam27. * Individuals whose audiological examination results can not obtained.
Figure 2.
Pedigrees of families segregation novel non-sydromic hearing loss genes variants. The segregation of non-sydromic hearing loss genes was shown in the respective families. A: Fam21, B: Fam22, C: Fam23, D: Fam24, E: Fam25, F: Fam26, G: Fam27. * Individuals whose audiological examination results can not obtained.
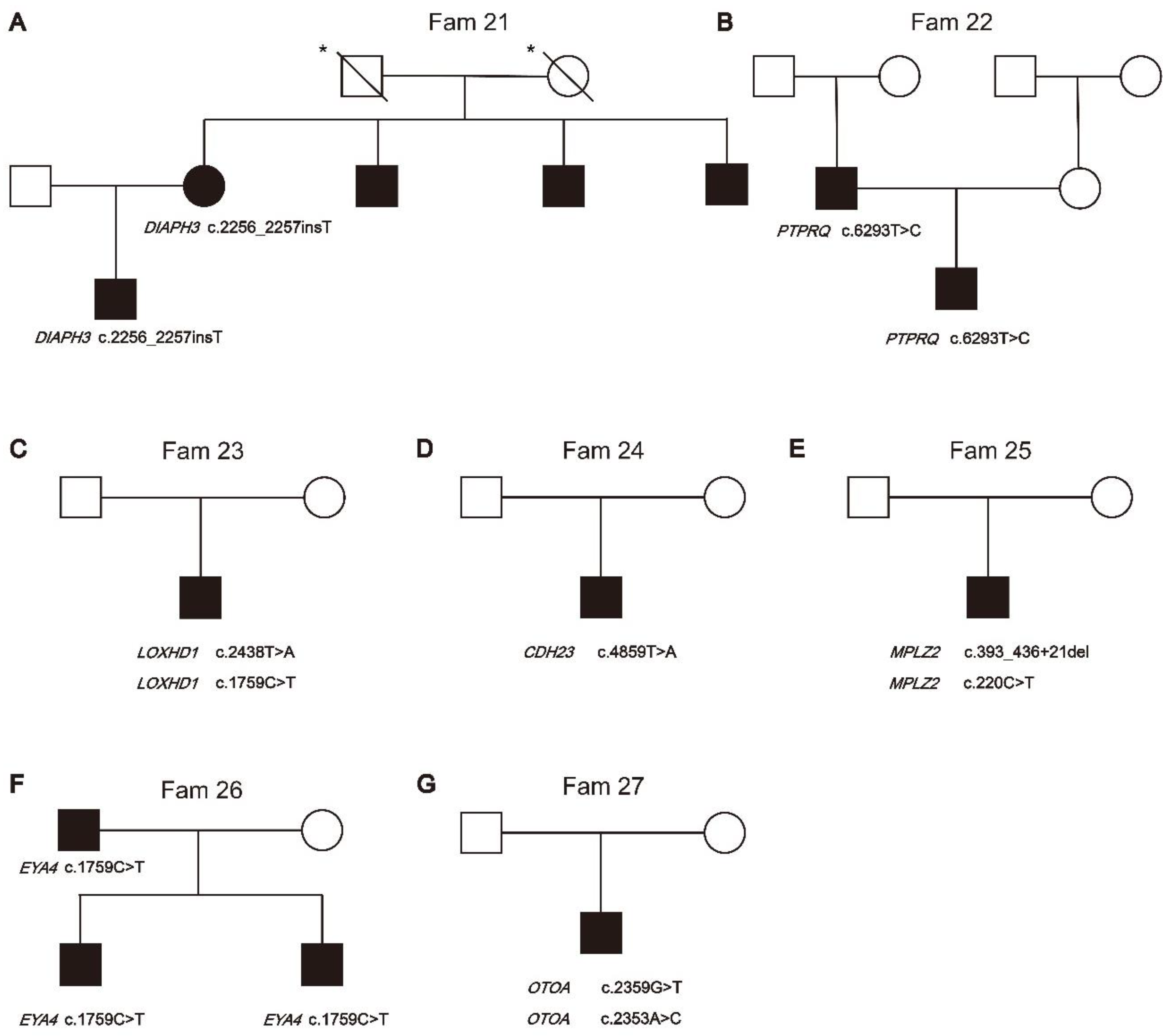
Figure 3.
Pedigrees of families segregation sydromic hearing loss genes variants. The segregation of sydromic hearing loss genes was shown in the respective families. A: Fam28, B: Fam29, C: Fam30, D: Fam31, E: Fam33, F: Fam34.
Figure 3.
Pedigrees of families segregation sydromic hearing loss genes variants. The segregation of sydromic hearing loss genes was shown in the respective families. A: Fam28, B: Fam29, C: Fam30, D: Fam31, E: Fam33, F: Fam34.
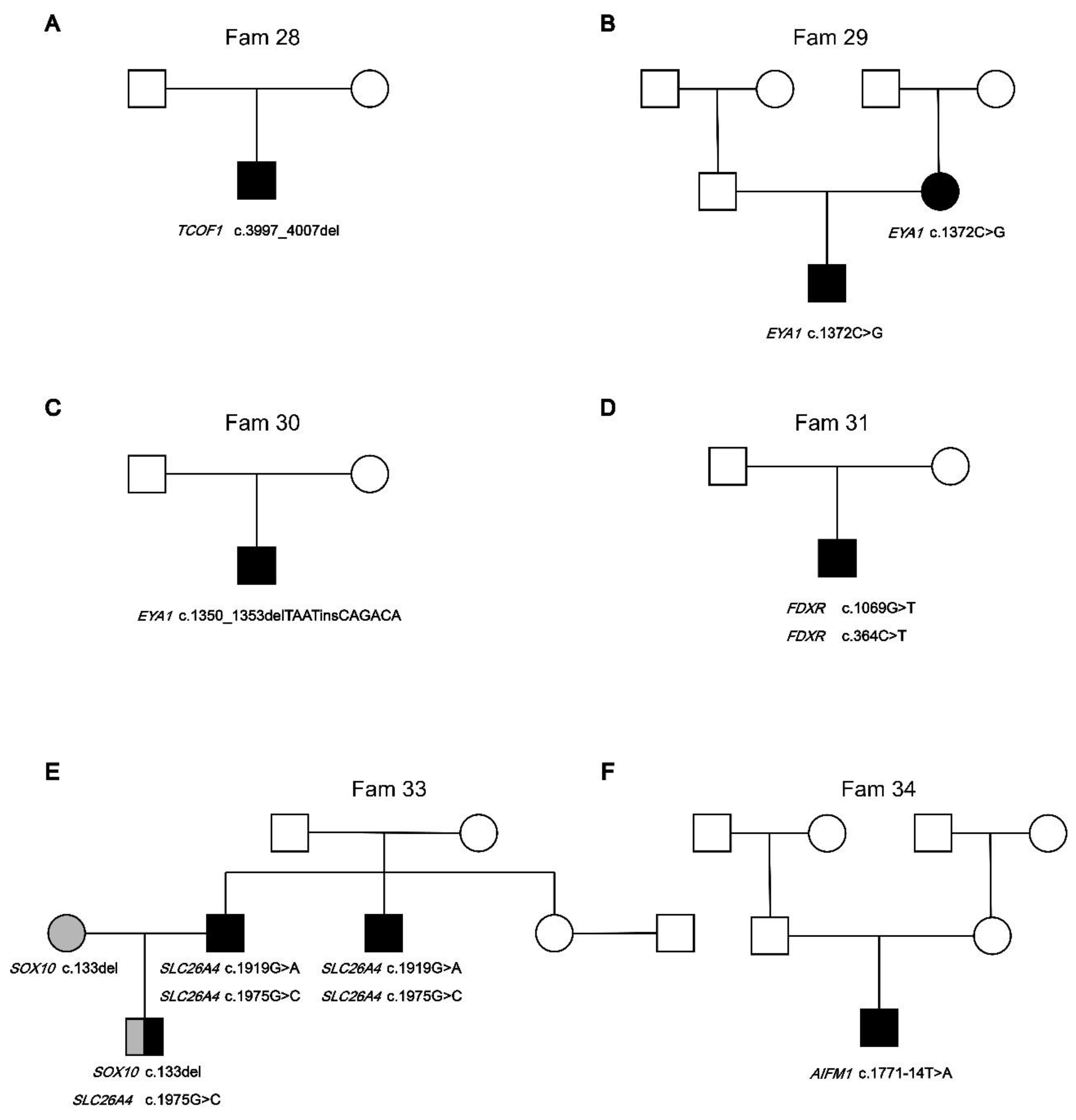
Figure 4.
Pedigrees of Fam32 segregation ATP7B variants and expression of novel candidate gene ATP7B in the development and adult cochlear. A: Pedigrees of Fam32 segregation ATP7B variants. B: ATP7B mRNA expression in the hair cells(HC) and surrounding cells(SC) of mouse cochlea and utricle during four different developmental stages: E16, P0, P4, and P7. RNA-sequencing data of hair cells (GFP+) and surrounding cells (GFP−) from the cochlear and utricles of mouse expressing EGFP under the Pou4f3 promoter during developmental stages. C: ATP7B mRNA expression in the inner ear cells of adult CBA/J mice. ATP7B mRNA expression in Deiters’ cells (DC), Pillar cells (PC), Inner Hair Cells (IHC), and Outer Hair Cells (OHC) were evaluated. The y-axis represents the gene expression normalized to transcripts per million (TPM). Data for (A,B) were obtained from Gene Expression Omnibus (GEO) database and SHIELD (Shared Harvard Inner-Ear La-boratory Database).
Figure 4.
Pedigrees of Fam32 segregation ATP7B variants and expression of novel candidate gene ATP7B in the development and adult cochlear. A: Pedigrees of Fam32 segregation ATP7B variants. B: ATP7B mRNA expression in the hair cells(HC) and surrounding cells(SC) of mouse cochlea and utricle during four different developmental stages: E16, P0, P4, and P7. RNA-sequencing data of hair cells (GFP+) and surrounding cells (GFP−) from the cochlear and utricles of mouse expressing EGFP under the Pou4f3 promoter during developmental stages. C: ATP7B mRNA expression in the inner ear cells of adult CBA/J mice. ATP7B mRNA expression in Deiters’ cells (DC), Pillar cells (PC), Inner Hair Cells (IHC), and Outer Hair Cells (OHC) were evaluated. The y-axis represents the gene expression normalized to transcripts per million (TPM). Data for (A,B) were obtained from Gene Expression Omnibus (GEO) database and SHIELD (Shared Harvard Inner-Ear La-boratory Database).
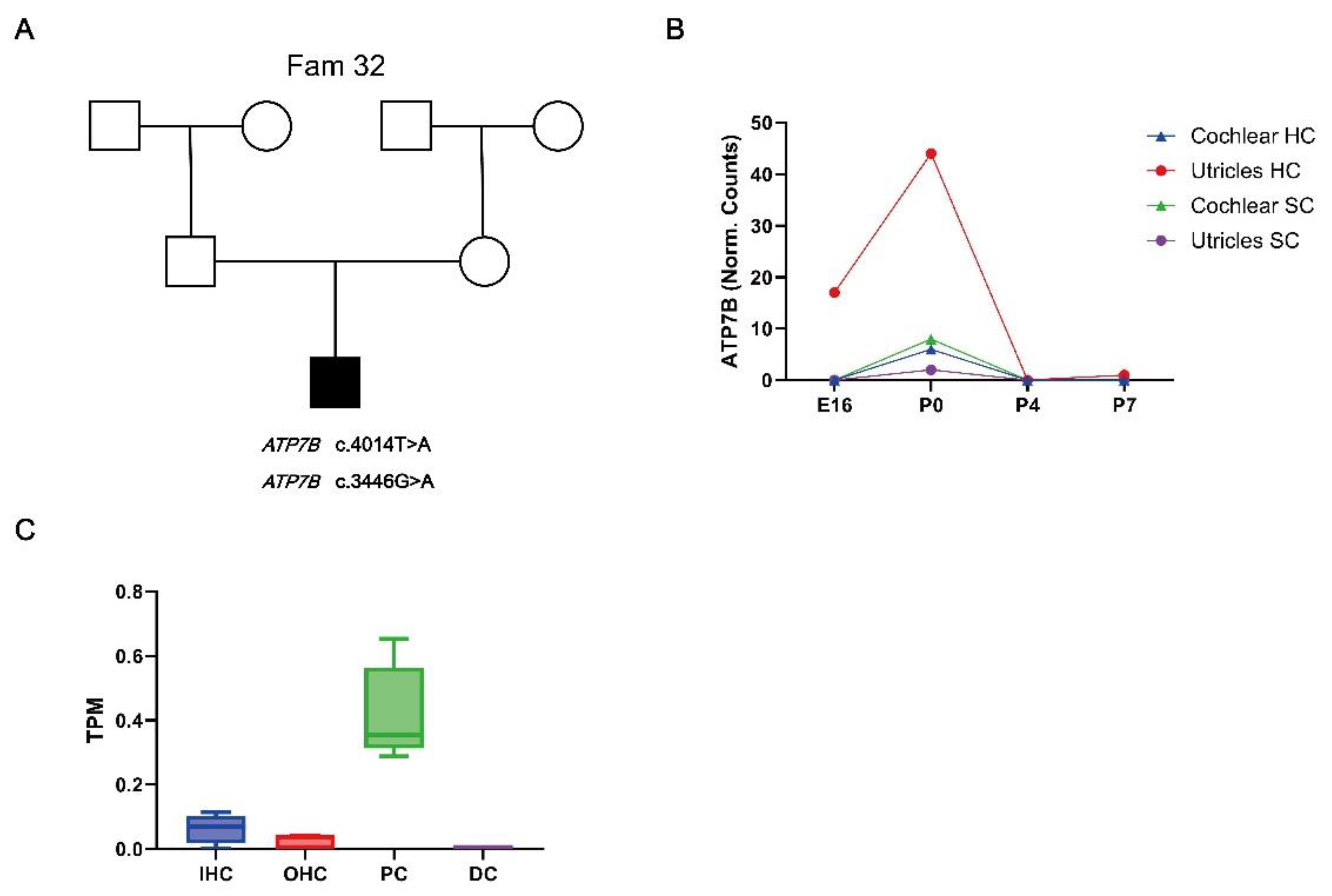
Figure 5.
Immunostaining of ATP7B in the cochlear of C57BL/6 mice at P18. A-C: Whole mount immunostaining of the cochlear of P18 C57BL/6 mice. Immunostaining of ATP7B (Green) is observed in both spiral ganglions cell and organ of Corti. Scale bar: 100μm. D-L: Immunostaining of ATP7B (Green) in regions of the OC at the apical, medial and basal turns of the cochlea specimens of C57BL/6 mice at P18. Immunostaining of ATP7B (Green) is observed in both IHCs, OHCs, DCs, PCs, and claudius cell. Scale bar: 100μm. M-O: Immunostaining of ATP7B (Green) in cytoplasmic region of the cochlear outer hair cells of C57BL/6 mice at P18. Nucleus was stained with DAPI (Blue). F-actin was stained with phalloidin (red). Scale bar:10μm.
Figure 5.
Immunostaining of ATP7B in the cochlear of C57BL/6 mice at P18. A-C: Whole mount immunostaining of the cochlear of P18 C57BL/6 mice. Immunostaining of ATP7B (Green) is observed in both spiral ganglions cell and organ of Corti. Scale bar: 100μm. D-L: Immunostaining of ATP7B (Green) in regions of the OC at the apical, medial and basal turns of the cochlea specimens of C57BL/6 mice at P18. Immunostaining of ATP7B (Green) is observed in both IHCs, OHCs, DCs, PCs, and claudius cell. Scale bar: 100μm. M-O: Immunostaining of ATP7B (Green) in cytoplasmic region of the cochlear outer hair cells of C57BL/6 mice at P18. Nucleus was stained with DAPI (Blue). F-actin was stained with phalloidin (red). Scale bar:10μm.
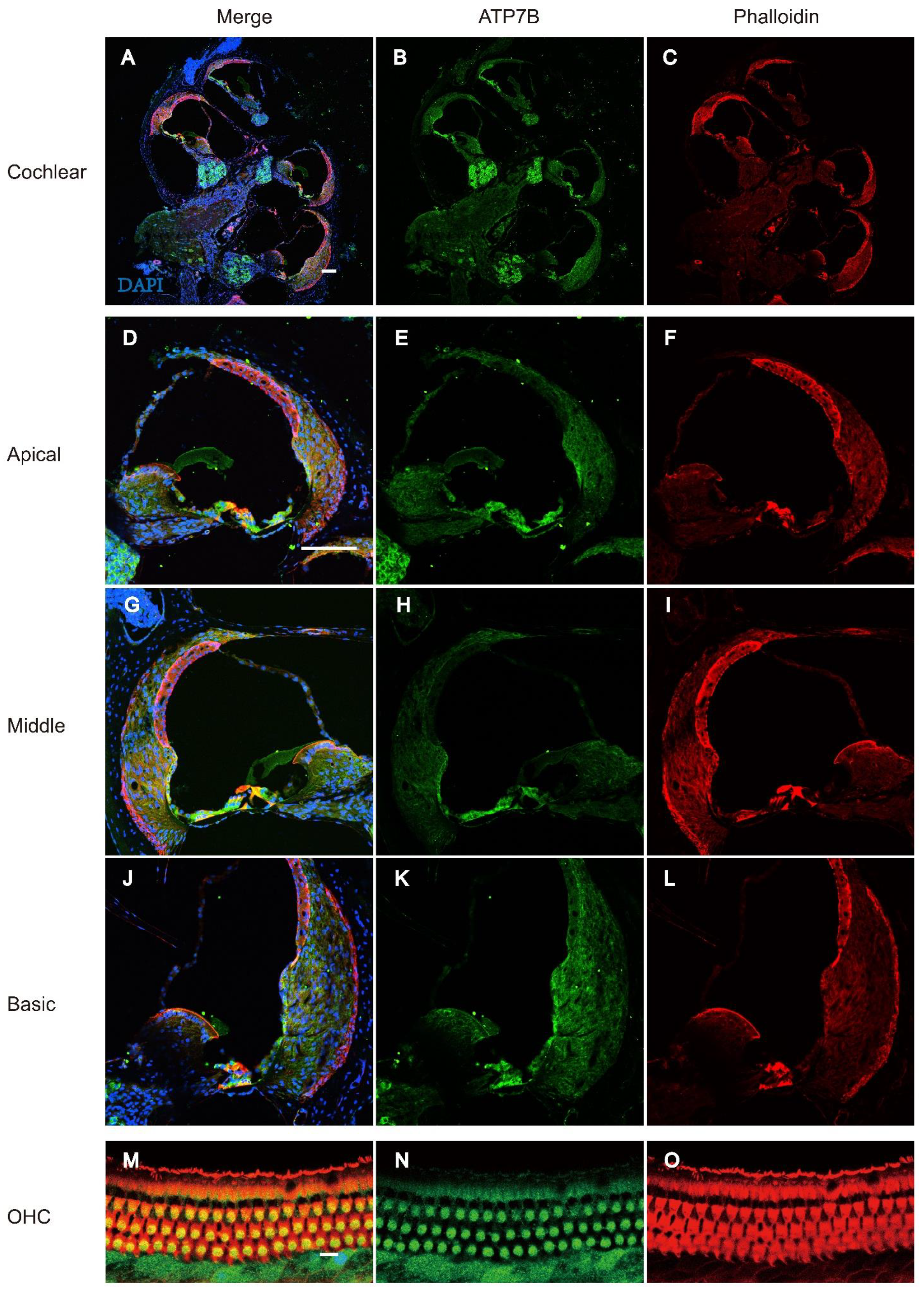
Figure 6.
Immunostaining of ATP7B in the spiral ganglion cell of C57BL/6 mice at P18. A-H: Whole mount immunostaining of ATP7B in the spiral ganglion cell of P18 C57BL/6 mice. Nucleus was stained with DAPI (Blue). F-actin was stained with phalloidin (red). Immunostaining of ATP7B (Green) is observed in spiral ganglions cell at apical, middle and basic turn. Scale bar: 50μm.
Figure 6.
Immunostaining of ATP7B in the spiral ganglion cell of C57BL/6 mice at P18. A-H: Whole mount immunostaining of ATP7B in the spiral ganglion cell of P18 C57BL/6 mice. Nucleus was stained with DAPI (Blue). F-actin was stained with phalloidin (red). Immunostaining of ATP7B (Green) is observed in spiral ganglions cell at apical, middle and basic turn. Scale bar: 50μm.
4. Discussion
In this study, we investigated HL in 44 China families that segregate bilateral sensorineural HL. Totally, we identified variants in 15 known HL genes. Genetic causes account for at least 79.1% of the studied families, which is higher than the earlier result [21]. Via the present data, we re-evaluated the contribution of GJB2 to 32.6% (14/43) among all families investigated, while the contribution of SLC26A4 to 11.6% (5/43), which suggests the prioritization of GJB2 and SLC26A4 in clinical practice of NIHL. This is consistent with the results in the previous Chinese HL population cohort [22,23,24].
83.7% family (36 /43) family was diagnosed NSHL. It should be noted that patients with hereditary hearing loss may only have isolated hearing loss phenotypes in childhood, which are often misdiagnosed as non syndromic hearing loss in clinical practice, ignoring other systemic diseases that may occur after puberty or adulthood [25,26,27]. Genetic diagnosis helps detect additional phenotypes earlier or alert patients to potential risks. MYO7A variants may causes either NSHI or Usher syndrome (USH). Liu et al reported a NSHL proband carried MYO7A heterozygous variants of c.1183C>T and c.1496T>C [28]. Xia et al reported the co-segregation of the homozygous MYO7A c.3696_3706del variant with the phenotype of deafness and progressive visual loss in the USH family [29]. Until now, the proband in Fam 20 carried MYO7A heterozygous variants of c.1183C>T and c.3696_3706del has not shown any ophthalmic symptoms. Similarly, PTPRQ variants could cause autosomal recessive or autosomal dominant congenital sensorineural hearing loss, with or without vestibular dysfunction in infancy or early childhood [30]. It cannot be ruled out that patients currently diagnosed with NSHL may develop late-onset syndrome symptoms in the future. We will closely follow up with the proband in order to intervene in time.
Accurate genetic diagnosis could help us develop targeted and efficient strategies for SHL patients [31,32,33,34]. In this study, SHL patients found in seven families (16.2%). It has been reported that prolonged treatment with riboflavin may result in some mild improvement in the ataxia caused by AIFM1 mutation. Ghezzi et al. reported that riboflavin supplementation could partially correct increased cell death and severe respiratory-chain deficiency [35,36]. After the proband was identified as carrying the AIFM1 variant, riboflavin therapy has been recommended. So far, no significant improvement in the patient's symptoms has been observed.
ATP7B encode a copper transporter. Variant in the ATP7B resulte in a rare autosomal recessive disorder of copper metabolism, Wilson disease (WD), also called progressive lenticular degeneration [37,38,39]. Copper excessive accumulaties in the WD patient’s body and its toxic effects causes significant damage on various organs and systemsm (most characteristically in brain and liver), leading to variability of symptoms such as liver dysfunction, neuropsychiatric disorders, Kayser–Fleischer rings on the cornea, and hemolysis caused by acute liver failure [40,41,42,43]. The age range of onset of WD is wide, and symptoms of WD can appear at any age [44,45]. In adult, ATP7B protein express in OC and SGN cells. We reported a patient with sensorineural hearing loss carrying a compound heterozygous variant c.4014T>A and c.3446G>A of ATP7B. Variant ATP7B c.4014T>A could cause exon skipping in the ATP7B mRNA transcript, and the ATP7B mutant may cause gradually affects the function and survival of HC and SGN cells in a dose and time-dependent manner.
Author Contributions
Y.S. and W.C. designed the project provided clinical examinations for the participants. Q.Z., Y.X., Y.H., and X.H.H collected and analyzed the data. Y.J. and Z.X.L., wrote the article. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Key Program of the National Natural Science Foundation of China (No. 82430035), the Foundation for Innovative Research Groups of Hubei Province (No. 2023AFA038), the National Key Research and Development Program of China (Nos. 2021YFF0702303, 2023YFE0203200), the Fundamental Research Funds for the Central Universities (No. 2024BRA019), and the National Natural Science Foundation of China (No. 82071058).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Korver, A.M. , et al., Congenital hearing loss. Nat Rev Dis Primers, 2017. 3: p. 16094.
- Cunningham, L.L. and D.L. Tucci, Hearing Loss in Adults. N Engl J Med, 2017. 377(25): p. 2465-2473.
- Wonkam, A.; Adadey, S.M.; Schrauwen, I.; Aboagye, E.T.; Wonkam-Tingang, E.; Esoh, K.; Popel, K.; Manyisa, N.; Jonas, M.; Dekock, C.; et al. Exome sequencing of families from Ghana reveals known and candidate hearing impairment genes. Commun. Biol. 2022, 5, 369. [Google Scholar] [CrossRef] [PubMed]
- Van Camp, G.; Smith, R.J.H. Hereditary Hearing Loss Homepage. 2018. Available online: http://hereditaryhearingloss.org (accessed on 17 December 2021).
- Jin, Y.; Liu, X.; Chen, S.; Xiang, J.; Peng, Z.; Sun, Y. Analysis of the Results of Cytomegalovirus Testing Combined with Genetic Testing in Children with Congenital Hearing Loss. J. Clin. Med. 2022, 11, 5335. [Google Scholar] [CrossRef] [PubMed]
- Dai, P.; Yu, F.; Han, B.; Liu, X.; Wang, G.; Li, Q.; Yuan, Y.; Liu, X.; Huang, D.; Kang, D.; et al. GJB2 mutation spectrum in 2063 Chinese patients with nonsyndromic hearing impairment. J. Transl. Med. 2009, 7, 26–26. [Google Scholar] [CrossRef]
- Dai, P.; Yu, F.; Han, B.; Yuan, Y.; Li, Q.; Wang, G.; Liu, X.; He, J.; Huang, D.; Kang, D.; et al. The prevalence of the 235delC GJB2 mutation in a Chinese deaf population. Anesthesia Analg. 2007, 9, 283–289. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, A.R.; Han, J.H.; Kim, S.D.; Kim, S.H.; Koo, J.-W.; Oh, S.H.; Choi, B.Y. Outcome of Cochlear Implantation in Prelingually Deafened Children According to Molecular Genetic Etiology. Ear Hear. 2017, 38, e316–e324. [Google Scholar] [CrossRef]
- Carlson, R.J.; Walsh, T.; Mandell, J.B.; Aburayyan, A.; Lee, M.K.; Gulsuner, S.; Horn, D.L.; Ou, H.C.; Sie, K.C.Y.; Mancl, L.; et al. Association of Genetic Diagnoses for Childhood-Onset Hearing Loss With Cochlear Implant Outcomes. Arch. Otolaryngol. Neck Surg. 2023, 149, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Tropitzsch, A.; Schade-Mann, T.; Gamerdinger, P.; Dofek, S.; Schulte, B.; Schulze, M.; Fehr, S.; Biskup, S.; Haack, T.B.; Stöbe, P.; et al. Variability in Cochlear Implantation Outcomes in a Large German Cohort With a Genetic Etiology of Hearing Loss. Ear Hear. 2023, 44, 1464–1484. [Google Scholar] [CrossRef]
- Vivero, R.J.; Fan, K.; Angeli, S.; Balkany, T.J.; Liu, X.Z. Cochlear implantation in common forms of genetic deafness. Int. J. Pediatr. Otorhinolaryngol. 2010, 74, 1107–1112. [Google Scholar] [CrossRef]
- Lin, P.-H.; Wu, H.-P.; Wu, C.-M.; Chiang, Y.-T.; Hsu, J.S.; Tsai, C.-Y.; Wang, H.; Tseng, L.-H.; Chen, P.-Y.; Yang, T.-H.; et al. Cochlear Implantation Outcomes in Patients with Auditory Neuropathy Spectrum Disorder of Genetic and Non-Genetic Etiologies: A Multicenter Study. Biomedicines 2022, 10, 1523. [Google Scholar] [CrossRef]
- Xing, G.; Yao, J.; Wu, B.; Liu, T.; Wei, Q.; Liu, C.; Lu, Y.; Chen, Z.; Zheng, H.; Yang, X.; et al. Identification of OSBPL2 as a novel candidate gene for progressive nonsyndromic hearing loss by whole-exome sequencing. Anesthesia Analg. 2015, 17, 210–218. [Google Scholar] [CrossRef]
- Morgan, A.; Vuckovic, D.; Krishnamoorthy, N.; Rubinato, E.; Ambrosetti, U.; Castorina, P.; Franzè, A.; Vozzi, D.; La Bianca, M.; Cappellani, S.; et al. Next-generation sequencing identified SPATC1L as a possible candidate gene for both early-onset and age-related hearing loss. Eur. J. Hum. Genet. 2019, 27, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, T.; Schrauwen, I.; Rehman, S.; Liaqat, K.; Acharya, A.; Giese, A.P.J.; Nouel-Saied, L.M.; Nasir, A.; Everard, J.L.; Pollock, L.M.; et al. ADAMTS1, MPDZ, MVD, and SEZ6: candidate genes for autosomal recessive nonsyndromic hearing impairment. Eur. J. Hum. Genet. 2022, 30, 22–33. [Google Scholar] [CrossRef]
- Liu, M.; Liang, Y.; Huang, B.; Sun, J.; Chen, K. Report of rare and novel mutations in candidate genes in a cohort of hearing-impaired patients. Mol. Genet. Genom. Med. 2022, 10, e1887. [Google Scholar] [CrossRef]
- Liu, X.-Z.; Jin, Y.; Chen, S.; Xu, K.; Xie, L.; Qiu, Y.; Wang, X.-H.; Sun, Y.; Kong, W.-J. F-Actin Dysplasia Involved in Organ of Corti Deformity in Gjb2 Knockdown Mouse Model. Front. Mol. Neurosci. 2022, 14, 808553. [Google Scholar] [CrossRef]
- Wang, Y.; Jin, Y.; Zhang, Q.; Xiong, Y.; Gu, X.; Zeng, S.; Chen, W. Research progress in delineating the pathological mechanisms of GJB2-related hearing loss. Front. Cell. Neurosci. 2023, 17, 1208406. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Ni, W.; Chen, W.-J.; Wan, B.; Zhao, G.-X.; Shi, Z.-Q.; Zhang, Y.; Wang, N.; Yu, L.; Xu, J.-F.; et al. Spectrum and Classification of ATP7B Variants in a Large Cohort of Chinese Patients with Wilson's Disease Guides Genetic Diagnosis. Theranostics 2016, 6, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhou, W.; Huang, Y.; Yin, H.; Jin, Y.; Jia, Z.; Zhang, A.; Liu, Z.; Zheng, B. Presumed missense and synonymous mutations in ATP7B gene cause exon skipping in Wilson disease. Liver Int. 2018, 38, 1504–1513. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.; Jin, Y.; Song, N.; Chen, S.; Shen, J.; Xie, W.; Sun, X.; Peng, Z.; Sun, Y. Comprehensive genetic testing improves the clinical diagnosis and medical management of pediatric patients with isolated hearing loss. BMC Med Genom. 2022, 15, 1–10. [Google Scholar] [CrossRef]
- Ma, J.; Ma, X.; Lin, K.; Huang, R.; Bi, X.; Ming, C.; Li, L.; Li, X.; Li, G.; Zhao, L.; et al. Genetic screening of a Chinese cohort of children with hearing loss using a next-generation sequencing panel. Hum. Genom. 2023, 17, 1–10. [Google Scholar] [CrossRef]
- Wu, J.; Cao, Z.; Su, Y.; Wang, Y.; Cai, R.; Chen, J.; Gao, B.; Han, M.; Li, X.; Zhang, D.; et al. Molecular diagnose of a large hearing loss population from China by targeted genome sequencing. J. Hum. Genet. 2022, 67, 643–649. [Google Scholar] [CrossRef]
- nbsp; Wu, C. -C.; Tsai, C.-Y.; Lin, Y.-H.; Chen, P.-Y.; Lin, P.-H.; Cheng, Y.-F.; Wu, C.-M.; Lin, Y.-H.; Lee, C.-Y.; Erdenechuluun, J.; et al. Genetic Epidemiology and Clinical Features of Hereditary Hearing Impairment in the Taiwanese Population. Genes 2019, 10, 772. [Google Scholar] [CrossRef]
- Chen, S.; Jin, Y.; Xie, L.; Xie, W.; Xu, K.; Qiu, Y.; Bai, X.; Zhang, H.-M.; Liu, X.-Z.; Wang, X.-H.; et al. A Novel Spontaneous Mutation of the SOX10 Gene Associated with Waardenburg Syndrome Type II. Neural Plast. 2020, 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Liu X, Chen S, Sun Y, Kong W. Lin Chuang Er Bi Yan Hou Tou Jing Wai Ke Za Zhi. 2021;35(3):229-237. [CrossRef]
- Shi, X.; Liu, X.; Zong, Y.; Zhao, Z.; Sun, Y. Novel compound heterozygous variants in MARVELD2 causing autosomal recessive hearing loss in two Chinese families. Mol. Genet. Genom. Med. 2024, 12, e2502. [Google Scholar] [CrossRef] [PubMed]
- Liu J, Ding Y, Hu Y. Lin Chuang Er Bi Yan Hou Tou Jing Wai Ke Za Zhi. 2022;36(1):27-31. [CrossRef]
- Xia, H.; Hu, P.; Yuan, L.; Xiong, W.; Xu, H.; Yi, J.; Yang, Z.; Deng, X.; Guo, Y.; Deng, H. A homozygous MYO7A mutation associated to Usher syndrome and unilateral auditory neuropathy spectrum disorder. Mol. Med. Rep. 2017, 16, 4241–4246. [Google Scholar] [CrossRef]
- Jin, Y.; Liu, X.-Z.; Xie, L.; Xie, W.; Chen, S.; Sun, Y. Targeted Next-Generation Sequencing Identified Novel Compound Heterozygous Variants in the PTPRQ Gene Causing Autosomal Recessive Hearing Loss in a Chinese Family. Front. Genet. 2022, 13, 884522. [Google Scholar] [CrossRef]
- Liu, X.; Zhao, Z.; Shi, X.; Zong, Y.; Sun, Y. The Effects of Viral Infections on the Molecular and Signaling Pathways Involved in the Development of the PAOs. Viruses 2024, 16, 1342. [Google Scholar] [CrossRef]
- Corriols-Noval, P.; Simón, E.C.L.; Cadiñanos, J.; Diñeiro, M.; Capín, R.; Aguado, R.G.; Marcos, M.C.; Angulo, C.M.; Farpón, R.C. Clinical Impact of Genetic Diagnosis of Sensorineural Hearing Loss in Adults. Otol. Neurotol. 2022, 43, 1125–1136. [Google Scholar] [CrossRef]
- Sloan-Heggen, C.M.; Bierer, A.O.; Shearer, A.E.; Kolbe, D.L.; Nishimura, C.J.; Frees, K.L.; Ephraim, S.S.; Shibata, S.B.; Booth, K.T.; Campbell, C.A.; et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 2016, 135, 441–450. [Google Scholar] [CrossRef]
- Liu, C.; Huang, Y.; Zhang, Y.; Ding, H.; Yu, L.; Wang, A.; Wang, Y.; Zeng, Y.; Liu, L.; Liu, Y.; et al. Next-generation sequencing facilitates genetic diagnosis and improves the management of patients with hearing loss in clinical practice. Int. J. Pediatr. Otorhinolaryngol. 2022, 161, 111258. [Google Scholar] [CrossRef]
- Ghezzi, D.; Sevrioukova, I.; Invernizzi, F.; Lamperti, C.; Mora, M.; D'Adamo, P.; Novara, F.; Zuffardi, O.; Uziel, G.; Zeviani, M. Severe X-Linked Mitochondrial Encephalomyopathy Associated with a Mutation in Apoptosis-Inducing Factor. Am. J. Hum. Genet. 2010, 86, 639–649. [Google Scholar] [CrossRef]
- Wooton-Kee, C.R. Therapeutic implications of impaired nuclear receptor function and dysregulated metabolism in Wilson's disease. Pharmacol. Ther. 2023, 251, 108529. [Google Scholar] [CrossRef] [PubMed]
- Huster, D.; Kühne, A.; Bhattacharjee, A.; Raines, L.; Jantsch, V.; Noe, J.; Schirrmeister, W.; Sommerer, I.; Sabri, O.; Berr, F.; et al. Diverse Functional Properties of Wilson Disease ATP7B Variants. Gastroenterology 2012, 142, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.J.; Yi, F.; Dayuha, R.; Duong, P.; Horslen, S.; Camarata, M.; Coskun, A.K.; Houwen, R.H.; Pop, T.L.; Zoller, H.; et al. Direct Measurement of ATP7B Peptides Is Highly Effective in the Diagnosis of Wilson Disease. Gastroenterology 2021, 160, 2367–2382. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Ni, W.; Chen, W.-J.; Wan, B.; Zhao, G.-X.; Shi, Z.-Q.; Zhang, Y.; Wang, N.; Yu, L.; Xu, J.-F.; et al. Spectrum and Classification of ATP7B Variants in a Large Cohort of Chinese Patients with Wilson's Disease Guides Genetic Diagnosis. Theranostics 2016, 6, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Hua, R.; Hua, F.; Jiao, Y.; Pan, Y.; Yang, X.; Peng, S.; Niu, J. Mutational analysis of ATP7B in Chinese Wilson disease patients. . 2016, 8, 2851–61. [Google Scholar]
- Zhang, S.; Yang, W.; Li, X.; Pei, P.; Dong, T.; Yang, Y.; Zhang, J. Clinical and genetic characterization of a large cohort of patients with Wilson’s disease in China. Transl. Neurodegener. 2022, 11, 1–11. [Google Scholar] [CrossRef]
- Bandmann, O.; Weiss, K.H.; Kaler, S.G. Wilson's disease and other neurological copper disorders. Lancet Neurol. 2015, 14, 103–113. [Google Scholar] [CrossRef]
- Xie, J.-J.; Wu, Z.-Y. Wilson’s Disease in China. Neurosci. Bull. 2017, 33, 323–330. [Google Scholar] [CrossRef]
- Wungjiranirun, M.; Sharzehi, K. Wilson's Disease. Semin. Neurol. 2023, 43, 626–633. [Google Scholar] [CrossRef]
- Gouider-Khouja, N. Wilson's disease. Park. Relat. Disord. 2009, 15, S126–S129. [Google Scholar] [CrossRef]

Table 1.
Variants identified in the non-syndromic hearing loss families.
| Family | Gene | Nucleotide change | Protein change | Inh | GT | Mutation type |
|---|---|---|---|---|---|---|
| Fam1 | GJB2 | c.235del | p.Leu79CysfsTer3 | AR | Hom | Deletion |
| Fam2 | GJB2 | c.99del | p.Met34Ter | AR | Het | Deletion |
| c.299_300del | p.His100ArgfsTer14 | AR | Het | Deletion | ||
| Fam3 | GJB2 | c.235del | p.Leu79CysfsTer3 | AR | Hom | Deletion |
| Deletion | ||||||
| Fam4 | GJB2 | c.109G>A | p.Val37Ile | AR | Hom | Missense |
| Fam5 | GJB2 | c.139G>T | p.Glu47Ter | AR | Het | Nonsense |
| c.176_191del | p.Gly59AlafsTer18 | AR | Het | Deletion | ||
| Fam6 | GJB2 | c.109G>A | p.Val37Ile | AR | Hom | Missense |
| Fam7 | GJB2 | c.235del | p.Leu79CysfsTer3 | AR | Hom | Deletion |
| Fam8 | GJB2 | c.235del | p.Leu79CysfsTer3 | AR | Het | Deletion |
| c.176_191del | p.Gly59AlafsTer18 | AR | Het | Deletion | ||
| Fam9 | GJB2 | c.109G>A | p.Val37Ile | AR | Hom | Missense |
| Fam10 | GJB2 | c.109G>A | p.Val37Ile | AR | Hom | Missense |
| Fam11 | GJB2 | c.235del | p.Leu79CysfsTer3 | AR | Het | Deletion |
| c.299_300del | p.His100ArgfsTer14 | AR | Het | Deletion | ||
| Fam12 | GJB2 | c.235del | p.Leu79CysfsTer3 | AR | Hom | Deletion |
| Deletion | ||||||
| Fam13 | GJB2 | c.257C>G | p.Thr86Arg | AR | Het | Missense |
| c.109G>A | p.Val37Ile | AR | Het | Missense | ||
| Fam14 | GJB2 | c.299_300delAT | p.His100ArgfsTer14 | AR | Het | Deletion |
| c.176_191del | p.Gly59AlafsTer18 | AR | Het | Deletion | ||
| Fam15 | SLC26A4 | c.919-2A>G | - | AR | Het | Noncoding |
| c.281C>T | p.Thr94Ile | AR | Het | Missense | ||
| Fam16 | SLC26A4 | c.1594A>C | p.Ser532Arg | AR | Het | Missense |
| c.2168A>G | p.His723Arg | AR | Het | Missense | ||
| Fam17 | SLC26A4 | c.919-2A>G | - | AR | Hom | Noncoding |
| Fam18 | SLC26A4 | c.2009T>C | p.Val670Ala | AR | Hom | Missense |
| Fam19 | SLC26A4 | c.919-2A>G | - | AR | Het | Noncoding |
| c.2168A>G | p.His723Arg | AR | Het | Missense | ||
| Fam20 | MYO7A | c.1183C>T | p.Arg395Cys | AR | Het | Missense |
| c.3696_3706del | p.Arg1232SerTer72 | AR | Het | Deletion | ||
| Fam21 | DIAPH3 | c.2256_2257insT | p.Ser752SerfsTer12 | AD | Het | Deletion |
| Insertion | ||||||
| Fam22 | PTPRQ | c.6293T>C | p.Leu2098Ser | AD | Het | Missense |
| Fam23 | LOXHD1 | c.2438T>A | p.Leu813Ter | AR | Het | Nonsense |
| c.1759C>T | p.Arg587Trp | AR | Het | Missense | ||
| Fam24 | CDH23 | c.4859T>A | p.Val1620Glu | AR | Hom | Missense |
| Fam25 | MPZL2 | c.220C>T | p.Gln74Ter | AR | Het | Nonsense |
| c.393_436+21del | - | AR | Het | Noncoding | ||
| Fam26 | EYA4 | c.1759C>T | p.Arg587Ter | AD | Het | Nonsense |
| Fam27 | OTOA | c.2359G>T | p.Glu787Ter | AR | Het | Nonsense |
| c.2353A>C | p.Thr785Pro | AR | Het | Missense |
1 Inh mode of inheritance, hom homozygote, het heterozygote, GT patient genotype.
Table 2.
Variants identified in the syndromic hearing loss families.
| Family | Gene | Nucleotide change | Protein change | Inh | GT | Mutation type |
|---|---|---|---|---|---|---|
| Fam28 | TCOF1 | c.3997_4007del | p.Ser1333GlnfsTer16 | AD | Het | Deletion |
| Fam29 | EYA1 | c.1372C>G | p.Pro458Ala | AD | Het | Missense |
| Fam30 | EYA1 | c.1350_1353delTAATinsCAGACA | p.Asn451ArgfsTer18 | AD | Het | Deletion/Insertion |
| Fam31 | FDXR | c.1069G>T | p.Val357Leu | AR | Het | Missense |
| FDXR | c.364C>T | p.Arg122Cys | AR | Het | Missense | |
| Fam33 | SOX10 | C.133del | p.Gly38AlafsTer71 | AD | Het | Deletion |
| SLC26A4 | c.1975G>C | p.Val659Leu | AR | Het | Missense | |
| SLC26A4 | c.1919G>A | p.Trp640Ter | AR | Het | Nonsense | |
| Fam34 | AIFM1 | c.1771-14T>A | - | X-link | Het | Noncoding |
2 Inh mode of inheritance, hom homozygote, het heterozygote, GT patient genotype.
Table 3.
Hearing loss candidate variants segregated in Fam32.
| Family | Gene | Nucleotide change | Protein change | Inh | GT | Mutation type |
|---|---|---|---|---|---|---|
| Fam32 | ATP7B | c.4014T>A | p.Ile1338Ile | AR | Het | Synonymous |
| ATP7B | c.3446G>A | p.Gly1149Glu | AR | Het | Missense |
3 Inh mode of inheritance, hom homozygote, het heterozygote, GT patient genotype.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



