Submitted:
30 October 2024
Posted:
31 October 2024
You are already at the latest version
Abstract
Fungi play an increasingly important role in the biological control of insect pests. Aspergillus oryzae XJ1 is highly virulent to locusts, which are a destructive economic pest worldwide. Because of its host association with locusts, which is unique in Aspergillus, in this study we examined the genetic relationships of A. oryzae XJ1 within Aspergillus. We sequenced the genome of A. oryzae XJ1 and compared it with the genomes of other Aspergillus species. The complete genome of A. oryzae XJ1 is 37.9 Mb, comprising 11,720 putative genes, assembled into eight chromosomes. The genome size is similar to that of other A. oryzae strains. Phylogenomic analysis indicated that A. oryzae XJ1 was most closely related to A. flavus. Core/pan-genome analysis of A. oryzae XJ1 and other Aspergillus species revealed that A. oryzae XJ1 had 704 strain-specific genes, whereas A. flavus NRRL 3357, A. oryzae KDG 21, and A. parasiticus NRRL 2999 had 646, 955, and 779 unique genes, respectively. These results indicate that A. oryzae XJ1 is genetically distinct at the genome level from other Aspergillus species, including A. oryzae and A. flavus, and its recognition as a distinct species is supported.
Keywords:
Aspergillus
; genome
; comparison
; biological control
; locust pathogen
; core/pan-genome analysis
1. Introduction
Approximately 1000 known species of entomopathogenic fungi infect most insect species [1]. To date, ~13 species or subspecies of insect-pathogenic fungi have been formulated and registered as pesticides [2]. These products mainly comprise Metarhizium anisopliae and Beauveria bassiana. Reports of the development of Aspergillus spp. as biopesticides are extremely rare.
Recently, A. oryzae XJ1 was identified based on morphological characteristics and molecular analysis of the sequence of 18S rDNA and reported to be highly virulent to locusts, both in laboratory bioassays and large-scale field experiments [3,4]. Thus, this fungal strain shows promise for the biological control of locusts. The finding that A. oryzae XJ1 is an entomopathogenic fungus is in strong contrast to other strains of A. oryzae, which is a non-entomopathogenic species and has been used for centuries in the production of traditional fermented food products, such as soy sauce, soybean paste, and rice wine, in China, Japan, and other Asian countries [5]. Several reports indicate that A. flavus, A. fumigutus, and A. niger are capable of infecting certain locusts [6,7,8]. We want to reconsider the classification status of A. oryzae XJ1.
To date, at least 104 genomes of different A. oryzae strains have been sequenced [9,10,11,12]. Intraspecific genome comparisons between A. oryzae RIB326 and A. oryzae RIB40, and between A. oryzae and A. flavus and other species, have been performed previously [13,14]. In this study, to examine differences between A. oryzae XJ1 and other Aspergillus species, we conducted whole-genome sequencing and analyzed the genome profile of A. oryzae XJ1 in comparison with other Aspergillus species. The results revealed that A. oryzae XJ1 is genetically distinct from other strains of A. oryzae and A. flavus at the genome level, supporting its recognition as a distinct species.
2. Materials and Methods
2.1. Strain Culture
Aspergillus oryzae XJ1 was inoculated in potato dextrose agar liquid medium and cultured on a shaker at 28.0 ± 1.0 °C for 2 days, then the mycelium was collected for extraction of genomic DNA.
2.2. Genome Sequencing of A. oryzae XJ1
2.2.1. Extraction of Genomic DNA of A. oryzae XJ1
Genomic DNA of A. oryzae XJ1 was extracted using the sodium dodesyl sulfate method [15]. The purified DNA quality was checked by agarose gel electrophoresis and quantified with a Qubit® 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, USA).
2.2.2. Library Construction of A. oryzae XJ1
1) PacBio Sequel platform
Libraries for single-molecule real-time (SMRT) sequencing were constructed with an insert size of 20 kb using the SMRTbell™ Template Prep Kit 1.0. Briefly, the procedure comprised fragmentation and concentration of the DNA, damaged DNA was repaired and fragment ends were polished, blunt ligation of the fragment ends was performed, the SMRTbell templates were purified with 0.45× AMPure® PB Beads, size selection was conducted using the BluePippin™ System, and DNA damage was repaired after size selection. Finally, the library quality was assessed with a Qubit 2.0 Fluorometer and the insert fragment size was detected with an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, USA).
2) Illumina NovaSeq platform
A total amount of 1 μg DNA per sample was used as input material for the DNA sample preparations. Sequencing libraries were generated using the NEBNext® Ultra™ DNA Library Prep Kit for Illumina (New England Biolabs, Ipswich, USA) following the manufacturer’s recommendations and index codes were added to attribute sequences to each sample. Briefly, the DNA sample was fragmented by sonication into 350 bp fragments, and the fragments ends were polished, A-tailed, and ligated with the full-length adaptor for Illumina sequencing with further PCR amplification. Finally, the PCR products were purified using the AMPure XP system, and libraries were analyzed for size distribution with an Agilent 2100 Bioanalyzer and quantified using real-time PCR.
2.2.3. Sequencing
The whole genome of A. oryzae XJ1 was sequenced using the PacBio Sequel and Illumina NovaSeq PE150 platforms by the Beijing Novogene Bioinformatics Technology Co., Ltd.
2.2.4. Genome Assembly
2.2.4.1. Preliminary Assembly with SMRT Link
1) To ensure the reliability of the subsequent analysis, the low-quality reads (less than 500 bp) were discarded to obtain the clean data.
2) Using the automatic error correction function accessible on the SMRT Portal, the long reads (more than 6000 bp) were selected as the seed sequence, and the other shorter reads were aligned to the seed sequence with BLASR to enable the accuracy of the seed sequence to be improved further. This process yielded the preliminary assembly.
2.2.4.2. Correction of the Preliminary Assembly
Using the Structural Variant Calling module of the SMRT Link 5.0.1 software suite, the Arrow algorithm was used to correct and count the variant sites in the preliminary assembly.
2.2.5. Genome Component Prediction
Genome component prediction involved the prediction of the coding genes, repetitive sequences, and non-coding RNA. The steps were performed as follows.
1) By default, we used the Augustus 2.7 program to retrieve the relevant coding genes for fungi. If homologous reference gene sequences and transcript sequencing data were available, a complete annotation pipeline, PASA, as implemented at the Broad Institute, involved the following steps: (A) ab initio gene finding using a selection of the following software tools: GeneMarkHMM, FGENESH, Augustus, SNAP, and GlimmerHMM; (B) protein homology detection and intron resolution using the GeneWise software and the uniref90 non-redundant protein database; (C) alignment of known expressed sequence tags, full-length cDNAs, and Trinity RNA-Seq assemblies to the genome; (D) PASA alignment assemblies were based on overlapping transcript alignments from step (C); (E) use of EVidenceModeler (EVM) to compute weighted consensus gene structure annotations based on the preceding steps (A–D); and (F) use of PASA to update the EVM consensus predictions, adding untranslated region annotations and models for alternatively spliced isoforms (leveraging D and E).
2) The interspersed repetitive sequences were predicted using RepeatMasker (http://www.repeatmasker.org/). The tandem repeats were analyzed with the Tandem Repeats Finder program.
3) Transfer RNA (tRNA) genes were predicted with the tRNAscan-SE web server. Ribosomal RNA (rRNA) genes were analyzed using rRNAmmer. Small RNA (sRNA), small nuclear RNA (snRNA), and microRNAs (miRNA) were predicted by performing a BLAST search against the Rfam database.
2.2.6. Prediction of Gene Functions
We used seven databases to predict gene functions, namely, GO (Gene Ontology), KEGG (Kyoto Encyclopedia of Genes and Genomes), KOG (Clusters of Orthologous Groups), NR (Non-Redundant Protein Database), TCDB (Transporter Classification Database), P450, and Swiss-Prot. A whole-genome BLAST search (E-value < 1e−5, minimal alignment length percentage > 40%) was performed against each of the databases. Secretory proteins were predicted with the Signal P database. We analyzed the secondary metabolism gene clusters using antiSMASH. For pathogenic fungi, we added pathogenicity and drug resistance information using the PHI (Pathogen Host Interactions) and DFVF (Database of Fungal Virulence Factors) databases. Carbohydrate-active enzymes were predicted with the CAZy (Carbohydrate-Active enZYmes) Database.
2.3. Comparative Genomic Analysis
2.3.1. Gene Family Analysis
Gene families were constructed with the genes extracted from the genome assemblies for Aspergillus oryzae XJ1, Aspergillus oryzae RIB40 (GCA_000184455.3), Aspergillus fumigatus Af293 (GCA_000002655.1), Aspergillus niger CBS 513.88 (GCA_000002855.2), Aspergillus terreus NIH2624 (GCA_000149615.1), Aspergillus luchuensis IFO 4308 (GCA_016861625.1), Aspergillus flavus NRRL3357 (GCA_014117465.1), Aspergillus nidulans FGSC A4 (GCA_000149205.2), Aspergillus parasiticus ASM95608v1 (GCA_000956085.1), and Saccharomyces cerevisiae S288C (GCA_000146045.2). BLAST was used for pairwise alignment of all genes and redundancy was eliminated using SOLAR. Gene-family clustering was based on the alignment results obtained with the hcluster_sg software.
2.3.2. Phylogenomic tree Reconstruction Based on Genome Alignment
Alignment of the A. oryzae XJ1 genome and the aforementioned nine reference genomes was performed using the MUMmer and LASTZ tools. A phylogenomic tree was constructed using the maximum likelihood algorithm with TreeBeST. The reliability of the tree topology was assessed by performing a bootstrap analysis was 1000 replicates.
2.3.3. Core/Pan-Genome Analysis
Core genes and strain-specific genes between the A. oryzae XJ1 genome and nine reference genomes [Aspergillus oryzae RIB40 (GCA_000184455.3), Aspergillus oryzae KDG21 (GCA_018140735.1), Aspergillus fumigatus Af293 (GCA_000002655.1), Aspergillus niger CBS 513.88 (GCA_000002855.2), Aspergillus terreus NIH2624 (GCA_000149615.1), Aspergillus luchuensis IFO 4308 (GCA_016861625.1), Aspergillus flavus NRRL3357 (GCA_014117465.1), Aspergillus nidulans FGSC A4 (GCA_000149205.2), and Aspergillus parasiticus NRRL 2999 (GCA_012897115.1)] were analyzed using the CD-HIT software for rapid clustering of similar proteins with a threshold of 50% pairwise identity and 0.7 length difference cutoff. A Venn diagram was generated to visualize the proportions of core and unique genes among the strains.
2.3.4. Genomic Synteny Analysis
Genomic synteny between A. oryzae XJ1 and A. oryzae RIB40, and between A. oryzae XJ1 and A. flavus NRRL3357 was analyzed based on the aforementioned genome alignment.
3. Results
3.1. General Information on the A. oryzae XJ1 Genome
The complete genome assembly for A. oryzae XJ1 comprised 37.874054 Mbp and encoded 11,720 predicted genes with an average length of 1557 bp (Table 1). The total length of genes was 18.250593 Mbp, comprising 48.19% of the total genome length, whereas the gene internal length was 19.623461 Mbp. The GC content of the genes was 52.09%, compared with 42.88% for the gene internal GC content (Table 1). The number of genes of length ≥ 2500 bp was 1579, much more than that of others, which comprised 13.47% of the total number of genes (Figure 1).
A total of 10 scaffolds were identified and assigned to chromosomes 1 to 8 (Figure 2). The length of each chromosome was predicted as follows: C1, 6,525,071 bp; C2, 6,309,536 bp; C3, 5,185,625 bp; C4, 4,749,098 bp; C5, 4,562,565 bp; C6, 4,149,163 bp; C7, 3,211,970 bp; C8, 3,161,472 bp; C9, 14,541 bp; and C10, 5013 bp. Because C9 and C10 were so short, they are not included in the genome structure in Figure 2.
3.2. Analysis of Predicted Gene Functions in A. oryzae XJ1
The predicted genes in the A. oryzae XJ1 genome were annotated with pathway information from the KEGG database. Of the annotated genes, 596 genes were predicted to be associated with unaffected pathogenicity, 660 genes with reduced virulence, 131 genes with loss of pathogenicity, 67 genes with lethality, 36 genes with increased virulence, 6 genes were effectors, and 5 genes were associated with resistance to chemicals. The functions of 123 genes were not predicted (Figure 3A).
Signaling pathways and toxins may be important in interactions between the host and the pathogen. In the A. oryzae XJ1 genome, 1206 genes encoded signaling proteins, 54 genes were receptors, 971 genes encoded secreted proteins, and 2536 genes encoded TMHMM proteins (Figure 3B). Toxin and virulence factors, as lethality-related factors, may play important roles in the death of the host. Regarding virulence-related factors in the A. oryzae XJ1 genome, 114 genes encoded toxin-related proteins and 531 genes encoded virulence factors.
3.3. Evolutionary Analysis of the A. oryzae XJ1 Genome
The total number of genes in the A. oryzae XJ1 genome was less than that of A. flavus NRRL3357 and A. oryzae RIB40 (Figure 4A). The numbers of single-copy orthologs, multiple-copy orthologs, and non-clustered genes of A. oryzae XJ1 were similar to those of A. flavus NRRL3357. The numbers of single-copy orthologous genes were extremely similar among the 10 fungal strains analyzed, whereas for other gene clusters, such as multiple-copy orthologs, unique paralogs, other orthologs, and non-clustered genes, the number of genes varied among these species.
Comparison of the proportions of the different groups of orthologous genes revealed that the pattern in A. oryzae XJ1 was very similar to that of A. flavus NRRL3357, A. niger CBS 513.88, and A. parasiticus ASM95608v1, and less so to that of A. oryzae RIB40, A. fumigatus Af293, and A. terreus NIH2624 (Figure 4B).
To analyze phylogenomic relationships, a phylogenetic tree was constructed from an alignment of the genomes for the 10 strains. The number of substitutions/site for A. flavus NRRL3357 (0.000746995), A. oryzae XJ1 (0.00116078), A. oryzae RIB40 (0.00223712), A. parasiticus ASM95608v1 (0.00904747), A. terreus NIH2624 (0.0871863), A. luchuensis IFO 4308 (0.013228), A. niger CBS 513.88 (0.0130487), A. fumigatus Af293 (0.0974948), A. nidulans FGSC.A4 (0.109048), and S. cerevisiae S288C (0.301509) indicated that A. oryzae XJ1 was most closely related to A. flavus NRRL3357 and slightly more distantly related to A. oryzae RIB40 (Figure 5).
The core/pan-genome analysis revealed that the A. oryzae XJ1 genome included 704 unique genes, whereas A. flavus NRRL 3357, A. oryzae KDG21, and A. parasiticus NRRL 2999 had 646, 955, and 779 strain-specific genes, respectively. The genomes of the remaining six fungal species had more than 1000 strain-specific genes and 1595 common genes (Figure 6).
The A. oryzae XJ1 genome showed structural differences compared with the genomes of A. oryzae RIB40 and A. flavus NRRL3357 (Figure 7). The A. oryzae XJ1 genome contained three contigs with an almost colinear forward chain (brown color and pink color) and one contig with translocation genes (darker green color) compared with both A. flavus NRRL3357 and A. oryzae RIB40. A greater number of reverse chains were present in A. oryzae XJ1 compared with A. oryzae RIB40 than in comparison with A. flavus NRRL3357 (blue color). A greater number of inversion genes (saffron color) and translocation+inversion genes (green color) were detected in A. oryzae XJ1 compared with A. oryzae RIB40 than in comparison with A. flavus NRRL3357.
4. Discussion
A previous study indicated that A. oryzae XJ1 was most similar to strains of A. oryzae in morphology [3]. In the present study, comparison of the A. oryzae XJ1 genome to those of nine Aspergillus strains indicated that A. oryzae XJ1 was genetically distinct at the genome level from other Aspergillus species, including two A. oryzae strains and A. flavus. Although the genome size of A. oryzae XJ1 was similar to those of A. oryzae RIB40 and A. flavus NRRL 3357, at approximately 37 Mb, the numbers of putative genes differed, i.e., 11,720, 12,074, and 13,485, respectively [16,17]. The proportions of the different groups of orthologous genes in A. oryzae XJ1 were very similar to the patterns observed for A. flavus NRRL3357 and A. oryzae RIB40. However, the core/pan-genome analysis, phylogenomic reconstruction, and genomic synteny analysis indicated that A. oryzae XJ1 was genetically distinct from A. flavus and A. oryzae.
Aspergillus flavus and A. oryzae are known to be very similar. Numerous studies have sought to distinguish them, including on the basis of morphological characteristics [18], amplified fragment length polymorphisms [19], restriction fragment length polymorphisms [20], sequences of the nuclear ribosomal internal transcribed spacer (ITS) region [21], assessment of aflatoxin gene clusters [22], analysis of the cyp51A gene [23], and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry analysis [24], in addition to a study integrating selective culture methods, microscopic observations, secondary metabolite profiles, and ITS sequences [25]. However, these approaches have yet to provide an unambiguous means of differentiation between A. flavus and A. oryzae. Recent research suggests that A. flavus and A. oryzae may be conspecific based on analysis of phylogenetic, genomic, and metabolic homogeneity [26]. Aspergillus oryzae has been suggested to be a domesticated ecotype of A. flavus that is unable to produce the most potent natural carcinogen, aflatoxin, and any other carcinogenic metabolites [27,28]. Aspergillus oryzae is classified as Generally Recognized as Safe (GRAS) by the Food and Drug Administration in the USA, which is also supported by the World Health Organization [29,30,31]. It has occasionally been reported that A. oryzae shows pathogenicity to animals. However, surprisingly, our previous bioassay experiments showed that A. oryzae XJ1 is highly virulent to nymphs and adults of Locusta migratoria and other species of locusts [3,4]. Given the present disagreement in the classification of A. flavus and A. oryzae based on morphological, genomic, metabolic, and functional characteristics, the host specificity of virulence (representing a unique environmental niche) should be a crucial consideration in the recognition of species in the genus. Aspergillus oryzae XJ1 has the specific ability to kill locusts. Therefore, we propose that recognition of A. oryzae XJ1 as a new species distinct from A. flavus and A. oryzae is warranted, with specific virulence to acridids similar to Metarhizium acridum [32,33].
Author Contributions
L.Z. and Y.Y. conceived and designed research. Y.Y., X.X., H.L. and L.Z. conducted the experiments; Y.Y. and L.Z. wrote the manuscript. All authors read and approved the manuscript.
Funding
This work was supported by Shandong Provincial Natural Science Foundation (ZR2022MC117) and Agricultural scientific and technological innovation project of Shandong Academy of Agricultural Sciences (CXGC2024F05, CXGC2024D05).
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Acknowledgments
This work was supported by Shandong Provincial Natural Science Foundation (ZR2022MC117) and Agricultural scientific and technological innovation project of Shandong Academy of Agricultural Sciences (CXGC2024F05, CXGC2024D05). We thank Robert McKenzie, PhD, from Liwen Bianji (Edanz) (www.liwenbianji.cn) for editing a draft of this manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Lomer, C.J., Bateman, R.P., Johnson, D.L., Langewald, J., Thomas, M. Biological control of locusts and grasshoppers. Annu. Rev. Entomol. 2001, 46, 667–702. [CrossRef]
- de Faria, M.R., Wraight, S.P. Mycoinsecticides and Mycoacaricides: a comprehensive list with worldwide coverage and international classification of formulation types. Biol. Control. 2007, 43, 237–256. [CrossRef]
- Zhang, P., You, Y., Song, Y., Wang, Y., Zhang, L. First record of Aspergillus oryzae (Eurotiales: Trichocomaceae) as an entomopathogenic fungus of the locust, Locusta migratoria (Orthoptera: Acrididae). Biocontrol Sci. Techn. 2015, 25: 1285–1298. [CrossRef]
- You, Y., An, Z., Zhang, X., Liu, H., Yang, W., Yang, M., Wang, T., Xie, X., Zhang, L. Virulence of the fungal pathogen, Aspergillus oryzae XJ-1 to adult locusts (Orthoptera: Acrididae) in both laboratory and field trials. Pest Manag. Sci. 2023, 79, 3767–3772. [CrossRef]
- Zhong, Y., Lu, X., Xing, L., Ho, S.W.A., Kwan, H.S. Genomic and transcriptomic comparison of Aspergillus oryzae strains: a case study in soy sauce koji fermentation. J. Ind. Microbiol. Biotechnol. 2018, 45, 839–853. [CrossRef]
- Venkatesh, M.V., Joshi, K.R., Harjai, S.C., Ramdeo, I.N. Aspergillosis in desert locust (Schistocerka gregaria Forsk). Mycopathologia 1975, 57, 135–138. [CrossRef]
- Kumar, S., Slutana, R., Wagan, M.S. Pathogenic application of Aspergillus species for the control of agricultural important grasshoppers. J. Biodivers. Environ. Sci. 2013, 3, 223–229.
- Sultana, R., Wagan, Y.S., Naeem, M., Wagan, M.S., Khatri, I. Susceptibility of three Hieroglyphus species (Hemiacridinae: Acrididae: Orthoptera) to some strains of the entomopathogenic fungi from Pakistan. Can. J. Pure & Appl. Sci. 2013, 7, 2325–2332.
- He, B., Tu, Y., Jiang, C., Zhang, Z., Li, Y., Zeng, B. Functional genomics of Aspergillus oryzae: strategies and progress. Microorganisms 2019, 7, 103. [CrossRef]
- Watarai, N., Yamamoto, N., Sawada, K., Yamada, T., Watarai, N., Yamamoto, N., Sawada, K., Yamada, T. Evolution of Aspergillus oryzae before and after domestication inferred by large-scale comparative genomic analysis. DNA Res. 2019, 26, 465–472. [CrossRef]
- Jeon, J., Kim, J.A., Park, S.Y., Kim, G.W., Park, C.S., Kim, C., Park, H.Y., Yeo, J.H., Lee, Y.H., Kim, S. Draft genome sequence of Aspergillus oryzae BP2-1, isolated from traditional malted rice in South Korea. Microbiol. Resour. Announc. 2020, 9, e01405–19. [CrossRef]
- Chacón-Vargas, K., McCarthy, C.O., Choi, D., Wang, L., Yu, J.H., Gibbons, J.G. Comparison of two Aspergillus oryzae genomes from different clades reveals independent evolution of alpha-amylase duplication, variation in secondary metabolism genes, and differences in primary metabolism. Front. Microbiol. 2021, 12: 691296. [CrossRef]
- Umemura, M., Koike, H., Yamane, N., Koyama, Y., Satou, Y., Kikuzato, I., Teruya, M., Tsukahara, M., Imada, Y., Wachi, Y., Miwa, Y., Yano, S., Tamano, K., Kawarabayasi, Y., Fujimori, K.E., Machida, M., Hirano, T. Comparative genome analysis between Aspergillus oryzae strains reveals close relationship between sites of mutation localization and regions of highly divergent genes among Aspergillus species. DNA Res. 2012, 19, 375–382. [CrossRef]
- Kjærbølling, I., Vesth, T., Frisvad, J.C., Nybo, J.L., Theobald, S., Kildgaard, S., Petersen, T.I., Kuo, A., Sato, A., Lyhne, E.K., Kogle, M.E., Wiebenga, A., Kun, R.S., Lubbers, R.J.M., Mäkelä, M.R., Barry, K., Chovatia, M., Clum, A., Daum, C., Haridas, S., He, G., LaButti, K., Lipzen, A., Mondo, S., Pangilinan, J., Riley, R., Salamov, A., Simmons, B.A., Magnuson, J.K., Henrissat, B., Mortensen, U.H., Larsen, T.O., de Vries, R.P., Grigoriev, I.V., Machida, M., Baker, S.E., Andersen, M.R. A comparative genomics study of 23 Aspergillus species from section Flavi. Nat. Commun. 2020, 11, 1106. [CrossRef]
- Lim, H.J, Lee, E.-H, Yoon Y, Chua B., Son, A. Portable lysis apparatus for rapid single-step DNA extraction of Bacillus subtilis. J. Appl. Microbiol. 2016, 120, 379–387. [CrossRef]
- Machida, M., Asai, K., Sano, M., Tanaka, T., Kumagai, T., Terai, G., Kusumoto, K., Arima, T., Akita, O., Kashiwagi, Y., Abe, K., Gomi, K., Horiuchi, H., Kitamoto, K., Kobayashi, T., Takeuchi, M., Denning, D.W., Galagan, J.E., Nierman, W.C., Yu, J., Archer, D.B., Bennett, J.W., Bhatnagar, D., Cleveland, T.E., Fedorova, N.D., Gotoh, O., Horikawa, H., Hosoyama, A., Ichinomiya, M., Igarashi, R., Iwashita, K., Juvvadi, P.R., Kato, M., Kato, Y., Kin, T., Kokubun, A., Maeda, H., Maeyama, N., Maruyama, J., Nagasaki, H., Nakajima, T., Oda, K., Okada, K., Paulsen, I., Sakamoto, K., Sawano, T., Takahashi, M., Takase, K., Terabayashi, Y., Wortman, J.R., Yamada, O., Yamagata, Y., Anazawa, H., Hata, Y., Koide, Y., Komori, T., Koyama, Y., Minetoki, T., Suharnan, S., Tanaka, A., Isono, K., Kuhara, S., Ogasawara, N., Kikuchi, H. Genome sequencing and analysis of Aspergillus oryzae. Nature 2005, 438, 1157–1161. [CrossRef]
- Nierman, W.C., Yu, J., Fedorova-Abrams, N.D., Losada, L., Cleveland, T.E., Bhatnagar, D., Bennett, J.W., Dean, R., Payne, G.A. Genome sequence of Aspergillus flavus NRRL 3357, a strain that causes aflatoxin contamination of food and feed. Genome Announc. 2015, 3, e00168–15. [CrossRef]
- Jørgensen, T.R. Identification and toxigenic potential of the industrially important fungi, Aspergillus oryzae and Aspergillus sojae. J. Food Prot. 2007, 70, 2916–2934. [Google Scholar] [CrossRef] [PubMed]
- Montiel, D., Dickinson, M.J., Lee, H.A., Dyer, P.S., Jeenes, D.J., Roberts, I.N., James, S., Fuller, L.J., Matsuchima, K., Archer, D.B. Genetic differentiation of the Aspergillus section Flavi complex using AFLP fingerprints. Mycol. Res. 2003, 107(Pt 12), 1427–1434. [CrossRef]
- Abastabar, M., Shabanzadeh, S., Valadan, R., Mayahi, S., Haghani, I., Khojasteh, S., Nargesi, S., Seyedmousavi, S., Hedayati, M.T. Development of RFLP method for rapid differentiation of Aspergillus flavus and Aspergillus oryzae, two species with high importance in clinical and food microbiology. J. Mycol. Med. 2022, 32, 101274. [CrossRef]
- Kumeda, Y., Asao, T. Heteroduplex panel analysis, a novel method for genetic identification of Aspergillus Section Flavi strains. Appl. Environ. Microbiol. 2001, 67, 4084–4090. [CrossRef]
- Chang, P.K., Ehrlich, K.C., Hua, S.S. Cladal relatedness among Aspergillus oryzae isolates and Aspergillus flavus S and L morphotype isolates. Int. J. Food Microbiol. 2006, 108, 172–177. [CrossRef]
- Nargesi, S., Abastabar, M., Valadan, R., Mayahi, S., Youn, J.H., Hedayati, M.T., Seyedmousavi, S. Differentiation of Aspergillus flavus from Aspergillus oryzae targeting the cyp51A gene. Pathogens 2021, 10, 1279. [CrossRef]
- Hedayati, M.T., Taghizadeh-Armaki, M., Zarrinfar, H., Hoseinnejad, A., Ansari, S., Abastabar, M., Er, H., Özhak, B., Öğünç, D., Ilkit, M., Seyedmousavi, S. Discrimination of Aspergillus flavus from Aspergillus oryzae by matrix-assisted laser desorption/ionisation time-of-flight (MALDI-TOF) mass spectrometry. Mycoses 2019, 62, 1182–1188. [CrossRef]
- Suleiman, W.B. A multi-aspect analysis of two analogous aspergillus spp. belonging to section Flavi: aspergillus flavus and aspergillus oryzae. BMC Microbiol. 2023, 23, 71. [CrossRef]
- Han, D.M., Baek, J.H., Choi, D.G., Jeon, M.S., Eyun, S.I., Jeon, C.O. Comparative pangenome analysis of Aspergillus flavus and Aspergillus oryzae reveals their phylogenetic, genomic, and metabolic homogeneity. Food Microbiol. 2024, 119, 104435. [CrossRef]
- Barbesgaard, P., Heldt-Hansen, H.P., Diderichsen, B. On the safety of Aspergillus oryzae: a review. Appl. Microbiol. Biotechnol. 1992, 36, 569–572. [CrossRef]
- Geiser, D.M., Pitt, J.I., Taylor, J.W. Cryptic speciation and recombination in the aflatoxin–producing fungus Aspergillus flavus. Proc. Natl. Acad. Sci. USA 1998, 95, 388–393. [CrossRef]
- Tailor, M.J., Richardson, T. Applications of microbial enzymes in food systems and in biotechnology. Adv. Appl. Microbiol. 1979, 25, 7–35. [CrossRef]
- FAO_WHO. Committee on Food Additives 31. World Health Organization Technical Report Series: Geneva, 1987.
- Abe, K., Gomi, K., Hasegawa, F., Machida, M. Impact of Aspergillus oryzae genomics on industrial production of metabolites. Mycopathologia 2006, 162, 143–153. [CrossRef] [PubMed]
- Driver, F., Milner, R.J., Trueman, J.W..H. A taxonomic revision of Metarhizium based on a phylogenetic analysis of rDNA sequence data. Mycol. Res. 2000, 104, 134–150. [CrossRef]
- Bischoff, J.F., Rehner, S.A., Humber, R.A. A multilocus phylogeny of the Metarhizium anisopliae lineage. Mycologia 2009, 101, 512–530. [CrossRef] [PubMed]
Figure 1.
Distribution of gene length of A. oryzae XJ1.
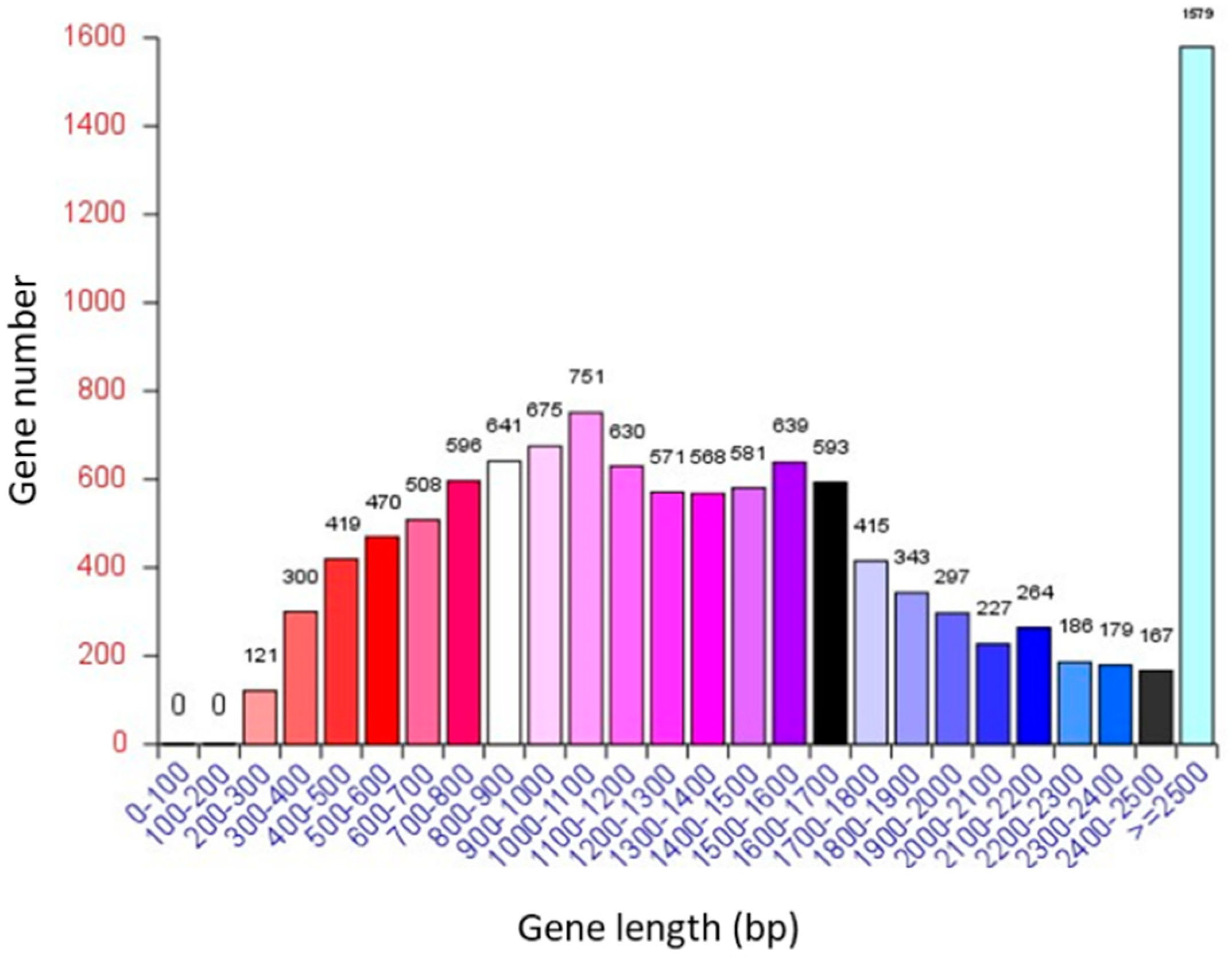
Figure 2.
Circular representation of the complete genome of Aspergillus oryzae XJ1. DNA sequencing revealed that the genome of A. oryzae XJ1 comprised eight chromosomes. The outermost circle shows the chromosome size (kb) for each chromosome (C1–C8). The second circle from the outside shows the GC content for each chromosome; inward blue boxes indicate a lower GC content than the average, purple boxes indicate a higher GC content than the average; the higher the peak, the larger the difference from the mean. The third circle from the outside represents GC skew values (G−C/G+C); inward green boxes indicate a mean G content lower than C, pink boxes indicate a mean G content higher than C. The fourth circle from the outside indicates the distribution of coding genes. The fifth circle shows the distribution of pathogenicity genes. The sixth circle indicates the genes associated with metabolism. The innermost circle shows chromosome duplication.
Figure 2.
Circular representation of the complete genome of Aspergillus oryzae XJ1. DNA sequencing revealed that the genome of A. oryzae XJ1 comprised eight chromosomes. The outermost circle shows the chromosome size (kb) for each chromosome (C1–C8). The second circle from the outside shows the GC content for each chromosome; inward blue boxes indicate a lower GC content than the average, purple boxes indicate a higher GC content than the average; the higher the peak, the larger the difference from the mean. The third circle from the outside represents GC skew values (G−C/G+C); inward green boxes indicate a mean G content lower than C, pink boxes indicate a mean G content higher than C. The fourth circle from the outside indicates the distribution of coding genes. The fifth circle shows the distribution of pathogenicity genes. The sixth circle indicates the genes associated with metabolism. The innermost circle shows chromosome duplication.
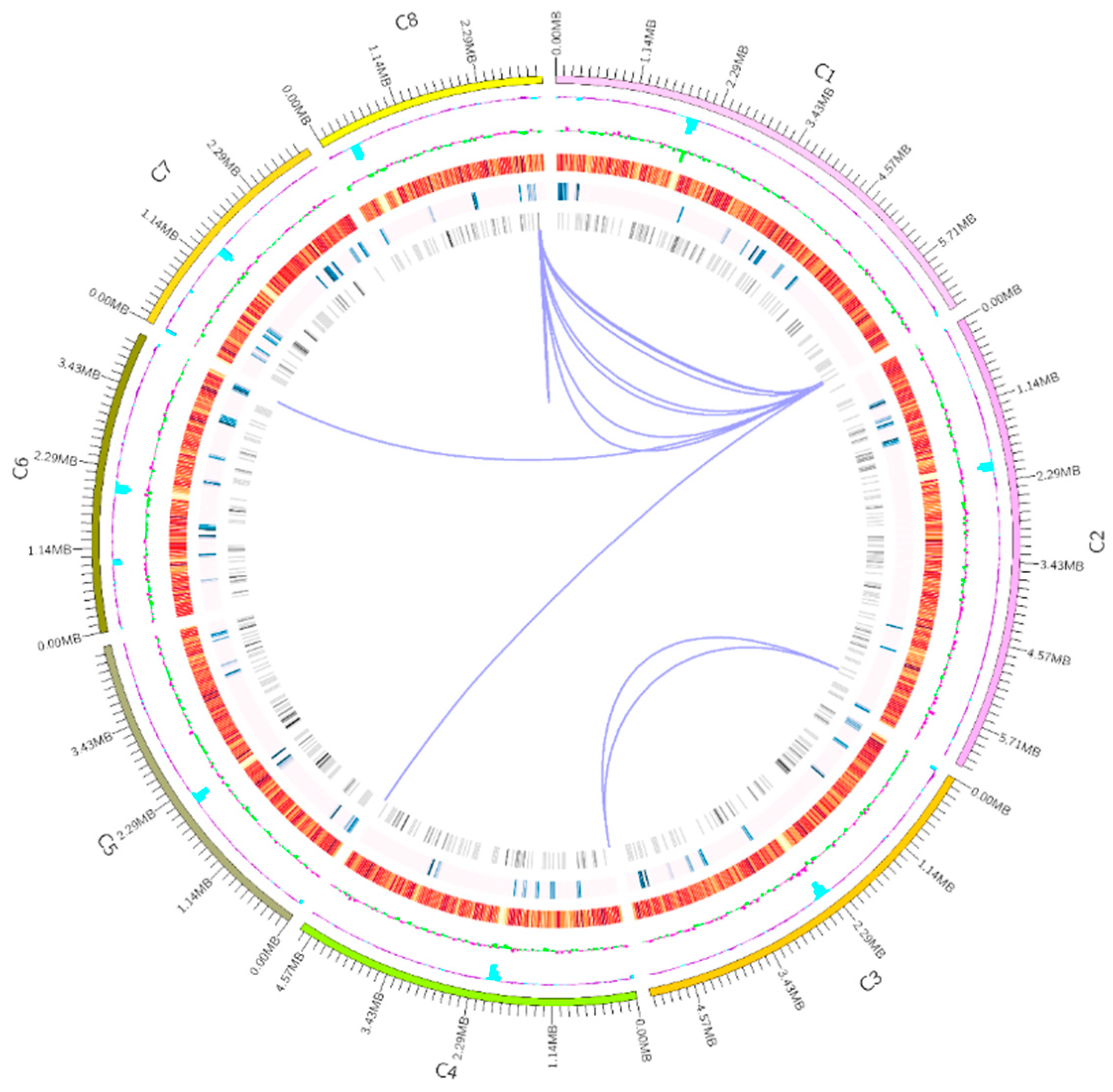
Figure 3.
Genes involved in host–pathogen interactions in signaling pathways (A) and virulence factors (B) in the Aspergillus oryzae XJ1 genome.
Figure 3.
Genes involved in host–pathogen interactions in signaling pathways (A) and virulence factors (B) in the Aspergillus oryzae XJ1 genome.
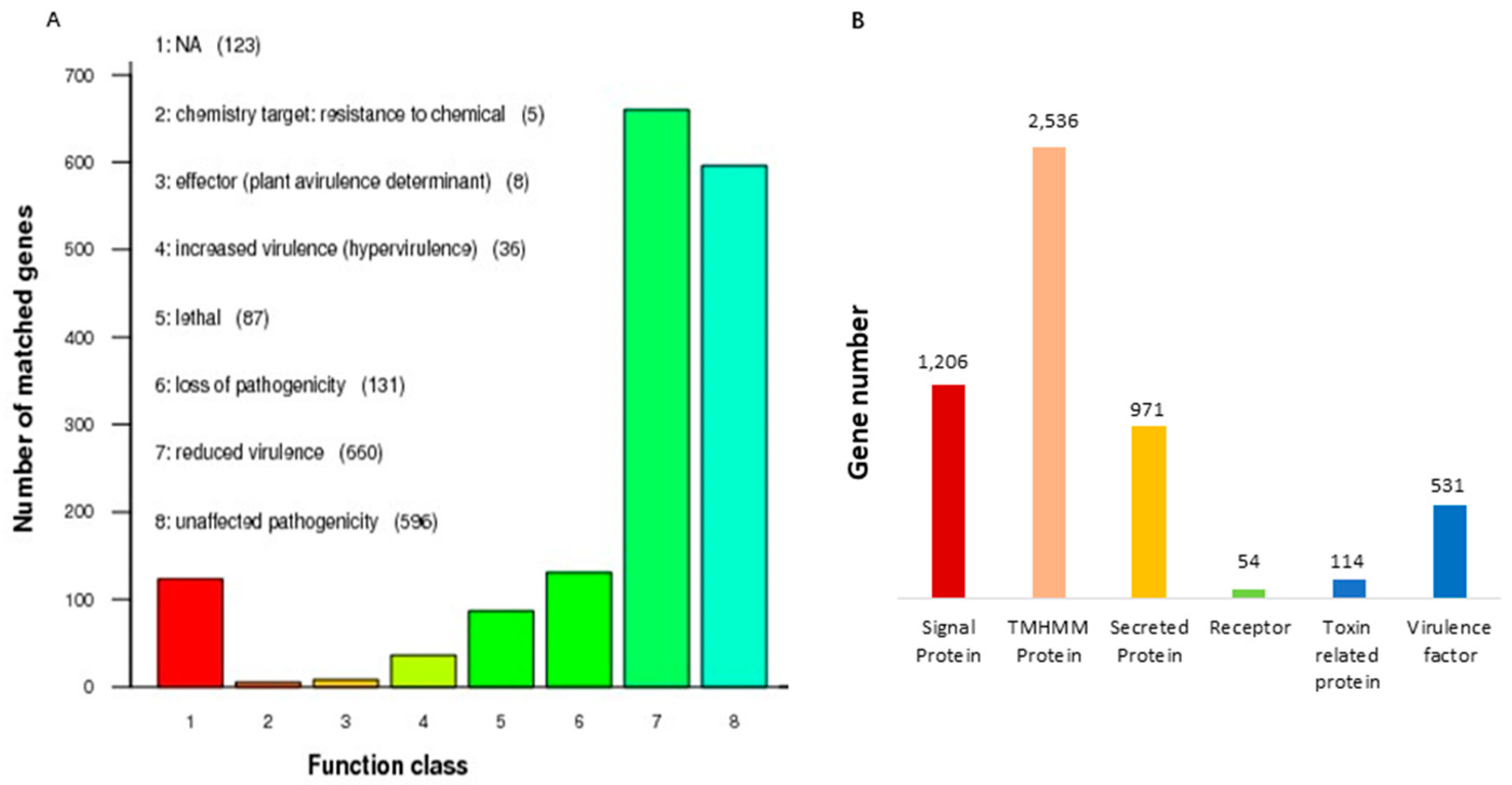
Figure 4.
Number (A) and proportion (B) of orthologous genes in nine Aspergillus strains compared with Saccharomyces cerevisiae.
Figure 4.
Number (A) and proportion (B) of orthologous genes in nine Aspergillus strains compared with Saccharomyces cerevisiae.
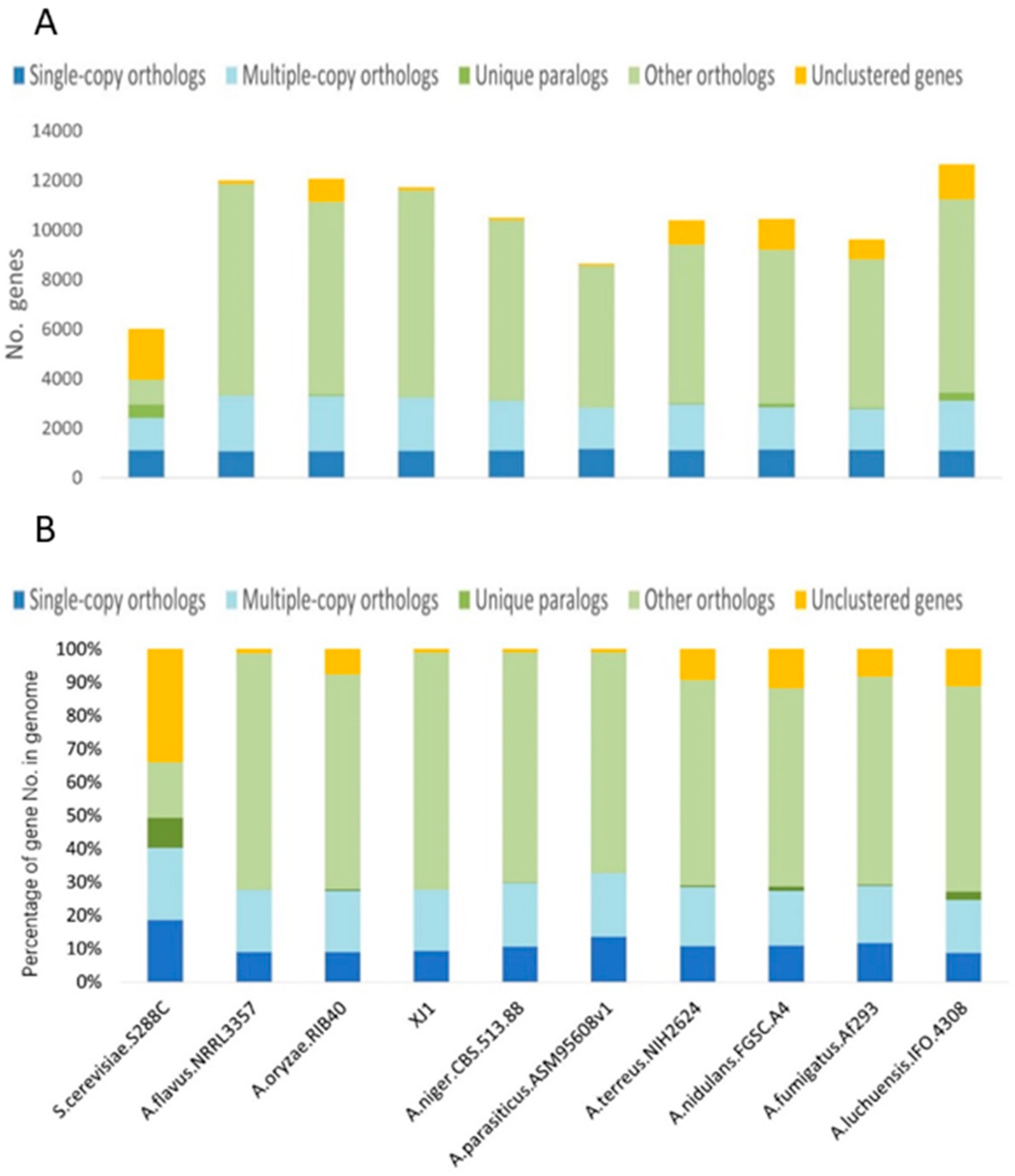
Figure 5.
Phylogenomic relationships of Aspergillus oryzae XJ1 within the Aspergillus genus.
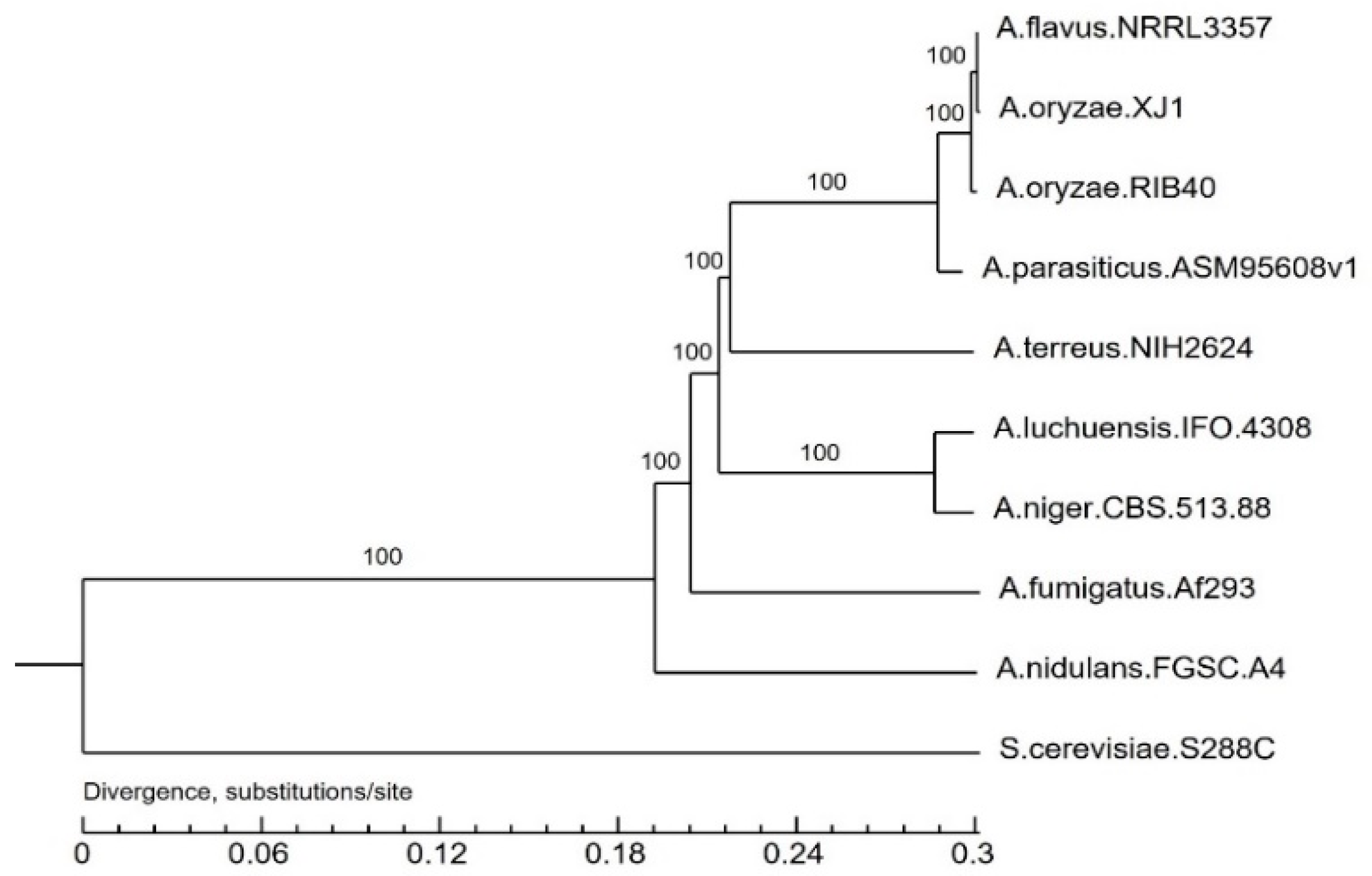
Figure 6.
Core/pan-genome analysis of genes in the genomes of eight species of Aspergillus.
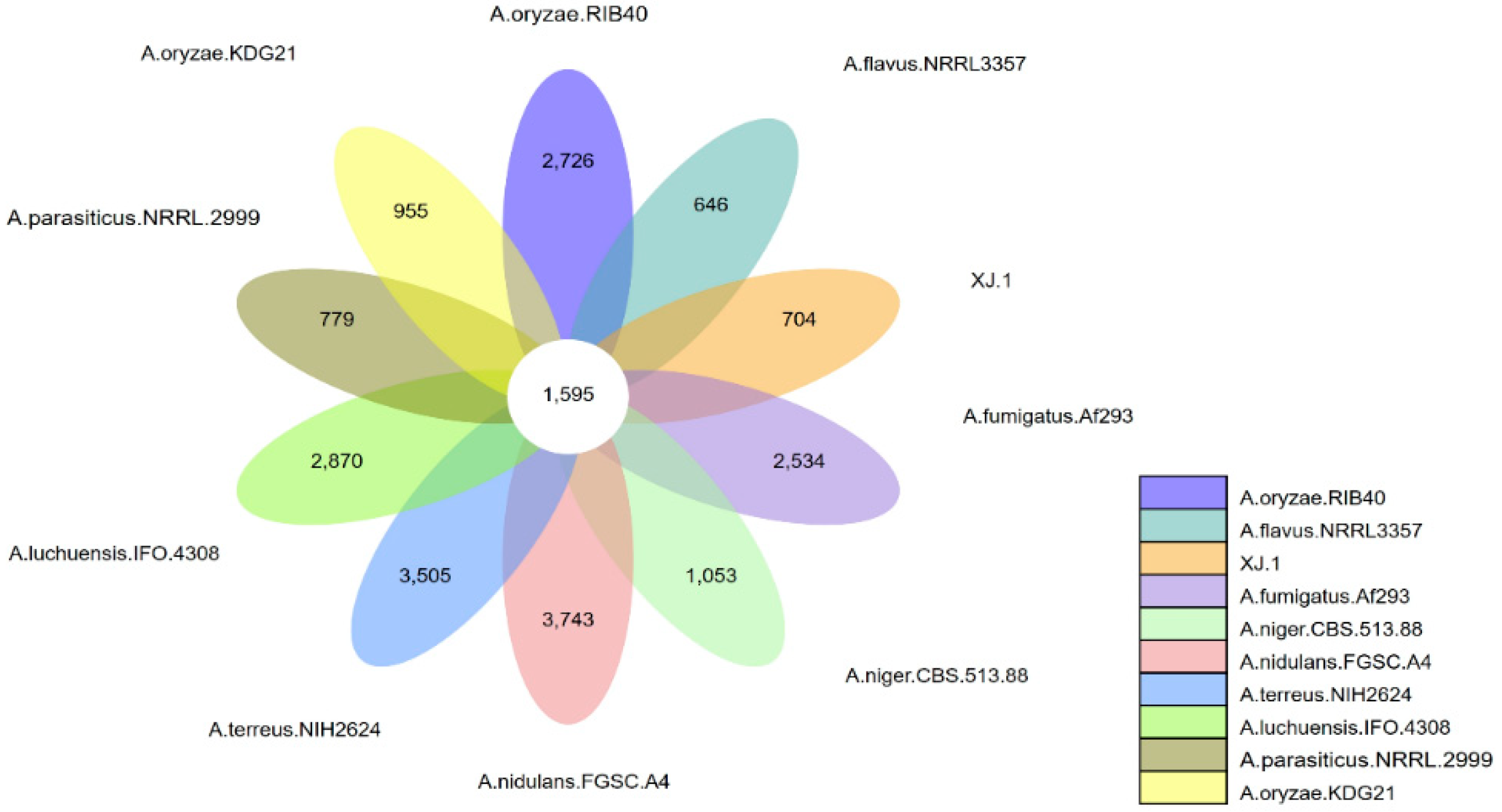
Figure 7.
Analysis of core genes in the genomes of Aspergillus oryzae XJ1, A. flavus NRRL3357, and A. oryzae RIB40. The shading color in boxes indicates the degree of similarity between the genomes; the deeper the color, the higher the similarity.
Figure 7.
Analysis of core genes in the genomes of Aspergillus oryzae XJ1, A. flavus NRRL3357, and A. oryzae RIB40. The shading color in boxes indicates the degree of similarity between the genomes; the deeper the color, the higher the similarity.
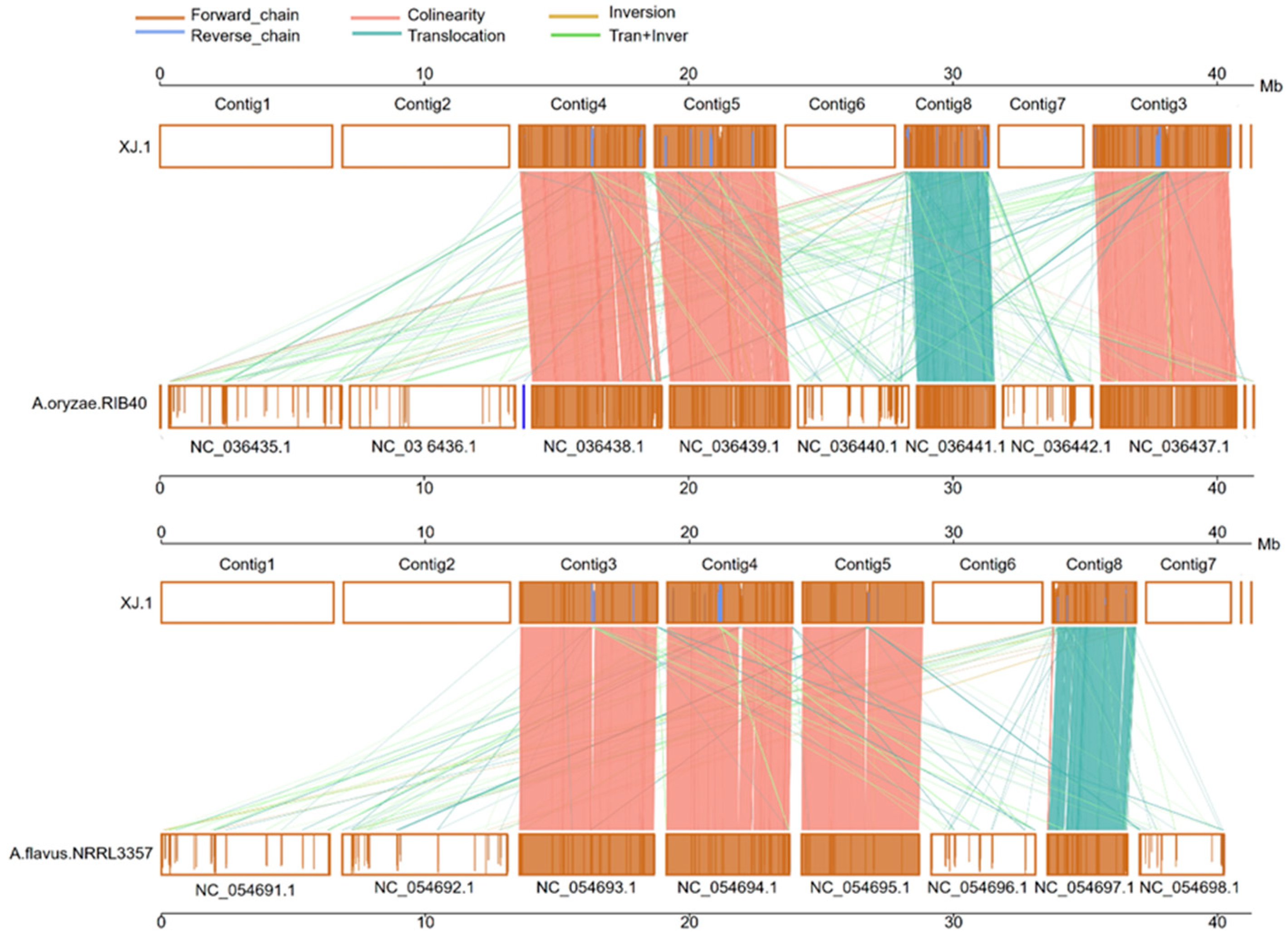

Table 1.
General information of A. oryzae XJ1 genome.
| Genome information summary | |
|---|---|
| Genome size (bp) | 37,874,054 |
| GC content of genome (%) | 47.31 |
| Gene number | 11,720 |
| Gene total length (bp) | 18,250,593 |
| GC content of gene (%) | 52.09 |
| Gene length/genome (%) | 48.19 |
| Gene average length (bp) | 1,557 |
| Gene internal length (bp) | 19,623,461 |
| Gene internal GC content (%) | 42.88 |
| Gene internal length/genome (%) | 51.81 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



