
Submitted:
29 October 2024
Posted:
31 October 2024
You are already at the latest version
Abstract
This study examines the chemical recycling of polymethylmethacrylate (PMMA) dental waste via pyrolysis in a semi-batch fixed-bed reactor, aiming to convert it into the valuable monomer methyl methacrylate (MMA). Experiments were conducted in a 2L reactor at temperatures of 425 °C, 450 °C, and 475 °C to understand the effects of heating rate and temperature on product yield and composition. Results show that at 425 °C, MMA was the primary liquid component, with minimal by-products, suggesting that lower temperatures enhance monomer recovery. Higher temperatures, however, increased gas yields and reduced MMA yield due to intensified thermal cracking. The study also highlights that char formation and non-condensable gases increase with reactor scale, indicating that heat transfer limitations can influence MMA purity and yield. These findings emphasize that for effective MMA recovery, lower temperatures and controlled heating rates are optimal, especially in larger reactors where heat transfer issues are more prominent. This research contributes to scaling up PMMA recycling processes, supporting industrial applications to achieve efficient monomer recovery from waste.
Keywords:
PMMA waste
; process design
; thermogravimetry
; reaction mechanism
; semi-batch reactors
; depolymerization
; pyrolysis
; char formation
; kinetics
1. Introduction
Chemical recycling of polymers refers to the transformation of polymeric waste into useful products to human society, being the most prominent alternative for treatment of waste plastic on a large scale [1]. Since the 1950s, world plastic production increased exponentially from 2 million tons to over 450 million tons [2] due to low production costs and technical possibilities displayed by polymers in the range of material properties as chemical and mechanical resistance, ductility, hardness and others. In this context, plastics are a problem as waste due to the same characteristics: their low cost facilitates the inadequate disposal and characteristics as chemical and mechanical resistance reduces its degradability in the environment [3,4].
Only a small percentage of produced plastics are recycled (9% each year) and world production increases rapidly (9% each year) [2], turning the development and application of recycling strategies a must. The main difficulties are related to obtention and separation of plastic waste and the recycling process itself that depends upon the type of waste recycled. The main strategies for treatment of plastic wastes are the following: disposal in landfills; energy recovery (incineration); mechanical recycling; and chemical recycling [5].
Disposal in landfills should be avoided for plastics once they are not biodegradable and incineration is a process demanding large investment and operational costs for production of a resource (energy) readily available through other materials. Incineration of mixed plastic waste produce varied contaminants that must be treated on site, increasing processing costs [6].
The most used process for recycling of plastics is mechanical recycling and it is usually applied to pre-treated wastes (clean, decontaminated, dry) partially or totally separated from other plastics, as in the case of mechanical recycling of polyethylene (PE) and polypropylene (PP). The waste is milled and is thermally molded (pressed or extruded) into new items. A maximum level of contamination is demanded, impeding its perpetual recycling and restricting its use in sanitary, food and health applications [1]. Some technical parameters are also reduced such as mechanical resistance, brightness and opacity, reducing its application for engineering plastics such as acrylic polymers, made from methacrylic acid or polymethylmethacrylate (PMMA) [7].
| PMMA type | Process Scale | T (°C) | Liquid Yield (wt.%) | % MMA | Investigation | Main Results | Reference |
| ModelPMMA (PM=350000 g/mol) | Lab (1.5 g) | 450 | 99.3 | 99.0 | Polymerization of recycled monomer | Contaminants in liquid phase slow down polymerization and it is necessary purification by fractional distillation | [22] |
| Commercial PMMA | Lab (1.5 g) | 450 | 98.1 | 96.8 | |||
| Process description and characterization of recycled polymer including test in technical scale. | Polymerization was possible after fractional distillation. In technical scale, liquid phase yields were lower due to poor heat flow and diffusion of products in reaction kinetics, more evidently shown in larger scale. | [23] | |||||
| Homopolymer PMMA | Lab (30 g) | 400 | 90.0 | 92.8 | |||
| Dental waste | Lab (30g) | 400 | 90.0 | 92.8 | |||
| Dental waste | Technical (2L) | 400 | 66.3 | 76.4 | |||
| Dental waste | Pilot (143 L) | 405 | 48.7 | 98.4 | Process description in pilot scale. | With a larger fixed bed of polymer, it is more evident the effect of poor heat transfer in products yields. | [24] |
| Artificial marble powder (PMMA) | Lab (50g) | 400 | 88.8 | 76.9 | Process investigation of pyrolysis of filled PMMA | Experiments showed liquid phase yields increasing with temperature, but MMA % decreased. The process showed large variation with heating rate demonstrating that semi-batch reactors are heavily dependent upon the residence time of vapors inside the reactor. It is important to note that the reactor presented large dimensions when compared to the weight of feed (i.d.=150 mm and L=600 mm). | [26] |
For complete recycling of polymers, chemical recycling must be used. Polymer molecules are converted into smaller ones through chemical reactions. Varied processes are described in academic literature as pyrolysis, gasification, hydrotreatment, viscosity breaking, steam and catalytic cracking [8]. Ideally, it is achieved full depolymerization of the waste, i.e., the conversion to its monomer and, after purification, can be used for fabrication of completely new polymers.
PMMA is a transparent thermoplastic used as engineering plastic, diverse applications where some characteristics are demanded as mechanical resistance, non-toxicity, chemical resistance, transparency and many others [9,10,11,12]. It is used as acrylic lens, dental cement, prosthetics, automobile parts and many more with an annual production estimated into more than 3.9 million tons [13]. PMMA can be recycled by pyrolysis, i.e., thermal decomposition of the polymer in oxygen-deficient atmosphere. Chemical recycling of PMMA is normally done through fluidized bed pyrolysis, where PMMA particles are fluidized along inert particles (sand) by an inert gas (nitrogen or gases generated by pyrolysis process itself). The heated particles and heated inert gas thermally decompose he polymer, generating over 90 wt.% of liquid phase containing over 90 wt.% of monomer methyl methacrylate (MMA) [7,14,15,16,17,18,19,20,21].
Pyrolysis in fluidized bed is a mature process for solid waste treatment and its conversion to fuels and/or monomers but it demands considerable investment costs with a complicated operation and other reaction types and modes are researched [22,23,24,25,26,27]. Most of those are focused in using semi-batch reactors, where the initial feed is loaded previously inside the reactor but formed vapors are allowed to flow freely out of the reactor. They comprise the simplest form of pyrolysis reactors with low investment and ease of operation. Table 1 comprises the main findings of those works and reveals that PMMA can be adequately depolymerized in semi-batch mode with liquid yields in the range of 50-99 wt.% and MMA concentration higher than 75 wt.%. The biggest problems are related when one tries to scale-up the process because of poor heat conduction in the fixed bed, increasing the decomposition of polymer instead of only depolymerization to its monomer, augmenting gas yields and reducing MMA concentration [23,27].
The semi-batch fixed bed reactor is of simple construction and operation and it is of interest to analyze the problems concerned with its scale-up process. Lab scale reactors show it is possible to obtain over 90 wt.% liquid phase with high MMA concentration (over 90 wt.%). The poor heat transfer associated with larger fixed beds of polymer particles tend to increase rim temperature and reduce liquid phase yields and MMA concentration due to further or side reactions. Also, one impending factor to not consider the use of semi-batch reactors, even in small scale, is the existing dynamics of the process, making it harder to analyze and optimize reactor conditions. This can be explained by the fact that in every given moment, the feed changes its composition with the reaction time and the same can be said of its vapor products. Variables known to affect product composition and yields, such as temperature and heating rate, have to be carefully examined and evaluated considering two factors: 1 – the residence time of vapors; 2 – the reaction time. As far as we know, there is no work available in academic literature where this discussion is present.
In previous works, we described the process dynamics of PMMA depolymerization in a pilot reactor [24] and also the effect of increasing process scale [27], but we did not analyze the effects of reaction time in products yields and composition. In this work, we conducted depolymerization of PMMA waste in a technical scale (2L) fixed bed semi-batch reactor and collected samples of liquid phase according to reaction time (time-fractioned samples) in order to evaluate how the process evolves dynamically. In this manner, it is thought that process variables and reactor design can be optimized for depolymerization of PMMA, minimizing the intrinsic problems related to semi-batch reactors.
2. Materials and Methods
In order to evaluate the effects of reaction time in depolymerization of PMMA waste by pyrolysis in semi-batch reactors, it was applied similar methodology used in previous works, concerning the catalytic pyrolysis of waste fats in semi-batch reactor [28,29]. Summarizing, the PMMA waste is acquired and prepared by milling it to 9 mm mean particle size. The milled PMMA particles are entirely loaded into a 2L semi-batch stainless-steel reactor heated by a 3500 W electrical heater. The reactor is heated to different temperatures (425, 450 and 475 °C) and, as soon as vapor starts to form inside the reactor, a timer is initiated and samples of liquid phase are collected and weighted every 10 minutes until reaction subsides. Samples are designated in a time-fraction manner and are not mixed together, being evaluated for their physical-chemical characteristics (density, viscosity and acid value) and chemical composition by GC-MS. By weighting and analyzing each fraction independently, it is possible to observe and evaluate process evolution in semi-batch pyrolysis processes, which presents considerable variation in feed and vapor composition from start to reaction finish. Below, a detailed presentation of each step follows.
The PMMA waste was supplied by Dentisply Indústria e Comércio (RJ, Brazil) and are composed 94 wt.% of PMMA, 5 wt.% of a crosslinking agent (ethylene glycol dimethacrylate – EGDMA) and 1 wt.% of titanium dioxide.
The pyrolysis unit used was well described in previous works [28,29] and it is composed by a 2L stainless-steel cylindrical reactor with x mm of wall thickness. A 3500 W refractory collar heater is coupled to the reactor and it is insulated by a glass wool jacket. The vapor outlet of the reactor is connected to a stainless-steel tube condenser, refrigerated by a thermostatic bath using water as cooling medium and temperature is maintained at 15 °C. The condenser outlet is coupled to a cylindrical drum with an upper and bottom outlet, used to separate the gas and liquid fractions, respectively. The liquid phase samples are collected through the bottom outlet at every 10 minutes of reaction until no more vapor or liquid condenses. The power used by the electrical heater is determined by a feedback controller located in the control panel of the unit and it is based in temperature measurement from a k-type thermocouple installed in a thermal well located in the pyrolysis reactor. This reactor also has an axial impeller for agitation and mixing, but in the case of solid particles of PMMA, it was not used. A process schematic of the pyrolysis unit is shown in Figure 1.
A typical depolymerization experiment is done by weighting 500-750 g of dental waste and loading in the open reactor. The reactor is then coupled to its cover fixed on the pyrolysis unit framework. Eight sets of nuts and bolts and a graphite gasket are used to close the reactor and guarantee that no vapors leak during the experiment. The nuts and bolts are tightened in pairs diametrically opposed to each other, in order to guarantee the correct fixation of the reactor and no leaks. After that, the cooling and circulation of water is started in the cooling water thermostatic bath and it is started the circulation of water in the water seal of the mechanical impeller of the reactor.
The heating is done in manual mode, where the temperature setpoint is chosen by the operator in the PID controller in order to obtain different heating rates. It was observed empirically that the power applied in the electrical heater is defined by the difference of setpoint to the actual temperature in the reactor and different heating modes are used depending on the error value (Te) between setpoint and actual reactor temperature. In the case of this controller, this value is set to 20 °C, supplying power to the heating element differently depending on this value.
1 – Te < 10 °C → Heating element provides power to increase slowly the temperature and maintain it once reaction starts;
2 – 10 °C < Te < 20 °C → Heating element provides power more frequently, increasing reaction rate;
3 – Te > 20 °C → Heating element is always on;
In order to differentiate between the experiments, it was used different setpoints for each different temperature (425, 450 and 475 °C) to obtain Te of 10, 20 and 30 °C, respectively. For each experiment, the final temperatures were also different. It is possible to set the controller to automatic mode where heating rate is defined previously but due to the weight of the stainless-steel reactor, there is considerable lag between the setpoint and actual temperature of the reactor, generating overshoots and high reaction rate. Also, the automatic mode also does not take in consideration the presence of chemical reactions, especially endothermic ones as in the case of depolymerization, increasing the error difference and generating high reaction rates.
As soon as vapors start to form, a timer is started and the liquid phase is collected every 10 minutes of the reaction in the bottom tap of the separating drum. The time-fractioned samples are weighted and allowed to rest in a separating funnel in order to separate possible aqueous phases. It is then weighted again and conditioned in amber glass bottles.
GC-MS analysis was done for each sample by diluting 1 µL of sample in 1 mL of acetone and injected in gas chromatographer (Agilent Technologies, Model CG-7890B) couple to a quadrupole mass spectrometer (MS-5977A) in a capillary column of fused silica SLBTM-5ms (30 m x 0.25 mm x 0.25 mm). Gathered spectra was compared with NIST database and no internal standard was used, the chemical composition reported in area.% of chromatograms.
3. Results
3.1. Product Yields
Table 1 presents the main findings in respect to product yields obtained by depolymerization considering different temperatures. It can be seen that liquid phase yields tend to decrease with increase in temperature, gas phase yields tend to increase and solid phase tend to decrease until a minimum is reached. Similar findings are reported in academic literature [22,23,24,26,27]. Achilias et al. reported that increasing temperature above 450 °C increased gas yields and lowered liquid yields [22]. Braido et. al. found only a minor increase to gas yields when polymerizing crosslinked PMMA from dental waste when increasing process temperature [23]. The presence of crosslinking in PMMA is said to increase gas formation and unwanted carbon [17] and Braido and collaborators reported that crosslinked PMMA did not melt during pyrolysis while the homopolymer tested did it, but they attributed the small increase in gas yields with process temperature due to the excellent properties of heat transfer of the small-scale reactor. In the larger-scale technical unit, considerable amount of char was produced (~25 wt.%) and reduced liquid yields (~65 wt.%) when compared with laboratory-scale reaction (~90 wt.% liquid yield) [23]. Santos et al. pyrolyzed dental waste in a pilot-scale 143 L fixed-bed reactor and reported that liquid yield shows a first-order decay function with temperature and found increased gas yields (~30 wt.%) when compared to the results obtained by Braido et al. in technical scale (~6 wt.% gas yields). The char yields obtained by Santos et al. were in the range of 11-14 wt.%, lower than Braido et. al, but higher than the ones obtained in this work for a technical-scale unit, where higher temperatures were used for conversion, char yields of ~2-3 wt.% was obtained for temperatures higher than 425 °C [24]. Braido et. al. commented that the depolymerization was not complete in technical-scale after 1 hour of reaction and a higher number of impurities was found in the reactor in technical scale when compared to lab-scale reactor, indicating that higher temperatures favors charring (polymerization to higher molecular weight hydrocarbons) of the crosslinked polymer and even though it is possible to increase liquid yield, it is not from the monomer MMA but of higher molecular weight products, such as aromatic hydrocarbons. This is not observed in lab-scale where heat transfer problems are negligible but become considerable when increasing process scale. The formed char can be further pyrolyzed by increase in process temperature, generating products in the gas phase, as it is observed in this work where higher temperatures of depolymerization (425-475 °C) was investigated when compared to Braido et al. (400 °C) [23]. This effect is further augmented when a larger fixed bed is used as it was showed by Santos et. al, where large gas quantities and lower char yields were obtained in lower temperatures (345-420 °C) [24]. Poudel et al. pyrolyzed artificial marble powder composed of filled PMMA and reported that increased temperature reduces the recovery of MMA from liquid-phase due to the presence of impurities, even though an increase in liquid yield occurs with temperature, pointing to the necessity of homogeneous temperature distribution in the reactor for maximization of the recovery of monomer [26]. For fluidized bed reactors, wher
It is interesting to note that current knowledge of reaction kinetics and mechanism of PMMA depolymerization focus heavily in the type of polymerization and initiation, depropagation and termination steps of depolymerization for production of monomer, but little attention is given to why a char residue may be left in the reactor [30]. It is known in academic literature that PMMA fabricated via a free-radical polymerization mechanism (the most common) decomposes via chain-end initiation and/or mid-chain random scission and depropagate to the monomer until a termination reaction occurs via disproportionation, recombination or chain-transfer to the solvent [30,31,32,33,34]. The average number of depropagation events between an initiation and termination event is called the zip length and it depends on the ratio of initiation and termination to propagation reaction rates constants and can be expressed as the probability p of a depropagation event over a termination event after initiation [30] defined by Equation (2). The zip length (1/ε) can be expressed in terms of this probability as in Equation (3).
For depolymerization reactions where the zip length is larger than the degree of polymerization, the polymer chain reacts fully, leaving no smaller species behind and consequently no char. If the zip length is lower, then a longer polymer residue is left behind. Depending on the dispersity of molecular mass of the reacting polymer and type of termination (disproportionation or recombination), this residue may be collected in solid, liquid or gas phases [30,32]. But this mechanism does not take into account competitive side reactions, such as polymer charring in the case of thermal decomposition of PMMA. The overall reaction represents a complex sequence of reactions occurring during high-temperature pyrolysis of PMMA, generating solid carbon and gases such as aromatic hydrocarbons, CO, CO2 and H2. These reactions occur between pyrolysis products, mainly through aromatization and condensation reactions but also gasification and reduction reactions in high-temperature zones. To some extent, may occur with any carbonaceous feedstock in the reactor [35,36,37,38]. This phenomenon changes the composition of the polymeric chain during semi-batch depolymerization and virtually stops the unzipping and depropagation to the monomer when a certain temperature is reached. This could explain why some polymers tend to stop the depolymerization reaction before even the monomer equilibrium concentration, i.e., the point where propagation and depropagation reactions reach the same reaction rate [30]. The mechanism of thermal decomposition of PMMA, composed of initiation, depropagation and termination reactions is shown in Figure 2, Figure 3 and Figure 4, respectively. The most popular charring reactions are shown in Figure 5. It is important to note that termination of reaction via chain-transfer to the solvent is not possible during thermal depolymerization of PMMA in solid phase and it is not shown in mechanism of Figure 4.
The initiation reactions are shown in increasing bond energy. A head-to-head linkage shown as (a) in Figure 2, is the lower in bond energy and it is the most likely to break during thermal decomposition. It is formed by termination by recombination of the polymer chain and it happens 20% of the time during FRP polymerization [38]. FRP-made polymers degrade in lower temperatures (140-200 °C) but this type of termination is not present in anionic polymerized PMMA, where propagation occurs almost exclusively by H-T propagation and termination only occurs deliberately and no H-H linkages are formed [38]. The mechanism of anionic polymerization is shown in Figure 5. The second most likely bond cleavage is at vinyl chain-ends, formed during termination by disproportionation, also not present in anionic polymerization. This type of initiation (Figure 2b) usually occurs around 300-323 °C, lower than for random H-T fission (327-402 °C) (Figure 2c), where usually most of the decomposition is observed in mass loss of thermogravimetry experiment.
Independently of the type of polymerization used to fabricate the polymer, due to the different energies present in different linkages, it is possible to obtain an approximate proportion of linkage types in determined polymer by thermogravimetry experiments, where a controlled mass of polymer is degraded in different temperatures at an inert atmosphere and it is observed the mass loss (or its derivative) at each respective temperature of decomposition (TGA/DTG). Due to the small mass used (~5 mg), heat transfer and diffusion effects are negligible in thermal decomposition and chemical effects are maximized, i.e., the parameters observed are largely due to linkages and substances present in the sample [39]. Figure 6 shows the weight loss of PMMA dental waste obtained by conducting a TGA/DTG experiment using 10 °C/min of heating rate, nitrogen atmosphere and maximum temperature of 600 °C. The DTG curve shows a peak of maximum decomposition at temperature of 376 °C indicating that most of the thermal degradation happens because of random H-T fission. Indeed, for FRP or anionic polymerization, most of the bonds in high molecular weights polymer chains are H-T linkages. But, from the weight loss curve, one can observe that thermal degradation started as early as 300 °C, pointing to the presence of vinyl chain-ends formed due to disproportionation in FRP, supposedly not present for anionic polymerization. Nevertheless, no decomposition was observed before this temperature, indicating that no H-H linkages were present due to termination by recombination, supposedly happening around 20% of the time with FRP. This effect could be suppressed by the presence of crosslinking agent EGDMA, giving thermal stability to the linear chain of polymer [31].
Da Ros et al. modelled the kinetics of thermal depolymerization of PMMA dental waste containing 5 wt.% EGDMA as crosslinking agent. They compared thermograms with pure PMMA and concluded that EGDMA-crosslinked PMMA degrades mainly through random H-T fission, suppressing early degradation by H-H fission. While pure PMMA showed DTG curves with 3 clearly distinct peaks, for EGDMA-PMMA only two peaks of degradation were observed, a small peak between 250-300 °C (vinyl chain-end fission) and a much higher peak at 376 °C (random H-T fission) and the kinetics of depolymerization could be modelled using a consecutive 2-step reaction, dominated by the random H-T fission mechanism [31]. This correlates well with the necessity of high degree of polymerization, i.e., a high molecular weight polymer, in dental cement to obtain a polymer with desired mechanical properties [40,41,42]. In such a polymer, most of the bonds in the branched polymer chain are H-T bonds formed during monomer propagation. In fact, Da Ros et al. could model the depolymerization kinetics even when considering a one-step reaction due to dominance of random scission in the mechanism [31]. When observing the thermogram from Figure 6 (this work), one can observe that for this dental waste cement, only a clear distinct peak was observed but it was observed thermal degradation of 22 wt.% before a temperature of 330 °C was reached (temperature of vinyl chain-ends fission), indicating that most termination reactions in the FRP process of crosslinked PMMA was due to disproportionation. It seems that the branching of the polymer chain minimizes the formation of H-H linkages formation due to recombination reactions (or at least suppress its early breaking in thermal decomposition) and allows that only 22 % of the polymerization reactions are terminated and 78 % occurs until all polymers is consumed in the polymerization approximating the process of depolymerization of FRP PMMA to anionic polymerized PMMA and conferring thermal stability to the polymer. This makes it possible that the branched chain of PMMA decomposes through the depropagation mechanism (unzipping of the polymer chain) until all the polymer is converted in monomer. Since most of the depolymerization occurs due to random fission, it is speculated that the FRP allied to crosslinking minimizes the formation of chain-end groups. Figure 7 shows how the EGDMA inserts itself and create the increased elasticity and strength PMMA polymer [40,41,42].
Indeed, branching of the polymer chain increases its molecular weight and improve properties such as low solubility in organic solvents and improved strength and elasticity [41]. As it was suggested by thermal analysis and application of PMMA as dental cement, a high degree of polymerization is expected for this polymer, since most of the decomposition is happens due to H-T fission of the polymeric chain, indicating a high polymerization degree. This parameter (equation 4) is the number of monomer units in a polymer or oligomer molecule and it can be represented by the ratio of the number-average molecular weight of the polymer () to the molecular weight of the monomer unit (M0). Dental resins are usually prepared by FRP of prepolymerized PMMA beads with benzoyl peroxide as initiator and liquid MMA as monomer. The prepolymerized beads usually presents high molecular weight (120000-996000) that is further increased after repolymerization [41]. Since molecular weight of MMA is 100.177 g/mol, dental resins usually present degrees of polymerization higher than 120. For saturated chains of PMMA polymer, degradation rate (mass loss) by random scission is described by equations (5) and (6) for zip lengths higher and lower than the degree of polymerization, respectively. Ks is the rate constant for random chain scission (H-T fission) and γ is the reciprocal of the average zip length between initiation and termination reactions [43].
If the zip length is lower than the degree of polymerization, it means that depropagation of polymer into monomer does not happen to completion, i.e., it terminates forming intermediary products of depolymerization. Observation of the thermogram from Figure 6 reveals that virtually all PMMA dental waste is converted to gases (99.5% of weight loss) suggesting that, even if zip length of this depolymerization process is lower than DP, all terminated products are converted to gases before a temperature of 450 °C. If the zip length is lower than DP, the vapors composition should change from an enriched-MMA vapor to the pyrolysis products of the terminated products of the depolymerization reaction at later stages (higher temperatures). Beyond that, there is also the possibility of side-reactions between depolymerization products [33] and charring reactions [35,36,37,38].
When one compares the results obtained in thermal analysis with the yields of products of depolymerization of PMMA dental waste in the technical unit of IME, some char is formed in the larger-scale unit (~2 wt.%), indicating that the overall depolymerization mechanism includes more reactions than what it is initially supposed for PMMA thermal degradation. Indeed, mechanisms of depolymerization are usually kinetically controlled and depends upon heat transfer and diffusion coefficients, being influenced by sample thickness [33]. This means that other reactions could be involved and be determinant to final product distribution and composition. Experimentation with pyrolysis of carbonaceous materials usually point to higher char yields when using low temperature and heating rates [35,36,37,38]. Although not fully known, it is estimated that char is formed due to inter- and intramolecular rearrangements reactions, forming a polycyclic aromatic structure as in Figure 8. Further reactions include hydrogen abstraction events, forming hydrogen gas and graphitic carbon. Due to the lower actual heating rate through the polymeric bed in the technical scale reactor, more char is formed than in the small-scale thermal reactor of TGA analysis.
Figure 9, Figure 10 and Figure 11 shows the temperature profile of reactor and vapor temperature according to reaction time for PMMA dental waste depolymerization at 425, 450 and 475 °C, respectively. In order to effectively analyze the semi-batch depolymerization process, it was also included in the graphs of Figure 9, Figure 10 and Figure 11, the vapor flow formed during the heated depolymerization reaction for each temperature. It can be observed that vapors started to form and condense in considerably lower temperature than what was observed in thermal analysis of Figure 6, where thermal degradation (weight loss) started above 200 °C and for the technical scale unit, vapors started to form and condense in liquid state at 75 °C. We believe it to be an effect of the greater dimensions of the particles bed of polymer inside the technical reactor when compared to the thermobalance of the TGA analysis. Since the PMMA dental waste present poor heat transfer properties, a larger fixed bed of polymer creates a temperature gradient between the rim and center of the reactor, i.e., when the center presents a certain temperature, it is expected that the rims of the fixed bed are at higher temperatures. Since the reactor thermocouple is located at the center of the reactor, it probably showed lower temperatures at reaction beginning, stabilizing at a correct value in higher reaction times. Ribeiro et al. conducted PMMA depolymerization at different production scales and concluded that this effect is augmented for larger semi-batch reactors [27]. By observing Table 1, one can perceive that it also affects the products distribution in different phases. Even though it is possible to obtain high liquid phase yields in lab-scale (30g) PMMA dental waste depolymerization [22,23], only 81 wt.% liquid phase yield could be obtained for temperature of 425 °C in the technical scale unit and for higher temperatures (450 and 475 °C), this yield was much lower, around 62 wt.%.
This behavior seems correlated to the different regimes of heating used for each experiment. Even though temperature profiles of the three experiments were alike, including reaction time, condensate flowrates reduced with temperature increase due to formation of gaseous non-condensable substances, reducing liquid phase yield and increasing gaseous yields, as in Table 1. It can be observed that for endothermic reactions being conducted in semi-batch reactors, almost no temperature increase will occur when an endothermic process is in place, in fact, added energy will be used to increase the rate of the endothermic processes, such as the depolymerization reaction and vaporization of formed products [28,29]. Only after those processes occur to completion, temperature starts to rise again to desired setpoint. In fact, the different temperatures used in the experiments actually represent different heating rates and final temperatures of reaction. In Figure 9, Figure 10 and Figure 11, the setpoint curve represents the temperature chosen by the operator to indirectly set the heating rate used. This is done by choosing setpoints with different temperature errors from actual reactor temperature, of 10, 20 and 30 °C for the experiments of 425, 450 and 475 °C, respectively. This was done in order to achieve different profiles of heating from the heating element providing the driving force of the chemical reaction happening. From the relay indication of the control panel, it is possible to visualize if power is supplied to the heating element. The amount of power of the heating element is controlled by the amount of time it receives electrical current and due to the configuration of the controller, is set to operate at different levels depending the temperature error difference (Te) between setpoint and actual reactor temperature. For the technical scale reactor, this error difference was set to 20 °C, causing it to operate under three different modes:
1 – Te < 10 °C → Heating element provides power to increase slowly the temperature and maintain it once reaction starts;
2 – 10 °C < Te < 20 °C → Heating element provides power more frequently, increasing reaction rate;
3 – Te > 20 °C → Heating element is always on;
In Figure 9, Figure 10 and Figure 11, liquid condensate flowrate reduced with increased processing temperature but the actual flowrate of vapors probably increased due to formation of more non-condensable gases. Increased heating rate was able to further react the MMA formed during PMMA dental waste depropagation into different reaction products, mostly in the gas phase, probably in the form of carbon dioxide and light hydrocarbons. Further testing of the composition of the gas phase is needed in order to obtain detailed characteristics of these secondary reactions.
3.2. Chemical Composition Analysis
Figure 12, Figure 13 and Figure 14 presents graphs of how chemical composition of the liquid product formed during PMMA dental waste depolymerization changed over reaction time in the three temperatures of depolymerization. Initially, liquid product is composed by the monomer MMA, being the only product of depolymerization in low temperature and reaction times, corroborating the established depolymerization mechanisms of PMMA, where most of the vapor product formed is due to the depropagation reaction, producing monomer MMA. As the reaction proceeds, since it is a semi-batch reactor, the degree of polymerization of the remaining feed in the reactor reduces over time but also the zip length due to the diminishing length of the polymer chain. Besides, the polymer chains are also transformed into other products due to condensation and aromatization reactions, turning into char and other cracking products of the char pyrolysis. Higher heating rates also can increase or accelerate some side-reactions, increasing product complexity and distribution. These reactions compete with the depropagation of the polymer chain into monomer and eventually other products are formed in the reactor and vaporize, condensing into the liquid product of depolymerization. This can be seen in Figure 13 and Figure 14 for the reactions conducted at 450 and 475 °C, respectively. After 20 minutes of liquid condensation, the composition starts to change from 100 area.% of MMA to side products as Methyl isobutyrate (MIB), aromatic hydrocarbons as benzene, toluene, xylene (BTEX) and other complex compounds that could not be accurately identified, supposedly hydrocarbons and oxygenated compounds with long chains of carbons (char and tar precursors) [35].
As it was seen in the previous section, the mechanism of depolymerization is affected by the heating rate used and it is related to the final temperature of reaction in the technical scale unit. By observing the temperature profiles of Figure 10 and Figure 11, higher heating rates produced lower liquid condensate yields but the chemical composition of the obtained liquid fraction shows that for most of the time, it was majorly composed of MMA, indicating that, initially, most of the secondary products formed were gases, being in the non-condensable gases phase and were formed due to secondary cracking reactions of the diffusing MMA through the heated polymeric bed. For the experiment conducted at 425 °C, liquid product was composed of 100 area.% of MMA and no side products were formed in liquid phase with a char yield of 9.44 wt.%. For the experiments using higher heating rate and final temperature of Figure 13 and Figure 14 (450 and 475 °C), lower char yields were obtained (~2.5 wt.%) but increased presence of side-products, showing that both the final temperature and heating rate influences the formation of char and side-products.
The presence of these side-products in the vapor composition of PMMA dental waste depolymerization can also be visualized by the vapor temperature showed in Figure 9, Figure 10 and Figure 11. It is possible to observe that vapor temperature tends to maintain its value near the boiling point of the vaporized mixture of substances coming out of the heated reactor. Since most of the liquid condensate is MMA, values around 101 °C should be observed (boiling point of MMA). For the experiment of Figure 9 (425 °C), maximum vapor temperature was 114 °C (70 minutes of reaction), but most of the liquid was formed under 100 °C. For Figure 10 (450 °C) and Figure 11 (475 °C), vapor temperature increased to over 110 °C at 30 and 40 minutes of reaction, respectively and maximum temperatures were 120 and 124 °C. Chemical composition of Figure 13 and Figure 14 reveal that vapor temperature increased due to the presence of side-products in liquid condensate, formed after 30 minutes of reaction.
The mechanism of depolymerization is also affected by the existing kinetics in the reactor, i.e., the heat transfer and diffusion of formed products through the polymer particle’s bed. In small-scale and miniaturized reactors (such as the thermobalance of TGA/DTG analysis), the geometry of the particle’s bed is small compared to the heat transfer and diffusion needed. As the size of the reactor increases, the effect of heat transfer and diffusion also increases, changing reaction rates and product composition [27]. Since the temperature controller thermocouple is located in a thermal well near the center of the particle bed, there is probably a temperature gradient from the rim to the center of the cylindrical reactor, meaning that the actual temperature at the rims should be higher than the setpoint. This can explain why semi-batch fixed bed reactors produce more gas and less liquid phase yields when compared to small-scale semi-batch reactors. The higher temperature at the rim can further crack the products of depolymerization, forming non-condensable gases such as CO, CO2, H2 and light hydrocarbons (CH4, C2H2 and others), increasing gas yields and reducing the actual recovery of MMA. Since formed products are part of the gas phase, there is no change of chemical composition of the liquid phase until most of the feed converts into char and the char pyrolysis dominates the products distribution and composition.
4. Discussion
In this work was detailed for the first time an accurate analysis of the semi-batch depolymerization process of EGDMA-crosslinked PMMA dental waste when considering the influence of reaction time, kinetics and chemical composition variation under high temperature of pyrolysis. Through thermal analysis, we could characterize and categorize the type of waste according to its decomposition profile and verify that crosslinking increase thermal stability of the polymer and reduces its degradation at low temperatures, favoring main chain random scission mechanism while suppressing the formation of H-H linkages in the polymer, causing it to degrade at low temperatures. In past studies, we did not observe this influence in the mechanism of thermal degradation [24,27], in fact, we could verify the conclusions obtained by Da Ros et al. [31], who modelled the chemical recycling of EGDMA-crosslinked PMMA. Thermogravimetry analysis proves once more to be a valuable tool in the analysis of pyrolysis reactions and mechanisms and study of thermal degradation of determined feedstock is paramount to the design of pyrolysis reactors. We expect that further studies of thermogravimetry profiles of plastic waste could be used to infer some characteristics such as overall polymerization mechanism, i.e., we could use thermal analysis to obtain valuable information about the waste, making it easier to process it in a hypothetical recycling factory.
Semi-batch reactors are cheap and easy to construct but its correct operation depends upon a considerable knowledge of the process in question due to its transient nature: the chemical composition of feed and vapors is ever-changing with reaction time. In some processes, such as PMMA depolymerization, a semi-stationary state is obtained due to the depropagation of the polymeric chain into monomer MMA. Our results showed that liquid condensate of PMMA depolymerization is over 90% composed of it but this composition is influence by reaction time, temperature and, if the dimensions of the bed of polymer particles is considerable, heating rate. A large bed of polymer particles increases temperature gradients between the rim and center of the reactor, overheating a part of the polymer bed and further reacting the diffusing formed MMA out of the bed, forming gaseous products. This observation is in accord with other researchers, who also identified condensate losses and increased gas yields for high temperature depolymerization in pilot scale [24,27,31]. The results show that it is possible to obtain high liquid phase yields while maintaining high composition of MMA using semi-batch reactors with low processing temperature.
The work further elaborates on the formation of char and secondary products of PMMA depolymerization in temperatures of 450 and 475 °C. While most plastics tend to thermally decompose forming almost no char, the mechanism of PMMA depolymerization clearly shows that depolymerization can be terminated before all polymeric chain is consumed [43] and that, in a competitive manner, there is char formation reactions due to condensation, aromatization and hydrogen abstraction event of the formed depolymerization products, producing aromatic hydrocarbons, identified in the liquid phase of experiments at higher temperatures. This formed char cracks at higher temperatures and experiment at 425 °C produced the highest amount of char but composition of liquid phase was composed only of MMA. Higher temperatures reduced formed char yields but increased the presence of aromatic hydrocarbons in liquid phase, indicating that the formed char further cracked to those products. Higher temperatures improve the extent of cracking and depolymerization but can also pollute the liquid phase with undesired products. If one desires to maximize the recovery of MMA, one should aim to use the lower possible temperature with low heating rates for fixed bed reactors. High heating rates and high final temperatures would provoke low MMA recoveries due to increased gaseous phase yields (reducing liquid phase yields) and lower purity of liquid phase (lower MMA content). By conducting it in low heating rate and final temperature, higher char yields are obtained (not that bad considering you can use it for heat) but higher MMA recoveries.
References
- Dai, L.; Zhou, N.; Lv, Y.; Cheng, Y.; Wang, Y.; Liu, Y.; Cobb, K.; Chen, P.; Lei, H.; Ruan, R. Pyrolysis Technology for Plastic Waste Recycling: A State-of-the-Art Review. Prog Energy Combust Sci 2022, 93, 101021. [Google Scholar] [CrossRef]
- Global Plastics Outlook; OECD, 2022; ISBN 9789264654945.
- Gigault, J.; Halle, A. ter; Baudrimont, M.; Pascal, P.Y.; Gauffre, F.; Phi, T.L.; El Hadri, H.; Grassl, B.; Reynaud, S. Current Opinion: What Is a Nanoplastic? Environmental Pollution 2018, 235, 1030–1034. [Google Scholar] [CrossRef] [PubMed]
- Rillig, M.C.; Lehmann, A. Microplastic in Terrestrial Ecosystems. Science (1979) 2020, 368, 1430–1431. [Google Scholar] [CrossRef] [PubMed]
- Nayanathara Thathsarani Pilapitiya, P.G.C.; Ratnayake, A.S. The World of Plastic Waste: A Review. Cleaner Materials 2024, 11, 100220. [Google Scholar] [CrossRef]
- Thiounn, T.; Smith, R.C. Advances and Approaches for Chemical Recycling of Plastic Waste. Journal of Polymer Science 2020, 58, 1347–1364. [Google Scholar] [CrossRef]
- Kaminsky, W.; Franck, J. Monomer Recovery by Pyrolysis of Poly(Methyl Methacrylate) (PMMA). J Anal Appl Pyrolysis 1991, 19, 311–318. [Google Scholar] [CrossRef]
- Maisels, A.; Hiller, A.; Simon, F. Chemisches Recycling Für Kunststoffe: Status Und Perspektiven. Chemie Ingenieur Technik 2021, 93, 1742–1750. [Google Scholar] [CrossRef]
- Ali, U.; Karim, K.J.B.A.; Buang, N.A. A Review of the Properties and Applications of Poly (Methyl Methacrylate) (PMMA). Polymer Reviews 2015, 55, 678–705. [Google Scholar] [CrossRef]
- Frazer, R.Q.; Byron, R.T.; Osborne, P.B.; West, K.P. PMMA: An Essential Material in Medicine and Dentistry. J Long Term Eff Med Implants 2005, 15, 629–639. [Google Scholar] [CrossRef]
- Gozum, N.; Safgonul Unal, E.; Altan-Yaycioglu, R.; Gucukoglu, A.; Ozgun, C. Visual Performance of Acrylic and PMMA Intraocular Lenses. Eye (Lond) 2003, 17, 238–242. [Google Scholar] [CrossRef]
- Spasojevic, P.; Zrilic, M.; Panic, V.; Stamenkovic, D.; Seslija, S.; Velickovic, S. The Mechanical Properties of a Poly(Methyl Methacrylate) Denture Base Material Modified with Dimethyl Itaconate and Di-n-Butyl Itaconate. Int J Polym Sci 2015, 2015. [Google Scholar] [CrossRef]
- Sponchioni, M.; Altinok, S. Poly(Methyl Methacrylate): Market Trends and Recycling. In; 2022; pp. 269–287.
- Grause, G.; Predel, M.; Kaminsky, W. Monomer Recovery from Aluminium Hydroxide High Filled Poly(Methyl Methacrylate) in a Fluidized Bed Reactor. J Anal Appl Pyrolysis 2006, 75, 236–239. [Google Scholar] [CrossRef]
- Kaminsky, W. Thermal Recycling of Polymers. J Anal Appl Pyrolysis 1985, 8, 439–448. [Google Scholar] [CrossRef]
- KAMINSKY, W. Recycling of Polymers by Pyrolysis. Le Journal de Physique IV 1993, 03, C7-1543–C7-1552. [Google Scholar] [CrossRef]
- Kaminsky, W.; Eger, C. Pyrolysis of Filled PMMA for Monomer Recovery. J Anal Appl Pyrolysis 2001, 58–59, 781–787. [Google Scholar] [CrossRef]
- Kaminsky, W.; Predel, M.; Sadiki, A. Feedstock Recycling of Polymers by Pyrolysis in a Fluidised Bed. Polym Degrad Stab 2004, 85, 1045–1050. [Google Scholar] [CrossRef]
- Kang, B.S.; Kim, S.G.; Kim, J.S. Thermal Degradation of Poly(Methyl Methacrylate) Polymers: Kinetics and Recovery of Monomers Using a Fluidized Bed Reactor. J Anal Appl Pyrolysis 2008, 81, 7–13. [Google Scholar] [CrossRef]
- Sasaki, A.; Tsuji, T. POLY(METHYL METHACRYLATE) PYROLYSIS BY TWO FLUIDIZED BED PROCESS.
- Smolders, K.; Baeyens, J. Thermal Degradation of PMMA in Fluidised Beds. Waste Manag 2004, 24, 849–857. [Google Scholar] [CrossRef]
- Achilias, D.S. Chemical Recycling of Poly(Methyl Methacrylate) by Pyrolysis. Potential Use of the Liquid Fraction as a Raw Material for the Reproduction of the Polymer. Eur Polym J 2007, 43, 2564–2575. [Google Scholar] [CrossRef]
- Braido, R.S.; Borges, L.E.P.; Pinto, J.C. Chemical Recycling of Crosslinked Poly(Methyl Methacrylate) and Characterization of Polymers Produced with the Recycled Monomer. J Anal Appl Pyrolysis 2018, 132, 47–55. [Google Scholar] [CrossRef]
- Dos Santos, P.B.; da Silva Ribeiro, H.J.; Ferreira, A.C.; Ferreira, C.C.; Bernar, L.P.; da Costa Assunção, F.P.; de Castro, D.A.R.; Santos, M.C.; Duvoisin, S.; Borges, L.E.P.; et al. Process Analysis of PMMA-Based Dental Resins Residues Depolymerization: Optimization of Reaction Time and Temperature. Energies 2022, Vol. 15, Page 91 2021, 15, 91. [Google Scholar] [CrossRef]
- Lopez, G.; Artetxe, M.; Amutio, M.; Elordi, G.; Aguado, R.; Olazar, M.; Bilbao, J. Recycling Poly-(Methyl Methacrylate) by Pyrolysis in a Conical Spouted Bed Reactor. Chemical Engineering and Processing: Process Intensification 2010, 49, 1089–1094. [Google Scholar] [CrossRef]
- Poudel, J.; Lee, Y.M.; Kim, H.J.; Oh, S.C. Methyl Methacrylate (MMA) and Alumina Recovery from Waste Artificial Marble Powder Pyrolysis. J Mater Cycles Waste Manag 2021, 23, 214–221. [Google Scholar] [CrossRef]
- Ribeiro, H.J.da S.; Ferreira, A.C.; Ferreira, C.C.; Pereira, L.M.; Santos, M.C.; Guerreiro, L.H.H.; Assunção, F.P. da C.; da Mota, S.A.P.; Castro, D.A.R. de; Duvoisin, S.; et al. Depolymerization of PMMA-Based Dental Resin Scraps on Different Production Scales. Energies (Basel) 2024, 17, 1196. [Google Scholar] [CrossRef]
- Ferreira, C.C.; Bernar, L.P.; de Freitas Costa, A.F.; da Silva Ribeiro, H.J.; Santos, M.C.; Moraes, N.L.; Costa, Y.S.; Baia, A.C.F.; Mendonça, N.M.; da Mota, S.A.P.; et al. Improving Fuel Properties and Hydrocarbon Content from Residual Fat Pyrolysis Vapors over Activated Red Mud Pellets in Two-Stage Reactor: Optimization of Reaction Time and Catalyst Content. Energies (Basel) 2022, 15, 5595. [Google Scholar] [CrossRef]
- Bernar, L.P.; Ferreira, C.C.; Costa, A.F.de F.; Ribeiro, H.J. da S.; Santos, W.G. dos; Pereira, L.M.; Pereira, A.M.; Moraes, N.L.; Assunção, F.P. da C.; Mota, S.A.P. da; et al. Catalytic Upgrading of Residual Fat Pyrolysis Vapors over Activated Carbon Pellets into Hydrocarbons-like Fuels in a Two-Stage Reactor: Analysis of Hydrocarbons Composition and Physical-Chemistry Properties. Energies (Basel) 2022, 15, 1–26. [Google Scholar] [CrossRef]
- Lohmann, V.; Jones, G.R.; Truong, N.P.; Anastasaki, A. The Thermodynamics and Kinetics of Depolymerization: What Makes Vinyl Monomer Regeneration Feasible? Chem Sci 2024, 15, 832–853. [Google Scholar] [CrossRef]
- Da Ros, S.; Braido, R.S.; de Souza e Castro, N.L.; Brandão, A.L.T.; Schwaab, M.; Pinto, J.C. Modelling the Chemical Recycling of Crosslinked Poly (Methyl Methacrylate): Kinetics of Depolymerisation. J Anal Appl Pyrolysis 2019, 144, 104706. [Google Scholar] [CrossRef]
- Barlow, A.; Lehrle, R.S.; Robb, J.C.; Sunderland, D. Polymethylmethacrylate Degradation—Kinetics and Mechanisms in the Temperature Range 340° to 460°C. Polymer (Guildf) 1967, 8, 537–545. [Google Scholar] [CrossRef]
- Manring, L.E. Thermal Degradation of Poly(Methyl Methacrylate). 4. Random Side-Group Scissiont. Macromolecules 1991, 24, 3304–3309. [Google Scholar] [CrossRef]
- Manring, L.E. Thermal Degradation of Poly(Methyl Methacrylate). 2. Vinyl-Terminated Polymer. A. Rev. Plast. Mod 1969, 19, 1515. [Google Scholar] [CrossRef]
- Chang, C.-Chu.; Wan, S.-Wu. China’s Motor Fuels from Tung Oil. Ind Eng Chem 1947, 39, 1543–1548. [Google Scholar] [CrossRef]
- Benson, T.J.; Hernandez, R.; French, W.T.; Alley, E.G.; Holmes, W.E. Elucidation of the Catalytic Cracking Pathway for Unsaturated Mono-, Di-, and Triacylglycerides on Solid Acid Catalysts. J Mol Catal A Chem 2009, 303, 117–123. [Google Scholar] [CrossRef]
- Speight, J.G. Handbook of Gasification Technology; Wiley, 2020; ISBN 9781118773536. [Google Scholar]
- Moens, E.K.C.; De Smit, K.; Marien, Y.W.; Trigilio, A.D.; Van Steenberge, P.H.M.; Van Geem, K.M.; Dubois, J.L.; D’hooge, D.R. Progress in Reaction Mechanisms and Reactor Technologies for Thermochemical Recycling of Poly(Methyl Methacrylate). Polymers 2020, Vol. 12, Page 1667 2020, 12, 1667. [Google Scholar] [CrossRef]
- Hirata, T.; Kashiwagi, T.; Brown, J.E. Thermal and Oxidative Degradation of Poly(Methyl Methacrylate): Weight Loss. Macromolecules 1985, 18, 1410–1418. [Google Scholar] [CrossRef]
- Zafar, M.S. Prosthodontic Applications of Polymethyl Methacrylate (PMMA): An Update. Polymers (Basel) 2020, 12, 2299. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Lassila, L.V.J.; Tokue, A.; Takahashi, Y.; Vallittu, P.K. Influence of Molecular Weight of Polymethyl(Methacrylate) Beads on the Properties and Structure of Cross-Linked Denture Base Polymer. J Mech Behav Biomed Mater 2011, 4, 1846–1851. [Google Scholar] [CrossRef]
- Yuan, M.; Huang, D.; Zhao, Y. Development of Synthesis and Application of High Molecular Weight Poly(Methyl Methacrylate). Polymers (Basel) 2022, 14, 2632. [Google Scholar] [CrossRef]
- Manring, L.E. Thermal Degradation of Saturated Poly (Methyl Methacrylate). Macromolecules 1988, 21, 528–530. [Google Scholar] [CrossRef]
Figure 1.
Process schematic of pyrolysis unit.
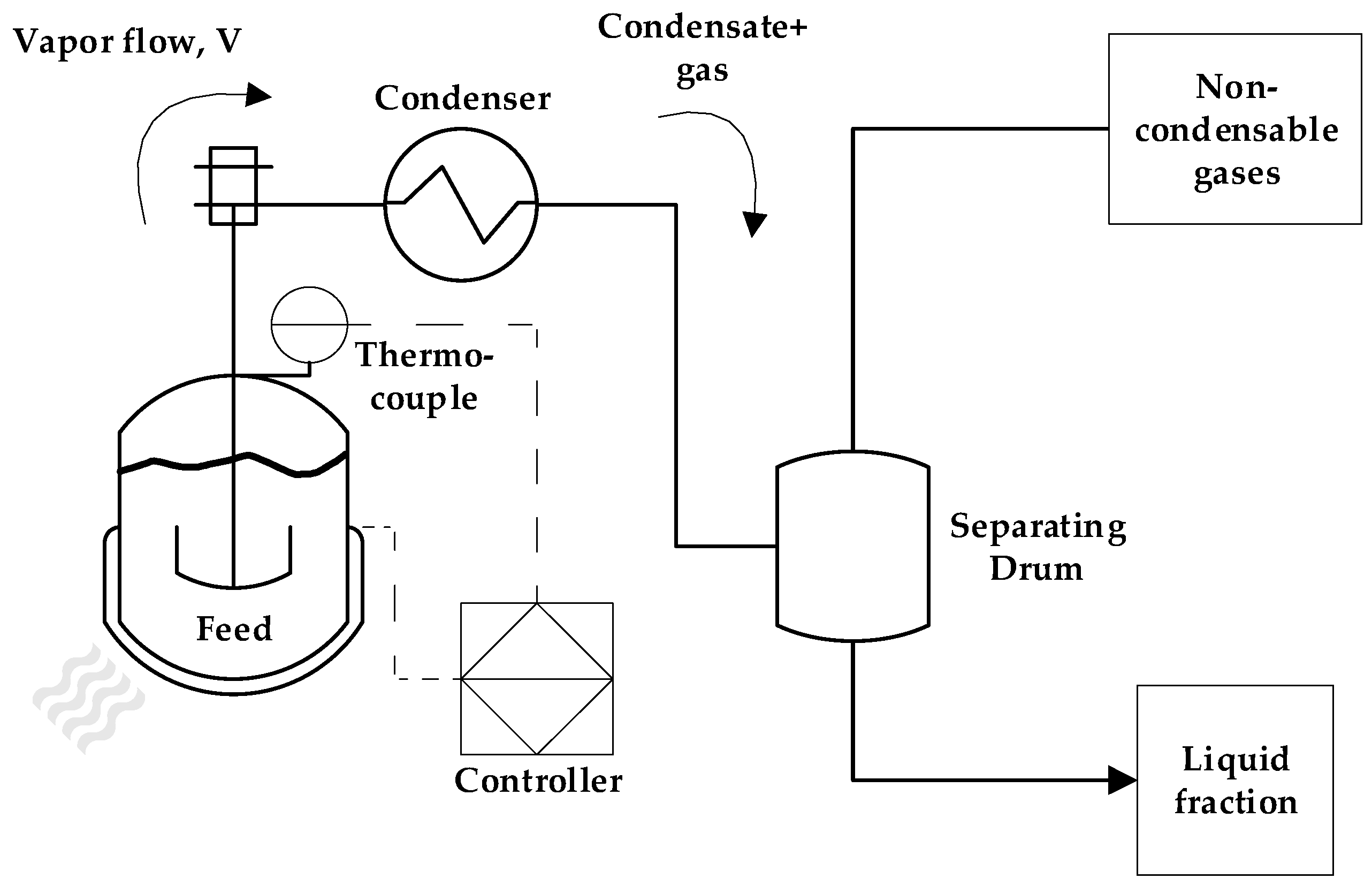
Figure 2.
Initiation reactions of PMMA depolymerization.
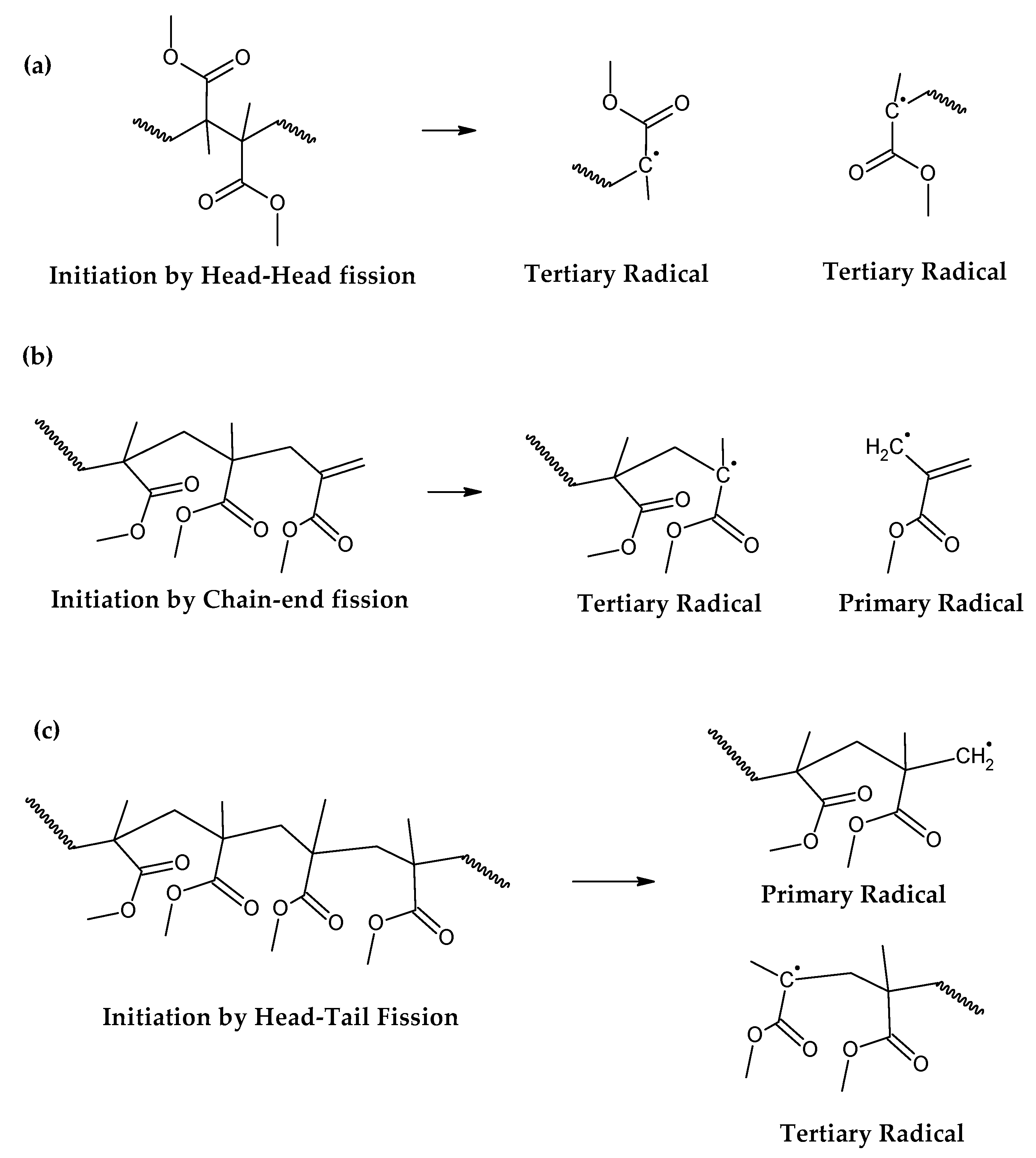
Figure 3.
Depropagation to the monomer MMA.
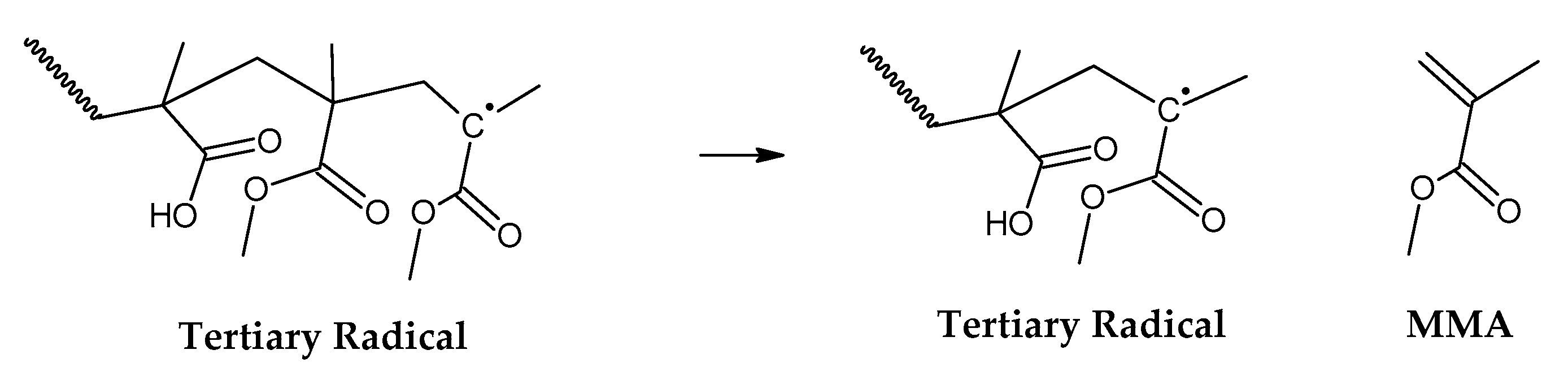
Figure 4.
Termination of depolymerization reactions of PMMA.
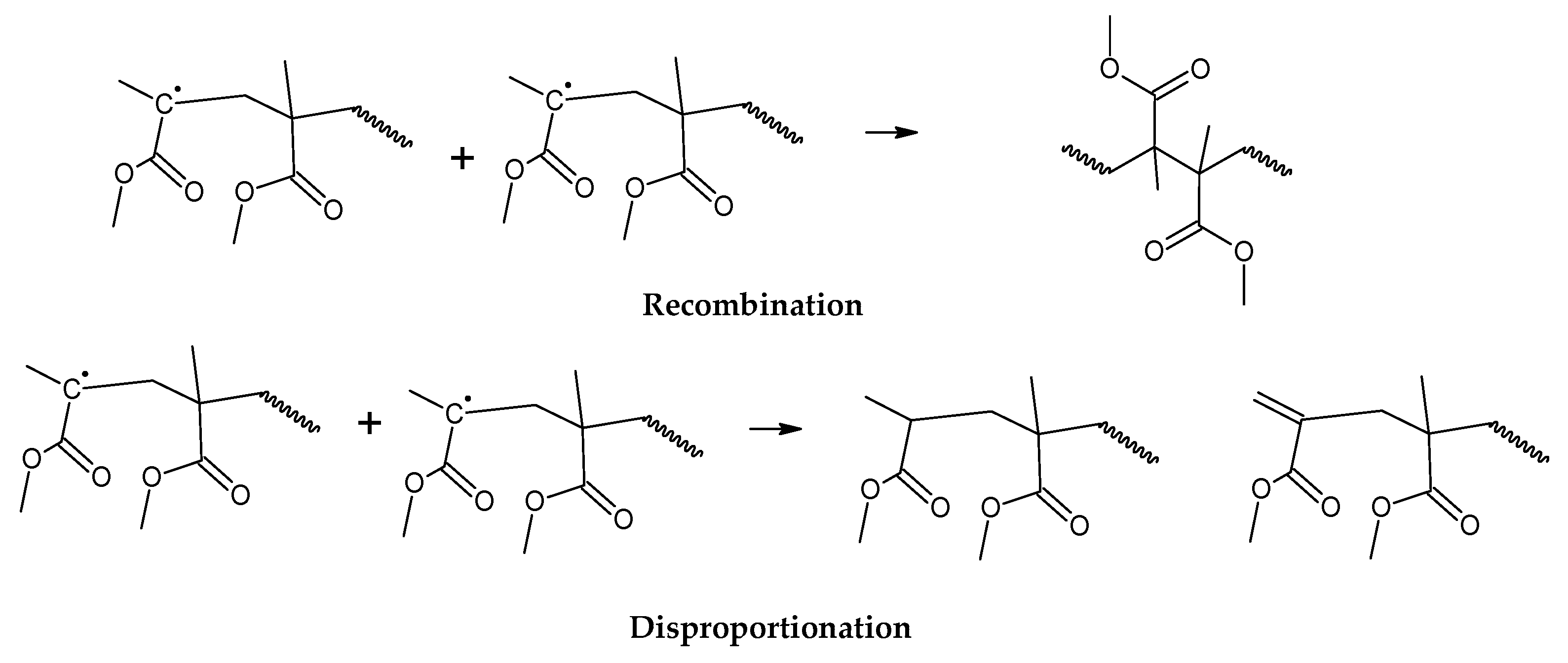
Figure 5.
Anionic polymerization mechanism of PMMA.
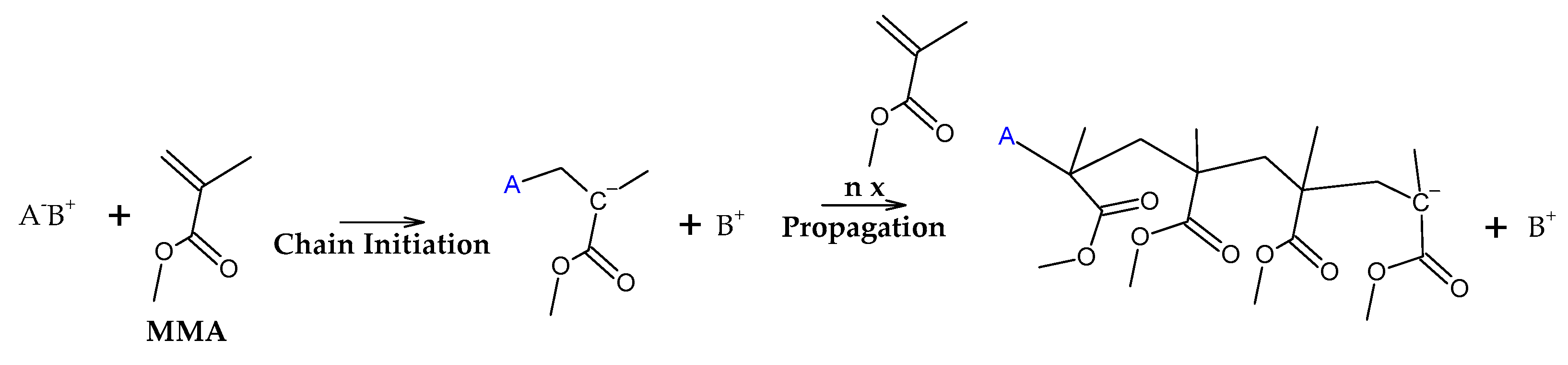
Figure 6.
TGA/DTG profile of PMMA dental waste.
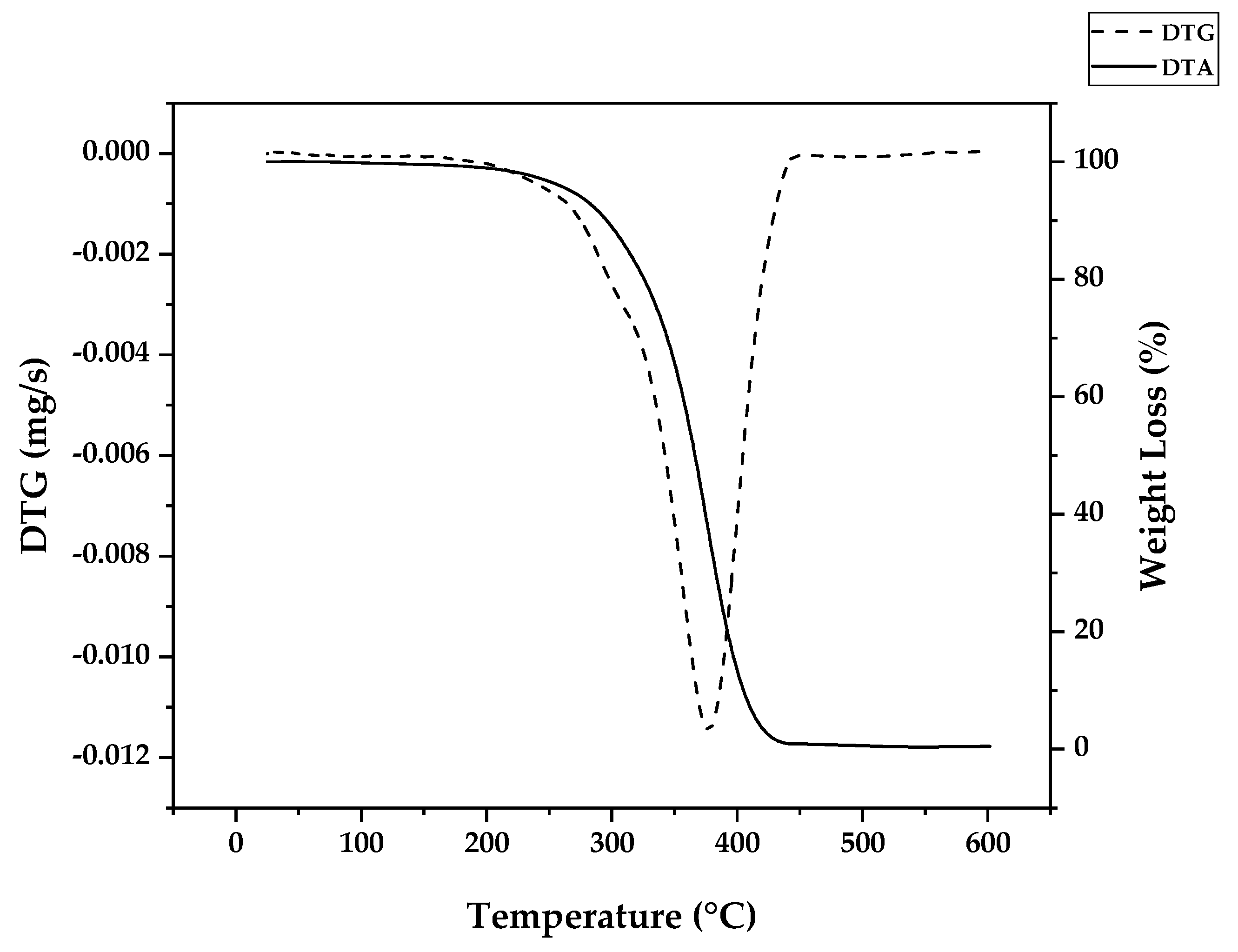
Figure 7.
Branched chain of EGDMA-PMMA polymer.
Figure 8.
Charring reactions of PMMA.
Figure 9.
Temperature profile of PMMA dental waste depolymerization at 425 °C.
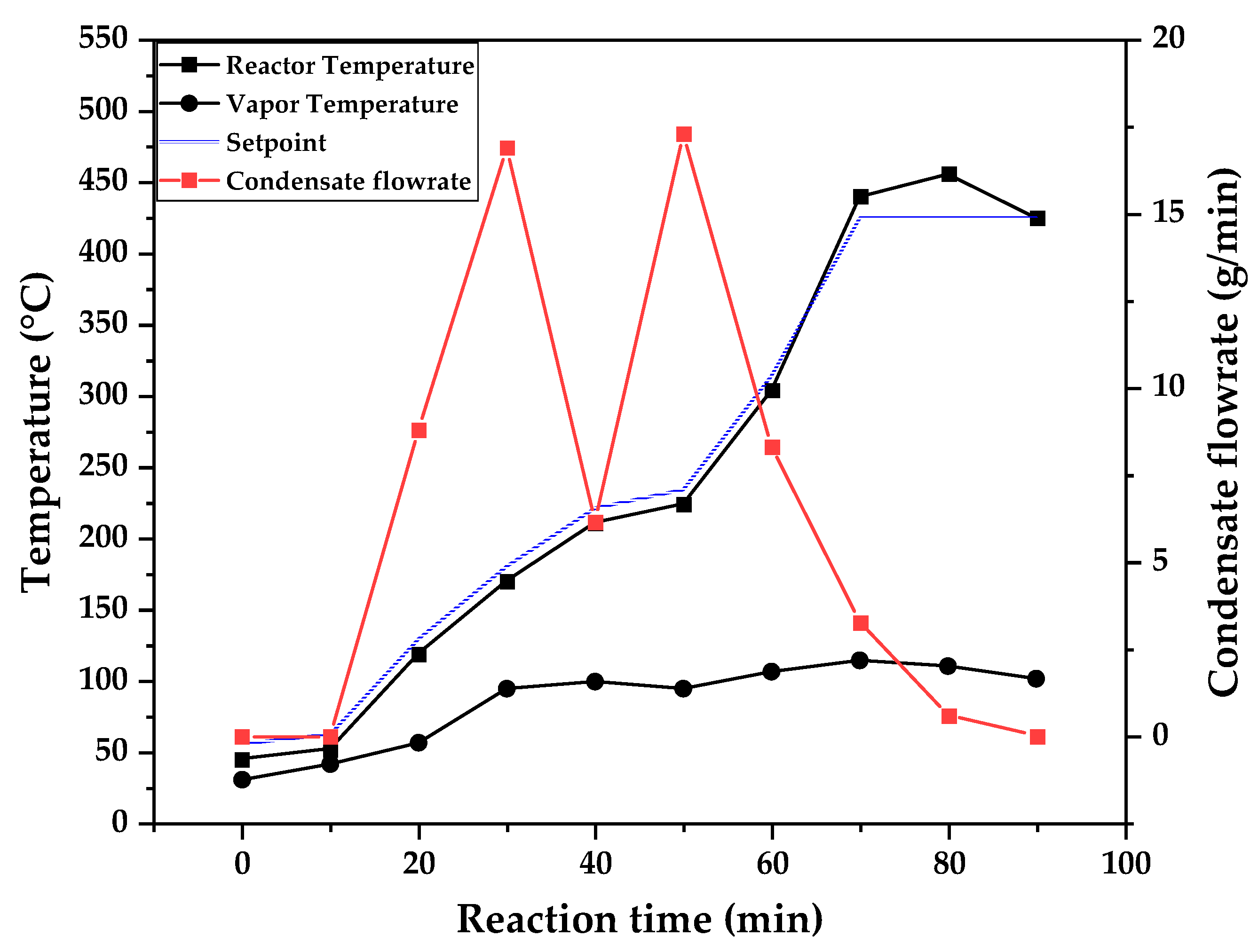
Figure 10.
Temperature profile of PMMA dental waste depolymerization at 450 °C.
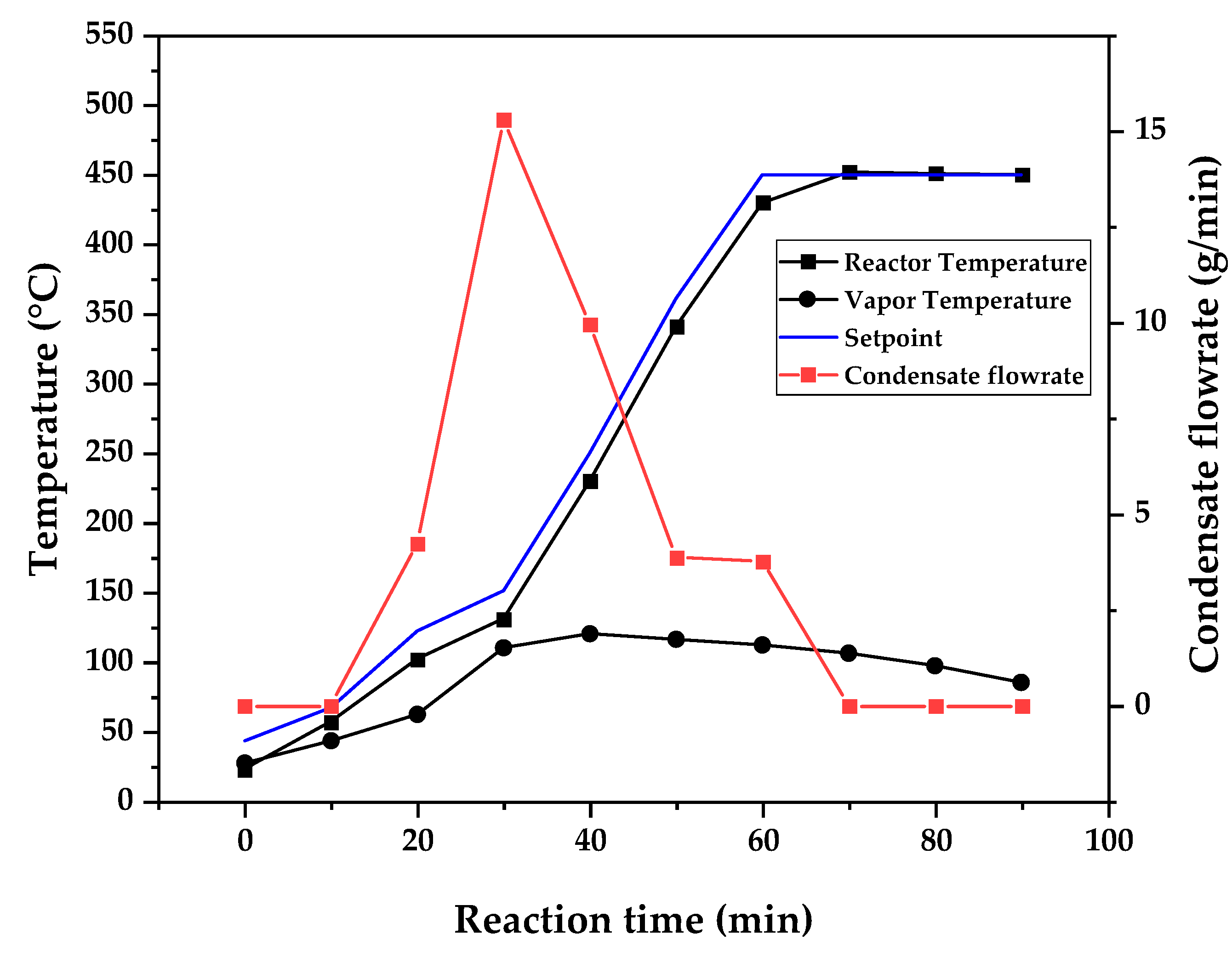
Figure 11.
Temperature profile of PMMA dental waste depolymerization at 475 °C.
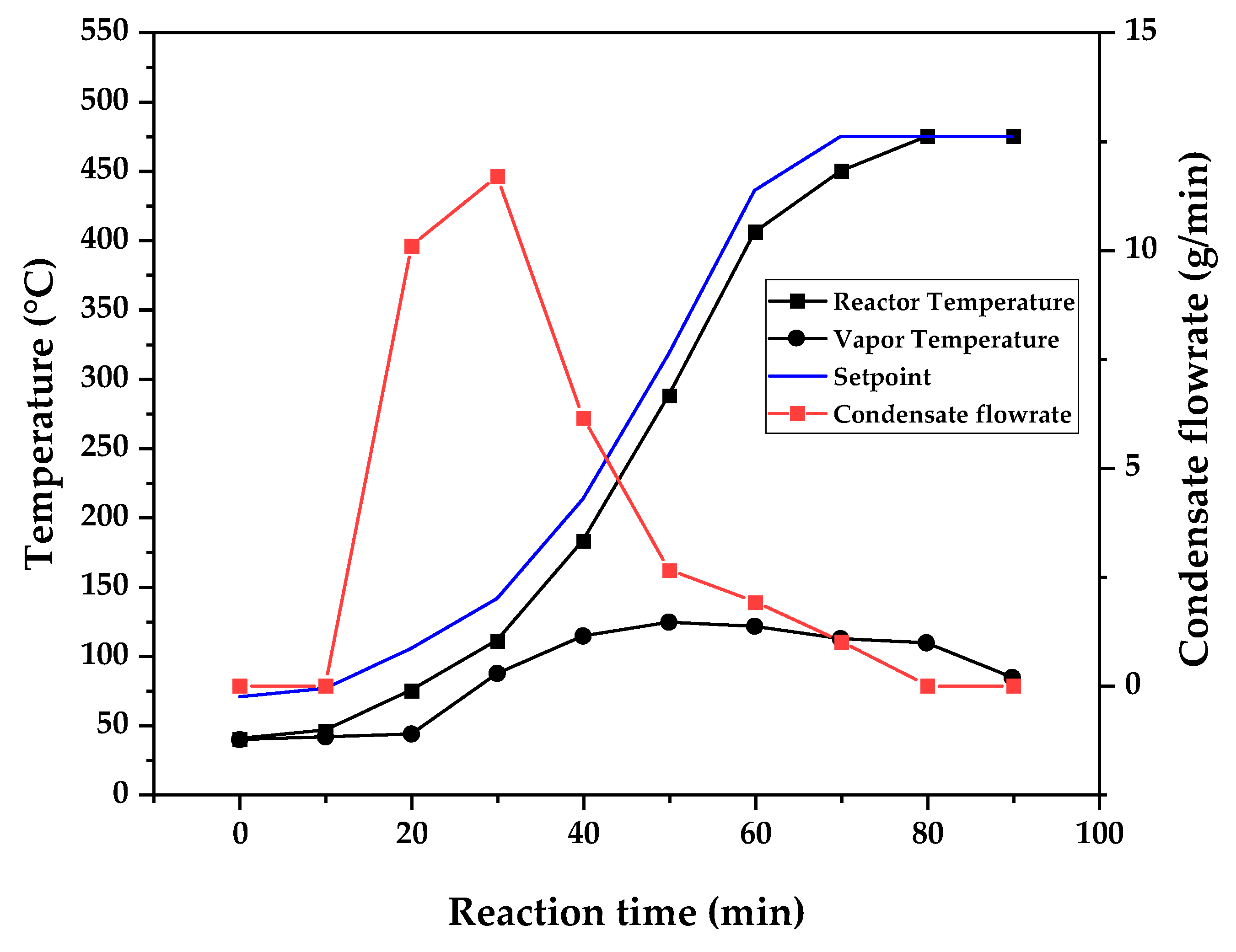
Figure 12.
Chemical composition according to reaction time of liquid product of PMMA dental waste depolymerization at 425 °C.
Figure 12.
Chemical composition according to reaction time of liquid product of PMMA dental waste depolymerization at 425 °C.
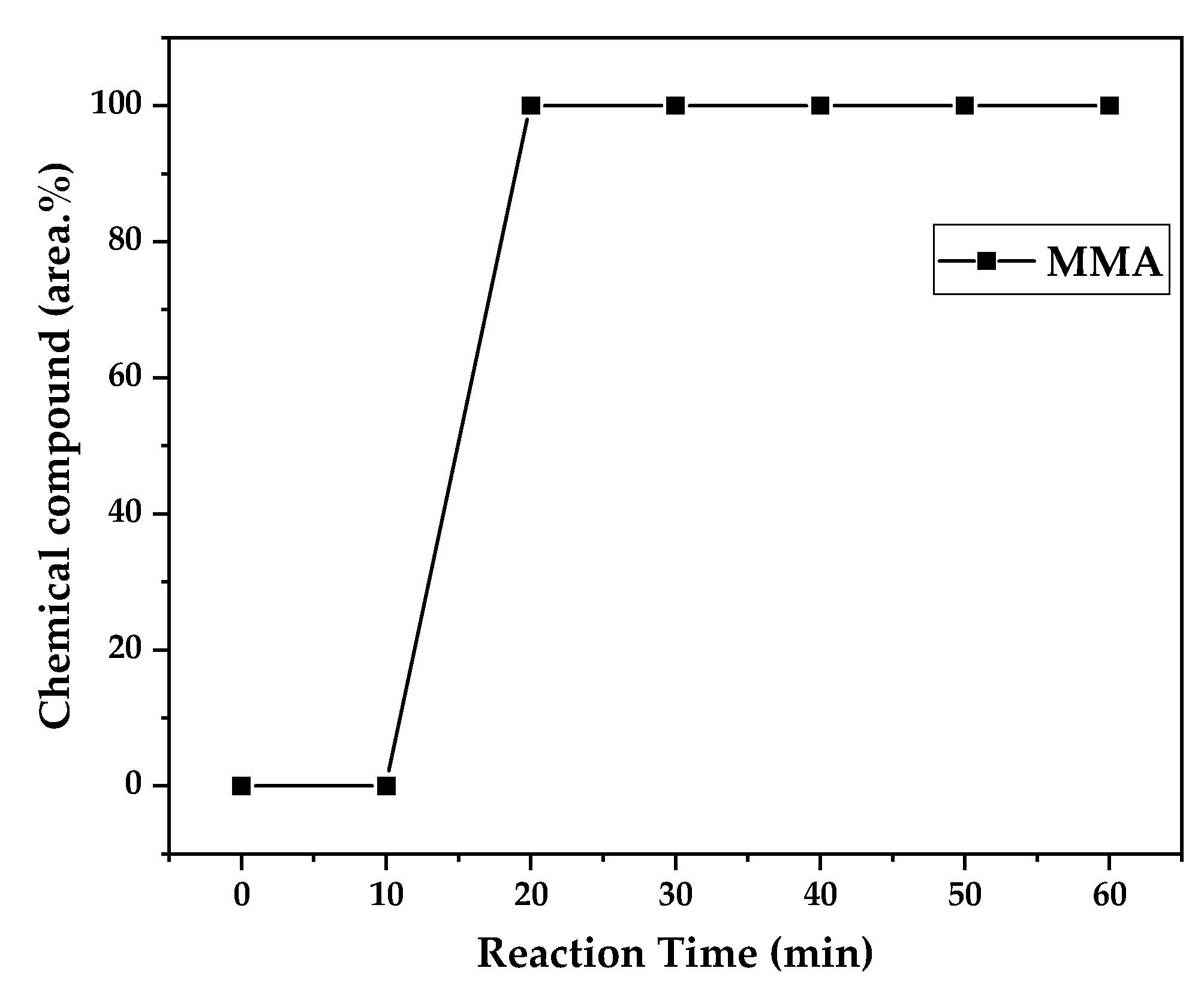
Figure 13.
Chemical composition according to reaction time of liquid product of PMMA dental waste depolymerization at 450 °C.
Figure 13.
Chemical composition according to reaction time of liquid product of PMMA dental waste depolymerization at 450 °C.
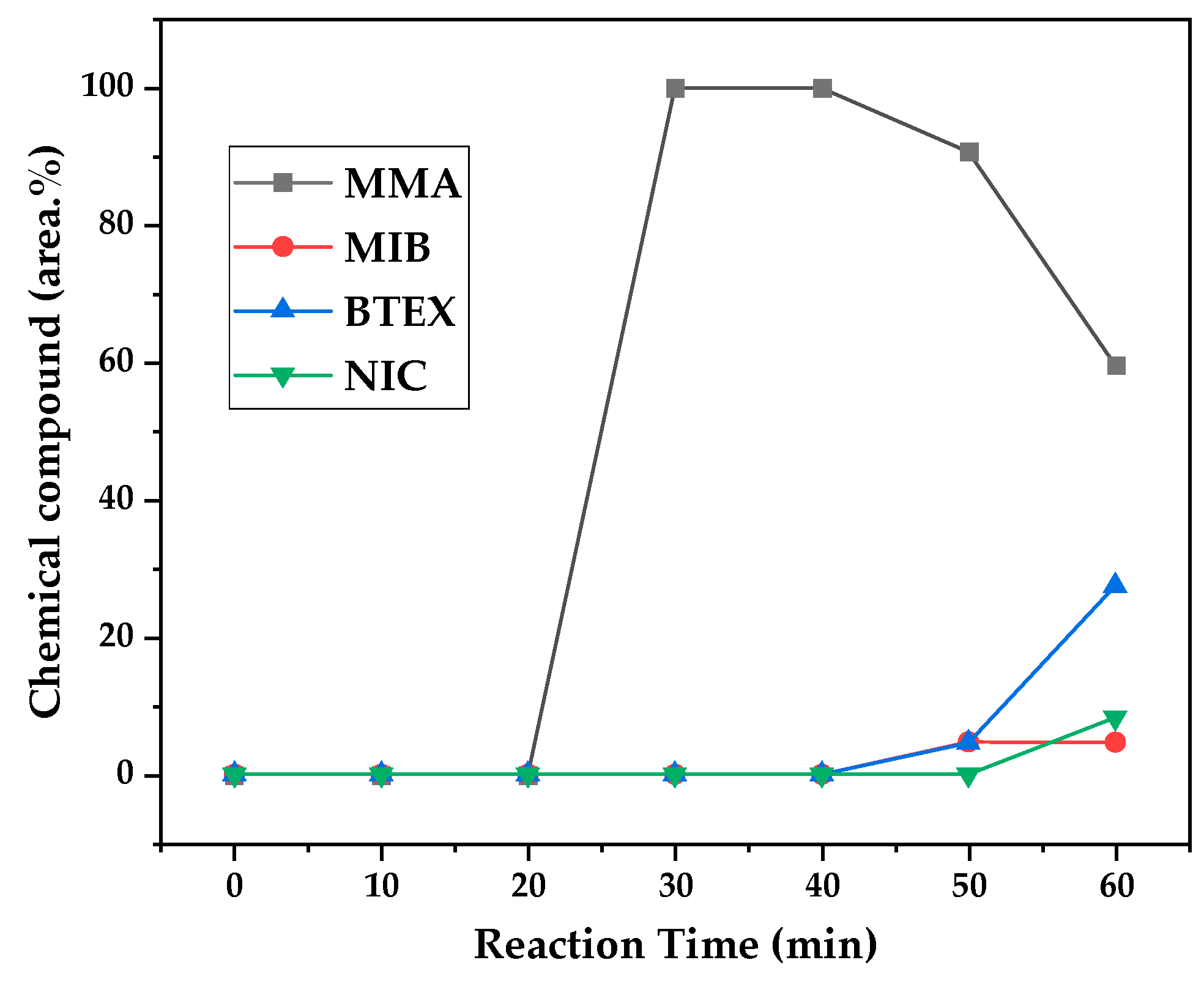
Figure 14.
Chemical composition according to reaction time of liquid product of PMMA dental waste depolymerization at 475 °C.
Figure 14.
Chemical composition according to reaction time of liquid product of PMMA dental waste depolymerization at 475 °C.
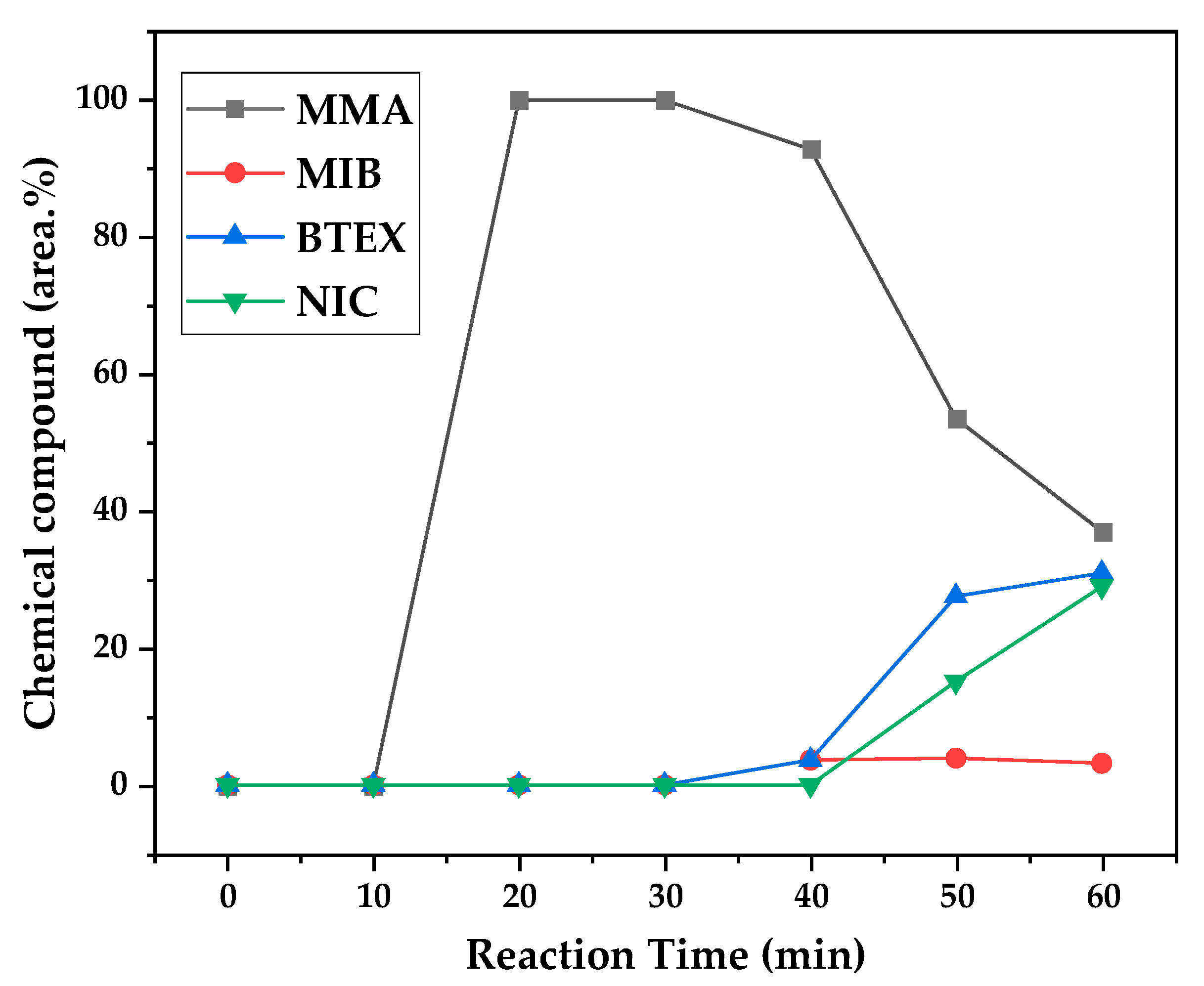

Table 1.
Product yields of PMMA dental waste depolymerization at 425-475°C.
| Process Parameter | 425 °C | 450 °C | 475 °C |
|---|---|---|---|
| Feed weight (g) | 750.23 | 623.75 | 580.38 |
| Total reaction time (min) | 120 | 120 | 110 |
| Initial condensation temperature (°C) | 119 | 109 | 75 |
| Final reaction temperature (°C) | 440 | 455 | 475 |
| Liquid phase Yield (wt.%) | 81.63 | 61.67 | 61.32 |
| Solid phase Yield (wt.%) | 9.44 | 2.43 | 2.81 |
| Gas phase Yield (wt.%) | 8.93 | 35.90 | 35.86 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



