
Submitted:
31 October 2024
Posted:
01 November 2024
You are already at the latest version
Abstract
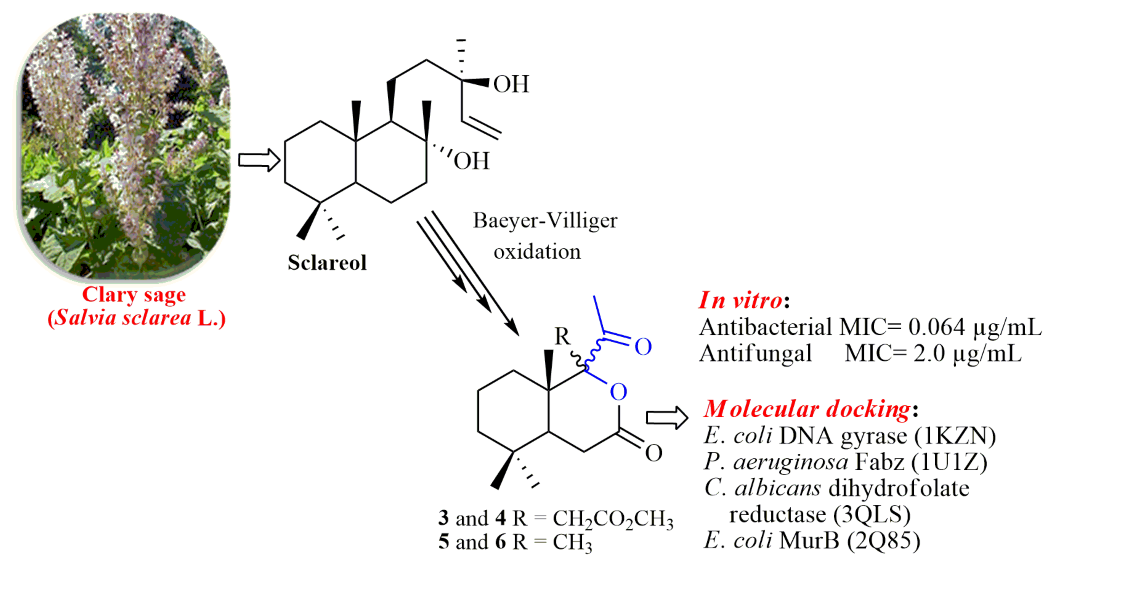
The synthesis of tetra- and pentanorlabdane compounds with rearranged cycle B based on commercially available (+)-sclareolide is reported. Desired compounds were prepared from intermediate ketones via Baeyer-Villiger oxidation. The structures of synthesized compounds were confirmed by spectral IR, 1D (1H, 13C and 15N, DEPT) and 2D (H-COSY, H,C-HSQC, H,C-HMBC, H,N-HMBC, NOESY) NMR analyses, mass-spectrometry and single crystal X-rays diffraction. Two of the four obtained compounds showed high antifungal and antimicrobial activity, comparable and exceeding that of standards Caspofungin and Kanamycin. DFT calculations revealed the stability of compounds in gas, DCM, water and methanol. Molecular docking to four targets (1KZN, 1U1Z, 3QLS, 2Q85) showed good agreement with the results of in vitro evaluation and confirmed the biological activity of compounds 3 and 4, with binding affinities comparable and for some targets exceeding that of Caspofungin and Kanamycin.
Keywords:
1. Introduction
2. Results and Discussion
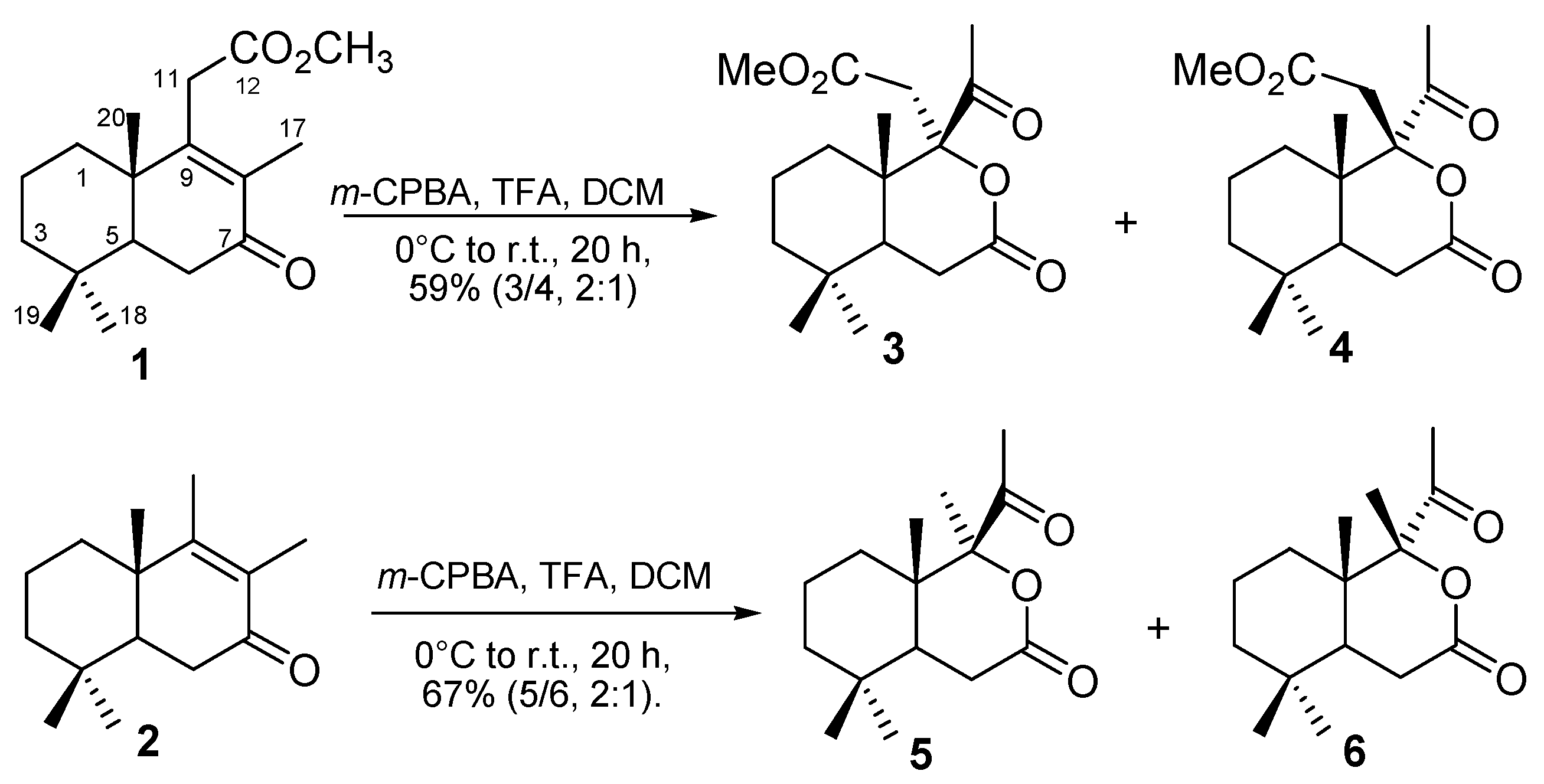
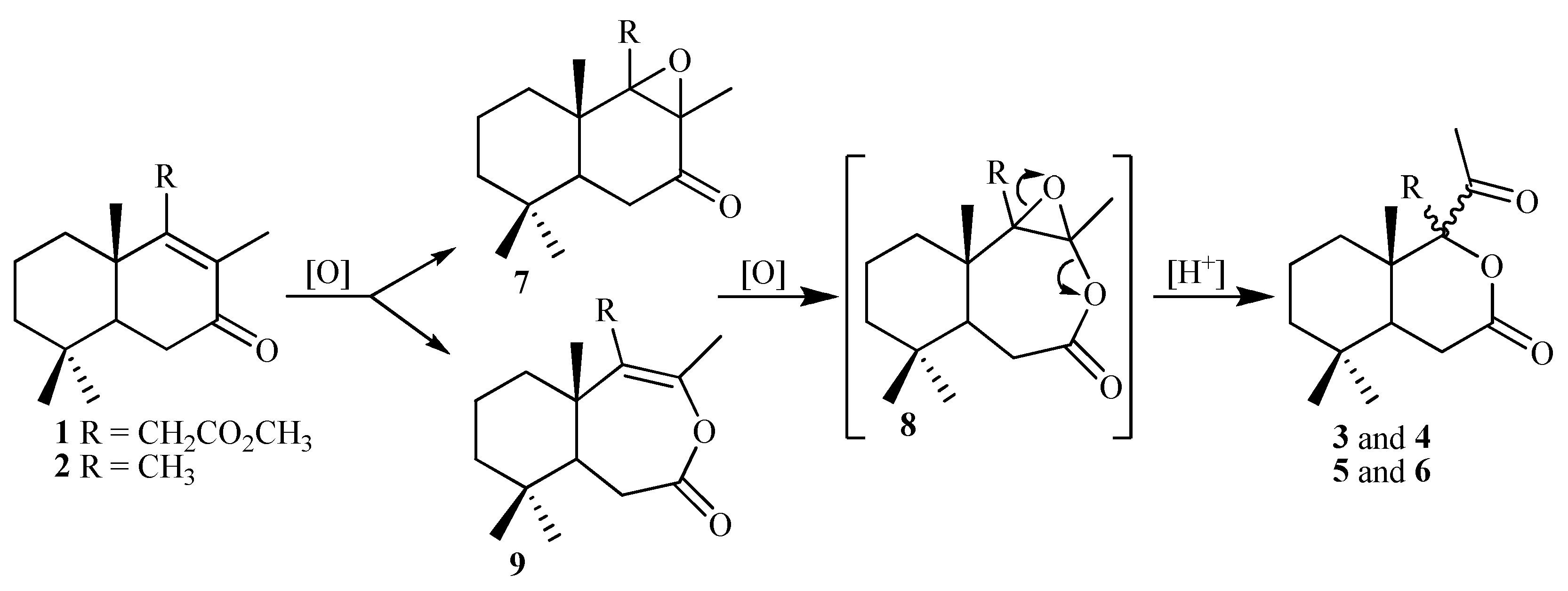
2.1. Synthesis
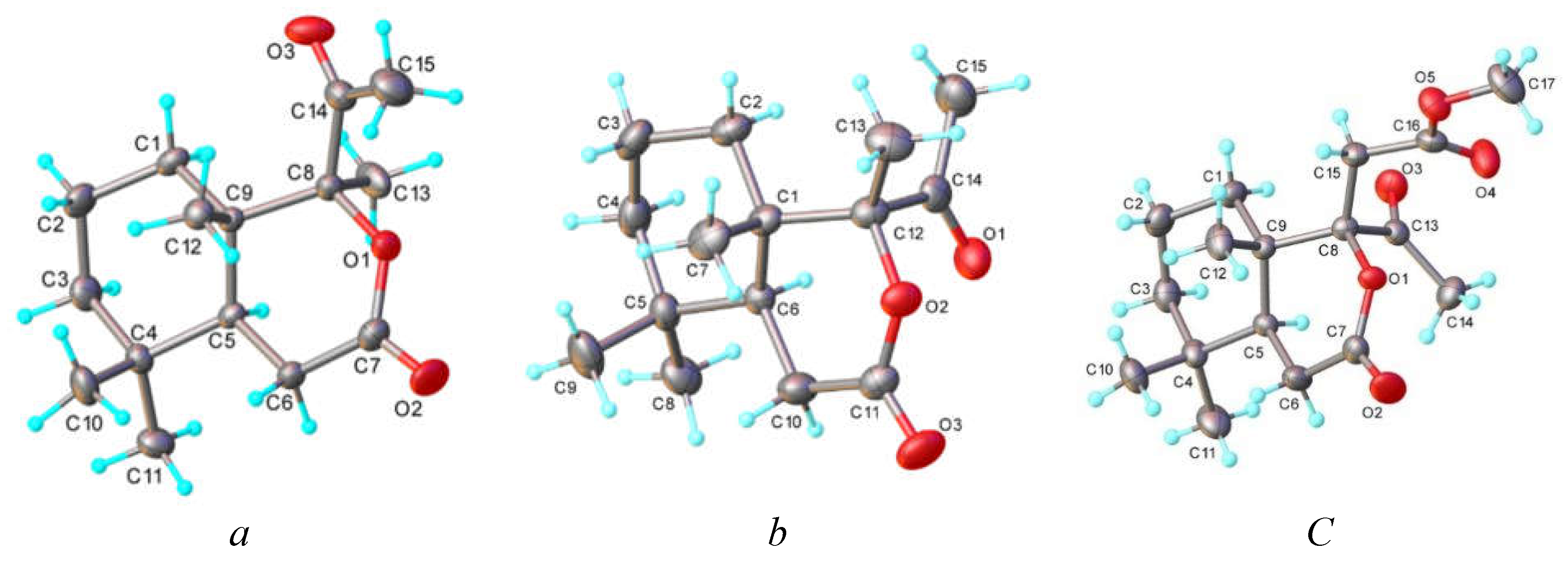
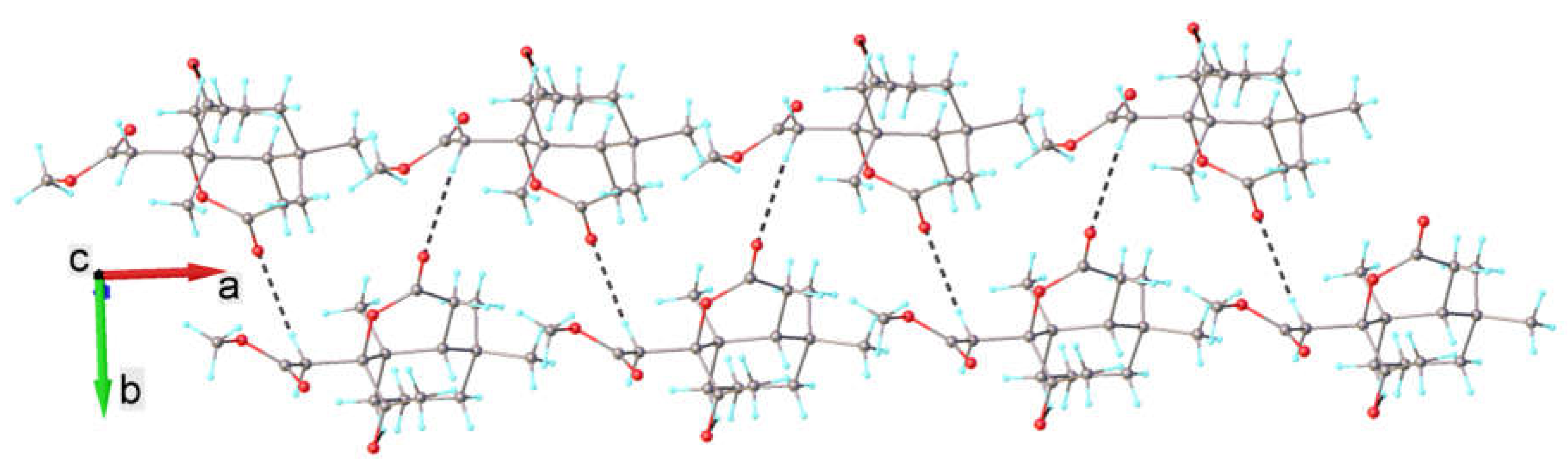
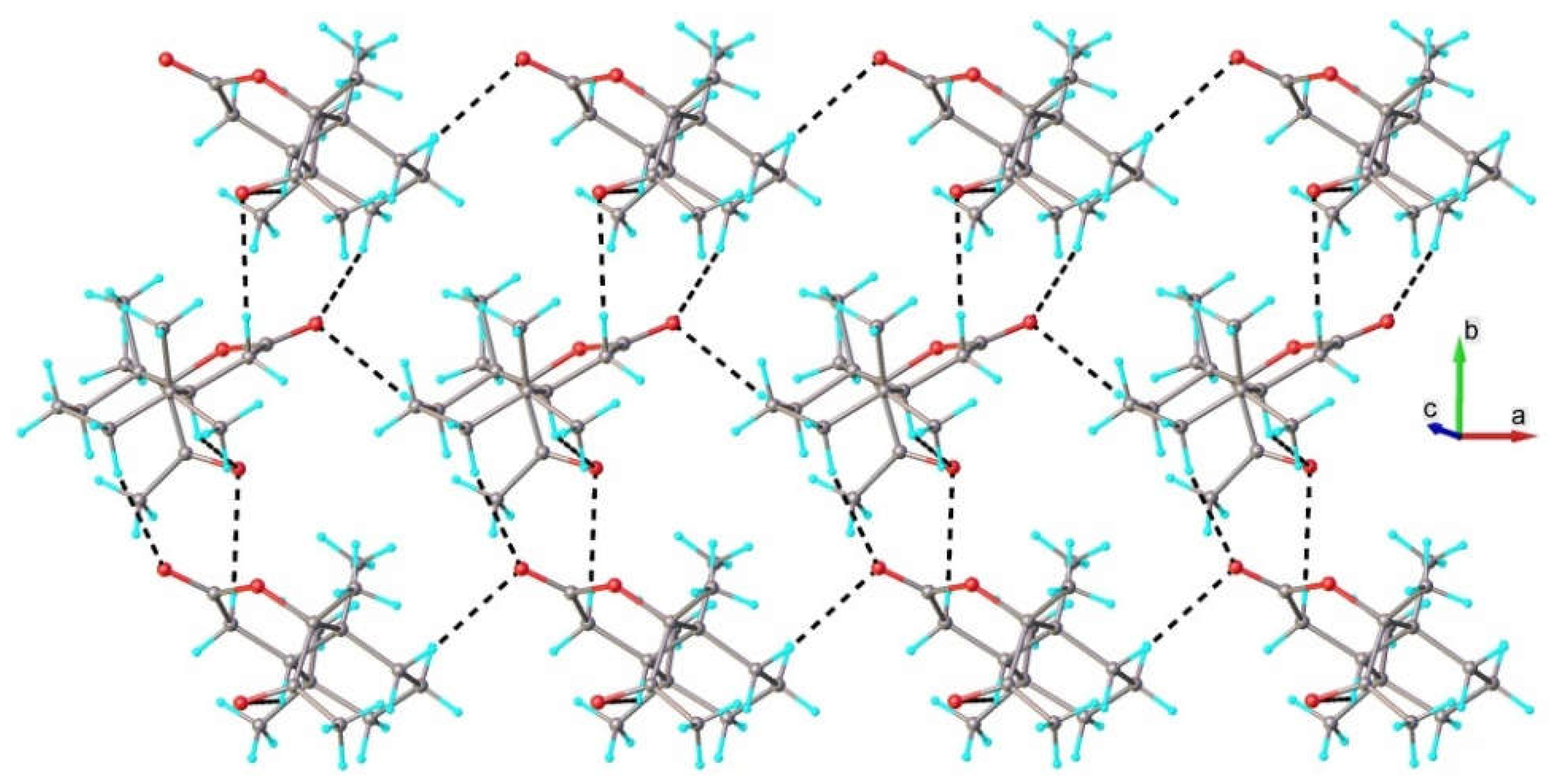
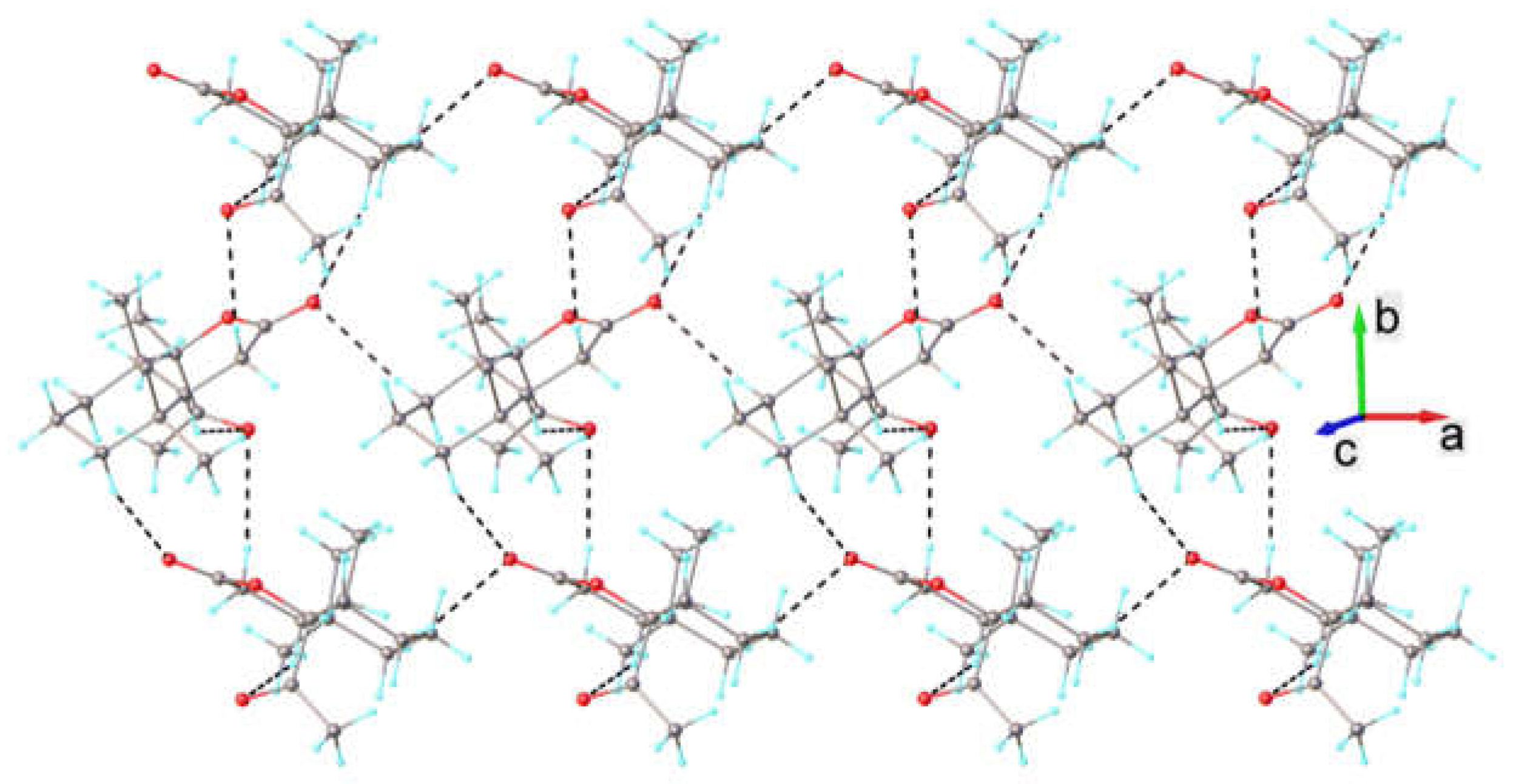
2.2. Single-Crystal X-Ray Diffraction Study
2.3. Antimicrobial Activity
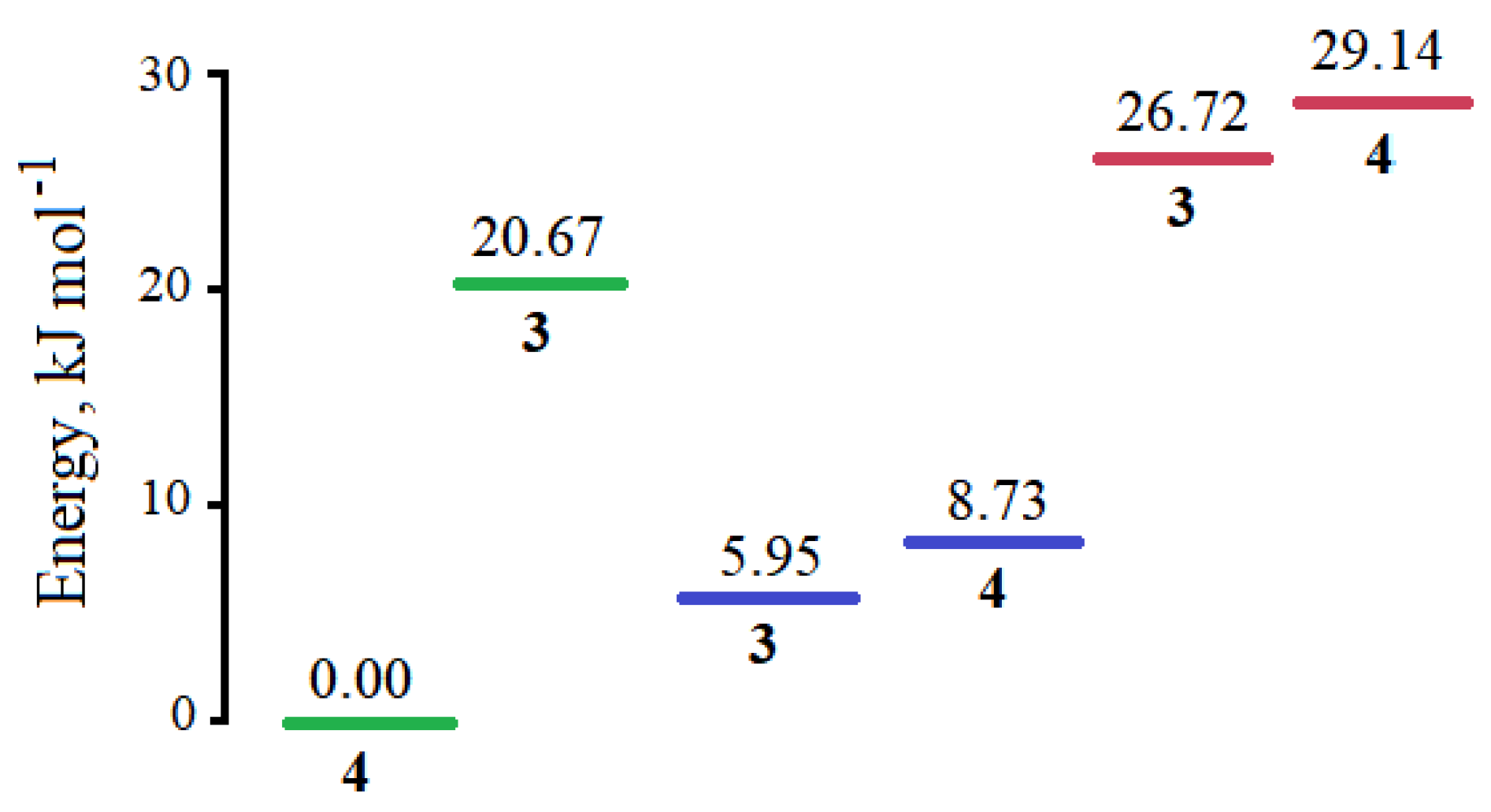
2.4. DFT Study of the Structure of Compounds 3 and 4
2.5. Molecular Docking of Compounds 3 and 4
3. Materials and Methodst
3.1. General Procedure of Baeyer-Villiger Oxidation of Ketones 1 and 2
3.2. Crystallographic Studies
3.3. Antifungal and Antibacterial Activity Assay
3.4. DFT Calculations
3.5. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fraga, B.M. Natural sesquiperpenpids. Nat. Prod. Rep. 2013, 30(9), 1226–1264. [Google Scholar] [CrossRef] [PubMed]
- Barrero, A.F., Manzaneda, E.A., Altarejos, J., Salido, S., Ramos, J.M., Simmonds, M.S.J., Blaney, W.M. Synthesis of biologically active drimanes and homodrimanes from (−)-sclareol. Tetrahedron 1995, 51(27), 7435-7450. [CrossRef]
- Koltsa, M.N., Mironov, G.N., Malinovskii, S.T., Vlad, P.F. Synthesis of drim-8(9)-en-7-one, drima-5,8(9)-dien-7-one and their 11,12-dibromo derivatives from norambreinolide. Russ. Chem. Bull. 1996, 45, 208-214. [CrossRef]
- D’Ambrosio, M., Guerriero, A., Deharo, E., Debitus, C., Munoz, V., Pietra, F. New Types of Potentially Antimalarial Agents: Epidioxy-substituted norditerpene and norsesterpenes from the marine sponge Diacarnus levii. Helv. Chim. Acta 1998, 81(5-8), 1285-1292. [CrossRef]
- Vlad, P.F., Popa, D.P., Gorincioi, E.K., Coltsa, M.N., Mironov, G.N. Synthesis of 11-hydroxydrim-8(9)-en-7-one and 11,12-dihydroxydrim-8(9)-en-7-one from drim-8(9)-en-7-one. Russ. Chem. Bull. 2000, 49(1), 98-101. [CrossRef]
- Vlad, P.F., Gorincioi, E.K., Coltsa, M.N. Synthesis of 5,6-dehydro-7-oxoisodrimenin from drim-8-en-7-one. Russ. Chem. Bull. 2003, 52(2), 502-504. [CrossRef]
- Vlad, P.F., Aryku, A.N., Ciocarlan, A.G. Synthesis of (+)-drim-9(11)-en-8-ol from sclareol. Russ. Chem. Bull. 2004, 53, 443–446. [CrossRef]
- Vlad, P.F., Coltsa, M.N., Aricu, A.N., Ciocarlan, A.G., Gorincioi, E.C., Edu, C.G., Deleanu, C. Photooxidative dehydrogenation of Δ8-drimen-and Δ8-11-homodrimen-7-ones into α,α′-dienones. Russ. Chem. Bull. 2006, 55, 703–707. [CrossRef]
- Vlad, P.F., Ciocarlan, A.G., Coltsa, M.N., Baranovsky, A.V., Khripach, N.B. Synthesis of drim-7,9(11)-diene and its hydroxylated derivatives. Synth. Commun. 2008, 38(22), 3960-3972. [CrossRef]
- Vlad, P.F., Edu, K., Koltsa, M.N., Chocyrlan, A., Nikolescu, A., Delyanu, K. Enantioselective synthesis of 11-homodrim-7-en-9α,12,13-triol. Chem. Nat. Compd. 2011, 47(4), 574-578. [CrossRef]
- Ciocarlan, A., Aricu, A., Lungu, L., Edu, C., Barba, A., Shova, S., Mangalagiu, I.I., D’Ambrosio, M., Nicolescu, A., Deleanu, C., Vornicu, N. Synthesis of novel tetranorlabdane derivatives with unprecedented carbon skeleton. Synlett 2017, 28(5), 565–571. [CrossRef]
- Aricu, A., Ciocarlan, A., Lungu, L., Shova, S., Zbancioc, Gh., Mangalagiu, I.I., D’Ambrosio, M., Vornicu, N. Synthesis, antibacterial, and antifungal activities of new drimane sesquiterpenoids with azaheterocyclic units. Med. Chem. Res. 2016, 25(10), 2316-2323. [CrossRef]
- Lungu, L., Cucicova, C., Blaja, S., Ciocarlan, A., Dragalin, I., Barba, A., Vornicu, N., Geana. E.-I., Mangalagiu, I.I., Aricu, A. Synthesis of homodrimane sesquiterpenoids earing 1,3-benzothiazole unit and their antimicrobial activity evaluation. Molecules 2022, 27(16), 5082-5094. [CrossRef]
- Lungu, L., Blaja, S., Cucicova, C., Ciocarlan, A., Barba, A., Kulcițki, V., Shova, S., Vornicu, N., Geana, E.-I., Mangalagiu, I.I., Aricu, A. Synthesis and antimicrobial activity evaluation of homodrimane sesquiterpenoids with a benzimidazole Unit. Molecules 2023, 28(3), 933–946. [CrossRef]
- Secara, E. Synthesis of new drimane and homodrimane lactams by beckmann rearrangement of some ketoximes. Chem. J. Mold. 2016, 11(1), 50–54. [Google Scholar] [CrossRef] [PubMed]
- Rigaku Oxford Diffraction. CrysAlisPro Software System, Version 1.171.41.64; Rigaku Corporation: Oxford, UK (2015).
- Sheldrick, G.M. SHELXT – Integrated space-group and crystal-structure determination. Acta Cryst. Section A 2015, 71(1), 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. Section C 2015, 71(1), 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V., Bourhis, L.J., Gildea, R.J., Howard, J.A.K., Puschmann, H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42(2), 339-341. [CrossRef]
- Russell, A.D. Antibiotic and biocide resistance in bacteria: Introduction. J. Appl. Microbiol. 2002, 92(s1), 1S–3S. [Google Scholar] [CrossRef]
- NCCLS guidelines (National Committee on Clinical Laboratory Standards (NCCLS) Antimicrobial Susceptibility Standards (ATS) 2003, for M7 (CMI) and M100).
- Neese, F., Wennmohs, F., Becker, U., Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152(22), 224108. [CrossRef] [PubMed]
- Grimme, S., Hansen, A., Brandenburg, J.G., Bannwarth, C. Dispersion-Corrected Mean-Field Electronic Structure Methods. Chem. Rev. 2016, 116(9), 5105-5154. [CrossRef]
- Forli, S., Huey, R., Pique, M.E., Sanner, M.F., Goodsell, D.S., Olson, A. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. J. Nat. Protoc. 2016, 11(5), 905-919. [CrossRef]
- Trott, O., Olson, A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem., 2010, 31(2), 455-461. [CrossRef]
- Aasen, A.J., Vogt, C.H.G., Enzel, C.R. Tobaco Chemistry. 28. Structure and synthesis of drim-8-en-7-one, a new tobacco constituent. Acta Chem. Scand. 1975, B29, 51–55.
- Vlad, P.F., Vorob’eva, E.A. Synthesis of drim-8-en-7-one. Chem. Nat. Compd. 1983, 19, 139–141. [CrossRef]
- Ciocarlan, A., Lungu, L., Blaja, S., Dragalin, I., Aricu, A. The use of some non-conventional methods in chemistry of bicyclohomofarnesenic methyl esters. Chem. J. Mold. 2020, 15(20), 69-77. [CrossRef]
- Renz, M., Meunier, B. 100 Years of Baeyer–Villiger Oxidations. Euro. J. Org. Chem. 1999, 4, 737–750.
- Koch, S.S.C., Chamberlin, A.R. Modified conditions for efficient Baeyer-Villiger oxidation with m-CPBA. Synth. Commun. 1989, 19(5-6), 829-833. [CrossRef]
- Mendelovich, M., Glotter, E. Epoxidation and Baeyer–Villiger oxidation of γ-hydroxy-α,β-unsaturated ketones on exposure to m-chloroperbenzoic acid. J. Chem. Soc., Perkin Trans. 1. 1992, 1, 1735–1740. [CrossRef]

| Sample | MIC (µg/mL) | ||||
|---|---|---|---|---|---|
| Aspergillus niger | Penicillium frequentans | Alternaria alternata | Bacillus polymyxa | Pseudomonas aeruginosa | |
| 1/2 | >128 | >128 | >128 | >128 | >128 |
| 3/4 | 0.064 (±0.035) |
0.064 (±0.035) |
0.064 (±0.035) |
2.0 (±0.060) |
2.0 (±0.060) |
| Caspofungin* | 0.38 | 0.38 | 0.38 | - | - |
| Kanamycin* | - | - | - | 2.0 | 2.0 |
| Compound | Calculated binding energy (kcal/mol) | |||
|---|---|---|---|---|
|
E. coli DNA girase, 1KZN |
P. aeruginosa FabZ, 1U1Z |
C. albicans dihidrofolate reductase, 3QLS |
E. coli MurB, 2Q85 |
|
| 3 | -6.6 | -7.7 | -6.7 | -7.1 |
| 4 | -6.2 | -7.7 | -6.4 | -6.4 |
| Caspofungin | -6.1 | -7.3 | -7.7 | -6.8 |
| Kanamycin | -6.8 | -8.0 | -7.6 | -8.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



