
Submitted:
30 October 2024
Posted:
01 November 2024
You are already at the latest version
Abstract
Antimicrobial resistance (AMR) is a growing global threat, affecting the effectiveness of antimicrobials and posing significant challenges in the treatment of bacterial and viral infections. The emergence of the coronavirus in 2019 and the ongoing menace of infectious diseases such as HIV/AIDS, tuberculosis, and hepatitis remind us of the impact infections have on economic stability and global health, and therefore, the need for a more holistic and long-term solution as opposed to antibiotics - measures that were previously relied on. This review explores recent advances in immunotherapeutic strategies, particularly cytokine-based therapies, adoptive cell therapy, monoclonal antibodies, and immune checkpoint inhibitors, in the control of AMR. It focuses on pathogens with high clinical relevance, including Staphylococcus aureus, Escherichia coli, Pseudomonas aeruginosa, and Mycobacterium tuberculosis, and viral threats such as HIV and SARS-CoV-2. New inventive approaches such as CAR T cell therapy and MAIT cells were discussed in the context of bacterial and viral infections, highlighting promising results from clinical trials and addressing the challenges of toxicity, immune evasion, and therapy resistance inherent in these diseases. Future priorities include optimizing combination therapies and exploring new immunomodulatory targets to improve these interventions' effectiveness in treating AMR and other infectious diseases.
Keywords:
Antimicrobial Resistance (AMR)
; immunotherapy
; cytokine-based therapies
; adoptive cell therapy
; monoclonal antibodies
; immune checkpoint inhibitors
Background
One of the biggest threats to public health in the twenty-first century is antimicrobial resistance (AMR), which is the result of bacterial mutations that make antibiotics less effective [1]. AMR, which originated directly after the introduction of antimicrobials, has grown in recent years, threatening the entire cornerstone of modern medicine [2]. The emergence of drug-resistant pathogens has outpaced the development of new antimicrobials, leaving us with a dwindling arsenal against bacterial and viral diseases [3]. Resistance affects efficacy; hence, the next series of strategies in combating drug resistance must be made [1]. Viral diseases like HIV, SARS-CoV-2, and other clinically significant viruses, as well as bacterial diseases like tuberculosis, gram-positive (like Staphylococcus and Streptococcus) and gram-negative bacteria (Escherichia coli, Pseudomonas aeruginosa, Actinobacter spp.), and other clinically important bacterial pathogens, are all serious concerns when dealing with drug resistance. Resistance to currently accessible antimicrobial agents has become an important public health issue of the twenty-first century, posing a threat to the effective diagnosis and treatment of an ever-expanding spectrum of diseases caused by different pathogenic microorganisms that are no longer susceptible to commonly used antimicrobials [2]. Serious preventative and control actions are needed to address the AMR issue.
People have long used antimicrobial compounds to treat infections; in ancient times, they utilized several natural extracts for their therapeutic qualities [4]. Numerous antibacterial drugs have been produced since Fleming discovered penicillin in 1929, and these have had a significant impact on global human mortality rates as well as global health [5]. Unfortunately, the abuse and overuse of these drugs have sped up the development of drug resistance, making several diseases that were once curable incurable. AMR is a worldwide threat that necessitates quick response; it is not limited to any one area [2]. Pathogens such as HIV-1 and HIV-2, the 1918 influenza virus, the Middle East respiratory disease coronavirus, and SARS-CoV-2 have repeatedly emerged in human populations from domestic or wild animal reservoirs in recent decades [6]. The vulnerability of the health care systems, particularly in underdeveloped nations, has been blatantly revealed by these re-emerging pathogens. Furthermore, the need for novel therapeutic approaches has increased due to the emergence of drug-resistant tuberculosis, gram-negative and gram-positive bacteria, HIV-1 and HIV-2, the 1918 influenza virus, the Middle East respiratory disease coronavirus, and SARS-CoV-2. As a result, developing creative approaches and procedures is important to solve the problem of growing AMR [7].
Immunotherapeutic approaches in immunocompromised patients are an excellent and innovative way to boost host defence and, consequently, an essential way to tackle the problem of growing AMR caused by several opportunistic infections [7]. Immunotherapeutic techniques have grown increasingly important in the broader context of disease prevention and control because of recent discoveries in the treatment of illnesses, including AIDS, malaria, tuberculosis, and, most recently, COVID-19 [7]. The current trend towards immunotherapies has had a big impact on the development of new tactics to fight AMR. Immunotherapy is becoming more popular for treating a variety of diseases. Immunotherapy-based treatments efficiently modulate the host's innate and adaptive immune responses, hence assisting in the management of several harmful microbial diseases [8]. In this context, immunotherapy has become a viable strategy for treating viral and bacterial illnesses [8]. Since they are so effective at treating various malignancies, antibodies that target immunological checkpoints, such as programmed death receptor-1 and cytotoxic T-lymphocyte antigen 4, have recently attracted a lot of attention [8]. A better understanding of immune system mechanisms, along with advances in immunomodulatory methods and technologies, considerably improves the prospects for creating successful immunotherapies against bacterial and viral illnesses. This review aims to explore the recent advances, challenges, and future perspectives in immunotherapeutic strategies for bacterial and viral diseases, with a focus on HIV, SARS-CoV-2, tuberculosis, Staphylococcus, Streptococcus, Escherichia coli, Pseudomonas aeruginosa, Actinobacter spp., and other pathogens of clinical importance. By analyzing the current state of the field and identifying areas of further research, we hope to shed further light on the role of immunotherapy in combating the growing threat of AMR and new infectious illnesses, as shown in Figure 1.
Cytokine-Based Therapies
Cytokines are small proteins essential for cell signalling within the immune system. These proteins play critical roles in regulating immune responses, inflammation, and haematopoiesis [9]. The primary cytokines used in these therapies include interleukins (ILs), interferons (IFNs), tumour necrosis factors (TNFs), and colony-stimulating factors (CSFs). Cytokine-based therapies represent a powerful tool in the treatment of bacterial and viral diseases. Despite challenges such as toxicity and delivery issues, recent advances and combination strategies offer promising solutions. For bacterial diseases, granulocyte-macrophage colony-stimulating factor (GM-CSF) has demonstrated effectiveness in stimulating the activation and proliferation of granulocytes and macrophages, thus boosting the immune response against bacterial pathogens. Currently, it is undergoing clinical trials for sepsis and bacterial pneumonia, with promising initial outcomes [10]. For instance, an in vivo study revealed that a newly developed albumin-fused GM-CSF exhibited improved biostability and increased dendritic cell populations, which are key to initiating a strong immune response against Mycobacterium tuberculosis (MTB) [11]. Moreover, recombinant human interleukin-2 (rhIL-2) is being tested as an adjunctive immunotherapy in patients with multidrug-resistant tuberculosis (MDR-TB) to improve treatment efficacy and shorten the treatment duration [12]. Additionally, the cytokine IL-7, which supports immune haematopoiesis, is being evaluated in several clinical trials for treating lymphopenia in sepsis patients suffering from excessive inflammation, known as a cytokine storm [13].
In viral diseases, interferon-based therapies have shown significant promise. Interferon-alpha (IFN-α) has been extensively used in treating chronic viral infections such as hepatitis B and C [14]. Recent clinical studies have demonstrated its efficacy in significantly reducing viral loads and improving liver function, thereby offering substantial therapeutic benefits. Interferon-beta (IFN-β), traditionally used in the management of multiple sclerosis, is now being investigated for its potential in treating COVID-19 [15]. Early clinical trials indicate that IFN-β may reduce viral replication and modulate inflammatory responses, providing a promising therapeutic avenue for SARS-CoV-2 infections. Similarly, Interleukin-7 (IL-7) is emerging as a potent therapeutic agent in viral infections, primarily through its ability to enhance T-cell recovery and function. Current clinical trials are evaluating its efficacy in treating HIV and post-viral fatigue syndromes, suggesting broader applications in viral immunotherapy [16].
The efficacy of cytokine therapies can be significantly enhanced through combination with other treatment modalities. For example, combining IFN-α with ribavirin has substantially improved outcomes in hepatitis C treatment, demonstrating the synergistic effects of cytokine and antiviral combinations [17]. Similarly, IFN-β combined with remdesivir is being investigated for its synergistic potential against SARS-CoV-2, presenting a promising therapeutic strategy for COVID-19 [18,19,20]. Furthermore, cytokines are paired with monoclonal antibodies or immune checkpoint inhibitors to amplify anti-tumor and anti-infective immune responses. These combinations are being examined in various clinical settings to optimize the effectiveness of immunotherapies. Interleukin-6 (IL-6) inhibitors, such as Tocilizumab, have also shown potential in managing severe bacterial infections by regulating the inflammatory response [21]. This highlights the therapeutic versatility of cytokine inhibitors in tackling complex bacterial diseases.
However, cytokine therapies face several challenges and disadvantages. High doses of cytokines can result in severe side effects, including systemic inflammation, cytokine release syndrome, and organ damage. Efficiently delivering cytokines to target tissues is also challenging due to rapid degradation and off-target effects. Additionally, prolonged use of cytokines can lead to immune tolerance or resistance, diminishing their efficacy over time [22].
Possible solutions to these challenges include the development of nanoparticle-based delivery systems to protect cytokines from degradation and enhance their targeted delivery. These advanced delivery systems have the potential to significantly improve the therapeutic efficacy of cytokine therapies. Efforts are also being made to design modified cytokines with enhanced stability and reduced toxicity. These engineered cytokines offer promising solutions to the current challenges in cytokine therapy [23]. Ongoing research aims to determine the optimal dosing and timing of cytokine administration to maximize efficacy while minimizing side effects. Optimizing these regimens is crucial for improving therapeutic outcomes. Additionally, combining cytokines with other immunomodulatory agents or therapies can enhance their efficacy and reduce the required dose, thereby mitigating side effects. This combinational approach offers a promising strategy to overcome the limitations of current cytokine therapies [24]. Continued research and clinical trials are essential to fully harnessing the potential of cytokines in infectious disease therapy.
Adoptive Cell Therapy
Adoptive cell therapy involves boosting immune cell numbers or modifying immune cell function to treat disease conditions. We achieve this by expanding autologous or allogeneic immune cell numbers and then infusing or genetically engineering immune cells to enhance their function [25]. Adoptive cell therapy, most particularly chimeric antigen receptor (CAR) T cell therapy, has gained popularity in haematological malignancies therapy, with six CAR T cell therapy FDA approvals to date [26]. The relative success of adoptive cell therapy in haematological malignancies has prompted the feasibility of adopting this strategy for chronic infectious diseases, infections due to a dysfunctional or suppressed immune system, and multidrug-resistant infections [27]. Hematopoietic stem cell transplantation is used as a treatment option for various disorders, but it comes at the cost of an immune-deficient phase where the patient is susceptible to opportunistic viral infections such as cytomegalovirus, Epstein-Barr virus, and adenovirus infections [28]. Transfusion of virus-specific T cells (VSTs) is effective in treating these infections, as garnered from around twenty completed phase I/II clinical trials and over thirty ongoing clinical trials [28,29]. VSTs are currently in clinical use against post-transplantation viral infections on a compassionate use basis; posoleucel was expected to receive FDA approval; however, it failed to satisfy the primary endpoints in a phase III clinical trial [30]. Tabelecleucel for patients with EBV-associated post-transplant lymphoproliferative disease is another VST in a phase III clinical trial (NCT03394365), which enthusiasts are hoping will clinch an FDA approval [31]. Genetically engineering VSTs with CAR to increase their lifespan and efficacy is already underway in studies targeting human immunodeficiency virus (HIV), hepatitis B virus (HBV), hepatitis C virus (HCV), and coronavirus [32]. CAR T cell strategies gained more prominence in HIV studies compared to other viruses, considering the formidable challenge of developing a cure for HIV [33]. Preclinical and clinical trials (NCT04648046 and NCT03240328) targeting viral proteins—majorly gp-120—employing CD4 and/or CD8 CAR T cells depicted significant suppression of HIV replication and destruction of HIV infected cells; however, total elimination of HIV-infected cells has not yet been achieved with this approach due to low surface HIV antigen expression on the infected cell membrane and poor CAR T cell infiltration [34,35]. Intermittent co-administration with vaccine peptides or antigen-presenting cells has been shown to sustain CAR T cell expansion and boost its immune responses, considering their poor persistence in tissues [36]. Additionally, Schreiber et al. reported the efficacy of CAR T cells transduced with HBV-specific antibody fragments in murine studies, demonstrating the plausibility of CAR T cell therapy for infectious diseases. Studies employing a genetically modified TCR to target a specific bacterial antigen are also another form of adoptive cell therapy [37]. Kalinina et al. transduced naïve T cells with a TCR targeting S. Typhimurium antigen; the T cells demonstrated a higher capacity for bacterial elimination after transfer into infected mice when compared to normal T cells [38]. Similar outcomes were observed when monocyte-derived macrophages were transferred to treat multidrug-resistant (MDR) bacterial infection in murine models [39] and when macrophage lysosomes were loaded with photosensitizers to treat MDR Staphylococcus aureus and Acinetobacter baumannii in mice [40]. CAR T cell therapy for Mycobacterium tuberculae infections is currently being evaluated, considering the increased cases of drug resistance and its chronic proclivities [27]. Adoptive T-cell therapy has been widely explored, and scientists are beginning to pay more attention to the adoptive transfer of other immune cell types as a treatment option in the last couple of years. Chung et al. showed an increase in antibody population and fall in viral load when virus-specific B cells targeting lymphocytic choriomeningitis virus were infused in mice [41]. The variety of microbial antigen and its potential for mutation, which dampens CAR efficacy, the cost of CAR T cell production, and safety concerns are some drawbacks of this strategy that are being addressed with better sequencing tools and gene editing technologies [36]. The rise of superbugs, chronic infection, and therapy-induced immunosuppression makes adoptive cell therapy a viable alternative to other less effective therapeutic strategies [26].
Tumour Infiltrating Lymphocytes
The development of immunotherapies for diseases depends on a thorough understanding of immune modifiers within the tumour immune microenvironment [42]. Tumour-infiltrating lymphocytes (TILs) are specialised mononuclear immune cells that migrate from circulation into tumours and can recognize and kill disease-causing cells [43]. Tumour-infiltrating lymphocyte (TIL) therapy involves extracting these immune cells from patients' tumours, expanding them in vitro to increase their numbers, and reinfusing them into the patient to target and destroy tumour cells [44]. TIL therapy offers significant advantages because the cells are directly sourced from the patient's body, without genetic modification. This reduces the risk of adverse reactions and enhances the specificity and efficacy of the tumor-targeting effect [45].
TILs exhibit significant heterogeneity and dynamism within the tumour microenvironment, greatly influencing cancer progression, metastasis, and the response to bacterial and viral infections [46]. Bacteria inherently stimulate the host's immune system and have been recognized as potent agents for immunotherapy [47]. Historically, efforts to treat cancer using bacteria, such as inactivated Streptococcus pyogenes and Serratia marcescens, through the production of Coley’s toxin, laid the foundation for modern cancer immunotherapy [48]. Oncolytic bacteria, such as Gram-negative Salmonella strains (e.g., S. typhimurium YS8211 and YS1629, and the non-pathogenic E. coli strain Nissle 1917), and Gram-positive bacteria (e.g., Clostridium butyricum M−55, Lactococcus lactis, and Bifidobacterium), typically accumulate at tumour sites following intravenous administration [49]. Their potential as therapeutic agents against tumours have been recognized, and genetic engineering has been employed to modify their biological behaviour [50]. Despite the demonstrated potential of many modified bacteria in immunotherapy, a systematic classification of their contributions is still lacking [51]. Specifically, TILs have been observed to enhance host immunity in mycobacterial infections, complementing antibacterial drug therapy [52].
In viral infections, TILs often interact with cytokine-based therapies, not only to eliminate the infection but also to prevent permanent damage to tumor tissues. In hepatitis and hepatocellular carcinoma, interactions between various TIL subsets and clinicopathologic parameters have been well documented. A high proportion of CD8+ TILs has been associated with noncirrhotic livers and large tumours. In contrast, low alpha-fetoprotein levels, a high percentage of CD4+ TILs, and a high CD4 to CD8 ratio have been linked to nonviral aetiology and direct-acting antiviral therapy. Additionally, a high proportion of CTLA-4 in tumour cells correlates with numerous lesions and lower tumour grades, whereas a high percentage of CTLA-4-positive TILs is often associated with high-grade hepatocellular carcinoma (HCC) [53].
As with all immune checkpoint inhibitors, selecting patients who are most likely to benefit from TIL therapy is crucial, with the greatest benefit observed in those with slowly progressing soft tissue disease [54]. Prognostic factors such as disease burden, viral load, lactate dehydrogenase serum levels, and specific infected organs can help identify patients who may benefit more from TIL therapy in bacterial and viral infections [54]. The time required to expand and harvest the T-cell population is one of the biggest obstacles to TIL therapy, often causing delays in patient intervention [55]. Thus, reducing the time between the clinical decision to treat with TILs and the product's availability for infusion is essential. One strategy is to harvest TILs early in the process, before they are urgently needed. Since tumour banking methods are not yet fully developed, TILs could be maintained as a manufacturing intermediary or as a frozen tumour sample [56]. Finally, the cost of TIL administration and production presents a significant challenge for payers and healthcare providers. As TIL therapy is typically a customised, one-time treatment strategy, the overhead costs are substantial [57].
Monoclonal Antibodies
Antibodies are naturally found existing in the human blood and cells. Another type of immunity could be observed in the case of invasive immunity, which is imposed with the use of synthetically manufactured antibodies that mimic the system of the body, otherwise known as monoclonal antibodies (mAbs). Some mAbs are used as immunotherapeutic agents that work synchronously with cells to attack foreign bodies and diseases. They can equally target and block signals that cause abnormal multiplication or division of cells, as seen in cancer cells [58].
Monoclonal antibodies are more effective than polyclonal antibodies in most immunotherapeutic remedies and show promise in managing various cancers with fewer side effects because of their high specificity, leading to innovative cancer treatment strategies [59]. This result is attributed to its affinity for targeting a specific antigen. mAbs are categorically made based on the composites used: murine, chimeric, humanised, and human [60]. Although antibodies have been used in a wide variety of disease treatments, only a few can be attributed to the treatment of viral and bacterial infections [61]. Even though mAbs have some effect when host species and receptor species differ, they are observable in the adverse excessive immune response induced occasionally when mouse mAbs are administered to humans. To address this, a chimeric antibody has been made to suppress the immunogenicity caused by using mAbs in humans [62].
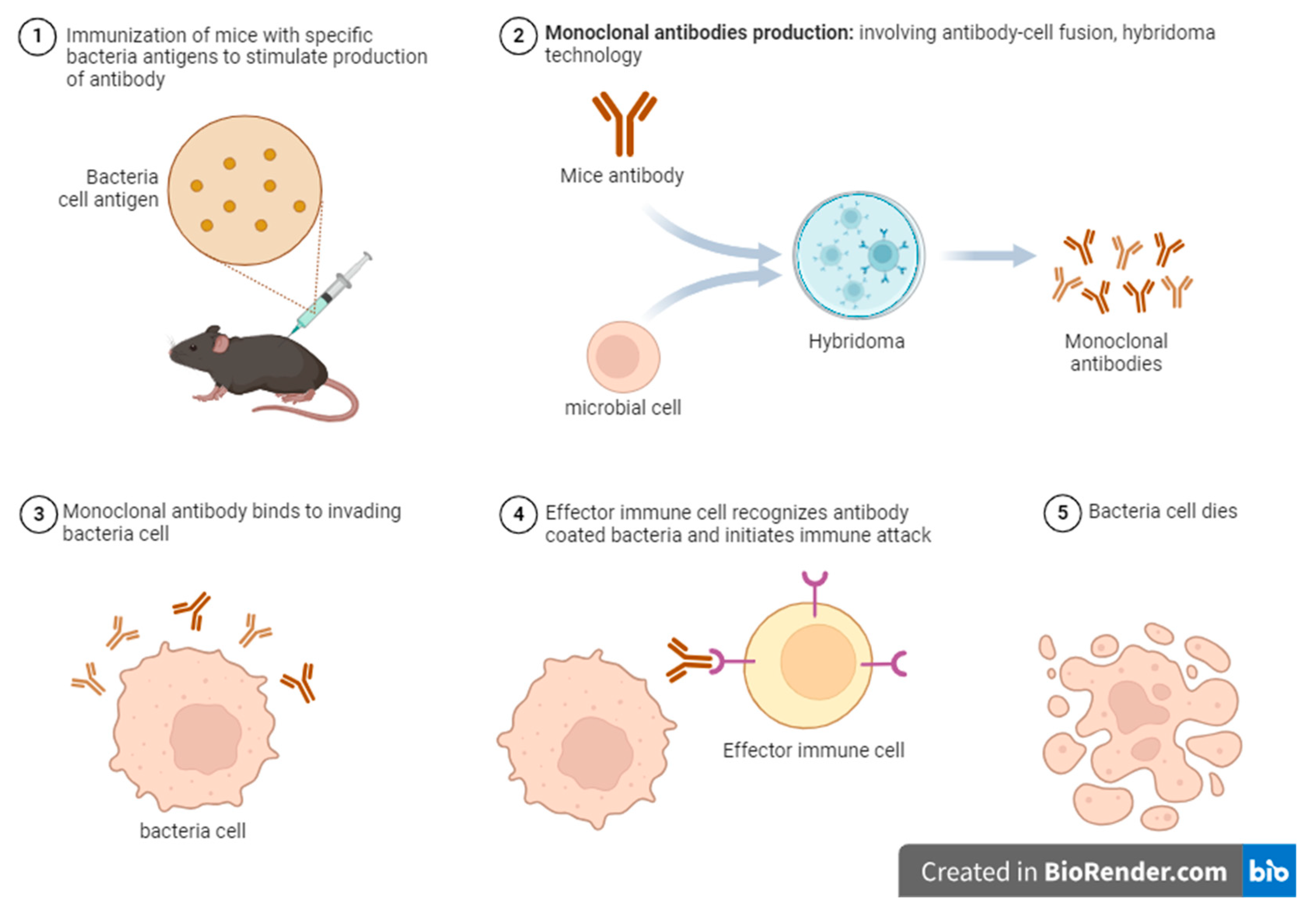
The curbing of SARS-CoV-2 entry was remedied through an immunotherapeutic mechanism identifying the interaction between angiotensin-converting enzyme (ACE) and viral spike glycoprotein, which could be blocked by antibodies targeting the spike viral domain, thus inhibiting viral infection [61]. Monoclonal antibody-based immunotherapy effectively targets tumour cells and promotes long-lasting antitumor immune responses, thereby improving cancer treatment strategies [63]. These protective effects could be employed in bacterial and viral pathogens as observed in tumour cell (see Figure 2). A previous study demonstrated that evaluating antibody-coated bacteria (ACB) in endotracheal aspirate samples significantly improves the specificity of ventilator-associated pneumonia (VAP) diagnosis by ensuring a clear distinction between viral colonisation and non-infectious conditions, thereby reducing overtreatment resulting in antibody resistance [64].
Monoclonal antibodies offer several advantages over traditional serum-derived immunoglobulin treatments, including greater specificity and potency by targeting specific epitopes, reduced risk of pathogen transmission, more consistent antibody content between batches, and an ability to engineer for extended half-life. Human antibodies with unprecedented activities could become the principal tool for managing future viral and bacterial epidemics, with potential applications in preventing and treating severe human infections [65].
Immune checkpoint inhibitors
Over the years, the use of immune checkpoint inhibitors has shown remarkable efficacy in the treatment of haematological malignancies and solid tumours. Checkpoint inhibition therapy utilises mabs to disrupt the interactions between immunosuppressive receptors and their ligands [66]. They primarily target proteins like cytotoxic T-lymphocyte-associated protein 4 (CTLA4), mucin domain-containing protein 3 (TIM3), programmed cell death 1 ligand 1 (PD-L1), etc. [67], thereby improving effector T-cell activation [66]. However, their use in the treatment of microbial diseases including human immunodeficiency virus (HIV), Hepatitis B (HBV), Hepatitis C (HCV) is poorly studied [68].
One of the unique features through which several chronic viral infections such as HIV, hepatitis, SARS-CoV-2 bypass immune responses is T cell exhaustion as a result of loss of T-cell effector function [69]. Apart from this, infectious agents also possess pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMP) [70]. Understanding the immune checkpoint molecule’s function is crucial for reversing T-cell exhaustion and mounting a strong immune response. Blockade of PD-1 has prompted that T-cell exhaustion is redeemable via restoration of CD8+ T cell function by reducing viral load in a murine model of induced chronic Lymphocytic choriomeningitis viral infection [71]. Cao et al. demonstrated that the activity of hepatitis B virus- (HBV-) specific CD8+ T cells in the peripheral and intrahepatic niche could be boosted by inhibiting the CTLA-4 checkpoint molecule in chronic hepatitis B patients [72]. Nonetheless, inhibition of PD-1 could improve host immune alertness by promoting the production of interferon-gamma in HIV and HBV-specific cytotoxic T cells [73]. However, in a preclinical study, blocking PD-1 activity in vitro or ex vivo has been shown to potentiate latent HIV reversal [74,75]. Another murine study indicated a potential adjuvant role of anti-CTLA-4 as CTLA-4 blockade during HIV immunisation in mice led to increased CD4+ T-cell activation, expansion of HIV-specific follicular helper T-cell (Tfh) cells, altered HIV-specific B-cell-responses-and significantly increased anti-HIV antibodies with higher avidity and antibody-dependent cellular cytotoxicity capabilities [76]. In addition, only three published studies of immune checkpoint inhibitors have focused on people living with HIV in the absence of malignancy and on antiretroviral therapy. Out of these, two were abruptly dismissed due to toxicities and complications in the study population [67]. These immune-related adverse effects still pose a major setback to immune checkpoint blockade clinical usage. In the case of COVID-19, several studies have suggested the likelihood of ICI therapy being beneficial [77]. Clinical trials on the safety and efficacy of ICI in COVID-19 patients are still being explored [78], with no published results to validate ICIs antiviral properties.
Among all bacterial infections of clinical importance, Mycobacterium tuberculosis remains a public health concern. Immune checkpoint inhibitors have not shown effectiveness against TB in recent times. A group of researchers using a 3D microsphere model of human TB indicated that PD-1 inhibition promotes Mtb growth and survival by enhancing cytokines production and tumour necrosis factor-alpha (TNF-alpha) [79]. In a recent case report in a child, active TB development is correlated with inherited PD-1 deficiency [80]. The emergence of TB has also been indicated in cancer patients receiving anti-PD-1/PD-L1 treatment [81,82]. To resolve this, combining immune checkpoint inhibitors with other treatment regimens could potentially curb TB pathogenesis with positive outcomes [69]. In a study, combining antibodies against LAG-3, CTLA-4, and TIGIT exhibited an additive impact in stimulating cytokine production by HIV-specific T cells. However, combinations with anti-PD-1 therapy did not have the same outcome [83].
Several limitations impact the clinical usage of ICIs, ranging from safety due to immune-related adverse events (irAEs) and immune checkpoint expression on other immune cell types such as gamma delta T-cells and Tregs, NK cells, and monocytes [67,84]. Due to this, other immune checkpoint molecules, such as A2AR, B7-H4, BTLA, KIR, NOX2, HO-1, and SIGLEC7 [85], could be extensively explored toward the treatment of acute and chronic microbial infections apart from the commonly explored immune checkpoints.
Vaccines (Adjuvants)
The effectiveness and efficacy of vaccines against viral and bacterial infections are enhanced by adding adjuvants; therefore, the use of adjuvants is important in vaccine production. They are usually added to bolster the immune responses and improve protection against diseases. To achieve this function, it is necessary to understand the different types of adjuvants and their mechanisms of action. Adjuvants are proteins or polysaccharides like tiny substances in vaccines that facilitate the elicitation of robust immune responses in inactivated vaccines and may have lesser immunogenicity than live-attenuated or whole-killed vaccines [86]. Adjuvants are divided into several classes based on their mode of action and composition. The most widely used adjuvant is the Aluminium salt, which has been used for more than seven decades. It functions by eliciting local immune responses, which enhance antigen presentation [87]. Other classes of adjuvants produced by bacteria effectively stimulate immune responses, such as lipopolysaccharides or toxins. The toxins from bacteria have been studied as effective adjuvants and can improve mucosal immune responses [88,89]. Recent adjuvants like the CpG motifs precisely target the immune cells to enhance the immune response by mimicking the bacterial DNA. MatrixM is included in the Novavax Covid-19 vaccine, and it boosts the immune system by combining lipids and saponins [87]. Another special adjuvant, MF59, used in the influenza vaccine also boosts the immunological response by using a distinctive formulation that boosts and improves antigen delivery and immune cell absorption [90].
Adjuvants work by provoking the immunological responses at the innate level, followed by influencing or steering the adaptive immune system. Antigen-presenting cells (APCs) are activated, and cytokines are produced as a result of their targeting of pattern recognition receptors (PRRs) on immune cells. This mechanism enhances vaccine antigen recognition as well as boosts a powerful and targeted, focused immune response [88,89]. Incorporation of adjuvants is vital in vaccine production, especially for certain diseases for which conventional vaccines don't offer effective and sufficient immunity. Adjuvants offer several benefits in vaccine production but must be used with caution to reduce any potential side effects.
There is current development in the development of adjuvants, which have substantially improved the prevention of infectious diseases and have led to the production of efficient vaccines that offer robust and long-lasting immune responses. For instance, TLR agonists such as monophosphoryl lipid A (MPL) are a potential class of adjuvants that activate innate immunity, thereby enhancing the immune response to viral antigens [91]. They have been used in hepatitis B and SARS-CoV-2 vaccines, substantially improving both the antibody and T-cell responses [91]. Similarly, TLR7/8 agonists such as imiquimod are promising adjuvants in HIV and influenza vaccines by evoking a sturdy cellular immune response, thereby making them efficient in producing a powerful and long-lasting immune response [91]. Additionally, cholera toxin (CT) and heat-labile enterotoxin (LT) from Escherichia coli, which are engineered bacterial enterotoxins, have developed into strong mucosal adjuvants [89]. Mutant LT (dmLT), when used in influenza, hepatitis and HIV vaccines, boosts the antibody and T-cell responses when used intranasally or orally. The influenza vaccine is effective when administered intranasally in animal models [89]. Another class of adjuvants is adjuvant systems (AS), which are a combination of both novel and traditional adjuvants and have enhanced the productivity of effective vaccines [92]. For example, the AS01, which is a combination of MPL and QS-21, has potential in the HIV and tuberculosis vaccines. AS04 is used in hepatitis B and human papillomavirus vaccines to induce long-lasting immunity, significantly improving their effectiveness [92].
Moreover, immune-stimulating complexes (ISCOMs) are cage-like structures containing saponin adjuvants that can encapsulate viral antigens and are promising in boosting the delivery and presentation of antigens to the immune system, thereby enhancing immune response and vaccine efficiency [92]. The bacteria-derived adjuvants like lipopolysaccharides (LPS) can stimulate robust immune responses. For example, monophosphoryl Lipid A (MPL), a derivative of LPS, has been utilised in various vaccines, such as hepatitis B and human papillomavirus. It can activate Toll-like receptor 4 (TLR4) to enhance the Th1 immune responses while minimising toxicity compared to full LPS [93]. Another potential bacterial component is muramyl dipeptide (MDP), which influences cytokine production, modulates the immune response, and enhances the effectiveness of vaccines against bacterial pathogens such as Mycobacterium tuberculosis [94]. A combination of adjuvants with therapeutic approaches like vaccines and antimicrobials will substantially enhance treatment efficiency by leveraging the effects of multiple agents. Likewise, they'll enhance vaccine-induced immune responses, resulting in a robust, longer-lasting immunity vital for effective viral elimination [95].
Invariant Killer T-Cells (INKT)
The cells of the immune system are conventionally cells of innate or adaptive immunity [96], although some cells are more prepared to switch between the two functions of innate immunity and adaptive immunity, and one of the best-equipped cells is the invariant killer T cells [97]. Invariant killer T cells, also called cytotoxic innate lymphoid cells (ILCs), are a subset of cells endowed with a molecular memory of surface markers [98]. An invariant αβ T cell receptor (TCR) associated with the Class I major histocompatibility complex, class I-related protein CD1d, reacts to glycolipid antigens presented to it on the surface of antigen-presenting cells [99]. They are activated in many infectious diseases and inflammatory conditions and rapidly produce large amounts of cytokines that influence other immune cells [100].
iNKTs recognize lipid-derived determinants on the cell surface, supporting its weaponization as an antitumor, viral, or bacterial therapy [101]. They are also explicitly equipped with compact exosome production that packages and expresses eomesodermin (Eomes) [102]. These eomesodermin-containing exosomes exhibit antitumor properties that can be compared to the NK cells and respond to various intracellular pathogens such as bacteria and viruses [103,104]. Exosomes found in iNKTs are also rich in cytotoxic proteins such as perforin and granzymes, as well as death receptor ligands such as FasL and TRAIL [105], which enable exosomes to induce apoptosis in cancer cells and cells infected by viral or bacterial pathogens such as Mycobacterium tuberculosis [106] and SARS-CoV-2 [107], effectively mimicking the cytotoxic and cell dissolving effects of NK cells but without requiring direct cell-to-cell contact [108].
Advantageously, the use of NK cell-derived exosomes in immunotherapy also helps generate a broader network of immune responses that can penetrate tissue and dissipate deep tumour cells or tissue-invading pathogens that have systemic effects [109,110], so that more giant cells cannot get there without the risk of an autoimmune reaction [111]. This particular property is a revolutionary point for the use of iNKT in immunotherapy and allows pre-administration, re-administration, and re-dosing of the cellular components in clinical trials until therapeutic interventions are achieved [112].
Previously, effector cells that mediate antitumor, viral, or bacterial therapy immunity were αβ T cells and invariant natural killer T cells [113]. Recently, it has been shown that γδ T cells are the complementary element by which tumour cells are rejected and are adapted to defend against the invasion of highly pathogenic organisms into cells and tissue systems [114]. The limitation of iNKT cells is due to the lack of ability to enhance the ability of γδ T cells in antigen presentation, regulatory functions, and induction of antitumor responses to excessively malignant tumours, as well as in the treatment of inflammatory conditions, caused mainly by the invasion caused by pathogens [115]. The weaponization of iNKTs by molecular mechanisms that allow them to express the functions of γδ T cells by switching intracellularly and producing compact cytokines that will enable them to express the functions of both iNKTs and γδ T cells to demonstrate can potentially revolutionise their use in immunotherapy, molecular immunology, and vaccinology [116].
Mucosal-Associated Invariant T-Cells (MAIT)
Mucosal-associated invariant T (MAIT) cells are a unique subset of innate-like T cells that play a crucial role in recognizing and responding to microbial-derived products. These cells utilise a distinct antigen recognition mechanism, which is dependent on their T cell receptor (TCR) and facilitated by the interaction with MR-1, a highly conserved MHC class I-related molecule [117]. Through this TCR-dependent mechanism, MAIT cells are able to recognize and respond to a wide range of microbial-derived products, including bacteria, viruses, and fungi, making them an important component of the immune system's defence against infection [117,118]. MAIT cells can be specifically activated and stimulated by the ligand 5-OP-RU, a microbial-derived riboflavin metabolite, which serves as a potent agonist for these cells. This activation is a result of the specific recognition of 5-OP-RU by the MAIT cell receptor, leading to a robust immune response [117,118]. In addition to TCR-dependent activation, MAIT cells can also be activated through a TCR-independent mechanism by various cytokines, including interleukins (IL)-7, IL-12, IL-15, and IL-18. This alternative activation pathway expands the modes of activation and enables MAIT cells to target a broader range of cell types, further emphasizing their versatility and importance in the immune response [119]. MAIT cells possess the ability to engage with and influence a wide range of immune cell types, including B cells, neutrophils, and monocytes, demonstrating their capacity for intercellular communication and immunomodulation. This interactive capability enables MAIT cells to coordinate and fine-tune the immune response, fostering a collaborative environment among various immune cell populations [120].
The protective effects of MAIT cells against pulmonary infections have been clinically demonstrated, showing efficacy in preventing respiratory tract infections caused by diverse bacterial pathogens. This highlights the potential of MAIT cells as a therapeutic target for the development of innovative treatments aimed at enhancing immune defence against pulmonary infections [121]. MAIT cells can be activated in viral infections through an antigen-independent mechanism, which is distinct from their activation in bacterial infections. This activation is mediated by the cytokines IL-12 and IL-18, which are produced by antigen-presenting cells in response to viral pathogens. As a result, MAIT cells can orchestrate an inflammatory response even in the absence of bacterial infection, contributing to the immune response against viral infections. This highlights the versatility and importance of MAIT cells in responding to various types of pathogens. Furthermore, MAIT cells have a special talent - they can spot specific molecules (called MR1) that are found in many types of bacteria. This allows them to quickly respond to bacterial infections [122]. They do not only recognize and respond to bacterial infections but also play a crucial role in orchestrating a coordinated adaptive immune response. They achieve this by modulating the activity of various immune cells, either through direct cell-to-cell contact or by releasing signalling molecules [122].
Conclusions
Immunotherapeutic strategies offer promising alternatives for addressing the growing threat of AMR and other infectious diseases. Advances in cytokine-based therapies, adoptive cell therapy, monoclonal antibodies, and immune checkpoint inhibitors have shown significant potential in modulating immune responses and improving patient outcomes. However, challenges such as toxicity, delivery mechanisms, and immune resistance remain. By leveraging novel technologies, such as nanoparticle-based delivery systems and genetic modifications, the therapeutic efficacy of these approaches can be enhanced. Future research should focus on optimising combination therapies and exploring new immunomodulatory targets to develop more effective, durable treatments against AMR and emerging infectious diseases.
Ethical Approval and Consent to Participate
Not Applicable.
Consent for publication
Not Applicable.
Availability of Data and Materials
Not Applicable.
Competing interest
All authors declare that they have no conflicts of interest.
Funding
Not Applicable.
Authors’ contribution
All authors contributed to the conceptualization of the review. Writing of manuscript: AIA, OAA, AMI, BIO, CMI, II, PYN, EKP, EDO. All authors contributed to the manuscript revision and final approval of the manuscript draft.
Acknowledgements
We thank everyone involved for their efforts in the writing and critical review of this manuscript. The authors also thank the Biorender platform (biorender.com) for support in drawing schematics.
List of Abbreviations
| ACB | Antibody-coated bacteria |
| ACE | Angiotensin-converting enzyme |
| AMR | Antimicrobial resistance |
| CAR | Chimeric antigen receptor |
| CTLA-4 | Cytotoxic T-lymphocytic associated protein -4 |
| DAMP | Damage-associated molecular patterns |
| GM-CSF | Granulocyte Macrophage-stimulating factor |
| HCV | Hepatitis C virus |
| HIV | Human immune deficiency virus |
| ICIs | Immune checkpoint inhibitors |
| ILC | Innate lymphoid cells |
| IL | Interleukin |
| iNKT | Invariant killer T cells |
| ISCOMS | Immune-stimulating complexes |
| LPS | Lipopolysaccharide |
| Mabs | Monoclonal antibodies |
| MAIT | Mucosa-associated invariant T-cells |
| MDP | Muramyl dipeptide |
| MDR-TB | Multi-drug resistant tuberculosis |
| MPL | Monophosphoryl lipid A |
| PAMP | Pathogen-associated molecular patterns |
| PD-L1 | Programmed cell death ligand -1 |
| PRR | Pattern recognition receptors |
| TIL | Tumour infiltrating lymphocytes |
References
- Murray, C.J.L.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Aguilar, G.R.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- Haldar, J. Confronting the Rising Threat of Antimicrobial Resistance: A Global Health Imperative. ACS Infect. Dis. 2023, 10, 1–2. [Google Scholar] [CrossRef] [PubMed]
- CDC. Antimicrobial Resistance. 2024 [cited 2024 Jul 20]. Antimicrobial Resistance Threats in the United States, 2021-2022. Available from: https://www.cdc.gov/antimicrobial-resistance/data-research/threats/update-2022.
- Muteeb, G.; Rehman, T.; Shahwan, M.; Aatif, M. Origin of Antibiotics and Antibiotic Resistance, and Their Impacts on Drug Development: A Narrative Review. Pharmaceuticals 2023, 16, 1615. [Google Scholar] [CrossRef] [PubMed]
- Coico, R. Gram staining. Curr Protoc Microbiol. 2005 Oct;Appendix 3:Appendix 3C.
- Baker, R.E.; Mahmud, A.S.; Miller, I.F.; Rajeev, M.; Rasambainarivo, F.; Rice, B.L.; Takahashi, S.; Tatem, A.J.; Wagner, C.E.; Wang, L.-F.; et al. Infectious disease in an era of global change. Nat. Rev. Microbiol. 2022, 20, 193–205. [Google Scholar] [CrossRef]
- Qadri, H.; Shah, A.H.; Alkhanani, M.; Almilaibary, A.; Mir, M.A. Immunotherapies against human bacterial and fungal infectious diseases: A review. Front. Med. 2023, 10, 1135541. [Google Scholar] [CrossRef]
- Mir MA, Hamdani SS, Qadri H. Chapter 6 - Significance of immunotherapy for human bacterial diseases and antibacterial drug discovery. In: Mir MA, editor. Human Pathogenic Microbes [Internet]. Academic Press; 2022 [cited 2024 Jul 20]. p. 129–61. (Developments in Microbiology). Available from: https://www.sciencedirect.com/science/article/pii/B9780323961271000048.
- The Editors of Encyclopaedia Britannica. Cytokines. In 2024. Available from: https://www.britannica.com/science/cytokine.
- Petrina, M.; Martin, J.; Basta, S. Granulocyte macrophage colony-stimulating factor has come of age: From a vaccine adjuvant to antiviral immunotherapy. Cytokine Growth Factor Rev. 2021, 59, 101–110. [Google Scholar] [CrossRef]
- Chuang, Y.-M.; He, L.; Pinn, M.L.; Tsai, Y.-C.; Cheng, M.A.; Farmer, E.; Karakousis, P.C.; Hung, C.-F. Albumin fusion with granulocyte-macrophage colony-stimulating factor acts as an immunotherapy against chronic tuberculosis. Cell. Mol. Immunol. 2020, 18, 2393–2401. [Google Scholar] [CrossRef]
- Sheng, L.; Li, X.; Weng, F.; Wu, S.; Chen, Y.; Lou, L. Efficacy and Safety of Adjunctive Recombinant Human Interleukin-2 for Patients with Pulmonary Tuberculosis: A Meta-Analysis. J. Trop. Med. 2022, 2022, 1–18. [Google Scholar] [CrossRef]
- Karki R, Kanneganti TD. The ‘cytokine storm’: molecular mechanisms and therapeutic prospects. Trends Immunol [Internet]. 2021 Aug;42(8):681–705. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1471490621001150.
- Niloufar R. Khanna; Valerie Gerriets. Interferon [Internet]. 2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK555932/.
- Calabrese, L.H.; Lenfant, T.; Calabrese, C. Interferon therapy for COVID-19 and emerging infections: Prospects and concerns. Clevel. Clin. J. Med. 2020. [Google Scholar] [CrossRef]
- Gunst JD, Goonetilleke N, Rasmussen TA, Søgaard OS. Immunomodulation with IL-7 and IL-15 in HIV-1 infection. J Virus Erad [Internet]. 2023 Sep;9(3):100347. Available from: https://linkinghub.elsevier.com/retrieve/pii/S205566402300033X.
- Morillas RM, Masnou H. Gastroenterología y Hepatología. 2017;40(10).
- Wagoner, J.; Herring, S.; Hsiang, T.-Y.; Ianevski, A.; Biering, S.B.; Xu, S.; Hoffmann, M.; Pöhlmann, S.; Gale, M.; Aittokallio, T.; et al. Combinations of Host- and Virus-Targeting Antiviral Drugs Confer Synergistic Suppression of SARS-CoV-2. Microbiol. Spectr. 2022, 10, e0333122. [Google Scholar] [CrossRef]
- Choi, M.H.; Wan, E.Y.F.; Wong, I.C.K.; Chan, E.W.Y.; Chu, W.M.; Tam, A.R.; Yuen, K.Y.; Hung, I.F.N. Comparative effectiveness of combination therapy with nirmatrelvir–ritonavir and remdesivir versus monotherapy with remdesivir or nirmatrelvir–ritonavir in patients hospitalised with COVID-19: a target trial emulation study. Lancet Infect. Dis. 2024. [Google Scholar] [CrossRef] [PubMed]
- Bojkova D, Stack R, Rothenburger T, Kandler JD, Ciesek S, Wass MN, et al. Synergism of interferon-beta with antiviral drugs against SARS-CoV-2 variants. J Infect [Internet]. 2022 Nov;85(5):573–607. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0163445322004558.
- Aliyu, M.; Zohora, F.T.; Anka, A.U.; Ali, K.; Maleknia, S.; Saffarioun, M.; Azizi, G. Interleukin-6 cytokine: An overview of the immune regulation, immune dysregulation, and therapeutic approach. Int. Immunopharmacol. 2022, 111, 109130. [Google Scholar] [CrossRef] [PubMed]
- Nicholas P McAndrew, MD, MSCE, Noelle Frey, MD, MSCE and David C Fajgenbaum, MD Ms. Cytokine Release Syndrome [Internet]. 2023. Available from: https://emedicine.medscape.com/article/2500111-overview.
- Deckers, J.; Anbergen, T.; Hokke, A.M.; de Dreu, A.; Schrijver, D.P.; de Bruin, K.; Toner, Y.C.; Beldman, T.J.; Spangler, J.B.; de Greef, T.F.A.; et al. Engineering cytokine therapeutics. Nat. Rev. Bioeng. 2023, 1, 286–303. [Google Scholar] [CrossRef]
- Pires, I.S.; Hammond, P.T.; Irvine, D.J. Engineering Strategies for Immunomodulatory Cytokine Therapies: Challenges and Clinical Progress. Adv. Ther. 2021, 4, 2100035. [Google Scholar] [CrossRef]
- Walti, C.S.; Stuehler, C.; Palianina, D.; Khanna, N. Immunocompromised host section: Adoptive T-cell therapy for dsDNA viruses in allogeneic hematopoietic cell transplant recipients. Curr. Opin. Infect. Dis. 2022, 35, 302–311. [Google Scholar] [CrossRef]
- Karsten, H.; Matrisch, L.; Cichutek, S.; Fiedler, W.; Alsdorf, W.; Block, A. Broadening the horizon: potential applications of CAR-T cells beyond current indications. Front. Immunol. 2023, 14, 1285406. [Google Scholar] [CrossRef]
- Morte-Romea, E.; Pesini, C.; Pellejero-Sagastizábal, G.; Letona-Giménez, S.; Martínez-Lostao, L.; Aranda, S.L.; Toyas, C.; Redrado, S.; Dolader-Ballesteros, E.; Arias, M.; et al. CAR Immunotherapy for the treatment of infectious diseases: a systematic review. Front. Immunol. 2024, 15, 1289303. [Google Scholar] [CrossRef]
- Motta, C.M.; Keller, M.D.; Bollard, C.M. Applications of virus-specific T cell therapies post-BMT. Semin. Hematol. 2023, 60, 10–19. [Google Scholar] [CrossRef]
- Keller, M.D.; Hanley, P.J.; Chi, Y.-Y.; Aguayo-Hiraldo, P.; Dvorak, C.C.; Verneris, M.R.; Kohn, D.B.; Pai, S.-Y.; Saldaña, B.J.D.; Hanisch, B.; et al. Antiviral cellular therapy for enhancing T-cell reconstitution before or after hematopoietic stem cell transplantation (ACES): a two-arm, open label phase II interventional trial of pediatric patients with risk factor assessment. Nat. Commun. 2024, 15, 1–14. [Google Scholar] [CrossRef]
- Pfeiffer, T.; Tzannou, I.; Wu, M.; Ramos, C.; Sasa, G.; Martinez, C.; Lulla, P.; Krance, R.A.; Scherer, L.; Ruderfer, D.; et al. Posoleucel, an Allogeneic, Off-the-Shelf Multivirus-Specific T-Cell Therapy, for the Treatment of Refractory Viral Infections in the Post-HCT Setting. Clin. Cancer Res. 2023, 29, 324–330. [Google Scholar] [CrossRef]
- Mahadeo, K.M.; Baiocchi, R.; Beitinjaneh, A.; Chaganti, S.; Choquet, S.; Dierickx, D.; Dinavahi, R.; Duan, X.; Gamelin, L.; Ghobadi, A.; et al. Tabelecleucel for allogeneic haematopoietic stem-cell or solid organ transplant recipients with Epstein–Barr virus-positive post-transplant lymphoproliferative disease after failure of rituximab or rituximab and chemotherapy (ALLELE): a phase 3, multicentre, open-label trial. Lancet Oncol. 2024, 25, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Seif, M.; Einsele, H.; Löffler, J. CAR T Cells Beyond Cancer: Hope for Immunomodulatory Therapy of Infectious Diseases. Front. Immunol. 2019, 10, 2711. [Google Scholar] [CrossRef] [PubMed]
- Rothemejer, F.H.; Lauritsen, N.P.; Søgaard, O.S.; Tolstrup, M. Strategies for enhancing CAR T cell expansion and persistence in HIV infection. Front. Immunol. 2023, 14, 1253395. [Google Scholar] [CrossRef]
- Mazzi, M.T.; Hajdu, K.L.; Ribeiro, P.R.; Bonamino, M.H. CAR-T cells leave the comfort zone: current and future applications beyond cancer. Immunother. Adv. 2020, 1, ltaa006. [Google Scholar] [CrossRef] [PubMed]
- Zmievskaya, E.; Valiullina, A.; Ganeeva, I.; Petukhov, A.; Rizvanov, A.; Bulatov, E. Application of CAR-T Cell Therapy beyond Oncology: Autoimmune Diseases and Viral Infections. Biomedicines 2021, 9, 59. [Google Scholar] [CrossRef]
- Maldini, C.R.; Ellis, G.I.; Riley, J.L. CAR T cells for infection, autoimmunity and allotransplantation. Nat. Rev. Immunol. 2018, 18, 605–616. [Google Scholar] [CrossRef]
- Schreiber, S.; Dressler, L.S.; Loffredo-Verde, E.; Asen, T.; Färber, S.; Wang, W.; Groll, T.; Chakraborty, A.; Kolbe, F.; Kreer, C.; et al. CARs derived from broadly neutralizing, human monoclonal antibodies identified by single B cell sorting target hepatitis B virus-positive cells. Front. Immunol. 2024, 15, 1340619. [Google Scholar] [CrossRef]
- Kalinina, A.A.; Nesterenko, L.N.; Bruter, A.V.; Balunets, D.V.; Chudakov, D.M.; Izraelson, M.; Britanova, O.V.; Khromykh, L.M.; Kazansky, D.B. Adoptive Immunotherapy Based on Chain-Centric TCRs in Treatment of Infectious Diseases. iScience 2020, 23, 101854. [Google Scholar] [CrossRef]
- Tacke, R.; Sun, J.; Uchiyama, S.; Polovina, A.; Nguyen, D.G.; Nizet, V. Protection Against Lethal Multidrug-Resistant Bacterial Infections Using Macrophage Cell Therapy. Infect. Microbes Dis. 2019, 1, 61–69. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, A.; Cheng, W.; Li, Y.; Li, D.; Wang, L.; Zhang, X.; Xiao, Y. Adoptive macrophage directed photodynamic therapy of multidrug-resistant bacterial infection. Nat. Commun. 2023, 14, 1–16. [Google Scholar] [CrossRef]
- Chung, Y.R.; Dangi, T.; Palacio, N.; Sanchez, S.; Penaloza-MacMaster, P. Adoptive B cell therapy for chronic viral infection. Front. Immunol. 2022, 13, 908707. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, Z.; Yang, Y.; Cui, J.; Sun, J.; Liu, Y. The prognostic and biology of tumour-infiltrating lymphocytes in the immunotherapy of cancer. Br. J. Cancer 2023, 129, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Zhang L, Ding J, Li HY, Wang ZH, Wu J. Immunotherapy for advanced hepatocellular carcinoma, where are we?. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer. 2020 Dec 1;1874(2):188441.
- Yeo, D.; Giardina, C.; Saxena, P.; Rasko, J.E. The next wave of cellular immunotherapies in pancreatic cancer. Mol. Ther. - Oncolytics 2022, 24, 561–576. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Du, L.; Li, H.; Zhu, X.; Cui, L.; Li, X. Tumor-infiltrating lymphocytes: Warriors fight against tumors powerfully. Biomed. Pharmacother. 2020, 132, 110873. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Li, F.; Zhang, Y.; Yu, X.; Li, J. Metabolic diversity of tumor-infiltrating T cells as target for anti-immune therapeutics. Cancer Immunol. Immunother. 2023, 72, 3453–3460. [Google Scholar] [CrossRef]
- Fan, J.-X.; Niu, M.-T.; Qin, Y.-T.; Sun, Y.-X.; Zhang, X.-Z. Progress of engineered bacteria for tumor therapy. Adv. Drug Deliv. Rev. 2022, 185, 114296. [Google Scholar] [CrossRef]
- Xu, W.; Ren, D.; Yu, Z.; Hou, J.; Huang, F.; Gan, T.; Ji, P.; Zhang, C.; Ma, L.; Hu, Y. Bacteria-mediated tumor immunotherapy via photothermally-programmed PD1 expression. Nanoscale Adv. 2022, 4, 1577–1586. [Google Scholar] [CrossRef]
- Jia, X.; Guo, J.; Guo, S.; Zhao, T.; Liu, X.; Cheng, C.; Wang, L.; Zhang, B.; Meng, C.; Jia, H.; et al. Antitumor effects and mechanisms of CpG ODN combined with attenuated Salmonella-delivered siRNAs against PD-1. Int. Immunopharmacol. 2020, 90, 107052. [Google Scholar] [CrossRef]
- Sieow, B.F.-L.; Wun, K.S.; Yong, W.P.; Hwang, I.Y.; Chang, M.W. Tweak to Treat: Reprograming Bacteria for Cancer Treatment. Trends Cancer 2020, 7, 447–464. [Google Scholar] [CrossRef]
- Chen, H.; Zhu, Y.; Zhang, C.; Hu, L.; Yang, K. Engineered bacteria in tumor immunotherapy. Cancer Lett. 2024, 589, 216817. [Google Scholar] [CrossRef]
- Young, C.; Walzl, G.; Du Plessis, N. Therapeutic host-directed strategies to improve outcome in tuberculosis. Mucosal Immunol. 2020, 13, 190–204. [Google Scholar] [CrossRef] [PubMed]
- El-Rebey, H.S.; Abdou, A.G.; Sultan, M.M.; Ibrahim, S.H.M.; Holah, N.S. The Profile and Role of Tumor-infiltrating Lymphocytes in Hepatocellular Carcinoma: An Immunohistochemical Study. Appl. Immunohistochem. Mol. Morphol. 2020, 29, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Michielin, O.; Atkins, M.B.; Koon, H.B.; Dummer, R.; Ascierto, P.A. Evolving impact of long-term survival results on metastatic melanoma treatment. J. Immunother. Cancer 2020, 8, e000948. [Google Scholar] [CrossRef] [PubMed]
- Zippel, D.; Friedman-Eldar, O.; Rayman, S.; Hazzan, D.; Nissan, A.; Schtrechman, G.; Markel, G.; Schachter, J.; Itzhaki, O.; Besser, M.J. Tissue Harvesting for Adoptive Tumor Infiltrating Lymphocyte Therapy in Metastatic Melanoma. Anticancer. Res. 2019, 39, 4995–5001. [Google Scholar] [CrossRef] [PubMed]
- Gastman, B.; Agarwal, P.K.; Berger, A.; Boland, G.; Broderick, S.; Butterfield, L.H.; Byrd, D.; E Fecci, P.; Ferris, R.L.; Fong, Y.; et al. Defining best practices for tissue procurement in immuno-oncology clinical trials: consensus statement from the Society for Immunotherapy of Cancer Surgery Committee. J. Immunother. Cancer 2020, 8, e001583. [Google Scholar] [CrossRef]
- Khushalani, N.I.; Truong, T.-G.; Thompson, J.F. Current Challenges in Access to Melanoma Care: A Multidisciplinary Perspective. Am. Soc. Clin. Oncol. Educ. Book 2021, 41, e295–e303. [Google Scholar] [CrossRef]
- Cancer Research UK. Monoclonal antibodies (MABs). Immunotherapy 2022. https://www.cancerresearchuk.org/about-cancer/treatment/immunotherapy/types/monoclonal-antibodies#:~:text=Examples%20of%20MABS%20that%20work,and%20head%20and%20neck%20cancer.
- Iqbal, T.; Choudhary, P.; Raza, K. Monoclonal Antibodies in Cancer Therapy. Int. J. Multidiscip. Res. 2023, 5. [Google Scholar] [CrossRef]
- American Cancer Society. Monoclonal Antibodies and Their Side Effects. 2022. https://www.cancer.org/cancer/managing-cancer/treatment-types/immunotherapy/monoclonal-antibodies.html.
- Pantaleo, G.; Correia, B.; Fenwick, C.; Joo, V.S.; Perez, L. Antibodies to combat viral infections: development strategies and progress. Nat. Rev. Drug Discov. 2022, 21, 676–696. [Google Scholar] [CrossRef]
- Otsubo, R.; Yasui, T. Monoclonal antibody therapeutics for infectious diseases: Beyond normal human immunoglobulin. Pharmacol. Ther. 2022, 240, 108233–108233. [Google Scholar] [CrossRef]
- Zahavi, D.; Weiner, L. Monoclonal Antibodies in Cancer Therapy. Antibodies 2020, 9, 34. [Google Scholar] [CrossRef]
- Ranzani, O.T.; Forte, D.N.; Forte, A.C.; Mimica, I.; Forte, W.C.N. The value of antibody-coated bacteria in tracheal aspirates for the diagnosis of ventilator-associated pneumonia: a case-control study. J. Bras. de Pneumol. 2016, 42, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Crowe, J.E. Human Antibodies for Viral Infections. Annu. Rev. Immunol. 2022, 40, 349–386. [Google Scholar] [CrossRef] [PubMed]
- Qadri, H.; Shah, A.H.; Alkhanani, M.; Almilaibary, A.; Mir, M.A. Immunotherapies against human bacterial and fungal infectious diseases: A review. Front. Med. 2023, 10, 1135541. [Google Scholar] [CrossRef] [PubMed]
- Gubser, C.; Chiu, C.; Lewin, S.R.; Rasmussen, T.A. Immune checkpoint blockade in HIV. EBioMedicine 2022, 76, 103840. [Google Scholar] [CrossRef]
- Shah, N.J.; Della Pia, A.; Wu, T.; Williams, A.; Weber, M.; Sinclaire, B.; Paleoudis, E.G.; Alaoui, A.; Lev-Ari, S.; Adams, S.; et al. Clinical Outcomes of Immune Checkpoint Inhibitors in Unique Cohorts Underrepresented in Clinical Trials. Cancers 2024, 16, 2223. [Google Scholar] [CrossRef]
- Wykes, M.N.; Lewin, S.R. Immune checkpoint blockade in infectious diseases. Nat. Rev. Immunol. 2017, 18, 91–104. [Google Scholar] [CrossRef]
- Vance, R.E.; Eichberg, M.J.; Portnoy, D.A.; Raulet, D.H. Listening to each other: Infectious disease and cancer immunology. Sci. Immunol. 2017, 2. [Google Scholar] [CrossRef]
- Saeidi, A.; Zandi, K.; Cheok, Y.Y.; Saeidi, H.; Wong, W.F.; Lee, C.Y.Q.; Cheong, H.C.; Yong, Y.K.; Larsson, M.; Shankar, E.M. T-Cell Exhaustion in Chronic Infections: Reversing the State of Exhaustion and Reinvigorating Optimal Protective Immune Responses. Front. Immunol. 2018, 9, 2569. [Google Scholar] [CrossRef]
- Cao, H.; Zhang, R.; Zhang, W. CTLA-4 interferes with the HBV-specific T�cell immune response (Review). Int. J. Mol. Med. 2018, 42, 703–712. [Google Scholar] [CrossRef]
- Jubel, J.M.; Barbati, Z.R.; Burger, C.; Wirtz, D.C.; Schildberg, F.A. The Role of PD-1 in Acute and Chronic Infection. Front. Immunol. 2020, 11, 487. [Google Scholar] [CrossRef]
- Fromentin, R.; DaFonseca, S.; Costiniuk, C.T.; El-Far, M.; Procopio, F.A.; Hecht, F.M.; Hoh, R.; Deeks, S.G.; Hazuda, D.J.; Lewin, S.R.; et al. PD-1 blockade potentiates HIV latency reversal ex vivo in CD4+ T cells from ART-suppressed individuals. Nat. Commun. 2019, 10, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Van der Sluis, R.M.; Kumar, N.A.; Pascoe, R.D.; Zerbato, J.M.; Evans, V.A.; Dantanarayana, A.I.; Anderson, J.L.; Sékaly, R.P.; Fromentin, R.; Chomont, N.; et al. Combination Immune Checkpoint Blockade to Reverse HIV Latency. J. Immunol. 2020, 204, 1242–1254. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.E.; Poteet, E.C.; Liu, D.; Chen, C.; LaBranche, C.C.; Stanfield-Oakley, S.A.; Montefiori, D.C.; Ferrari, G.; Yao, Q. CTLA-4 Blockade, during HIV Virus-Like Particles Immunization, Alters HIV-Specific B-Cell Responses. Vaccines 2020, 8, 284. [Google Scholar] [CrossRef] [PubMed]
- Mellinghoff, S.C.; Vanshylla, K.; Dahlke, C.; Addo, M.M.; Cornely, O.A.; Klein, F.; Persigehl, T.; Rybniker, J.; Gruell, H.; Bröckelmann, P.J. Case Report: Clinical Management of a Patient With Metastatic Non-Small Cell Lung Cancer Newly Receiving Immune Checkpoint Inhibition During Symptomatic COVID-19. Front. Immunol. 2021, 12, 798276. [Google Scholar] [CrossRef]
- Pan, Y.; Tan, J.; Li, J.; Li, T.; Li, J.; Cao, Y.; Yang, L.; Lin, X.; Li, M.; Liang, X. Immune checkpoint inhibitors in cancer patients with COVID-19. Open Life Sci. 2023, 18, 20220641. [Google Scholar] [CrossRef]
- Tezera, L.B.; Bielecka, M.K.; Ogongo, P.; Walker, N.F.; Ellis, M.; Garay-Baquero, D.J.; Thomas, K.; Reichmann, M.T.; A Johnston, D.; Wilkinson, K.A.; et al. Anti-PD-1 immunotherapy leads to tuberculosis reactivation via dysregulation of TNF-α. eLife 2020, 9. [Google Scholar] [CrossRef]
- Ogishi, M.; Yang, R.; Aytekin, C.; Langlais, D.; Bourgey, M.; Khan, T.; Al Ali, F.; Rahman, M.; Delmonte, O.M.; Chrabieh, M.; et al. Inherited PD-1 deficiency underlies tuberculosis and autoimmunity in a child. Nat. Med. 2021, 27, 1646–1654. [Google Scholar] [CrossRef]
- Barber, D.L.; Sakai, S.; Kudchadkar, R.R.; Fling, S.P.; Day, T.A.; Vergara, J.A.; Ashkin, D.; Cheng, J.H.; Lundgren, L.M.; Raabe, V.N.; et al. Tuberculosis following PD-1 blockade for cancer immunotherapy. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Anand, K.; Sahu, G.; Burns, E.; Ensor, A.; Ensor, J.; Pingali, S.R.; Subbiah, V.; Iyer, S.P. Mycobacterial infections due to PD-1 and PD-L1 checkpoint inhibitors. ESMO Open 2020, 5, e000866. [Google Scholar] [CrossRef]
- Chiu, C.Y.; Chang, J.J.; Dantanarayana, A.I.; Solomon, A.; Evans, V.A.; Pascoe, R.; Gubser, C.; Trautman, L.; Fromentin, R.; Chomont, N.; et al. Combination Immune Checkpoint Blockade Enhances IL-2 and CD107a Production from HIV-Specific T Cells Ex Vivo in People Living with HIV on Antiretroviral Therapy. J. Immunol. 2021, 208, 54–62. [Google Scholar] [CrossRef]
- Gay, C.L.; Bosch, R.J.; McKhann, A.; Moseley, K.F.; Wimbish, C.L.; Hendrickx, S.M.; Messer, M.; Furlong, M.; Campbell, D.M.; Jennings, C.; et al. Suspected Immune-Related Adverse Events With an Anti-PD-1 Inhibitor in Otherwise Healthy People With HIV. Am. J. Ther. 2021, 87, e234–e236. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Zhan, H.; Ye, Y.; Yang, J.; Zhang, M.; Li, J.; Zhuang, Y. Current Progress and Future Perspectives of Immune Checkpoint in Cancer and Infectious Diseases. Front. Genet. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Jin, S.; Gilmartin, L.; Toth, I.; Hussein, W.M.; Stephenson, R.J. Advances in Infectious Disease Vaccine Adjuvants. Vaccines 2022, 10, 1120. [Google Scholar] [CrossRef]
- Zhao, T.; Cai, Y.; Jiang, Y.; He, X.; Wei, Y.; Yu, Y.; Tian, X. Vaccine adjuvants: mechanisms and platforms. Signal Transduct. Target. Ther. 2023, 8, 1–24. [Google Scholar] [CrossRef]
- Manriquez, G.G.G.; Tuero, I. Adjuvants: friends in vaccine formulations against infectious diseases. Hum. Vaccines Immunother. 2021, 17, 3539–3550. [Google Scholar] [CrossRef]
- Crothers, J.W.; Norton, E.B. Recent advances in enterotoxin vaccine adjuvants. Curr. Opin. Immunol. 2023, 85, 102398–102398. [Google Scholar] [CrossRef]
- Centres for Disease Control and Prevention. Vaccine safety: adjuvants and vaccines. Available from: https://www.cdc.gov/vaccinesafety/concerns/adjuvants.html.
- Kayesh, M.E.H.; Kohara, M.; Tsukiyama-Kohara, K. TLR agonists as vaccine adjuvants in the prevention of viral infections: an overview. Front. Microbiol. 2023, 14, 1249718. [Google Scholar] [CrossRef]
- Lee, S.; Nguyen, M.T. Recent Advances of Vaccine Adjuvants for Infectious Diseases. Immune Netw. 2015, 15, 51–7. [Google Scholar] [CrossRef]
- Creative Biolabs. Vaccine Analytics and Qualifications: Bacteria-derived Adjuvant. Available from: https://www.creative-biolabs.com/vaccine/bacteria-derived-adjuvant.htm.
- Creative Biogene. Bacteria-Derived Adjuvant. Available from: https://www.creative-biogene.com/products/bacteria-derived-adjuvant.html.
- Dhanda, G.; Acharya, Y.; Haldar, J. Antibiotic Adjuvants: A Versatile Approach to Combat Antibiotic Resistance. ACS Omega 2023, 8, 10757–10783. [Google Scholar] [CrossRef]
- InformedHealth.org [Internet]. Cologne, Germany: Institute for Quality and Efficiency in Health Care (IQWiG); 2006-. In brief: The innate and adaptive immune systems. [Updated 2023 Aug 14]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK279396/.
- Van Kaer, L.; Parekh, V.V.; Wu, L. Invariant natural killer T cells: bridging innate and adaptive immunity. Cell Tissue Res. 2010, 343, 43–55. [Google Scholar] [CrossRef]
- Jeong, D.; Woo, Y.D.; Chung, D.H. Invariant natural killer T cells in lung diseases. Exp. Mol. Med. 2023, 55, 1885–1894. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Van Kaer, L. Natural killer T cells in health and disease. Front. Biosci. 2011, S3, 236–251. [Google Scholar] [CrossRef]
- Al-Qahtani, A.A.; Alhamlan, F.S.; Al-Qahtani, A.A. Pro-Inflammatory and Anti-Inflammatory Interleukins in Infectious Diseases: A Comprehensive Review. Trop. Med. Infect. Dis. 2024, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Tan, Y.; Wu, J.; Ren, Z. Lipid droplets: a cellular organelle vital in cancer cells. Cell Death Discov. 2023, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Lee, J.S.; Gartlan, K.H.; Schuster, I.S.; Comerford, I.; Varelias, A.; Ullah, M.A.; Vuckovic, S.; Koyama, M.; Kuns, R.D.; et al. Eomesodermin promotes the development of type 1 regulatory T (TR1) cells. Sci. Immunol. 2017, 2, eaah7152. [Google Scholar] [CrossRef]
- Batista, I.A.; Quintas, S.T.; Melo, S.A. The Interplay of Exosomes and NK Cells in Cancer Biology. Cancers 2021, 13, 473. [Google Scholar] [CrossRef]
- Razizadeh, M.H.; Zafarani, A.; Taghavi-Farahabadi, M.; Khorramdelazad, H.; Minaeian, S.; Mahmoudi, M. Natural killer cells and their exosomes in viral infections and related therapeutic approaches: where are we? Cell Commun. Signal. 2023, 21, 1–22. [Google Scholar] [CrossRef]
- Wen, C.; Seeger, R.C.; Fabbri, M.; Wang, L.; Wayne, A.S.; Jong, A.Y. Biological roles and potential applications of immune cell-derived extracellular vesicles. J. Extracell. Vesicles 2017, 6, 1400370. [Google Scholar] [CrossRef]
- Sada-Ovalle, I., Chiba, A., Gonzales, A., Brenner, M. B. & Behar, S. M. Innate invariant NKT cells recognize Mycobacterium tuberculosis-infected macrophages, produce interferon-gamma, and kill intracellular bacteria. PLoS Pathog. 4, e1000239 (2008).
- Chaudhari, P.; Ghate, V.; Nampoothiri, M.; Lewis, S. Multifunctional role of exosomes in viral diseases: From transmission to diagnosis and therapy. Cell. Signal. 2022, 94, 110325–110325. [Google Scholar] [CrossRef]
- Fiore, P.F.; Di Pace, A.L.; Conti, L.A.; Tumino, N.; Besi, F.; Scaglione, S.; Munari, E.; Moretta, L.; Vacca, P. Different effects of NK cells and NK-derived soluble factors on cell lines derived from primary or metastatic pancreatic cancers. Cancer Immunol. Immunother. 2022, 72, 1417–1428. [Google Scholar] [CrossRef]
- Hatami, Z.; Hashemi, Z.S.; Eftekhary, M.; Amiri, A.; Karpisheh, V.; Nasrollahi, K.; Jafari, R. Natural killer cell-derived exosomes for cancer immunotherapy: innovative therapeutics art. Cancer Cell Int. 2023, 23, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Prokopeva, A.E.; Emene, C.C.; Gomzikova, M.O. Antitumor Immunity: Role of NK Cells and Extracellular Vesicles in Cancer Immunotherapy. Curr. Issues Mol. Biol. 2023, 46, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.M.; Coupland, S.E.; Aittokallio, T.; Figueiredo, C.R. Resistance to immune checkpoint therapies by tumour-induced T-cell desertification and exclusion: key mechanisms, prognostication and new therapeutic opportunities. Br. J. Cancer 2023, 129, 1212–1224. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, G.; Chai, D.; Dang, Y.; Zheng, J.; Li, H. iNKT: A new avenue for CAR-based cancer immunotherapy. Transl. Oncol. 2022, 17, 101342. [Google Scholar] [CrossRef]
- Tognarelli, E.I.; Gutiérrez-Vera, C.; Palacios, P.A.; Pasten-Ferrada, I.A.; Aguirre-Muñoz, F.; Cornejo, D.A.; González, P.A.; Carreño, L.J. Natural Killer T Cell Diversity and Immunotherapy. Cancers 2023, 15, 5737. [Google Scholar] [CrossRef]
- Hu, Y.; Hu, Q.; Li, Y.; Lu, L.; Xiang, Z.; Yin, Z.; Kabelitz, D.; Wu, Y. γδ T cells: origin and fate, subsets, diseases and immunotherapy. Signal Transduct. Target. Ther. 2023, 8, 1–38. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, Y.; Chen, H.; Zhang, J.; He, W. Novel insights based on the plasticity of γδ T cells in the tumor microenvironment. Explor. Immunol. 2022, 2, 98–132. [Google Scholar] [CrossRef]
- A Revesz, I.; Joyce, P.; Ebert, L.M.; A Prestidge, C. Effective γδ T-cell clinical therapies: current limitations and future perspectives for cancer immunotherapy. Clin. Transl. Immunol. 2024, 13, e1492. [Google Scholar] [CrossRef]
- Mak, J.Y.W.; Liu, L.; Fairlie, D.P. Chemical Modulators of Mucosal Associated Invariant T Cells. Accounts Chem. Res. 2021, 54, 3462–3475. [Google Scholar] [CrossRef]
- Hagel J, Garner L, Bilton M, Mehta H, Leng T, Hackstein C, Phalora P, Amini A, Akther H, Provine N, Edmans M, Willberg C, Klenerman P. Human MAIT Cell Activation. Methods in Moelcular Biology. 2020;2098:97–124.
- Rha, M.-S.; Han, J.W.; Kim, J.H.; Koh, J.-Y.; Park, H.J.; Kim, S.I.; Kim, M.S.; Lee, J.G.; Lee, H.W.; Lee, D.H.; et al. Human liver CD8+ MAIT cells exert TCR/MR1-independent innate-like cytotoxicity in response to IL-15. J. Hepatol. 2020, 73, 640–650. [Google Scholar] [CrossRef]
- Ioannidis, M.; Cerundolo, V.; Salio, M. The Immune Modulating Properties of Mucosal-Associated Invariant T Cells. Front. Immunol. 2020, 11, 1556. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Liu, M.; Wang, J.; Fan, H.; Yang, D.; Zhang, L.; Gu, X.; Nie, J.; Chen, Z.; Corbett, A.J.; et al. IL-17 production by tissue-resident MAIT cells is locally induced in children with pneumonia. Mucosal Immunol. 2020, 13, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Horigane-Konakai, Y.; Ishii, Y.; Sugimoto, C.; Wakao, H. Mucosal-associated invariant T cells repress group 2 innate lymphoid cells in Alternaria alternata-induced model of allergic airway inflammation. Front. Immunol. 2022, 13, 1005226. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
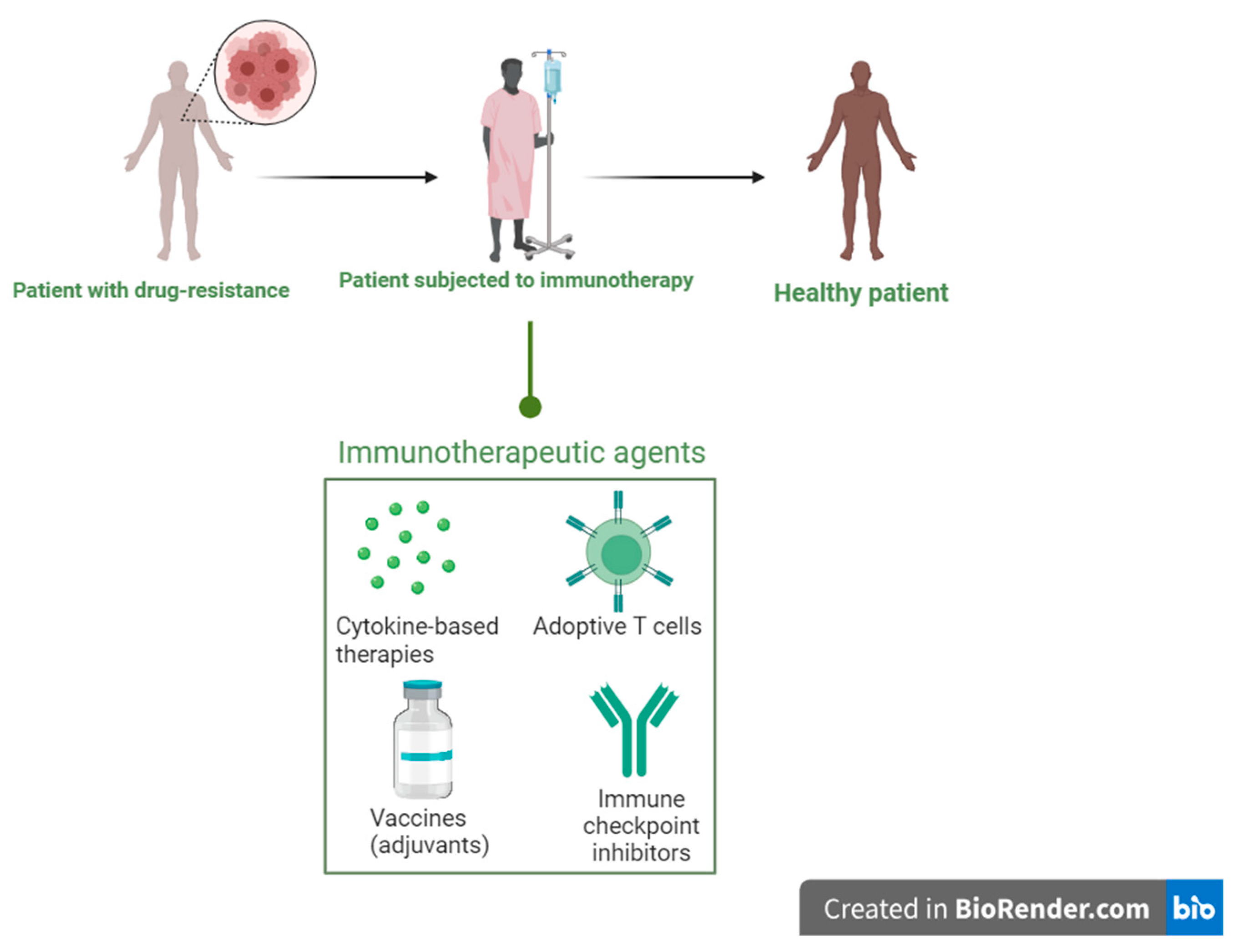
Figure 2.
Mechanism of action of monoclonal antibodies simulated on bacterial antigens using mouse infection model.
Figure 2.
Mechanism of action of monoclonal antibodies simulated on bacterial antigens using mouse infection model.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



