Submitted:
03 November 2024
Posted:
04 November 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
The Spike subunit S1 of SARS-CoV-2 was discovered to be free in our organism, during and after covid and after vaccination. One of its properties is that of interacting one-to-one with human proteins. S1 interacts one-to-one with 12 specific human proteins in the liver. We used these proteins as seeds to extract from the human proteome their functional relationships by enrichment. The interactome representing the set of metabolic activities in which they are involved shows several molecular processes (KEGG) connected with HBV (hepatitis B) and HCC (hepatocellular carcinoma). The number of genes/proteins involved is quite high. Literature data report many cases of patients with COVID where HBV was activated or progressed to cancer. Therefore, we analyzed the interactome with several approaches to understand whether the two pathologies have inde-pendent progressions or a common progression. All our efforts consistently showed that the mo-lecular processes involving both HBV and hepatocellular carcinoma are significantly present in all approaches we used, making it difficult to extract any useful information about their fate. All this has led us to consider that S1-induced molecular changes do not operate in isolation. The interac-tome shows the viral potentialities that, to be expressed or not, must interact with the specific bio-logical context of the patient's phenotype. A series of suggestions arises for using this knowledge in patient management from these considerations.
Keywords:
Spike subunit S1 of SARS-CoV-2
; covid-19
; liver pathologies
; interactomics
1. Introduction
In a recent article [1],
starting from experimental data in the literature on the liver of patients with
covid, we discriminated, through interactomic analysis and reverse engineering,
626 human proteins that interact physically and functionally with SARS-CoV-2
proteins. These interactions are specific to the liver, despite the broad
organotropism of the virus [2]. What surprised
us was that the S1 subunit of Spike can directly act with the human proteome,
through one-to-one interactions, after its uptake on the cell surface. S1 has
also been found in far organs [2,3,4] for
lengthy periods [5,6], and its interaction
with single human proteins seems organ-specific. After all, there are many
reports on the long permanence of the virus [7]
and there are also recent confirmations that even concern sperm cells [8]. The virus affects sperm count and gamete
quality even after 3 months and although the virus is not present in the
infected men’s semen; it is intracellularly present in spermatozoa. Although
this is a small study, it supports the many reports that the virus also affects
distant organs such as the reproductive system [9].
We also highlighted that the modes of interaction
between viral and human proteins are of two types [1,10].
Most times, groups of different viral proteins attack single human proteins, or
single proteins of molecular complexes. Unfortunately, the general lack of
space-temporal information, i.e., where this phenomenon occurs and with what
chronology severely limits description and identification of the molecular
mechanisms involved in these multi-to-one processes [10].
It is important to note that while the viral
proteins involved in these one-to-one interactions also take part in
multi-to-one attacks together with other viral proteins, human proteins
involved in one-to-one reactions are specifically involved only in this type of
interaction. Among the various viral proteins involved in this peculiar
interaction, we noted that in liver, the Spike S1 subunit protein interacted
specifically with 12 human proteins, but we did not investigate further.
However, this finding makes this set of viral and human proteins valuable
because the information that is drawn is specific and reflects the viral
strategy. In another our article [11],
starting from 146 proteins highly significant for physical interactions
experimentally proven with the Spike S1 subunit protein, through interactomics
and reverse engineering, we isolated 27 human proteins that interact one-to-one
with S1. These interactions are not because of a specific organ but appear to
be widespread in the human organism.
The S1 protein is peculiar because researchers have
found it free for long periods in the human organism, both during and after
covid, but also after vaccination [12,13,14,15]. The
mRNA of this protein is used to prepare vaccines, and even if modified, it
accesses the same cellular mechanisms that the SARS genome accesses after its
entry, producing the same protein with the same molecular process. Therefore,
this study focused on the autonomous activities of S1 that occur in the liver,
even because the protein appears to be involved in molecular processes related
to HBV and HCC. We have used the 12 human proteins interacting with S1 in the
liver as seed to extract their functional relationships by proteome enrichment.
While the interactome of the other 27 human proteins, also found interacting
with S1, was used to compare results. Results surprisingly showed that S1
induced the expression of many human proteins underlying the molecular
processes involving HBV and HCC. These two diseases show some molecular
processes in common. Evolutionary considerations led us to think that, although
the viral strategy induces the expression of the molecular components of these
two pathologies, it is the set of biological interactions with the individual
human phenotypes that favors, or hinders, their progression. All this also
suggests how to use these additional aspects.
2. Materials and Methods
2.1. BioGRID
BioGRID (https://thebiogrid.org/) is an important
biomedical database that collects curated protein and genetic interactions,
only from experimental studies and living cells [16].
Therefore, it represents a fundamental and unique resource for obtaining data
on certified functional interactions in biological contexts. Through a specific
Project (BioGRID COVID-19 Coronavirus Curation Project), BioGRID maintains
complete and continuous coverage of protein interaction data between human
proteins and all SARS-CoV-2 proteins. The Project is still active
(https://thebiogrid.org/project/3) and provides comprehensive datasets of
curated direct interactions for the viral proteins encoded by SARS-CoV-2. We
accessed the area SARS-CoV-2 Protein Interactions in October 2024.
BioGRID manages and integrates interaction data
from low- and high-workflow experiments through a data curation and
standardization process. This involves the analysis and validation of data from
both types of experiments to ensure the quality and reliability of the
information in the database.
2.2. STRING
STRING (Search Tool for the Retrieval of
Interacting Genes/Proteins database) (https://string-db.org/) Version 12.0 is a
database of predicted interactions for different organisms [17,18]. The interactions include direct (physical)
and indirect (functional) associations; they stem from computational
prediction, from knowledge transfer between organisms, and from interactions
aggregated from other (primary) databases. STRING is a database of known and
predicted protein-protein interactions. The interactions include direct
(physical) and indirect (functional) associations; they stem from computational
prediction, from knowledge transfer between organisms, and from interactions
aggregated from other (primary) databases. It considers conserved genomic
neighborhood, gene fusion events and co-occurrence of genes across genomes, as
well as information about orthologs. STRING quantifies the strength of the
evidence supporting each interaction by assigning it a confidence score. This
score is a combination of several sub-scores (based on seven channels of
evidence), each of which is calculated in a personalized and source-specific
way.
2.3. Protein Enrichment
It relies to some extent on prior knowledge, and
statistical enrichment of annotated features may not be an intrinsic property
of the input. To get a statistically valid enrichment test from STRING, we
input the entire set of enriched proteins into STRING, ensuring that “first
shell” and “second shell” are both set to “none”. To confirm the correctness of
the procedure, we also checked the STRING annotation, which disappears when the
analysis is performed correctly. Next, we introduce new interaction partners to
the network to expand the interaction neighborhood according to the desired
confidence score. We used 0.9 as the confidence score. We always added 1st
order proteins (direct interactions) first and then 2nd order proteins
(indirect interactions), when necessary.
2.4. Cytoscape and Network Topology Analysis
Cytoscape [19,20]
through Network Analyzer was used to analyze the topological parameters of
networks. Using Cytoscape software (Version 3.10.1), we visualized and analyzed
PPI networks, which offer diverse plugins for multiple analyses. Cytoscape
represents PPI networks as graphs with nodes illustrating proteins and edges
depicting associated inter-actions. We examined network architecture for
topological parameters such as clustering coefficient, centralization, density,
network diameter, and so on. Our analysis included undirected edges for every
network. We termed the number of connected neighbors of a node in a network as
the degree of a node. P(k) is used to describe distributing node degrees, which
counts the number of nodes with degree k where k = 0, 1, 2, …. We calculated
the power law of distribution of node degrees, which is one of the most crucial
network topological characteristics. The coefficient R-squared value (R2), also
known as the coefficient of determination, gives the proportion of variability
in the dataset. We also examined other network parameters, including the
distribution of various topological features. We performed a calculation of
high ranked nodes based on relevant topological parameters [21].
2.5. Highlighting the Nodes of a STRING Network Involved in the Same Biological Process (GO)
STRING makes visible all the nodes involved in the
same biological process evidenced through its databases mapped onto the
proteins (GO, KEGG, REACTOME, and so on) by activating the process itself with
a click of the cursor on the process line. Activation means that all nodes
involved in the same metabolic process have the same color. Nodes involved in
multiple processes receive multiple colors. This tool is very useful when one
wants to analyze involving multiple nodes in many metabolic processes, distinguishing
the effect of different processes between nodes, and identifying which nodes
represent the crossing points. If individual nodes do not show any coloration
after clicking, this identifies certain components of a path, or group, that a
specific activated process does not influence. The relationships that determine
the coloring of the nodes depend on the knowledge base that STRING organizes
for a specific network by extracting data and information from the scientific
literature in PubMed.
2.6. Enrichment Analysis
When using STRING for enrichment analysis
(Biological Process (GO), KEGG Pathways and Reactome Pathways), changing the
independent variable to either gene count or signal strength can lead to
differences because each parameter emphasizes distinct aspects of the data.
Gene Count: Using gene count as the main parameter
emphasizes the quantity of genes involved in each process. This approach often
identifies broad processes or pathways that involve many genes. Higher gene
counts can sometimes show more general biological processes or pathways, as
they involve multiple players to cover broader functional areas.
Signal Strength: signal strength highlights
processes based on the "weight" or impact of the connections within
the network, often considering the interaction confidence and association
strength. This parameter is likely to favor pathways or processes where
interactions are more robustly supported by data, even if they involve fewer
genes. Signal-based analyses can thus highlight more specialized pathways or
those with high relevance because of strong interactions. Because each approach
weights aspects of the data differently, the resulting enriched terms may vary
despite both analyses being statistically significant. This divergence often
highlights how certain biological processes can be more statistically
associated either with a higher number of contributing genes or with highly
specific, intense interactions.
The analyses were set according to the following
parameters: Similarity: > 0.9; Maximum FD shown: 0.0001; Minimum count in
the network: 3; Minimum intensity shown: 0.75; Minimum signal shown: 0.5. All
functions shown by STRING are significant, having a p-value < 1x10−27.
2.7. Data Merging
Data Merging is a process in data management, used
to coalesce multiple related datasets into one. The data-merging approach pools
all data together and then estimates statistics on the resulting dataset of GO
terms. The merging process enables the use of this combined data for more
effective analysis, particularly for extensive sets [22].
Data Merging merges disparate data sources, such as databases, or experiments
data, into a unified dataset. We have used Excel for calculations. It aids in
improving the accuracy of statistical data analysis, filling missing values in
datasets, identifying correlations between variables, and making the data
cleaning process more efficient. This procedure also presents some challenges.
These include handling large datasets, ensuring the correct alignment of merged
data, and dealing with ambiguities when datasets have similar identifiers.
These issues, if not dealt with carefully, can lead to data inconsistency or
incorrect data interpretation. We have used this approach in integrating
diverse data from various interactome analyses and data sources. The performance
depends on the size of the datasets being merged and the computational
resources available. With adequate resources, it is usually efficient and
quick, providing a unified data view in relatively little time. We have used a
storage repository that holds a vast amount of raw data in its native format
(Data Lake).
3. Results
3.1. Starting Conditions
For this study, we used both the 12 human proteins
got from our previous liver paper [1] and, as
a reference, the interactomic data of the 27 human proteins got from the recent
whole-organism S1 study [11]. These human
proteins have in common the one-to-one interaction with S1. All these
interactions are not speculative, because they are based on physical
interactions got from experimental studies, including cryo-EM structures,
co-immunoprecipitation assays and functional validation, which revealed the
full set of virus-host interactions. Curators collected these interactions in
vivo from various model cell systems and visualized them individually in
BioGRID (https://thebiogrid.org/search.php?search=SARS-CoV-2*&organism=2697049).
The S1 dataset is part of the “BioGRID COVID-19 Coronavirus Curation Project -
Severe acute breathing syndrome coronavirus 2”, which covers all viral proteins
(32 results), each organized by significance levels. The full set covers 41,683
protein-protein physical interactions (October 2024), where the S1 protein
alone shows 3,840 interactions with 2,012 specific interactors and 41 PTM
sites. These characteristics give the S1 protein an enormous capacity for
interaction within the human proteome.
The comparison (Table 1S in Supplements) shows that only 4 of them are in common between the two sets. Both the numerical difference and the few proteins in common suggest that the two sets derive from different metabolic contexts. This is characteristic of the wide variety of metabolic scale relationships that exist in a complex metabolic system such as the human organism, where each relationship depends on the metabolic context in which events occur [23]. We used this set of 12 proteins as a functional seed to extract from the human proteome the relationships in which they are involved.
The aim of this study is to find out whether S1 can reproduce the same conditions found for free S1 also in the liver, regarding the molecular components that characterize the pathological processes of HBV and HCC. The liver is the major organ where both pathologies occur. We want to find out whether both pathologies coexist with an independent development or whether there is an overlap of similar molecular components that would lead to the development of only one of them.
3.1.1. Interactome-12
Figure 1 reports the interactome resulting from the enrichment of the 12 human proteins that interact specifically with S1 in the liver.
Only ten of the twelve proteins are involved in the interactome, which from now on we will call interactome-12. We pruned the two non-interacting proteins (S100A8 and TMPRSS2). However, the relatively low number of total nodes (676) compared to an enrichment of 500 first order plus 500 second order proteins is a concrete sign that not all interactions have a robust experimental basis to ensure the reliability of their role in cellular functions [24,25]. This prompted us to increase the statistical significance of the results by operating on the enrichment parameters setting FDR: <0.0001 and strength: 0.75. This action reduces the number of insignificant and nonspecific terms in protein-protein interactions [26]. While in the last years the mention of PPIs as a topic in scientific articles has increased exponentially, the number of articles presenting real demonstrations of interactions using appropriate technologies has increased very little and only linearly [27].
3.1.2. Main Features of the Interactome-12
Excel file 1 shows node-degrees and PRKACB with 122 links is the main hub. According to Barabasi et al., [21,28,29], we can consider as the highest-ranking nodes or Hub-nodes about fifteen nodes, degrading up to 70-60 links. They are all kinases of various families. The scope of action of PRKACB is functionally very broad. Its activation regulates diverse cellular processes (cell proliferation, cell cycle, differentiation, and regulation of microtubule dynamics), chromatin condensation and decondensation, nuclear envelope disassembly, and reassembly, as well as regulation of intracellular transport mechanisms and ion flux [30]. So, we can find this protein (cAMP-dependent protein kinase catalytic subunit beta) in the cytoplasm, cell membrane, and nucleus. These are all signs of extensive metabolic activity (https://www.proteinatlas.org/ENSG00000142875-PRKACB). We can say the same about the various families of kinases that appear as high-ranking nodes of interactome-12. This interactome follows a scale free power law (figure 1S, Supplements).
3.1.3. Analysis of KEGG Terms hsa 05161-Hepatitis B and hsa 05225-Hepatocellular Carcinoma
The category expressing KEGG terms shows some of them involved in HBV and HCC. These terms are KEGG specific. However, these terms appeared also in the liver interactome [1]. HBV and HCC signals were already present in the KEGG terms of liver interactome, but the primary aim of that paper was to define the biological validity of that interactome and its topological features. In the present results, HBV (hsa05161 Hepatitis B; 90 of 158 network genes; strength: 1.22; signal: 6.91; FDR: 2.37x10-67) and HCC (hsa05225 Hepatocellular carcinoma; 54 of 161 network genes; strength: 0.99; signal: 3.69; FDR: 1.38x10-31) involve a high overall number of genes (144), when compared to the total number of genes in the interactome (676). 31 of these genes are in common and represent 57.4% of those of HCC and 34.4% of HBV. Table 2S (Supplements) shows these genes. It is difficult to make predictions on their physio-pathological trends because both terms are highly significant even if the hepatitis genes seem more many. This significant overlap between HBV and HCC-related genes suggests that both processes share molecular pathways. The asymmetry in the overlap (57.4% HCC genes common with HBV vs. 30% HBV genes common with HCC) points to the possibility that HCC may leverage molecular mechanisms initially triggered by HBV, but with additional factors specific to cancer progression. These topics are of extraordinary importance because of their intrinsic and multiple implications on the health of infected people [31,32,33,34,35,36,37] and for the long-term consequences after covid [38,39,40,41].
Several possibilities could explain these results:
- Common Pathways to Divergent Outcomes: HBV and HCC may share early molecular triggers, particularly related to inflammation, immune evasion, or cell survival [42]. However, HCC would require additional oncogenic events (mutations, dysregulated signaling) that go beyond the viral impact, resulting in its independent progression.
- Staged Evolution of Disease: It's possible that HBV creates a favorable environment for HCC development [43,44], with S1 inducing early changes that lead to hepatitis but also laying the groundwork for carcinogenesis in susceptible cells. The shared genes might represent pathways involved in liver damage, inflammation, and immune signaling that predispose cells to oncogenic transformation.
- Independent Evolution of Overlapping Pathways: Though HBV and HCC share pathways, they may develop independently once started [45,46]. HBV may follow a chronic inflammatory or immune-evasion route, while HCC could progress through mutations and other cancer-related alterations, despite initial similarity in gene expression patterns.
Both KEGG pathways involve immune modulation, apoptosis, and cell proliferation. We find genes like TP53 [47], RB1 [48], and STAT3 [49] that are crucial in both inflammation (hepatitis) and cancer progression. But we also find oncogenes and tumor suppressors like CTNNB1, GADD45A/B, and EGFR [50], which are more linked to cell proliferation, mutation repair failure, and cancer progression. But we also find genes like TLR3, IFNA1, or JAK1 [51] that are heavily involved in immune response pathways specifically for HBV. They reflect roles in antiviral response, immune system activation, or liver-specific inflammation.
Figures 2S (Supplements) show the impact of nodes related to HBV and HCC in the interactome-12. The nodes are a large and integral part of the central interactomic core. Many are involved in both pathologies. This makes any analysis very complex because of the many overlaps that generate an intricate metabolic system. The interactome-814 is shown in Figure 3S and table 3S similarly to the presentation of the interactome-12. The tables show both interactomes display overall quite similar genes. Even the genes shared between the two different interactomes are similar.
By comparing the functional results of these interactomes for HBV and HCC genes, we will probably be able to gain a clearer picture of whether both processes develop independently or whether one prevails. In conclusion, considering the genes involved in HBV and HCC of both interactomes, we see quite similar results, although with some differences. There is no typical data that can reliably suggest the possible evolution of the molecular processes involved in one or both pathologies.
3.2. Comparisons Between Enrichment Analysis Terms
To clarify these results, in Table 4S (Supplements) we show a comparison between the enrichment analyses got from the three (GO) terms and the KEGG term of each interactome. The number of genes is a macroscopic functional parameter that could identify the general trend of the two pathologies without bringing into play similar specific processes. By restricting some enrichment parameters, the analysis achieved a greater significance in the results, even if it eliminated some processes. In addition, we used the number of genes contributing to the same terms as the abscissa. To get this result, we used the new semantic analysis function of STRING (similarity > 0.8), which groups all the genes of the many processes that perform a similar functional activity. Human cellular systems often operate in a coordinated manner in the 3D cellular space, to manage similar metabolic actions in various tissues, essential for their physiological function [52].
The comparison of the three GO terms still shows similar biological, molecular and cellular actions for the two interactomes. However, this was to be expected since the human cellular systems of the various tissues perform quite similar metabolic/functional management actions. The higher total number of its nodes can explain an average difference of around 15% in the number of genes involved in the various processes in favor of the interactome-814 (814 nodes vs. 676).
The comparison between KEGG pathways highlights the strong and significant presence of tumor gene pathways that in this analysis appear both in terms of number of genes involved and in statistical terms preponderant compared to that expressed by HBV. It is important to note that the semantic-based analysis groups similar functionally genes scattered in each single term, thus highlighting the most important functions. During the reworking phase of the interactomes (pruning), and in the phase of functional analyses for categories, the operations of changing the enrichment display settings (eliminating those with lower values) decreased the number of nodes, as well as interactions, and terms in the categories, while increasing the overall significance. We therefore conclude that based on these interactomic models the processes related to HCC seem to be preponderant and probably with a greater propensity to progression compared to those of HBV. In fact, both the PI3K-AKT signaling pathway and MAPK signaling, involved in cancer progression, are significantly present. However, HBV is also based on the involvement of many genes (over 100) with an appreciable statistic (FDR < 1.0x10−69) that strongly reduces its distance from HCC but does not exclude it.
In conclusion, the functional analyses attempted so far show unclear results because of the intrinsic complexity of the interacting systems, making it difficult to discriminate between overlapping processes and components involved. Probably a way to have clear and reliable answers is not to look at the components at play in the various molecular processes, but to look at the final actions that these pathologies can determine, the various cell death that hepatitis and cancer involve.
3.3. Analysis of the Cell Death Present in the Interactomes Examined
One way to distinguish which pathophysiological process is prevalent is by evaluating the dominant modes of cell death and their implications. Cell death can occur through various pathways, each with a distinct pathophysiological significance. Each mode of cell death plays a unique role in health and disease. Understanding these mechanisms can provide insights into various pathological conditions but also help us understand the viral strategy. The molecular complexity of cell death in HCC and HBV infection is partly common (apoptosis) but often accompanied by different and well-differentiated molecular events [53,54]. The expected Type of Cell Death for HCC are Apoptosis, Necrosis, and autophagy [55,56], while for HBV infection we should expect apoptosis, necroptosis, and pyroptosis as types of cell death [57,58]. If one pathway is compromised, the pathways tightly cross-regulate apoptosis, necrosis, and pyroptosis, leading to coordinated cell death [59,60,61,62]. Sometimes, HCC cells may activate autophagy as a survival mechanism to manage stress and nutrient deprivation. Therefore, the ability of HCC to evade apoptosis and regulate autophagy can contribute to cancer development [63]. Tumors can also experience necrosis because of rapid growth outpacing blood supply, leading to ischemia and cell death. Necrosis leads to nonspecific systemic inflammation [64]. Although in cancer genetic alterations play an important role in the virus-host interaction, metabolism and immunology determine the fate of the viral action, because the viral strategy depends on the host and its phenotype [65,66]. With HBV infection, hepatocytes can undergo apoptosis, often mediated by the immune response as the body attempts to clear the virus [67]. This is the common response in acute HBV. If apoptosis is inhibited, causing inflammatory responses and liver damage, necroptosis may also occur in cases of chronic HBV infection [68]. Separately, we must consider anoikis, a crucial biological process that counteracts cancer metastasis, since it promotes apoptosis of cells that detach from the extracellular matrix [69]. Tumor cells employ mechanisms to overcome anoikis, promoting invasiveness and metastasis. These mechanisms may include cellular acidosis and stromal changes [70]. However, anoikis plays a crucial role in cancer progression, as tumor cells employ various strategies to circumvent this form of cell death and gain metastatic capability [71].
Figures 4S and 5S and Tables 5S and 6S (Supplements) show both interactomes with the proteins involved in the various programmed death implemented by tissue cellular processes following the action of S1. The analysis shows a variety of statistically significant cell deaths, although not yet attributable with certainty to a single pathology. Surprisingly, the interactome-12 shows a richer presence of terms related to cell death processes, mainly among Biological Processes (GO). Many of them have a relation to autophagy. The two networks show, in color, the genes involved, which are almost all in the central core where the main metabolic activities take place, and the speed of reactions is greatest.
These representations are still at a rather high level to draw reliable conclusions. Both HBV and HCC involve a high number of genes, and significant gene redundancy is expected. In fact, both networks show many single nodes involved in multiple cell death processes, a phenomenon that produces gene redundancy.
3.4. Data Merging
The two interactomes originate from different metabolic contexts and, as discussed, show different functional enrichments for each category. Previous analyses have shown that cell deaths essentially concentrate their effects on three categories: Biological Processes (GO), KEGG pathways, and Reactome Pathways. However, multiple and overlapping interactions within each interactome generate complex regulatory nets, where some genes can directly or indirectly influence many other biological processes. We compared these categories of the two interactomes through a Data Merging approach (details in Methods), combining the different datasets into one (see Excel file 2, sheets 1, 2, and 3). Data Merging is used to evaluate interaction parameters, add observations, and find repetitions. Therefore, the merging is the logic we used to distinguish common processes (coupled processes) from individual processes (uncoupled processes) of each interactome within examined categories. Merging optimizes the process by collecting all the data into a single set, maximizing the ability to extract and analyze critical information with thoroughness.
The Excel file 2 shows the merge among Biological Processes, KEGG Pathways, and Reactomes of the two interactomes. This file displays the terms of the tree categories in three different sheets. Data are all highly significant because of the parametric restrictions applied. The various sheets report for each category all the genes involved in the individual terms. Apoptosis is the prevalent programmed death of all cell types but accompanied by abundant phenomena of cell necrosis and autophagy, which are characteristic of cell death by cancer. Since both apoptosis and autophagy can play a dual role in cancer, either promoting or inhibiting tumor progression, understanding their co-occurrence with necrosis could reveal important insights into disease mechanisms. This approach could help clarify whether the observed processes are distinct or part of a continuum that contributes to a single pathology. Considering the interaction between these different cell deaths could provide valuable insights into how the S1 subunit affects liver cells and could guide therapeutic strategies. In both interactomes, only one KEGG term (hsa04217 Necroptosis) refers to necroptosis but with numerically limited genes that are also in common with other processes (they have multiple staining). Referring specifically to the liver, the total number of genes involved in the various cell deaths is high (963 genes), but with many redundancies. Therefore, we eliminated the redundancies, getting 220 genes that should be those specifically controlling the various cell death in liver tissue.
3.5. Genes That Control Cell Death in the Liver
The Data Merging approach allowed us to isolate 220 genes (see table 7S, Supplements) specifically involved in the control and implementation of how the liver manages the death of its cells during covid. The genes that program cancer death appear to be more many than those of HBV. However, even if with variable and different relationships, genes of both diseases always appear to be reciprocally involved.
Figure 2 shows the interactome calculated by STRING for these genes. The interactome is statistically very reliable, with a significant number of interactions, but it appears not very compact. Despite being well connected, gene sub-domains are clearly distinguished. Three nodes were not connected, so we pruned them. These general characteristics seem to further support the coexistence of the two pathological states in liver cells under the action of S1. However, we analyzed Biological Processes, KEGG Pathways, and Reactome pathways also for this network.
When using STRING for enrichment analysis, changing the independent variable to either gene count or signal strength can lead to differences because each parameter emphasizes distinct aspects of the data. Using gene count as the main parameter emphasizes the quantity of genes involved in each process. This approach often identifies broad processes or pathways that involve many genes. Signal strength highlights processes based on the "weight" of the connections within the network, considering a weighted average between the interaction confidence and association strength. This parameter is likely to favor processes where interactions are more robustly supported by data, even if they involve fewer genes. Signal-based analyses can thus highlight more specialized pathways or those with high relevance because of strong interactions. Because each approach weights aspects of the data differently, the resulting enriched terms may vary despite both analyses being statistically significant. This divergence often highlights how certain biological processes can be more statistically associated either with a higher number of contributing genes or with highly specific, intense interactions [72,73,74].
This dual approach allows for a more comprehensive understanding of the underlying biology. Table 2 shows the comparison among Biological Process (GO), KEGG Pathways, and Reactome Pathways.
Table 2. - Comparison among enriched terms of the 220 nodes interactome.
The terms being compared are: Biological Process (GO), KEGG Pathways, Reactome Pathways.
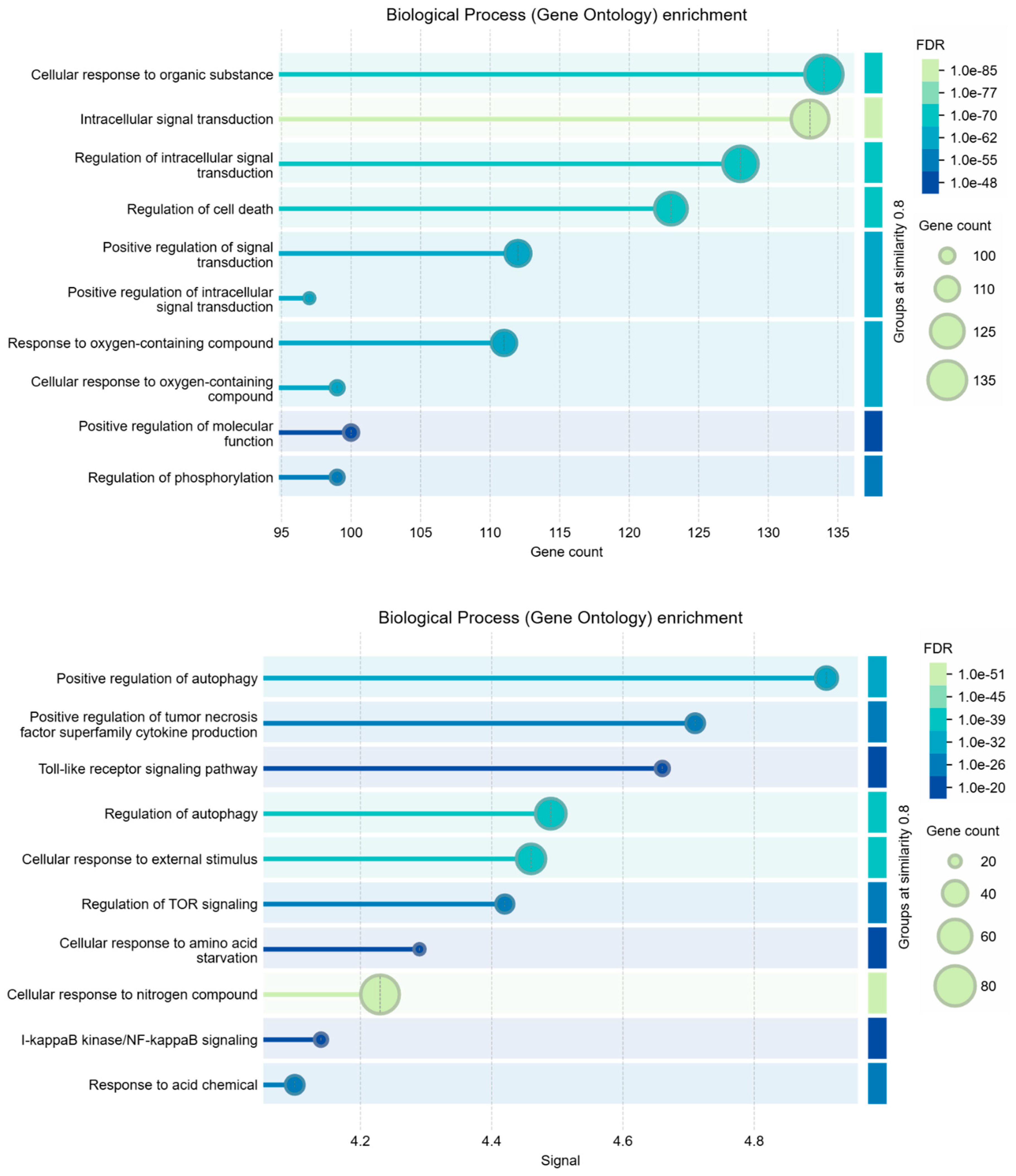
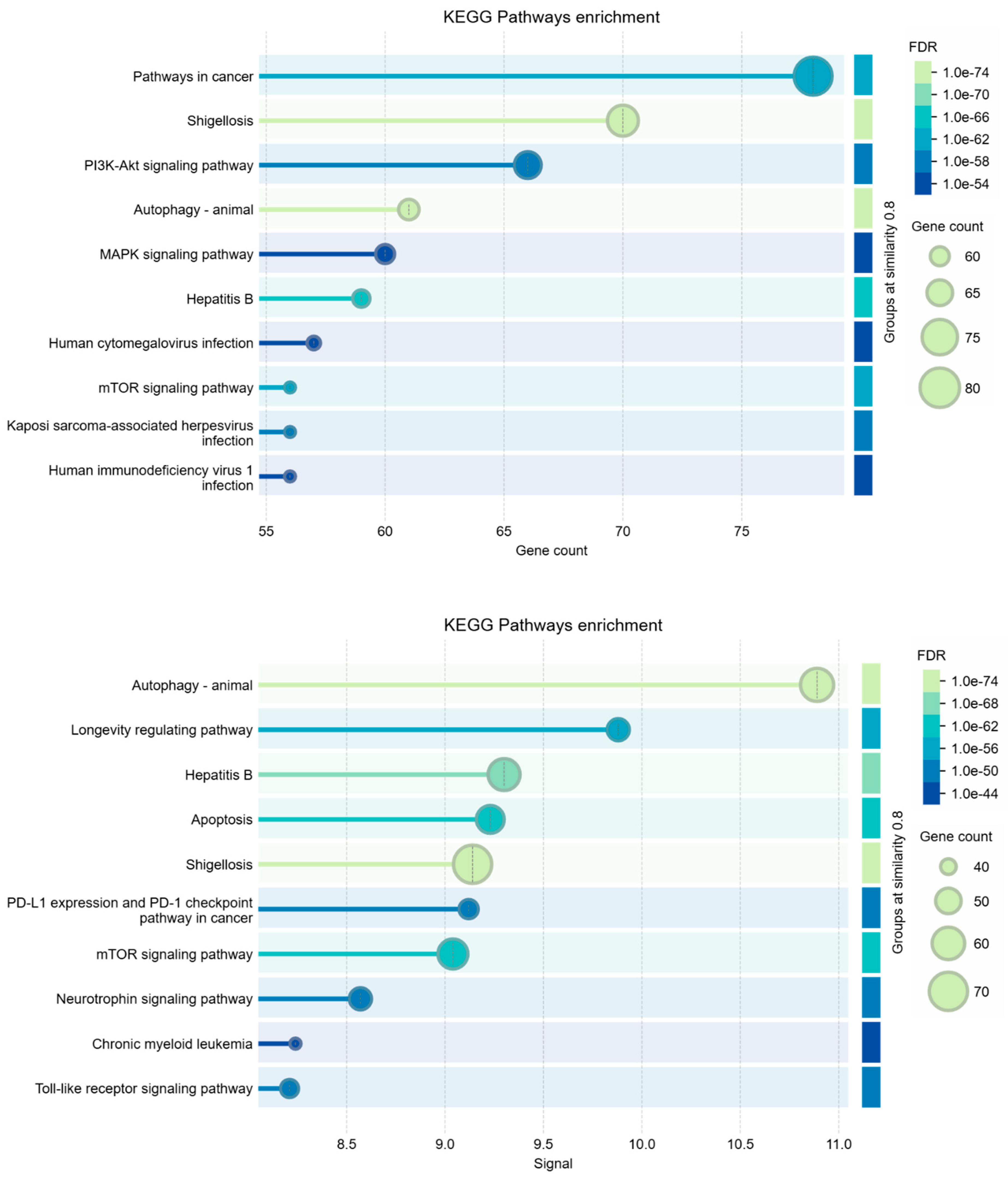
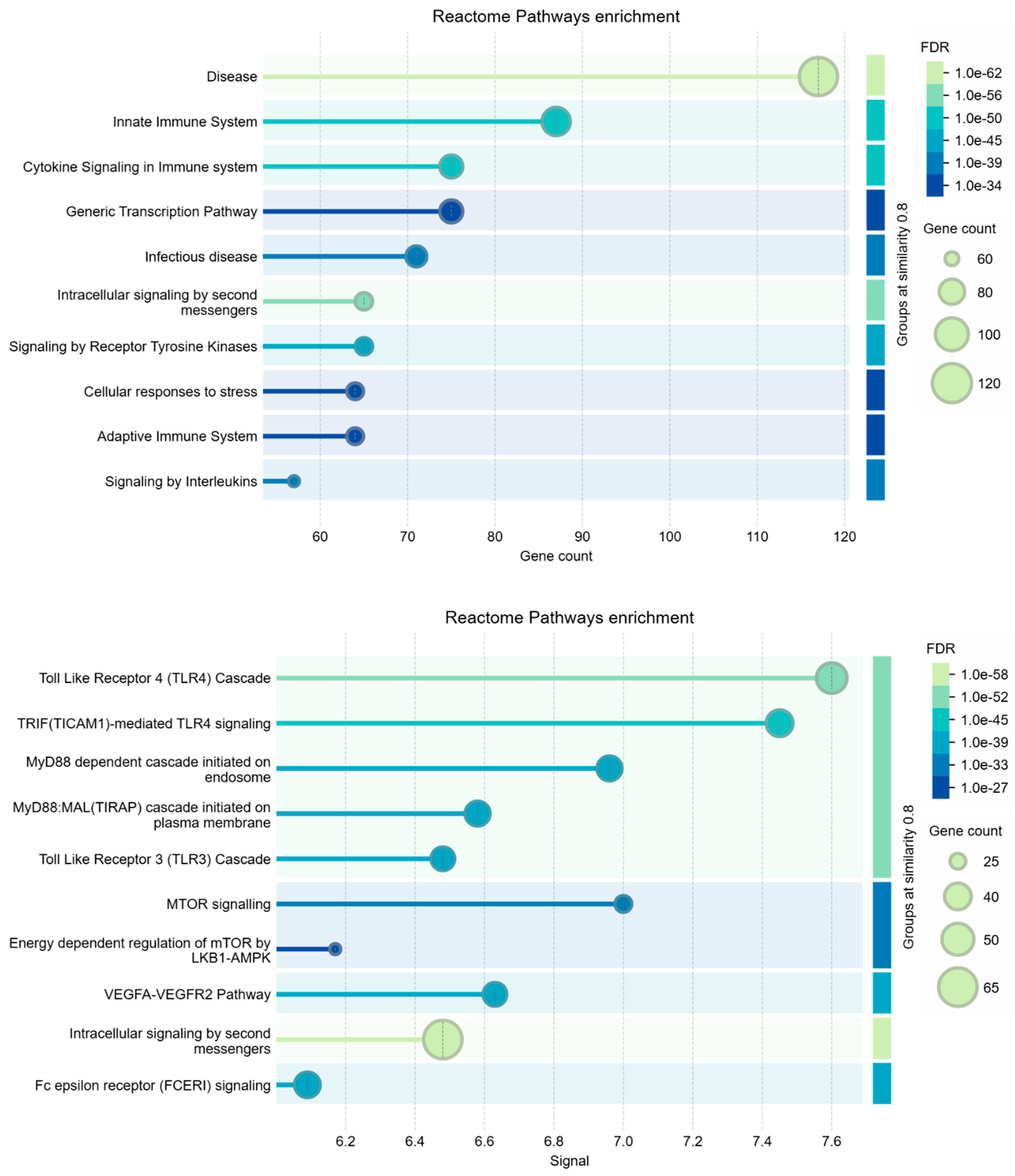
Analysis parameters: Similarity: > 0.9; Maximum FDR shown: 0.0001; Minimum count in network: 3; Minimum strength shown: 0.75; Minimum signal shown: 0.5.
The insights gained from gene count and signal strength enrichments provide a nuanced perspective, as each metric captures different facets of interactions within our datasets. We compared the enriched term lists from the analyses to identify processes that appear in both gene count-based and signal-based approaches, but also to identify overlapping processes. We found no overlap between processes, but there are processes that are broadly involved (as observed by gene count) and strongly supported by evidence for interaction. But there are also unique terms that appear in only one analysis (as observed by signal). Terms identified by gene count alone may represent core processes where many genes are loosely involved, providing broader functional support [75,76]. These may reveal more general biological environments or contexts. Terms identified by signal strength alone are more likely to show specific, potentially critical, high-confidence pathways with strong involvement [77,78,79]. They may reflect highly active or essential mechanisms. However, this integrated approach not only provides us with a clear picture of the essential processes for both pathogeneses observed in SARS-CoV-2-related liver diseases, but also confirms that these molecular components always coexist in any situation. Indeed, whatever the approach used, the genes and proteins responsible for HBV and HCC were always present, all well identifiable with high significance, even if with variable ratio depending on the analysis. All this suggests that traditional mechanistic models may not fully understand very complex interplays. This shows a need for a more integrative perspective that considers a broader biological context and is no longer linked to a mechanistic vision through the calculation of mathematical models represented by networks. Therefore, the need arises for these phenomena to be represented and understood in a much broader context of biological interactions, whose explanation should come from different approaches, as we will try to explain in the discussion.
4. Discussion
These results, taken together, show that molecular processes involving both HBV and hepatocellular carcinoma are consistently. and significantly, present in all approaches used. Many components (genes or proteins) of these pathological processes are common to both diseases, other components are specific. However, they represent most of the interactomes that we have considered. This issue is important because clinicians themselves state that although the attack of SARS2 on the liver causes non-serious symptoms, there have been many reported cases in the literature of patients who fell ill with HCC after having had covid [40,46,66]. Many hypotheses have emerged in these cases as researchers search for the causes. However, we believe it is important to frame clearly what an interactomic approach means and what information it can provide. This helps to explain the results.
Our evaluations made through interactomics are “aseptic” because we use interaction data obtained from experiments conducted in different laboratories on various cellular models (BioGRID). In these experiments, researchers extract single molecules from ground cells, establish direct physical interactions, purify them, and then characterize them. With them, we biologically validated each interaction studied. Although the BioGRID experimental data derive from both infected and uninfected model systems, they are “aseptic” references because they lack molecular data from actual patient cases. Therefore, any conclusions drawn from interactomic data are potential when applied to nonspecific clinical scenarios. While some anomalies can come from the functional data, collected with various methodologies, curated and archived in databases [80,81,82]. The certainty of the information connected to the archived interactions is the basis of protein networks and is crucial for having reliable and real results [1,83,84]. But this is not always the case, so, by implementing parametric pruning, we can manage and reduce these anomalies, resulting in a much more accurate and significant yield and a decrease in the total number of interactions. Using parametric pruning to handle data anomalies is crucial, as it ensures more reliable interaction networks and reduces noise.
It is not appropriate to compare interactomics results with macroscopic evidence such as laboratory tests or patient symptoms, since the microscopic molecular world and the meso-macroscopic world do not have linear correlation but multi-to-one, or multi-multi correlations [85,86,87,88]. This renders all the discussions that could be made between the complex intersection of viral pathogenesis, cellular mechanisms, and disease processes at least questionable [87]. If components related to both pathologies appear in the interactome, we cannot attribute them to actual cases or make hypothetical speculations. Instead, we must explain them solely at the molecular level because they detect them through similar molecular mechanisms. The System Biology approach shows that a single molecule can take part in dozens of different processes [88,89,90,91,92]. Only the numerical completeness of the components of the process and its significance identify which process that specific molecule belongs to [1,11]. All this excludes from the interactome analyses the hypotheses that a person with previous liver diseases can develop towards cancer, or vice versa, simply because we have not analyzed molecular data of patients of this type. The absence of specific molecular data from COVID patients in our analyses prevents us from discussing the relevance of this or that pathological event. The interactome merely describes, aseptically, the potentialities that virus can express in human phenotypes. However, the complexity of disease progression, especially with chronic infections like HBV, may require a more integrative approach that considers both molecular and clinical data when establishing connections. Our results show that S1 protein of SARS-CoV-2 induces microscopic conditions in the liver that may develop in various ways according to the specific physio-pathological state of each phenotype. The relationships that are drawn are correlative and not necessarily causal. Our results show the potential scenario, but the observed associations require more direct evidence to assert causality in a specific clinical scenario, such as patient genomics.
The interactions we studied derive from controlled in vivo studies in different cellular models, without direct links to specific clinical phenotypes. Model cell systems mimic an organism but are not the organism [93,94,95,96]. This reinforces the idea that molecular interactions potentially reflect mechanistic processes rather than direct clinical outcomes. The multi-to-one or multi-multi correlations between molecular mechanisms and phenotypic expressions further complicate the assumption that molecular evidence can directly explain macroscopic disease processes like the progression from hepatitis to HCC.
In short, SARS-CoV-2 S1 protein may induce microscopic conditions that develop differently based on individual phenotypic states is in line with the complexity of viral pathogenesis. This reflects the non-deterministic nature of interactome data—capturing potentialities rather than predicting phenotypic outcomes. We propose an approach to interpreting molecular data that avoids overgeneralization in linking it to clinical disease progression. We could use our molecular results as a gold standard against molecular data from specific patients, to identify whether the metabolic system of the infected patient is implementing those molecular mechanisms that drive the typical effects of a HBV infection, or even the progression to cancer. These processes should not be present in a healthy person. But they could also be a clinically useful signal of the level of severity reached by the viral infection in a patient with previous morbidity.
We can now discuss in more detail some aspects of our results through our interpretation of the interactomic approach used by exploring the different levels at which molecular variability could influence the results.
1. Phenotypic Heterogeneity in Liver Cells. Even without pre-existing liver diseases, subtle variations in liver cell signaling pathways or receptor expression could lead to diverse responses to the S1 protein. For instance, different tissues and even liver cell types variably express ACE2, a key receptor involved in SARS-CoV-2 entry [97]. If liver cells from two individuals express ACE2 at differing levels, the downstream signaling pathways activated by S1 binding will differ, leading to heterogeneity in the subsequent cellular response (e.g., stress, apoptosis, or immune modulation).
2. S1's Influence on Liver-Specific Signaling Pathways. The S1 protein might activate or inhibit liver-specific pathways [98]. For example: 1) JAK/STAT Pathway: This pathway plays a role in inflammation and cell growth. If S1 interacts with this pathway, it could lead to either a protective immune response or a maladaptive one, depending on how the individual's cellular environment modulates these signals. 2) MAPK Pathway: MAPK is crucial in regulating cell differentiation, proliferation, and stress responses. Variability in how liver cells manage oxidative stress, especially in the presence of external stressors (e.g., same S1 protein), could lead to apoptosis in some cells or survival in others, thus influencing outcomes like fibrosis or the progression toward HCC.
3. Metabolic Variations. SARS-CoV-2 influences host metabolism [99] as well factors highly individualized influence liver metabolism, such as diet, alcohol consumption, and metabolic disorders as non-alcoholic fatty liver disease [100]. If S1 protein influences metabolic pathways (e.g., by inducing oxidative stress or mitochondrial dysfunction), these pathways may respond differently depending on the metabolic state of the liver cells. Therefore, a person with a predisposition to oxidative stress may experience more severe mitochondrial dysfunction, leading to increased cell damage or a pro-fibrotic environment that could predispose them to HCC.
4. Epigenetic Modifications. Epigenetics plays a critical role in regulating gene expression in response to environmental stimuli. The chronic exposure to stressors (e.g., inflammation, viral infections) can lead to epigenetic changes in liver cells, altering their response to future stimuli [101]. If the S1 protein induces a specific molecular process (e.g., a stress response or an inflammatory reaction), cells with pre-existing epigenetic marks may amplify or dampen this response. This could explain why some individuals experience more severe liver pathologies in SARS-CoV-2 infection, while others do not. Some key genes from the 220 gene list can be epigenetically modified and play roles in HBV and HCC. Table 3 shows the key genes that undergo epigenetic phenomena among both the genes involved in HCC and HBV. From the table, we can appreciate the high epigenetic potential present in the set of 200 genes, both for HBV and HCC.
5. Cytokine Profiles and Immune Response. Cytokines, which can dictate the type and severity of immune responses, heavily influence the liver [126]. IL-6, TNF-α, and TGF-β are examples of cytokines that mediate inflammation, fibrogenesis, and carcinogenesis. The S1 protein can provoke immune dysregulation, potentially altering the cytokine environment. In individuals with a predisposition to an exaggerated cytokine response [127,128], this could lead to excessive fibrosis or even cellular transformation. In others, a more regulated response might limit tissue damage and maintain liver homeostasis.
6. Pre-existing Cellular Microenvironments. Even without prior liver disease, individual variability in cellular microenvironments (e.g., hypoxia, local inflammatory status) could affect how liver cells respond to S1-induced signaling. Hypoxia-inducible factors (HIFs), for example, are transcription factors that respond to low oxygen levels and play a role in cellular metabolism and survival [129]. If S1 alters cellular oxygen consumption or influences HIFs, it could exacerbate or mitigate processes like fibrosis or apoptosis, depending on the pre-existing microenvironment.
7. Autophagy and Apoptosis Pathways. The S1 protein might interfere with cellular mechanisms that regulate autophagy (a process that recycles damaged cellular components) and apoptosis [130,131]. Variations in the efficiency of these pathways in different individuals can lead to different outcomes: A) Efficient autophagy could protect cells by clearing damaged components, preventing fibrosis or cancer development. B) Dysregulated autophagy might contribute to the accumulation of damaged proteins or organelles, promoting liver cell death or fibrosis, particularly in individuals with pre-existing metabolic imbalances.
8. Cross-talk with Hepatic Stellate Cells. Hepatic stellate cells (HSCs) are central to liver fibrosis. If S1-induced signaling pathways lead to HSC activation, this could trigger a fibrotic response [132]. However, the threshold for HSC activation varies from individual to individual based on previous exposure of the liver to fibrotic stimuli. In those with a "primed" fibrotic response (because of genetic predispositions or environmental factors), the fibrosis triggered by S1 may be more severe, potentially setting the stage for HCC.
9. MicroRNA (miRNA) Regulation. miRNAs are small, non-coding RNAs that regulate gene expression post-transcriptionally and are involved in processes like cell growth, differentiation, and apoptosis [133,134]. The S1 protein might influence miRNA profiles, leading to differential gene regulation. For instance, changes in miR-122, which is liver-specific and plays a role in liver homeostasis, could cause altered cellular responses that favor fibrosis or tumorigenesis, depending on the individual’s miRNA regulatory network.
5. Conclusions: Integrating These Mechanisms
The interplay between these pathways shows that S1-induced molecular changes do not operate in isolation. Each individual's phenotypic state, shaped by genetic, metabolic, and environmental factors, will dictate how these molecular changes manifest at the cellular and tissue levels. For example, in a healthy liver with efficient autophagy and low baseline inflammation, S1 might induce transient stress that resolves without long-term damage without activating molecular processes capable of developing. Or even, in a liver with a history of subclinical inflammation or metabolic stress, S1 may exacerbate fibrosis or activate pre-cancerous pathways, leading to disease progression.
While the S1 protein can start certain molecular processes across liver phenotypes, the extent and type of pathological outcome depend on the individual's specific molecular landscape. These molecular interactions represent a set of potentialities, with the eventual phenotypic manifestation, depending on how individual liver cells and the broader tissue environment interpret and respond to these signals.
From all these considerations, other reflections arise. The same viral stimulus of S1 protein can lead to widely different outcomes in patients because of the heterogeneity in molecular and phenotypic responses. While some individuals may experience mild or reversible liver damage, others could progress to more severe conditions, like fibrosis or even hepatocellular carcinoma (HCC). The variability in immune response, genetic predisposition, and metabolic status within the population means that some individuals may recover with minimal liver damage. But others, especially those with underlying conditions (e.g., metabolic syndrome, mild hepatic inflammation), may face a greater risk of fibrosis or cancer. All this strongly supports the idea of using interactomic standards as a reference. But it also underscores the challenge of making population-wide predictions based on molecular data alone, as individual responses to the same viral insult can vary dramatically.
Given the differences in how individuals' molecular pathways respond to viral stimuli, stratifying patients into risk categories based on their genetic, metabolic, and inflammatory profiles becomes essential. For example, High-risk patients (those with metabolic dysfunction, chronic inflammation, or pre-existing liver stress) may require closer monitoring for signs of fibrosis or liver damage during and after COVID-19. While low-risk patients (those with healthy liver function and no metabolic dysregulation) might have a lower likelihood of developing serious liver-related complications. This suggests that implementing targeted screening and intervention strategies, focusing on those more likely to experience severe liver outcomes, is more effective than applying broad measures to the entire population.
As we emphasized earlier, there is no linear correlation between the molecular changes induced by SARS-CoV-2 and the clinical manifestations seen in patients. Therefore, clinicians should be cautious in making direct clinical inferences. For example, observing an upregulation of fibrosis-related pathways in molecular studies does not imply that every patient will develop fibrosis. Instead, medical professionals should use a combination of molecular findings, patient history, and real-time clinical data, such as liver function tests, imaging, and biopsy results, to inform clinical decisions but also longitudinal studies that collect molecular data from patients.
These considerations may also flow into different sectors with Public Health Implications that are outside the scope of this article. However, future research should focus on identifying biomarkers that can predict which patients are most likely to develop liver complications post-COVID. Clinical trials should explore interventions aimed at finding out specific molecular pathways activated by the S1 protein, particularly in high-risk groups, but also of other viral proteins capable of one-to-one interactions. Even studying how long-term liver outcomes vary across different demographic and genetic populations should provide more precise data for guiding clinical practice by personalized care. These are the activities we must aim for if we want to activate a personalized molecular medicine.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: title; Table S1: title; Video S1: title.
Funding
“This research received no external funding”
Institutional Review Board Statement
“Not applicable”
Informed Consent Statement
“Not applicable.”
Acknowledgments
The author acknowledges Drs. Rosario Della Santa and Vincenzo Saviano for the excellent technical support provided by the computer systems.
Conflicts of Interest
“The author declares no conflicts of interest.”
References
- Colonna, G. (2024). Understanding the SARS-CoV-2–Human Liver Interactome Using a Comprehensive Analysis of the Individual Virus–Host Interactions. Livers, 4(2), 209-239.
- Trypsteen, W., Van Cleemput, J., Snippenberg, W. V., Gerlo, S., & Vandekerckhove, L. (2020). On the whereabouts of SARS-CoV-2 in the human body: A systematic review. PLoS pathogens, 16(10), e1009037.
- Letarov, A. V., Babenko, V. V., & Kulikov, E. E. (2021). Free SARS-CoV-2 spike protein S1 particles may play a role in the pathogenesis of COVID-19 infection. Biochemistry (Mos-cow), 86, 257-261.
- Kopańska, M., Barnaś, E., Błajda, J., Kuduk, B., Łagowska, A., & Banaś-Ząbczyk, A. (2022). Effects of SARS-CoV-2 inflammation on selected organ systems of the human body. Inter-national Journal of Molecular Sciences, 23(8), 4178.
- Iyer, A. S., Jones, F. K., Nodoushani, A., Kelly, M., Becker, M., Slater, D., Mills, R., Teng, E., Kamruzzaman, M., Charles, R. C. (2020). Persistence and decay of human antibody responses to the receptor binding domain of SARS-CoV-2 spike protein in COVID-19 patients. Science immunology, 5(52), eabe0367.
- Frank, M. G., Ball, J. B., Hopkins, S., Kelley, T., Kuzma, A. J., Thompson, R. S., Fleshner, M., Maier, S. F. (2024). SARS-CoV-2 S1 subunit produces a protracted priming of the neuroinflammatory, physiological, and behavioral responses to a remote immune challenge: A role for corticosteroids. Brain, Behavior, and Immunity, 121, 87-103.
- Al-Aly, Z., Davis, H., McCorkell, L., Soares, L., Wulf-Hanson, S., Iwasaki, A., & Topol, E. J. (2024). Long COVID science, research and policy. Nature Medicine, 1-17.
- Hallak J., Caldini, E.G., Teixeira, T.A., Mendes Correa, M., Duarte-Neto, A.N., Zambrano, F., Taubert, A., Hermosilla, C., Drevet, J., Dolhnikoff, M., Sanchez, R., Saldiva, P.H.N. Trasmission electron microscopy reveals the presence of SARS-CoV-2 in human spermatozoa associated with an ETosis-like response. Andrology, (2024); 1-9. DOI: 10.1111/andr.13612.
- Madjunkov, M., Dviri, M., & Librach, C. (2020). A comprehensive review of the impact of COVID-19 on human reproductive biology, assisted reproduction care and pregnancy: a Canadian perspective. Journal of ovarian research, 13(1), 140.
- Mansueto, G., Fusco, G., & Colonna, G. (2024). A Tiny Viral Protein, SARS-CoV-2-ORF7b: Functional Molecular Mechanisms. Biomolecules, 14(5), 541.
- Colonna, G.--. 2024 "Interactomic Analyses and a Reverse Engineering Study Identify Spe-cific Functional Activities of One-to-One Interactions of the S1 Subunit of the SARS-CoV-2 Spike Protein with the Human Proteome" Submitted to Biomolecules - Preprints. [CrossRef]
- Cosentino, M., & Marino, F. (2022). The spike hypothesis in vaccine-induced adverse effects: questions and answers. Trends in molecular medicine, 28(10), 797-799.
- Tan, X., Lin, C., Zhang, J., Khaing Oo, M. K., & Fan, X. (2020). Rapid and quantitative detection of COVID-19 markers in micro-liter sized samples. BioRxiv, 2020-04.
- Bošnjak, B., Stein, S. C., Willenzon, S., Cordes, A. K., Puppe, W., Bernhardt, G., Ravens, I., Ritter, C., Schultze-Florey, C., Godecke, N., et al (2021). Low serum neutralizing an-ti-SARS-CoV-2 S antibody levels in mildly affected COVID-19 convalescent patients re-vealed by two different detection methods. Cellular & molecular immunology, 18(4), 936-944.
- Yonker, L. M., Swank, Z., Bartsch, Y. C., Burns, M. D., Kane, A., Boribong, B. P., Davis, JP., Loiselle, M., Novak, T., Senussi, Y., et al., (2023). Circulating spike protein detected in post–COVID-19 mRNA vaccine myocarditis. Circulation, 147(11), 867-876.
- Oughtred R, Rust J, Chang C, Breitkreutz BJ, Stark C, Willems A, Boucher L, Leung G, Kolas N, Zhang F, Dolma S, Coulombe-Huntington J, Chatr-Aryamontri A, Dolinski K, Tyers M. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021 Jan;30(1):187-200. doi: 10.1002/pro.3978. Epub 2020 Nov 23. PMID: 33070389; PMCID: PMC7737760.
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2020, 49, D605–D612; Erratum in: Nucleic Acids Res. 2021, 49, 10800. [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2022, 51, D638–D646. [CrossRef]
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J. Proteome Res. 2018, 18, 623–632. [CrossRef]
- Chung, F.; Lu, L.; Dewey, T.G.; Galas, D.J. Duplication Models for Biological Networks. J. Comput. Biol. 2003, 10, 677–687. [CrossRef]
- Barabási, A.-L. Network Science, 1st ed.; Cambridge University Press: Cambridge, UK, 2016.
- Arakawa, K., Tomita, M. (2013). Merging Multiple Omics Datasets In Silico: Statistical Analyses and Data Interpretation. In: Alper, H. (eds) Systems Metabolic Engineering. Methods in Molecular Biology, vol 985. Humana Press, Totowa, NJ. [CrossRef]
- Glazier, D. S. (2014). Metabolic scaling in complex living systems. Systems, 2(4), 451-540.
- De Las Rivas J, Fontanillo C. Protein-protein interactions essentials: key concepts to building and analyzing interactome networks. PLoS Comput Biol. 2010 Jun 24;6(6):e1000807. doi: 10.1371/journal.pcbi.1000807. PMID: 20589078; PMCID: PMC2891586.
- G., Grassmann, M., Miotto, F. Desantis, L. Di Rienzo, G.G. Tartaglia, A. Pastore, G., Ruocco, M., Monti, E., Milanetti. Computational Approaches to Predict Protein–Protein Interactions in Crowded Cellular Environments. Chem. Rev. 2024, 124, 7, 3932–3977. [CrossRef]
- Shuping Xing, Niklas Wallmeroth, Kenneth W. Berendzen, Christopher Grefen, Techniques for the Analysis of Protein-Protein Interactions in Vivo, Plant Physiology, Volume 171, Issue 2, June 2016, Pages 727–758, . [CrossRef]
- Lite TV, Grant RA, Nocedal I, Littlehale ML, Guo MS, Laub MT. Uncovering the basis of protein-protein interaction specificity with a combinatorially complete library. Elife. 2020 Oct 27;9:e60924. doi: 10.7554/eLife.60924. PMID: 33107822; PMCID: PMC7669267.
- Chen, D., Lü, L., Shang, M. S., Zhang, Y. C., & Zhou, T. (2012). Identifying influential nodes in complex networks. Physica a: Statistical mechanics and its applications, 391(4), 1777-1787.
- Barabási, A. L. (2013). Network science. Philosophical Transactions of the Royal Soci-ety A: Mathematical, Physical and Engineering Sciences, 371(1987), 20120375.
- Guthrie CR, Skâlhegg BS, McKnight GS. Two novel brain-specific splice variants of the murine beta gene of cAMP-dependent protein kinase. J Biol Chem. 1997 Nov 21;272(47):29560-5. doi: 10.1074/jbc.272.47.29560. PMID: 9368018.
- Alqahtani, S. A., & Buti, M. (2020). COVID-19 and hepatitis B infection. Antiviral therapy, 25(8), 389-397.
- Song, C. I., Lv, J., Liu, Y., Chen, J. G., Ge, Z., Zhu, J., Dai, J., Du, LB., Yu, C., Guo, Y., et al., (2019). Associations between hepatitis B virus infection and risk of all cancer types. JAMA network open, 2(6), e195718-e195718. doi:10.1001/jamanetworkopen.2019.5718.
- Li, Y., Li, C., Wang, J., Zhu, C., Zhu, L., Ji, F., Liu, L., Xu, T., Zhang, B., Xue, L., et al., (2020). A case series of COVID-19 patients with chronic hepatitis B virus infection. Journal of medical virology, 92(11), 2785-2791.
- Yu, Y., Li, X., & Wan, T. (2023). Effects of hepatitis B virus infection on patients with COVID-19: A meta-analysis. Digestive diseases and sciences, 68(4), 1615-1631.
- Chung, J., Yu, J., Cheon, M., & Tak, S. (2024). Evaluation of the acute hepatitis B surveillance system in the Republic of Korea following the transition to mandatory surveillance. Osong Public Health and Research Perspectives, 15(4), 353.
- Essam, S., Hassany, M., Maged, A., Mannaa, M., Sayed, A., Essam, S., & Magdy, M. Assessment of Hepatocellular Carcinoma Patients Infected with Covid-19 Infection during the Pandemic. International Journal of Chemical and Biochemical Sciences (IJCBS), 25(19) (2024): 1065-1069. Doi # . [CrossRef]
- Nasir, N., Khanum, I., Habib, K., Wagley, A., Arshad, A., & Majeed, A. (2024). Insight into COVID-19 associated liver injury: Mechanisms, evaluation, and clinical implications. In Hepatology Forum (Vol. 5, No. 3, p. 139). Turkish Association for the Study of the Liver.
- Mihai, N., Olariu, M. C., Ganea, O. A., Adamescu, A. I., Molagic, V., Aramă, Ș. S., Aramă, V. et al., (2024). Risk of Hepatitis B Virus Reactivation in COVID-19 Patients Receiving Immunosuppressive Treatment: A Prospective Study. Journal of Clinical Medicine, 13(20), 6032.
- Chang, H. C., Su, T. H., Huang, Y. T., Hong, C. M., Sheng, W. H., Hsueh, P. R., & Kao, J. H. (2024). Liver dysfunction and clinical outcomes of unvaccinated COVID-19 patients with and without chronic hepatitis B. Journal of Microbiology, Immunology and Infection, 57(1), 55-63.
- Mushtaq, M., Colletier, K., & Moghe, A. (2024). Hepatitis B Reactivation and Liver Failure Because of COVID-19 Infection. ACG Case Reports Journal, 11(7), e01397.
- Wu, H. Y., Su, T. H., Liu, C. J., Yang, H. C., Tsai, J. H., Wei, MH., Chen, CC., Tung, CC., Kao, JH., Chen, P. J. (2024). Hepatitis B reactivation: A possible cause of coronavirus disease 2019 vaccine induced hepatitis. Journal of the Formosan Medical Association, 123(1), 88-97.
- D'souza, S., Lau, K. C., Coffin, C. S., & Patel, T. R. (2020). Molecular mechanisms of viral hepatitis induced hepatocellular carcinoma. World journal of gastroenterology, 26(38), 5759.
- Pollicino, T., Saitta, C., & Raimondo, G. (2011). Hepatocellular carcinoma: the point of view of the hepatitis B virus. Carcinogenesis, 32(8), 1122-1132.
- Liu, W. B., Wu, J. F., Du, Y., & Cao, G. W. (2016). Cancer Evolution–Development: experience of hepatitis B virus–induced hepatocarcinogenesis. Current oncology, 23(1), e49.
- Guerrero, R. B., & Roberts, L. R. (2005). The role of hepatitis B virus integrations in the pathogenesis of human hepatocellular carcinoma. Journal of hepatology, 42(5), 760-777.
- Xie, Y. (2017). Hepatitis B virus-associated hepatocellular carcinoma. Infectious Agents Associated Cancers: Epidemiology and Molecular Biology, 11-21.
- Ham, S. W., Jeon, H. Y., Jin, X., Kim, E. J., Kim, J. K., Shin, Y. J., Lee, Y., Kim, S.H., Lee, S.Y., Seo, S., Park, M.G., Kim, H., Nam, D., Kim, H. (2019). TP53 gain-of-function mutation promotes inflammation in glioblastoma. Cell Death & Differentiation, 26(3), 409-425. [CrossRef]
- Zhou, P., Lu, S., Luo, Y., Wang, S., Yang, K., Zhai, Y., Sun, G., Sun, X. (2017). Attenuation of TNF-α-induced inflammatory injury in endothelial cells by ginsenoside Rb1 via inhibiting NF-κB, JNK and p38 signaling pathways. Frontiers in pharmacology, 8, 464. [CrossRef]
- Yu, H., Pardoll, D., & Jove, R. (2009). STATs in cancer inflammation and immunity: a leading role for STAT3. Nature reviews cancer, 9(11), 798-809. [CrossRef]
- Huebner, K. (2023). The role of the Activating Transcription Factor 2 (ATF2) in colorectal carcinogenesis. Doctoral Thesis, Friedrich-Alexander-Universitaet Erlangen-Nuernberg (Germany). https://nbn-resolving.org/urn:nbn:de:bvb:29-opus4-167132.
- Ji, L., Li, T., Chen, H., Yang, Y., Lu, E., Liu, J., ... & Chen, H. (2023). The crucial regulatory role of type I interferon in inflammatory diseases. Cell & Bioscience, 13(1), 230. [CrossRef]
- Moysidou, C. M., Barberio, C., & Owens, R. M. (2021). Advances in engineering human tissue models. Frontiers in bioengineering and biotechnology, 8, 620962.
- Tan YJ. Hepatitis B virus infection and the risk of hepatocellular carcinoma. World J Gastroenterol. 2011 Nov 28;17(44):4853-7. doi: 10.3748/wjg.v17.i44.4853. PMID: 22171125; PMCID: PMC3235627.
- Arbuthnot, P., & Kew, M. (2001). Hepatitis B virus and hepatocellular carcinoma. International journal of experimental pathology, 82(2), 77-100.
- Kouroumalis E, Tsomidis I, Voumvouraki A. Pathogenesis of Hepatocellular Carcinoma: The Interplay of Apoptosis and Autophagy. Biomedicines. 2023 Apr 13;11(4):1166. doi: 10.3390/biomedicines11041166. PMID: 37189787; PMCID: PMC10135776.
- De Re V, Rossetto A, Rosignoli A, Muraro E, Racanelli V, Tornesello ML, Zompicchiatti A, Uzzau A. Hepatocellular Carcinoma Intrinsic Cell Death Regulates Immune Response and Prognosis. Front Oncol. 2022 Jul 7;12:897703. doi: 10.3389/fonc.2022.897703. PMID: 35875093; PMCID: PMC9303009.
- Luedde, T., Kaplowitz, N., & Schwabe, R. F. (2014). Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology, 147(4), 765-783.
- Imre, G. (2020). Cell death signaling in virus infection. Cellular signaling, 76, 109772.
- Bertheloot, D., Latz, E. & Franklin, B.S. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol 18, 1106–1121 (2021). HCC cells may undergo apoptosis due to various factors such as immune response, or the activation of pro-apoptotic signals. [CrossRef]
- Wu X, Cao J, Wan X, Du S. Programmed cell death in hepatocellular carcinoma: mechanisms and therapeutic perspectives. Cell Death Discov. 2024 Aug 8;10(1):356. doi: 10.1038/s41420-024-02116-x. PMID: 39117626; PMCID: PMC11310460.
- García-Pras, E., Fernández-Iglesias, A., Gracia-Sancho, J., & Pérez-del-Pulgar, S. (2021). Cell death in hepatocellular carcinoma: pathogenesis and therapeutic opportunities. Cancers, 14(1), 48. However, many HCC cells can evade apoptosis, contributing to tumor progression.
- Fabregat I. Dysregulation of apoptosis in hepatocellular carcinoma cells. World J Gastroenterol. 2009 Feb 7;15(5):513-20. doi: 10.3748/wjg.15.513. PMID: 19195051; PMCID: PMC2653340.
- Wang, G., Jiang, X., Torabian, P., & Yang, Z. (2024). Investigating autophagy and intricate cellular mechanisms in hepatocellular carcinoma: Emphasis on cell death mechanism crosstalk. Cancer Letters, 216744.
- Gregory, C. D., Ford, C. A., & Voss, J. J. (2016). Microenvironmental effects of cell death in malignant disease. Apoptosis in Cancer Pathogenesis and Anti-cancer Therapy: New Perspectives and Opportunities, 51-88.
- Luo, G., & Liu, N. (2019). An integrative theory for cancer. International Journal of Molecular Medicine, 43(2), 647-656.
- Osuchowski, M. F., Winkler, M. S., Skirecki, T., Cajander, S., Shankar-Hari, M., Lachmann, G., ... & Rubio, I. (2021). The COVID-19 puzzle: deciphering pathophysiology and phenotypes of a new disease entity. The Lancet Respiratory Medicine, 9(6), 622-642.
- Cao, L., Quan, X. B., Zeng, W. J., Yang, X. O., & Wang, M. J. (2016). Mechanism of hepatocyte apoptosis. Journal of cell death, 9, JCD-S39824.
- Schwabe, R. F., & Luedde, T. (2018). Apoptosis and necroptosis in the liver: a matter of life and death. Nature reviews Gastroenterology & hepatology, 15(12), 738-7.
- Wang J, Luo Z, Lin L, Sui X, Yu L, Xu C, Zhang R, Zhao Z, Zhu Q, An B, Wang Q, Chen B, Leung EL, Wu Q. Anoikis-Associated Lung Cancer Metastasis: Mechanisms and Therapies. Cancers (Basel). 2022 Sep 30;14(19):4791. doi: 10.3390/cancers14194791. PMID: 36230714; PMCID: PMC9564242.
- Adeshakin FO, Adeshakin AO, Afolabi LO, Yan D, Zhang G, Wan X. Mechanisms for Modulating Anoikis Resistance in Cancer and the Relevance of Metabolic Reprogramming. Front Oncol. 2021 Mar 29;11:626577. doi: 10.3389/fonc.2021.626577. PMID: 33854965; PMCID: PMC8039382.
- Paoli, P., Giannoni, E., & Chiarugi, P. (2013). Anoikis molecular pathways and its role in cancer progression. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research, 1833(12), 3481-3498.
- Jin, L., Zuo, X. Y., Su, W. Y., Zhao, X. L., Yuan, M. Q., Han, L. Z., ... & Rao, S. Q. (2014). Pathway-based analysis tools for complex diseases: a review. Genomics, Proteomics and Bioinformatics, 12(5), 210-220.
- Brazhnik, P., De La Fuente, A., & Mendes, P. (2002). Gene networks: how to put the function in genomics. TRENDS in Biotechnology, 20(11), 467-472.
- Hecker, M., Lambeck, S., Toepfer, S., Van Someren, E., & Guthke, R. (2009). Gene regulatory network inference: data integration in dynamic models—a review. Biosystems, 96(1), 86-103.
- Carpenter, A. E., & Sabatini, D. M. (2004). Systematic genome-wide screens of gene function. Nature Reviews Genetics, 5(1), 11-22.
- Kelley, R., & Ideker, T. (2005). Systematic interpretation of genetic interactions using protein networks. Nature biotechnology, 23(5), 561-566.
- Kabir, M. H., Patrick, R., Ho, J. W., & O’Connor, M. D. (2018). Identification of active signaling pathways by integrating gene expression and protein interaction data. BMC systems biology, 12, 77-87.
- Kuenzi, B. M., & Ideker, T. (2020). A census of pathway maps in cancer systems biology. Nature Reviews Cancer, 20(4), 233-246.
- Jaeger, S., Min, J., Nigsch, F., Camargo, M., Hutz, J., Cornett, A.,Cleaver, S., Buckler, A., Jenkins, J. L. (2014). Causal network models for predicting compound targets and driving pathways in cancer. Journal of biomolecular screening, 19(5), 791-802.
- Buneman, P., Chapman, A., & Cheney, J. (2006, June). Provenance management in curated databases. In Proceedings of the 2006 ACM SIGMOD international conference on Management of data (pp. 539-550).
- Goudey, B., Geard, N., Verspoor, K., & Zobel, J. (2022). Propagation, detection and correction of errors using the sequence database network. Briefings in bioinformatics, 23(6), bbac416.
- Azeroual, O. (2020). Data wrangling in database systems: purging of dirty data. Data, 5(2), 50.
- Li, C., Liakata, M., & Rebholz-Schuhmann, D. (2014). Biological network extraction from scientific literature: state of the art and challenges. Briefings in bioinformatics, 15(5), 856-877.
- Klein, B., Hoel, E., Swain, A., Griebenow, R., & Levin, M. (2021). Evolution and emergence: higher order information structure in protein interactomes across the tree of life. Integrative Biology, 13(12), 283-294.
- Wautelet, M. (2001). Scaling laws in the macro-, micro-and nanoworlds. European Journal of Physics, 22(6), 601.
- Haken, H., & Haken, H. (1988). From the Microscopic to the Macroscopic World. Information and Self-Organization: A Macroscopic Approach to Complex Systems, 36-52.
- Bizzarri, M., Palombo, A., & Cucina, A. (2013). Theoretical aspects of systems biology. Progress in biophysics and molecular biology, 112(1-2), 33-43.
- Gosak, M., Markovič, R., Dolenšek, J., Rupnik, M. S., Marhl, M., Stožer, A., & Perc, M. (2018). Network science of biological systems at different scales: A review. Physics of life reviews, 24, 118-135.
- Moerner, W. E. (2002). A dozen years of single-molecule spectroscopy in physics, chemistry, and biophysics. The Journal of Physical Chemistry B, 106(5), 910-927.
- Muller, H. J. (1922). Variation due to change in the individual gene. The American Naturalist, 56(642), 32-50.
- El-Hani, C. N. (2007). Between the cross and the sword: the crisis of the gene concept. Genetics and molecular biology, 30, 297-307.
- Kirschner, Marc W. The Meaning of Systems Biology. Cell, (2005), Volume 121, Issue 4, 503 - 504.
- Salehi-Reyhani, A., Ces, O., & Elani, Y. (2017). Artificial cell mimics as simplified models for the study of cell biology. Experimental Biology and Medicine, 242(13), 1309-1317.
- Joyce, A. R., & Palsson, B. Ø. (2006). The model organism as a system: integrating'omics' data sets. Nature reviews Molecular cell biology, 7(3), 198-210.
- Hunter, P. (2008). The paradox of model organisms: the use of model organisms in research will continue despite their shortcomings. EMBO reports, 9(8), 717-720.
- Bándi, G., & Ramsden, J. J. (2011). Emulating biology: the virtual living organism. J Biol Phys Chem, 11, 97-106.
- Hatmal, M. M. M., Alshaer, W., Al-Hatamleh, M. A., Hatmal, M., Smadi, O., Taha, M. O., Oweida, AJ., Boer, J., Mohamud, R., & Plebanski, M. (2020). Comprehensive structural and molecular comparison of spike proteins of SARS-CoV-2, SARS-CoV and MERS-CoV, and their interactions with ACE2. Cells, 9(12), 2638.
- Arbuthnot, P., Capovilla, A., & Kew, M. (2000). Putative role of hepatitis B virus X protein in hepatocarcinogenesis: effects on apoptosis, DNA repair, mitogen-activated protein kinase and JAK/STAT pathways. Journal of gastroenterology and hepatology, 15(4), 357-368.
- Moolamalla, S. T. R., Balasubramanian, R., Chauhan, R., Priyakumar, U. D., & Vinod, P. K. (2021). Host metabolic reprogramming in response to SARS-CoV-2 infection: A systems biology approach. Microbial pathogenesis, 158, 105114.
- Juanola, O., Martínez-López, S., Francés, R., & Gómez-Hurtado, I. (2021). Non-alcoholic fatty liver disease: metabolic, genetic, epigenetic and environmental risk factors. International journal of environmental research and public health, 18(10), 5227.
- Hlady, R. A., & Robertson, K. D. (2024). Epigenetic memory of environmental exposures as a mediator of liver disease. Hepatology, 80(2), 451-464.
- Miller, J. L., & Grant, P. A. (2012). The role of DNA methylation and histone modifications in transcriptional regulation in humans. Epigenetics: Development and Disease, 289-317.
- Esteller, M., & Herman, J. G. (2002). Cancer as an epigenetic disease: DNA methylation and chromatin alterations in human tumours. The Journal of Pathology: A Journal of the Pathological Society of Great Britain and Ireland, 196(1), 1-7.
- Ozyerli-Goknar, E., & Bagci-Onder, T. (2021). Epigenetic deregulation of apoptosis in cancers. Cancers, 13(13), 3210.
- Gao, A., Zuo, X., Song, S., Guo, W., & Tian, L. (2011). Epigenetic modification involved in benzene-induced apoptosis through regulating apoptosis-related genes expression. Cell Biology International, 35(4), 391-396.
- Yang, S., Pang, L., Dai, W., Wu, S., Ren, T., Duan, Y., ... & Kong, J. (2021). Role of forkhead box O proteins in hepatocellular carcinoma biology and progression. Frontiers in Oncology, 11, 667730.
- Gong, Z., Yu, J., Yang, S., Lai, P. B., & Chen, G. G. (2020). FOX transcription factor family in hepatocellular carcinoma. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer, 1874(1), 188376.
- Kishor Roy, N., Bordoloi, D., Monisha, J., Padmavathi, G., Kotoky, J., Golla, R., & B. Kunnumakkara, A. (2017). Specific targeting of Akt kinase isoforms: Taking the precise path for prevention and treatment of cancer. Current drug targets, 18(4), 421-435.
- Sun, E. J., Wankell, M., Palamuthusingam, P., McFarlane, C., & Hebbard, L. (2021). Targeting the PI3K/Akt/mTOR pathway in hepatocellular carcinoma. Biomedicines, 9(11), 1639.
- Dong, Y., & Wang, A. (2014). Aberrant DNA methylation in hepatocellular carcinoma tumor suppression. Oncology letters, 8(3), 963-968.
- Guerrieri, F., Belloni, L., Pediconi, N., & Levrero, M. (2013, May). Molecular mechanisms of HBV-associated hepatocarcinogenesis. In Seminars in liver disease (Vol. 33, No. 02, pp. 147-156). Thieme Medical Publishers.
- Zeisel, M. B., Guerrieri, F., & Levrero, M. (2021). Host epigenetic alterations and hepatitis B virus-associated hepatocellular carcinoma. Journal of Clinical Medicine, 10(8), 1715.
- Elpek, G. O. (2021). Molecular pathways in viral hepatitis-associated liver carcinogenesis: An update. World Journal of Clinical Cases, 9(19), 4890.
- Sun, E. J., Wankell, M., Palamuthusingam, P., McFarlane, C., & Hebbard, L. (2021). Targeting the PI3K/Akt/mTOR pathway in hepatocellular carcinoma. Biomedicines, 9(11), 1639.
- Tian, L. Y., Smit, D. J., & Jücker, M. (2023). The role of PI3K/AKT/mTOR signaling in hepatocellular carcinoma metabolism. International Journal of Molecular Sciences, 24(3), 2652.
- Zeisel, M. B., Guerrieri, F., & Levrero, M. (2021). Host epigenetic alterations and hepatitis B virus-associated hepatocellular carcinoma. Journal of Clinical Medicine, 10(8), 1715.
- Rybicka, M., Verrier, E. R., Baumert, T. F., & Bielawski, K. P. (2023). Polymorphisms within DIO2 and GADD45A genes increase the risk of liver disease progression in chronic hepatitis b carriers. Scientific Reports, 13(1), 6124.
- Huebner, K., Procházka, J., Monteiro, A. C., Mahadevan, V., & Schneider-Stock, R. (2019). The activating transcription factor 2: an influencer of cancer progression. Mutagenesis, 34(5-6), 375-389.
- Rajan, P. K., Udoh, U. A., Sanabria, J. D., Banerjee, M., Smith, G., Schade, M. S., ... & Sanabria, J. (2020). The role of histone acetylation-/methylation-mediated apoptotic gene regulation in hepatocellular carcinoma. International journal of molecular sciences, 21(23), 8894.
- Huang, G., Chen, J., Zhou, J., Xiao, S., Zeng, W., Xia, J., & Zeng, X. (2021). Epigenetic modification and BRAF gene mutation in thyroid carcinoma. Cancer cell international, 21, 1-14.
- Kim, H. C., Choi, K. C., Choi, H. K., Kang, H. B., Kim, M. J., Lee, Y. H., ... & Yoon, H. G. (2010). HDAC3 selectively represses CREB3-mediated transcription and migration of metastatic breast cancer cells. Cellular and molecular life sciences, 67, 3499-3510.
- Groner, B., & von Manstein, V. (2017). Jak Stat signaling and cancer: Opportunities, benefits and side effects of targeted inhibition. Molecular and cellular endocrinology, 451, 1-14.
- Masliah-Planchon, J., Garinet, S., & Pasmant, E. (2016). RAS-MAPK pathway epigenetic activation in cancer: miRNAs in action. Oncotarget, 7(25), 38892.
- Xie, Z., Wang, Q., & Hu, S. (2021). Coordination of PRKCA/PRKCA-AS1 interplay facilitates DNA methyltransferase 1 recruitment on DNA methylation to affect protein kinase C alpha transcription in mitral valve of rheumatic heart disease. Bioengineered, 12(1), 5904-5915.
- Meng, X. M., Nikolic-Paterson, D. J., & Lan, H. Y. (2016). TGF-β: the master regulator of fibrosis. Nature Reviews Nephrology, 12(6), 325-338.
- Zimmermann, H. W., Trautwein, C., & Tacke, F. (2012). Functional role of monocytes and macrophages for the inflammatory response in acute liver injury. Frontiers in physiology, 3, 56.
- Hildebrand, F., Pape, H. C., van Griensven, M., Meier, S., Hasenkamp, S., Krettek, C., & Stuhrmann, M. (2005). Genetic predisposition for a compromised immune system after multiple trauma. Shock, 24(6), 518-522.
- Hazeldine, J., & Lord, J. M. (2020). Immunesenescence: a predisposing risk factor for the development of COVID-19? Frontiers in immunology, 11, 573662.
- Lee, P., Chandel, N. S., & Simon, M. C. (2020). Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nature reviews Molecular cell biology, 21(5), 268-283.
- Mukhopadhyay, S., Panda, P. K., Sinha, N., Das, D. N., & Bhutia, S. K. (2014). Autophagy and apoptosis: where do they meet?. Apoptosis, 19, 555-566.
- Das, S., Shukla, N., Singh, S. S., Kushwaha, S., & Shrivastava, R. (2021). Mechanism of interaction between autophagy and apoptosis in cancer. Apoptosis, 1-22.
- Moreira, R. K. (2007). Hepatic stellate cells and liver fibrosis. Archives of pathology & laboratory medicine, 131(11), 1728-1734.
- Hu, W., Alvarez-Dominguez, J. R., & Lodish, H. F. (2012). Regulation of mammalian cell differentiation by long non-coding RNAs. EMBO reports, 13(11), 971-983.
- Zhang, X., Wang, W., Zhu, W., Dong, J., Cheng, Y., Yin, Z., & Shen, F. (2019). Mechanisms and functions of long non-coding RNAs at multiple regulatory levels. International journal of molecular sciences, 20(22), 5573.
- Melkonian M, Juigné C, Dameron O, Rabut G, Becker E. Towards a reproducible interactome: semantic-based detection of redundancies to unify protein-protein interaction databases. Bioinformatics. 2022 Mar 4;38(6):1685-1691. doi: 10.1093/bioinformatics/btac013. PMID: 35015827.
- Arnold R, Boonen K, Sun MG, Kim PM. Computational analysis of interactomes: current and future perspectives for bioinformatics approaches to model the host-pathogen interaction space. Methods. 2012 Aug;57(4):508-18. doi: 10.1016/j.ymeth.2012.06.011. Epub 2012 Jun 28. PMID: 2750305; PMCID: PMC7128575.
Figure 1.
– Protein interactome extracted from [1] and calculated by STRING. Setting parameters: Confidence score: 0.900; without Text Mining; enrichment: 500 first order + 500 second order proteins. Topological parameters: number of nodes: 676; number of edges: 5679; average node degree: 16.8; avg. local clustering coefficient: 0.535; expected number of edges: 1924; PPI enrichment p-value: <1.0e-16; connected components: 1.
Figure 1.
– Protein interactome extracted from [1] and calculated by STRING. Setting parameters: Confidence score: 0.900; without Text Mining; enrichment: 500 first order + 500 second order proteins. Topological parameters: number of nodes: 676; number of edges: 5679; average node degree: 16.8; avg. local clustering coefficient: 0.535; expected number of edges: 1924; PPI enrichment p-value: <1.0e-16; connected components: 1.

Figure 2.
– Interactome of the 220 genes specifically involved in the control of liver cell death during covid. The three genes in the top right were pruned and excluded from the graph calculation. Graph parameters - number of nodes: 217; number of edges: 1655; expected number of edges: 283; PPI enrichment p-value:< 1.0e-16. Confidence score: 0.700; NO Text Mining. A confidence score of 0.7 was used to capture more information without introducing too much noise. Topological parameters: average node degree: 15.3; avg. local clustering coefficient: 0.508; network diameter: 6; characteristic path length: 2.621; network density: 0.075; Network heterogeneity: 0.745; connected components: 1. (calculated by Cytoscape).
Figure 2.
– Interactome of the 220 genes specifically involved in the control of liver cell death during covid. The three genes in the top right were pruned and excluded from the graph calculation. Graph parameters - number of nodes: 217; number of edges: 1655; expected number of edges: 283; PPI enrichment p-value:< 1.0e-16. Confidence score: 0.700; NO Text Mining. A confidence score of 0.7 was used to capture more information without introducing too much noise. Topological parameters: average node degree: 15.3; avg. local clustering coefficient: 0.508; network diameter: 6; characteristic path length: 2.621; network density: 0.075; Network heterogeneity: 0.745; connected components: 1. (calculated by Cytoscape).


Table 1.
shows the various functional enrichments in quantitative terms. All enriched terms account for 5,936 items in 15 categories.
Table 1.
shows the various functional enrichments in quantitative terms. All enriched terms account for 5,936 items in 15 categories.
| Table 1 - Enrichment analysis of interactome-12 | |
| Biological Process (Gene Ontology) | 2015 GO-terms significantly enriched; |
| Molecular Function (Gene Ontology) | 276 GO-terms significantly enriched; |
| Cellular Component (Gene Ontology) | 217 GO-terms significantly enriched; |
| Reference Publications (PubMed) | 10,000 publications significantly enriched; |
| Local Network Cluster (STRING) | 193 clusters significantly enriched; |
| KEGG Pathways | 199 pathways significantly enriched; |
| Reactome Pathways | 802 pathways significantly enriched; |
| WikiPathways | 388 pathways significantly enriched; |
| Disease-gene Associations (DISEASES) | 137 diseases significantly enriched; |
| Tissue Expression (TISSUES) | 162 tissues significantly enriched; |
| Subcellular Localization (COMPARTMENTS) | 205 compartments significantly enriched; |
| Human Phenotype (Monarch) | 1013 phenotypes significantly enriched; |
| Annotated Keywords (UniProt) | 87 keywords significantly enriched; |
| Protein Domains (Pfam) | 9 domains significantly enriched; |
| Protein Domains and Features (InterPro) | 187 domains significantly enriched; |
| Protein Domains (SMART) | 46 domains significantly enriched; |
| All enriched terms (without PubMed) | 5,936 enriched terms in 15 categories; |
Table 3.
Occurrence of epigenetic phenomena among key genes of the 200 gene-set.
| Key Genes Linked to Epigenetic Phenomena in HCC | Key Genes Linked to Epigenetic Phenomena in HBV | ||||
| name | function | bibliography | name | function | bibliography |
| TP53 | DNA methylation patterns and histone modifications | [102,103] | TP53 | also plays a critical role in HBV-associated carcinogenesis | [111,112] |
| BAX, BCL2 | Apoptosis-related genes which undergo epigenetic regulation of their expression. | [104,105] | BAX, BCL2 | also involved in HBV-related apoptosis regulation influenced by viral-mediated epigenetic modifications | [113] |
| FOXO1, FOXO3 | Members of the FOXO family are involved in histone modifications and can influence cell proliferation in HCC. | [106,107] | AKT1, AKT2, AKT3 | Epigenetically regulated in response to HBV infection, these genes modulate survival and proliferation pathways | [114,115] |
| AKT1, AKT2, AKT3 | AKT isoforms are involved in signaling pathways that are epigenetically regulated, particularly in cancer processes such as HCC. | [108,109] | PTEN | PTEN is often epigenetically silenced via methylation in HBV. | [116] |
| PTEN | Tumor suppressor gene, regulated via promoter methylation in HCC. | [110] | GADD45A, GADD45B, GADD45G | These genes are involved in DNA repair and can influence epigenetic modifications through their role in response to cellular stress. | [117] |
| Other key genes usually linked to epigenetic phenomena | |||||
| ATF2: This gene is involved in regulating gene expression through chromatin remodelling and can influence cancer progression [118]. | |||||
| BCL2L1: While primarily known for its role in apoptosis, it may also have implications in epigenetic regulation through interactions with chromatin-modifying complexes [119]. | |||||
| BRAF: Known for its role in cell signaling, its mutations are also associated with epigenetic changes in various cancers [120]. | |||||
| CREB3: Involved in transcriptional regulation linked to epigenetic modifications in various cancers [121]. | |||||
| GADD45A, GADD45B, GADD45G: These genes are involved in DNA repair and can influence epigenetic modifications through their role in response to cellular stress [117]. | |||||
| JAK2: While primarily part of the signaling pathway, it can influence gene expression and epigenetic modifications indirectly [122] | |||||
| MAPK1 and MAPK3: These genes are part of signaling pathways that can lead to changes in gene expression and implicated in epigenetic modifications [123]. | |||||
| NRAS: Like KRAS and BRAF, it is involved in signaling pathways that can lead to epigenetic alterations [123]. | |||||
| PRKCA: Plays a role in various signaling pathways and can influence epigenetic changes by modulating gene expression [124]. | |||||
| SMAD3: Involved in TGF-β signaling, which can lead to epigenetic modifications related to fibrosis and cancer progression [125]. | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



