
Submitted:
04 November 2024
Posted:
05 November 2024
You are already at the latest version
Abstract
Ischemic stroke (IS) remains a leading cause of mortality and long-term disability worldwide, with limited therapeutic options available. Despite the success of early interventions such as tissue-type plasminogen activator administration and mechanical thrombectomy, many patients continue to experience persistent neurological deficits. The pathophysiology of IS is multifaceted, encompassing excitotoxicity, oxidative and nitrosative stress, inflammation, and blood-brain barrier disruption, all of which contribute to neural cell death, further complicating the treatment of IS. Recently, extracellular vesicles (EVs) secreted naturally by various cell types have emerged as promising therapeutic agents because of their ability to facilitate selective cell-to-cell communication, neuroprotection, and tissue regeneration. Furthermore, engineered EVs, designed to enhance targeted delivery and therapeutic cargo, hold potential to improve their therapeutic benefits by mitigating neuronal damage and promoting neurogenesis and angiogenesis. This review summarizes the characteristics of EVs, the molecular mechanisms underlying IS pathophysiology, and the emerging role of EVs in IS treatment at the molecular level. This review also explores the recent advancements in EV engineering, including the incorporation of specific proteins, RNAs, or pharmacological agents into EVs to enhance their therapeutic efficacy.
Keywords:
Ischemic stroke
; Pathophysiology
; Extracellular vesicles
; Extracellular vesicle engineering
1. Introduction
Stroke is a neurological disorder caused by acute focal injury of the central nervous system (CNS) with a vascular etiology [1]. According to the World Health Organization, stroke is the second leading cause of death and the fourth leading cause of disability worldwide [2,3]. Ischemic stroke (IS), which accounts for approximately 87% of all strokes, has a steadily increasing incidence and is responsible for nearly half of all stroke-related mortality [4]. IS is defined by neurological dysfunction resulting from focal cerebral, spinal, or retinal infarction, most commonly due to cardioembolism or atherosclerosis in the aortic arch or cervical arteries [1,5]. In clinical practice, treatment of IS primarily involves intravenous administration of alteplase, the only tissue-type plasminogen activator (tPA) approved by the USFDA for thrombolysis, mechanical thrombectomy, or a combination of both, as quickly as possible after the onset of ischemic events [5,6,7].
Although advancements in infrastructure and treatment technologies have enabled rapid intervention for IS, more than one-third of patients continue to experience temporary or permanent disabilities, such as motor function impairment or dementia [8,9]. These post-stroke complications are closely correlated with both the severity and the frequency of the stroke [10,11]. The primary mechanism of complications arises from damage to brain parenchymal cells due to nutrient-depleted hypoxia and reperfusion injury [12]. The underlying pathophysiological processes include excitotoxicity, oxidative stress, inflammatory responses, disruption of the blood-brain barrier (BBB), and apoptosis [12,13,14]. Since the 1960s, numerous neuroprotective agents have entered clinical trials, but most candidates failed to demonstrate sufficient efficacy, likely due to a lack of preclinical models that accurately mimic the complex
pathology of stroke, particularly in elderly populations [14,15,16]. Furthermore, clinical trials involving cell therapies aimed at tissue replacement or modulation of inflammation have faced significant challenges, including low therapeutic efficacy and unexpected adverse events [17,18,19,20].
Over the last decade, increasing attention has been directed toward the potential of extracellular vesicles (EVs) as an alternative therapeutic strategy for IS [21]. EVs secreted by nearly all cell types facilitate intercellular communication by delivering their luminal cargos to target cells [22]. Moreover, EVs derived from various cell types have been shown to enhance neurogenesis [23], promote angiogenesis [24], attenuate inflammation [25,26], and reduce oxidative stress [27], all of which are critical for the treatment of IS (Table 1). Additionally, the luminal cargos of EVs from damaged tissues reflect disease progression, allowing the EVs to serve as potential biomarkers in various neurological disorders, including stroke [28,29]. Because they can cross the BBB, and their properties and cargos can be modified relatively easily, EVs have emerged as attractive candidates for therapeutic applications in CNS-related disease [30,31].
In this review, we briefly summarize the general characteristics of EVs, describe the molecular mechanisms underlying the pathophysiology of IS, and discuss the naïve EV cargos that target these mechanisms. We also discuss recent advances in EV engineering that aim to improve the therapeutic efficacy of EVs.
2. General Characteristics of EVs
EVs are non-replicative, membrane-bound particles enclosed by a lipid bilayer that are naturally released by cells [32]. EVs are generally categorized into three main types based on their size and biogenesis: exosomes (40–200 nm), microvesicles (200–1,000 nm), and apoptotic bodies (500–2,000 nm) [33]. Exosomes are formed by inward budding of the endosomal membrane, leading to the generation of intraluminal vesicles. The intraruminal vesicles are released into the extracellular space through the fusion of multivesicular bodies, also known as late endosomes, with the plasma membrane [34]. Microvesicles and apoptotic bodies are produced and released by outward budding of the plasma membrane in normal and apoptotic cells, respectively [35].
EV biogenesis involves several key pathways, one of which requires the endosomal sorting complexes required for transport (ESCRT) protein complex. This complex plays a crucial role in cargo recruitment and sorting during EV formation [36]. EVs can also be produced by ESCRT-independent mechanisms, such as the ceramide-based sphingomyelinase (SMase) pathway or the tetraspanin-dependent pathway [37,38]. Tetraspanins (e.g., CD63, CD81, and CD9), ESCRT-associated proteins (e.g., Alix, TSG101, and Syntenin), and heat shock protein 70 (HSP70) are incorporated into EVs during biogenesis and serve as conventional markers for identifying EVs [32]. EVs are also capable of encapsulating a variety of bioactive molecules, including proteins, lipids, and nucleic acids (double-stranded DNAs, mRNAs, and microRNAs [miRNAs]), which they subsequently deliver to target cells [39]. In the CNS, EVs secreted by stimulated cortical neurons are preferentially taken up by neurons rather than glial cells [40]. Additionally, glioblastoma-derived EVs can modulate the local immune environment by transferring the miRNAs miR-451 and miR-21 to monocytes and macrophages in the brain [41].
Once secreted, EVs interact with recipient cells either by being taken up directly or by activating receptors on the plasma membrane [42,43]. Direct uptake of EVs can occur by various mechanisms, including clathrin- or caveolin-mediated endocytosis, phagocytosis, micropinocytosis, lipid raft-mediated endocytosis, and membrane fusion [44]. Specific EV surface proteins, such as lactadherin and CD209, play critical roles in facilitating EV uptake by interacting with corresponding surface proteins on recipient cells [45,46]. EV surface proteins can also prevent uptake by non-target cells; for example, EVs expressing CD47 can evade clearance by monocytes, allowing them to persist and function in an endocrine manner [47].
3. The Molecular Pathophysiology of IS and the Therapeutic Potential of EVs
The complications associated with IS arise from pathophysiological changes within the CNS triggered by restricted nutrient and oxygen supply, resulting in activation of glial cells and irreversible loss of brain parenchymal cells [12]. Omics data analyses have demonstrated that EVs, particularly those derived from mesenchymal stem cells (MSCs), carry a diverse array of anti-inflammatory or anti-apoptotic cargos while simultaneously promoting neurogenesis and angiogenesis for tissue repair [48]. In this section, we explore the molecular mechanisms driving IS pathologies and discuss the therapeutic potential of naïve EV cargos to mitigate these processes.
3.1. Excitotoxicity
Exitotoxicity refers to nerve cell damage or death caused by excessive stimulation by neurotransmitters. IS begins with a reduction in cerebral blood flow, leading to a decrease in ATP levels that disrupts transmembrane ionic gradients (Figure 1A). This disruption triggers anoxic depolarization and excessive release of neurotransmitters into the extracellular space [49]. The released neurotransmitters cannot be sufficiently cleared due to the insufficient ATP supply, resulting in their continued accumulation in the extracellular environment. Glutamate, an excitatory neurotransmitter, induces calcium ion (Ca2+) influx by activating N-methyl-D-aspartate receptors (NMDARs), kainate receptors, and α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptors (AMPARs) [50]. The Ca2+ overload is further exacerbated by Ca2+ release from the endoplasmic reticulum through the mGluR–PLC pathway, calcium-dependent protease (e.g., calpain)-mediated cleavage of the sodium-calcium exchanger (NCX), and activation of other Ca2+-permeable channels such as acid-sensing ion channels (ASICs) and TRPM7 [51,52,53,54]. Excessive Ca2+ accumulation generates free radicals and initiates cell death pathways, ultimately leading to neuronal cell death [55]. Notably, NMDARs containing the GluN2B subunit can directly activate various cell death signals in neurons [55].
The potential use of EVs to treat IS by reducing extracellular glutamate concentrations has not been fully investigated. Recent studies showed that EVs derived from microglia and neurons deliver miR-124 to astrocytes, leading to upregulation of astrocytic glutamate transporter-1 (GLT-1) expression, which enhances the clearance of extracellular glutamate in normal conditions and in glioma [56,57] (Figure 1A). Additionally, EVs originating from astrocytes were found to contain glutamate transporters, suggesting a potential role in scavenging extracellular glutamate [58]. Although investigations of therapeutic EVs remain in the early stages, the results so far suggest that EVs might be useful in treating IS by reducing extracellular glutamate levels.
3.2. Oxidative/Nitrosative Stress
Oxidative stress in IS typically manifests in three distinct phases [59]. Initially, oxygen and glucose deprivation leads to decoupling of the mitochondrial respiratory chain, resulting in the accumulation of reduced intermediates and the generation of reactive oxygen species (ROS) (Figure 1B). The second phase involves the production of hydrogen peroxide (H2O2) as a result of xanthine oxidase (XO) activation. The final phase generally occurs during reoxygenation and is associated with NADPH oxidase (NOX) activation and a subsequent increase in Ca2+ concentrations. Concurrently, nitric oxide (NO) production, driven by the activation of the NMDARs (especially those containing the GluN2B subunit)–PSD95–neuronal nitric oxide synthase (nNOS) complex, further contributes to oxidative stress in the third phase [60]. Oxidative stress can also arise during excitotoxicity, primarily by ROS generation due to NOX and phospholipase A2 (PLA2) activation in response to Ca2+ influx [61,62]. The ROS and reactive nitrogen species (RNS) generated during IS can directly damage DNA, proteins, membranes, and other cellular compounds ultimately leading to cell death [63]. For instance, NO and superoxide anions produced by nNOS and NOX activation combine to form highly reactive peroxynitrite, which induces DNA fragmentation, lipid peroxidation, and disruption of the BBB [64].
Oxidative stress in IS can be mitigated by overexpression and nuclear translocation of the nuclear factor-erythroid 2-related factor 2 (Nrf2) transcription factor. In the nucleus, Nrf2 binds to the antioxidant response element (ARE), promoting the expression of antioxidant enzymes such as heme oxygenase (HO-1), glutathione peroxidase (GPx), catalase (CAT), and superoxide dismutase (SOD) [65,66]. Recent studies showed that neural stem cell (NSC)-derived EVs can stimulate Nrf2 translocation and increase the expression of SOD1, CAT, and GPx-1, thereby reducing intracellular ROS levels in neuronal hypoxia/reperfusion models [67] (Figure 1B). Additionally, EVs from mini-pig adipose-derived MSCs (AD-MSCs) were shown to decrease the expression of iNOS, NOX-1, and NOX-2 and reduce oxidized proteins in a rat model of acute IS [68]. Furthermore, human AD-MSC-derived EVs were found to lower ROS levels and restore mitochondrial respiratory chain function in H2O2-treated endothelial cells by delivering miR-146a-5p [69]. Recent studies demonstrated that EVs from young human donors are enriched with GST or NAMPT and can ameliorate age-related tissue damage by enhancing antioxidant capacity [70,71]. These findings highlight the potential of stem cell-derived EVs in IS therapy by restoring antioxidant balance and counteracting oxidative stress.
3.3. Inflammation and Ischemia/Reperfusion (I/R) Injury
As IS progresses, excitotoxicity and excessive ROS/RNS generation occurs from the center of the infarction (ischemic core). This process escalates with reperfusion, leading to brain cell death and the release of various danger-associated molecular patterns (DAMPs) such as ATP, heat shock protein (HSP), and high mobility group box 1 (HMGB1) [72]. Microglia and astrocytes are subsequently activated by DAMPs, oxidative stress, and other inflammatory signals. Along with other brain cells such as neurons and endothelial cells, the activated microglia and astrocytes release pro-inflammatory cytokines (e.g., TNF-α, IL-1β, and IL-6), chemokines (e.g., MCP-1 and MIP-1a), matrix metalloproteinases (MMPs), ROS, and NO, which collectively induce inflammation (Figure 1C) [72,73,74]. These released molecules stimulate the expression of cell adhesion molecules (e.g., ICAM-1 and E/P-selectin) in endothelial cells, initiating infiltration of peripheral immune cells, including leukocytes, monocytes, and lymphocytes [75]. Neutrophils, the first leukocytes to infiltrate, release large amounts of pro-inflammatory mediators and neutrophil extracellular traps, which further exacerbate brain injury by intensifying inflammation, releasing ROS/NO, and disrupting the BBB [76]. For instance, MMP-9 contributes to BBB disruption by degrading endothelial cell tight junction proteins (e.g., claudin-5) and cerebrovascular basal lamina proteins (e.g., collagen-4), resulting in detrimental effects including brain edema and hemorrhagic transformation [77]. The complement cascade also plays a role in this process; for example, C3a, an anaphylatoxin, and the C1 protein complex exacerbate I/R injury by promoting leukocyte infiltration and activating endothelial cells [78,79].
The inflammatory reactions in IS are mediated by various signaling pathways. Members of the mitogen-activated protein kinase (MAPK) family, including extracellular signal-regulated kinase 1/2 (ERK1/2), c-Jun N-terminal kinase (JNK), and p38, as well as the NF-κB subunits p65/RelA and p50, are activated in response to external signals such as DAMPs, ROS, and inflammatory cytokines, leading to upregulation of pro-inflammatory mediators that exacerbate IS pathology [80,81,82,83,84] (Figure 1C). Additionally, Toll-like receptors (TLRs), which serve as upstream regulators of MAPKs and NF-κB, induce inflammation by responding to DAMPs [85].
Recent findings suggest that EVs secreted by various cell types may target inflammatory pathways and offer therapeutic benefits in IS. For example, MSC-derived EVs reduced inflammation in a rodent model of transient middle cerebral artery occlusion (tMCAO), potentially by reversing CysLT2R-ERK1/2–mediated M1 polarization of microglia or by inhibiting immune cell infiltration from the blood [86,87]. Furthermore, astrocyte-derived EVs carrying miR-34c alleviated I/R injury in in vitro models of oxygen-glucose deprivation/reperfusion and in vivo tMCAO models by downregulating TLR7, NF-κB, and MAPK pathways [88] (Figure 1C). Anti-inflammatory effects can also be achieved by activating regulatory T cells that secrete anti-inflammatory cytokines such as IL-10 and TGF-β. Embryonic stem cells also release EVs containing TGF-β, Smad2, and Smad4 and were shown to reduce peripheral immune cell infiltration and neuroinflammation by promoting regulatory T cell expansion [89].
It is important to note that certain molecules and cells that promote inflammation and I/R injury might paradoxically play beneficial roles in IS under specific conditions. For example, ERK1/2 activated by brain-derived neurotrophic factor (BDNF) was shown to inhibit apoptosis by reducing caspase-3 activity in hypoxic-ischemic brain injury [90]. Additionally, MMP-9 and proliferating microglia were shown to contribute to neurovascular remodeling during the later stages of cerebral ischemia [91,92]. Therefore, the therapeutic application of EVs harboring immunomodulatory agents in IS requires careful consideration of the target signaling molecules and the time window for treatments.
3.4. Ischemic Brain Cell Death
Various molecular mechanisms contribute to ischemic brain cell death (Figure 2) [93]. One major pathway involves either activation of the Ca2+-dependent protease calpain [94,95] or induction of the mitochondrial permeability transition due to Ca2+ overload and oxidative stress [96,97]. These processes activate the intrinsic apoptosis pathway by causing the release of cytochrome C or apoptosis inducing factor (AIF) from mitochondria. Additionally, signaling molecules such as death-associated protein kinase 1 (DAPK1), JNKs, p38, and Notch, which are activated during IS, can induce p53 activation [98]. Once activated, p53 promotes the transcription of proapoptotic genes such as PUMA and NOXA, which can directly interact with Bcl-xL to permeabilize the outer mitochondrial membrane, ultimately triggering mitochondrial apoptosis [99,100,101].
Extracellular factors released during inflammatory responses, including TNF-α [102], Fas ligand (FasL) [103,104], and TNF-related apoptosis-inducing ligand (TRAIL) [104,105], can induce caspase-8 activation through ligand-receptor interactions, initiating both the extrinsic and the mitochondrial apoptosis pathways [106,107] (Figure 2). However, caspase activity may also be reduced under IS conditions [108,109]. In such cases, external signals, particularly TNF-α, prompt formation of the necrosome complex, which consists of receptor-interacting protein kinase (RIPK) 1, RIPK3, and mixed lineage kinase domain-like pseudokinase (MLKL), and induces necroptosis [109,110,111].
Under the acidic conditions of the ischemic brain, the intracellular free iron level increases as a result of ferritinophagy and iron dissociation from transferrin [112,113]. The accumulation of free iron, combined with ROS, enhances lipoxygenase-mediated lipid peroxidation, resulting in the formation of lipid hydroperoxides that trigger ferroptosis [114]. Additionally, excessive intracellular Ca2+ activates cytosolic PLA2, promoting the production of arachidonic acid (AA) [115] (Figure 2). The AA is then esterified into phosphatidylethanolamines by acyl-CoA synthetase long-chain family member 4 (ACSL4), further contributing to ferroptosis [116]. Conversely, GPx-4 utilizes glutathione (GSH) as a substrate to reduce lipid hydroperoxides, thereby inhibiting an iron-dependent cell death ferroptosis [117]. Excessive extracellular glutamate in IS inhibits the cystine/glutamate antiporter (System Xc-), impairing the uptake of cystine, an essential precursor of GSH, thereby reducing GPx-4 activity and increasing susceptibility to ferroptosis [118] (Figure 2). Moreover, DNA damage caused by excitotoxicity and ROS/RNS activates poly(ADP-ribose) polymerase 1 (PARP1), initiating a series of events including cytoplasmic translo-cation of poly(ADP-ribose), AIF release from mitochondria, AIF/macrophage migration inhibitory factor (MIF) translocation, and DNA cleavage, ultimately resulting in the induction of a cell death pathway known as parthanatos [119] (Figure 2). Cell death can also be mediated by the inflammasome, a multi-protein complex composed of a single type of sensor protein, such as NLRP or AIM2, along with the adaptor ASC and pro-caspase-1. This complex activates caspase-1 and eventually induces pyroptosis, a type of inflammatory cell death [120,121]. Alternatively, exposure of phosphatidylserine (PS) on the outer leaflet of the plasma membrane and expression of proteins that mediate PS recognition (e.g., MerTK and MFG-e8) can trigger phagoptosis, the cell death induced by phagocytosis [122].
Although controversial findings exist, excessive autophagy can exacerbate IS by promoting neuron death [123]. In IS, inhibition of the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) pathway [124] and activation of ERK1/2 [125] both trigger autophagy-related cell death by inhibiting mammalian target of rapamycin complex 1 (mTORC1) activation (Figure 2). Additionally, activation of AMP-activated protein kinase (AMPK), either due to a high AMP/ATP ratio [126,127] or by calmodulin-dependent protein kinase kinase β (CaMKKβ) [128], causes autophagy-induced cell death by inhibiting mTORC1. Hypoxia-inducible factor 1 (HIF-1) is also activated in response to hypoxia, leading to upregulation of p53 and Bcl-2/adenovirus E1B 19-kDa-interacting protein 3 (BNIP3) [129,130], which contributes to excessive autophagy by promoting the release of beclin-1 from the Bcl-2/beclin-1 complex [131] or by inhibiting mTORC1 activation through its binding to Ras homolog enriched in brain (Rheb) [132] (Figure 2). In addition, p53 enhances expression of the damage-regulated autophagy modulator (DRAM) [133], potentially leading to excessive autophagy. Furthermore, the forkhead box O (FOXO) family proteins, which are regulated by Sirt1, AKT, and signal inducer and activator of transcription 3 (STAT3), can worsen IS outcomes by increasing the expression of autophagy-related proteins such as ATG7 [134,135,136,137,138]. This dysregulated autophagy can further exacerbate neuronal impairments in IS.
Recent studies suggest that the various forms of cell death observed in IS can be mitigated by EVs. MSC-derived EVs were shown to alleviate both extrinsic and intrinsic apoptosis [139,140]. Specifically, miR-134 delivered by exosomes inhibited apoptotic cell death of oligodendrocytes by targeting caspase-8 [140] (Figure 2). In addition, circBBS2 and miR-760-3p enriched in MSC-EVs targeted miR-494 and glutathione-specific gamma-glutamylcyclotransferase 1, respectively, thereby inhibiting ferroptosis by increasing System Xc- activity and boosting GSH levels [141,142] (Figure 2). Furthermore, EVs derived from bone marrow MSCs were shown to inhibit pyroptosis by reducing the expression of NLRP3, ASC, gasdermin D, and mature IL-1β [143]. Neuronal EVs containing miR-98 were also reported to inhibit phagoptosis by reducing the expression of platelet-activating factor receptor (PAFR) [144]. Moreover, EVs from AD-MSCs and induced pluripotent stem cell-derived MSCs were shown to inhibit autophagy-associated cell death by modulating p53–BNIP3 signaling and STAT3 expression, respectively [24,145] (Figure 2). Although direct studies in IS models are limited, inhibition of RIPK1/3 and PARP1 expression by MSC-derived EVs in other injury models suggests that these EVs hold potential for inhibiting necroptosis and parthanatos [146,147,148].
4. EV Engineering Methods
Advances in understanding the molecular mechanisms underlying IS pathophysiology have led to the development of strategies to target these mechanisms using bioactive molecules. The low permeability of the BBB poses a significant obstacle to non-invasive drug delivery for IS treatment; however, EVs exhibit a range of beneficial effects in IS-related pathologies and can cross the BBB, maintain circulation stability, and protect internal cargos with their lipid bilayer. These advantages, along with the therapeutic efficacy of EVs in IS, can be enhanced by modifications that enable EVs to precisely regulate signaling pathways in specific target cells (Table 2) [149].
Generally, EV engineering methods can be categorized depending on whether modifications are made before or after EV isolation (Figure 3). Pre-isolation modifications involve various pretreatments or gene transfection of EV-producing cells, whereas post-isolation modifications involve passive methods, such as co-incubation with desired target molecules, as well as active methods that use physicochemical stimulation to enable loading of exogenous factors [31].
4.1. Pre-Isolation Modification: Pretreatment and Gene Transfection
Various pretreatments of EV-producing cells can enhance the efficacy of EVs for IS treatment by altering the composition of the EV cargo. In vitro treatments with cytokines [150,151], metallic compounds [152], magnetic nanoparticles [153], drugs [154], and therapeutic biomolecules [155] under normal or hypoxic culture conditions [156] have been shown to modulate the activity or polarization state of EV-producing cells, including NSCs, MSCs, and macrophages (Figure 3A). These methods either facilitate the loading of extrinsic therapeutic agents into EVs or upregulate the intrinsic levels of neurotrophic, angiogenic, and anti-inflammatory cytokines or cell survival-related miRNAs within the EVs. These modifications potentiate the ability of EVs to inhibit ischemic brain damage while promoting tissue regeneration and neurological recovery.
Enrichment of TGF-β, miR-124, and miR-133a in EVs, achieved by hypoxic conditioning or inflammatory cytokine treatment of EV-producing cells, has been shown to reduce neural cell death in tMCAO animal models [150,151,154]. In addition, systemic administration of EVs derived from magnetic nanoparticle-treated MSCs improved the efficiency of EV delivery to IS lesions in a rat MCAO model [153]. Furthermore, exosomes from melatonin-treated rat plasma inhibited microglial and neuronal pyroptosis, partially by downregulating the TLR4/NF-κB pathway [157]. Additionally, mice that underwent moderate exercise before MCAO surgery exhibited increased levels of miR-126 in EVs obtained from circulating endothelial progenitor cells, which enhanced neurogenesis and angiogenesis by promoting BDNF secretion and PI3K/AKT signaling activation [158]. Exercise also elevated miR-484 levels in skeletal muscle-derived EVs, which targeted ACSL4 and thereby inhibited neuronal ferroptosis in a rat model of I/R injury [159].
Gene transfection of EV-producing cells is another commonly used method to modify the contents of EVs. Overexpression to increase the cytosolic levels of therapeutic proteins or RNAs in EV-producing cells can lead to enrichment of these molecules within EVs [160,161,162,163] (Figure 3A). Alternatively, specific proteins can be selectively loaded into EVs by creating EV-related fusion proteins [164], using the protein loading platform known as the exosomes for protein loading via optically reversible protein-protein interaction(EXPLORs) [165], or employing VSV-G and split GFP complementation [166]. In addition, specific RNAs can be loaded into EVs by incorporating specific motif sequences (e.g. hnRNPA2B1 binding motif [167] and Zip code-like sequence [168]) in the RNAs or by using the Targeted and Modular EV Loading (TAMEL) platform [169].
4.2. Post-Isolation Modification: Passive and Active Methods
One of the simplest methods for engineering EVs is to incubate isolated EVs with molecules that enhance the therapeutic potential of the EVs. Molecules conjugated to hydrophobic lipid derivatives, such as cholesterol and phospholipid-polyethylene glycol, can integrate into the EV membrane during simple incubations [170,171] (Figure 3B). Non-hydrophobic molecules that have an affinity for EV surface molecules can also be attached to the periphery of EVs by incubation. Additionally, physical stimulation allows incorporation of non-hydrophobic molecules in the luminal space of EVs by creating micropores in the EV membrane. Sonication or electroporation of a mixture of EVs and therapeutic molecules is the most common method for actively loading desired cargos into EVs via micropores. Repeated freeze-thaw cycles provide another physical way to actively load molecules into EVs. Cargos can also be loaded into EVs by extrusion, dialysis, or permeabilization with surfactants (e.g., saponin, Triton X-100, or Tween-80) [172,173,174,175]. Recent studies demonstrated that saponin-mediated cargo loading achieved an 11-fold higher efficiency for loading hydrophilic porphyrins compared with passive loading [174]. Furthermore, EVs that were actively loaded with catalase using saponin achieved higher neuron survival rates and more effective ROS removal compared with passively loaded EVs [173].
The surface of EVs can be chemically modified by carbodiimide coupling or copper-catalyzed azide-alkyne cycloaddition (click chemistry). Compounds with carbodiimide functional groups, such as 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride, act as crosslinkers by catalyzing linkages between carboxyl and amino groups. Using this chemical method, EV surfaces can be conjugated with antibodies that bind to specific molecules highly expressed in target cells or tissues under pathological conditions (Figure 3B). Additionally, carbodiimide chemistry was used to introduce alkyne groups onto EV surface proteins, which were then conjugated with azide-fluor 545 by click chemistry for fluorescent labeling of the EVs [176].
4.3. EV Engineering for IS Treatment
The therapeutic potential of EVs in IS can be enhanced by modifying the luminal or surface cargo molecules in EVs before or after their isolation using several approaches (Figure 3C). Recent studies demonstrated that increasing the levels of therapeutic proteins, mRNAs, miRNAs, and circRNAs within EVs can enhance the therapeutic efficacy of the EVs for treating IS. For example, induction of BDNF overexpression in MSCs by lentiviral transfection led to BDNF enrichment in MSC-derived EVs, which reduced apoptosis and inflammation and promoted behavioral recovery and neural repair by activating BDNF–tropomyosin receptor kinase B (TrkB) signaling [160] (Figure 3C). Furthermore, EVs derived from MSCs induced to overexpress the miR-17-92 cluster by electroporation and lentiviral infection were shown to increase neuronal plasticity, promote neurogenesis and oligodendrogenesis, and improve behavioral outcomes. This effect was achieved by remodeling the cortico-spinal tract and enhancing neuronal innervation via PTEN downregulation and PI3K/AKT/mTOR signaling activation [161,162]. In another study, EVs derived from circSCMH1-overexpressing HEK293T cells enhanced neuronal plasticity, reduced glial activation, and decreased peripheral immune cell infiltration in a mouse model of IS. These therapeutic benefits were mediated by direct binding between circSCMH1 and methyl CpG binding protein 2 (MeCP2), which inhibited the nuclear localization of MeCP2 [163]. Notably, circSCMH1-containing EVs also facilitated functional recovery in a nonhuman primate model of stroke [163].
Pre-incubation of MSC-derived EVs with cholesterol-conjugated miR-210 promoted vascular endothelial growth factor (VEGF) expression and angiogenesis after administration into a mouse MCAO model [170]. Additionally, bone marrow stromal cell-derived EVs that were loaded with the hydrophobic anti-inflammatory agent curcumin caused significant reduction of pro-inflammatory cytokine secretion and apoptosis after intravenous administration in a mouse model of IS [177]. Strong free radical scavengers quercetin and baicalin, which were incorporated into EVs by creating micropores using sonication, enhanced the innate potential of the EVs to inhibit ROS generation through the Nrf2/heme oxygenase pathway [172,178]. Additionally, EVs loaded with miR-124 or HMGB1 siRNA by electroporation enhanced neurogenesis and reduced TNF-α expression and apoptosis, respectively [179,180]. Furthermore, EVs that encapsulated the antioxidant drug edaravone by sonication improved the safety and bioavailability of the drug, thereby intensifying its neuroprotective effect [181]. Mouse embryonic stem cell-derived EVs enriched with curcumin via two or three rapid freeze-thaw cycles enhanced abilities to reduce inflammation, glial activation, and loss of vascular integrity in a mouse model of IS [182].
The expression of specific peptides on the EV surface can enhance the targeted uptake of EVs by ischemic brain lesions. For instance, the ischemic brain can be targeted by fusing the Lamp2b EV membrane protein with the rabies viral glycoprotein (RVG) peptide, which specifically binds to acetylcholine receptors [163] (Figure 3C). Lamp2b can also be fused with a peptide (RBP) that binds to the receptor for advanced glycation end products (RAGE), which is highly expressed in hypoxic cells of the ischemic brain, providing the additional benefit of potentially alleviating DAMP-induced inflammation by directly blocking RAGE [183]. Additionally, a fusion protein consisting of the PS-binding domain (C1C2) of MFG-e8 and the Arg-Gly-Asp (RGD)-4C peptide (ACDCRGDCFC) was attached to EVs by incubation, facilitating targeted delivery of the EVs to ischemic brain [184]. During the incubation, the C1C2-RGD fusion protein bound to PS, which is abundant on the EV surface. After systemic injection, the interaction between the RGD peptide on EVs and integrin αvβ3, which is highly expressed on reactive cerebral vascular endothelial cells in brain ischemia, enhanced the EV targeting efficiency. Sustained EV delivery was also achieved by directly mixing embryonic NSC-derived EVs with a glucose/ROS dual-responsive hydrogel [185]. Transplantation of this mixture into the cortex of infarcted brain hemispheres enhanced angiogenesis and improved neurobehavioral recovery. Carbodiimide coupling can be used to conjugate EV surface proteins with an antibody targeting growth-associated protein-43 (GAP43), which shows increased neuronal expression in a rat model of IS [172] (Figure 3C). This modification effectively enhanced targeted delivery of the EVs to ischemic brain tissues. Furthermore, EVs treated with dibenzocyclooctyne-sulfo-N-hydroxysuccinimidyl ester (DBCO-sulfo-NHS) can be reacted with the azide-containing cyclo(Arg-Gly-Asp-D-Tyr-Lys) peptide [c(RGDyK)], which has a high affinity for integrin αvβ3 [177]. This copper-free click chemistry method covalently attached c(RGDyK) to the EV surface, thereby facilitating targeted delivery of EVs to the lesion area in the ischemic brain.
5. Discussion
EVs have emerged as important messengers of intercellular crosstalk and gained attention for their crucial roles in diverse physiological and pathological processes across many organs. Numerous studies have explored the application of EVs for the treatment of various diseases. EV-based therapies offer several advantages over traditional cell therapies by addressing issues such as low cell viability, thromboembolism following intravenous injection, and vascular occlusion due to cell entrapment [186]. EVs also provide benefits of low toxicity, high stability in circulation, scalable production and storage, ability to cross the BBB, and enhanced targeting efficiency [186]. Furthermore, recent findings have demonstrated that administration of EVs in IS can achieve therapeutic outcomes comparable to those of direct cell injections [187]. Engineered EVs in particular have shown improved efficiency in targeting ischemic brain lesions, resulting in reduced inflammation and cell death and enhanced neurogenesis and angiogenesis compared with naïve EVs, leading to safer and more effective functional recovery.
Another promising application of EVs is the rapid diagnosis of IS. Patients with IS often experience slow disease progression due to high collateral blood flow capacity and overall cerebral ischemic tolerance [188]. Alteplase, the only drug approved by the USFDA for IS treatment, is a recombinant tPA that is most effective when administered early, ideally within 4¬–5 hours of IS onset, highlighting the importance of rapid diagnosis [6]. EVs derived from blood or cerebrospinal fluid of patients with IS or animal models show altered levels of non-coding RNAs and proteins, offering potential diagnostic markers [189]. For example, circulating EVs from patients with IS and tMCAO rat models exhibit increased levels of miR-20b-5p and miR-93-5p [190]. Inflammatory proteins such as C-reactive protein (CRP) are also elevated in EVs derived from the serum of patients with acute IS [191]. Furthermore, 67 miRNAs in blood-circulating EVs were found to differ significantly between the ischemic and hemorrhagic subtypes of stroke [192]. This suggests that EVs may help diagnose disease progression and distinguish between major stroke subtypes, enabling more rapid and effective treatments.
The limited blood flow in ischemic regions can be restored by tPA-induced clot lysis; however, tPA may also promote neutrophil degranulation and MMP-9 release, potentially increasing the risk of hemorrhagic transformation [193]. Therefore, several clinical studies are currently investigating EVs as promising alternative treatments for IS (NCT06138210, NCT03384433, and NCT05326724) [194]. A phase 1 clinical trial (NCT06138210) conducted by Xuanwu Hospital is assessing the safety and preliminary efficacy of intravenous administration of exosomes derived from human induced pluripotent stem cells (GD-iExo-003) in patients with acute IS. Another phase 1/2 trial (NCT03384433) by Isfahan University of Medical Sciences is investigating the therapeutic effects of allogenic MSC-derived exosomes transfected with miR-124 in patients with acute IS. A pilot study involving five participants with IS who received allogenic placental MSC-derived exosomes showed no adverse effects [195].
The therapeutic application of EVs is still in early stages, and several challenges must be addressed before EVs can be used as therapeutic agents for IS [186,196]. One critical challenge is the reproducible production and quality control of EVs. The characteristics and purity of EVs can vary depending on their source, the environment of the EV-producing cells, and the techniques used for EV isolation and storage. Therefore, it is essential to establish precise and strictly monitored conditions for EV harvesting, along with the use of appropriate markers and analytical methods for quality control. Additionally, a detailed understanding of the in vivo biological activity of administered EVs is needed. Because of the diversity of EV cargos, EV application can affect not only the ischemic brain environment but also the overall physiological conditions of the body. Furthermore, different modes of action may be required depending on whether IS is in the acute or chronic phase. Therefore, therapeutic parameters including administration routes, biodistribution profiles, comprehensive biological functions of EV cargos, optimal treatment timing and dosages, and cytotoxicity profiles must be thoroughly studied to advance the application of EVs as therapeutic agents for IS.
Author Contributions
Conceptualization, J.L.; writing—original draft preparation, J.L.; writing—review and editing, J.L., D.G., D.H.P. and J.H.K.; visualization, J.L.; supervision, J.L., D.G., D.H.P. and J.H.K. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Research Foundation of Korea under grant No. RS-2023-00280923 and by the Korean Fund for Regenerative Medicine under grant No. 22A0204L1 from the Ministry of Science & ICT and the Ministry of Health & Welfare.
Data Availability Statement
Details are available from authors.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Sacco, R.L.; Kasner, S.E.; Broderick, J.P.; Caplan, L.R.; Connors, J.J.; Culebras, A.; Elkind, M.S.; George, M.G.; Hamdan, A.D.; Higashida, R.T.; et al. An updated definition of stroke for the 21st century: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2013, 44, 2064–2089. [Google Scholar] [CrossRef]
- Collaborators, G.B.D.S. Global, regional, and national burden of stroke and its risk factors, 1990-2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurol 2021, 20, 795–820. [Google Scholar] [CrossRef]
- Ferrari, A.J.; Santomauro, D.F.; Aali, A.; Abate, Y.H.; Abbafati, C.; Abbastabar, H.; Abd ElHafeez, S.; Abdelmasseh, M.; Abd-Elsalam, S.; Abdollahi, A.; et al. Global incidence, prevalence, years lived with disability (YLDs), disability-adjusted life-years (DALYs), and healthy life expectancy (HALE) for 371 diseases and injuries in 204 countries and territories and 811 subnational locations, 1990–2021: a systematic analysis for the Global Burden of Disease Study 2021. The Lancet 2024, 403, 2133–2161. [Google Scholar] [CrossRef]
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Anderson, C.A.M.; Arora, P.; Avery, C.L.; Baker-Smith, C.M.; Beaton, A.Z.; Boehme, A.K.; Buxton, A.E.; et al. Heart Disease and Stroke Statistics-2023 Update: A Report From the American Heart Association. Circulation 2023, 147, e93–e621. [Google Scholar] [CrossRef]
- Campbell, B.C.V.; Khatri, P. Stroke. Lancet 2020, 396, 129–142. [Google Scholar] [CrossRef]
- Emberson, J.; Lees, K.R.; Lyden, P.; Blackwell, L.; Albers, G.; Bluhmki, E.; Brott, T.; Cohen, G.; Davis, S.; Donnan, G.; et al. Effect of treatment delay, age, and stroke severity on the effects of intravenous thrombolysis with alteplase for acute ischaemic stroke: a meta-analysis of individual patient data from randomised trials. The Lancet 2014, 384, 1929–1935. [Google Scholar] [CrossRef]
- Mistry, E.A.; Mistry, A.M.; Nakawah, M.O.; Chitale, R.V.; James, R.F.; Volpi, J.J.; Fusco, M.R. Mechanical Thrombectomy Outcomes With and Without Intravenous Thrombolysis in Stroke Patients: A Meta-Analysis. Stroke 2017, 48, 2450–2456. [Google Scholar] [CrossRef]
- Hankey, G.J.; Spiesser, J.; Hakimi, Z.; Bego, G.; Carita, P.; Gabriel, S. Rate, degree, and predictors of recovery from disability following ischemic stroke. Neurology 2007, 68, 1583–1587. [Google Scholar] [CrossRef]
- Pendlebury, S.T.; Rothwell, P.M.; Oxford Vascular, S. Incidence and prevalence of dementia associated with transient ischaemic attack and stroke: analysis of the population-based Oxford Vascular Study. Lancet Neurol 2019, 18, 248–258. [Google Scholar] [CrossRef]
- Park, J.H.; Ovbiagele, B. Relationship of functional disability after a recent stroke with recurrent stroke risk. Eur J Neurol 2016, 23, 361–367. [Google Scholar] [CrossRef]
- Koton, S.; Pike, J.R.; Johansen, M.; Knopman, D.S.; Lakshminarayan, K.; Mosley, T.; Patole, S.; Rosamond, W.D.; Schneider, A.L.C.; Sharrett, A.R.; et al. Association of Ischemic Stroke Incidence, Severity, and Recurrence With Dementia in the Atherosclerosis Risk in Communities Cohort Study. JAMA Neurol 2022, 79, 271–280. [Google Scholar] [CrossRef]
- Campbell, B.C.V.; De Silva, D.A.; Macleod, M.R.; Coutts, S.B.; Schwamm, L.H.; Davis, S.M.; Donnan, G.A. Ischaemic stroke. Nat Rev Dis Primers 2019, 5, 70. [Google Scholar] [CrossRef]
- George, Paul M.; Steinberg, Gary K. Novel Stroke Therapeutics: Unraveling Stroke Pathophysiology and Its Impact on Clinical Treatments. Neuron 2015, 87, 297–309. [Google Scholar] [CrossRef]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci 1999, 22, 391–397. [Google Scholar] [CrossRef]
- O'Collins, V.E.; Macleod, M.R.; Donnan, G.A.; Horky, L.L.; van der Worp, B.H.; Howells, D.W. 1,026 Experimental treatments in acute stroke. Annals of Neurology 2006, 59, 467–477. [Google Scholar] [CrossRef]
- Paul, S.; Candelario-Jalil, E. Emerging neuroprotective strategies for the treatment of ischemic stroke: An overview of clinical and preclinical studies. Experimental Neurology 2021, 335. [Google Scholar] [CrossRef]
- Hess, D.C.; Wechsler, L.R.; Clark, W.M.; Savitz, S.I.; Ford, G.A.; Chiu, D.; Yavagal, D.R.; Uchino, K.; Liebeskind, D.S.; Auchus, A.P.; et al. Safety and efficacy of multipotent adult progenitor cells in acute ischaemic stroke (MASTERS): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol 2017, 16, 360–368. [Google Scholar] [CrossRef]
- Savitz, S.I.; Yavagal, D.; Rappard, G.; Likosky, W.; Rutledge, N.; Graffagnino, C.; Alderazi, Y.; Elder, J.A.; Chen, P.R.; Budzik, R.F., Jr.; et al. A Phase 2 Randomized, Sham-Controlled Trial of Internal Carotid Artery Infusion of Autologous Bone Marrow-Derived ALD-401 Cells in Patients With Recent Stable Ischemic Stroke (RECOVER-Stroke). Circulation 2019, 139, 192–205. [Google Scholar] [CrossRef]
- Kondziolka, D.; Steinberg, G.K.; Wechsler, L.; Meltzer, C.C.; Elder, E.; Gebel, J.; Decesare, S.; Jovin, T.; Zafonte, R.; Lebowitz, J.; et al. Neurotransplantation for patients with subcortical motor stroke: a phase 2 randomized trial. J Neurosurg 2005, 103, 38–45. [Google Scholar] [CrossRef]
- Savitz, S.I.; Dinsmore, J.; Wu, J.; Henderson, G.V.; Stieg, P.; Caplan, L.R. Neurotransplantation of Fetal Porcine Cells in Patients with Basal Ganglia Infarcts: A Preliminary Safety and Feasibility Study. Cerebrovascular Diseases 2005, 20, 101–107. [Google Scholar] [CrossRef]
- Chen, J.; Chopp, M. Exosome Therapy for Stroke. Stroke 2018, 49, 1083–1090. [Google Scholar] [CrossRef]
- H. Rashed, M.; Bayraktar, E.; K. Helal, G.; Abd-Ellah, M.; Amero, P.; Chavez-Reyes, A.; Rodriguez-Aguayo, C. Exosomes: From Garbage Bins to Promising Therapeutic Targets. International Journal of Molecular Sciences 2017, 18. [Google Scholar] [CrossRef]
- Xin, H.; Li, Y.; Cui, Y.; Yang, J.J.; Zhang, Z.G.; Chopp, M. Systemic administration of exosomes released from mesenchymal stromal cells promote functional recovery and neurovascular plasticity after stroke in rats. J Cereb Blood Flow Metab 2013, 33, 1711–1715. [Google Scholar] [CrossRef]
- Xia, Y.; Ling, X.; Hu, G.; Zhu, Q.; Zhang, J.; Li, Q.; Zhao, B.; Wang, Y.; Deng, Z. Small extracellular vesicles secreted by human iPSC-derived MSC enhance angiogenesis through inhibiting STAT3-dependent autophagy in ischemic stroke. Stem Cell Res Ther 2020, 11, 313. [Google Scholar] [CrossRef]
- Yue, K.-Y.; Zhang, P.-R.; Zheng, M.-H.; Cao, X.-L.; Cao, Y.; Zhang, Y.-Z.; Zhang, Y.-F.; Wu, H.-N.; Lu, Z.-H.; Liang, L.; et al. Neurons can upregulate Cav-1 to increase intake of endothelial cells-derived extracellular vesicles that attenuate apoptosis via miR-1290. Cell Death & Disease 2019, 10. [Google Scholar] [CrossRef]
- Huang, S.; Ge, X.; Yu, J.; Han, Z.; Yin, Z.; Li, Y.; Chen, F.; Wang, H.; Zhang, J.; Lei, P. Increased miR-124-3p in microglial exosomes following traumatic brain injury inhibits neuronal inflammation and contributes to neurite outgrowth via their transfer into neurons. FASEB J 2018, 32, 512–528. [Google Scholar] [CrossRef]
- de Godoy, M.A.; Saraiva, L.M.; de Carvalho, L.R.P.; Vasconcelos-Dos-Santos, A.; Beiral, H.J.V.; Ramos, A.B.; Silva, L.R.P.; Leal, R.B.; Monteiro, V.H.S.; Braga, C.V.; et al. Mesenchymal stem cells and cell-derived extracellular vesicles protect hippocampal neurons from oxidative stress and synapse damage induced by amyloid-beta oligomers. J Biol Chem 2018, 293, 1957–1975. [Google Scholar] [CrossRef]
- Simpson, R.J.; Lim, J.W.; Moritz, R.L.; Mathivanan, S. Exosomes: proteomic insights and diagnostic potential. Expert Rev Proteomics 2009, 6, 267–283. [Google Scholar] [CrossRef]
- Colombo, E.; Borgiani, B.; Verderio, C.; Furlan, R. Microvesicles: novel biomarkers for neurological disorders. Front Physiol 2012, 3, 63. [Google Scholar] [CrossRef]
- Yang, T.; Martin, P.; Fogarty, B.; Brown, A.; Schurman, K.; Phipps, R.; Yin, V.P.; Lockman, P.; Bai, S. Exosome delivered anticancer drugs across the blood-brain barrier for brain cancer therapy in Danio rerio. Pharm Res 2015, 32, 2003–2014. [Google Scholar] [CrossRef]
- Garcia-Manrique, P.; Matos, M.; Gutierrez, G.; Pazos, C.; Blanco-Lopez, M.C. Therapeutic biomaterials based on extracellular vesicles: classification of bio-engineering and mimetic preparation routes. J Extracell Vesicles 2018, 7, 1422676. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. Journal of Extracellular Vesicles 2018, 7. [Google Scholar] [CrossRef]
- Shao, H.; Im, H.; Castro, C.M.; Breakefield, X.; Weissleder, R.; Lee, H. New Technologies for Analysis of Extracellular Vesicles. Chem Rev 2018, 118, 1917–1950. [Google Scholar] [CrossRef]
- Kowal, J.; Tkach, M.; Théry, C. Biogenesis and secretion of exosomes. Current Opinion in Cell Biology 2014, 29, 116–125. [Google Scholar] [CrossRef]
- Doyle, L.M.; Wang, M.Z. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8. [Google Scholar] [CrossRef]
- Hurley, J.H. ESCRTs are everywhere. EMBO J 2015, 34, 2398–2407. [Google Scholar] [CrossRef]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brugger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef]
- van Niel, G.; Charrin, S.; Simoes, S.; Romao, M.; Rochin, L.; Saftig, P.; Marks, M.S.; Rubinstein, E.; Raposo, G. The tetraspanin CD63 regulates ESCRT-independent and -dependent endosomal sorting during melanogenesis. Dev Cell 2011, 21, 708–721. [Google Scholar] [CrossRef]
- Meldolesi, J. Exosomes and Ectosomes in Intercellular Communication. Current Biology 2018, 28, R435–R444. [Google Scholar] [CrossRef]
- Chivet, M.; Javalet, C.; Laulagnier, K.; Blot, B.; Hemming, F.J.; Sadoul, R. Exosomes secreted by cortical neurons upon glutamatergic synapse activation specifically interact with neurons. J Extracell Vesicles 2014, 3, 24722. [Google Scholar] [CrossRef]
- van der Vos, K.E.; Abels, E.R.; Zhang, X.; Lai, C.; Carrizosa, E.; Oakley, D.; Prabhakar, S.; Mardini, O.; Crommentuijn, M.H.; Skog, J.; et al. Directly visualized glioblastoma-derived extracellular vesicles transfer RNA to microglia/macrophages in the brain. Neuro Oncol 2016, 18, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Thery, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat Cell Biol 2019, 21, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jiao, Y.; Pan, Y.; Zhang, L.; Gong, H.; Qi, Y.; Wang, M.; Gong, H.; Shao, M.; Wang, X.; et al. Fetal Dermal Mesenchymal Stem Cell-Derived Exosomes Accelerate Cutaneous Wound Healing by Activating Notch Signaling. Stem Cells Int 2019, 2019, 2402916. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J Extracell Vesicles 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Véron, P.; Segura, E.; Sugano, G.; Amigorena, S.; Théry, C. Accumulation of MFG-E8/lactadherin on exosomes from immature dendritic cells. Blood Cells, Molecules, and Diseases 2005, 35, 81–88. [Google Scholar] [CrossRef]
- Naslund, T.I.; Paquin-Proulx, D.; Paredes, P.T.; Vallhov, H.; Sandberg, J.K.; Gabrielsson, S. Exosomes from breast milk inhibit HIV-1 infection of dendritic cells and subsequent viral transfer to CD4+ T cells. AIDS 2014, 28, 171–180. [Google Scholar] [CrossRef]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef]
- Varderidou-Minasian, S.; Lorenowicz, M.J. Mesenchymal stromal/stem cell-derived extracellular vesicles in tissue repair: challenges and opportunities. Theranostics 2020, 10, 5979–5997. [Google Scholar] [CrossRef]
- Obrenovitch, T.P.; Urenjak, J.; Richards, D.A.; Ueda, Y.; Curzon, G.; Symon, L. Extracellular neuroactive amino acids in the rat striatum during ischaemia: comparison between penumbral conditions and ischaemia with sustained anoxic depolarisation. J Neurochem 1993, 61, 178–186. [Google Scholar] [CrossRef]
- Choi, D.W. Excitotoxicity: Still Hammering the Ischemic Brain in 2020. Front Neurosci 2020, 14, 579953. [Google Scholar] [CrossRef]
- Pin, J.P.; Duvoisin, R. The metabotropic glutamate receptors: structure and functions. Neuropharmacology 1995, 34, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Bano, D.; Young, K.W.; Guerin, C.J.; Lefeuvre, R.; Rothwell, N.J.; Naldini, L.; Rizzuto, R.; Carafoli, E.; Nicotera, P. Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell 2005, 120, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.G.; Zhu, X.M.; Chu, X.P.; Minami, M.; Hey, J.; Wei, W.L.; MacDonald, J.F.; Wemmie, J.A.; Price, M.P.; Welsh, M.J.; et al. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell 2004, 118, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Aarts, M.; Iihara, K.; Wei, W.L.; Xiong, Z.G.; Arundine, M.; Cerwinski, W.; MacDonald, J.F.; Tymianski, M. A key role for TRPM7 channels in anoxic neuronal death. Cell 2003, 115, 863–877. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Progress in Neurobiology 2014, 115, 157–188. [Google Scholar] [CrossRef] [PubMed]
- Serpe, C.; Monaco, L.; Relucenti, M.; Iovino, L.; Familiari, P.; Scavizzi, F.; Raspa, M.; Familiari, G.; Civiero, L.; D’Agnano, I.; et al. Microglia-Derived Small Extracellular Vesicles Reduce Glioma Growth by Modifying Tumor Cell Metabolism and Enhancing Glutamate Clearance through miR-124. Cells 2021, 10. [Google Scholar] [CrossRef]
- Men, Y.; Yelick, J.; Jin, S.; Tian, Y.; Chiang, M.S.R.; Higashimori, H.; Brown, E.; Jarvis, R.; Yang, Y. Exosome reporter mice reveal the involvement of exosomes in mediating neuron to astroglia communication in the CNS. Nat Commun 2019, 10, 4136. [Google Scholar] [CrossRef]
- Gosselin, R.D.; Meylan, P.; Decosterd, I. Extracellular microvesicles from astrocytes contain functional glutamate transporters: regulation by protein kinase C and cell activation. Front Cell Neurosci 2013, 7, 251. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Scorziello, A.; Duchen, M.R. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci 2007, 27, 1129–1138. [Google Scholar] [CrossRef]
- Zhou, L.; Li, F.; Xu, H.-B.; Luo, C.-X.; Wu, H.-Y.; Zhu, M.-M.; Lu, W.; Ji, X.; Zhou, Q.-G.; Zhu, D.-Y. Treatment of cerebral ischemia by disrupting ischemia-induced interaction of nNOS with PSD-95. Nature Medicine 2010, 16, 1439–1443. [Google Scholar] [CrossRef]
- Reynolds, I.J.; Hastings, T.G. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J Neurosci 1995, 15, 3318–3327. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, K.; Li, R.C.; Zhang, J.; Klaus, J.A.; Kibler, K.K.; Dore, S.; Koehler, R.C.; Sapirstein, A. Cytosolic phospholipase A2 alpha amplifies early cyclooxygenase-2 expression, oxidative stress and MAP kinase phosphorylation after cerebral ischemia in mice. J Neuroinflammation 2010, 7, 42. [Google Scholar] [CrossRef] [PubMed]
- Orellana-Urzua, S.; Rojas, I.; Libano, L.; Rodrigo, R. Pathophysiology of Ischemic Stroke: Role of Oxidative Stress. Curr Pharm Des 2020, 26, 4246–4260. [Google Scholar] [CrossRef]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhu, X.; Kim, Y.; Li, J.; Huang, S.; Saleem, S.; Li, R.C.; Xu, Y.; Dore, S.; Cao, W. Histone deacetylase inhibition activates transcription factor Nrf2 and protects against cerebral ischemic damage. Free Radic Biol Med 2012, 52, 928–936. [Google Scholar] [CrossRef]
- Duan, J.; Cui, J.; Yang, Z.; Guo, C.; Cao, J.; Xi, M.; Weng, Y.; Yin, Y.; Wang, Y.; Wei, G.; et al. Neuroprotective effect of Apelin 13 on ischemic stroke by activating AMPK/GSK-3beta/Nrf2 signaling. J Neuroinflammation 2019, 16, 24. [Google Scholar] [CrossRef]
- Liu, Q.; Tan, Y.; Qu, T.; Zhang, J.; Duan, X.; Xu, H.; Mu, Y.; Ma, H.; Wang, F. Therapeutic mechanism of human neural stem cell-derived extracellular vesicles against hypoxia-reperfusion injury in vitro. Life Sci 2020, 254, 117772. [Google Scholar] [CrossRef]
- Chen, K.H.; Chen, C.H.; Wallace, C.G.; Yuen, C.M.; Kao, G.S.; Chen, Y.L.; Shao, P.L.; Chen, Y.L.; Chai, H.T.; Lin, K.C.; et al. Intravenous administration of xenogenic adipose-derived mesenchymal stem cells (ADMSC) and ADMSC-derived exosomes markedly reduced brain infarct volume and preserved neurological function in rat after acute ischemic stroke. Oncotarget 2016, 7, 74537–74556. [Google Scholar] [CrossRef]
- Xiao, X.; Xu, M.; Yu, H.; Wang, L.; Li, X.; Rak, J.; Wang, S.; Zhao, R.C. Mesenchymal stem cell-derived small extracellular vesicles mitigate oxidative stress-induced senescence in endothelial cells via regulation of miR-146a/Src. Signal Transduct Target Ther 2021, 6, 354. [Google Scholar] [CrossRef]
- Yoshida, M.; Satoh, A.; Lin, J.B.; Mills, K.F.; Sasaki, Y.; Rensing, N.; Wong, M.; Apte, R.S.; Imai, S.I. Extracellular Vesicle-Contained eNAMPT Delays Aging and Extends Lifespan in Mice. Cell Metab 2019, 30, 329–342 e325. [Google Scholar] [CrossRef]
- Fafián-Labora, J.A.; Rodríguez-Navarro, J.A.; O’Loghlen, A. Small Extracellular Vesicles Have GST Activity and Ameliorate Senescence-Related Tissue Damage. Cell Metabolism 2020, 32, 71–86.e75. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Anrather, J. The immunology of stroke: from mechanisms to translation. Nat Med 2011, 17, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Gülke, E.; Gelderblom, M.; Magnus, T. Danger signals in stroke and their role on microglia activation after ischemia. Therapeutic Advances in Neurological Disorders 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Astrocyte Reactivity: Subtypes, States, and Functions in CNS Innate Immunity. Trends Immunol 2020, 41, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Frijns, C.J.; Kappelle, L.J. Inflammatory cell adhesion molecules in ischemic cerebrovascular disease. Stroke 2002, 33, 2115–2122. [Google Scholar] [CrossRef]
- Jickling, G.C.; Liu, D.; Ander, B.P.; Stamova, B.; Zhan, X.; Sharp, F.R. Targeting Neutrophils in Ischemic Stroke: Translational Insights from Experimental Studies. Journal of Cerebral Blood Flow & Metabolism 2015, 35, 888–901. [Google Scholar] [CrossRef]
- McColl, B.W.; Rothwell, N.J.; Allan, S.M. Systemic Inflammation Alters the Kinetics of Cerebrovascular Tight Junction Disruption after Experimental Stroke in Mice. The Journal of Neuroscience 2008, 28, 9451–9462. [Google Scholar] [CrossRef]
- Wu, F.; Zou, Q.; Ding, X.; Shi, D.; Zhu, X.; Hu, W.; Liu, L.; Zhou, H. Complement component C3a plays a critical role in endothelial activation and leukocyte recruitment into the brain. J Neuroinflammation 2016, 13, 23. [Google Scholar] [CrossRef]
- De Simoni, M.G.; Rossi, E.; Storini, C.; Pizzimenti, S.; Echart, C.; Bergamaschini, L. The powerful neuroprotective action of C1-inhibitor on brain ischemia-reperfusion injury does not require C1q. Am J Pathol 2004, 164, 1857–1863. [Google Scholar] [CrossRef]
- Maddahi, A.; Edvinsson, L. Cerebral ischemia induces microvascular pro-inflammatory cytokine expression via the MEK/ERK pathway. J Neuroinflammation 2010, 7, 14. [Google Scholar] [CrossRef]
- Shvedova, M.; Anfinogenova, Y.; Atochina-Vasserman, E.N.; Schepetkin, I.A.; Atochin, D.N. c-Jun N-Terminal Kinases (JNKs) in Myocardial and Cerebral Ischemia/Reperfusion Injury. Front Pharmacol 2018, 9, 715. [Google Scholar] [CrossRef] [PubMed]
- Barone, F.C.; Irving, E.A.; Ray, A.M.; Lee, J.C.; Kassis, S.; Kumar, S.; Badger, A.M.; Legos, J.J.; Erhardt, J.A.; Ohlstein, E.H.; et al. Inhibition of p38 mitogen-activated protein kinase provides neuroprotection in cerebral focal ischemia. Med Res Rev 2001, 21, 129–145. [Google Scholar] [CrossRef] [PubMed]
- Ridder, D.A.; Schwaninger, M. NF-κB signaling in cerebral ischemia. Neuroscience 2009, 158, 995–1006. [Google Scholar] [CrossRef] [PubMed]
- Fann, D.Y.; Lim, Y.A.; Cheng, Y.L.; Lok, K.Z.; Chunduri, P.; Baik, S.H.; Drummond, G.R.; Dheen, S.T.; Sobey, C.G.; Jo, D.G.; et al. Evidence that NF-kappaB and MAPK Signaling Promotes NLRP Inflammasome Activation in Neurons Following Ischemic Stroke. Mol Neurobiol 2018, 55, 1082–1096. [Google Scholar] [CrossRef] [PubMed]
- Marsh, B.J.; Williams-Karnesky, R.L.; Stenzel-Poore, M.P. Toll-like receptor signaling in endogenous neuroprotection and stroke. Neuroscience 2009, 158, 1007–1020. [Google Scholar] [CrossRef]
- Zhao, Y.; Gan, Y.; Xu, G.; Yin, G.; Liu, D. MSCs-Derived Exosomes Attenuate Acute Brain Injury and Inhibit Microglial Inflammation by Reversing CysLT2R-ERK1/2 Mediated Microglia M1 Polarization. Neurochem Res 2020, 45, 1180–1190. [Google Scholar] [CrossRef]
- Wang, C.; Borger, V.; Sardari, M.; Murke, F.; Skuljec, J.; Pul, R.; Hagemann, N.; Dzyubenko, E.; Dittrich, R.; Gregorius, J.; et al. Mesenchymal Stromal Cell-Derived Small Extracellular Vesicles Induce Ischemic Neuroprotection by Modulating Leukocytes and Specifically Neutrophils. Stroke 2020, 51, 1825–1834. [Google Scholar] [CrossRef]
- Wu, W.; Liu, J.; Yang, C.; Xu, Z.; Huang, J.; Lin, J. Astrocyte-derived exosome-transported microRNA-34c is neuroprotective against cerebral ischemia/reperfusion injury via TLR7 and the NF-kappaB/MAPK pathways. Brain Res Bull 2020, 163, 84–94. [Google Scholar] [CrossRef]
- Xia, Y.; Hu, G.; Chen, Y.; Yuan, J.; Zhang, J.; Wang, S.; Li, Q.; Wang, Y.; Deng, Z. Embryonic Stem Cell Derived Small Extracellular Vesicles Modulate Regulatory T Cells to Protect against Ischemic Stroke. ACS Nano 2021, 15, 7370–7385. [Google Scholar] [CrossRef]
- Han, B.H.; Holtzman, D.M. BDNF protects the neonatal brain from hypoxic-ischemic injury in vivo via the ERK pathway. J Neurosci 2000, 20, 5775–5781. [Google Scholar] [CrossRef]
- Zhao, B.-Q.; Wang, S.; Kim, H.-Y.; Storrie, H.; Rosen, B.R.; Mooney, D.J.; Wang, X.; Lo, E.H. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nature Medicine 2006, 12, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Lalancette-Hebert, M.; Gowing, G.; Simard, A.; Weng, Y.C.; Kriz, J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci 2007, 27, 2596–2605. [Google Scholar] [CrossRef] [PubMed]
- Tuo, Q.Z.; Zhang, S.T.; Lei, P. Mechanisms of neuronal cell death in ischemic stroke and their therapeutic implications. Med Res Rev 2022, 42, 259–305. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; He, H.; Zhan, S.; Krajewski, S.; Reed, J.C.; Gottlieb, R.A. Bid is cleaved by calpain to an active fragment in vitro and during myocardial ischemia/reperfusion. J Biol Chem 2001, 276, 30724–30728. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Xing, J.; Xiao, X.; Liou, A.K.; Gao, Y.; Yin, X.M.; Clark, R.S.; Graham, S.H.; Chen, J. Critical role of calpain I in mitochondrial release of apoptosis-inducing factor in ischemic neuronal injury. J Neurosci 2007, 27, 9278–9293. [Google Scholar] [CrossRef]
- Schinzel, A.C.; Takeuchi, O.; Huang, Z.; Fisher, J.K.; Zhou, Z.; Rubens, J.; Hetz, C.; Danial, N.N.; Moskowitz, M.A.; Korsmeyer, S.J. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A 2005, 102, 12005–12010. [Google Scholar] [CrossRef]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef]
- Datta, A.; Sarmah, D.; Mounica, L.; Kaur, H.; Kesharwani, R.; Verma, G.; Veeresh, P.; Kotian, V.; Kalia, K.; Borah, A.; et al. Cell Death Pathways in Ischemic Stroke and Targeted Pharmacotherapy. Translational Stroke Research 2020, 11, 1185–1202. [Google Scholar] [CrossRef]
- Nijboer, C.H.; Heijnen, C.J.; van der Kooij, M.A.; Zijlstra, J.; van Velthoven, C.T.J.; Culmsee, C.; van Bel, F.; Hagberg, H.; Kavelaars, A. Targeting the p53 pathway to protect the neonatal ischemic brain. Annals of Neurology 2011, 70, 255–264. [Google Scholar] [CrossRef]
- Niizuma, K.; Endo, H.; Nito, C.; Myer, D.J.; Chan, P.H. Potential Role of PUMA in Delayed Death of Hippocampal CA1 Neurons After Transient Global Cerebral Ischemia. Stroke 2009, 40, 618–625. [Google Scholar] [CrossRef]
- Endo, H.; Kamada, H.; Nito, C.; Nishi, T.; Chan, P.H. Mitochondrial translocation of p53 mediates release of cytochrome c and hippocampal CA1 neuronal death after transient global cerebral ischemia in rats. J Neurosci 2006, 26, 7974–7983. [Google Scholar] [CrossRef] [PubMed]
- Badiola, N.; Malagelada, C.; Llecha, N.; Hidalgo, J.; Comella, J.X.; Sabriá, J.; Rodríguez-Alvarez, J. Activation of caspase-8 by tumour necrosis factor receptor 1 is necessary for caspase-3 activation and apoptosis in oxygen–glucose deprived cultured cortical cells. Neurobiology of Disease 2009, 35, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Wu, Z.; Xu, J.; Xu, Y. Calreticulin Binds to Fas Ligand and Inhibits Neuronal Cell Apoptosis Induced by Ischemia-Reperfusion Injury. BioMed Research International 2015, 2015, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Martin-Villalba, A.; Herr, I.; Jeremias, I.; Hahne, M.; Brandt, R.; Vogel, J.; Schenkel, J.; Herdegen, T.; Debatin, K.M. CD95 ligand (Fas-L/APO-1L) and tumor necrosis factor-related apoptosis-inducing ligand mediate ischemia-induced apoptosis in neurons. J Neurosci 1999, 19, 3809–3817. [Google Scholar] [CrossRef] [PubMed]
- Cantarella, G.; Pignataro, G.; Di Benedetto, G.; Anzilotti, S.; Vinciguerra, A.; Cuomo, O.; Di Renzo, G.F.; Parenti, C.; Annunziato, L.; Bernardini, R. Ischemic tolerance modulates TRAIL expression and its receptors and generates a neuroprotected phenotype. Cell Death Dis 2014, 5, e1331. [Google Scholar] [CrossRef]
- Benchoua, A.; Guegan, C.; Couriaud, C.; Hosseini, H.; Sampaio, N.; Morin, D.; Onteniente, B. Specific caspase pathways are activated in the two stages of cerebral infarction. J Neurosci 2001, 21, 7127–7134. [Google Scholar] [CrossRef]
- Li, H.; Zhu, H.; Xu, C.J.; Yuan, J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef]
- Ferrari, D.; Stepczynska, A.; Los, M.; Wesselborg, S.; Schulze-Osthoff, K. Differential regulation and ATP requirement for caspase-8 and caspase-3 activation during CD95- and anticancer drug-induced apoptosis. J Exp Med 1998, 188, 979–984. [Google Scholar] [CrossRef]
- Vieira, M.; Fernandes, J.; Carreto, L.; Anuncibay-Soto, B.; Santos, M.; Han, J.; Fernandez-Lopez, A.; Duarte, C.B.; Carvalho, A.L.; Santos, A.E. Ischemic insults induce necroptotic cell death in hippocampal neurons through the up-regulation of endogenous RIP3. Neurobiol Dis 2014, 68, 26–36. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, M.; Li, X.; Zhang, H.; Wang, L.; Wu, X.; Zhang, H.; Luo, Y. Catalytically inactive RIP1 and RIP3 deficiency protect against acute ischemic stroke by inhibiting necroptosis and neuroinflammation. Cell Death Dis 2020, 11, 565. [Google Scholar] [CrossRef]
- Chen, A.Q.; Fang, Z.; Chen, X.L.; Yang, S.; Zhou, Y.F.; Mao, L.; Xia, Y.P.; Jin, H.J.; Li, Y.N.; You, M.F.; et al. Microglia-derived TNF-alpha mediates endothelial necroptosis aggravating blood brain-barrier disruption after ischemic stroke. Cell Death Dis 2019, 10, 487. [Google Scholar] [CrossRef] [PubMed]
- Lipscomb, D.C.; Gorman, L.G.; Traystman, R.J.; Hurn, P.D. Low molecular weight iron in cerebral ischemic acidosis in vivo. Stroke 1998, 29, 487–492, discussion 493. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Sun, G.; Chen, B.; Xu, L.; Ye, Y.; He, J.; Bao, Z.; Zhao, P.; Miao, Z.; Zhao, L.; et al. Nuclear receptor coactivator 4-mediated ferritinophagy contributes to cerebral ischemia-induced ferroptosis in ischemic stroke. Pharmacol Res 2021, 174, 105933. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, S.; Li, Q.; Sun, H.; Wang, H. Pharmacological Inhibition of Ferroptosis as a Therapeutic Target for Neurodegenerative Diseases and Strokes. Advanced Science 2023, 10. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, J.V.; Huang, Z.; Taheri, M.R.; O'Leary, E.; Li, E.; Moskowitz, M.A.; Sapirstein, A. Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature 1997, 390, 622–625. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic Biol Med 2020, 152, 175–185. [Google Scholar] [CrossRef]
- Ratan, R.R. The Chemical Biology of Ferroptosis in the Central Nervous System. Cell Chem Biol 2020, 27, 479–498. [Google Scholar] [CrossRef]
- Liu, S.; Luo, W.; Wang, Y. Emerging role of PARP-1 and PARthanatos in ischemic stroke. J Neurochem 2022, 160, 74–87. [Google Scholar] [CrossRef]
- Poh, L.; Kang, S.W.; Baik, S.H.; Ng, G.Y.Q.; She, D.T.; Balaganapathy, P.; Dheen, S.T.; Magnus, T.; Gelderblom, M.; Sobey, C.G.; et al. Evidence that NLRC4 inflammasome mediates apoptotic and pyroptotic microglial death following ischemic stroke. Brain Behav Immun 2019, 75, 34–47. [Google Scholar] [CrossRef]
- Ye, X.; Shen, T.; Hu, J.; Zhang, L.; Zhang, Y.; Bao, L.; Cui, C.; Jin, G.; Zan, K.; Zhang, Z.; et al. Purinergic 2X7 receptor/NLRP3 pathway triggers neuronal apoptosis after ischemic stroke in the mouse. Exp Neurol 2017, 292, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Neher, J.J.; Emmrich, J.V.; Fricker, M.; Mander, P.K.; Thery, C.; Brown, G.C. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc Natl Acad Sci U S A 2013, 110, E4098–4107. [Google Scholar] [CrossRef] [PubMed]
- Ajoolabady, A.; Wang, S.; Kroemer, G.; Penninger, J.M.; Uversky, V.N.; Pratico, D.; Henninger, N.; Reiter, R.J.; Bruno, A.; Joshipura, K.; et al. Targeting autophagy in ischemic stroke: From molecular mechanisms to clinical therapeutics. Pharmacol Ther 2021, 225, 107848. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.M.; Zhang, M.; Feng, Y.S.; Xing, Y.; Tan, Z.X.; Li, W.B.; Dong, F.; Zhang, F. Electroacupuncture Inhibits Neuronal Autophagy and Apoptosis via the PI3K/AKT Pathway Following Ischemic Stroke. Front Cell Neurosci 2020, 14, 134. [Google Scholar] [CrossRef]
- Xu, D.; Kong, T.; Zhang, S.; Cheng, B.; Chen, J.; Wang, C. Orexin-A protects against cerebral ischemia-reperfusion injury by inhibiting excessive autophagy through OX1R-mediated MAPK/ERK/mTOR pathway. Cell Signal 2021, 79, 109839. [Google Scholar] [CrossRef]
- Liang, J.; Shao, S.H.; Xu, Z.X.; Hennessy, B.; Ding, Z.; Larrea, M.; Kondo, S.; Dumont, D.J.; Gutterman, J.U.; Walker, C.L.; et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol 2007, 9, 218–224. [Google Scholar] [CrossRef]
- Nowosad, A.; Jeannot, P.; Callot, C.; Creff, J.; Perchey, R.T.; Joffre, C.; Codogno, P.; Manenti, S.; Besson, A. p27 controls Ragulator and mTOR activity in amino acid-deprived cells to regulate the autophagy-lysosomal pathway and coordinate cell cycle and cell growth. Nat Cell Biol 2020, 22, 1076–1090. [Google Scholar] [CrossRef]
- Sun, B.; Ou, H.; Ren, F.; Huan, Y.; Zhong, T.; Gao, M.; Cai, H. Propofol inhibited autophagy through Ca2+/CaMKKβ/AMPK/mTOR pathway in OGD/R-induced neuron injury. Molecular Medicine 2018, 24. [Google Scholar] [CrossRef]
- Chen, C.; Hu, Q.; Yan, J.; Yang, X.; Shi, X.; Lei, J.; Chen, L.; Huang, H.; Han, J.; Zhang, J.H.; et al. Early inhibition of HIF-1alpha with small interfering RNA reduces ischemic-reperfused brain injury in rats. Neurobiol Dis 2009, 33, 509–517. [Google Scholar] [CrossRef]
- Barteczek, P.; Li, L.; Ernst, A.S.; Bohler, L.I.; Marti, H.H.; Kunze, R. Neuronal HIF-1alpha and HIF-2alpha deficiency improves neuronal survival and sensorimotor function in the early acute phase after ischemic stroke. J Cereb Blood Flow Metab 2017, 37, 291–306. [Google Scholar] [CrossRef]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-Induced Autophagy Is Mediated through Hypoxia-Inducible Factor Induction of BNIP3 and BNIP3L via Their BH3 Domains. Molecular and Cellular Biology 2023, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Y.; Kim, E.; Beemiller, P.; Wang, C.Y.; Swanson, J.; You, M.; Guan, K.L. Bnip3 mediates the hypoxia-induced inhibition on mammalian target of rapamycin by interacting with Rheb. J Biol Chem 2007, 282, 35803–35813. [Google Scholar] [CrossRef] [PubMed]
- Crighton, D.; Wilkinson, S.; O'Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Yu, M.; Bu, Z.; He, P.; Feng, J. FKBP5 Exacerbates Impairments in Cerebral Ischemic Stroke by Inducing Autophagy via the AKT/FOXO3 Pathway. Front Cell Neurosci 2020, 14, 193. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Yu, M.; He, X.; Wen, L.; Bu, Z.; Feng, J. KCNQ1OT1 promotes autophagy by regulating miR-200a/FOXO3/ATG7 pathway in cerebral ischemic stroke. Aging Cell 2019, 18, e12940. [Google Scholar] [CrossRef]
- Mei, Z.G.; Huang, Y.G.; Feng, Z.T.; Luo, Y.N.; Yang, S.B.; Du, L.P.; Jiang, K.; Liu, X.L.; Fu, X.Y.; Deng, Y.H.; et al. Electroacupuncture ameliorates cerebral ischemia/reperfusion injury by suppressing autophagy via the SIRT1-FOXO1 signaling pathway. Aging (Albany NY) 2020, 12, 13187–13205. [Google Scholar] [CrossRef]
- Xu, S.; Huang, P.; Yang, J.; Du, H.; Wan, H.; He, Y. Calycosin alleviates cerebral ischemia/reperfusion injury by repressing autophagy via STAT3/FOXO3a signaling pathway. Phytomedicine 2023, 115, 154845. [Google Scholar] [CrossRef]
- Xie, W.; Zhu, T.; Zhang, S.; Sun, X. Protective effects of Gypenoside XVII against cerebral ischemia/reperfusion injury via SIRT1-FOXO3A- and Hif1a-BNIP3-mediated mitochondrial autophagy. Journal of Translational Medicine 2022, 20. [Google Scholar] [CrossRef]
- Mathew, B.; Ravindran, S.; Liu, X.; Torres, L.; Chennakesavalu, M.; Huang, C.-C.; Feng, L.; Zelka, R.; Lopez, J.; Sharma, M.; et al. Mesenchymal stem cell-derived extracellular vesicles and retinal ischemia-reperfusion. Biomaterials 2019, 197, 146–160. [Google Scholar] [CrossRef]
- Xiao, Y.; Geng, F.; Wang, G.; Li, X.; Zhu, J.; Zhu, W. Bone marrow-derived mesenchymal stem cells-derived exosomes prevent oligodendrocyte apoptosis through exosomal miR-134 by targeting caspase-8. J Cell Biochem 2019, 120, 2109–2118. [Google Scholar] [CrossRef] [PubMed]
- Hong, T.; Zhao, T.; He, W.; Xia, J.; Huang, Q.; Yang, J.; Gu, W.; Chen, C.; Zhang, N.; Liu, Y.; et al. Exosomal circBBS2 inhibits ferroptosis by targeting miR-494 to activate SLC7A11 signaling in ischemic stroke. FASEB J 2023, 37, e23152. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Niu, H.; Li, L.; Han, J.; Liu, Z.; Chu, M.; Sha, X.; Zhao, J. Anti-CHAC1 exosomes for nose-to-brain delivery of miR-760-3p in cerebral ischemia/reperfusion injury mice inhibiting neuron ferroptosis. Journal of Nanobiotechnology 2023, 21. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, M.; Liu, H.; Zhu, R.; He, H.; Zhou, Y.; Zhang, Y.; Li, C.; Liang, D.; Zeng, Q.; et al. Bone marrow mesenchymal stem cell-derived exosomes attenuate cerebral ischemia-reperfusion injury-induced neuroinflammation and pyroptosis by modulating microglia M1/M2 phenotypes. Experimental Neurology 2021, 341. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Cao, L.L.; Wang, X.P.; Guo, W.; Guo, R.B.; Sun, Y.Q.; Xue, T.F.; Cai, Z.Y.; Ji, J.; Cheng, H.; et al. Neuronal extracellular vesicle derived miR-98 prevents salvageable neurons from microglial phagocytosis in acute ischemic stroke. Cell Death Dis 2021, 12, 23. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Y.; Zheng, X.; Zhang, L.; Ai, X.; Venkataramani, V.; Kilic, E.; Hermann, D.M.; Majid, A.; Bahr, M.; Doeppner, T.R. Adipose-derived mesenchymal stem cells reduce autophagy in stroke mice by extracellular vesicle transfer of miR-25. J Extracell Vesicles 2020, 10, e12024. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhu, X.; Zhang, L.; Ferguson, C.M.; Song, T.; Jiang, K.; Conley, S.M.; Krier, J.D.; Tang, H.; Saadiq, I.; et al. Mesenchymal Stem/Stromal Cells and their Extracellular Vesicle Progeny Decrease Injury in Poststenotic Swine Kidney Through Different Mechanisms. Stem Cells Dev 2020, 29, 1190–1200. [Google Scholar] [CrossRef]
- Xie, W.; Luo, T.; Ma, Z.; Xue, S.; Jia, X.; Yang, T.; Song, Z. Tumor Necrosis Factor Alpha Preconditioned Umbilical Cord Mesenchymal Stem Cell-Derived Extracellular Vesicles Enhance the Inhibition of Necroptosis of Acinar cells in Severe Acute Pancreatitis. Tissue Eng Part A 2023, 29, 607–619. [Google Scholar] [CrossRef]
- Chen, J.; Ma, S.; Luo, B.; Hao, H.; Li, Y.; Yang, H.; Zhu, F.; Zhang, P.; Niu, R.; Pan, P. Human umbilical cord mesenchymal stromal cell small extracellular vesicle transfer of microRNA-223-3p to lung epithelial cells attenuates inflammation in acute lung injury in mice. J Nanobiotechnology 2023, 21, 295. [Google Scholar] [CrossRef]
- Khan, H.; Pan, J.-J.; Li, Y.; Zhang, Z.; Yang, G.-Y. Native and Bioengineered Exosomes for Ischemic Stroke Therapy. Frontiers in Cell and Developmental Biology 2021, 9. [Google Scholar] [CrossRef]
- Song, Y.; Li, Z.; He, T.; Qu, M.; Jiang, L.; Li, W.; Shi, X.; Pan, J.; Zhang, L.; Wang, Y.; et al. M2 microglia-derived exosomes protect the mouse brain from ischemia-reperfusion injury via exosomal miR-124. Theranostics 2019, 9, 2910–2923. [Google Scholar] [CrossRef]
- Zhang, G.; Zhu, Z.; Wang, H.; Yu, Y.; Chen, W.; Waqas, A.; Wang, Y.; Chen, L. Exosomes derived from human neural stem cells stimulated by interferon gamma improve therapeutic ability in ischemic stroke model. J Adv Res 2020, 24, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Haupt, M.; Zheng, X.; Kuang, Y.; Lieschke, S.; Janssen, L.; Bosche, B.; Jin, F.; Hein, K.; Kilic, E.; Venkataramani, V.; et al. Lithium modulates miR-1906 levels of mesenchymal stem cell-derived extracellular vesicles contributing to poststroke neuroprotection by toll-like receptor 4 regulation. Stem Cells Transl Med 2021, 10, 357–373. [Google Scholar] [CrossRef]
- Kim, H.Y.; Kim, T.J.; Kang, L.; Kim, Y.J.; Kang, M.K.; Kim, J.; Ryu, J.H.; Hyeon, T.; Yoon, B.W.; Ko, S.B.; et al. Mesenchymal stem cell-derived magnetic extracellular nanovesicles for targeting and treatment of ischemic stroke. Biomaterials 2020, 243, 119942. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhao, L.; Shi, Y.; Liang, J. Edaravone-Loaded Macrophage-Derived Exosomes Enhance Neuroprotection in the Rat Permanent Middle Cerebral Artery Occlusion Model of Stroke. Mol Pharm 2020, 17, 3192–3201. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Jiang, Y.; Shi, Y.; Liang, J.; Zhao, L. Curcumin-laden exosomes target ischemic brain tissue and alleviate cerebral ischemia-reperfusion injury by inhibiting ROS-mediated mitochondrial apoptosis. Mater Sci Eng C Mater Biol Appl 2020, 117, 111314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wei, W.; Ai, X.; Kilic, E.; Hermann, D.M.; Venkataramani, V.; Bahr, M.; Doeppner, T.R. Extracellular vesicles from hypoxia-preconditioned microglia promote angiogenesis and repress apoptosis in stroke mice via the TGF-beta/Smad2/3 pathway. Cell Death Dis 2021, 12, 1068. [Google Scholar] [CrossRef]
- Wang, K.; Ru, J.; Zhang, H.; Chen, J.; Lin, X.; Lin, Z.; Wen, M.; Huang, L.; Ni, H.; Zhuge, Q.; et al. Melatonin Enhances the Therapeutic Effect of Plasma Exosomes Against Cerebral Ischemia-Induced Pyroptosis Through the TLR4/NF-kappaB Pathway. Front Neurosci 2020, 14, 848. [Google Scholar] [CrossRef]
- Wang, J.; Liu, H.; Chen, S.; Zhang, W.; Chen, Y.; Yang, Y. Moderate exercise has beneficial effects on mouse ischemic stroke by enhancing the functions of circulating endothelial progenitor cell-derived exosomes. Experimental Neurology 2020, 330. [Google Scholar] [CrossRef]
- Huang, M.; Cheng, S.; Li, Z.; Chen, J.; Wang, C.; Li, J.; Zheng, H. Preconditioning Exercise Inhibits Neuron Ferroptosis and Ameliorates Brain Ischemia Damage by Skeletal Muscle–Derived Exosomes via Regulating miR-484/ACSL4 Axis. Antioxidants & Redox Signaling 2024. [Google Scholar] [CrossRef]
- Zhou, X.; Deng, X.; Liu, M.; He, M.; Long, W.; Xu, Z.; Zhang, K.; Liu, T.; So, K.F.; Fu, Q.L.; et al. Intranasal delivery of BDNF-loaded small extracellular vesicles for cerebral ischemia therapy. J Control Release 2023, 357, 1–19. [Google Scholar] [CrossRef]
- Xin, H.; Katakowski, M.; Wang, F.; Qian, J.Y.; Liu, X.S.; Ali, M.M.; Buller, B.; Zhang, Z.G.; Chopp, M. MicroRNA cluster miR-17-92 Cluster in Exosomes Enhance Neuroplasticity and Functional Recovery After Stroke in Rats. Stroke 2017, 48, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.; Liu, Z.; Buller, B.; Li, Y.; Golembieski, W.; Gan, X.; Wang, F.; Lu, M.; Ali, M.M.; Zhang, Z.G.; et al. MiR-17-92 enriched exosomes derived from multipotent mesenchymal stromal cells enhance axon-myelin remodeling and motor electrophysiological recovery after stroke. J Cereb Blood Flow Metab 2021, 41, 1131–1144. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Han, B.; Zhang, Z.; Wang, S.; Bai, Y.; Zhang, Y.; Tang, Y.; Du, L.; Xu, L.; Wu, F.; et al. Extracellular Vesicle-Mediated Delivery of Circular RNA SCMH1 Promotes Functional Recovery in Rodent and Nonhuman Primate Ischemic Stroke Models. Circulation 2020, 142, 556–574. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol 2011, 29, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Yim, N.; Ryu, S.W.; Choi, K.; Lee, K.R.; Lee, S.; Choi, H.; Kim, J.; Shaker, M.R.; Sun, W.; Park, J.H.; et al. Exosome engineering for efficient intracellular delivery of soluble proteins using optically reversible protein-protein interaction module. Nat Commun 2016, 7, 12277. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, Q.; Zi, Z.; Liu, Z.; Wan, C.; Crisman, L.; Shen, J.; Liu, X. Programmable Extracellular Vesicles for Macromolecule Delivery and Genome Modifications. Dev Cell 2020, 55, 784–801 e789. [Google Scholar] [CrossRef]
- Villarroya-Beltri, C.; Gutierrez-Vazquez, C.; Sanchez-Cabo, F.; Perez-Hernandez, D.; Vazquez, J.; Martin-Cofreces, N.; Martinez-Herrera, D.J.; Pascual-Montano, A.; Mittelbrunn, M.; Sanchez-Madrid, F. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun 2013, 4, 2980. [Google Scholar] [CrossRef]
- Bolukbasi, M.F.; Mizrak, A.; Ozdener, G.B.; Madlener, S.; Strobel, T.; Erkan, E.P.; Fan, J.B.; Breakefield, X.O.; Saydam, O. miR-1289 and "Zipcode"-like Sequence Enrich mRNAs in Microvesicles. Mol Ther Nucleic Acids 2012, 1, e10. [Google Scholar] [CrossRef]
- Hung, M.E.; Leonard, J.N. A platform for actively loading cargo RNA to elucidate limiting steps in EV-mediated delivery. J Extracell Vesicles 2016, 5, 31027. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, J.; Wu, J.; Fan, Q.; Zhou, J.; Wu, J.; Liu, S.; Zang, J.; Ye, J.; Xiao, M.; et al. Exosome-mediated targeted delivery of miR-210 for angiogenic therapy after cerebral ischemia in mice. J Nanobiotechnology 2019, 17, 29. [Google Scholar] [CrossRef]
- Kooijmans, S.A.A.; Fliervoet, L.A.L.; van der Meel, R.; Fens, M.; Heijnen, H.F.G.; van Bergen En Henegouwen, P.M.P.; Vader, P.; Schiffelers, R.M. PEGylated and targeted extracellular vesicles display enhanced cell specificity and circulation time. J Control Release 2016, 224, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Huang, Z.; Huang, L.; Liang, J.; Wang, P.; Zhao, L.; Shi, Y. Surface-modified engineered exosomes attenuated cerebral ischemia/reperfusion injury by targeting the delivery of quercetin towards impaired neurons. J Nanobiotechnology 2021, 19, 141. [Google Scholar] [CrossRef] [PubMed]
- Haney, M.J.; Klyachko, N.L.; Zhao, Y.; Gupta, R.; Plotnikova, E.G.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V.; et al. Exosomes as drug delivery vehicles for Parkinson's disease therapy. Journal of Controlled Release 2015, 207, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, G.; Serio, A.; Mazo, M.; Nair, R.; Stevens, M.M. Active loading into extracellular vesicles significantly improves the cellular uptake and photodynamic effect of porphyrins. J Control Release 2015, 205, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Song, Y.; Wang, Q.; Chen, J.; Gao, J.; Tan, H.; Li, S.; Wu, Y.; Yang, H.; Huang, H.; et al. Engineering extracellular vesicles with platelet membranes fusion enhanced targeted therapeutic angiogenesis in a mouse model of myocardial ischemia reperfusion. Theranostics 2021, 11, 3916–3931. [Google Scholar] [CrossRef]
- Smyth, T.; Petrova, K.; Payton, N.M.; Persaud, I.; Redzic, J.S.; Graner, M.W.; Smith-Jones, P.; Anchordoquy, T.J. Surface functionalization of exosomes using click chemistry. Bioconjug Chem 2014, 25, 1777–1784. [Google Scholar] [CrossRef]
- Tian, T.; Zhang, H.-X.; He, C.-P.; Fan, S.; Zhu, Y.-L.; Qi, C.; Huang, N.-P.; Xiao, Z.-D.; Lu, Z.-H.; Tannous, B.A.; et al. Surface functionalized exosomes as targeted drug delivery vehicles for cerebral ischemia therapy. Biomaterials 2018, 150, 137–149. [Google Scholar] [CrossRef]
- Huang, Z.; Guo, L.; Huang, L.; Shi, Y.; Liang, J.; Zhao, L. Baicalin-loaded macrophage-derived exosomes ameliorate ischemic brain injury via the antioxidative pathway. Mater Sci Eng C Mater Biol Appl 2021, 126, 112123. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, X.; Chen, X.; Wang, L.; Yang, G. Exosome Mediated Delivery of miR-124 Promotes Neurogenesis after Ischemia. Mol Ther Nucleic Acids 2017, 7, 278–287. [Google Scholar] [CrossRef]
- Kim, M.; Kim, G.; Hwang, D.W.; Lee, M. Delivery of High Mobility Group Box-1 siRNA Using Brain-Targeting Exosomes for Ischemic Stroke Therapy. J Biomed Nanotechnol 2019, 15, 2401–2412. [Google Scholar] [CrossRef]
- Guo, L.; Pan, J.; Li, F.; Zhao, L.; Shi, Y. A novel brain targeted plasma exosomes enhance the neuroprotective efficacy of edaravone in ischemic stroke. IET Nanobiotechnology 2021, 15, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Kalani, A.; Chaturvedi, P.; Kamat, P.K.; Maldonado, C.; Bauer, P.; Joshua, I.G.; Tyagi, S.C.; Tyagi, N. Curcumin-loaded embryonic stem cell exosomes restored neurovascular unit following ischemia-reperfusion injury. Int J Biochem Cell Biol 2016, 79, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Lee, Y.; Lee, M. Hypoxia-specific anti-RAGE exosomes for nose-to-brain delivery of anti-miR-181a oligonucleotide in an ischemic stroke model. Nanoscale 2021, 13, 14166–14178. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Cao, L.; He, C.; Ye, Q.; Liang, R.; You, W.; Zhang, H.; Wu, J.; Ye, J.; Tannous, B.A.; et al. Targeted delivery of neural progenitor cell-derived extracellular vesicles for anti-inflammation after cerebral ischemia. Theranostics 2021, 11, 6507–6521. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Wang, R.; Wang, C.; Guo, Y.; Xu, T.; Zhang, Z.; Yang, G.Y.; Xu, H.; Tang, Y. Brain Microenvironment Responsive and Pro-Angiogenic Extracellular Vesicle-Hydrogel for Promoting Neurobehavioral Recovery in Type 2 Diabetic Mice After Stroke. Adv Healthc Mater 2022, 11, e2201150. [Google Scholar] [CrossRef]
- Bang, O.Y.; Kim, E.H. Mesenchymal Stem Cell-Derived Extracellular Vesicle Therapy for Stroke: Challenges and Progress. Front Neurol 2019, 10, 211. [Google Scholar] [CrossRef]
- Doeppner, T.R.; Herz, J.; Gorgens, A.; Schlechter, J.; Ludwig, A.K.; Radtke, S.; de Miroschedji, K.; Horn, P.A.; Giebel, B.; Hermann, D.M. Extracellular Vesicles Improve Post-Stroke Neuroregeneration and Prevent Postischemic Immunosuppression. Stem Cells Transl Med 2015, 4, 1131–1143. [Google Scholar] [CrossRef]
- Rocha, M.; Jovin, T.G. Fast Versus Slow Progressors of Infarct Growth in Large Vessel Occlusion Stroke: Clinical and Research Implications. Stroke 2017, 48, 2621–2627. [Google Scholar] [CrossRef]
- Ollen-Bittle, N.; Roseborough, A.D.; Wang, W.; Wu, J.-l.D.; Whitehead, S.N. Mechanisms and Biomarker Potential of Extracellular Vesicles in Stroke. Biology 2022, 11. [Google Scholar] [CrossRef]
- van Kralingen, J.C.; McFall, A.; Ord, E.N.J.; Coyle, T.F.; Bissett, M.; McClure, J.D.; McCabe, C.; Macrae, I.M.; Dawson, J.; Work, L.M. Altered Extracellular Vesicle MicroRNA Expression in Ischemic Stroke and Small Vessel Disease. Translational Stroke Research 2019, 10, 495–508. [Google Scholar] [CrossRef]
- Couch, Y.; Akbar, N.; Davis, S.; Fischer, R.; Dickens, A.M.; Neuhaus, A.A.; Burgess, A.I.; Rothwell, P.M.; Buchan, A.M. Inflammatory Stroke Extracellular Vesicles Induce Macrophage Activation. Stroke 2017, 48, 2292–2296. [Google Scholar] [CrossRef] [PubMed]
- Kalani, M.Y.S.; Alsop, E.; Meechoovet, B.; Beecroft, T.; Agrawal, K.; Whitsett, T.G.; Huentelman, M.J.; Spetzler, R.F.; Nakaji, P.; Kim, S.; et al. Extracellular microRNAs in blood differentiate between ischaemic and haemorrhagic stroke subtypes. J Extracell Vesicles 2020, 9, 1713540. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, E.; Ortega, L.; Hernandez-Guillamon, M.; Penalba, A.; Fernandez-Cadenas, I.; Rosell, A.; Montaner, J. Tissue plasminogen activator (t-PA) promotes neutrophil degranulation and MMP-9 release. J Leukoc Biol 2008, 84, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Dhir, N.; Medhi, B.; Prakash, A.; Goyal, M.K.; Modi, M.; Mohindra, S. Pre-clinical to Clinical Translational Failures and Current Status of Clinical Trials in Stroke Therapy: A Brief Review. Curr Neuropharmacol 2020, 18, 596–612. [Google Scholar] [CrossRef] [PubMed]
- Dehghani, L.; Khojasteh, A.; Soleimani, M.; Oraee-Yazdani, S.; Keshel, S.H.; Saadatnia, M.; Saboori, M.; Zali, A.; Hashemi, S.M.; Soleimani, R. Safety of Intraparenchymal Injection of Allogenic Placenta Mesenchymal Stem Cells Derived Exosome in Patients Undergoing Decompressive Craniectomy Following Malignant Middle Cerebral Artery Infarct, A Pilot Randomized Clinical Trial. Int J Prev Med 2022, 13, 7. [Google Scholar] [CrossRef]
- Lener, T.; Gimona, M.; Aigner, L.; Borger, V.; Buzas, E.; Camussi, G.; Chaput, N.; Chatterjee, D.; Court, F.A.; Del Portillo, H.A.; et al. Applying extracellular vesicles based therapeutics in clinical trials - an ISEV position paper. J Extracell Vesicles 2015, 4, 30087. [Google Scholar] [CrossRef]
Figure 1.
Schematic overview of disease progression and molecular pathophysiology in ischemic stroke. As cerebral blood flow (CBF) decreases, the supply of nutrients and oxygen to brain tissue is reduced, leading to a decrease in intracellular ATP levels and the formation of reactive oxygen species (ROS). The reduction in ATP levels causes membrane depolarization, resulting in excessive glutamate release at synapses and triggering excitotoxicity (A). Concurrently, mitochondrial ROS formation due to reduced oxygen, along with intracellular calcium overload from excitotoxicity and nNOS activation via N-methyl-D-aspartate receptors (NMDARs), induces oxidative/nitrosative stress. Notably, peroxynitrite (ONOO-) generated in this process causes severe cellular damage through DNA fragmentation and lipid peroxidation (B). Activated astrocytes and microglia secrete pro-inflammatory cytokines such as IL-1β and TNF-α, and danger-associated molecular patterns (DAMPs) released from dead cells further trigger inflammation. Oxidative/nitrosative stress and inflammation result in damage to brain endothelial cells, facilitating infiltration of peripheral immune cells, which exacerbates the pathological process (C). Reperfusion of CBF contributes to the worsening pathology via similar mechanisms. Key molecules or pathways targeted by the extracellular vesicles (EV) are highlighted in red color. Green boxes indicate molecules with enzymatic functions. Schematic illustration was created with BioRender.com.
Figure 1.
Schematic overview of disease progression and molecular pathophysiology in ischemic stroke. As cerebral blood flow (CBF) decreases, the supply of nutrients and oxygen to brain tissue is reduced, leading to a decrease in intracellular ATP levels and the formation of reactive oxygen species (ROS). The reduction in ATP levels causes membrane depolarization, resulting in excessive glutamate release at synapses and triggering excitotoxicity (A). Concurrently, mitochondrial ROS formation due to reduced oxygen, along with intracellular calcium overload from excitotoxicity and nNOS activation via N-methyl-D-aspartate receptors (NMDARs), induces oxidative/nitrosative stress. Notably, peroxynitrite (ONOO-) generated in this process causes severe cellular damage through DNA fragmentation and lipid peroxidation (B). Activated astrocytes and microglia secrete pro-inflammatory cytokines such as IL-1β and TNF-α, and danger-associated molecular patterns (DAMPs) released from dead cells further trigger inflammation. Oxidative/nitrosative stress and inflammation result in damage to brain endothelial cells, facilitating infiltration of peripheral immune cells, which exacerbates the pathological process (C). Reperfusion of CBF contributes to the worsening pathology via similar mechanisms. Key molecules or pathways targeted by the extracellular vesicles (EV) are highlighted in red color. Green boxes indicate molecules with enzymatic functions. Schematic illustration was created with BioRender.com.
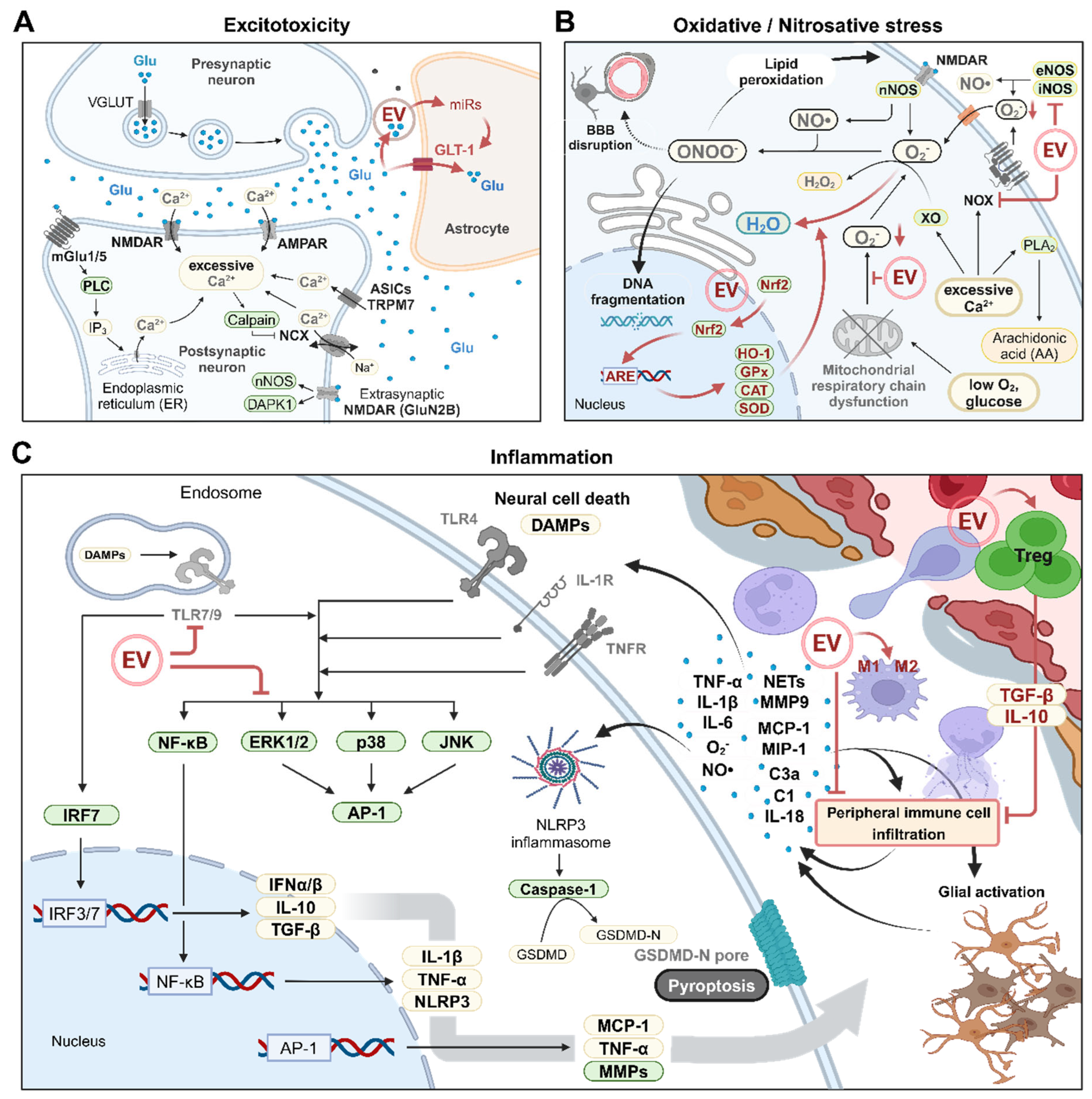
Figure 2.
Schematic overview of brain cell death in ischemic stroke. Reduced cerebral blood flow (CBF) leads to decreased nutrient and oxygen supply, triggering excitotoxicity, inflammation, and oxidative/nitrosative stress, which ultimately result in brain cell death. The molecular mechanisms underlying various forms of brain cell death in ischemic stroke are illustrated in the figure. Excitotoxicity-induced receptor activation and intracellular calcium overload contribute to autophagy-related cell death and apoptosis via DAPK1, CaMKKβ, and calpain activation, while also triggering ferroptosis through PLA2 activation, which provides arachidonic acid (AA). Oxidative/nitrosative stress induces mitochondrial outer membrane permeabilization, DNA damage, and lipid peroxidation, driving apoptosis, parthanatos, and ferroptosis. Furthermore, external signals, including pro-inflammatory cytokines, induce apoptosis and autophagy-related cell death through MAPK and NOTCH signaling, caspase-8 activation, and mTORC1 inhibition. Under conditions of ATP depletion, rather than activating caspase-8, cell death signaling induced by TNF-α and FasL promotes necroptosis by forming a necrosome complex comprising RIPK1/3 and MLKL. Key molecules or pathways targeted by the extracellular vesicles (EV) are highlighted in red color. Green boxes indicate molecules with enzymatic functions. Schematic illustration was created with BioRender.com.
Figure 2.
Schematic overview of brain cell death in ischemic stroke. Reduced cerebral blood flow (CBF) leads to decreased nutrient and oxygen supply, triggering excitotoxicity, inflammation, and oxidative/nitrosative stress, which ultimately result in brain cell death. The molecular mechanisms underlying various forms of brain cell death in ischemic stroke are illustrated in the figure. Excitotoxicity-induced receptor activation and intracellular calcium overload contribute to autophagy-related cell death and apoptosis via DAPK1, CaMKKβ, and calpain activation, while also triggering ferroptosis through PLA2 activation, which provides arachidonic acid (AA). Oxidative/nitrosative stress induces mitochondrial outer membrane permeabilization, DNA damage, and lipid peroxidation, driving apoptosis, parthanatos, and ferroptosis. Furthermore, external signals, including pro-inflammatory cytokines, induce apoptosis and autophagy-related cell death through MAPK and NOTCH signaling, caspase-8 activation, and mTORC1 inhibition. Under conditions of ATP depletion, rather than activating caspase-8, cell death signaling induced by TNF-α and FasL promotes necroptosis by forming a necrosome complex comprising RIPK1/3 and MLKL. Key molecules or pathways targeted by the extracellular vesicles (EV) are highlighted in red color. Green boxes indicate molecules with enzymatic functions. Schematic illustration was created with BioRender.com.
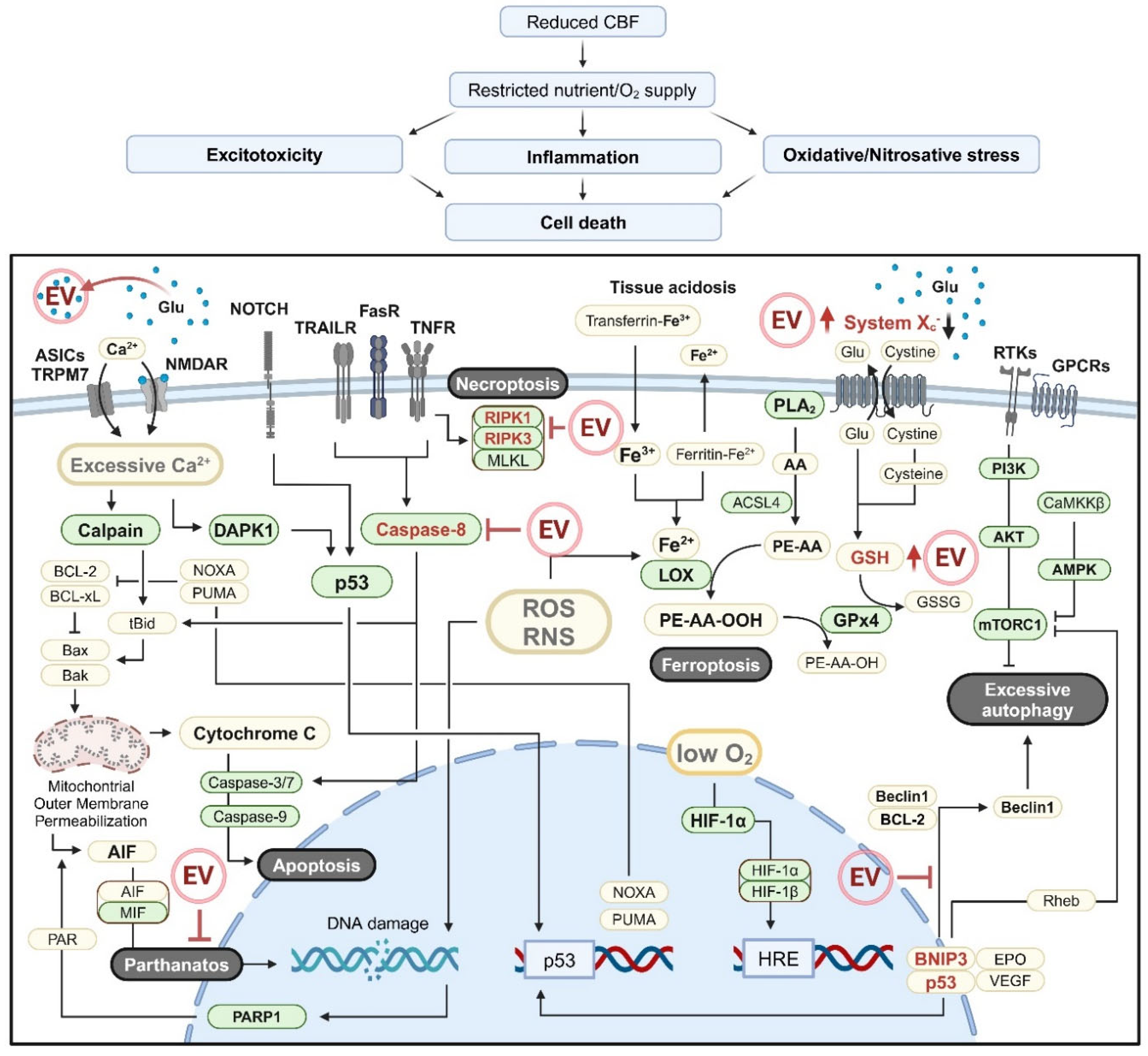
Figure 3.
Schematic overview of extracellular vesicle (EV) engineering. Various methods of EV engineering can enhance the therapeutic efficacy of ischemic stroke (IS) treatment. (A) Pre-isolation EV engineering involves modification of the EV-producing cells. Intracellular expression patterns are modulated by altering extracellular conditions or by treating the cells with cytokines, hydrophobic molecules, or molecules capable of cellular uptake, resulting in changes in EV cargos. Desired molecules can be directly loaded into EVs by transfection of EV-producing cells, the creation of EV-related fusion proteins, or using RNAs containing specific motifs. (B) EV engineering is also possible after EV isolation. Hydrophobic molecules or molecules that interact with the EV surface are loaded into EVs by simple co-incubation. Other molecules must be conjugated with hydrophobic agents such as cholesterol or EV surface-interacting molecules before co-incubation. Additional post-isolation engineering methods include microporation of the EV membrane by ultrasonication, electroporation, or freeze-thaw cycles and the use of active loading techniques such as click chemistry or carbodiimide coupling. (C) Different approaches of EV engineering for IS treatment. Schematic illustration was created with BioRender.com.
Figure 3.
Schematic overview of extracellular vesicle (EV) engineering. Various methods of EV engineering can enhance the therapeutic efficacy of ischemic stroke (IS) treatment. (A) Pre-isolation EV engineering involves modification of the EV-producing cells. Intracellular expression patterns are modulated by altering extracellular conditions or by treating the cells with cytokines, hydrophobic molecules, or molecules capable of cellular uptake, resulting in changes in EV cargos. Desired molecules can be directly loaded into EVs by transfection of EV-producing cells, the creation of EV-related fusion proteins, or using RNAs containing specific motifs. (B) EV engineering is also possible after EV isolation. Hydrophobic molecules or molecules that interact with the EV surface are loaded into EVs by simple co-incubation. Other molecules must be conjugated with hydrophobic agents such as cholesterol or EV surface-interacting molecules before co-incubation. Additional post-isolation engineering methods include microporation of the EV membrane by ultrasonication, electroporation, or freeze-thaw cycles and the use of active loading techniques such as click chemistry or carbodiimide coupling. (C) Different approaches of EV engineering for IS treatment. Schematic illustration was created with BioRender.com.
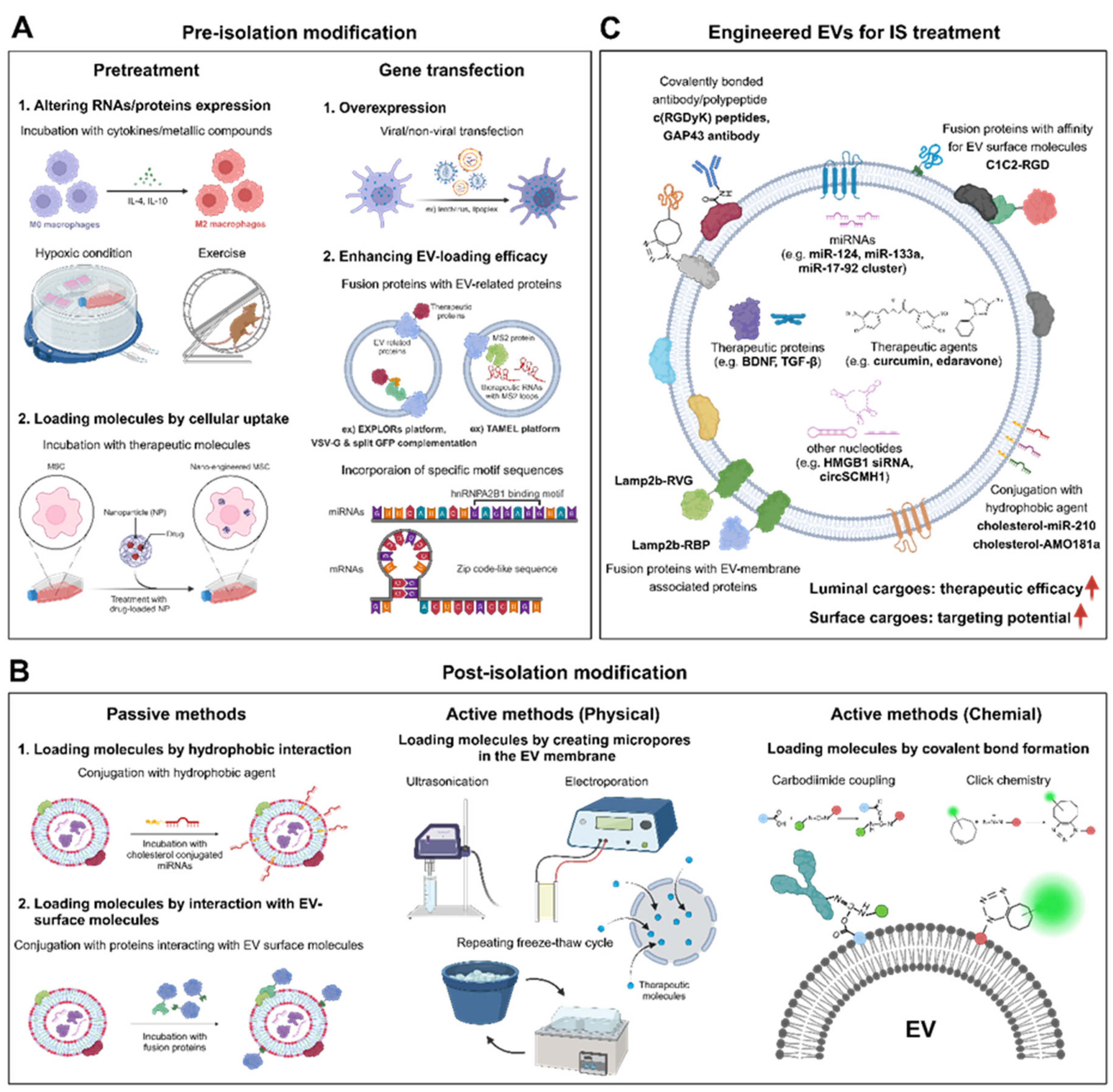

Table 1.
Recent research on the use of naïve extracellular vesicles (EVs) for treatment of ischemic stroke (IS).
Table 1.
Recent research on the use of naïve extracellular vesicles (EVs) for treatment of ischemic stroke (IS).
| EV source | Major cargo molecules | In vitro stroke model | In vivo stroke model | Major targeted molecules/pathway | Outcome | Reference | |||
|---|---|---|---|---|---|---|---|---|---|
| animal model | administration route | dosage | Time point of administration | ||||||
| rat BM-MSCs | - | - | rat tMCAO model (2h) | tail vein | 100μg | 24h | - | enhanced neurite remodeling enhanced neurogenesis & angiogenesis |
[23] |
| human iPSC-derived MSCs | - | OGD/R-HUVECs (8h) | rat tMCAO model (2h) | tail vein | 1 x 10^11 particles | 4h | STAT3 | enhanced angiogenesis reduced autophagy |
[24] |
| HUVECs | miR-1290 | OGD/R-neurons (1.5h) | mouse tMACO model (1h) | Intracranial (AP: 2.0 mm, ML: 1.7 mm, DV: 1.35 mm) |
5μg | Immediately (0h) |
- | reduced apoptosis | [25] |
| human NSCs | - | glucose-free H/R model (1.5h) | - | - | - | - | - | reduced apoptosis and oxidative stress enhanced axonal elongation enhanced angiogenesis |
[67] |
| rat BM-MSCs | - | OGD/R-microglia (1 - 5h) | rat tMCAO model (1.5h) | tail vein | 120μg | 2h | cysLT2R ERK1/2 |
mitigated microglia M1 polarization | [86] |
| human BM-MSCs | - | - | mouse tMACO model (0.5h) | tail vein | released by 2×10^6 MSCs | Immediately (0h) |
- | reducing apoptosis reduced peripheral immune cell inflitration |
[87] |
| astrocytes | miR-34c | OGD/R-N2a cells (-) | rat tMACO model (-) | tail vein | - | - | TLR7 NF-κB/MAPK pathway |
reduced apoptosis and inflammation | [88] |
| human ESCs | TGF-β & Smad2 & Smad4 |
- | mouse tMACO model (1h) | tail vein | 1 x 10^9 particles | 2h and day 1, 2 (3 times) |
TGF-β/Smad pathway | reduced apoptosis and inflammation reduced peripheral immune cell inflitration |
[89] |
| UC-MSCs | circBBS2 | H/R model of SH-SY5Y cells (4h) | rat tMCAO model (2h) | tail vein | 50μg | 4h and day 1, 2 (3 times) |
miR-494 | reduced ferroptosis by upregulation of SLC7A11 | [141] |
| mouse AD-MSCs | miR-760-3p | OGD/R-N2a cells (4h) | mouse tMACO model (1h) | intranasal | 10μg | day 1, 3, 5 (3 times) |
CHAC1 | reduced ferroptosis | [142] |
| rat BM-MSCs | - | OGD/R-BV2 & PC12 cells (6h) | rat tMCAO model (2h) | tail vein | 80μg (low) 100μg (medium) 120μg (high) |
2h | - | shift of microglial polarization state toward M2 phenotype reduced pyroptosis and inflammation |
[143] |
| mouse AD-MSCs | miR-25-3p | OGD/R-neurons (10h) | mouse tMACO model (1h) | femoral vein | 10μg | Immediately (0h) or 12h |
p53-BNIP3 signaling | reduced autophagy | [145] |
| human BM-MSCs | - | - | mouse tMACO model (0.5h) | femoral vein | EVs released by 2 x 10^6 MSCs | day 1, 3, 5 (3 times) |
- | neuroprotection enhanced neurogenesis & angiogenesis modulated peripheral immune response |
[187] |
Table 2.
Recent research in the use of engineered extracellular vesicles (EVs) for treatment of ischemic stroke (IS).
Table 2.
Recent research in the use of engineered extracellular vesicles (EVs) for treatment of ischemic stroke (IS).
| EV source | Modification | Major cargo molecules | In vitro stroke model | In vivo stroke model | Major targeted molecules/pathway | Outcome | Reference | |||
|---|---|---|---|---|---|---|---|---|---|---|
| animal model | administration route | dosage | Time point of administration | |||||||
| BV2 cells | IL-4 pretreatment | miR-124 | OGD/R-neurons (45min) | mouse tMACO model (1h) | tail vein | 100μg | 0h and day 1, 2 (3 times) |
USP14 | reduced apoptosis | [150] |
| human NSCs | INF-γ pretreatment | miR-206 & miR-133a-3p & miR-3656 |
- | rat tMACO model (-) |
intracranially (striatum) | 4 x 10^9 particles | 24h | - | reduced apoptosis and oxidative stress | [151] |
| mouse BM-MSCs | lithium pretreatment | miR-1906 | OGD/R-neurons (1h) OGD/R-microglia (8h) OGD/R-astrocytes (12h) |
mouse tMACO model (1h) | femoral vein (day 1) retro-orbital vein (day 3, 5) |
13.5μg | day 1, 3, 5 (3 times) |
TLR4/NF-κB pathway | reduced apoptosis enhanced neurogenesis & angiogenesis reduced peripheral immune cell inflitration |
[152] |
| human BM-MSCs | iron oxide nanoparticle (IONP) pretreatment | IONP & various growth factors |
LPS treated hypoxia-PC12 or rBMDM cells (24h) | rat tMACO model (1h) |
tail vein | 200μg | Immediately (0h) |
- | enhanced neurogenesis & angiogenesisreduced apoptosis and inflammationshift of macrophage polarization state toward M2 phenotype | [153] |
| RAW264.7 cells | edaravone pretreatment | edaravone | - | rat pMACO model | tail vein | - | day 1-7 (7 times) |
- | Neuroprotection shift of microglial polarization state toward M2 phenotype |
[154] |
| RAW264.7 cells | curcumin pretreatment | curcumin | - | rat tMACO model (2h) |
tail vein | - | Immediately (0h) |
- | reduced oxidative stress and apoptosisneuroprotection attenuated BBB damage |
[155] |
| mouse microglia | OGD/R preconditioning | TGF-β | OGD/R-neurons (6h) OGD/R-microglia (4h) OGD/R-bEnd.3 (16h) |
mouse tMACO model (1h) | femoral vein | 10μg | 0h, 6h (2 times) |
TGF-β/Smad2/3 pathway | promotion of endothelial cell survival and migration reduced neuronal apoptosis enhanced angiogenesis shift of microglial polarization state toward M2 phenotype |
[156] |
| rat plasma | Melatonin pretreatment | various miRNAs | - | rat pMACO model | tail vein | 100μg | 1h, 12h, 36h (3 times) |
NLRP3-mediated pathway & TLR4/NF-κB pathway |
reduced pyroptosis and inflammation | [157] |
| circulating endothelial progenitor cells | treadmill exercise | miR-126 | H/R-N2a cells (-) | mouse pMACO model | - | - | - | BDNF & PI3k/Akt pathway |
reduced apoptosis enhanced neurogenesis & angiogenesis |
[158] |
| rat skeletal muscle | treadmill exercise | miR-484 | OGD/R-PC12 cells (4h) | rat tMACO model (1h) |
tail vein | - | 2h before operation | ACSL4 | reduced ferroptosis | [159] |
| human iPSC-derived MSCs | transfection (BDNF) | BDNF | - | mouse tMACO model (45min) | intranasally | 1 x 10^10 particles | 2h, 24h, 48h (3 times) |
BDNF/TrkB signaling | reduced apoptosis and inflammation enhanced neurogenesis & angiogenesisneuroprotection |
[160] |
| rat BM-MSCs | transfection (miR-17-92 cluster) | miR-17-92 cluster | - | rat tMACO model (2h) |
intravenously | 100μg (3 x 10^11 particles) |
24h | PTEN & PI3k/Akt/mTOR pathway |
enhanced neurite remodeling and neuronal plasticity enhanced neurogenesis & oligodendrogenesis enhanced cortico-spinal tract axonal remodeling |
[161] |
| HEK293T cells | transfection (RGV-Lamp2b, circSCMH1) | RGV-Lamp2b & circSCMH1 |
OGD/R-neurons (3h) | mouse photothrombosis (PT) model mouse dMCAO/tMCAO (1h) model rhesus monkey PT stroke model |
mouse : tail veinrhesus monkey : hind limb vein | mouse : 12mg/kg rhesus monkey : 3mg/kg |
mouse : 24h rhesus monkey : 24h, 48h (2 times) |
MeCP2 | enhanced neuronal plasticity reduced glial activation reduced peripheral immune cell infiltration |
[163] |
| mouse BM-MSCs | passive loading (miR-210-cholesterol) click chemistry (c(RGDyK) peptides) |
miR-210 & c(RGDyK) peptides |
- | mouse tMACO model (0.5h or 1h) | tail vein | 100μg | 24h | VEGF | enhanced angiogenesis | [170] |
| rat blood | active loading-ultrasonication (quercetin) carbodiimide coupling (GAP43 antibody) |
Quercetin & GAP43 antibody |
OGD/R-SH-SY5Y cells (1h) | rat tMACO model (2h) |
tail vein | - | 24h | GAP43 & Nrf2/HO-1 pathway |
reduced apoptosis and oxidative stress | [172] |
| mouse BM-MSCs | passive loading (curcumin) click chemistry (c(RGDyK) peptides) |
Curcumin & c(RGDyK) peptides |
- | mouse tMACO model (1h) | tail vein | 300μg | 12h | NF-κB | reduced apoptosis and inflammation | [177] |
| RAW264.7 cells | active loading-ultrasonication (baicalin) | baicalin | OGD/R-SH-SY5Y cells (1h) | rat pMCAO/tMACO (2h) model |
tail vein | 1.6mg baicalin | Immediately (0h) |
Nrf2/HO-1 pathway | reduced apoptosis and oxidative stress | [178] |
| mouse BM-MSCs | transfection (RVG-Lamp2b)active loading-electroporation (miR-124) | miR-124 | - | mouse PT stroke model | tail vein | - | day 1 | Gli3 &Stat3 |
enhanced neurogenesis | [179] |
| HEK293T cells | transfection (RVG-Lamp2b)active loading-electroporation (HMGB1 siRNA) | HMGB1 siRNA | - | rat tMACO model (1h) |
tail vein | 30μg siRNAs | 18h before operation | HMGB1 | reduced apoptosis and inflammation | [180] |
| rat plasma | active loading-ultrasonication (edaravone) | edaravone | - | rat pMACO model | tail vein | 10mg/kg edaravone | day 1-7 (7 times) |
- | neuroprotection | [181] |
| mouse ESCs | active loading-freeze-thawing (curcumin) | curcumin | - | mouse tMACO model (40min) | intranasally | - | day 0-7 (twice a day) |
- | reduced oxidative stress and inflammationreduced glial activation and loss of vascular integrity | [182] |
| HEK293T cells | transfection (RBP-Lamp2b)passive loading (AMO181a-cholesterol) | RBP-Lamp2b &AMO181a |
hypoxia-Neuro2A cells (24h) | rat tMACO model (1h) |
intranasally | 75μg | 1h | RAGE &miR-181a |
reduced apoptosis and inflammation | [183] |
| human NPCs (ReN cells) | passive loading (RGD-C1C2) | RGD-C1C2 &various miRNAs |
- | mouse tMACO model (1h) | tail vein | 300μg | 12h | p38 MAPK pathway | reduced inflammation | [184] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



