
Submitted:
05 November 2024
Posted:
06 November 2024
You are already at the latest version
Abstract
Heart failure with preserved ejection fraction (HFpEF) is increasing at an alarming rate worldwide, with limited effective therapeutic interventions in patients. Sudden cardiac death (SCD) and ventricular arrhythmias present substantial risks for the prognosis of these patients. Obesity is a risk factor for HFpEF and life-threatening arrhythmias. Obesity and its associated metabolic dysregulation, leading to metabolic syndrome, are an epidemic that poses a significant public health problem. More than one-third of the world population is overweight or obese, leading to an enhanced risk of incidence and mortality due to cardiovascular disease (CVD). Obesity predisposes patients to atrial fibrillation and ventricular and supraventricular arrhythmias—conditions that are caused by dysfunction in the electrical activity of the heart. To date, current therapeutic options for cardiomyopathy of obesity are limited, suggesting that there is considerable room for the development of therapeutic interventions with novel mechanisms of action that will help normalize sinus rhythms in obese patients. Emerging candidates for modulation by obesity are cardiac ion channels and Ca-handling proteins. However, the underlying molecular mechanisms of the impact of obesity on these channels/Ca-handling proteins remain incompletely understood. Obesity is marked by the accumulation of adipose tissue, which is associated with a variety of adverse adaptations, including dyslipidemia (or abnormal systemic levels of free fatty acids), increased secretion of pro-inflammatory cytokines, fibrosis, hyperglycemia, and insulin resistance, which cause electrical remodeling and, thus, predispose patients to arrhythmias. Furthermore, adipose tissue is also associated with the accumulation of subcutaneous and visceral fat, which is marked by distinct signaling mechanisms. Thus, there may also be functional differences in the outcome of regional distribution of fat deposits on ion channel/Ca-handling protein expression. Evaluating alterations in their functional expression in obesity will lead to progress in the knowledge about the mechanisms responsible for obesity-related arrhythmias. These advances are likely to reveal new targets for pharmacological modulation. The objective of this study is to review cardiac ion channel/Ca-handling protein remodeling in the predisposition to metabolic HFpEF and arrhythmias. Understanding how obesity and related mechanisms lead to cardiac electrical remodeling is likely to have a significant medical and economic impact.
Keywords:
obesity
; diabetes
; heart failure
; sudden cardiac death
; ventricular
; arrhythmias
; ion channel remodeling
Introduction
Heart failure (HF) with preserved ejection fraction (HFpEF), defined as a state of diastolic dysfunction and myocardial stiffness with a left ventricular ejection fraction (LVEF) exceeding 50%, is a complex clinical condition and is the most prominent form of HF among the elderly [1]. Despite the substantial increase in HFpEF at a rate of 1% a year, there is a critical paucity of rational therapeutic interventions, suggesting that the increasing burden for HF/HFpEF will likely continue to increase globally [1,2]. This lack of progress may be because HF/HFpEF is heterogeneous, and existing preclinical animal models may not fully capture the complexity of the cellular remodeling processes involved in HF [2]. Therefore, there is an unmet clinical need to improve our current understanding of the underlying mechanisms of HF.
There is increasing evidence in the extant literature that HF/HFpEF mortality in patients occurs in 53% to 74% of all HFpEF cases in the first 5 years of diagnosis [1]. Randomized clinical trials of HFpEF patients have shown an increased risk of sudden cardiac death (SCD) in patients [3]. However, the underlying mechanisms involved are unknown. The major drivers of SCD are known to be lethal cardiac arrhythmias. While ventricular arrhythmias (VAs) are the most common cause of SCD in HF with reduced ejection fraction (HFrEF), their role and incidence in HFpEF need further investigation [4]. Ventricular arrhythmias predispose HF patients to an increased number of hospitalizations, in-hospital cardiac arrest, and overall higher rates of morbidity [5]. Clinical studies, despite involving a relatively small patient population, have observed a high prevalence of non-sustained ventricular tachycardia (NSVT) in HFpEF patients [6,7], accompanied by a probability of SCD that is 2.3 times higher [6]. Although the concurrent occurrence of VA fails to surpass that of HFrEF in a pooled analysis of large clinical HFpEF trials, its presence remains significantly linked to poor overall prognosis and higher overall mortality rates [8].
The putative mechanisms governing the development of VA in HFpEF include deteriorated conduction velocity and re-entry circuits stemming from hypertrophic and fibrotic ventricles, delayed repolarization, and altered excitation–contraction coupling [9]. Major cardiac currents play pivotal roles in maintaining the integrity of the electrical activity of the heart. This electrical activity can be defined by the spatiotemporal patterns of action potential phenotypes, as well as cardiac refractoriness [10].
Ion channel dysfunction remains one of the crucial factors in VT initiation. The normal ventricular cardiac action potential (AP) is defined as follows: Phase 0, due to a large inward sodium current (INa), followed by currents due to voltage-gated L-type calcium (Ca) (ICa,L) and the Na-Ca exchanger (INCX) channels [11]. Repolarization is controlled by the delayed rectifier current (IK), which comprises rapid (IKr) and slow (IKs) components. The resting membrane potential is controlled by the inward rectifier K current (IK1) [12]. Thus, decreases in outward currents [13,14] or increases in depolarizing mechanisms [15] delay repolarization, resulting in the prolongation of the heart-rate-corrected QT interval (QTc), which predisposes patients to fatal VT, such as torsades de pointes (TdP) [16] and SCD [17,18].
IKr remains the dominant channel of clinical cardiotoxicity; the FDA and European Medicines Agency require screening against human ether-a-go-go-related gene (hERG) for all new drugs being evaluated [19,20]. Therefore, decreases in IKr prolong the ventricular action potential duration (APD) [14,21,22,23] and predispose patients to an elevated risk for early afterdepolarizations (EADs) [24,25], a prolonged QTc interval, and TdP [26]. In addition, impaired function of IKs, INa (peak and late), and ICa currents contributes to arrhythmia risk [27,28,29,30], highlighting the importance of multi-ion-channel analyses, which may inform the rational development of safer approaches (with reduced cardiotoxic effects) to antiarrhythmic monotherapy and polytherapy for patients.
Growing evidence suggests that obesity and associated pathologies (metabolic syndrome and insulin resistance) are important contributors to the development of HFpEF in patients [31]. Furthermore, 24.3% of obese patients have a prolonged QTc interval compared with patients with normal weight [32,33]. Preclinical studies have shown that the downregulation of K currents and delayed repolarization have imperative roles in the increased susceptibility to VA and, subsequently, SCD in rats fed with a high-salt diet who developed HFpEF [34,35]. In that regard, pathological changes in ionic currents or channelopathies may prominently contribute to an increased risk of VA/SCD in obese/HFpEF patients. Therefore, it would be interesting to discern the impact of a functional interplay between obesity triggers and pathological ion channel functional expression (Figure), as well as how they affect disease outcomes in patients. In this regard, an improved understanding of the cellular proarrhythmic mechanisms of remodeled ion channels may be a key component for the rational development of safer and more effective interventions in obese patients with HF and, particularly, HFpEF.
Obesity moderates diastolic dysfunction by triggering core pathophysiological factors of HFpEF, such as oxidative stress and inflammation [36,37]. Obesity and metabolic syndrome bring about a state of metabolic inflammation where damaged hypertrophic, hyperplastic, and hypoxic adipocytes provoke the accentuated production and release of inflammatory cytokines and adipokines and promote macrophage polarization towards the M1 pro-inflammatory phenotype [36,37]. It should be noted that although increased levels of cytokines may be initially cardioprotective, they become detrimental as inflammation persists [36].
Obesity has been related to VA development through heightened levels of free fatty acids (FFAs), inducing dysregulation in ventricular repolarization and resulting in a prolonged Q-T interval [38]. Guinea pig models fed with a high-fat diet (HFD) serve as an inflammatory preclinical model and have shown increased susceptibility to VA [39]. HFD-fed preclinical models have been extensively used to study the effects of obesity on ion channels and currents. Dysregulations in Na, Ca, and K currents in the context of obesity and high FFAs have been linked to altered cardiac excitability, long QT intervals, and VA, particularly in response to an enhanced sympathetic trigger, in mice and guinea pigs [39,40,41]. Moreover, epicardial adipose tissue (EAT) has recently been identified as a possible promoter of VA in addition to its already established involvement in atrial arrhythmias [42]. Although advances have been made in identifying obesity-related VA-inducing factors, much remains to be discovered regarding the precise mechanism of how major cardiac ion channels are modulated in metabolically disrupted HFpEF. A thorough comprehension of pathways binding obesity with HFpEF and VA is crucial for filling the gap in these patients, and this will have clinical and pharmaceutical implications. This review aims to provide an overview of factors linking HFpEF, obesity, and ventricular arrhythmias by exploring the underlying mechanisms of disturbances in electrical currents and channelopathies (Table 1). Lastly, we further summarize the role of ventricular assistance devices in HFpEF.
For the purpose of this review, search engines that included the PubMed Central and Google Scholar databases were used to search for studies published in the English language. Our searches were not limited by date restrictions, were free texts, and included the following keywords: “heart failure”, “heart failure with preserved ejection fraction”, “diastolic dysfunction”, “sudden cardiac death”, “obesity”, “lipid mediators”, “leukotrienes”, “ventricular arrhythmias”, “dyslipidemia”, “cardiac calcium channel”, “cardiac potassium channel”, “cardiac sodium channel”, “Ca handling proteins”, “SERCA2a”, “RyR2”, “phospholamban”, “pro-inflammatory cytokines”, “interleukin signaling”, “leukotrienes B4 signaling”, “cBIN1”, and “LVAD”.
Pathophysiology and Promotion of Molecular Changes in Cardiac Ionic Currents By Va
Cardiac AP occurs due to finely tuned interactions between intracellular structures and ionic currents that stimulate the excitation and contraction of ventricular cardiomyocytes [43]. A prolonged AP is a prominent phenomenon observed in HFpEF and ventricular arrhythmias [34]. Assessment of AP using patch-clamp electrophysiology [44] and optical mapping with voltage-sensitive dyes [45,46] has become an important tool in studying cardiac electrophysiology and arrhythmogenicity in preclinical animal models. In the clinical setting, action potential phases are demonstrated as an electrocardiogram (ECG) [47]. The QRS complex and Q-T interval constitute ventricular depolarization and repolarization, respectively, and their variations serve as indicators of the imminent onset of ventricular arrhythmias [47]. The ventricular tissue’s triggered activity manifests as EAD and delayed (DAD) afterdepolarizations [47]. While an increase in ICa,L current is thought to be the main contributor in both, they differ in timing, as EADs occur in phase 2 (prolonged AP duration ) or 3 (abbreviated AP duration), and DADs take place in the resting stage of the action potential [48,49]. Furthermore, afterdepolarizations and abnormalities in calcium handling may promote the formation of reentry circuits, which are arrhythmogenic conditions where an AP repeatedly moves in a closed-circle manner [50]. Dispersion in action potentials throughout the heart is defined as varying repolarization times in different regions of the heart and is believed to be able to cause reentry circuits and eventually VA [51]. Despite the significant role of ionic changes in triggering VA, a definite therapeutic option has yet to be proposed, which prompts a deeper investigation into the molecular basis of ionic changes.
Voltage-Gated Sodium Channels and Sodium Currents
Voltage-gated Na channels play a pivotal role in cardiac excitation and are key modulators of the AP. Na channels consist of one pore-forming α-subunit and non-pore-forming auxiliary beta-subunits [52]. Cardiac Na channels are composed of various isoforms, with the sodium channel protein type 5 subunit alpha (Nav1.5) being the most abundant [53]. The Nav1.5 channel is encoded by the SCN5A gene and is highly expressed in the conduction tissues of atria, ventricles, and Purkinje fibers [54]. Nav1.5 channels are responsible for the rapid inward sodium currents entering the cell at phase 0 of the AP [54]. While this rapid influx of Na and its subsequent upstroke in AP constitute the largest component of Na currents, another current called the late Na+ current (late INa), although smaller in magnitude, greatly influences the duration and morphology of the AP due to the sustained depolarization it creates in phases 2 and 3 [55,56]. Late Na currents occur due to the late physiological inactivation of a number of Nav1.5 channels, and their density is increased in HFpEF [57]. Although dysregulations in both peak and late Na currents contribute to cardiac arrhythmias [58], an increase or “gain of function” in late INa is accountable for the development of cardiac diastolic dysfunction by reversing the function of the NCX, allowing Ca to enter the cell, inducing a decline in myocyte relaxation, and triggering DADs [59,60]. Subsequently, these alterations lead to a delay in repolarization and a prolonged AP and Q-T interval, creating an arrhythmogenic environment [61], which has been demonstrated to underly the incidence of lethal VA in rat models of HFpEF [9,34].
The pivotal role of late INa in HFpEF and associated dysrhythmias is further confirmed by the ability of late INa-inhibiting drugs such as ranolazine[62,63,64] to decrease NCX expression [59] and enhance diastolic function in HFpEF patients [65]. However, it should be noted that, although echocardiographic changes in these patients were significantly in favor of enhanced diastolic function, conflicting evidence exists regarding the effect of ranolazine in reversing the elongated QT interval in HFpEF [65]. More recently, sodium–glucose cotransporter-2 (SGLT2) inhibitors such as empagliflozin and dapagliflozin have emerged as promising treatment options in HFpEF patients, exhibiting lower overall hospitalization and CVD rates [66,67]. Moreover, empagliflozin has demonstrated protective clinical effects against VA in diabetic patients [68,69]. To delve deeper, preclinical animal and in vitro investigations have been conducted to evaluate their roles in late INa and VA [57,70], demonstrating a reversal in late INa in long QT syndrome mutations of Nav1.5 with empagliflozin, dapagliflozin, and canagliflozin [70], as well as proarrhythmic AP changes in HFpEF models, particularly with empagliflozin [57]. Notably, these three SGLT2 inhibitor drugs display selective inhibition of late INa, meaning that they display few to no effects on peak INa [70]. Philippaert et al. also confirmed the ability of empagliflozin to diminish late INa current in an HF mouse model that had undergone transverse aortic constriction (TAC) [70]. Their study, however, does not indicate whether the animals had reduced or preserved EF [70]. In addition, it is known that although the pressure overload caused by TAC can result in hypertrophy, it is not a promising model for HFpEF [71]. Adding subcutaneous deoxycorticosterone acetate (DOCA), a mineralocorticoid agent, just after TAC surgery in mice can lead to a successful mimic of HFpEF at both the structural and physiological levels [72]. Considering the significance of the selective inhibition of late INa in HFpEF outcomes, it can be proposed that future studies should assess the effects of dapagliflozin and canagliflozin in HFpEF-mimicking animal models, such as DOCA+TAC mice [72] or HFD-fed guinea pigs [39]. Nevertheless, much remains unknown regarding proarrhythmic changes in HFpEF, and the identification of the underlying molecular mechanisms regulating late INa currents to better understand the risk factors precipitating resistance or sensitivity to medications is crucial.
Phosphorylation of Nav1.5 channels by kinases is a crucial modulator of late INa physiology [61]. The calcium-calmodulin kinase (CaMK) II family and their cardiac predominant isoform, CaMKIIδc, are of paramount significance in this phosphorylation given their overexpression in human and animal HF models [73]. In vivo, overexpression of CaMKII has been linked to shortened effective refractory periods, prolonged repolarization duration, and increased inclination for ventricular tachycardia [74]. Furthermore, the application of the autocamtide-2-related inhibitory peptide (AIP, a CaMKII inhibitor) reportedly reverses AP prolongation by inhibiting CaMKII in obese HFpEF mice [57], supporting the arrhythmogenic behavior of CaMKII. Notably, the same experiment demonstrated that myocytes with CaMKII proteins carrying oxidative resistant mutations (MM281/282VV) are less influenced by H2O2-induced late INa [57].
There is an intricate relationship among intracellular Na and Ca levels, reactive oxygen species (ROSs), and CaMKII, where an elevated intracellular Na+ level leads to ROS production in mitochondria, which eventually activates and stimulates CaMKII and ryanodine receptors (RyRs), thereby repeating the cycle [75]. Thus, oxidative stress and an inflammatory state may precipitate VA through intracellular Na, Ca, and late INa modulation [75]. The microvascular structure in HFpEF exhibits coronary microvascular endothelial inflammation and signs of oxidative stress, providing constant stimulation through elevated inflammatory cytokines [36]. Eventually, this systemic microvascular inflammation causes endothelial dysfunction, which was well demonstrated in an investigation on a rat model of HFpEF metabolic risk and HFpEF patients, which revealed the presence of enhanced monomeric endothelial nitric oxide synthesis (eNOS) and NO uncoupling, along with a decline in NO bioavailability in endothelial cells and macrophages under oxidative stress [76]. Most of these inflammation-induced endothelial dysfunctions are attributed to pro-inflammatory changes in obesity and metabolic syndrome [75]. Adipocytes in the obese state release pro-inflammatory cytokines, such as interleukins 1, 6, and 23 (IL-1, IL-6, and IL-23), tumor necrosis factor-alpha (TNF-α), and transforming growth factor-β (TGF-β)[36,77], instigating local and systemic inflammatory responses that precipitate oxidative stress [78]. These changes may be the reason behind the prolonged late INa observed in obese mouse models of HFpEF in the study by Hegyi et al. [57]. Still, no alterations were observed in Na currents in rats with metabolic syndrome [79], contradicting this hypothesis. Nevertheless, unlike other ions and currents, less is known about the mechanisms underlying how these cytokines influence Na concentration and currents.
Voltage-Gated l-Type Calcium Channels, Calcium Currents, and Calcium-Handling Proteins
The coordination between voltage-gated L-type calcium channels and calcium-handling proteins is crucial for the proper functioning of the excitation–contraction (EC) coupling machinery [80]. The alpha-conducting subunit of Cav1.2, which is the dominant form of voltage-gated L-type calcium channels in cardiac muscle, is encoded by the CACNAC1 gene [81]. Calcium-handling proteins, including RyRs and sarcoplasmic reticulum Ca-ATPase (SERCA), play a vital role in regulating intracellular calcium concentrations (Table 2) [82]. The ICa,L-current-mediated increase in calcium concentrations initiates the opening of the RyR2 channel, leading to the release of calcium from the sarcoplasmic reticulum (SR), a phenomenon known as Ca-induced Ca release (CICR) [83]. This calcium release activates Ca-sensing troponin C, initiating contraction. Relaxation occurs as cytosolic calcium levels decrease, facilitated by both the SERCA-mediated influx of calcium back into the SR and the NCX-mediated efflux of calcium to the extracellular space [84]. Dysregulation in Ca EC coupling is one of the main pillars of VA pathophysiology in HFpEF patients [9]. The alterations in EC and intracellular Ca concentrations are typically characterized by either an increased release of Ca into the sarcoplasm through Cav1.2, transient receptor potential (TRP), RyR2, or inositol 1,4,5-trisphosphate receptor type 2 (IP3R2) or the impaired uptake of Ca by NCX or SERCA2a [85]. Although the mechanisms behind EC changes are well established in HFrEF, the parts that play more important roles in HFpEF are not well understood.
In contrast to HFrEF, Ca transient amplitudes are generally heightened in HFpEF [86,87]. While overexpression of Cav1.2 proteins was identified in DAHL salt-sensitive HFpEF rat models by Kilfoil et al. [88], Ca recruitment by RyR2 is deemed to be a more prominent and observed moderator of Ca influx into the cytosol in other HFpEF models [86,87,89]. RyR2-mediated Ca leakage into the cytosol instigates diastolic Ca waves known as Ca sparks [87]. This enhanced extraction of Ca from the SR by RyR2 channels may be due to the increased or maintained density of transverse tubules (T-tubules) in HFpEF [90], contrasting with HFrEF [91]. T-tubules are invaginations of the surface membrane that are predominantly found in ventricular myocytes and are abundant in proteins involved in EC and Ca handling [92]. A higher density of T-tubules has been linked to higher intracellular Ca concentrations [92] and underlies the adaptive mechanism of HFpEF towards a stiff myocardium [90,91]. An elevated density of T-tubules is negatively correlated with the distance to the sarcoplasmic reticulum, contributing to faster Ca release, which may explain the reduced latency attributed to Ca release exhibited in the high-salt-fed rat models of HFpEF in a study by Kilfoil et al. [86,90]. Additionally, RyR2 phosphorylation by either protein kinase A (PKA) or CaMKII may also be an underlying feature leading to the aberrant release of Ca into the cytosol; however, debate persists as to which of these kinases are responsible for this phenomenon and to what extent [91,93]. Nevertheless, dantrolene, a muscle relaxant that has shown preventive behavior towards CPVT in a CPVT mutant knock-in mouse model [94], has been identified as facilitating the affinity of calmodulin, a calcium-sensitive protein that inhibits RyR2 activity, to RyR2 in mild hypertensive HFpEF rats, hence attenuating Ca release[89].
Cellular calcium transient kinetic phenotypes (including amplitude and decay) are another fundamental mediators pathological Ca responses and associated arrhythmogenesis. Phospholamban (PLB) serves as a critical and reversible modulator of SERCA, which inhibits SERCA2a in its dephosphorylated state, thereby reducing its affinity for Ca ions and curbing Ca decay by the SR [95]. This mechanism is, however, reversed by PKA- or CaMKII-mediated phosphorylation of PLB [96]. Disruptions in the PLB–SERCA2a relationship are portrayed as a dysregulated SERCA2a–PLB ratio in favor of SERCA2a inhibition, leading to perturbed calcium uptake by the SR and accumulated cytosolic Ca [97]. Agreeing with this notion, the hypertensive HFpEF rats presented by Rouhana et al. exhibited a prolonged Ca decay time and an increased PLB to-SERCA2a ratio, which led to enhanced diastolic Ca levels, in addition to enhanced contractility performance was demonstrated by longer sarcomere length (SL) shortening time and lower IK1 currents in phase 4 [87]. Nonetheless, the electrophysiological mechanism of Ca-handling systems is complex, and not one HFpEF model can fill all gaps. In particular, SERCA2a promotion may also lead to a long QRS interval and VA as a result of the release of Ca into the cytosol [98]. Interestingly, in a normotensive hypertrophic rat model with diastolic dysfunction, while both SERCA2a and PLB proteins were found to be expressed at a low level, both were phosphorylated at the CaMKII site, indicating SERCA2a activation and PLB inhibition [98]. This augmented Ca decay, therefore, prompted heightened Ca sensitivity and release—probably through RyR2 channels [98].
The contributions of obesity and metabolic syndrome to Ca current alterations, which elevate susceptibility to VA, are more prominent than the contributions to Na current alterations. Obesity exerts Ca-handling remodeling through various pathways and mechanisms, mainly encompassing PLB [99,100,101,102] and mitochondrial Ca cycling [100,103] alterations. Obesity not only hinders PKA-mediated PLB phosphorylation [99,104] but is also associated with elevated PLB mRNA expression in obese Wistar rats [101]. However, contrasting data exist regarding PLB protein expression [105]. The influence of obesity on Ca-handling proteins and the L-type calcium channel is seemingly more pronounced at the mRNA level rather than the post-translational levels [99,101,102,105,106]. Mitochondrial Ca cycling in HFpEF is distinct from that in HFrEF, as mitochondrial Ca concentrations are increased, unlike the decreased values in systolic dysfunction [100]. In the obese diabetic and hypertensive rat models in a study by Miranda-Silva et al., Ca mitochondrial levels, as well as cytosolic concentrations, were elevated both during contraction and at rest [100]. Therefore, it can be concluded that a close relationship exists between mitochondrial and cytosolic Ca concentrations in HFpEF. High mitochondrial Ca levels likely compensate for the high contractility performance through ATP production, as indicated by the slow sarcomere shortening [100]. However, persistently elevated levels of Ca in the mitochondria open the apoptotic-inducing mitochondrial permeability transition pores (mPTPs), which, in turn, can stimulate the release of Ca from the SR into the cytosol. This state of Ca overload will eventually trigger the formation of Ca waves, where each Ca efflux into the cytosol triggers a rapid Ca reuptake by SERCA and then a greater Ca efflux by RyR2 [107,108]. Ca waves can potentially lead to DADs and cardiac arrhythmias [107]. Gordan et al. discovered that mice with diminished mPTP function exhibited a lower incidence of Ca waves and were protected against cardiac arrhythmias [109]. Moreover, there is evidence that a high-fat diet induces mPTP opening [110] Hence, the inhibition of these pores may present a viable solution for VA in metabolism-associated HFpEF.
Voltage-Gated Potassium Channels and Potassium Currents
Several distinct classes of K channels are present in the ventricular tissue, and they are responsible for the flow of associated K currents and shaping the AP in different phases. Voltage-gated K channels, which are responsible for rapid repolarization and transient outward K currents (Ito), are encoded by KCND2/3 and KCNA4 [111,112]. The hERG, KCNQ1, and KCNE1 genes encode α-subunits of voltage-gated K channels controlling the final repolarization of the membrane potential through rapid (IKr) and slow (IKs) K currents (KCNQ1 and KCNE1)[112,113,114]. Lastly, the inward rectifier K currents (IK1), responsible for setting the resting membrane potentials are encoded by the KCNJ12/14/4 genes [112,115].
A decline in K currents is reportedly linked to HFpEF and associated VA. An in vitro investigation in high-salt-fed HFpEF rat cardiomyocytes revealed delayed repolarization and reduced Ito, IKr, and IK1, leading to prolonged QTc and APD, which was confirmed with ex vivo optical mapping [34]. Moreover, HFpEF rat models were more inclined to develop VT, ventricular fibrillation (VF), premature ventricular contractions (PVCs), and SCD, which is believed to be the result of reentry circuits [34]. Reentry circuits are the result of delayed repolarization due to decreased K currents [34]. Delayed repolarization creates an environment of varying refractoriness in the heart, allowing for opportunistic functional reentrant signals [34]. Similarly, a reduction in IK1 and Ito was observed in obese diabetic mice who received subcutaneous aldosterone infusions and eventually demonstrated HFpEF [116]. Isolated ventricular cardiomyocytes of these animals were further treated with empagliflozin, AIP, and vericiguat (an FDA-approved medication for severe HFrEF that acts by promoting the soluble guanylate cyclase pathway [117]), leading to reduced AP prolongation and APD alternans among all HFpEF mice exposed to empagliflozin and AIP, as well as in female mice treated with vericiguat [116]. Given the promising results in alleviating these arrhythmogenic changes, future studies can be proposed to investigate their effects on K currents and channels.
Disruptions in voltage-gated K channels predispose the heart to arrhythmias and increase the likelihood of abnormal electrical patterns [118]. Mutations in KCNQ1 and the subsequent dysregulation in slow delayed rectifier channels are closely linked with long QT syndrome [119,120]. The activation and deactivation of slow delayed rectifier K currents are slow at rest; however, once stimulated, their activation becomes faster, while deactivation remains slow [121]. This allows their associated mutations and downregulations to be closely linked to lethal VA, such as TdP [121]. However, this phenomenon is limited to sympathetic stimulation; therefore, medications targeting IKs have not yet yielded any success [122].
While all ventricular K currents exhibit decreased transients in preclinical models of HFpEF [34,87], the most prominent ion channel activity change observed in HFpEF is the functional downregulation of the Ito current, which is critical in determining early repolarization [34,123,124]. However, exogenous activation of Ito currents, although beneficial in reversing QTc prolongation, has failed to attenuate arrhythmias in HFpEF, which may be due to the downregulation of the cardiac channel responsible for Ito (Kv4.3) found in cardiomyocytes [34]. The simultaneous reduction in KCND2/3 and Kv4.3 highlights the significance of investigating K channel protein-encoding genes at both the transcriptional and protein expression levels [34].
There is a paucity of studies on how obesity regulates pathological K current functional expression and ultimately continues to VA in HFpEF. However, there is evidence that obesity regulates K currents through lipotoxicity and cytokine release [114]. Palmitic acid (PA) is a saturated fatty acid and a promoter of lipotoxicity that induces the secretion of pro-inflammatory cytokines, including IL-1β, IL-6, and TNFα [125]. Assessments of ventricular myocytes from guinea pigs fed with high-fat diets that included PA showed prolonged AP and QT intervals, likely through individual or multiple combinations of proinflammatory cytokines, including IL-18, IL-1β, and IL-6, as well as TNFα−induced cardiac remodeling [39,125]. As reduced expression of KCNQ1, KCNE1, and ERG-1a (IKr channel encoded by hERG) was detected in these myocytes, it was evident that lipotoxicity and obesity led to alterations in potassium channels, allowing for lower IKr and IKs densities [39,125]. Notably, preincubation of non-obese guinea pig ventricular myocytes with IL-6 and IL-6/IL-6 receptors (IL-6R) showed a similar decline in ERG expression, with the latter having a more prominent decrease [21]. Corbin et al. further elucidated that the inhibition of IKs in obese guinea pigs resulted in a higher risk of a more severe VA once exposed to isoproterenol [39]. However, investigations by Haim et al. on ventricular myocytes from palmitate-fed mice yielded contrasting results, showing elevation in all outward K currents [126]. This contrast likely lies in the major differences in major repolarizing K currents between mice and humans, prompting the search for ideal preclinical cardiometabolic models that can accurately mimic human conditions [127].
Beyond voltage-gated K currents, other cardiac K currents may play a role in the development of HFpEF. Intriguingly, inhibition of TWIK-related potassium channels (TREK-1), members of the cardiac two-pore domain K channels (K2P), in fibroblasts exhibited a protective effect against pressure overload [128,129,130]. Further research is needed to assess the role of TREK-1 in VA, as fibrosis is believed to induce reentry circuits by disrupting normal electrical conduction [9,131].
Inflammation and Structural Changes in Hfpef
The complex pathophysiology of HFpEF is the fundamental reason underlying the failure to reach suitable therapeutic options and is why the medications proven beneficial in HFrEF fail to provide significant outcomes in HFpEF [132,133,134]. Cardiometabolic disturbances and associated comorbidities drive HFpEF by disrupting the endothelial structure. Indeed, HFpEF is not a single disease summarized by a preserved systolic function. Thus, the therapeutic approach to HFpEF also needs to cover both structural and functional aspects of the disease [135]. Among the various aspects shaping the pathophysiology of HFpEF, inflammation and Ca-handling disturbances in cardiomyocytes are of great importance.
Interleukin 6 Signaling in Heart Failure
IL-6 is a pleiotropic cytokine (downstream of IL-1β action) [136] that serves as the core of cardiometabolic cytokine signaling [137], and it is a powerful predictor of the severity of heart diseases [138,139,140]. IL-6 has been associated with the development of atherosclerosis [141], angiotensin-2-mediated formation of unstable atherosclerotic plaques and unstable angina [142,143], increased myocardial infarct size 4 months post-ST elevated myocardial infarction (STEMI) [144], and elevated incidence of major adverse events and mortality in patients with coronary diseases [145]. Although less clinical evidence exists regarding the role of IL-6 in HFpEF, recent studies have demonstrated that IL-6 seems to be playing a crucial role in the prognosis of HFpEF and is positively associated with higher readmission rates and mortality [146,147]. Moreover, observations suggest that a marked elevation of IL-6 holds an intricate link to excess body fat, body mass index (BMI), and obesity in patients with HFpEF [148,149,150], further agreeing with the proinflammatory consequences of lipotoxicity[151].
Classical IL-6 signaling occurs through its membrane-bound receptor (IL-6Rα)–glycoprotein 130 (gp130 receptor complex), and it mediates homeostasis and regenerative functions [139,152]. The soluble IL-6R (sIL-6R) is generated through extracellular shedding or alternative processing of the mRNA encoding IL-6R [153]. The proinflammatory effects IL-6 are mediated via trans-signaling, whereby IL-6 binds to sIL-6R [154,155] and engages gp130 in target cells [156], leading to the activation of the downstream JAK-STAT and SHP2/ERK signaling pathways [157,158]. However, the potential therapeutic benefits and efficacy of selectively targeting IL-6 trans-signaling for the prevention of HF are unknown. There is, therefore, a critical need to define the cellular mechanisms of IL-6 and determine the therapeutic potential of selective anti-IL-6 trans-signaling in preclinical models of HF. Without such information, the promise of a novel class of anti-IL-6 therapies for the treatment of human HF will likely remain unfulfilled.
Notably, in a study by Hagiwara et al., cardiomyocytes derived from SHP2-dysregulated gp130 mutant mice that had been pre-incubated in an IL-6- and IL-6R-containing medium did not show increased Ca current density in a whole-cell patch-clamp analysis in comparison with wild-type mice in the same environment [158]. In vitro studies have elucidated that IL-6 may play a role in cardiac diastolic dysfunction through the downregulation of SERCA2 [151,152,153] and can lead to increased ICa, L-type current density, and Ca transients [154]. Treatment of HEK-hERG cells with a JAK1 inhibitor prevented IL-6 from decreasing IKr [21]. Increased expression of the JAK2 gene has been found in obese guinea pig models fed with a PA-containing HFD [39]. Injection of a JAK2 agonist, coumermycin, in the same models led to an enhanced risk of VA [39]. In addition to JAKs, phosphorylation and nuclear translocation of their downstream proteins called STATs may also be involved in cardiometabolic arrhythmogenic changes, as evidenced by the enhanced phosphorylation and nuclear translocation of STAT4 in these animal models, which has been found to be strongly associated with the enhanced VA/SCD frequency observed in them [39]. Therefore, understanding the pathophysiology of IL-6 is imperative for targeting IL-6 pathways for therapeutic means.
FFAs exert a proinflammatory response by promoting IL-6 expression and the initiation of downstream IL-6 signaling [159]. Two crucial mechanisms by which FFAs initiate this signaling cascade are through interrelated pathways, including Toll-like receptors (TLRs) and the NACHT, leucine-rich repeat (LRR)-, and pyrin domain (PYD)-containing protein 3 (NLRP3) inflammasome; both of these are part of the pattern recognition class of proteins belonging to the first line of defense in the body, which is known as innate immunity [159,160]. TLRs are transmembrane pattern recognition receptors that are able to respond and bind to pathogen-associated molecular patterns (PAMPs), and they are known to instigate an inflammatory state in cardiomyocytes by activating nuclear factor kappa B (NF-κB) [161]. While cardiomyocytes express most TLRs, TLR2 and TLR4 are commonly influenced by FFAs [159,161]. Qian et al. identified that inhibition of TLR2 by C29 (a TLR2 inhibitor) or TLR2-knock-out high-fat-diet-fed mice successfully reversed the elevated creatine kinase MB (CK-MB ) and cytokine (IL-6, IL-1β, and TNF-α) levels, and it attenuated cardiac hypertrophy and myocardial fibrosis, confirming the role of TLR2 in obesity-mediated cardiomyopathy [162]. In addition, immunoblot analysis revealed that the decline in TLR2 expression did indeed result in NF-κB inhibition [162]. Further, immunoprecipitation assays showed that lipotoxicity augments TLR2 myeloid differentiation primary response 88 (MYD88) complex levels in a TLR4-independent manner rather than TLR2 alone [162]. The hypertrophic changes induced by TLR2 through NF-κB and IL-1β may be due to adaptive responses to pressure overload, as smaller cardiomyocytes, lower heart-weight-to-body-weight ratio, and lower fibrosis were observed in Tlr2-/- mice in a study by Higashikuni et al. [163]. Therefore, it can be concluded that TLR2 may be a potential therapeutic target in diastolic HF. Similarly, inhibition of TLR4 also mitigates metabolic-associated cardiomyopathy [164,165,166,167,168]. TLR4 and co-receptor myeloid differentiation protein 2 (MD2) mostly share a downstream pathway similar to that of TLR2, as they signal through the TLR/MYD88/NF-κB pathway [169]. TLR4 can additionally be activated independently from MYD88, which also leads to NF-κB activation [169].
Activation of NF-κB by pattern recognition receptors such as TLRs can lead to the “priming” of NLRP3. NLRP3 is an inflammasome modulating the production of active IL-1β and IL-18. The activation of NLRP3 comprises two distinct steps called priming and triggering, which are responsible for the transcription and reorganization of NLRP3 components, respectively [160]. Priming of the NLRP3 components (NLRP3, pro-IL-1β, and pro-IL-18) leads to an increase in caspase-1, which, in turn, positively stimulates IL-1β [160]. Elevated NLRP3 levels are observed in obesity-related vascular dysfunction [170]. Moreover, in a study by Ralston et al., it was demonstrated that extracellular ATP accumulation activates NLRP3 through P2X purinoceptor 7 (P2X7) [171], which is a protein known for inducing cellular K depletion and K efflux [172]. In addition to K, NLRP3 also holds an intricate relationship with Ca, as revealed by cytosolic Ca increases either through P2X7/K exchange or from the SR [160]. These findings indicate that a deeper exploration of the relationship between IL-6, K, and Ca ionic properties and NLRP3 is warranted to gain a full understanding of the role of IL6 in HF.
Leukotriene B4 Signaling in Heart Failure
Leukotriene B4 (LTB4), a member of the leukotriene family, originates from the oxidation of arachidonic acid by 5-lipoxygenase (5-LO) [173]. LTB4 is a proinflammatory lipid mediator and leukocyte chemoattractant that is known for its significant role in insulin resistance and metabolic inflammation. Elevated levels thereof are extensively found in diabetic preclinical models [174,175]. LTB4 signaling plays a key role in cardiovascular diseases, and there is increasing evidence in the extant literature that LTB4 signaling serves as a contributing factor and a potential biomarker in a wide range of cardiovascular diseases, including atherosclerosis, MI, and acute coronary syndrome [176,177,178,179,180,181]. Intriguingly, a recent study by Corbin et al. demonstrated an increased propensity of spontaneous arrhythmias along with a poor response to isoproterenol challenge, which included severe electrical conduction abnormalities, resulting in heart block and asystole in LTB4-challenged guinea pigs [175]. In vitro administration of LTB4 to guinea pig ventricular myocytes further demonstrated a significant decline in IKr current and a reduction in hERG1a surface expression levels, suggesting that LTB4 might be a potent modulator of cardiac electrical activity, making this lipid mediator a possible therapeutic target in metabolism-induced cardiac arrhythmias [175].
Interestingly, the inhibition of the LTB4 receptor (LTBR) instigates glucose tolerance, anti-inflammatory macrophage polarization, and cardioprotective effects against arrhythmogenic QT prolongations [175,182,183]. The cardioprotective benefits of LTB4 inhibition are further confirmed in a recent study on mice exposed to doxorubicin, which is a cardiotoxic chemotherapeutic agent with established alterations in Ca handling as a consequence [184]. Pre-treatment with empagliflozin in these mouse models increased the EF and reduced LTB4 production by arachidonic acid [185]. Thus, investigating other LTB4 inhibitors may provide promising targets for reversing cardiac remodeling. Moreover, a pronounced increase in LTB4 is evident in adipose tissue [182]. It is well documented that elevated adipose tissue levels are accountable for ventricular remodeling in HFpEF [186]. In particular, changes in EAT, due to its proximity to the ventricles and positive correlation with BMI values, greatly influence AP disturbances, fibrosis, and the overall outcome in HFpEF [187]. Furthermore, it is known that EAT partly exerts its effect by releasing proinflammatory cytokines such as IL-6, IL-1β, and TNFα [186], thus emphasizing the importance of investigating the LTB4 pathway and its mechanisms in metabolic inflammation.
The LTB4 pathway is initiated by LTB4 binding to its G-protein-coupled receptors, BLT1 and BLT2 [188]. Despite the high BLT2 gene expression observed in patients with atherosclerosis in a study by Sanchez-Galan et al. [189], BLT2 is generally more correlated with non-cardiovascular inflammation and holds a lower affinity for LTB4 than BLT1 [188,190]. On the other hand, the LTB4/BLT1 axis is directly involved in cardiac lipotoxicity and inflammation [183]. Flow cytometry analysis in high-fat-diet-fed mice showed an increased surface expression of BLT1, specifically on monocytes [182]. The same study revealed that BLT1-deficient mice demonstrated fewer M1 macrophages and lower IL-6 levels [182]. These findings indicate that LTB4 may serve as a chemoattractant for monocytes in a state of obesity. Additionally, studies investigating the role of the LTB4/BLT1 axis in atherosclerosis have demonstrated that LTB4 can also stimulate the production of monocyte chemoattractant protein-1 (MCP-1/CCL2) [176,191]. It is known that in obesity, monocytes, as part of the innate immune system, are recruited to adipocytes from the bloodstream and are subsequently polarized into M1 macrophages, which initiates a cascade leading to pro-inflammatory cytokine secretion and eventually insulin resistance [182,192,193]. Therefore, it can be concluded that monocyte trafficking, a crucial step in obesity-mediated lipotoxicity, is partly dependent on LTB4 [192]. Intriguingly, the inhibition of BLT1 in obese mouse models resulted in lower levels of M1 macrophages and proinflammatory cytokines, including IL-6 [183].
Another path by which LTB4 instigates the release of inflammatory cytokines is by enhancing the activity of TLRs [194,195]. Although it was found by Gaudreault et al. that LTB4 does not have a direct effect on the expression of TLRs [194], LTB4 influences the TLR/MyD88/ NF-κB pathway by either increasing the expression of MyD88 through the positive regulation of miR-155 [195] or the phosphorylation of TGF-β-activated kinase 1 (TAK1), a downstream protein of TLR/MyD88, promoting cytokine production through the overexpression of NF-κB [183,194]. Moreover, TAK1 phosphorylation by LTB4 further contributes to cytokine infiltration through the mitogen-activated protein kinase (MAPK) pathway [183,194].
The notable correlations of LTB4 with insulin resistance and lipotoxicity suggest a role as a biomarker and a therapeutic role for LTB4 in metabolic HFpEF. Furthermore, higher LTB4 levels have been found in diabetic patients with cardiovascular autonomic neuropathy (CAN) [196]. CAN constitutes a state of dysfunctional nerve fibers serving the cardiovascular system [197]. CAN is a severe and common complication of diabetes that is strongly associated with long QT intervals, cardiac arrhythmias, and SCD in diabetic patients [197]. It is now widely believed that CAN is more a consequence of chronic inflammation associated with metabolic syndrome rather than merely a complication of a hyperglycemic state [198]. In addition to inflammatory cytokines, factors indicating endothelial dysfunction, such as eNOS, NO, and NO bioavailability, are also linked with CAN incidence and outcomes in diabetic patients [199].
The elevated levels of LTB4 in patients with CAN and the high incidence of cardiac arrhythmias in these patients suggest that LTB4 inhibition may be a possible treatment for ventricular arrhythmias in these patients and, consequently, for diabetic patients developing HFpEF. Administration of zileuton, an LTB4 and cysteinyl leukotrienes inhibitor, has successfully shortened the duration of VAs in rats undergoing ischemia–reperfusion injury [200]. Conversely, montelukast, a cysteinyl leukotriene inhibitor, did not have such an effect [200]. Therefore, LTB4 stands as a potent target for VA treatment. Nevertheless, extensive investigation is needed to assess how LTB4 modulates the expression of cardiac ion channels and their resulting ion currents. This information is crucial to attaining a comprehensive evidence-based approach to utilizing LTB4 inhibitors for the treatment of VAs in metabolic HFpEF [201].
Cardiac Bridging Integrator 1 Signaling in Heart Failure
Bridging integrator 1 (BIN1) is a BAR-domain-containing protein that is widely expressed in various tissues throughout the body [202]. Alternative splicing of the BIN1 gene results in tissue-specific BIN1 proteins [202]. The most frequently observed isoform in the heart is BIN1+13, which is responsible for cell proliferation [203]. However, the BIN1+13+17 isoform, which is formed by the combination of the cardiac alternatively spliced +13 and the ubiquitously alternatively spliced +17, is the cardiac isoform located on the T-tubules [204], and it mediates their densely folded morphology [204].
BIN1+13+17 is also known as cardiac bridging integrator 1 (cBIN1) and is a membrane scaffolding protein that creates micromembrane domains in cardiomyocytes; it is one of the critical structural proteins constituting cardiac dyads [205]. cBIN1 mediates T-tubule functions and cardiac Ca machinery by forming microfolds in T-tubules [204,206], aiding in the clustering and forward-trafficking of the L-type calcium channel (LTCC) or Cav1.2 to T-tubules [207,208], recruiting RyRs to dyads [209], and eventually leading to RyR-Cav1.2 coupling at the dyads [202,209].
Due to its reported low expression in HF and re-tubulization ability, it has been postulated that cBIN1 has the potential to be used as a gene therapy agent for HFpEF [135,208,210,211,212]. cBIN1 gene therapy in diabetic mice with HFpEF has shown promising results by reversing T-tubule disarrangements and lowering glucose levels [213]. Chronic exposure to sympathetic stress by isoproterenol (beta agonist medication) induces concentric hypertrophy and disrupted Ca handling [210]. Exogenous cBIN1 replacement in mice undergoing 4 weeks of isoproterenol administration reversed hypertrophy in a study by Liu et al. [210]. The same study also indicated that cBIN1 gene therapy further attenuates the effects of pressure overload and mitigates diastolic dysfunction [210]. Loss of cBIN1 is also linked with arrhythmogenic alterations in failing cardiomyocytes [204]. Administration of augmented ventricular pacing to mice carrying heterozygous and homozygous deletions of BIN1 showed a higher prevalence of VF and VT arrhythmias compared with the control wild-type group, with a more severe VT being attributed to mice bearing the homozygous deletion of BIN1 [204]. Agreeing with these preclinical findings are the results of a clinical study by Hong et al. [211]. Utilizing an enzyme-linked immunosorbent assay (ELISA), Hong et al. found that a marked decrease (<30) in plasma BIN1 levels in patients with arrhythmogenic right ventricular cardiomyopathy can predict arrhythmia incidence with 82% and 79% sensitivity and accuracy, respectively [211].
As cBIN1 is a major factor influencing cardiac T-tubule function, disruptions in T-tubule folding are the primary reasons behind the cardiomyopathic effects of cBIN1 loss. While reduced cBIN1 may not change the expression level of Cav1.2, it leads to dysregulated trafficking of the channels to T-tubules [210]. A reduction in cBIN1 levels does not result in smaller T-tubules but rather causes diminished T-tubule folding and invaginations, creating an enlarged round lumen [204,210]. The diminished T-tubule folds disrupt the “fuzzy space” created by cBIN1 [204]. Fuzzy spaces are diffusion barriers that slow the ion diffusion taking place in T-tubules, thereby stabilizing myocytes during ionic alterations [204]. Disruption of diffusion barriers prompts heightened sensitivity to sympathetic stimulations and susceptibility to lethal arrhythmias [204]. In contrast, one study by Frisk et al. assessed ventricular tissues of patients undergoing coronary artery bypass graft surgery, and they found that although T-tubules were dilated and increased, BIN1 gene expression was not changed in HFpEF patients [90]. However, since BIN1 is alternatively spliced, a precise PCR isoform identification with real-time PCR confirmation is crucial for investigating the cBIN1 patterns in HFpEF [204,214].
Transmission electron microscopy findings by Xu et al. have revealed the presence of cBIN1-containing micro-particles (MPs) in mouse plasma [215]. Further analysis by Xu et al. indicated that these cBIN1-containing MPs originate from T-tubule microfolds of ventricular cardiomyocytes [215]. Since MPs, like other vesicles, are subject to destruction when faced with adverse changes in osmolarity, Xu et al. induced hypotonic shock through the serial dilution of human plasma samples from heart failure patients, which led to a significant increase in the plasma concentrations of cBIN1 [215]. Lastly, the same study elucidated a strong and negative correlation between plasma cBIN1 levels and heart failure, evidenced by the reduced concentrations in HF patients compared with those in patients with normal heart conditions [215]. Given the availability of cBIN1 in plasma, it may serve as a potent biomarker in HFpEF patients. To this end, a normalized inverse index of the value of plasma cBIN1 levels called the cBIN1 score has been formulated [216]. The cBIN1 score is shown to be higher in HFpEF patients and is a promising diagnostic and prognostic biomarker for HFpEF [216].
Mechanical Circulatory Support Devices in Hfpef
Despite the advances in precision medicine and the early diagnosis and treatment of heart failure, many patients proceed to the late stages, where mechanical support is needed, and this is more pronounced in patients with HFpEF [217,218]. Left ventricular assistance devices (LVAD) were initially proposed as a bridging therapy for patients waiting for cardiac transplantation, but after the success of the landmark REMATCH trial, LVADs became FDA-approved as a definite therapy in patients with severe heart failure [217,219]. Currently, LVADs are viable therapeutic options in HFrEF patients, and most studies evaluating LVADs are in the HFrEF population.
Surprisingly, the prevalence of VA in patients undergoing LVAD surgery, particularly in the first month after the implantation of a device, is considerable and increases early mortality rates significantly [220]. The probable mechanism behind the incidence of VA is the alterations caused by the mechanical unloading of the left ventricle [221]. Ito et al. performed a heterotopic cardiac transplantation to the abdominal aorta in preclinical rat models to assess the impact of severe unloading on cardiac muscle function [221]. The results indicated that the contractile reserve and strength were significantly reduced after 5 weeks post-unloading, and this was accompanied by a significant delay in Ca transient decay [221]. Western blot analysis further demonstrated that the prolonged Ca transient decay was likely due to elevated PLB and reduced SERCA2a concentrations [221], underscoring the role of SR Ca release in post-LVAD cardiac function. Agreeing with this are the results from confocal microscopy of paced human cardiomyocytes from patients undergoing LVAD implantation or heart transplantation despite previous LVAD surgery, which showed an increased number of Ca sparks at the time of the device’s implantation and heart transplant, both of which were well inhibited through the administration of AIP [222]. Intriguingly, cardiomyocytes derived from patients undergoing heart transplants exhibited a more pronounced diastolic Ca release [222]. These findings indicate that not only is SR Ca release of significant importance in cardiac function, but marked dysregulations of this mechanism are seen after LVAD transplants [222].
Given the crucial role of Ca handling in EC coupling and pro-arrhythmogenic changes in the heart, the mentioned changes may be the reason behind the prevalence of VA after LVADs. Additionally, an increase in NCX function and a reduction in Kv4.3 Ito channels have also been reported in a subgroup of patients with LVAD and VA [223]. However, contradicting data exist regarding this matter, with studies reporting reduced RyR2 activity [224], increased SR Ca storage [225], and a slight decrease in NCX proteins [226] after LVAD implantation. Terracciano et al. demonstrated that while elevated SR Ca storage and reduced AP duration were evident in patients who recovered after concomitant LVAD implantation and medical therapies (as demonstrated by improved echocardiographic parameters), no such effect was seen in patients that did not recover after this dual-treatment approach [227,228]. Moreover, patients with no recovery after LVAD implantation exhibited a longer baseline QRS duration [229]. Pre-LVAD QRS durations have also shown good predictive accuracy for post-LVAD recovery [229]. Thus, it is also crucial to determine factors that can be used as predictive biomarkers of future VA in patients receiving LVADs.
Nevertheless, patients with HFpEF do not benefit much from LVADs in comparison with HFrEF patients [230]. Patients with concentric heart failure, which is common in HFpEF, do not demonstrate the same outcome with LVADs due to their reduced left ventricular volume [218]. Furthermore, LVAD implantation showed an increased susceptibility to right heart failure, as demonstrated by right atrial pressure, in patients with restricted and hypertrophic cardiomyopathies [231]. In addition to structural changes in the heart in HFpEF, the accompanying metabolic conditions may also be the reason for LVADs not being a suitable option in HFpEF. It has been reported that obese patients undergoing LVAD surgery demonstrate dysregulation in their immune system and are more prone to post-LVAD infections [232,233]. Additionally, investigation of adipose tissue using an image segmentation technique for pre-LVAD computed tomography (CT) images showed that adipose tissue is strongly and positively correlated with higher mortality rates and increased incidence of infections [234]. Likewise, a higher baseline hemoglobin A1c (HbA1c) level, a biomarker of the severity of hyperglycemia, is associated with elevated incidence of post-LVAD adverse events, including infections, cerebral hemorrhages, and thrombosis [235].
Since the left atrium is enlarged in most cases of HFpEF, left atrial decompression may pose a potent solution for providing more efficient blood flow [218,236]. In vitro, prototypes of left atrial assistance devices (LAADs) have been positioned at the mitral valve for HFpEF patients [237,238]. With the LAAD pumping blood from the left atrium to the left ventricle, a decline in aortic pressure and an increase in cardiac output were observed, without adverse effects on the left ventricular pressure [237,238]. However, it is important to investigate whether left atrial unloading leads to cardiac arrhythmias as well. Still, much remains unknown about the use of LAADs in HFpEF and whether they can modulate ion channels to decrease VA in these patients.
Conclusions and Future Directions
In conclusion, since the prevalence of HFpEF is increasing, and the development of an effective treatment approach remains elusive, particular attention to this disease is warranted. Sudden cardiac death through VA poses a significant threat to HFpEF patients. An obesity-linked HFpEF phenotype is particularly common, with obesity not only being a comorbidity but also a significant risk factor for HFpEF; consequently, this prompts an examination of how obesity and metabolic inflammation may exacerbate the underlying pathophysiology of VA. Given the evident role of metabolic inflammation in HFpEF, exploring ion channel modulations in this context may provide novel insights into potential treatment approaches.
This study focuses on how cardiac ion channels and currents are influenced during lipotoxicity, inflammation, and HFpEF. It is postulated that identifying the specific currents and channels that are dysregulated in HFpEF paves the way for therapeutic advances in this field. This hypothesis is further supported by the inhibition of late INa current by empagliflozin, AIP, and vericiguat, as well as the inhibition of NCX by ranolazine, in HFpEF [57,59,116]. The data are, however, still limited in this regard. It would be beneficial to investigate the utilization of these medications in animal models that more accurately mimic the expression of ion channels in humans. As we have discussed, mice differ from humans in most K currents, including IKr [127]. This proposition is highlighted by pharmaceutical guidelines deeming hERG cardiotoxicity screening to be essential for new drugs [239].
Moreover, it is crucial to investigate cardiac currents and channels in the context of metabolic inflammation, leading to our discussion on IL-6 and LTB4. As demonstrated in this study, LTB4 is involved in insulin resistance, a phenomenon that is crucial in the formation of diabetes, which may also drive arrhythmogenic changes in diabetic patients and eventually lead to HF. Diabetes and obesity compose an inflammatory state leading to heightened secretion of cytokines such as IL-6 and eventually the formation of HFpEF. Thus, it could be intriguing to know the following: (1) how LTB4 and IL-6 trans-signaling would affect cardiomyocytes and cardiac channels; (2) if the administration of an LTB4 inhibitor would reverse cardiac remodeling in obese and diabetic patients; (3) the differential expression of LTB4 and IL-6 in various adipose tissues and macrophages; (4) whether LTB4 could potentially influence myocardial fibrosis and instigate reentry circuits and fatal arrhythmias.
To advance our comprehension of these molecular mechanisms, there is an urgent need for preclinical models that can bridge existing gaps in the extant literature. If successful, these studies have the potential to make HFpEF more manageable for both clinicians and patients dealing with this condition.

Table 1.
Altered functional expression of ion channels in HFpEF and/or obese animal models.
| Current | Gene | mRNA | Protein | Current Density | Animal Model | HFpEF/Obese | Cardiac Tissue | QTC | Drug Tx (effect) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| late INa | SCN5A | NR | NR | ↑ | Mouse (C57BL/6J) |
+/+ | Ventricle | NR | Pre-incubation with Empagliflozin (↓ current density) |
[57] |
| NR | NR | ↔ | Rat (SD) | -/+ | Ventricle | ↔ (↑QRS) | NR | [79] | ||
| NR | NR | ↑ (male˃ female) |
Mouse (db/db+Aldo) | +/+ | Ventricle | NR | Empagliflozin (↓ APD prolongation, male + female) AIP (↓ APD prolongation, male + female) Vericiguat (↓ APD prolongation, only female) |
[116] | ||
| Peak INa | SCN5A | NR | NR | ↔ | Rat (SD) | -/+ | Ventricle | ↔ (↑QRS) | NR | [79] |
| ↔ | NR | ↑* | Rat (WR) | -/+ | Ventricle | NR | NR | [106] | ||
| ICa,L | CACNA1c | ↔ | ↑ | ↑ | Rat (Dahl/SS) | +/- | Ventricle | NR | NR | [86] |
| ↑ | NR | ↑* | Rat (WR) | -/+ | Ventricle | NR | NR | [106] | ||
| NR | ↔ | ↑ | Rat (WR) | +/- | Ventricle | NR | [88] | |||
| NR | NR | ↑ | Rat (HHR) | +/- | Ventricle | ↔ (↑QRS) | NR | [98] | ||
| NR | ↔ | NR | Rat (WR) | -/+ | Ventricle | NR | NR | [99] | ||
| ↓ | NR | NR | Rat (WR, 15 weeks) | -/+ | Ventricle | NR | NR | [102] | ||
| ↑ | NR | NR | Rat (WR, 30 weeks) | -/+ | Ventricle | NR | NR | [102] | ||
| ↔ | NR | NR | Rat (WR, 45 weeks) | -/+ | Ventricle | NR | NR | [102] | ||
| NR | ↔ | NR | Rat (WR) | -/+ | Ventricle | NR | NR | [105] | ||
| NR | ↔ | NR | Mouse (db/db) | +/- | Ventricle | NR | cBIN1 | [213] | ||
| ↔ | NR | ↑ | Rat (Dahl/SS) | +/- | Ventricle | ↑ | NR | [34] | ||
| NR | NR | ↓ (only male) | Mouse (db/db+Aldo) | +/+ | Ventricle | NR | Empagliflozin (↓ APD prolongation, male + female) AIP (↓ APD prolongation, male + female) Vericiguat (↓ APD prolongation, only female) |
[116] | ||
| Ito | KCND2/3, KCNA4 | NR | NR | ↔ | Rat (WR) | +/- | Ventricle | NR | NR | [87] |
| ↔ | NR | ↑* | Rat (WR) | -/+ | Ventricle | NR | NR | [106] | ||
| NR | NR | ↓ (male + female) | Mouse (db/db+Aldo) | +/+ | Ventricle | NR | Empagliflozin (↓ APD prolongation, male + female) AIP (↓ APD prolongation, male + female) Vericiguat (↓ APD prolongation, only female) |
[116] | ||
| NR | NR | ↑ | Mouse (C57BL6) | -/+ | Ventricle | NR | NR | [126] | ||
| KCND2 | ↔ | ↔ | ↓ | Rat (Dahl/SS) | +/- | Ventricle | ↑ | NR | [34] | |
| KCND3 | ↓ | ↓ | ↓ | Rat (Dahl/SS) | +/- | Ventricle | ↑ | NR | [34] | |
| KCNA4 | ↔ | ↔ | ↓ | Rat (Dahl/SS) | +/- | Ventricle | ↑ | NR | [34] | |
| IKr | hERG | ↔ | ↔ | ↓ | Rat (Dahl/SS) | +/- | Ventricle | ↑ | NR | [34] |
| ↓ | NR | NR | Rat (WR) | -/+ | Ventricle | NR | NR | [106] | ||
| NR | ↓ | ↓ | Guinea pig (HFD) | -/+ | Ventricle | ↑ | NR | [39] | ||
| IKs | KCNQ1, KCNE1 |
↓ | NR | ↓ | Guinea pig (HFD) | -/+ | Ventricle | ↑ | NR | [39] |
| IK1 | KCNJ12/14/4 | ↔ | NR | ↓ | Rat (WR) | +/- | Ventricle | NR | NR | [87] |
| ↔ | ↔ | NR | Rat (Dahl/SS) | +/- | Ventricle | ↑ | NR | [34] | ||
| ↑ | NR | ↑* | Rat (WR) | -/+ | Ventricle | NR | NR | [106] | ||
| NR | NR | ↓ (male + female) | Mouse (db/db+Aldo) | +/+ | Ventricle | NR | Empagliflozin (↓ APD prolongation, male + female) AIP (↓ APD prolongation, male + female) Vericiguat (↓ APD prolongation, only female) |
[116] |
↑, Increased; ↓, decreased; ↔, no change; NR, not reported; Tx, treatment; SD, Sprague Dawley; WR, Wistar rats; Dahl/SS, Dahl salt sensitive; db/db, a genetically diabetic mouse; Aldo, aldosterone; HFD, high-fat diet; QTc, QT interval corrected for heart rate; AIP, autocamtide-2-related inhibitory peptide; APD, action potential duration. Note that any changes that were not found to be statistically significant are displayed as “no change”. *Computer simulations were used for prediction.
Table 2.
Altered functional expression of cardiac Ca-handling proteins in HFpEF and/or obese animal models.
Table 2.
Altered functional expression of cardiac Ca-handling proteins in HFpEF and/or obese animal models.
| Ca handling protein | mRNA | Protein | Animal Model | HFpEF/Obese | Cardiac Tissue | Drug Tx (effect) | Ref. |
|---|---|---|---|---|---|---|---|
| SERCA2a | ↑ | NR | Rat (WR) | -/+ | Ventricle | NR | [106] |
| ↓ | ↓ | Rat (Dahl/SS) | +/- | Ventricle | Intraperitoneal Ranolazine (↑ expression) | [59] | |
| NR | ↔ | Rat (WR) | +/- | Ventricle | NR | [87] | |
| NR | ↓ | Rat (WR) | +/- | Ventricle | NR | [88] | |
| NR | ↓ | Rat (HHR) | +/- | Ventricle | NR | [98] | |
| NR | ↔ | Rat (WR) | -/+ | Ventricle | NR | [99] | |
| ↓ | NR | Rat (WR, 15 weeks) | -/+ | Ventricle | NR | [102] | |
| ↑ | NR | Rat (WR, 30 weeks) | -/+ | Ventricle | NR | [102] | |
| ↓ | NR | Rat (WR, 45 weeks) | -/+ | Ventricle | NR | [102] | |
| NR | ↔ | Rat (WR) | -/+ | Ventricle | NR | [105] | |
| ↑ | NR | Rat (WR) | -/+ | Ventricle | NR | [101] | |
| NR | ↔ | Rat (ZSF1) | +/+ | Ventricle | NR | [100] | |
| NR | ↔ | Mouse (C57BL/6J) | *+/+ | NR | NR | [104] | |
| NR | ↓ | Mouse (db/db) | +/- | Ventricle | cBIN1 (↑expression) | [213] | |
| NR | ↓ | Rat (Dahl/SS) | +/- | Ventricle | NR | [90] | |
| NR | ↓ | Rat (ZSF1) | +/+ | Ventricle | NR | [90] | |
| RyR2 | ↑ | NR | Rat (WR) | -/+ | Ventricle | NR | [106] |
| NR | ↔ | Rat (WR) | +/- | Ventricle | NR | [87] | |
| NR | NR | Rat (SD) | +/- | Ventricle | Dantrolene (↑RyR2 inhibition) | [89] | |
| ↔ | NR | Rat (WR, 15 weeks) | -/+ | Ventricle | NR | [102] | |
| ↑ | NR | Rat (WR, 30 weeks) | -/+ | Ventricle | NR | [102] | |
| ↔ | NR | Rat (WR, 45 weeks) | -/+ | Ventricle | NR | [102] | |
| ↑ | NR | Rat (WR) | -/+ | Ventricle | NR | [101] | |
| NR | ↔ | Mouse (db/db) | +/- | Ventricle | cBIN1 | [213] | |
| NCX | ↑ | NR | Rat (WR) | -/+ | Ventricle | NR | [106] |
| ↑ | ↑ | Rat (Dahl/SS) | +/- | Ventricle | Intraperitoneal Ranolazine (↓ expression) | [59] | |
| NR | ↔ | Rat (WR) | +/- | Ventricle | NR | [87] | |
| NR | ↔ | Rat (WR) | +/- | Ventricle | NR | [88] | |
| ↔ | Rat (WR) | +/- | [98] | ||||
| ↓ | Rat (WR, 15 weeks) | -/+ | Ventricle | NR | [102] | ||
| ↑ | NR | Rat (WR, 30 weeks) | -/+ | Ventricle | NR | [102] | |
| ↓ | NR | Rat (WR, 45 weeks) | -/+ | Ventricle | NR | [102] | |
| NR | ↔ | Rat (Dahl/SS) | +/- | Ventricle | NR | [90] | |
| NR | ↓ | Rat (ZSF1) | +/+ | Ventricle | NR | [90] | |
| PLB | NR | ↔ | Rat (WR) | +/- | Ventricle | NR | [87] |
| NR | ↔ | Rat (WR) | +/- | Ventricle | NR | [88] | |
| NR | ↓ | Rat (HHR) | +/- | Ventricle | NR | [98] | |
| NR | ↔ | Rat (WR) | -/+ | Ventricle | NR | [99] | |
| ↓ | NR | Rat (WR, 15 weeks) | -/+ | Ventricle | NR | [102] | |
| ↑ | NR | Rat (WR, 30 weeks) | -/+ | Ventricle | NR | [102] | |
| ↔ | NR | Rat (WR, 45 weeks) | -/+ | Ventricle | NR | [102] | |
| NR | ↓ | Rat (WR) | -/+ | Ventricle | NR | [105] | |
| ↑ | NR | Rat (WR) | -/+ | Ventricle | NR | [101] | |
| NR | ↑ | Rat (ZSF1) | +/+ | Ventricle | NR | [100] | |
| NR | ↔ | Rat (Dahl/SS) | +/- | Ventricle | NR | [90] | |
| NR | ↔ | Rat (ZSF1) | +/+ | Ventricle | NR | [90] |
↑, Increased; ↓, decreased; ↔, no change; NR, not reported; Tx, treatment; WR, Wistar rats; Dahl/SS, Dahl salt sensitive; ZSF1, Zucker fatty; db/db, a genetically diabetic mouse; SD, Sprague Dawley; HRR, hypertrophic heart rat; QTc, QT interval corrected for heart rate; cBIN1, cardiac bridging integrator 1. Note that any changes that were not found to be statistically significant are displayed as “no change”. *Diastolic dysfunction with no systolic dysfunction.
Author Contributions
P.B. wrote and finalized the manuscript. K.A.A. undertook manuscript editing and approval and finalized the manuscript. A.S.A. obtained funding, conceived, and wrote the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the Nora Eccles Harrison Treadwell Foundation and the NIH (R01 HL147044-01; R01 HL174450-01 to A.S.A.).
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study. All of the relevant data are included within the paper itself.
Conflicts of Interest
The authors declare that they have no competing interests.
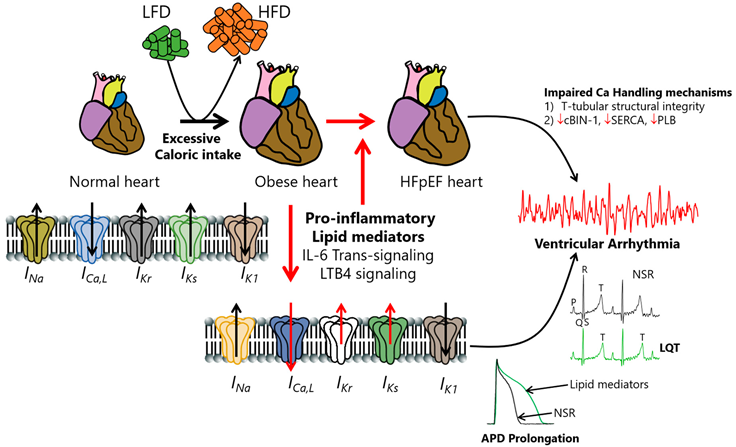
Cartoon illustration showing hypothesized obesity-linked channelopathies leading to pathological APD prolongation, LQT prolongation, ventricular arrhythmias, and ultimately, increased risk of sudden cardiac death in obese patients with HFpEF.
We speculate that overactivated obesity-linked heightened proinflammation—possibly through individual or multiple combinations of IL-6 trans-signaling or LTB4 pathways—contributes prominently to the pathogenesis of HFpEF. The exact cellular proarrhythmic effects of IL-6 and LTB4 involved are poorly understood. We suspect that overactivated IL-6- and LTB4-signaling-linked remodeling processes may occur through altered gene and protein expression of ion channel subunits and biophysical defects (subunit trafficking and/or gating defects). Whether and how INa and IK1 are modulated in obesity-linked IL-6 and LTB4 signaling or HFpEF is unknown. We further speculate that understanding the contributions of distinct cellular arrhythmia triggers that are sensitive to obesity/lipotoxic mechanisms is important for interpreting the mechanistic bases of how obesity channelopathies promote life-threatening VA/SCD risk and inform targeted anti-arrhythmia therapy in patients. HFpEF may promote structural changes in the organizational integrity of Ca-handling proteins (including cBin-1, SERCA, and PLB) and contribute to VA/SCD risk. Interestingly, there is a growing interest in the potential beneficial cardiovascular impact of cBIN1 gene therapy in preserving the functional and structural integrity of Ca-handling mechanisms in HF, with significant implications for HFpEF. The modulation of ventricular ion channels by cBIN-1 is currently unknown, suggesting that there is significant room for improving current therapeutic interventions in obese HF/HFpEF patients.
| Nonstandard Abbreviations and Acronyms | |
| HFpEF | Heart failure with preserved ejection fraction |
| HFrEF | Heart failure with reduced ejection fraction |
| SCD | Sudden cardiac death |
| EAT | Epicardial adipose tissue |
| INa | Sodium current |
| ICa, L | L-type calcium current |
| Ito | Transient outward potassium current |
| IKr | Rapid delayed rectifier potassium current |
| IKs | Slow delayed rectifier potassium current |
| IK1 | Inward rectifier potassium current |
| QTc | Corrected QT interval |
| NCX | Sodium/calcium exchanger |
| EAD | Early after depolarization |
| DAD | Delayed after depolarization |
| SGLT2 | Sodium–glucose cotransporter-2 |
| CamK | Calcium–calmodulin kinase |
| RyR | Ryanodine receptor |
| eNOS | Endothelial nitric oxide synthesis |
| NO | Nitric oxide |
| TNF | Tumor necrosis factor |
| TGF- β | Transforming growth factor-β |
| SERCA | Sarcoplasmic reticulum Ca+2-ATPase |
| PLB | Phospholamban |
| hERG | Human Ether-à-go-go-related gene |
| VT | Ventricular tachycardia |
| VF | Ventricular fibrillation |
| IL-6R | Interleukin 6 receptor |
| gp130 | Glycoprotein 130 |
| JAK/STAT | Janus kinase/signal transducers and activators of transcription |
| TLR | Toll-like receptor |
| NLRP3 | Leucine-rich repeat (LRR) and pyrin domain (PYD)-containing protein 3 |
| NF-κB | Nuclear factor kappa B |
| MyD88 | Myeloid differentiation primary response 88 |
| FFA | Free fatty acid |
| LTB4 | Leukotriene B4 |
| BLT1/2 | Leukotriene B4 receptor 1/2 |
| cBIN1 | Cardiac bridging integrator 1 |
| LVAD | Left ventricular assistance device |
| LAAD | Left atrial assistance device |
References
- Toth PP, Gauthier D: Heart failure with preserved ejection fraction: disease burden for patients, caregivers, and the health-care system. Postgraduate Medicine 2021, 133(2):140-145.
- Roh J, Houstis N, Rosenzweig A: Why Don’t We Have Proven Treatments for HFpEF? Circ Res 2017, 120(8):1243-1245.
- Vaduganathan M, Patel RB, Michel A, Shah SJ, Senni M, Gheorghiade M, Butler J: Mode of Death in Heart Failure With Preserved Ejection Fraction. J Am Coll Cardiol 2017, 69(5):556-569.
- Yuyun MF, Kinlay S, Singh JP, Joseph J: Are arrhythmias the drivers of sudden cardiac death in heart failure with preserved ejection fraction? A review. ESC Heart Fail 2023, 10(3):1555-1569.
- Hooks M, Downey MC, Joppa S, Beard A, Gravely A, Tholakanahalli V, Adabag S: Arrhythmic causes of in-hospital cardiac arrest among patients with heart failure with preserved ejection fraction. Heart Rhythm O2 2021, 2(6Part A):665-667.
- Gutierrez A, Ash J, Akdemir B, Alexy T, Cogswell R, Chen J, Adabag S: Nonsustained ventricular tachycardia in heart failure with preserved ejection fraction. Pacing Clin Electrophysiol 2020, 43(10):1126-1131.
- Cho JH, Leong D, Cuk N, Ebinger JE, Bresee C, Yoon SH, Ehdaie A, Shehata M, Wang X, Chugh SS et al: Delayed repolarization and ventricular tachycardia in patients with heart failure and preserved ejection fraction. PLoS One 2021, 16(7):e0254641.
- Curtain JP, Adamson C, Kondo T, Butt JH, Desai AS, Zannad F, Rouleau JL, Rohde LE, Kober L, Anand IS et al: Investigator-reported ventricular arrhythmias and mortality in heart failure with mildly reduced or preserved ejection fraction. Eur Heart J 2023, 44(8):668-677.
- Cho JH: Sudden Death and Ventricular Arrhythmias in Heart Failure With Preserved Ejection Fraction. Korean Circ J 2022, 52(4):251-264.
- Crespo-García T, Cámara-Checa A, Dago M, Rubio-Alarcón M, Rapún J, Tamargo J, Delpón E, Caballero R: Regulation of cardiac ion channels by transcription factors: Looking for new opportunities of druggable targets for the treatment of arrhythmias. Biochemical Pharmacology 2022, 204:115206.
- Bers DM, Despa S: Na+ transport in cardiac myocytes; Implications for excitation-contraction coupling. IUBMB Life 2009, 61(3):215-221.
- Varró A, Nánási PP, Lathrop DA: Potassium currents in isolated human atrial and ventricular cardiocytes. Acta Physiol Scand 1993, 149(2):133-142.
- Aromolaran AS, Subramanyam P, Chang DD, Kobertz WR, Colecraft HM: LQT1 mutations in KCNQ1 C-terminus assembly domain suppress IKs using different mechanisms. Cardiovasc Res 2014, 104(3):501-511.
- Puckerin A, Aromolaran KA, Chang DD, Zukin RS, Colecraft HM, Boutjdir M, Aromolaran AS: hERG 1a LQT2 C-terminus truncation mutants display hERG 1b-dependent dominant negative mechanisms. Heart Rhythm 2016, 13(5):1121-1130.
- Cheng EP, Yuan C, Navedo MF, Dixon RE, Nieves-Cintrón M, Scott JD, Santana LF: Restoration of Normal L-Type Ca2+ Channel Function During Timothy Syndrome by Ablation of an Anchoring Protein. Circulation Research 2011, 109(3):255-261.
- Wit AL: Afterdepolarizations and triggered activity as a mechanism for clinical arrhythmias. Pacing and Clinical Electrophysiology 2018, 41(8):883-896.
- Anderson A, Kulkarni K, Marron Fernandez de Velasco E, Carlblom N, Xia Z, Nakano A, Martemyanov KA, Tolkacheva EG, Wickman K: Expression and relevance of the G protein-gated K(+) channel in the mouse ventricle. Sci Rep 2018, 8(1):1192.
- Liu X, Shi J, Xiao P: Associations between common ion channel single nucleotide polymorphisms and sudden cardiac death in adults: A MOOSE-compliant meta-analysis. Medicine 2018, 97(38):e12428.
- Hancox JC, McPate MJ, El Harchi A, Zhang YH: The hERG potassium channel and hERG screening for drug-induced torsades de pointes. Pharmacol Ther 2008, 119(2):118-132.
- Garrido A, Lepailleur A, Mignani SM, Dallemagne P, Rochais C: hERG toxicity assessment: Useful guidelines for drug design. European Journal of Medicinal Chemistry 2020, 195:112290.
- Aromolaran AS, Srivastava U, Alí A, Chahine M, Lazaro D, El-Sherif N, Capecchi PL, Laghi-Pasini F, Lazzerini PE, Boutjdir M: Interleukin-6 inhibition of hERG underlies risk for acquired long QT in cardiac and systemic inflammation. PLoS One 2018, 13(12):e0208321.
- Sale H, Wang J, O’Hara TJ, Tester DJ, Phartiyal P, He JQ, Rudy Y, Ackerman MJ, Robertson GA: Physiological properties of hERG 1a/1b heteromeric currents and a hERG 1b-specific mutation associated with Long-QT syndrome. Circ Res 2008, 103(7):e81-95.
- Jones DK, Liu F, Vaidyanathan R, Eckhardt LL, Trudeau MC, Robertson GA: hERG 1b is critical for human cardiac repolarization. Proc Natl Acad Sci U S A 2014, 111(50):18073-18077.
- Zünkler BJ: Human ether-a-go-go-related (HERG) gene and ATP-sensitive potassium channels as targets for adverse drug effects. Pharmacol Ther 2006, 112(1):12-37.
- Wu L, Ma J, Li H, Wang C, Grandi E, Zhang P, Luo A, Bers DM, Shryock JC, Belardinelli L: Late sodium current contributes to the reverse rate-dependent effect of IKr inhibition on ventricular repolarization. Circulation 2011, 123(16):1713-1720.
- Weiss JN, Garfinkel A, Karagueuzian HS, Chen P-S, Qu Z: Early afterdepolarizations and cardiac arrhythmias. Heart rhythm 2010, 7(12):1891-1899.
- Remme CA, Bezzina CR: REVIEW: Sodium Channel (Dys)Function and Cardiac Arrhythmias. Cardiovascular Therapeutics 2010, 28(5):287-294.
- Ravens U, Cerbai E: Role of potassium currents in cardiac arrhythmias. EP Europace 2008, 10(10):1133-1137.
- Choi BR, Burton F, Salama G: Cytosolic Ca2+ triggers early afterdepolarizations and Torsade de Pointes in rabbit hearts with type 2 long QT syndrome. J Physiol 2002, 543(Pt 2):615-631.
- Paar V, Jirak P, Larbig R, Zagidullin NS, Brandt MC, Lichtenauer M, Hoppe UC, Motloch LJ: Pathophysiology of Calcium Mediated Ventricular Arrhythmias and Novel Therapeutic Options with Focus on Gene Therapy. Int J Mol Sci 2019, 20(21).
- Stienen S, Ferreira JP, Kobayashi M, Preud’homme G, Dobre D, Machu J-L, Duarte K, Bresso E, Devignes M-D, López N et al: Enhanced clinical phenotyping by mechanistic bioprofiling in heart failure with preserved ejection fraction: insights from the MEDIA-DHF study (The Metabolic Road to Diastolic Heart Failure). Biomarkers 2020, 25(2):201-211.
- Carella MJ, Mantz SL, Rovner DR, Willis 3rd PW, Gossain VV, Bouknight RR, Ferenchick GS: Obesity, adiposity, and lengthening of the QT interval: improvement after weight loss. International journal of obesity and related metabolic disorders: journal of the International Association for the Study of Obesity 1996, 20(10):938-942.
- Milovancev A, Stokic E: Corrected QT Interval and Corrected QT Dispersion in Obesity. CJournal of Clinical Cardiology and Diagnostics 2019, 2(1):1-4.
- Cho JH, Zhang R, Kilfoil PJ, Gallet R, de Couto G, Bresee C, Goldhaber JI, Marbán E, Cingolani E: Delayed Repolarization Underlies Ventricular Arrhythmias in Rats With Heart Failure and Preserved Ejection Fraction. Circulation 2017, 136(21):2037-2050.
- Cho JH, Zhang R, Aynaszyan S, Holm K, Goldhaber JI, Marbán E, Cingolani E: Ventricular Arrhythmias Underlie Sudden Death in Rats With Heart Failure and Preserved Ejection Fraction. Circ Arrhythm Electrophysiol 2018, 11(8):e006452.
- Aimo A, Castiglione V, Borrelli C, Saccaro LF, Franzini M, Masi S, Emdin M, Giannoni A: Oxidative stress and inflammation in the evolution of heart failure: From pathophysiology to therapeutic strategies. European Journal of Preventive Cardiology 2019, 27(5):494-510.
- Paulus WJ, Tschöpe C: A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 2013, 62(4):263-271.
- Omran J, Bostick BP, Chan AK, Alpert MA: Obesity and Ventricular Repolarization: a Comprehensive Review. Progress in Cardiovascular Diseases 2018, 61(2):124-135.
- Corbin A, Aromolaran KA, Aromolaran AS: STAT4 Mediates IL-6 Trans-Signaling Arrhythmias in High Fat Diet Guinea Pig Heart. Int J Mol Sci 2024, 25(14).
- Biet M, Morin N, Benrezzak O, Naimi F, Bellanger S, Baillargeon JP, Chouinard L, Gallo-Payet N, Carpentier AC, Dumaine R: Lasting alterations of the sodium current by short-term hyperlipidemia as a mechanism for initiation of cardiac remodeling. American Journal of Physiology-Heart and Circulatory Physiology 2013, 306(2):H291-H297.
- Sánchez G, Araneda F, Peña JP, Finkelstein JP, Riquelme JA, Montecinos L, Barrientos G, Llanos P, Pedrozo Z, Said M et al: High-Fat-Diet-Induced Obesity Produces Spontaneous Ventricular Arrhythmias and Increases the Activity of Ryanodine Receptors in Mice. Int J Mol Sci 2018, 19(2).
- Załęska-Kocięcka M, Wojdyńska Z, Kalisz M, Litwiniuk A, Mączewski M, Leszek P, Paterek A: Epicardial fat and ventricular arrhythmias. Heart Rhythm 2024, 21(2):206-212.
- Qu Z, Weiss JN: Mechanisms of ventricular arrhythmias: from molecular fluctuations to electrical turbulence. Annu Rev Physiol 2015, 77:29-55.
- Kornreich BG: The patch clamp technique: principles and technical considerations. J Vet Cardiol 2007, 9(1):25-37.
- O’Shea C, Winter J, Kabir SN, O’Reilly M, Wells SP, Baines O, Sommerfeld LC, Correia J, Lei M, Kirchhof P et al: High resolution optical mapping of cardiac electrophysiology in pre-clinical models. Scientific Data 2022, 9(1):135.
- Zhang R, Mesquita T, Cho Jae H, Li C, Sanchez L, Holm K, Akhmerov A, Liu W, Li Y, Ibrahim Ahmed G, Cingolani E: Systemic Delivery of Extracellular Vesicles Attenuates Atrial Fibrillation in Heart Failure With Preserved Ejection Fraction. JACC: Clinical Electrophysiology 2023, 9(2):147-158.
- Monitillo F, Leone M, Rizzo C, Passantino A, Iacoviello M: Ventricular repolarization measures for arrhythmic risk stratification. World J Cardiol 2016, 8(1):57-73.
- Weiss JN, Garfinkel A, Karagueuzian HS, Chen PS, Qu Z: Early afterdepolarizations and cardiac arrhythmias. Heart Rhythm 2010, 7(12):1891-1899.
- Tse G: Mechanisms of cardiac arrhythmias. J Arrhythm 2016, 32(2):75-81.
- Liu MB, de Lange E, Garfinkel A, Weiss JN, Qu Z: Delayed afterdepolarizations generate both triggers and a vulnerable substrate promoting reentry in cardiac tissue. Heart Rhythm 2015, 12(10):2115-2124.
- Clayton RH, Holden AV: Dispersion of cardiac action potential duration and the initiation of re-entry: A computational study. BioMedical Engineering OnLine 2005, 4(1):11.
- Bouza AA, Isom LL: Voltage-Gated Sodium Channel β Subunits and Their Related Diseases. Handb Exp Pharmacol 2018, 246:423-450.
- Dong C, Wang Y, Ma A, Wang T: Life Cycle of the Cardiac Voltage-Gated Sodium Channel NaV1.5. Frontiers in Physiology 2020, 11.
- András V, Tomek J, Nagy N, Virág L, Passini E, Rodriguez B, Baczkó I: Cardiac transmembrane ion channels and action potentials: cellular physiology and arrhythmogenic behavior. Physiological Reviews 2021, 101(3):1083-1176.
- Gintant GA, Datyner NB, Cohen IS: Slow inactivation of a tetrodotoxin-sensitive current in canine cardiac Purkinje fibers. Biophys J 1984, 45(3):509-512.
- Patlak JB, Ortiz M: Slow currents through single sodium channels of the adult rat heart. J Gen Physiol 1985, 86(1):89-104.
- Hegyi B, Mira Hernandez J, Shen EY, Habibi NR, Bossuyt J, Bers DM: Empagliflozin Reverses Late Na+ Current Enhancement and Cardiomyocyte Proarrhythmia in a Translational Murine Model of Heart Failure With Preserved Ejection Fraction. Circulation 2022, 145(13):1029-1031.
- Maier LS: A novel mechanism for the treatment of angina, arrhythmias, and diastolic dysfunction: inhibition of late I(Na) using ranolazine. J Cardiovasc Pharmacol 2009, 54(4):279-286.
- De Angelis A, Cappetta D, Piegari E, Rinaldi B, Ciuffreda LP, Esposito G, Ferraiolo FA, Rivellino A, Russo R, Donniacuo M et al: Long-term administration of ranolazine attenuates diastolic dysfunction and adverse myocardial remodeling in a model of heart failure with preserved ejection fraction. Int J Cardiol 2016, 217:69-79.
- Fink M, Noble PJ, Noble D: Ca²⁺-induced delayed afterdepolarizations are triggered by dyadic subspace Ca2²⁺ affirming that increasing SERCA reduces aftercontractions. Am J Physiol Heart Circ Physiol 2011, 301(3):H921-935.
- Iqbal SM, Lemmens-Gruber R: Phosphorylation of cardiac voltage-gated sodium channel: Potential players with multiple dimensions. Acta Physiol (Oxf) 2019, 225(3):e13210.
- Hale SL, Kloner RA: Ranolazine, an inhibitor of the late sodium channel current, reduces postischemic myocardial dysfunction in the rabbit. J Cardiovasc Pharmacol Ther 2006, 11(4):249-255.
- Belardinelli L, Shryock JC, Fraser H: Inhibition of the late sodium current as a potential cardioprotective principle: effects of the late sodium current inhibitor ranolazine. Heart 2006, 92(suppl 4):iv6-iv14.
- Undrovinas AI, Belardinelli L, Undrovinas NA, Sabbah HN: Ranolazine improves abnormal repolarization and contraction in left ventricular myocytes of dogs with heart failure by inhibiting late sodium current. Journal of cardiovascular electrophysiology 2006, 17:S169-S177.
- Cempaka Putri DKS, Andrianto A, Al-Farabi MJ, Saputra PBT, Nugraha RA: Efficacy of Ranolazine to Improve Diastolic Performance in Heart Failure with Preserved Ejection Fraction: A Systematic Review and Meta-analysis. Eur Cardiol 2023, 18:e02.
- Anker Stefan D, Butler J, Filippatos G, Ferreira João P, Bocchi E, Böhm M, Brunner–La Rocca H-P, Choi D-J, Chopra V, Chuquiure-Valenzuela E et al: Empagliflozin in Heart Failure with a Preserved Ejection Fraction. New England Journal of Medicine 2021, 385(16):1451-1461.
- Solomon Scott D, McMurray John JV, Claggett B, de Boer Rudolf A, DeMets D, Hernandez Adrian F, Inzucchi Silvio E, Kosiborod Mikhail N, Lam Carolyn SP, Martinez F et al: Dapagliflozin in Heart Failure with Mildly Reduced or Preserved Ejection Fraction. New England Journal of Medicine 2022, 387(12):1089-1098.
- Ozturk F, Tuner H, Atici A, Ali Barman H: Effect of Empagliflozin Treatment on Ventricular Repolarization Parameters. RCM 2024, 25(2):64-null.
- Fujiki S, Iijima K, Nakagawa Y, Takahashi K, Okabe M, Kusano K, Owada S, Kondo Y, Tsujita K, Shimizu W et al: Effect of empagliflozin on ventricular arrhythmias in patients with type 2 diabetes treated with an implantable cardioverter-defibrillator: the EMPA-ICD trial. Cardiovascular Diabetology 2024, 23(1):224.
- Philippaert K, Kalyaanamoorthy S, Fatehi M, Long W, Soni S, Byrne NJ, Barr A, Singh J, Wong J, Palechuk T et al: Cardiac Late Sodium Channel Current Is a Molecular Target for the Sodium/Glucose Cotransporter 2 Inhibitor Empagliflozin. Circulation 2021, 143(22):2188-2204.
- Withaar C, Lam CSP, Schiattarella GG, de Boer RA, Meems LMG: Heart failure with preserved ejection fraction in humans and mice: embracing clinical complexity in mouse models. European Heart Journal 2021, 42(43):4420-4430.
- Mohammed SF, Ohtani T, Korinek J, Lam CS, Larsen K, Simari RD, Valencik ML, Burnett JC, Jr., Redfield MM: Mineralocorticoid accelerates transition to heart failure with preserved ejection fraction via "nongenomic effects". Circulation 2010, 122(4):370-378.
- Beghi S, Furmanik M, Jaminon A, Veltrop R, Rapp N, Wichapong K, Bidar E, Buschini A, Schurgers LJ: Calcium Signalling in Heart and Vessels: Role of Calmodulin and Downstream Calmodulin-Dependent Protein Kinases. In: International Journal of Molecular Sciences. vol. 23; 2022.
- Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, Maier SK, Zhang T, Hasenfuss G, Brown JH et al: Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest 2006, 116(12):3127-3138.
- Hegyi B, Pölönen R-P, Hellgren KT, Ko CY, Ginsburg KS, Bossuyt J, Mercola M, Bers DM: Cardiomyocyte Na+ and Ca2+ mishandling drives vicious cycle involving CaMKII, ROS, and ryanodine receptors. Basic Research in Cardiology 2021, 116(1):58.
- Franssen C, Chen S, Unger A, Korkmaz HI, De Keulenaer GW, Tschöpe C, Leite-Moreira AF, Musters R, Niessen HW, Linke WA et al: Myocardial Microvascular Inflammatory Endothelial Activation in Heart Failure With Preserved Ejection Fraction. JACC Heart Fail 2016, 4(4):312-324.
- Kern PA, Saghizadeh M, Ong JM, Bosch RJ, Deem R, Simsolo RB: The expression of tumor necrosis factor in human adipose tissue. Regulation by obesity, weight loss, and relationship to lipoprotein lipase. J Clin Invest 1995, 95(5):2111-2119.
- Trayhurn P, Wood IS: Adipokines: inflammation and the pleiotropic role of white adipose tissue. Br J Nutr 2004, 92(3):347-355.
- Axelsen LN, Calloe K, Braunstein TH, Riemann M, Hofgaard JP, Liang B, Jensen CF, Olsen KB, Bartels ED, Baandrup U et al: Diet-induced pre-diabetes slows cardiac conductance and promotes arrhythmogenesis. Cardiovascular Diabetology 2015, 14(1):87.
- Jafri MS: Models of excitation-contraction coupling in cardiac ventricular myocytes. Methods Mol Biol 2012, 910:309-335.
- Buonarati OR, Henderson PB, Murphy GG, Horne MC, Hell JW: Proteolytic processing of the L-type Ca (2+) channel alpha (1)1.2 subunit in neurons. F1000Res 2017, 6:1166.
- Locatelli J, de Assis LV, Isoldi MC: Calcium handling proteins: structure, function, and modulation by exercise. Heart Fail Rev 2014, 19(2):207-225.
- Bers DM: Cardiac excitation-contraction coupling. Nature 2002, 415(6868):198-205.
- Valentim MA, Brahmbhatt AN, Tupling AR: Skeletal and cardiac muscle calcium transport regulation in health and disease. Biosci Rep 2022, 42(12).
- Saad NS, Mashali MA, Repas SJ, Janssen PML: Altering Calcium Sensitivity in Heart Failure: A Crossroads of Disease Etiology and Therapeutic Innovation. Int J Mol Sci 2023, 24(24).
- Kilfoil PJ, Lotteau S, Zhang R, Yue X, Aynaszyan S, Solymani RE, Cingolani E, Marbán E, Goldhaber JI: Distinct features of calcium handling and β-adrenergic sensitivity in heart failure with preserved versus reduced ejection fraction. J Physiol 2020, 598(22):5091-5108.
- Rouhana S, Farah C, Roy J, Finan A, Rodrigues de Araujo G, Bideaux P, Scheuermann V, Saliba Y, Reboul C, Cazorla O et al: Early calcium handling imbalance in pressure overload-induced heart failure with nearly normal left ventricular ejection fraction. Biochim Biophys Acta Mol Basis Dis 2019, 1865(1):230-242.
- Røe Å T, Aronsen JM, Skårdal K, Hamdani N, Linke WA, Danielsen HE, Sejersted OM, Sjaastad I, Louch WE: Increased passive stiffness promotes diastolic dysfunction despite improved Ca2+ handling during left ventricular concentric hypertrophy. Cardiovasc Res 2017, 113(10):1161-1172.
- Nawata J, Yamamoto T, Tanaka S, Yano Y, Uchida T, Fujii S, Nakamura Y, Suetomi T, Uchinoumi H, Oda T et al: Dantrolene improves left ventricular diastolic property in mineralcorticoid-salt-induced hypertensive rats. Biochem Biophys Rep 2023, 34:101449.
- Frisk M, Le C, Shen X, Røe ÅT, Hou Y, Manfra O, Silva GJJ, van Hout I, Norden ES, Aronsen JM et al: Etiology-Dependent Impairment of Diastolic Cardiomyocyte Calcium Homeostasis in Heart Failure With Preserved Ejection Fraction. Journal of the American College of Cardiology 2021, 77(4):405-419.
- Shou J, Huo Y: Changes of calcium cycling in HFrEF and HFpEF. Mechanobiology in Medicine 2023, 1(1):100001.
- Brette F, Orchard C: T-Tubule Function in Mammalian Cardiac Myocytes. Circulation Research 2003, 92(11):1182-1192.
- Benitah JP, Perrier R, Mercadier JJ, Pereira L, Gómez AM: RyR2 and Calcium Release in Heart Failure. Front Physiol 2021, 12:734210.
- Kobayashi S, Yano M, Uchinoumi H, Suetomi T, Susa T, Ono M, Xu X, Tateishi H, Oda T, Okuda S et al: Dantrolene, a Therapeutic Agent for Malignant Hyperthermia, Inhibits Catecholaminergic Polymorphic Ventricular Tachycardia in a RyR2R2474S/+ Knock-In Mouse Model. Circulation Journal 2010, 74(12):2579-2584.
- Koss KL, Kranias EG: Phospholamban: a prominent regulator of myocardial contractility. Circ Res 1996, 79(6):1059-1063.
- Mattiazzi A, Kranias E: The role of CaMKII regulation of phospholamban activity in heart disease. Frontiers in Pharmacology 2014, 5.
- Loffredo FS, Nikolova AP, Pancoast JR, Lee RT: Heart failure with preserved ejection fraction: molecular pathways of the aging myocardium. Circ Res 2014, 115(1):97-107.
- Curl CL, Danes VR, Bell JR, Raaijmakers AJA, Ip WTK, Chandramouli C, Harding TW, Porrello ER, Erickson JR, Charchar FJ et al: Cardiomyocyte Functional Etiology in Heart Failure With Preserved Ejection Fraction Is Distinctive-A New Preclinical Model. J Am Heart Assoc 2018, 7(11).
- Lima-Leopoldo AP, Leopoldo AS, da Silva DC, do Nascimento AF, de Campos DH, Luvizotto RA, de Deus AF, Freire PP, Medeiros A, Okoshi K, Cicogna AC: Long-term obesity promotes alterations in diastolic function induced by reduction of phospholamban phosphorylation at serine-16 without affecting calcium handling. J Appl Physiol (1985) 2014, 117(6):669-678.
- Miranda-Silva D, Wüst RCI, Conceição G, Gonçalves-Rodrigues P, Gonçalves N, Gonçalves A, Kuster DWD, Leite-Moreira AF, van der Velden J, de Sousa Beleza JM et al: Disturbed cardiac mitochondrial and cytosolic calcium handling in a metabolic risk-related rat model of heart failure with preserved ejection fraction. Acta Physiol (Oxf) 2020, 228(3):e13378.
- Lima-Leopoldo AP, Sugizaki MM, Leopoldo AS, Carvalho RF, Nogueira CR, Nascimento AF, Martinez PF, Luvizotto RA, Padovani CR, Cicogna AC: Obesity induces upregulation of genes involved in myocardial Ca2+ handling. Braz J Med Biol Res 2008, 41(7):615-620.
- Lima-Leopoldo AP, Leopoldo AS, Silva DC, Nascimento AF, Campos DH, Luvizotto Rde A, Oliveira Júnior SA, Padovani CR, Nogueira CR, Cicogna AC: Influence of long-term obesity on myocardial gene expression. Arq Bras Cardiol 2013, 100(3):229-237.
- Dia M, Gomez L, Thibault H, Tessier N, Leon C, Chouabe C, Ducreux S, Gallo-Bona N, Tubbs E, Bendridi N et al: Reduced reticulum–mitochondria Ca2+ transfer is an early and reversible trigger of mitochondrial dysfunctions in diabetic cardiomyopathy. Basic Research in Cardiology 2020, 115(6):74.
- Abdurrachim D, Ciapaite J, Wessels B, Nabben M, Luiken JJ, Nicolay K, Prompers JJ: Cardiac diastolic dysfunction in high-fat diet fed mice is associated with lipotoxicity without impairment of cardiac energetics in vivo. Biochim Biophys Acta 2014, 1842(10):1525-1537.
- Leopoldo AS, Lima-Leopoldo AP, Sugizaki MM, Nascimento AFd, de Campos DHS, Luvizotto RdAM, Castardeli E, Alves CAB, Brum PC, Cicogna AC: Involvement of L-type calcium channel and serca2a in myocardial dysfunction induced by obesity. Journal of Cellular Physiology 2011, 226(11):2934-2942.
- Ashrafi R, Yon M, Pickavance L, Yanni Gerges J, Davis G, Wilding J, Jian K, Zhang H, Hart G, Boyett M: Altered Left Ventricular Ion Channel Transcriptome in a High-Fat-Fed Rat Model of Obesity: Insight into Obesity-Induced Arrhythmogenesis. J Obes 2016, 2016:7127898.
- Zhao Z, Gordan R, Wen H, Fefelova N, Zang W-J, Xie L-H: Modulation of intracellular calcium waves and triggered activities by mitochondrial ca flux in mouse cardiomyocytes. PloS one 2013, 8(11):e80574.
- Landstrom AP, Dobrev D, Wehrens XHT: Calcium Signaling and Cardiac Arrhythmias. Circulation Research 2017, 120(12):1969-1993.
- Gordan R, Fefelova N, Gwathmey JK, Xie LH: Involvement of mitochondrial permeability transition pore (mPTP) in cardiac arrhythmias: Evidence from cyclophilin D knockout mice. Cell Calcium 2016, 60(6):363-372.
- Littlejohns B, Pasdois P, Duggan S, Bond AR, Heesom K, Jackson CL, Angelini GD, Halestrap AP, Suleiman MS: Hearts from mice fed a non-obesogenic high-fat diet exhibit changes in their oxidative state, calcium and mitochondria in parallel with increased susceptibility to reperfusion injury. PLoS One 2014, 9(6):e100579.
- Huo R, Sheng Y, Guo WT, Dong DL: The potential role of Kv4.3 K+ channel in heart hypertrophy. Channels (Austin) 2014, 8(3):203-209.
- Wulff H, Castle NA, Pardo LA: Voltage-gated potassium channels as therapeutic targets. Nature Reviews Drug Discovery 2009, 8(12):982-1001.
- Organ-Darling LE, Vernon AN, Giovanniello JR, Lu Y, Moshal K, Roder K, Li W, Koren G: Interactions between hERG and KCNQ1 α-subunits are mediated by their COOH termini and modulated by cAMP. Am J Physiol Heart Circ Physiol 2013, 304(4):H589-599.
- Aromolaran AS, Boutjdir M: Cardiac Ion Channel Regulation in Obesity and the Metabolic Syndrome: Relevance to Long QT Syndrome and Atrial Fibrillation. Front Physiol 2017, 8:431.
- Reisqs JB, Qu YS, Boutjdir M: Ion channel trafficking implications in heart failure. Front Cardiovasc Med 2024, 11:1351496.
- Mira Hernandez J, Shen EY, Ko CY, Hourani Z, Spencer ER, Smoliarchuk D, Bossuyt J, Granzier H, Bers DM, Hegyi B: Differential sex-dependent susceptibility to diastolic dysfunction and arrhythmia in cardiomyocytes from obese diabetic HFpEF model. Cardiovasc Res 2024.
- Armstrong Paul W, Pieske B, Anstrom Kevin J, Ezekowitz J, Hernandez Adrian F, Butler J, Lam Carolyn SP, Ponikowski P, Voors Adriaan A, Jia G et al: Vericiguat in Patients with Heart Failure and Reduced Ejection Fraction. New England Journal of Medicine 2020, 382(20):1883-1893.
- Chen L, Sampson KJ, Kass RS: Cardiac Delayed Rectifier Potassium Channels in Health and Disease. Card Electrophysiol Clin 2016, 8(2):307-322.
- Lu H, Ding W, Xiao H, Dai M, Xue Y, Jia Z, Guo J, Wu M, Shen B, Zhao R: Association of the P441L KCNQ1 variant with severity of long QT syndrome and risk of cardiac events. Frontiers in Cardiovascular Medicine 2022, 9.
- Wilson AJ, Quinn KV, Graves FM, Bitner-Glindzicz M, Tinker A: Abnormal KCNQ1 trafficking influences disease pathogenesis in hereditary long QT syndromes (LQT1). Cardiovascular Research 2005, 67(3):476-486.
- Aromolaran AS: Is there an emerging role for I(Ks) in aging-related ventricular arrhythmias? J Cell Physiol 2022, 237(12):4337-4338.
- Jost N, Virág L, Bitay M, Takács J, Lengyel C, Biliczki P, Nagy Z, Bogáts G, Lathrop DA, Papp JG, Varró A: Restricting excessive cardiac action potential and QT prolongation: a vital role for IKs in human ventricular muscle. Circulation 2005, 112(10):1392-1399.
- Tomaselli GF, Marbán E: Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res 1999, 42(2):270-283.
- He Q, Feng Y, Wang Y: Transient outward potassium channel: a heart failure mediator. Heart Fail Rev 2015, 20(3):349-362.
- Chowdhury MKH, Martinez-Mateu L, Do J, Aromolaran KA, Saiz J, Aromolaran AS: Macrophage-Dependent Interleukin-6-Production and Inhibition of I(K) Contributes to Acquired QT Prolongation in Lipotoxic Guinea Pig Heart. Int J Mol Sci 2021, 22(20).
- Haim TE, Wang W, Flagg TP, Tones MA, Bahinski A, Numann RE, Nichols CG, Nerbonne JM: Palmitate attenuates myocardial contractility through augmentation of repolarizing Kv currents. J Mol Cell Cardiol 2010, 48(2):395-405.
- Killeen MJ, Thomas G, Sabir IN, Grace AA, Huang CLH: Mouse models of human arrhythmia syndromes. Acta Physiologica 2008, 192(4):455-469.
- Abraham DM, Lee TE, Watson LJ, Mao L, Chandok G, Wang HG, Frangakis S, Pitt GS, Shah SH, Wolf MJ, Rockman HA: The two-pore domain potassium channel TREK-1 mediates cardiac fibrosis and diastolic dysfunction. J Clin Invest 2018, 128(11):4843-4855.
- Frangogiannis NG: Cardiac fibrosis. Cardiovasc Res 2021, 117(6):1450-1488.
- Sweeney M, Corden B, Cook SA: Targeting cardiac fibrosis in heart failure with preserved ejection fraction: mirage or miracle? EMBO Mol Med 2020, 12(10):e10865.
- Nguyen TP, Qu Z, Weiss JN: Cardiac fibrosis and arrhythmogenesis: the road to repair is paved with perils. J Mol Cell Cardiol 2014, 70:83-91.
- Pitt B, Pfeffer Marc A, Assmann Susan F, Boineau R, Anand Inder S, Claggett B, Clausell N, Desai Akshay S, Diaz R, Fleg Jerome L et al: Spironolactone for Heart Failure with Preserved Ejection Fraction. New England Journal of Medicine, 370(15):1383-1392.
- Solomon Scott D, McMurray John JV, Anand Inder S, Ge J, Lam Carolyn SP, Maggioni Aldo P, Martinez F, Packer M, Pfeffer Marc A, Pieske B et al: Angiotensin–Neprilysin Inhibition in Heart Failure with Preserved Ejection Fraction. New England Journal of Medicine 2019, 381(17):1609-1620.
- Peh ZH, Dihoum A, Hutton D, Arthur JSC, Rena G, Khan F, Lang CC, Mordi IR: Inflammation as a therapeutic target in heart failure with preserved ejection fraction. Front Cardiovasc Med 2023, 10:1125687.
- Correale M, Tricarico L, Fortunato M, Mazzeo P, Nodari S, Di Biase M, Brunetti ND: New Targets in Heart Failure Drug Therapy. Frontiers in Cardiovascular Medicine 2021, 8.
- Ridker PM, Libby P, MacFadyen JG, Thuren T, Ballantyne C, Fonseca F, Koenig W, Shimokawa H, Everett BM, Glynn RJ: Modulation of the interleukin-6 signalling pathway and incidence rates of atherosclerotic events and all-cause mortality: analyses from the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS). European heart journal 2018, 39(38):3499-3507.
- Libby P, Rocha VZ: All roads lead to IL-6: A central hub of cardiometabolic signaling. Int J Cardiol 2018, 259:213-215.
- Lazzerini PE, Laghi-Pasini F, Bertolozzi I, Morozzi G, Lorenzini S, Simpatico A, Selvi E, Bacarelli MR, Finizola F, Vanni F et al: Systemic inflammation as a novel QT-prolonging risk factor in patients with torsades de pointes. Heart 2017, 103(22):1821.
- Ridker PM, Rane M: Interleukin-6 Signaling and Anti-Interleukin-6 Therapeutics in Cardiovascular Disease. Circulation Research 2021, 128(11):1728-1746.
- Deokar SA, Dandekar SP, Shinde GA, Prabhu SS, Patawardhan M: Role of serum interleukin-6 in heart failure. International Journal of Advances in Medicine 2018, 5(4):936-940.
- Wassmann S, Stumpf M, Strehlow K, Schmid A, Schieffer B, Böhm M, Nickenig G: Interleukin-6 induces oxidative stress and endothelial dysfunction by overexpression of the angiotensin II type 1 receptor. Circ Res 2004, 94(4):534-541.
- Neri Serneri GG, Boddi M, Modesti PA, Coppo M, Cecioni I, Toscano T, Papa ML, Bandinelli M, Lisi GF, Chiavarelli M: Cardiac Angiotensin II Participates in Coronary Microvessel Inflammation of Unstable Angina and Strengthens the Immunomediated Component. Circulation Research 2004, 94(12):1630-1637.
- Schieffer B, Schieffer E, Hilfiker-Kleiner D, Hilfiker A, Kovanen PT, Kaartinen M, Nussberger J, Harringer W, Drexler H: Expression of Angiotensin II and Interleukin 6 in Human Coronary Atherosclerotic Plaques. Circulation 2000, 101(12):1372-1378.
- Groot HE, Al Ali L, van der Horst ICC, Schurer RAJ, van der Werf HW, Lipsic E, van Veldhuisen DJ, Karper JC, van der Harst P: Plasma interleukin 6 levels are associated with cardiac function after ST-elevation myocardial infarction. Clin Res Cardiol 2019, 108(6):612-621.
- Held C, White HD, Stewart RAH, Budaj A, Cannon CP, Hochman JS, Koenig W, Siegbahn A, Steg PG, Soffer J et al: Inflammatory Biomarkers Interleukin-6 and C-Reactive Protein and Outcomes in Stable Coronary Heart Disease: Experiences From the STABILITY (Stabilization of Atherosclerotic Plaque by Initiation of Darapladib Therapy) Trial. Journal of the American Heart Association, 6(10):e005077.
- Zhao Z, Qi D, Zhang Z, Du X, Zhang F, Ma R, Liang Y, Zhao Y, Gao Y, Yang Y: Prognostic Value of Inflammatory Cytokines in Predicting Hospital Readmissions in Heart Failure with Preserved Ejection Fraction. J Inflamm Res 2024, 17:3003-3012.
- Berger M, März W, Niessner A, Delgado G, Kleber M, Scharnagl H, Marx N, Schuett K: IL-6 and hsCRP predict cardiovascular mortality in patients with heart failure with preserved ejection fraction. ESC Heart Fail 2024.
- Alogna A, Koepp KE, Sabbah M, Espindola Netto JM, Jensen MD, Kirkland JL, Lam CSP, Obokata M, Petrie MC, Ridker PM et al: Interleukin-6 in Patients With Heart Failure and Preserved Ejection Fraction. JACC Heart Fail 2023, 11(11):1549-1561.
- Ratchford SM, Lee JF, Bunsawat K, Alpenglow JK, Zhao J, Ma CL, Ryan JJ, Khor LL, Wray DW: The impact of obesity on the regulation of muscle blood flow during exercise in patients with heart failure with a preserved ejection fraction. J Appl Physiol (1985) 2022, 132(5):1240-1249.
- Gui XY, Rabkin SW: C-Reactive Protein, Interleukin-6, Trimethylamine-N-Oxide, Syndecan-1, Nitric Oxide, and Tumor Necrosis Factor Receptor-1 in Heart Failure with Preserved Versus Reduced Ejection Fraction: a Meta-Analysis. Curr Heart Fail Rep 2023, 20(1):1-11.
- Ertunc ME, Hotamisligil GS: Lipid signaling and lipotoxicity in metaflammation: indications for metabolic disease pathogenesis and treatment. Journal of Lipid Research 2016, 57(12):2099-2114.
- Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S: The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta 2011, 1813(5):878-888.
- Lust JA, Donovan KA, Kline MP, Greipp PR, Kyle RA, Maihle NJ: Isolation of an mRNA encoding a soluble form of the human interleukin-6 receptor. Cytokine 1992, 4(2):96-100.
- Taga T, Kishimoto T: Gp130 and the interleukin-6 family of cytokines. Annu Rev Immunol 1997, 15:797-819.
- Fontes JA, Rose NR, Čiháková D: The varying faces of IL-6: From cardiac protection to cardiac failure. Cytokine 2015, 74(1):62-68.
- Collaboration IRGCERF: Interleukin-6 receptor pathways in coronary heart disease: a collaborative meta-analysis of 82 studies. The Lancet 2012, 379(9822):1205-1213.
- Garbers C, Heink S, Korn T, Rose-John S: Interleukin-6: designing specific therapeutics for a complex cytokine. Nat Rev Drug Discov 2018, 17(6):395-412.
- Hagiwara Y, Miyoshi S, Fukuda K, Nishiyama N, Ikegami Y, Tanimoto K, Murata M, Takahashi E, Shimoda K, Hirano T et al: SHP2-mediated signaling cascade through gp130 is essential for LIF-dependent ICaL, [Ca2+]i transient, and APD increase in cardiomyocytes. Journal of Molecular and Cellular Cardiology 2007, 43(6):710-716.
- Alí A, Boutjdir M, Aromolaran AS: Cardiolipotoxicity, Inflammation, and Arrhythmias: Role for Interleukin-6 Molecular Mechanisms. Front Physiol 2018, 9:1866.
- Toldo S, Mezzaroma E, Buckley LF, Potere N, Di Nisio M, Biondi-Zoccai G, Van Tassell BW, Abbate A: Targeting the NLRP3 inflammasome in cardiovascular diseases. Pharmacol Ther 2022, 236:108053.
- Boyd JH, Mathur S, Wang Y, Bateman RM, Walley KR: Toll-like receptor stimulation in cardiomyoctes decreases contractility and initiates an NF-κB dependent inflammatory response. Cardiovascular Research 2006, 72(3):384-393.
- Qian C, Xu D, Wang J, Luo Y, Jin T, Huang L, Zhou Y, Cai Z, Jin B, Bao H, Wang Y: Toll-like receptor 2 deficiency ameliorates obesity-induced cardiomyopathy via inhibiting NF-κB signaling pathway. Int Immunopharmacol 2024, 128:111551.
- Higashikuni Y, Tanaka K, Kato M, Nureki O, Hirata Y, Nagai R, Komuro I, Sata M: Toll-like receptor-2 mediates adaptive cardiac hypertrophy in response to pressure overload through interleukin-1β upregulation via nuclear factor κB activation. J Am Heart Assoc 2013, 2(6):e000267.
- Zheng X-y, Sun C-c, Liu Q, Lu X-y, Fu L-l, Liang G, Zhang X-h, Chen G-z: Compound LM9, a novel MyD88 inhibitor, efficiently mitigates inflammatory responses and fibrosis in obesity-induced cardiomyopathy. Acta Pharmacologica Sinica 2020, 41(8):1093-1101.
- Hu N, Zhang Y: TLR4 knockout attenuated high fat diet-induced cardiac dysfunction via NF-κB/JNK-dependent activation of autophagy. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2017, 1863(8):2001-2011.
- Wang S, Han Y, Liu R, Hou M, Neumann D, Zhang J, Wang F, Li Y, Zhao X, Schianchi F et al: Glycolysis-Mediated Activation of v-ATPase by Nicotinamide Mononucleotide Ameliorates Lipid-Induced Cardiomyopathy by Repressing the CD36-TLR4 Axis. Circ Res 2024, 134(5):505-525.
- Liu H, Jia W, Tang Y, Zhang W, Qi J, Yan J, Ding W, Cao H, Liang G, Zhu Z et al: Inhibition of MyD88 by LM8 Attenuates Obesity-Induced Cardiac Injury. J Cardiovasc Pharmacol 2020, 76(1):63-70.
- Xu Z, Kong X-Q: Bixin ameliorates high fat diet-induced cardiac injury in mice through inflammation and oxidative stress suppression. Biomedicine & Pharmacotherapy 2017, 89:991-1004.
- Zhang Y, Wu J, Dong E, Wang Z, Xiao H: Toll-like receptors in cardiac hypertrophy. Frontiers in Cardiovascular Medicine 2023, 10.
- Gao J, Xie Q, Wei T, Huang C, Zhou W, Shen W: Nebivolol Improves Obesity-Induced Vascular Remodeling by Suppressing NLRP3 Activation. J Cardiovasc Pharmacol 2019, 73(5):326-333.
- Ralston JC, Lyons CL, Kennedy EB, Kirwan AM, Roche HM: Fatty Acids and NLRP3 Inflammasome–Mediated Inflammation in Metabolic Tissues. Annual Review of Nutrition 2017, 37(Volume 37, 2017):77-102.
- Yaron J, Gangaraju S, Rao M, Kong X, Zhang L, Su F, Tian Y, Glenn H, Meldrum D: K+ regulates Ca2+ to drive inflammasome signaling: Dynamic visualization of ion flux in live cells. Cell Death & Disease 2015, 6.
- Saeki K, Yokomizo T: Identification, signaling, and functions of LTB4 receptors. Seminars in Immunology 2017, 33:30-36.
- Li P, Oh DY, Bandyopadhyay G, Lagakos WS, Talukdar S, Osborn O, Johnson A, Chung H, Mayoral R, Maris M: LTB4 promotes insulin resistance in obese mice by acting on macrophages, hepatocytes and myocytes. Nature medicine 2015, 21(3):239-247.
- Corbin A, Aromolaran KA, Aromolaran AS: Leukotriene B4 is elevated in diabetes and promotes ventricular arrhythmogenesis in guinea pig. Journal of Cellular Physiology 2024, n/a(n/a):e31467.
- Subbarao K, Jala VR, Mathis S, Suttles J, Zacharias W, Ahamed J, Ali H, Tseng MT, Haribabu B: Role of leukotriene B4 receptors in the development of atherosclerosis: potential mechanisms. Arterioscler Thromb Vasc Biol 2004, 24(2):369-375.
- Lefebvre B, Pépin JL, Baguet JP, Tamisier R, Roustit M, Riedweg K, Bessard G, Lévy P, Stanke-Labesque F: Leukotriene B4: early mediator of atherosclerosis in obstructive sleep apnoea? Eur Respir J 2008, 32(1):113-120.
- Molaie M, Lotfi R, Heidari Moghadam R, Rezaiemanesh A, Karaji AG, Salari F: Imbalanced serum levels of resolvin E1 (RvE1) and leukotriene B4 (LTB4) may contribute to the pathogenesis of atherosclerosis. Prostaglandins & Other Lipid Mediators 2023, 169:106781.
- Horii Y, Nakaya M, Ohara H, Nishihara H, Watari K, Nagasaka A, Nakaya T, Sugiura Y, Okuno T, Koga T et al: Leukotriene B4 receptor 1 exacerbates inflammation following myocardial infarction. The FASEB Journal 2020, 34(6):8749-8763.
- Xu S, Tang L, Mi Y, Jiang J, Zhu M, Chen B, Wang B, Li T, Xu C, Wang J et al: Clinical significance of leukotriene b4 and extracellular matrix metalloproteinase inducer in acute coronary syndrome. Clin Invest Med 2013, 36(6):E282-289.
- Sasaki K, Ueno A, Katori M, Kikawada R: Detection of leukotriene B4 in cardiac tissue and its role in infarct extension through leucocyte migration. Cardiovascular Research 1988, 22(2):142-148.
- Spite M, Hellmann J, Tang Y, Mathis SP, Kosuri M, Bhatnagar A, Jala VR, Haribabu B: Deficiency of the Leukotriene B4 Receptor, BLT-1, Protects against Systemic Insulin Resistance in Diet-Induced Obesity. The Journal of Immunology 2011, 187(4):1942-1949.
- Li P, Oh DY, Bandyopadhyay G, Lagakos WS, Talukdar S, Osborn O, Johnson A, Chung H, Mayoral R, Maris M et al: LTB4 promotes insulin resistance in obese mice by acting on macrophages, hepatocytes and myocytes. Nature Medicine 2015, 21(3):239-247.
- Shinlapawittayatorn K, Chattipakorn SC, Chattipakorn N: The effects of doxorubicin on cardiac calcium homeostasis and contractile function. Journal of Cardiology 2022, 80(2):125-132.
- Quagliariello V, De Laurentiis M, Rea D, Barbieri A, Monti MG, Carbone A, Paccone A, Altucci L, Conte M, Canale ML et al: The SGLT-2 inhibitor empagliflozin improves myocardial strain, reduces cardiac fibrosis and pro-inflammatory cytokines in non-diabetic mice treated with doxorubicin. Cardiovascular Diabetology 2021, 20(1):150.
- Borlaug BA, Jensen MD, Kitzman DW, Lam CSP, Obokata M, Rider OJ: Obesity and heart failure with preserved ejection fraction: new insights and pathophysiological targets. Cardiovasc Res 2023, 118(18):3434-3450.
- Aitken-Buck HM, Moharram M, Babakr AA, Reijers R, Van Hout I, Fomison-Nurse IC, Sugunesegran R, Bhagwat K, Davis PJ, Bunton RW et al: Relationship between epicardial adipose tissue thickness and epicardial adipocyte size with increasing body mass index. Adipocyte 2019, 8(1):412-420.
- Tager AM, Luster AD: BLT1 and BLT2: the leukotriene B(4) receptors. Prostaglandins Leukot Essent Fatty Acids 2003, 69(2-3):123-134.
- Sánchez-Galán E, Gómez-Hernández A, Vidal C, Martín-Ventura JL, Blanco-Colio LM, Muñoz-García B, Ortega L, Egido J, Tuñón J: Leukotriene B4 enhances the activity of nuclear factor-κB pathway through BLT1 and BLT2 receptors in atherosclerosis. Cardiovascular Research 2009, 81(1):216-225.
- Yokomizo T, Shimizu T: The leukotriene B receptors BLT1 and BLT2 as potential therapeutic targets. Immunological Reviews 2023, 317(1):30-41.
- Huang L, Zhao A, Wong F, Ayala JM, Struthers M, Ujjainwalla F, Wright SD, Springer MS, Evans J, Cui J: Leukotriene B4 Strongly Increases Monocyte Chemoattractant Protein-1 in Human Monocytes. Arteriosclerosis, Thrombosis, and Vascular Biology 2004, 24(10):1783-1788.
- Wenzl FA, Ambrosini S, Mohammed SA, Kraler S, Lüscher TF, Costantino S, Paneni F: Inflammation in Metabolic Cardiomyopathy. Frontiers in Cardiovascular Medicine 2021, 8.
- Liu Y, Lu X, Li X, Du P, Qin G: High-fat diet triggers obesity-related early infiltration of macrophages into adipose tissue and transient reduction of blood monocyte count. Molecular Immunology 2020, 117:139-146.
- Gaudreault É, Paquet-Bouchard C, Fiola S, Le Bel M, Lacerte P, Shio MT, Olivier M, Gosselin J: TAK1 contributes to the enhanced responsiveness of LTB4-treated neutrophils to Toll-like receptor ligands. International Immunology 2012, 24(11):693-704.
- Wang Z, Filgueiras LR, Wang S, Serezani AP, Peters-Golden M, Jancar S, Serezani CH: Leukotriene B4 enhances the generation of proinflammatory microRNAs to promote MyD88-dependent macrophage activation. J Immunol 2014, 192(5):2349-2356.
- Santos-Bezerra DP, Filgueiras LR, Monteiro MB, Admoni SN, Perez RV, Cavaleiro AM, Machado CG, Machado UF, Passarelli M, Jancar S, Correa-Giannella ML: Leukotriene Pathway Activation Associates with Poor Glycemic Control and with Cardiovascular Autonomic Neuropathy in Type 1 Diabetes. Mediators Inflamm 2020, 2020:5704713.
- Duque A, Mediano MFF, De Lorenzo A, Rodrigues LF, Jr.: Cardiovascular autonomic neuropathy in diabetes: Pathophysiology, clinical assessment and implications. World J Diabetes 2021, 12(6):855-867.
- Bakkar N-MZ, Dwaib HS, Fares S, Eid AH, Al-Dhaheri Y, El-Yazbi AF: Cardiac Autonomic Neuropathy: A Progressive Consequence of Chronic Low-Grade Inflammation in Type 2 Diabetes and Related Metabolic Disorders. In: International Journal of Molecular Sciences. vol. 21; 2020.
- Bhati P, Alam R, Moiz JA, Hussain ME: Subclinical inflammation and endothelial dysfunction are linked to cardiac autonomic neuropathy in type 2 diabetes. Journal of Diabetes & Metabolic Disorders 2019, 18(2):419-428.
- Gonca E: The Effects of Zileuton and Montelukast in Reperfusion-Induced Arrhythmias in Anesthetized Rats. Current Therapeutic Research 2013, 75:27-32.
- Aromolaran AS: Leukotriene B4 signaling in diabetic ventricular arrhythmias. J Cell Physiol 2024.
- Jiang X-X, Zhu Y-R, Liu H-M, Chen S-L, Zhang D-M: Effect of BIN1 on cardiac dysfunction and malignant arrhythmias. Acta Physiologica 2020, 228(3):e13429.
- Zhou K, Hong T: Cardiac BIN1 (cBIN1) is a regulator of cardiac contractile function and an emerging biomarker of heart muscle health. Sci China Life Sci 2017, 60(3):257-263.
- Hong T, Yang H, Zhang S-S, Cho HC, Kalashnikova M, Sun B, Zhang H, Bhargava A, Grabe M, Olgin J et al: Cardiac BIN1 folds T-tubule membrane, controlling ion flux and limiting arrhythmia. Nature Medicine 2014, 20(6):624-632.
- Mackrill JJ: Evolution of the cardiac dyad. Philosophical Transactions of the Royal Society B: Biological Sciences 2022, 377(1864):20210329.
- Lawless M, Caldwell JL, Radcliffe EJ, Smith CER, Madders GWP, Hutchings DC, Woods LS, Church SJ, Unwin RD, Kirkwood GJ et al: Phosphodiesterase 5 inhibition improves contractile function and restores transverse tubule loss and catecholamine responsiveness in heart failure. Sci Rep 2019, 9(1):6801.
- Hong T-T, Smyth JW, Gao D, Chu KY, Vogan JM, Fong TS, Jensen BC, Colecraft HM, Shaw RM: BIN1 localizes the L-type calcium channel to cardiac T-tubules. PLoS biology 2010, 8(2):e1000312.
- Hong T-T, Smyth JW, Chu KY, Vogan JM, Fong TS, Jensen BC, Fang K, Halushka MK, Russell SD, Colecraft H: BIN1 is reduced and Cav1. 2 trafficking is impaired in human failing cardiomyocytes. Heart rhythm 2012, 9(5):812-820.
- Fu Y, Shaw SA, Naami R, Vuong CL, Basheer WA, Guo X, Hong T: Isoproterenol Promotes Rapid Ryanodine Receptor Movement to Bridging Integrator 1 (BIN1)–Organized Dyads. Circulation 2016, 133(4):388-397.
- Liu Y, Zhou K, Li J, Agvanian S, Caldaruse A-M, Shaw S, Hitzeman Tara C, Shaw Robin M, Hong T: In Mice Subjected to Chronic Stress, Exogenous cBIN1 Preserves Calcium-Handling Machinery and Cardiac Function. JACC: Basic to Translational Science 2020, 5(6):561-578.
- Hong T-T, Cogswell R, James CA, Kang G, Pullinger CR, Malloy MJ, Kane JP, Wojciak J, Calkins H, Scheinman MM et al: Plasma BIN1 correlates with heart failure and predicts arrhythmia in patients with arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm 2012, 9(6):961-967.
- on AR, Nikolaev VO, Miragoli M, Sikkel MB, Paur H, Benard L, Hulot JS, Kohlbrenner E, Hajjar RJ, Peters NS et al: Plasticity of surface structures and β(2)-adrenergic receptor localization in failing ventricular cardiomyocytes during recovery from heart failure. Circ Heart Fail 2012, 5(3):357-365.
- Li J, Richmond B, Cluntun AA, Bia R, Walsh MA, Shaw K, Symons JD, Franklin S, Rutter J, Funai K et al: Cardiac gene therapy treats diabetic cardiomyopathy and lowers blood glucose. JCI Insight 2023, 8(18).
- Harvey SE, Cheng C: Methods for Characterization of Alternative RNA Splicing. Methods Mol Biol 2016, 1402:229-241.
- Xu B, Fu Y, Liu Y, Agvanian S, Wirka RC, Baum R, Zhou K, Shaw RM, Hong T: The ESCRT-III pathway facilitates cardiomyocyte release of cBIN1-containing microparticles. PLoS Biol 2017, 15(8):e2002354.
- Nikolova AP, Hitzeman TC, Baum R, Caldaruse A-M, Agvanian S, Xie Y, Geft DR, Chang DH, Moriguchi JD, Hage A et al: Association of a Novel Diagnostic Biomarker, the Plasma Cardiac Bridging Integrator 1 Score, With Heart Failure With Preserved Ejection Fraction and Cardiovascular Hospitalization. JAMA Cardiology 2018, 3(12):1206-1210.
- Marcel R, Meyer DM: An overview of approved and investigational left ventricular assist devices. Proc (Bayl Univ Med Cent) 2004, 17(4):407-410.
- Rosenblum H, Brener M, Burkhoff D: Theoretical considerations for a left atrial pump in heart failure with preserved ejection fraction. Heart Fail Rev 2023, 28(2):273-280.
- Rose Eric A, Gelijns Annetine C, Moskowitz Alan J, Heitjan Daniel F, Stevenson Lynne W, Dembitsky W, Long James W, Ascheim Deborah D, Tierney Anita R, Levitan Ronald G et al: Long-Term Use of a Left Ventricular Assist Device for End-Stage Heart Failure. New England Journal of Medicine, 345(20):1435-1443.
- Galand V, Flécher E, Auffret V, Pichard C, Boulé S, Vincentelli A, Rollin A, Mondoly P, Barandon L, Pernot M et al: Early Ventricular Arrhythmias After LVAD Implantation Is the Strongest Predictor of 30-Day Post-Operative Mortality. JACC: Clinical Electrophysiology 2019, 5(8):944-954.
- Ito K, Nakayama M, Hasan F, Yan X, Schneider MD, Lorell BH: Contractile reserve and calcium regulation are depressed in myocytes from chronically unloaded hearts. Circulation 2003, 107(8):1176-1182.
- Fischer TH, Kleinwächter A, Herting J, Eiringhaus J, Hartmann N, Renner A, Gummert J, Haverich A, Schmitto JD, Sossalla S: Inhibition of CaMKII Attenuates Progressing Disruption of Ca2+ Homeostasis Upon Left Ventricular Assist Device Implantation in Human Heart Failure. Artificial Organs 2016, 40(8):719-726.
- Refaat M, Chemaly E, Lebeche D, Gwathmey JK, Hajjar RJ: Ventricular arrhythmias after left ventricular assist device implantation. Pacing Clin Electrophysiol 2008, 31(10):1246-1252.
- Klotz S, Barbone A, Reiken S, Holmes JW, Naka Y, Oz MC, Marks AR, Burkhoff D: Left ventricular assist device support normalizes left and right ventricular beta-adrenergic pathway properties. J Am Coll Cardiol 2005, 45(5):668-676.
- Terracciano CM, Harding SE, Adamson D, Koban M, Tansley P, Birks EJ, Barton PJ, Yacoub MH: Changes in sarcolemmal Ca entry and sarcoplasmic reticulum Ca content in ventricular myocytes from patients with end-stage heart failure following myocardial recovery after combined pharmacological and ventricular assist device therapy. Eur Heart J 2003, 24(14):1329-1339.
- Chaudhary KW, Rossman EI, Piacentino V, 3rd, Kenessey A, Weber C, Gaughan JP, Ojamaa K, Klein I, Bers DM, Houser SR, Margulies KB: Altered myocardial Ca2+ cycling after left ventricular assist device support in the failing human heart. J Am Coll Cardiol 2004, 44(4):837-845.
- Terracciano CM, Hardy J, Birks EJ, Khaghani A, Banner NR, Yacoub MH: Clinical recovery from end-stage heart failure using left-ventricular assist device and pharmacological therapy correlates with increased sarcoplasmic reticulum calcium content but not with regression of cellular hypertrophy. Circulation 2004, 109(19):2263-2265.
- Yacoub MH: A novel strategy to maximize the efficacy of left ventricular assist devices as a bridge to recovery. Eur Heart J 2001, 22(7):534-540.
- Khan MS, Kyriakopoulos CP, Taleb I, Dranow E, Scott M, Ranjan R, Yin M, Tseliou E, Alharethi R, Caine W et al: Baseline QRS duration associates with cardiac recovery in patients with continuous-flow left ventricular assist device implantation. Am Heart J Plus 2022, 22:100211.
- Gude E, Fiane AE: Can mechanical circulatory support be an effective treatment for HFpEF patients? Heart Fail Rev 2023, 28(2):297-305.
- Topilsky Y, Pereira NL, Shah DK, Boilson B, Schirger JA, Kushwaha SS, Joyce LD, Park SJ: Left ventricular assist device therapy in patients with restrictive and hypertrophic cardiomyopathy. Circ Heart Fail 2011, 4(3):266-275.
- Klaeske K, Messer EK, Klein S, Sieg F, Eifert S, Haunschild J, Jawad K, Saeed D, Dashkevich A, Borger MA, Dieterlen MT: Body mass index-dependent immunological profile changes after left ventricular assist device implantation. Front Immunol 2023, 14:1256725.
- Messer EK, Meyer AL, Klaeske K, Sieg F, Eifert S, Schmiedel D, Haunschild J, Jawad K, Saeed D, Hildebrandt L et al: The Impact of Obesity on T and NK Cells after LVAD Implantation. Obes Facts 2023, 16(4):364-373.
- Just IA, Schoenrath F, Roehrich L, Heil E, Stein J, Auer TA, Fehrenbach U, Potapov E, Solowjowa N, Balzer F et al: Artificial intelligence-based analysis of body composition predicts outcome in patients receiving long-term mechanical circulatory support. J Cachexia Sarcopenia Muscle 2024, 15(1):270-280.
- Kyriakopoulos CP, Taleb I, Tseliou E, Sideris K, Hamouche R, Maneta E, Nelson M, Krauspe E, Selko S, Visker JR et al: Impact of Diabetes and Glycemia on Cardiac Improvement and Adverse Events Following Mechanical Circulatory Support. J Am Heart Assoc 2024, 13(14):e032936.
- Santos AB, Kraigher-Krainer E, Gupta DK, Claggett B, Zile MR, Pieske B, Voors AA, Lefkowitz M, Bransford T, Shi V et al: Impaired left atrial function in heart failure with preserved ejection fraction. Eur J Heart Fail 2014, 16(10):1096-1103.
- Fukamachi K, Horvath DJ, Karimov JH, Kado Y, Miyamoto T, Kuban BD, Starling RC: Left atrial assist device to treat patients with heart failure with preserved ejection fraction: Initial in vitro study. The Journal of Thoracic and Cardiovascular Surgery 2021, 162(1):120-126.
- Miyagi C, Kuban BD, Flick CR, Polakowski AR, Miyamoto T, Karimov JH, Starling RC, Fukamachi K: Left atrial assist device for heart failure with preserved ejection fraction: initial results with torque control mode in diastolic heart failure model. Heart Failure Reviews 2023, 28(2):287-296.
- Lester RM: Update on ICH E14/S7B Cardiac Safety Regulations: The Expanded Role of Preclinical Assays and the "Double-Negative" Scenario. Clin Pharmacol Drug Dev 2021, 10(9):964-973.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



