
Submitted:
28 November 2024
Posted:
29 November 2024
You are already at the latest version
Abstract
Ovarian cancer remains one of the leading causes of cancer-related deaths in women. There are several processes that are described to have a causal relationship in ovarian cancer development, progression, and metastasis formation, that occur both at the genetic and epigenetic level. One of the mechanisms involved in its pathogenesis and progression is estrogen signaling. Estrogen receptors ERα, ERβ and GPER1, in concert with various coregulators and pioneer transcription factors, mediate the effects of estrogens primarily by transcriptional regulation of estrogen responsive genes, thereby exerting pleiotropic effects including regulation of cellular proliferation and apoptosis. The expression and activity of estrogen receptors and their coregulators have been demonstrated to be regulated by epigenetic mechanisms like histone modifications and DNA methylation. Here, we intend to summarize and to provide an update on the current understanding of epigenetic mechanisms regulating estrogen signaling and their role in ovarian cancer. For this purpose, we reviewed publications on this topic listed in the Pubmed database. Finally, we provide an assessment to which extent drugs acting on the epigenetic level might be suitable for therapy of ovarian cancer.
Keywords:
epigenetics
; ovarian cancer
; methylation
; histone modification
; estrogen signaling
; ESR1
; ESR2
; GPER1
1. Introduction
Ovarian cancer (OC) represents the fourth leading cause of cancer-related deaths in women worldwide, being responsible for 4.4% of cancer-related deaths in the world [1,2]. OC is a heterogeneous group of diseases categorized primarily into type I and type II tumors, each with distinct characteristics, prevalence, and prognostic implications [3]. Type I tumors include low-grade serous, endometrioid, clear cell, mucinous carcinomas, and malignant Brenner tumors. These tumors are typically slow growing, localized, and low-grade. However, clear cell carcinomas, though part of this group, are considered high-grade due to their aggressive behaviour [3]. Type I ovarian cancers are frequently associated with endometriosis and are less likely to involve TP53 mutations. Common genetic alterations include mutations in KRAS, BRAF, PTEN, PIK3CA, and ARID1A [4]. Prognosis is generally favourable when detected early, as these tumors often progress slowly. Histological subtypes such as endometrioid, clear cell, and mucinous carcinomas are less frequent than type II tumors but exhibit regional variation; for instance, clear cell carcinoma is more common in East Asian populations. This type of OC is typically detected at early stages and has better outcomes due to its indolent nature. Type II tumors comprise high-grade serous carcinoma (HGSC), carcinosarcoma, and undifferentiated carcinoma. These tumors represent the majority of OC cases and are highly aggressive, with most originating from fallopian tube epithelial cells [5]. Type II OCs are characterized by widespread TP53 mutations and genomic instability. Defects in homologous recombination repair, such as BRCA1/2 mutations, are also prevalent. These tumors are frequently diagnosed at advanced stages, with rapid progression and poor prognosis despite aggressive treatment. HGSC accounts for approximately 70% of epithelial ovarian cancers, making it the most common and lethal subtype. The advanced presentation and aggressive behavior of type II tumors contribute to their poor prognosis, accounting for around 90% of ovarian cancer-related deaths [3]. The high mortality rate is caused by a variety of factors, such as the lack of symptoms that leads to late diagnosis, no screening tests available, development of drug resistance, and cancer recurrence [6,7]. The five-year survival rate largely depends on the tumor stage, reaching 89% at stage I, and is rapidly decreasing to 20% at stage IV. Unfortunately, most OC patients are diagnosed at the later stages of tumor development [8]. The standard of care includes cytoreductive therapy, platinum - based chemotherapy, as well as adjuvant intraperitoneal therapy [9]. Endocrine therapy, using tamoxifen or aromatase inhibitors, which has become a standard treatment for estrogen receptor α (ERα)-positive breast cancer, has been shown to have limited efficacy in ERα-positive OC [10]. Currently, various efforts are underway to evaluate the effects of treatment regimens combining tamoxifen with novel anti-cancer drugs [11]. For example, it has been recently shown that combination of tamoxifen and Gatipotuzumab has better outcomes than single drug treatment, and thus may provide a novel therapeutic strategy for OC [12]. After standard therapies, approximately 70% of OC patients have disease recurrence [13]. During the last twenty years, new technologies emerged that allowed a better understanding of the molecular changes that are related to OC development, progression, and therapy resistance [2]. These new techniques led to the definition of biomarkers with clinical use, i.e. BRCA1/2 mutations, which lack increases the effectiveness of the therapy with PARP inhibitors such as Olaparib [2]. Another implication of genomic findings can be the optimization of therapeutic strategies. As was postulated by Gu et al., the primary debulking surgery followed by chemotherapy during treatment of high-grade serous tubo-ovarian carcinoma can be more beneficial than chemotherapy treatment with surgery only after three to four cycles [14]. The authors of the cited work concluded that most patients carry chemo-resistant cancer cells at the time of diagnosis, and this strategy can better deplete these resistant cells. In addition to a growing number of molecular data that describe changes occurring throughout cancer development and progression at the level of classical gene regulation or DNA mutation there are also growing multi-omics and mathematical models that gather this information and present the holistic analysis of malfunction of cancer cells [15].
With the recognition of epigenetics' crucial role in regulating physiological and pathological processes, the relevance of mechanisms like histone modification and DNA methylation in cancer, including OC, increasingly came into the focus of investigations. It is now commonly known that epigenetic factors can drive various cellular processes that can lead to the initiation, development, and progression of cancer as well as their chemoresistance to cancer drugs [16]. While treatment strategies targeting epigenetic alterations with specific drugs currently appear unfeasible, a deeper understanding of the role of epigenetic changes in OC development and progression and particularly their trigger mechanisms might lead to novel strategies for OC prevention.
2. Estrogen Signaling
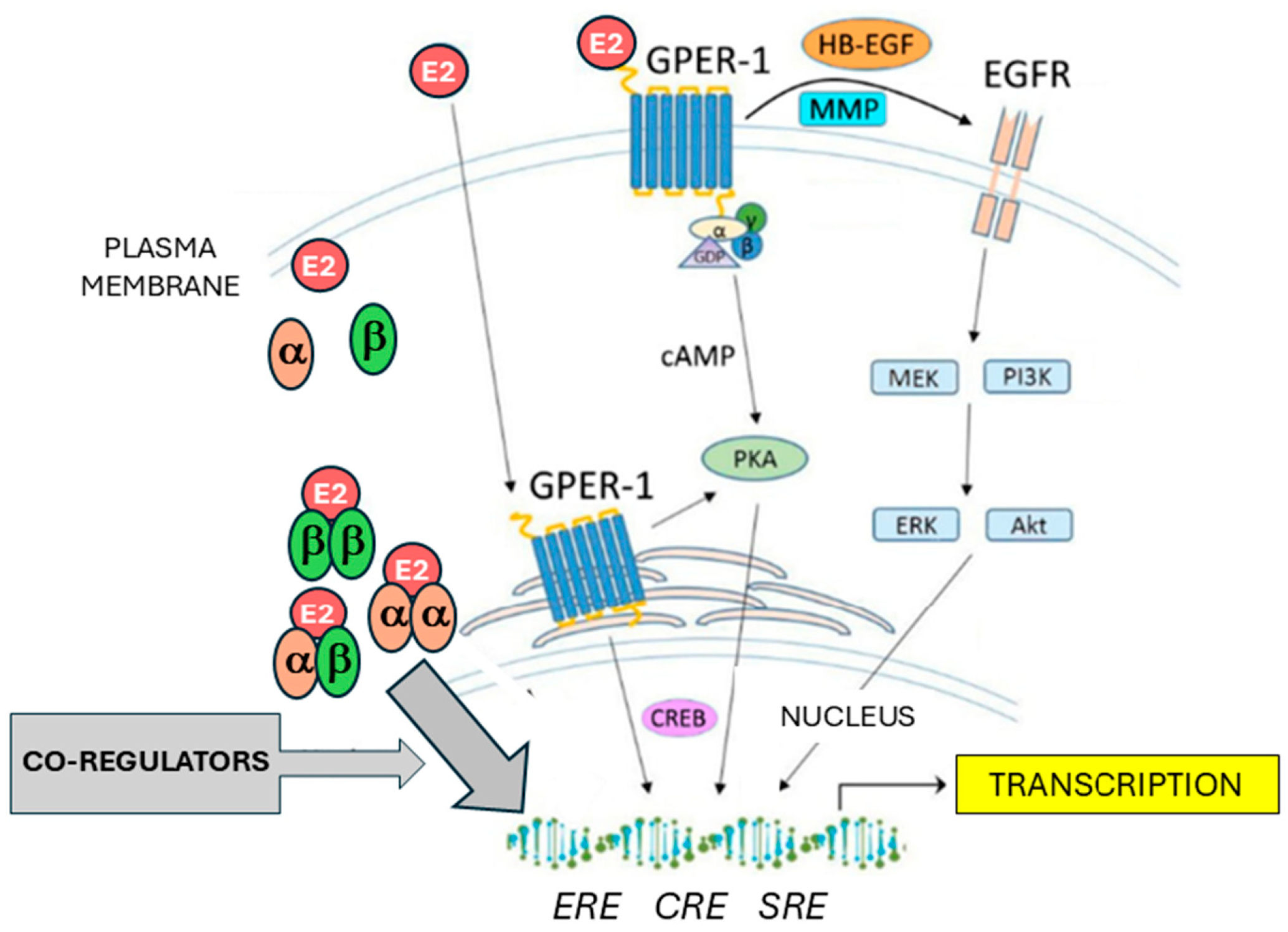
Estrogens represent one of the most important groups of sex steroid hormones in women. Among this group, the most widely distributed and having active functions in multiple tissues is 17β-estradiol (E2) [17]. Estrogen signaling is mediated through binding to estrogen receptors, among which the most important are the nuclear estrogen-activated transcription factors ERα and ERβ, coded by the genes ESR1 and ESR2, respectively, and the transmembrane G-protein coupled estrogen receptor (GPER1) (Figure 1). E2 was found to have a high binding affinity for ERα, ERβ and GPER1 [18]. Estrogens, upon binding to ERα or ERβ, trigger HSP90 dissociation and conformational changes enabling formation of active ER homodimer or ERα/ERβ heterodimer complexes which enter the cell nucleus. Interacting with ER-coactivators or corepressors and pioneer factors like FOXA1 or GATA3, they act as a ligand-inducible transcription factor [19]. The main target DNA sequence for ER dimers is the estrogen response element (ERE). The resulting complex acts as a transcription factor that regulate gene transcription [20]. Estrogen signaling is dependent on the action of several coactivators and corepressors, as well as ligand-independent activation of estrogen receptors [17]. These coregulators cannot bind to DNA directly, but they act via the interaction with DNA-bound estrogen receptor complexes. Coregulators can modify both histones and DNA thus altering its accessibility to transcription factors. So far more than 450 estrogen signaling coregulators have been discovered [21]. The best characterized transcriptional coactivators belong to the SRC/p160 family, consisting of SRC-1, SRC-2 and SCR-3 coactivators [22,23]. SRC proteins bound to estrogen receptor complexes can recruit further molecules – secondary coactivators [24]. Such secondary coactivators can be enzymes like histone methyltransferases or histone acetyltransferases [25]. The E2-driven regulation of gene transcription also depends on the interplay between coactivators and corepressors such as NCoR1 (nuclear receptor corepressor 1)/SMRT (silencing mediator of retinoic acid and thyroid hormone receptor) corepressor family [21,26]. In addition to ERα and β, the G protein-coupled estrogen receptor (GPER1) mediates estrogen effects not as transcription factor binding to EREs, but via non-genomic signaling. This seven-transmembrane receptor, formerly known as GPR30, has several mechanisms of action. On the one hand, it mobilizes calcium and initiates cAMP synthesis. On the other hand, it transactivates the epidermal growth factor receptor (EGFR) which induces PI3K and MAPK signaling pathways and other mechanisms. GPER1 signaling ultimately leads to gene regulation affecting cell-cycle progression as well as proliferation, differentiation, apoptosis, migration, and invasion, making it an important player in carcinogenesis [27].
2.1. Role of Estrogen Signaling in Ovarian Cancer
The ovaries are essential components of the female reproductive system, performing dual roles in gametogenesis and endocrine regulation. As endocrine glands, the ovaries synthesize and secrete steroid hormones, primarily estrogens and progesterone, which are critical for regulating the menstrual cycle, supporting pregnancy, and maintaining secondary sexual characteristics. Estrogen is produced predominantly by the granulosa cells of developing ovarian follicles. This hormone is vital for endometrial proliferation during the follicular phase of the menstrual cycle and contributes to the maintenance of bone density and cardiovascular health. The ovaries also produce small quantities of androgens, such as testosterone, which serve as precursors for estrogen synthesis through the aromatization process [28]. Estrogens do not only trigger physiological actions but have also been reported to play a role in development and progression of OC [29]. Particularly in epithelial OC (EOC), estrogens have been shown to affect various cellular pathways including those involved in regulation of proliferation, apoptosis, invasiveness and epithelial-to-mesenchymal transition (EMT) [30]. The estrogen receptors – ERα, ERβ, and GPER1 are expressed both in normal ovarian tissue and in ovarian cancer[28]. At the mRNA level, expression of ESR1 gene was detected in approximately 60% of OC tissues, whereas ESR2 transcripts were the predominant ones in normal ovary tissue [31]. The ESR1/ESR2 mRNA ratio was significantly increased in OC tissue. In a cell culture study employing primary cells, the ESR1/ESR2 mRNA ratio in primary OC cells was ten times higher than in normal ovarian surface epithelium cultures, a finding that was basically corroborated on the protein level [32].
Figure 1.
Schematic overview of estrogen signaling. E2: estradiol, α: ERα, β: ERβ, cAMP: cyclic adenosine monophosphate; PKA: protein kinase A; CREB: cAMP-response element binding protein; ERE: estrogen response element, CRE: cAMP-response element; SRE: serum response element; MMP: matrix metalloproteinase; HB-EGF: heparin-binding EGF-like growth factor; EGFR: epidermal growth factor receptor; MEK: Mitogen-activated protein kinase kinase; ERK: extracellular signal-regulated kinase; PI3K: Phosphoinositide 3-kinase; AKT: Protein kinase B (PKB). Further abbreviations are addressed in the text.
Figure 1.
Schematic overview of estrogen signaling. E2: estradiol, α: ERα, β: ERβ, cAMP: cyclic adenosine monophosphate; PKA: protein kinase A; CREB: cAMP-response element binding protein; ERE: estrogen response element, CRE: cAMP-response element; SRE: serum response element; MMP: matrix metalloproteinase; HB-EGF: heparin-binding EGF-like growth factor; EGFR: epidermal growth factor receptor; MEK: Mitogen-activated protein kinase kinase; ERK: extracellular signal-regulated kinase; PI3K: Phosphoinositide 3-kinase; AKT: Protein kinase B (PKB). Further abbreviations are addressed in the text.
After initial controversies mainly based on immunohistochemical (IHC) studies using unspecific antibodies, the tumor-suppressive role of ERβ protein in OC became more and more obvious. In their IHC-based study, Lindgren et al. analyzed 53 benign, borderline, and malignant ovarian tumors of different types and found a significantly lower ERβ expression in OC tissue compared to normal ovaries [33]. Moreover, the level of ERβ protein expression in ovarian cancers has an impact on the survival of the patients. In the IHC-based study published by Halon et al., a higher ERβ expression (>30% of cells) was associated with increased overall survival time and progression-free time (p = 0.00161 and p = 0.03255, respectively) compared to patients with lower ERβ expression [34]. The role of ERα in OC has been considered controversially. In a large IHC-based study published by Sieh et al. in 2013, patients with OC of the endometrioid subtype survived significantly longer when their tumors expressed ERα [35]. However, in high-grade serous OC (HGSOC), no clear association of ERα expression with survival was reported, which is thought to result from the fact that HGSOC is typically driven by mutations in genes like TP53 and has less reliance on hormone receptor signaling. Bogush et al. analyzed ER expression in serous OC by a quantitative immunofluorescence assay, and found that high expression levels of both ERα (≥25%) and ERβ (≥44%) predicted a significantly longer progression-free survival in patients after the first-line treatment of platinum and taxane-based adjuvant chemotherapy, which primarily might result from antitumoral ERβ action [36]. ERβ expression was found to be decreased not only in OC but also during tumorigenesis of breast, colon, and prostate cancer [37,38,39]. In nude mice injected with ERβ-expressing ovarian BG1 cells, ERβ was able to strongly reduce the development of orthotopic ovarian xenograft as well as the presence of tumor cells at the sites of metastasis, leading to an increased survival of the mice [40].
A multitude of in vitro studies employing OC cell lines revealed molecular mechanisms underlying the actions of ERα and ERβ in OC. The growth-promoting role of ERα, known from other cancer entities like breast cancer and mediated by activation of proliferation genes, was also observed in OC. Furthermore, activation of ERα was shown to trigger gene regulation patterns associated with invasion and metastasis of OC cells [41]. Other in vitro studies elucidated the role of ERβ in this cancer entity. In studies including several from our group, ERβ was shown to exert tumor-suppressive effects like the decrease of growth and motility, but activation of apoptosis of OC cells, and transcriptome alterations were identified underlying these actions [42,43]. Several highly specific ERβ agonists like ERB-041 and WAY200070 have been reported to trigger tumor suppressive responses in OC cells in vitro and could therefore be evaluated for efficacy in mouse models and clinical settings. In a recent study, another specific ERβ agonist was found to reduce the EMT and cancer stem cell (CSC) population in ovarian cancer. Given that ERα can trigger EMT and facilitates maintenance of CSCs, the ERβ agonist was suggested to limit the CSC subpopulation with the potential to increase survival of OC patients. In summary, regarding the two nuclear ERs, ERα is considered to mediate adverse E2 actions on OC cells, whereas ERβ is reported to predominantly exert tumor-suppressive actions in this cancer entity [44,45,46,47].
Studies on the role of the third ER, the non-nuclear, G protein-coupled estrogen receptor GPER1, in OC came to conflicting results, particularly those using IHC to detect this protein in OC tissues to correlate GPER1 protein levels with OC outcome. Several IHC-based studies suggested a tumor-promoting role of this receptor in OC [48,49]. In ovarian tissue samples, GPER1 was found to be broadly expressed in high-risk ovarian cancer, associated with lower 5-year survival rates [49]. Besides, its co-expression with EGFR was associated with shorter progression-free survival in OC patients [48]. However, there are also contrary data suggesting tumor-suppressive functions of GPER1 in ovarian malignancies. In a study by Ignatov et al., benign tumors and those of a low-malignant potential were found to have significantly higher GPER1 expression levels than investigated OCs [50]. Early stage and well differentiated cancers strongly expressed GPER1, which was found in 83.1% of all malignant tumors. Moreover, they observed significantly longer disease-free survival for patients with GPER1-expressing OCs compared to those with GPER1-negative tumors [50]. In line with this, a study from our group reported that OC patients with tumors expressing high GPER1 mRNA levels survived longer and had more lifetime without progression, when open-access mRNA and clinical data were analyzed [51]. A further study by Fraungruber et al. analyzing 156 OC samples supported a tumor suppressive role of GPER1. High co-expression of Dkk2 and GPER1 was associated with better overall survival in OC patients [52]. These data suggest a prognostic relevance of both pathways, and indicate that therapeutic interventions targeting both estrogen and Wnt signaling pathways may be successful in OC. Taken together, current data suggest a tumor-promoting role of ERα, but anti-tumoral effects of ERβ and GPER1 in OC.
In recent years, non-cancerous cells constituting the tumor microenvironment (TME) have been considered as critical mediators of tumor progression. Both ERα and ERβ as well as aromatase, the key enzyme in the production of estrogens, are expressed in cells of the TME, like cancer-associated fibroblasts (CAFs) and tumor-associated macrophages (TAMs). In ovarian cancer, CAFs have been shown to overexpress ERα, which promotes tumor progression via paracrine signaling pathways [53]. The role of ERs in TAMs is an area of growing research interest, especially in the context of ovarian cancer. TAMs are highly plastic immune cells within the TME, and their behavior is modulated by various signals, including those mediated by estrogen and its receptors. In the ovarian tumor microenvironment, estrogen may contribute to the polarization of TAMs toward a pro-tumoral M2-like phenotype. This polarization supports tumor progression by promoting angiogenesis, immune suppression, and extracellular matrix remodeling [54,55].
3. Epigenetic Mechanisms
Epigenetics refers to heritable changes in gene expression, that are primarily driven by mechanisms such as DNA methylation, histone modification, nucleosome positioning and chromatin remodeling [56] (Figure 2). Some authors also consider non-coding RNAs (ncRNAs) as part of the epigenetic machinery, since epigenetic regulation is often described as gene regulatory mechanism not altering the DNA sequence. However, there are compelling reasons not to classify ncRNAs as part of epigenetics. First, ncRNAs do not trigger heritable effects, but tend to function short-term, as observed with miRNAs and lncRNAs. Additionally, eukaryotic gene regulation, mediated by transcription factors or ncRNAs, does not involve changes to the DNA sequence. Therefore, ncRNAs can be considered as an additional mechanism of non-epigenetic gene regulation, primarily operating at the post-transcriptional level in eukaryotes [57,58].
DNA methylation is characterized by the addition of methyl groups to the 5th carbon of cytosine residues in CpG dinucleotides, typically within CpG islands. This methylation often occurs in the promoter regions of genes, where it plays a key role in regulating gene expression [59]. Several studies have reported differences in the methylation status between normal and tumor cells and tissues [60,61,62,63]. The methylation process is mediated by enzymes of the DNA methyltransferases group (DNMTs), and hypermethylation is usually related to transcriptional repression [64]. The opposite effect is induced by DNA demethylases, involving the Ten-Eleven Translocation Dioxygenases (TET), and leads to removal of the methyl group from DNA sequences, leading to hypomethylation activating gene expression [65]. The second major mechanism in epigenetics is the posttranslational modification of histones [66]. Such changes occur usually at the histone end and can be caused by acetylation, methylation, ubiquitination, phosphorylation, SUMOylation or ADP ribosylation. Histone modifications alter the configuration and density of chromatin, thereby influencing its accessibility to transcription factors and other components of the transcription machinery [67,68]. Among these modifications, those induced by histone methyltransferases (HMTs) and histone demethylases (HDMs) regulate histone methylation. Histone methylation can activate gene expression by addition of epigenetic marks like H3K4me3 or suppress transcription by posting marks like H3K9me3. Another common epigenetic modification involves the addition or removal of acetyl groups at lysine residues in histones. This process is regulated by acetyltransferases (HATs) and histone deacetylases (HDACs), respectively. HATs are able to loosen chromatin structure and activate transcription by adding epigenetic marks like H4K16ac and H3K27ac. HDACs are classically associated with transcriptional repression by removing acetyl groups from histones, like H4K16ac, leading to chromatin compaction [66]. In recent years, advancements in epigenetic research, driven by high-throughput technologies, have greatly expanded our understanding of the role of these enzymes in oncogenesis [68,69]. Among the various epigenetic modulators, one particularly noteworthy and reversible modification that has garnered significant research interest is m6A RNA modification, especially in the context of cancer development [70]. The m6A modification is not only implicated in cancer pathogenesis but also plays a role in drug response and immune modulation [71,72]. This methylation of adenosine at the N6 position has been identified as one of the most crucial mRNA modifications. Subsequent studies have underscored its importance in numerous biological processes, both under normal and pathological conditions [71,73]. It has been discovered that the function of m6A is regulated by specific RNA methyltransferases, referred to as "writers," and demethylases, known as "erasers." A balanced ratio between these regulators is essential for maintaining proper m6A modification levels. Dysregulation of this balance, resulting from mutations or altered expression levels, can contribute to the development of various diseases, including cancer [71]. In ovarian cancer, it has been reported that the m6A-related RNA signature holds potential as a prognostic factor [70]. A similar impact on immune modulation was observed in the study by Gu et al., which showed that a low level of m6A modification was associated with immune system activation and enhanced response to immunotherapy, while a high level of m6A modification correlated with tumor progression. A negative correlation was also found between the stage of OC and m6A modification levels. In OC patients with a low m6A modification score, high expression of genes involved in immune response modulation was observed, including genes related to human leukocyte antigens, immune activation, and immune checkpoint molecules [74]. Additionally, it has been demonstrated that m6A modification negatively regulates the expression of ERα by influencing RNA demethylases. When RNA demethylases are reduced, m6A levels increase, leading to enhanced methylation of ERα mRNA and consequently decreased ERα protein translation, as observed in cholangiocarcinoma. Similar findings, suggesting a role in immune response modulation, have been reported in osteosarcoma and breast cancer [75,76]. Although the precise role of m6A modifications in OC is not yet fully understood, existing data suggests that m6A modification regulators, by influencing epigenetic changes, could play a critical role in OC therapy, especially in the context of Immunotherapeutics. This may help identify patient subgroups most likely to benefit from such treatments [77].
3.1. Epigenetic Mechanisms Involved in Estrogen Signaling in Ovarian Cancer
Estrogen signaling can be regulated by epigenetic mechanisms such as DNA methylation and histone modifications. These epigenetic changes can influence the expression or activity of estrogen receptors (ERα, ERβ, GPER1), estrogen receptor co-regulators, and downstream effectors like cyclins, which in turn promote estrogen (E2)-dependent proliferation of OC cells [78].
3.1.1. Epigenetic Regulation of ERα Expression and Activity in Ovarian Cancer
ERα functions as a ligand-inducible transcription factor that mediates various biological processes in response to estrogen stimulation [79]. Upon binding to its ligand, ERα translocates to the nucleus, where it binds to estrogen response elements (EREs)—specific DNA sequences that are often subject to epigenetic modifications or particular histone modifications, which mark them as transcriptional enhancers [80]. After binding to EREs, ERα functions as a co-factor recruitment molecule, that targets the transcription of target genes [81]. Promoter hypermethylation, particularly in the context of DNA methyltransferase (DNMT) activity, is known to be associated with decreased expression of estrogen receptor alpha (ERα) in various cancers. DNA methylation in the promoter region of the ERα gene ESR1 can silence its expression, which is observed in several cancer types, including ovarian and endometrial cancers [82]. Studies have shown that overexpression of DNMT1 and DNMT3A, enzymes responsible for methylation, correlates with lower ERα levels and poor prognosis in cancer [83]. ESR1 silencing has also been observed in other cancers, including endometrial cancer, where ESR1 promoter methylation led to decreased ERα expression [84]. In vitro it was reported that ESR1 demethylation by 5-aza-2′-deoxycytidine (DAC) restored ERα expression in an ERα-negative cancer cell line [85]. Although this suggests that epigenetic silencing of ESR1 via DNA methylation is reversible, and demethylation can potentially restore ERα expression, a clinical use, e.g., aiming at re-sensitizing cancers to hormone therapies, is not possible, since demethylating drugs are totally unspecific and thus could lead to an increased expression of pathobiologically active genes like oncogenes.
In ovarian cancer, dysregulation of ERα via epigenetic modifications is increasingly recognized as a key factor influencing tumor progression, therapy resistance, and prognosis. Epigenetic changes like DNA methylation and histone modifications modulate ERα expression and activity in the tumor microenvironment. Hypermethylation of the ESR1 promoter is a hallmark of reduced ERα expression in ovarian cancer. However, the ESR1 methylation status plays different roles in high-grade OC and in low-grade OC. Although the majority of high-grade serous ovarian and endometrioid ovarian cancers express different, IHC-detectable levels of ERα, expression of this receptor is generally higher in low-grade OC. Both hypo- and hypermethylation of the ESR1 promoter were observed in OC. Hypermethylation of the ESR1 gene, which is also present in many OC cell lines, results in reduced gene expression, but does not necessarily lead to an ERα-negative IHC status, whereas ESR1 hypomethylation generally increases ERα levels [86]. A study comparing both the methylome and transcriptome of not further characterized OC samples and normal ovarian tissues identified ESR1 to be among the genes which were most significantly hypomethylated and overexpressed in OC. ESR1 hypomethylation leading to high ERα levels was associated with longer overall survival (OS) and post-progression survival (PPS) in OC patients [87]. In another study differentiating high- and low-grade OC, hypermethylation of ESR1 was associated with a poorer prognosis in low-grade OC, but not in HGSOC [88]. This observation is thought to result from the fact that low-grade OC generally is more hormone-dependent than HGSOC being primarily driven by other pathways. ESR1 hypermethylation resulting in reduced ERα levels is thought to be associated with a poorer prognosis particularly in LGOC, because it might trigger the usage of compensatory, more aggressive growth and survival pathways, which are already dominant in HGSOC. ERα-negative ovarian cancers often show increased activation of pathways like the PI3K/AKT/mTOR or NFκB signaling, contributing to enhanced proliferation, shorter survival, and resistance to apoptosis. Furthermore, studies indicate that ERα plays a role in maintaining epithelial characteristics of OC cells. Its loss due to ESR1 hypermethylation can promote epithelial-to-mesenchymal transition (EMT), a process associated with increased invasiveness, migration, and metastatic capacity. The histone methyltransferase DOT1L has been proposed to be a functional component of ERα signaling in OC. ERα and DOT1L were shown to be highly co-expressed in ERα-positive, chemotherapy-resistant OC cells. DOT1L controls chromatin functions involved in tumor initiation and progression and has been proposed as a prognostic OC biomarker. Inhibition of DOT1L or ERα exerted growth inhibitory effects in ERα-positive, hormone-dependent OC [89].
Another level of complexity in ERα epigenetics was added by identification of the MegaTrans (Mega Transcriptional) complex, which was demonstrated to modulate both ERα-activity and -expression. The MegaTrans complex epigenetically regulates the transcriptional activity of ERα by influencing chromatin accessibility and enhancer activity at its target genes. In estrogen-responsive cells, the MegaTrans complex amplifies the activity of ERα by enabling the formation of super-enhancers, clusters of highly active enhancers enriched in histone acetylation marks, which drive robust transcription of estrogen-responsive genes. Additionally, by epigenetic feedback, the MegaTrans complex activates the expression of ESR1 gene by histone acetylation at its regulatory regions. The MegaTrans complex is a large transcriptional regulatory assembly consisting of transcription factors, coregulators and histone modifying enzymes, that integrates signals from various pathways to modulate gene expression. It connects extracellular and intracellular signals to trigger epigenetic modifications such as histone acetylation to facilitate dynamic changes in chromatin structure and transcriptional activity. ERα is a critical component of the MegaTrans complex, which connects ERα signaling with other pathways, e.g., NFκB, enabling a synergistic regulation of histone acetylation and transcription. Upon estrogen binding, ERα recruits the MegaTrans complex, which includes histone acetyltransferases like CBP/p300. These enzymes deposit histone acetylation marks, such as H3K27ac, at promoter or enhancer regions, serving as important epigenetic signals for the recruitment of bromodomain-containing proteins, which regulate chromatin compaction and promote transcriptional activation. The MegaTrans complex also coordinates with other transcription factors, enabling the establishment of super-enhancers, regions of densely acetylated chromatin that amplify transcription of genes critical for cell proliferation and survival [90]. This mechanism is particularly relevant in estrogen-driven cancers, where dysregulation of ERα signaling and associated histone acetylation marks contribute to oncogenesis and therapy resistance. The MegaTrans complex, which has been shown to play an important role in aberrant ERα-regulated gene expression in breast cancer, also plays a major role in OC [91]. In a study aiming to discover new regulatory factors causing distinct biological properties of HGSOCs and serous borderline tumors (SBTs), several factors were identified which are known to cooperate with and predict the presence of ERα and which are known to form the MegaTrans complex. The results of this study implicated an estrogen-responsive regulatory network in the differential gene expression of HGSOC and SBT and was the first demonstrating the contribution of the MegaTrans complex in distinct biological trajectories of different OC subtypes [92].
3.1.2. Epigenetic Regulation of ERβ Expression and Transcriptional Activity in Ovarian Cancer
In ovarian cancer, like in other cancer entities, the expression of ESR2 gene coding for ERβ is reduced compared to normal tissues, and low levels of this receptor have been reported to be associated with a more aggressive cancer phenotype and with poor OC survival due to the role of ERβ as tumor suppressor [31,40,44,93]. The ESR2 gene is known to be epigenetically regulated both by promoter methylation and by histone modifications, affecting transcriptional activity and gene expression of ERβ. However, few studies addressed epigenetic regulation of ERβ in OC. Early studies on breast cancer tissues demonstrated a high level of ESR2 promoter methylation, leading to down-regulation or loss of ERβ expression, in invasive breast cancer, but not in the normal mammary gland [94]. Furthermore, ESR2-activity and -expression both are regulated by histone modifications, e.g., triggered by KDM6B histone demethylase [95]. In many in vitro studies, drugs like DNMT inhibitors exerting de-methylating effects or HDAC inhibitors reducing histone acetylation, leading to a more accessible chromatin structure allowing higher transcriptional activity, have been employed to indirectly examine and counteract epigenetic silencing of ERβ [96,97]. In OC, ERβ activity has been reported to be regulated by modulation of histone acetylation marks in the promoter of ERβ target genes [98]. In human OC cells, analysis of the ESR2 promoter revealed 13 CpG methylation sites. Demethylation of these sites could be triggered by treatment with 5-aza-2′-deoxycytidine (5-aza-dC), an inhibitor of DNA and RNA methyltransferases. In this study, it was shown that the treatment of OC cells by epigenetic agents - 5-AzaC and histone deacetylase inhibitor trichostatin A - leads to reactivation of ERβ expression and -activity and thereby triggers inhibition of ovarian cell proliferation. This study demonstrated that down-regulation of ERβ expression and -activity in OC cells results both from promoter hypermethylation and histone acetylation [99]. A deeper analysis of ESR2 methylation examining both promoter regions of the ESR2 gene, which are termed 0K and 0N, showed that promoter 0N was rarely methylated in normal ovarian tissues, but extensively methylated in OC cell lines and tissues, whereas the promoter 0K was unmethylated in both normal and malignant ovarian cells and tissues. The same study also examined expression of the ERβ splice variants, demonstrating that hypermethylation of the 0N promoter was associated with loss of expression of the variants ERβ1, ERβ2, and ERβ4 in ovarian carcinoma cells and tissues. Treatment with demethylating agent 5-aza-dC restored expression of these ERβ splice variants. However, ERβ5 expression was not decreased in clear cell adenocarcinoma when compared to the normal ovary and was not found to be associated with the methylation status [100]. The data of this study clearly support the functional connection of promoter hypermethylation with loss of ERβ expression, which is observed not only in OC, but also other cancer entities like breast and prostate cancer [101,102].
3.1.3. Epigenetic Regulation of GPER1 Expression and Activity in Ovarian Cancer
The G-protein-coupled estrogen receptor (GPER1) was found to be expressed in normal ovary and in OC and its function has crucial implications in both physiological and pathological processes [103,104]. GPER1 expression was reported to be lower in OC tissue than in benign and low-malignant ovarian tumors. However, as discussed above, conflicting data exist about the correlation of GPER1 expression in OC patients with survival [48,49,50]. Loss or reduction of GPER1 expression was reported to occur in 20-50% of OC cases [49,105]. A recent study using data from the cancer genome atlas (TCGA) and the genotype-tissue expression (GTEx) databases for analysing the expression profile of estrogen receptors through gene expression profiling interactive analysis (GEPIA) found GPER1 downregulation in OC, which, however, was not statistically significant. In contrast, in the majority of cancer entities including breast and endometrial cancer, GPER1 expression was significantly reduced and associated with hypermethylation of GPER1 gene promoter [106]. Other studies confirmed GPER1 expression to be reduced by hypermethylation in various cancers, like breast cancer [107,108]. In ovarian cancer, a recent study suggests that GPER1 expression is epigenetically activated by histone H3 trimethylation (H3K4me) (Table 1). This histone H3 modification is one of the most recognized epigenetic marks of active transcription [109]. Although the role of histone marks in various cell types has been extensively explored, it is still not fully understood in cancer. However, H3K4me3 marks are an epigenetic signal, which in normal cells is associated with increased transcription elongation and enhancer activity at tumor-suppressor genes like TP53 and PTEN. In cancer cells, these H3K4me3 marks and enhancer activity are reduced, leading to decreased transcription of tumor suppressor genes and elevated cancer cell proliferation [109,110,111]. In ovarian cancer, however, high levels of H3K4me3 marks were shown to be associated with high GPER1 expression, leading to a better prognosis of GPER1-positive OC patients. Given that GPER1 was suggested to act as tumor suppressor in the ovary, this observation might be explained by the known activation of tumor suppressor genes by H3K4me3 marks [112].
3.1.4. Epigenetic Regulation of Estrogen Receptor Target Genes in Ovarian Cancer
Particularly estrogen-driven activation of the nuclear receptors ERα and ERβ, which are ligand-inducible transcription factors, induces transcription of their target genes, which are mediators of estrogen signaling. In studies on breast cancer, but also OC cells, ERα itself was recognized as component of the epigenetic acting MegaTrans complex, which contains histone acetyltransferases (HAT), and deposits histone acetylation marks, such as H3K27ac, at promoter or enhancer regions, and such regulates chromatin compaction and activates transcription particularly of estrogen-dependent genes. This epigenetic mechanism was reported to activate transcription of estrogen target genes like TFF1 (PS2), FOXA1 and GREB1 [90]. Cyclin D1 coded by the gene CCND1 is an important target gene of ERα and mediator of estrogen-induced cellular proliferation. Recently it has been shown that CCND1 is regulated by epigenetic mechanisms. First, CCND1 expression is epigenetically activated by protein arginine methyltransferase 6 (PRMT6), which generates histone modification mark H3R2me2a, in conjunction with transcription factor LEF [113]. In ovarian cancer, CCND1 promoter was reported to be hypomethylated leading to enhanced gene expression and poor prognosis [114]. CDH1 gene coding for E-cadherin was described as estrogen target gene being downregulated by ERα in OC [115]. This gene was found to be hypermethylated and downregulated in OC patients, leading to enhanced metastasis [116,117]. While PTEN is not a direct target of ERα, estrogen signaling can affect its expression indirectly. Estrogen can downregulate PTEN via pathways that activate AKT or other signaling cascades promoting cell survival and proliferation. In certain cancers, reduced PTEN levels due to estrogen action are linked to enhanced tumorigenesis. Epigenetic mechanisms such as promoter methylation also modulate PTEN expression in cancers, including OC [118]. Antiapoptotic factor Bcl-2 is a direct target of ERα. Its expression is upregulated by estrogen, contributing to cell survival in hormone-sensitive cancers. In ovarian cancer, expression of Bcl-2 was demonstrated to be epigenetically suppressed by HDAC1, which deacetylates histone H3K9,14 marks across Bcl-2 regulatory regions, resulting in reduced Bcl-2 transcription [119]. FOXA1, a key determinant of estrogen receptor function, which enables ERα interactions with chromatin, was reported to induce epithelial OC tumorigenesis and progression [120]. Recently, FOXA1 was reported to be epigenetically modulated by Histone deacetylase HDAC3 in the progression of epithelial ovarian carcinoma [121].

Table 1.
Modulation of estrogen signaling by DNA methylation or histone modification in OC.
| Epigenetic mechanism | Gene | References |
|---|---|---|
| Promoter methylation |
ESR1 ESR2 GPER1 CDH1 CCND1 PTEN |
[86,87,88,89] [99,100] [59] [116,117] [114] [118] |
| Histone modification |
ESR1 ESR2 GPER1 TFF1 FOXA1 GREB1 CCND1 BCL2 |
[89], [91,92] [98,99] [109,112] [90] [90,121] [90] [113] [119] |
4. Drugs with Epigenetic Functions: Are they Promising Tools for OC Therapy?
The epigenetic changes occurring during the process of ovarian carcinogenesis, including those altering estrogen signaling, are increasingly understood and known. Given that epigenetic mechanisms play an important role in OC development and progression, e.g. activating the expression of oncogenes or reducing the activity of tumor-suppressor genes, they seem to be promising targets for OC therapy. The anti-tumoral effect of drugs with epigenetic functions has been thoroughly examined in in vitro studies employing cell lines of various cancer entities, including OC, or studies using mouse models, and often led to impressing results [122,123,124,125,126,127,128,129,130,131]. First clinical studies examined the effect of “epidrugs” like HDAC inhibitors, mostly in combination with standard treatments, on survival of patients with various cancer types [132,133].
However, the major problem is the totally unspecific action of “epidrugs”. They can change the expression of every gene in the human body. They can even have the potential to activate oncogenes or to suppress tumor suppressor genes. This lack of specificity of “epidrugs” thus can be expected to cause major side effects leading to a high toxicity or to the development of any disease, which, however, might come to light months or years after the treatment with such drugs, since the pathogenesis of many diseases takes time. Thus, prolonged studies on animal models are needed before the use of such substances can be considered to be ethically justifiable for treatment of cancer patients. A promising strategy to make “epidrugs” more specific could be the modification of their structure limiting their action to the target gene by addition of a short but specific DNA sequence which can guide the drug/DNA complex to the target site. However, novel insights in the epigenetic mechanisms involved in development and progression of various cancer entities, like OC, have the potential to open new avenues for novel treatment strategies of OC.
Author Contributions
Conceptualization, O.T.; resources, O.O. and L.A.; writing—original draft preparation, O.T., E.W., M.S.; writing—review and editing, M.S., L.A., O.T.; visualization, O.T.; supervision, O.T.; funding acquisition, L.A. and O.O. All authors have read and agreed to the published version of the manuscript
Funding
This research received no external funding
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Cabasag, C.J.; Fagan, P.J.; Ferlay, J.; Vignat, J.; Laversanne, M.; Liu, L.; van der Aa, M.A.; Bray, F.; Soerjomataram, I. OC today and tomorrow: A global assessment by world region and Human Development Index using GLOBOCAN 2020. Int J Cancer 2022. [Google Scholar] [CrossRef]
- Munoz-Galvan, S.; Carnero, A. Leveraging Genomics, Transcriptomics, and Epigenomics to Understand the Biology and Chemoresistance of Ovarian Cancer. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih Ie, M. The Dualistic Model of Ovarian Carcinogenesis: Revisited, Revised, and Expanded. Am J Pathol 2016, 186, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Rojas, V.; Hirshfield, K.M.; Ganesan, S.; Rodriguez-Rodriguez, L. Molecular Characterization of Epithelial Ovarian Cancer: Implications for Diagnosis and Treatment. Int J Mol Sci 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, I.; Matsuura, T. Screening and Prevention for High-Grade Serous Carcinoma of the Ovary Based on Carcinogenesis-Fallopian Tube- and Ovarian-Derived Tumors and Incessant Retrograde Bleeding. Diagnostics (Basel) 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Liberto, J.M.; Chen, S.Y.; Shih, I.M.; Wang, T.H.; Wang, T.L.; Pisanic, T.R., 2nd. Current and Emerging Methods for OC Screening and Diagnostics: A Comprehensive Review. Cancers (Basel) 2022, 14. [Google Scholar]
- Wang, Y.; Huang, Z.; Li, B.; Liu, L.; Huang, C. The Emerging Roles and Therapeutic Implications of Epigenetic Modifications in Ovarian Cancer. Front Endocrinol (Lausanne) 2022, 13, 863541. [Google Scholar] [CrossRef]
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. OC statistics, 2018. CA Cancer J Clin 2018, 68, 284–296. [Google Scholar] [CrossRef]
- Pignata, S.; Cannella, L.; Leopardo, D.; Pisano, C.; Bruni, G.S.; Facchini, G. Chemotherapy in epithelial ovarian cancer. Cancer Lett 2011, 303, 73–83. [Google Scholar] [CrossRef]
- Williams, C.; Simera, I.; Bryant, A. Tamoxifen for relapse of ovarian cancer. Cochrane Database Syst Rev 2010, 2010, CD001034. [Google Scholar] [PubMed]
- Borella, F.; Fucina, S.; Mangherini, L.; Cosma, S.; Carosso, A.R.; Cusato, J.; Cassoni, P.; Bertero, L.; Katsaros, D.; Benedetto, C. Hormone Receptors and Epithelial Ovarian Cancer: Recent Advances in Biology and Treatment Options. Biomedicines 2023, 11. [Google Scholar] [CrossRef] [PubMed]
- Heublein, S.; Page, S.; Mayr, D.; Schmoeckel, E.; Trillsch, F.; Marme, F.; Mahner, S.; Jeschke, U.; Vattai, A. Potential Interplay of the Gatipotuzumab Epitope TA-MUC1 and Estrogen Receptors in Ovarian Cancer. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef]
- Singh, N.; Jayraj, A.S.; Sarkar, A.; Mohan, T.; Shukla, A.; Ghatage, P. Pharmacotherapeutic treatment options for recurrent epithelial ovarian cancer. Expert Opin Pharmacother 2022. [Google Scholar] [CrossRef]
- Gu, S.; Lheureux, S.; Sayad, A.; Cybulska, P.; Hogen, L.; Vyarvelska, I.; Tu, D.; Parulekar, W.R.; Nankivell, M.; Kehoe, S.; Chi, D.S.; Levine, D.A.; Bernardini, M.Q.; Rosen, B.; Oza, A.; Brown, M.; Neel, B.G. Computational modeling of OC dynamics suggests optimal strategies for therapy and screening. Proc Natl Acad Sci U S A 2021, 118. [Google Scholar]
- Hira, M.T.; Razzaque, M.A.; Angione, C.; Scrivens, J.; Sawan, S.; Sarker, M. Integrated multi-omics analysis of OC using variational autoencoders. Sci Rep 2021, 11, 6265. [Google Scholar] [CrossRef]
- Zeller, C.; Dai, W.; Steele, N.L.; Siddiq, A.; Walley, A.J.; Wilhelm-Benartzi, C.S.; Rizzo, S.; van der Zee, A.; Plumb, J.A.; Brown, R. Candidate DNA methylation drivers of acquired cisplatin resistance in OC identified by methylome and expression profiling. Oncogene 2012, 31, 4567–4576. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Li, B.; Ou-Yang, L. Role of estrogen receptors in health and disease. Front Endocrinol (Lausanne) 2022, 13, 839005. [Google Scholar] [CrossRef]
- Russell, J.K.; Jones, C.K.; Newhouse, P.A. The Role of Estrogen in Brain and Cognitive Aging. Neurotherapeutics 2019, 16, 649–665. [Google Scholar] [CrossRef]
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. Adv Protein Chem Struct Biol 2019, 116, 135–170. [Google Scholar]
- Klinge, C.M. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res 2001, 29, 2905–2919. [Google Scholar] [CrossRef]
- Feng, Q.; O'Malley, B.W. Nuclear receptor modulation--role of coregulators in selective estrogen receptor modulator (SERM) actions. Steroids 2014, 90, 39–43. [Google Scholar] [CrossRef]
- Onate, S.A.; Tsai, S.Y.; Tsai, M.J.; O'Malley, B.W. Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science 1995, 270, 1354–1357. [Google Scholar] [CrossRef]
- Johnson, A.B.; O'Malley, B.W. Steroid receptor coactivators 1, 2, and 3: critical regulators of nuclear receptor activity and steroid receptor modulator (SRM)-based cancer therapy. Mol Cell Endocrinol 2012, 348, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Ma, H.; Hong, H.; Koh, S.S.; Huang, S.M.; Schurter, B.T.; Aswad, D.W.; Stallcup, M.R. Regulation of transcription by a protein methyltransferase. Science 1999, 284, 2174–2177. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lin, R.J.; Schiltz, R.L.; Chakravarti, D.; Nash, A.; Nagy, L.; Privalsky, M.L.; Nakatani, Y.; Evans, R.M. Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with P/CAF and CBP/p300. Cell 1997, 90, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.D.; Evans, R.M. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature 1995, 377, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Silva, C.D.; Villegas-Pineda, J.C.; Pereira-Suarez, A.L. Expression and Role of the G Protein-Coupled Estrogen Receptor (GPR30/GPER) in the Development and Immune Response in Female Reproductive Cancers. Front Endocrinol (Lausanne) 2020, 11, 544. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.L.; Huang, Z.Y.; Yu, K.; Li, J.; Fu, X.W.; Deng, S.L. Estrogen Biosynthesis and Signal Transduction in Ovarian Disease. Front Endocrinol (Lausanne) 2022, 13, 827032. [Google Scholar] [CrossRef] [PubMed]
- Longuespee, R.; Boyon, C.; Desmons, A.; Vinatier, D.; Leblanc, E.; Farre, I.; Wisztorski, M.; Ly, K.; D'Anjou, F.; Day, R.; Fournier, I.; Salzet, M. OC molecular pathology. Cancer Metastasis Rev 2012, 31, 713–732. [Google Scholar] [CrossRef]
- Koziel, M.J.; Piastowska-Ciesielska, A.W. Estrogens, Estrogen Receptors and Tumor Microenvironment in Ovarian Cancer. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Pujol, P.; Rey, J.M.; Nirde, P.; Roger, P.; Gastaldi, M.; Laffargue, F.; Rochefort, H.; Maudelonde, T. Differential expression of estrogen receptor-alpha and -beta messenger RNAs as a potential marker of ovarian carcinogenesis. Cancer Res 1998, 58, 5367–5373. [Google Scholar]
- Li, A.J.; Baldwin, R.L.; Karlan, B.Y. Estrogen and progesterone receptor subtype expression in normal and malignant ovarian epithelial cell cultures. Am J Obstet Gynecol 2003, 189, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, P.R.; Cajander, S.; Backstrom, T.; Gustafsson, J.A.; Makela, S.; Olofsson, J.I. Estrogen and progesterone receptors in ovarian epithelial tumors. Mol Cell Endocrinol 2004, 221, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Halon, A.; Nowak-Markwitz, E.; Maciejczyk, A.; Pudelko, M.; Gansukh, T.; Gyorffy, B.; Donizy, P.; Murawa, D.; Matkowski, R.; Spaczynski, M.; Lage, H.; Surowiak, P. Loss of estrogen receptor beta expression correlates with shorter overall survival and lack of clinical response to chemotherapy in OC patients. Anticancer Res 2011, 31, 711–718. [Google Scholar] [PubMed]
- Sieh, W.; Kobel, M.; Longacre, T.A.; Bowtell, D.D.; deFazio, A.; Goodman, M.T.; Hogdall, E.; Deen, S.; Wentzensen, N.; Moysich, K.B.; Brenton, J.D.; Clarke, B.A.; Menon, U.; Gilks, C.B.; Kim, A.; Madore, J.; Fereday, S.; George, J.; Galletta, L.; Lurie, G.; Wilkens, L.R.; Carney, M.E.; Thompson, P.J.; Matsuno, R.K.; Kjaer, S.K.; Jensen, A.; Hogdall, C.; Kalli, K.R.; Fridley, B.L.; Keeney, G.L.; Vierkant, R.A.; Cunningham, J.M.; Brinton, L.A.; Yang, H.P.; Sherman, M.E.; Garcia-Closas, M.; Lissowska, J.; Odunsi, K.; Morrison, C.; Lele, S.; Bshara, W.; Sucheston, L.; Jimenez-Linan, M.; Driver, K.; Alsop, J.; Mack, M.; McGuire, V.; Rothstein, J.H.; Rosen, B.P.; Bernardini, M.Q.; Mackay, H.; Oza, A.; Wozniak, E.L.; Benjamin, E.; Gentry-Maharaj, A.; Gayther, S.A.; Tinker, A.V.; Prentice, L.M.; Chow, C.; Anglesio, M.S.; Johnatty, S.E.; Chenevix-Trench, G.; Whittemore, A.S.; Pharoah, P.D.; Goode, E.L.; Huntsman, D.G.; Ramus, S.J. Hormone-receptor expression and OC survival: an Ovarian Tumor Tissue Analysis consortium study. Lancet Oncol 2013, 14, 853–862. [Google Scholar] [CrossRef]
- Bogush, T.A.; Basharina, A.A.; Bogush, E.A.; Ryabinina, O.M.; Tjulandina, A.S.; Tjulandin, S.A. Estrogen Receptors alpha and beta in Ovarian Cancer: Expression Level and Prognosis. Dokl Biochem Biophys 2018, 482, 249–251. [Google Scholar] [CrossRef]
- Fixemer, T.; Remberger, K.; Bonkhoff, H. Differential expression of the estrogen receptor beta (ERbeta) in human prostate tissue, premalignant changes, and in primary, metastatic, and recurrent prostatic adenocarcinoma. Prostate 2003, 54, 79–87. [Google Scholar] [CrossRef]
- Foley, E.F.; Jazaeri, A.A.; Shupnik, M.A.; Jazaeri, O.; Rice, L.W. Selective loss of estrogen receptor beta in malignant human colon. Cancer Res 2000, 60, 245–248. [Google Scholar] [PubMed]
- Park, B.W.; Kim, K.S.; Heo, M.K.; Ko, S.S.; Hong, S.W.; Yang, W.I.; Kim, J.H.; Kim, G.E.; Lee, K.S. Expression of estrogen receptor-beta in normal mammary and tumor tissues: is it protective in breast carcinogenesis? Breast Cancer Res Treat 2003, 80, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Bossard, C.; Busson, M.; Vindrieux, D.; Gaudin, F.; Machelon, V.; Brigitte, M.; Jacquard, C.; Pillon, A.; Balaguer, P.; Balabanian, K.; Lazennec, G. Potential role of estrogen receptor beta as a tumor suppressor of epithelial ovarian cancer. PLoS One 2012, 7, e44787. [Google Scholar] [CrossRef]
- O'Donnell, A.J.; Macleod, K.G.; Burns, D.J.; Smyth, J.F.; Langdon, S.P. Estrogen receptor-alpha mediates gene expression changes and growth response in OC cells exposed to estrogen. Endocr Relat Cancer 2005, 12, 851–866. [Google Scholar] [CrossRef]
- Treeck, O.; Pfeiler, G.; Mitter, D.; Lattrich, C.; Piendl, G.; Ortmann, O. Estrogen receptor beta1 exerts antitumoral effects on SK-OV-3 OC cells. J Endocrinol 2007, 193, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Schuler-Toprak, S.; Moehle, C.; Skrzypczak, M.; Ortmann, O.; Treeck, O. Effect of estrogen receptor beta agonists on proliferation and gene expression of OC cells. BMC Cancer 2017, 17, 319. [Google Scholar] [CrossRef]
- Bardin, A.; Hoffmann, P.; Boulle, N.; Katsaros, D.; Vignon, F.; Pujol, P.; Lazennec, G. Involvement of estrogen receptor beta in ovarian carcinogenesis. Cancer Res 2004, 64, 5861–5869. [Google Scholar] [CrossRef]
- Lazennec, G. Estrogen receptor beta, a possible tumor suppressor involved in ovarian carcinogenesis. Cancer Lett 2006, 231, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Langdon, S.P.; Hirst, G.L.; Miller, E.P.; Hawkins, R.A.; Tesdale, A.L.; Smyth, J.F.; Miller, W.R. The regulation of growth and protein expression by estrogen in vitro: a study of 8 human ovarian carcinoma cell lines. J Steroid Biochem Mol Biol 1994, 50, 131–135. [Google Scholar] [CrossRef]
- Langdon, S.P.; Crew, A.J.; Ritchie, A.A.; Muir, M.; Wakeling, A.; Smyth, J.F.; Miller, W.R. Growth inhibition of oestrogen receptor-positive human ovarian carcinoma by anti-oestrogens in vitro and in a xenograft model. Eur J Cancer 1994, 30A, 682–686. [Google Scholar] [CrossRef]
- Fujiwara, S.; Terai, Y.; Kawaguchi, H.; Takai, M.; Yoo, S.; Tanaka, Y.; Tanaka, T.; Tsunetoh, S.; Sasaki, H.; Kanemura, M.; Tanabe, A.; Yamashita, Y.; Ohmichi, M. GPR30 regulates the EGFR-Akt cascade and predicts lower survival in patients with ovarian cancer. J Ovarian Res 2012, 5, 35. [Google Scholar] [CrossRef]
- Smith, H.O.; Arias-Pulido, H.; Kuo, D.Y.; Howard, T.; Qualls, C.R.; Lee, S.J.; Verschraegen, C.F.; Hathaway, H.J.; Joste, N.E.; Prossnitz, E.R. GPR30 predicts poor survival for ovarian cancer. Gynecol Oncol 2009, 114, 465–471. [Google Scholar] [CrossRef]
- Ignatov, T.; Modl, S.; Thulig, M.; Weissenborn, C.; Treeck, O.; Ortmann, O.; Zenclussen, A.; Costa, S.D.; Kalinski, T.; Ignatov, A. GPER-1 acts as a tumor suppressor in ovarian cancer. J Ovarian Res 2013, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Schuler-Toprak, S.; Skrzypczak, M.; Ignatov, T.; Ignatov, A.; Ortmann, O.; Treeck, O. G protein-coupled estrogen receptor 1 (GPER-1) and agonist G-1 inhibit growth of OC cells by activation of anti-tumoral transcriptome responses: impact of GPER-1 mRNA on survival. J Cancer Res Clin Oncol 2020, 146, 3175–3188. [Google Scholar] [CrossRef] [PubMed]
- Fraungruber, P.; Kaltofen, T.; Heublein, S.; Kuhn, C.; Mayr, D.; Burges, A.; Mahner, S.; Rathert, P.; Jeschke, U.; Trillsch, F. G Protein-Coupled Estrogen Receptor Correlates With Dkk2 Expression and Has Prognostic Impact in OC Patients. Front Endocrinol (Lausanne) 2021, 12, 564002. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Chen, Z.; Wang, Y.; Zhao, H.; Du, Y. The Role of Cancer-Associated Fibroblasts in Ovarian Cancer. Cancers (Basel) 2022, 14. [Google Scholar] [CrossRef]
- Yousefzadeh, Y.; Hallaj, S.; Baghi Moornani, M.; Asghary, A.; Azizi, G.; Hojjat-Farsangi, M.; Ghalamfarsa, G.; Jadidi-Niaragh, F. Tumor associated macrophages in the molecular pathogenesis of ovarian cancer. Int Immunopharmacol 2020, 84, 106471. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, A.; Rothenberger, N.J.; Stabile, L.P. The Impact of Estrogen in the Tumor Microenvironment. Adv Exp Med Biol 2020, 1277, 33–52. [Google Scholar]
- Esteller, M. Epigenetics in cancer. N Engl J Med 2008, 358, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Peng, M.; Tan, S.; Oyang, L.; Lin, J.; Xia, L.; Wang, J.; Wu, N.; Jiang, X.; Peng, Q.; Zhou, Y.; Liao, Q. The roles and molecular mechanisms of non-coding RNA in cancer metabolic reprogramming. Cancer Cell Int 2024, 24, 37. [Google Scholar] [CrossRef] [PubMed]
- Good, D.J. Non-Coding RNAs in Human Health and Diseases. Genes (Basel) 2023, 14. [Google Scholar] [CrossRef]
- Esteller, M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet 2007, 8, 286–298. [Google Scholar] [CrossRef]
- Rossello-Tortella, M.; Bueno-Costa, A.; Martinez-Verbo, L.; Villanueva, L.; Esteller, M. DNA methylation-associated dysregulation of transfer RNA expression in human cancer. Mol Cancer 2022, 21, 48. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Yang, Y.; Li, L.; Tang, J.; Zhang, R. DNA methylation profiles in cancer: functions, therapy, and beyond. Cancer Biol Med 2023, 21, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Tajbakhsh, J.; Mortazavi, F.; Gupta, N.K. DNA methylation topology differentiates between normal and malignant in cell models, resected human tissues, and exfoliated sputum cells of lung epithelium. Front Oncol 2022, 12, 991120. [Google Scholar] [CrossRef] [PubMed]
- Davalos, V.; Esteller, M. Cancer epigenetics in clinical practice. CA Cancer J Clin 2022. [Google Scholar] [CrossRef] [PubMed]
- Mattei, A.L.; Bailly, N.; Meissner, A. DNA methylation: a historical perspective. Trends Genet 2022, 38, 676–707. [Google Scholar] [CrossRef]
- Chen, C.; Wang, Z.; Ding, Y.; Wang, L.; Wang, S.; Wang, H.; Qin, Y. DNA Methylation: From Cancer Biology to Clinical Perspectives. Front Biosci (Landmark Ed) 2022, 27, 326. [Google Scholar] [CrossRef]
- Zhao, S.; Allis, C.D.; Wang, G.G. The language of chromatin modification in human cancers. Nat Rev Cancer 2021, 21, 413–430. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Chi, P.; Allis, C.D.; Wang, G.G. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer 2010, 10, 457–469. [Google Scholar] [CrossRef]
- Zhuang, J.; Huo, Q.; Yang, F.; Xie, N. Perspectives on the Role of Histone Modification in Breast Cancer Progression and the Advanced Technological Tools to Study Epigenetic Determinants of Metastasis. Front Genet 2020, 11, 603552. [Google Scholar] [CrossRef] [PubMed]
- Geng, R.; Chen, T.; Zhong, Z.; Ni, S.; Bai, J.; Liu, J. The m6A-Related Long Noncoding RNA Signature Predicts Prognosis and Indicates Tumor Immune Infiltration in Ovarian Cancer. Cancers (Basel) 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Weng, H.; Chen, J. m(6)A Modification in Coding and Non-coding RNAs: Roles and Therapeutic Implications in Cancer. Cancer Cell 2020, 37, 270–288. [Google Scholar] [CrossRef]
- Shulman, Z.; Stern-Ginossar, N. The RNA modification N(6)-methyladenosine as a novel regulator of the immune system. Nat Immunol 2020, 21, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Frye, M.; Harada, B.T.; Behm, M.; He, C. RNA modifications modulate gene expression during development. Science 2018, 361, 1346–1349. [Google Scholar] [CrossRef]
- Gu, J.; Bi, F. Significance of N6-Methyladenosine RNA Methylation Regulators in Immune Infiltrates of Ovarian Cancer. Front Genet 2021, 12, 671179. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Meng, D.; Wan, M.; Xu, N.; Xu, Y.; Yuan, K.; Liu, P.; Fang, H.; Hu, H.; Lan, S. m6A-Related lncRNAs Predict Overall Survival of Patients and Regulate the Tumor Immune Microenvironment in Osteosarcoma. Comput Intell Neurosci 2022, 2022, 9315283. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Feng, J.; Wu, J.; Zhong, W.; Zouxu, X.; Huang, W.; Huang, X.; Yi, J.; Wang, X. Prognostic value of comprehensive typing based on m6A and gene cluster in TNBC. J Cancer Res Clin Oncol 2022. [Google Scholar] [CrossRef]
- Alam, S.; Giri, P.K. Emerging role of m6A modification in ovarian cancer: progression, drug resistance, and therapeutic prospects. Front Oncol 2024, 14, 1366223. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.; Cortez, V.; Vadlamudi, R.K. Epigenetics of estrogen receptor signaling: role in hormonal cancer progression and therapy. Cancers (Basel) 2011, 3, 1691–1707. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Shang, Y. Estrogen and cancer. Annu Rev Physiol 2013, 75, 225–240. [Google Scholar] [CrossRef]
- Carroll, J.S.; Meyer, C.A.; Song, J.; Li, W.; Geistlinger, T.R.; Eeckhoute, J.; Brodsky, A.S.; Keeton, E.K.; Fertuck, K.C.; Hall, G.F.; Wang, Q.; Bekiranov, S.; Sementchenko, V.; Fox, E.A.; Silver, P.A.; Gingeras, T.R.; Liu, X.S.; Brown, M. Genome-wide analysis of estrogen receptor binding sites. Nat Genet 2006, 38, 1289–1297. [Google Scholar] [CrossRef]
- Li, W.; Hu, Y.; Oh, S.; Ma, Q.; Merkurjev, D.; Song, X.; Zhou, X.; Liu, Z.; Tanasa, B.; He, X.; Chen, A.Y.; Ohgi, K.; Zhang, J.; Liu, W.; Rosenfeld, M.G. Condensin I and II Complexes License Full Estrogen Receptor alpha-Dependent Enhancer Activation. Mol Cell 2015, 59, 188–202. [Google Scholar] [CrossRef]
- Kirn, V.; Strake, L.; Thangarajah, F.; Richters, L.; Eischeid, H.; Koitzsch, U.; Odenthal, M.; Fries, J. ESR1-promoter-methylation status in primary breast cancer and its corresponding metastases. Clin Exp Metastasis 2018, 35, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Quintas-Granados, L.I.; Cortes, H.; Carmen, M.G.; Leyva-Gomez, G.; Bustamante-Montes, L.P.; Rodriguez-Morales, M.; Villegas-Vazquez, E.Y.; Lopez-Reyes, I.; Alcaraz-Estrada, S.L.; Sandoval-Basilio, J.; Soto-Reyes, E.; Sharifi-Rad, J.; Figueroa-Gonzalez, G.; Reyes-Hernandez, O.D. The high methylation level of a novel 151-bp CpG island in the ESR1 gene promoter is associated with a poor breast cancer prognosis. Cancer Cell Int 2021, 21, 649. [Google Scholar] [CrossRef] [PubMed]
- Giacinti, L.; Claudio, P.P.; Lopez, M.; Giordano, A. Epigenetic information and estrogen receptor alpha expression in breast cancer. Oncologist 2006, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bos, M.K.; Deger, T.; Sleijfer, S.; Martens, J.W.M.; Wilting, S.M. ESR1 Methylation Measured in Cell-Free DNA to Evaluate Endocrine Resistance in Metastatic Breast Cancer Patients. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Imura, M.; Yamashita, S.; Cai, L.Y.; Furuta, J.; Wakabayashi, M.; Yasugi, T.; Ushijima, T. Methylation and expression analysis of 15 genes and three normally-methylated genes in 13 OC cell lines. Cancer Lett 2006, 241, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Gong, G.; Lin, T.; Yuan, Y. Integrated analysis of gene expression and DNA methylation profiles in ovarian cancer. J Ovarian Res 2020, 13, 30. [Google Scholar] [CrossRef] [PubMed]
- Kirn, V.; Shi, R.; Heublein, S.; Knabl, J.; Guenthner-Biller, M.; Andergassen, U.; Fridrich, C.; Malter, W.; Harder, J.; Friese, K.; Mayr, D.; Jeschke, U. Estrogen receptor promoter methylation predicts survival in low-grade ovarian carcinoma patients. J Cancer Res Clin Oncol 2014, 140, 1681–1687. [Google Scholar] [CrossRef] [PubMed]
- Salvati, A.; Gigantino, V.; Nassa, G.; Giurato, G.; Alexandrova, E.; Rizzo, F.; Tarallo, R.; Weisz, A. The Histone Methyltransferase DOT1L Is a Functional Component of Estrogen Receptor Alpha Signaling in OC Cells. Cancers (Basel) 2019, 11. [Google Scholar]
- Liu, Z.; Merkurjev, D.; Yang, F.; Li, W.; Oh, S.; Friedman, M.J.; Song, X.; Zhang, F.; Ma, Q.; Ohgi, K.A.; Krones, A.; Rosenfeld, M.G. Enhancer activation requires trans-recruitment of a mega transcription factor complex. Cell 2014, 159, 358–373. [Google Scholar] [CrossRef]
- Yang, F.; Ma, Q.; Liu, Z.; Li, W.; Tan, Y.; Jin, C.; Ma, W.; Hu, Y.; Shen, J.; Ohgi, K.A.; Telese, F.; Liu, W.; Rosenfeld, M.G. Glucocorticoid Receptor:MegaTrans Switching Mediates the Repression of an ERalpha-Regulated Transcriptional Program. Mol Cell 2017, 66, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jaiswal, S.K.; Kaur, R.; Alsaadi, D.; Liang, X.; Drews, F.; DeLoia, J.A.; Krivak, T.; Petrykowska, H.M.; Gotea, V.; Welch, L.; Elnitski, L. Differential gene expression identifies a transcriptional regulatory network involving ER-alpha and PITX1 in invasive epithelial ovarian cancer. BMC Cancer 2021, 21, 768. [Google Scholar] [CrossRef]
- Schuler-Toprak, S.; Skrzypczak, M.; Grundker, C.; Ortmann, O.; Treeck, O. Role of Estrogen Receptor beta, G-Protein Coupled Estrogen Receptor and Estrogen-Related Receptors in Endometrial and Ovarian Cancer. Cancers (Basel) 2023, 15. [Google Scholar]
- Rody, A.; Holtrich, U.; Solbach, C.; Kourtis, K.; von Minckwitz, G.; Engels, K.; Kissler, S.; Gatje, R.; Karn, T.; Kaufmann, M. Methylation of estrogen receptor beta promoter correlates with loss of ER-beta expression in mammary carcinoma and is an early indication marker in premalignant lesions. Endocr Relat Cancer 2005, 12, 903–916. [Google Scholar] [CrossRef] [PubMed]
- Manente, A.G.; Pinton, G.; Zonca, S.; Tavian, D.; Habib, T.; Jithesh, P.V.; Fennell, D.; Nilsson, S.; Moro, L. KDM6B histone demethylase is an epigenetic regulator of estrogen receptor beta expression in human pleural mesothelioma. Epigenomics 2016, 8, 1227–1238. [Google Scholar] [CrossRef]
- Salahuddin, A.; Ghanem, H.; Omran, G.A.; Helmy, M.W. Epigenetic restoration and activation of ERbeta: an inspiring approach for treatment of triple-negative breast cancer. Med Oncol 2022, 39, 150. [Google Scholar] [CrossRef] [PubMed]
- Walton, T.J.; Li, G.; Seth, R.; McArdle, S.E.; Bishop, M.C.; Rees, R.C. DNA demethylation and histone deacetylation inhibition co-operate to re-express estrogen receptor beta and induce apoptosis in prostate cancer cell-lines. Prostate 2008, 68, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Venkata, P.P.; Jayamohan, S.; He, Y.; Alejo, S.; Johnson, J.D.; Palacios, B.E.; Pratap, U.P.; Chen, Y.; Liu, Z.; Zou, Y.; Lai, Z.; Suzuki, T.; Viswanadhapalli, S.; Weintraub, S.T.; Palakurthi, S.; Valente, P.T.; Tekmal, R.R.; Kost, E.R.; Vadlamudi, R.K.; Sareddy, G.R. Pharmacological inhibition of KDM1A/LSD1 enhances estrogen receptor beta-mediated tumor suppression in ovarian cancer. Cancer Lett 2023, 575, 216383. [Google Scholar] [CrossRef]
- Yap, O.W.; Bhat, G.; Liu, L.; Tollefsbol, T.O. Epigenetic modifications of the Estrogen receptor beta gene in epithelial OC cells. Anticancer Res 2009, 29, 139–144. [Google Scholar] [PubMed]
- Suzuki, F.; Akahira, J.; Miura, I.; Suzuki, T.; Ito, K.; Hayashi, S.; Sasano, H.; Yaegashi, N. Loss of estrogen receptor beta isoform expression and its correlation with aberrant DNA methylation of the 5'-untranslated region in human epithelial ovarian carcinoma. Cancer Sci 2008, 99, 2365–2372. [Google Scholar] [CrossRef] [PubMed]
- Dalal, H.; Dahlgren, M.; Gladchuk, S.; Brueffer, C.; Gruvberger-Saal, S.K.; Saal, L.H. Clinical associations of ESR2 (estrogen receptor beta) expression across thousands of primary breast tumors. Sci Rep 2022, 12, 4696. [Google Scholar] [CrossRef] [PubMed]
- Hartman, J.; Strom, A.; Gustafsson, J.A. Current concepts and significance of estrogen receptor beta in prostate cancer. Steroids 2012, 77, 1262–1266. [Google Scholar] [CrossRef]
- Heublein, S.; Lenhard, M.; Vrekoussis, T.; Schoepfer, J.; Kuhn, C.; Friese, K.; Makrigiannakis, A.; Mayr, D.; Jeschke, U. The G-protein-coupled estrogen receptor (GPER) is expressed in normal human ovaries and is upregulated in ovarian endometriosis and pelvic inflammatory disease involving the ovary. Reprod Sci 2012, 19, 1197–1204. [Google Scholar] [CrossRef]
- Heublein, S.; Mayr, D.; Friese, K.; Jarrin-Franco, M.C.; Lenhard, M.; Mayerhofer, A.; Jeschke, U. The G-protein-coupled estrogen receptor (GPER/GPR30) in ovarian granulosa cell tumors. Int J Mol Sci 2014, 15, 15161–15172. [Google Scholar] [CrossRef]
- Tirado-Garibay, A.C.; Falcon-Ruiz, E.A.; Ochoa-Zarzosa, A.; Lopez-Meza, J.E. GPER: An Estrogen Receptor Key in Metastasis and Tumoral Microenvironments. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Rong, J.; Xie, X.; Niu, Y.; Su, Z. Correlation between the RNA Expression and the DNA Methylation of Estrogen Receptor Genes in Normal and Malignant Human Tissues. Curr Issues Mol Biol 2024, 46, 3610–3625. [Google Scholar] [CrossRef]
- Manjegowda, M.C.; Gupta, P.S.; Limaye, A.M. Hyper-methylation of the upstream CpG island shore is a likely mechanism of GPER1 silencing in breast cancer cells. Gene 2017, 614, 65–73. [Google Scholar] [CrossRef]
- Weissenborn, C.; Ignatov, T.; Nass, N.; Kalinski, T.; Dan Costa, S.; Zenclussen, A.C.; Ignatov, A. GPER Promoter Methylation Controls GPER Expression in Breast Cancer Patients. Cancer Invest 2017, 35, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kim, G.W.; Kwon, S.H.; Lee, J.S. Broad domains of histone H3 lysine 4 trimethylation in transcriptional regulation and disease. FEBS J 2020, 287, 2891–2902. [Google Scholar] [CrossRef]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: from mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Chen, Z.; Wu, D.; Zhang, L.; Lin, X.; Su, J.; Rodriguez, B.; Xi, Y.; Xia, Z.; Chen, X.; Shi, X.; Wang, Q.; Li, W. Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor-suppressor genes. Nat Genet 2015, 47, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Han, N.; Heublein, S.; Jeschke, U.; Kuhn, C.; Hester, A.; Czogalla, B.; Mahner, S.; Rottmann, M.; Mayr, D.; Schmoeckel, E.; Trillsch, F. The G-Protein-Coupled Estrogen Receptor (GPER) Regulates Trimethylation of Histone H3 at Lysine 4 and Represses Migration and Proliferation of OC Cells In Vitro. Cells 2021, 10. [Google Scholar] [CrossRef]
- Schneider, L.; Herkt, S.; Wang, L.; Feld, C.; Wesely, J.; Kuvardina, O.N.; Meyer, A.; Oellerich, T.; Haupl, B.; Seifried, E.; Bonig, H.; Lausen, J. PRMT6 activates cyclin D1 expression in conjunction with the transcription factor LEF1. Oncogenesis 2021, 10, 42. [Google Scholar] [CrossRef] [PubMed]
- Ivan, C.; Hu, W.; Bottsford-Miller, J.; Zand, B.; Dalton, H.J.; Liu, T.; Huang, J.; Nick, A.M.; Lopez-Berestein, G.; Coleman, R.L.; Baggerly, K.A.; Sood, A.K. Epigenetic analysis of the Notch superfamily in high-grade serous ovarian cancer. Gynecol Oncol 2013, 128, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Cheung, L.W.; Wong, A.S.; Leung, P.C. Estrogen regulates Snail and Slug in the down-regulation of E-cadherin and induces metastatic potential of OC cells through estrogen receptor alpha. Mol Endocrinol 2008, 22, 2085–2098. [Google Scholar] [CrossRef] [PubMed]
- Moselhy, S.S.; Kumosani, T.A.; Kamal, I.H.; Jalal, J.A.; Jabaar, H.S.; Dalol, A. Hypermethylation of P15, P16, and E-cadherin genes in ovarian cancer. Toxicol Ind Health 2015, 31, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Sawada, K.; Mitra, A.K.; Radjabi, A.R.; Bhaskar, V.; Kistner, E.O.; Tretiakova, M.; Jagadeeswaran, S.; Montag, A.; Becker, A.; Kenny, H.A.; Peter, M.E.; Ramakrishnan, V.; Yamada, S.D.; Lengyel, E. Loss of E-cadherin promotes OC metastasis via alpha 5-integrin, which is a therapeutic target. Cancer Res 2008, 68, 2329–2339. [Google Scholar] [CrossRef]
- Ho, C.M.; Lin, M.C.; Huang, S.H.; Huang, C.J.; Lai, H.C.; Chien, T.Y.; Chang, S.F. PTEN promoter methylation and LOH of 10q22-23 locus in PTEN expression of ovarian clear cell adenocarcinomas. Gynecol Oncol 2009, 112, 307–313. [Google Scholar] [CrossRef]
- Zhao, R.; Han, C.; Eisenhauer, E.; Kroger, J.; Zhao, W.; Yu, J.; Selvendiran, K.; Liu, X.; Wani, A.A.; Wang, Q.E. DNA damage-binding complex recruits HDAC1 to repress Bcl-2 transcription in human OC cells. Mol Cancer Res 2014, 12, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.L.; Xiu, Y.L.; Chen, X.; Sun, K.X.; Chen, S.; Wu, D.D.; Liu, B.L.; Zhao, Y. The transcription factor FOXA1 induces epithelial OC tumorigenesis and progression. Tumour Biol 2017, 39, 1010428317706210. [Google Scholar]
- Lou, T.; Liu, C.; Qu, H.; Zhang, Z.; Wang, S.; Zhuang, H. FOXA1 can be modulated by HDAC3 in the progression of epithelial ovarian carcinoma. J Transl Med 2022, 20, 19. [Google Scholar] [CrossRef] [PubMed]
- Matthews, B.G.; Bowden, N.A.; Wong-Brown, M.W. Epigenetic Mechanisms and Therapeutic Targets in Chemoresistant High-Grade Serous Ovarian Cancer. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef]
- Wilson, A.J.; Lalani, A.S.; Wass, E.; Saskowski, J.; Khabele, D. Romidepsin (FK228) combined with cisplatin stimulates DNA damage-induced cell death in ovarian cancer. Gynecol Oncol 2012, 127, 579–586. [Google Scholar] [CrossRef]
- Helland, O.; Popa, M.; Bischof, K.; Gjertsen, B.T.; McCormack, E.; Bjorge, L. The HDACi Panobinostat Shows Growth Inhibition Both In Vitro and in a Bioluminescent Orthotopic Surgical Xenograft Model of Ovarian Cancer. PLoS One 2016, 11, e0158208. [Google Scholar] [CrossRef] [PubMed]
- Garrett, L.A.; Growdon, W.B.; Rueda, B.R.; Foster, R. Influence of a novel histone deacetylase inhibitor panobinostat (LBH589) on the growth of ovarian cancer. J Ovarian Res 2016, 9, 58. [Google Scholar] [CrossRef]
- Ma, Y.Y.; Lin, H.; Moh, J.S.; Chen, K.D.; Wang, I.W.; Ou, Y.C.; You, Y.S.; Lung, C.C. Low-dose LBH589 increases the sensitivity of cisplatin to cisplatin-resistant OC cells. Taiwan J Obstet Gynecol 2011, 50, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.J.; Gupta, V.G.; Liu, Q.; Yull, F.; Crispens, M.A.; Khabele, D. Panobinostat enhances olaparib efficacy by modifying expression of homologous recombination repair and immune transcripts in ovarian cancer. Neoplasia 2022, 24, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Ovejero-Sanchez, M.; Gonzalez-Sarmiento, R.; Herrero, A.B. Synergistic effect of Chloroquine and Panobinostat in OC through induction of DNA damage and inhibition of DNA repair. Neoplasia 2021, 23, 515–528. [Google Scholar] [CrossRef]
- Rodrigues Moita, A.J.; Bandolik, J.J.; Hansen, F.K.; Kurz, T.; Hamacher, A.; Kassack, M.U. Priming with HDAC Inhibitors Sensitizes OC Cells to Treatment with Cisplatin and HSP90 Inhibitors. Int J Mol Sci 2020, 21. [Google Scholar]
- Booth, L.; Roberts, J.L.; Rais, R.; Poklepovic, A.; Dent, P. Valproate augments Niraparib killing of tumor cells. Cancer Biol Ther 2018, 19, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Joshi, J.; Yeh, I.J.; Doughman, Y.; Blankenberg, D.; Wald, D.; Montano, M.M. Re-Expression of ERalpha and AR in Receptor Negative Endocrine Cancers via GSK3 Inhibition. Front Oncol 2022, 12, 824594. [Google Scholar] [CrossRef] [PubMed]
- Dizon, D.S.; Blessing, J.A.; Penson, R.T.; Drake, R.D.; Walker, J.L.; Johnston, C.M.; Disilvestro, P.A.; Fader, A.N. A phase II evaluation of belinostat and carboplatin in the treatment of recurrent or persistent platinum-resistant ovarian, fallopian tube, or primary peritoneal carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol 2012, 125, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Meteran, H.; Knudsen, A.O.; Jorgensen, T.L.; Nielsen, D.; Herrstedt, J. Carboplatin plus Paclitaxel in Combination with the Histone Deacetylate Inhibitor, Vorinostat, in Patients with Recurrent Platinum-Sensitive Ovarian Cancer. J Clin Med 2024, 13. [Google Scholar] [CrossRef]
Figure 2.
Schematic overview of DNA methylation and histone modifications modulating estrogen signaling. DNMTs: DNA methyltransferases, TET: Ten-Eleven Translocation Dioxygenases, HATs: Histon Acetyltransferases, BAF: BRG1/BRM-associated factor, HDM; Histone Demethylase, HMT: Histone Methyltransferase, HDACs: Histone Deacetylases, NuRD: Nucleosome Remodelling and Deacetylase Complex. TFs: Transcription factors, RNA Pol II: RNA Polymerase II, H3K9me3: Histone H3 lysine 9 trimethylation, H3K27me3: Histone H3 lysine 27 trimethylation, H4K20me3: Histone H4 lysine 20 trimethylation, H3K27ac: Histone H3 lysine 27 acetylation, H3K9ac: Histone H3 lysine 9 acetylation, H3K4me3: Histone H3 lysine 4 trimethylation, H3K4me1: Histone H3 lysine 4 monomethylation.
Figure 2.
Schematic overview of DNA methylation and histone modifications modulating estrogen signaling. DNMTs: DNA methyltransferases, TET: Ten-Eleven Translocation Dioxygenases, HATs: Histon Acetyltransferases, BAF: BRG1/BRM-associated factor, HDM; Histone Demethylase, HMT: Histone Methyltransferase, HDACs: Histone Deacetylases, NuRD: Nucleosome Remodelling and Deacetylase Complex. TFs: Transcription factors, RNA Pol II: RNA Polymerase II, H3K9me3: Histone H3 lysine 9 trimethylation, H3K27me3: Histone H3 lysine 27 trimethylation, H4K20me3: Histone H4 lysine 20 trimethylation, H3K27ac: Histone H3 lysine 27 acetylation, H3K9ac: Histone H3 lysine 9 acetylation, H3K4me3: Histone H3 lysine 4 trimethylation, H3K4me1: Histone H3 lysine 4 monomethylation.
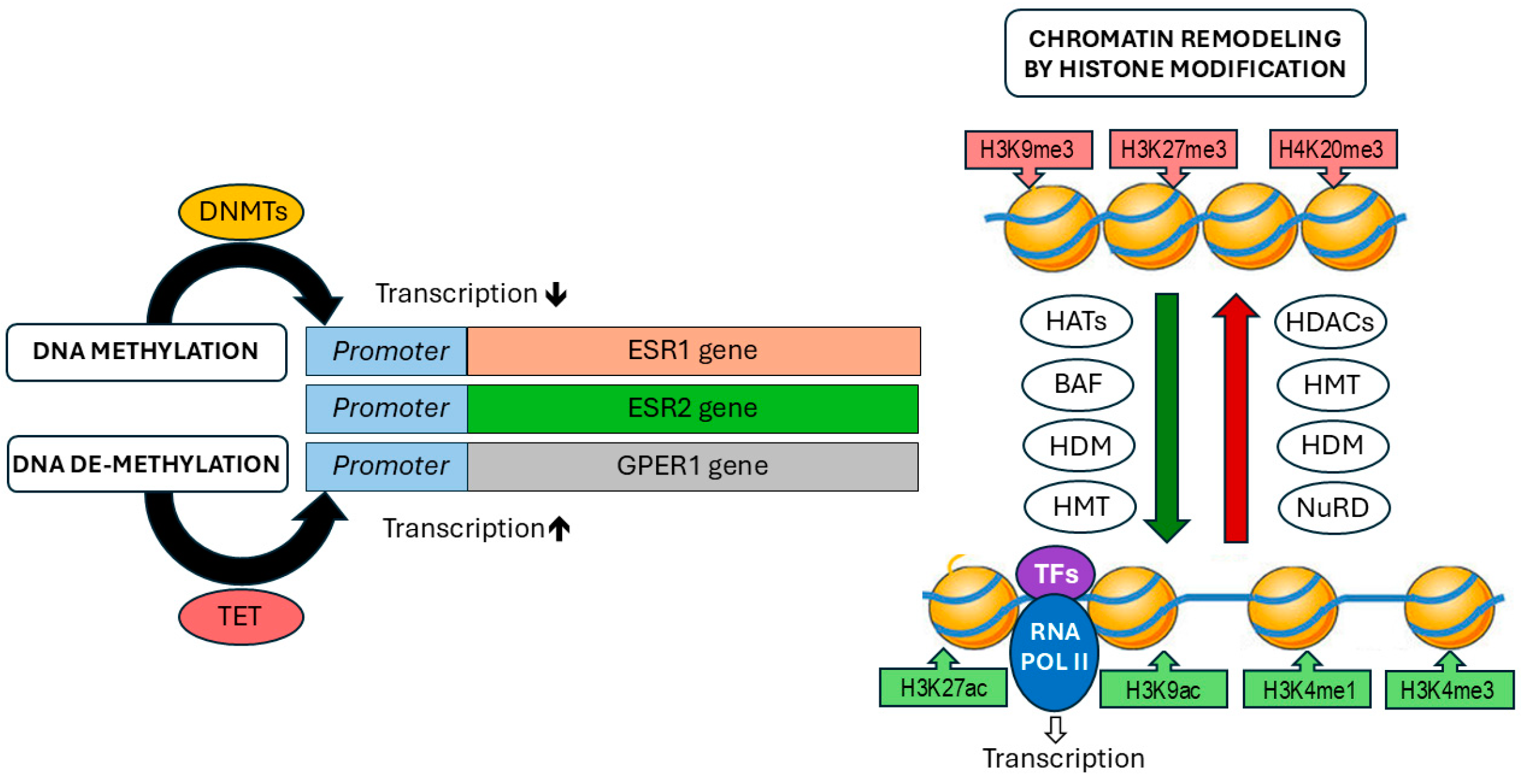
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



