
Submitted:
03 December 2024
Posted:
05 December 2024
You are already at the latest version
Abstract
Neurodegenerative disorders (NDs) lead to progressive neuronal loss in various brain regions and are a significant public health concern, with NDs expected to become the second-leading cause of death globally within the next two decades. Huntington's disease (HD) is a rare progressive ND that severely impacts patients and families due to its severe neuronal effects. HD is caused by an autosomal dominant mutation in the huntingtin (HTT) gene, which leads to progressive motor, cognitive, and psychiatric symptoms. Given the complex nature of HD, biomarkers are essential for early diagnosis, disease progression monitoring, and evaluation of treatment efficacy. However, the identification of consistent HD biomarkers is challenging due to HD's heterogeneous presentation and its multiple underlying biological pathways. To identify reliable HD biomarkers, integration of research efforts through longitudinal studies that track disease progression, new biomarker exploration to enhance diagnostic accuracy, and collaborative efforts to standardize protocols and ensure reproducibility are warranted. These efforts should also advance personalized treatment approaches and improve the overall management of HD. This study provides a comprehensive review and bibliometric analysis of HD biomarker research over the past decade, revealing key research trends and gaps. The findings could guide future investigations and contribute to improving personalized treatment approaches, clinical management, and informed decision-making in HD.
Keywords:
Huntington’s Disease
; Huntingtin protein
; Biomarkers
; Rare Diseases
; Neurodegenerative disorders
1. Introduction
1.1. Huntington’s Disease: An overview
Among the genetic neurodegenerative disorders (NDs), Huntington’s disease (HD, OMIM 143100, https://omim.org/entry/143100) is the primary focus of this review. HD is a monogenic autosomal dominant progressive neurodegenerative disorder, first described in 1872 by Dr. George Huntington. The disease results from a mutation in the huntingtin (HTT) gene, first located in 1993 to chromosome 4p16.3 [1]. The HTT gene mutation, marked by a variable expansion in the number of cytosine–adenine–guanine (CAG) repeats in exon1 of the HTT gene, causes the synthesis of abnormal huntingtin (HTT) protein. The expansion of CAG triplet repeats produces a toxic HTT protein with expanded polyglutamine (polyQ) amino acids [2]. Notably, the number of CAG repeats correlates with the risk of HD. CAG repeat lengths below 35 do not lead to the disease, and repeat lengths between 36-39 may express the disease with low penetrance, whereas repeat lengths exceeding 40 are considered pathogenic [1,2,3].
Huntington’s disease is a rare neurodegenerative disorder with an estimated incidence of 0.38 per 100,000 person-years and a global overall prevalence of 2.71 per 100,000 [3]. Though rare, its prevalence varies greatly (more than 10-fold) across geographical areas and populations [4,5]. Its highest prevalence is recorded within the Western/ White population (5-12 cases/ 100,000 individuals) [1]. HD has a higher prevalence in the USA (4.1-8.4/ 100,000 people) [3,4] and Europe (1.63-9.95/100,000 individuals) [6]. HD prevalence is lower in Asia (0.5 to 1.5 per 100,000) [4] and Africa (0.25 per 100,000) [3], although it increases in areas where intermarriage with Western individuals occurs [4]. China has the lowest HD prevalence (0.25 cases per 100,000 people). In Japan, the prevalence of the disorder is 0.5 cases per 100,000 people [4]. Huntington's disease rarity becomes evident when compared to other NDs. For instance, in the USA, 9.92 million Americans suffer from mild cognitive impairment due to Alzheimer’s Disease compared to merely 30,000 HD patients [7]. NDs impose a burden on governments worldwide; in the USA alone, the mean total annual cost per patient with early to late-stage HD ranges from $6113 to $27,904 among commercially insured patients [8].
HD is commonly classified into three stages: presymptomatic, prodromal, and manifest. In the presymptomatic stage, individuals carry the CAG expansion mutation but exhibit no signs or symptoms related to HD. The prodromal stage includes individuals with the CAG expansion who display nonspecific or possible motor abnormalities on examination and subtle yet clear cognitive changes[9]. Manifest HD is characterized by individuals with the CAG expansion who have a greater than 90% confidence of motor abnormalities accompanied by minor or major neurocognitive changes, or a greater than 99% probability of motor abnormalities with unchanged cognition. For individuals who have not undergone genetic testing but are clinically suspected of having HD, similar classifications are applied: at risk for HD but not manifest, clinically prodromal HD, and clinically manifest HD[3].
Individuals with HD usually lead a normal life until the prodromal stage initiates. Symptoms of the disease generally appear in middle age, although it can present at any time from infancy to old age [10]. The first manifestations of neurological symptoms include a change in personality, depression, anxiety, restlessness, and deficits in social cognition, leading to stigmatization. In addition to difficulties with detail retention, patients often experience learning, organization, and task-planning challenges. During this stage, diagnosis is typically established, and symptoms become progressively worse, ultimately resulting in deteriorating speech capabilities [11]. HD symptoms also encompass weight loss, which is attributed to the increased caloric demand resulting from incessant choreiform movements [1]. The main neuropsychiatric features of HD include deterioration of motor functions encompassing involuntary irregular and unpredicted muscle movement, known as chorea, and involuntary muscle contractions, known as dystonia, in addition to deterioration in coordination skills, cognitive decline, and behavioral changes. As motor and cognitive functions decline, complications such as falls, dysphagia, or aspiration may lead to fatal outcomes. Typically, patients have a survival span of 15 to 20 years following diagnosis [12]. The progression of the disease profoundly impacts both patients and their families, necessitating comprehensive support that includes medical, psychological, and social aspects [1,13]. The detailed pathophysiology of Huntington's disease is depicted in Figure 1.
The complex and progressive HD pathophysiology is underpinned by intricate molecular and cellular processes and signaling pathways (Figure 2).
1.2. Challenges in HD diagnosis and treatment
The mutant HD gene produces a protein with a toxic gain of function due to polyQ expansion [14]. HD, similar to other genetic diseases with polyQ expansion such as various types of spinocerebellar ataxia, dentatorubral-pallidoluysian atrophy, and spinobulbar muscular atrophy, has a direct relation between disease severity and polyQ repeat length and an inverse correlation between the repeat length and the age of disease onset [5]. Mutant huntingtin proteins (mHTT) with extended polyQ repeats undergo protein misfolding, leading to the accumulation of unmanageable protein aggregates, overwhelming cellular protein degradation via proteasomes and autophagic vacuolization [15]. Aggregation of mHTT impacts many nuclear and cytoplasmic proteins involved in vital functions such as transcription regulation, apoptosis, mitochondrial function, neurotransmitter release, and axonal transport. However, the exact neuronal function affected by mHTT misfolding and aggregation remains unclear [16].
Traditionally classified as a neuropsychiatric disease, recent research into the molecular mechanisms underlying HD suggests it is better described as a systemic illness, as autonomic symptoms often precede motor deficits by several years[10]. This shift in understanding highlights the complexity of HD, both in its clinical presentation and underlying pathophysiology. Despite significant research developments over the last two decades, there has been limited advancement in medical treatment for HD [13]. Various therapeutic options, including antisense oligonucleotide therapy, have been explored, but none have been proven effective in halting disease progression[17]. Currently, optimal care for HD patients typically involves multidisciplinary approaches primarily aiming to manage symptoms and enhance the patient’s quality of life. Medications used in such approaches include tetrabenazine for chorea control, selective serotonin reuptake inhibitors (SSRIs), such as citalopram and sertraline, for addressing depression and anxiety, atypical antipsychotics for managing psychosis, and sertraline for handling obsessive-compulsive behaviors in certain patients [1].
However, HD remains relatively understudied compared to other neurodegenerative disorders due to its rarity, which limits the number of patients available for clinical studies. For instance, a one-year PubMed search yields approximately 1,200 publications on HD, compared to 16,400 on Alzheimer’s disease and 9,900 on Parkinson’s disease [18]. This lack of extensive research has resulted in a limited understanding of HD pathophysiology, presenting challenges in diagnosis and the development of effective treatments. Consequently, current therapeutic approaches remain primarily symptomatic[19]. Compounding these challenges is the complexity and pleiotropy of HD clinical symptomatology, along with variations in cellular and neurochemical changes in the brain [20]. Studies suggest that the variability in symptoms cannot be fully explained by differences in CAG repeat numbers alone[21,22].
Adding to the complexity, brain atrophy and structural disruptions occur during the prodromal phase, before overt symptoms manifest. This period is characterized by intricate molecular and cellular changes that remain poorly defined [22]. Nevertheless, the premanifest stage offers a valuable window to explore novel therapeutic interventions that could delay HD progression before symptoms manifest [23]. These facts substantiate the search for new robust and sensitive diagnostic and prognostic HD biomarkers to improve knowledge of the disease pathophysiology and its underlying mechanisms, to better predict HD pathogenesis, progression, and age of symptoms onset, and to explore patient response to potential therapies.
HD is mainly diagnosed clinically based on the development of chorea in combination with other movement abnormalities and behavioral changes, which includes poor attention, cognitive rigidity, irritability, and dementia. Furthermore, A family history of autosomal dominant inheritance can support the clinical diagnosis based on observed symptoms. However, definitive diagnosis of HD can only be confirmed through genetic testing [3,24].
This study aims to perform a comprehensive bibliometric analysis of research on biomarkers in Huntington's disease over the past decade. By examining publication trends, the study seeks to identify significant progress and research gaps, providing insights into the evolution of biomarker research. These findings will provide insights into the evolution of HD biomarker research, reveal opportunities for future investigations, and offer recommendations to enhance understanding and development of biomarkers for effective diagnosis, progression monitoring, and therapeutic evaluation of emerging HD therapies.
2. Methods
Publications related to biomarkers of HD were sourced from PubMed through the Bibliometrix package in R software. Bibliometrix, an open-source R package widely utilized by researchers for bibliometric analysis, offers comprehensive tools for quantitative analysis of scientific publications, including data collection, preprocessing, visualization, and statistical modeling [25,26]. The search strategy included the following terms: ("biomarkers"[All Fields] OR "biomarkers"[MeSH Terms] OR "biomarker"[All Fields]) AND "Huntington's disease"[Title/Abstract] AND "English"[Language] AND "journal article"[Publication Type] AND ("2014/01/01"[Date - Publication] : "2024/12/31"[Date - Publication]). The search, conducted on 29 August 2024, covered various document types, such as original research articles, reviews, and conference proceedings, and was filtered to include only articles published in English.
Moreover, a literature review was conducted using PubMed to identify emerging biomarkers for Huntington's disease (HD). The search strategy included terms such as "Huntington's disease," "biomarkers," and related keywords. Articles published were screened for relevance based on titles and abstracts, and full texts were reviewed to extract data on novel biomarkers, including their diagnostic, prognostic, and therapeutic implications.
3. Results
3.1. Bibliometric Analysis
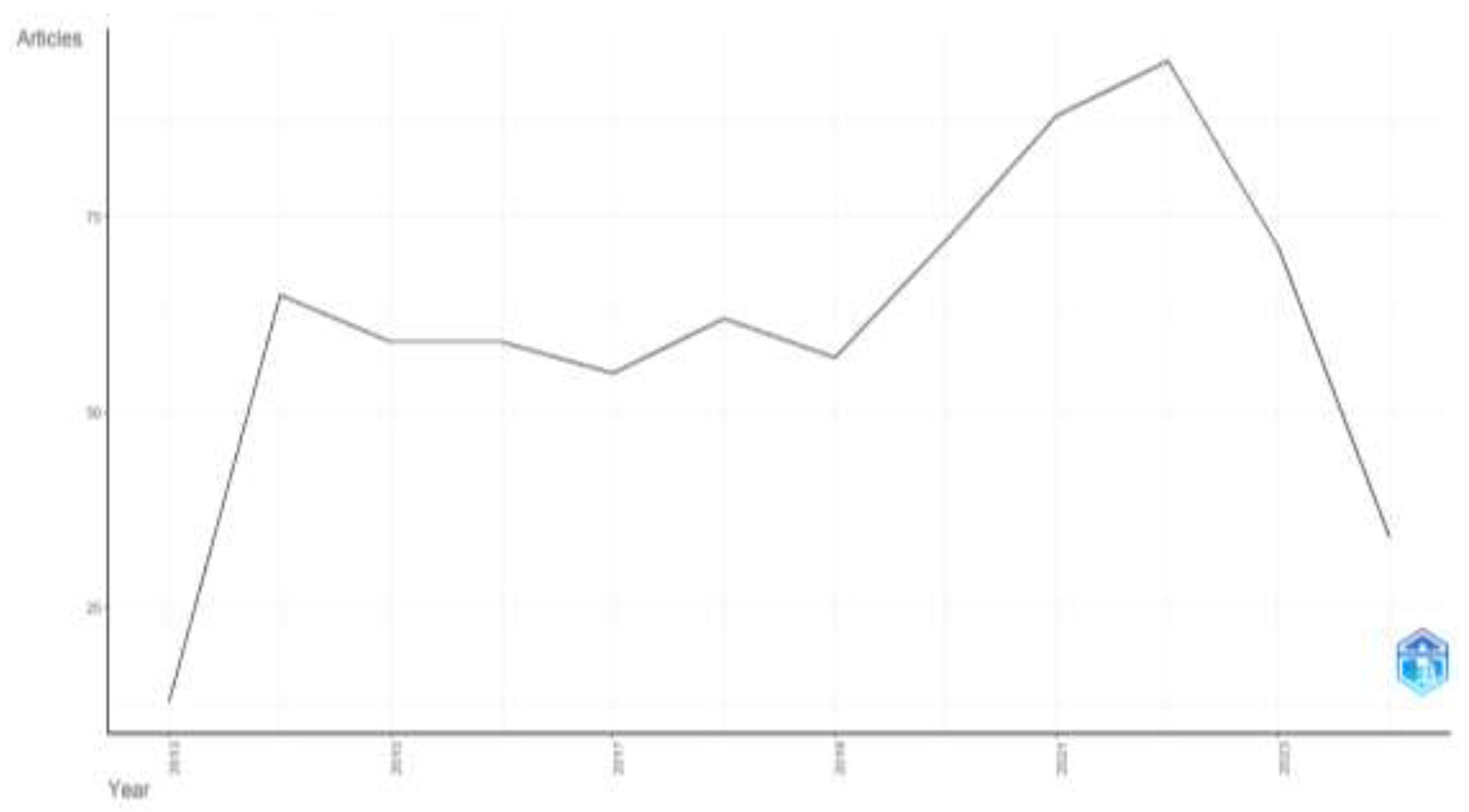
The analysis uncovered 730 articles related to biomarkers in Huntington's disease, published between 2014 and 2024. The annual growth rate of publications in this field was 9.13% (Table 1). n total, 3,959 authors from diverse countries contributed to these publications, with an average of 7 co-authors per article. A significant rise in publications was observed from 2019 to 2022, with 2022 marking the peak at 95 articles (Figure 3).
Figure 2.
Trends in publications over the years.
The articles were distributed across 319 different journals, with Journal of Huntington's Disease publishing the highest number (36 articles), followed by the International Journal of Molecular Sciences (35 articles). A summary of the top 10 journals in this research area is presented in Figure 3. In terms of international collaboration, the strongest partnerships were observed between researchers in the USA and Germany, followed by collaborations between the USA and Australia, and the USA and Canada (Figure 4).
Figure 3.
Most published journals.
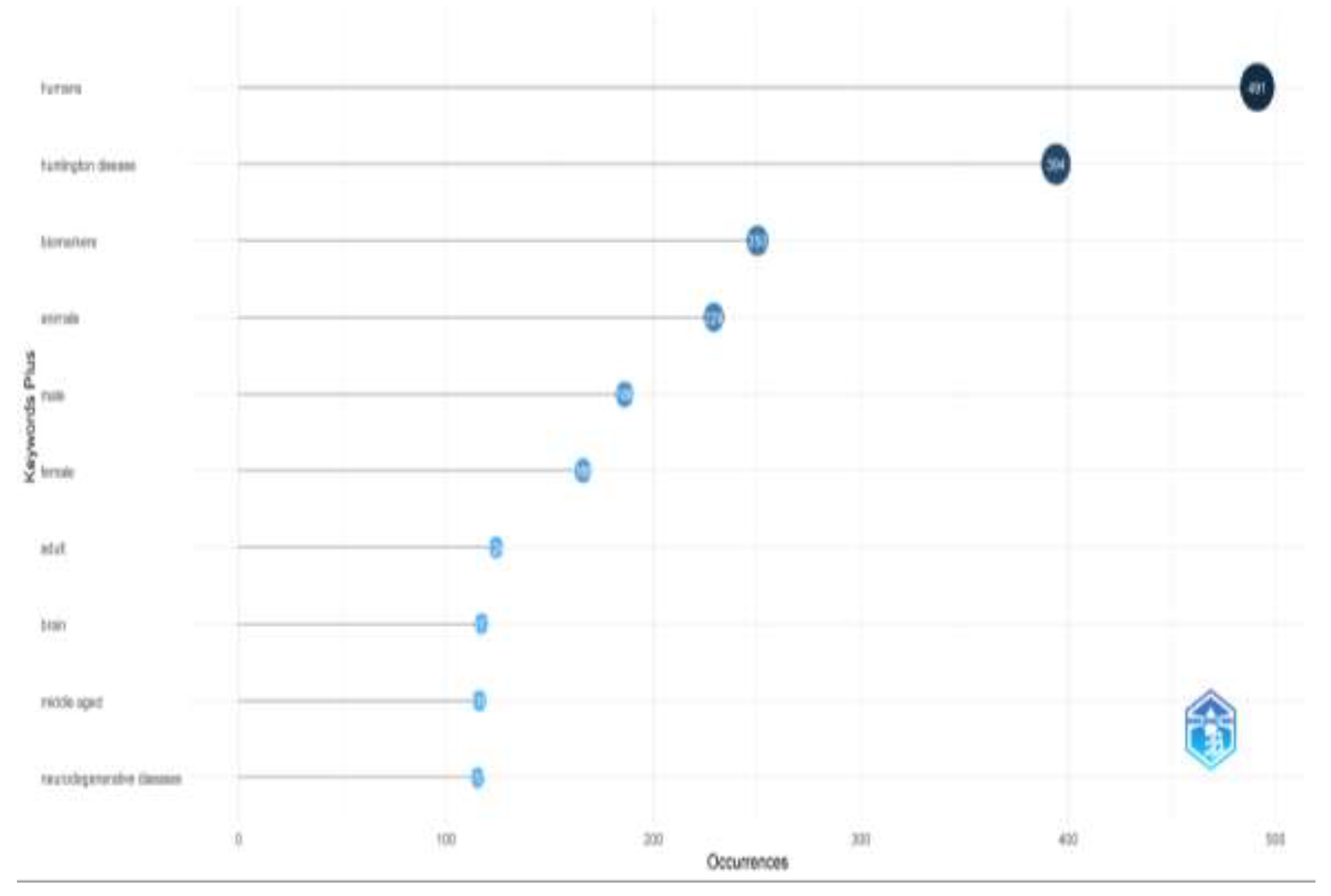
Through keyword analysis, we identified the top 10 most frequently occurring keywords. Of the 1,684 keywords analyzed, "humans" appeared most frequently (491 occurrences), followed by "Huntington's disease" with 394 occurrences (Figure 5). Several other keywords also emerged as significant. This increase in keyword frequency mirrored the overall rise in publications. The co-occurrence network of the top 50 keywords is shown in Figure 6, where node size represents keyword frequency and node color indicates clusters and relationships. The keywords clustered into three main groups. The largest cluster, shown in blue, includes terms such as "humans," "Huntington's disease," and "biomarkers." The red cluster, which contains the most nodes, features terms like "animals," "brain," "neurodegenerative diseases," and "disease models, animal." The green cluster includes keywords like "Alzheimer's disease," "Parkinson's disease," and "amyotrophic lateral sclerosis."
3.2. Emerging biomarkers in HD research
3.2.1. Tau protein
Tau is a microtubule-associated protein (MAP) that is known to be involved in neurogenesis, maintaining axons, and axonal transport. Tau has been extensively studied in Alzheimer's disease, Parkinson's disease, and Traumatic Brain.
Injury, with research demonstrating its strong association with cognitive impairments; these findings suggest that tau could also be a promising biomarker for Huntington's disease[27]. Emerging evidence implicates tau pathology in HD, akin to tauopathies, suggesting that therapeutic strategies targeting tau dysfunction, ranging from small molecules to gene modulation approaches, hold promise for addressing cognitive decline in HD[28]. A study found that age-adjusted cerebrospinal fluid (CSF) tau levels were significantly elevated in HD gene expansion carriers compared to healthy controls (p = 0.002), and these levels were correlated with disease progression. Specifically, CSF tau showed significant correlations with the Unified Huntington's Disease Rating Scale (UHDRS) total functional capacity (r = -0.29, p = 0.004) and total motor score (r = 0.32, p = 0.002), suggesting its potential as a biomarker for HD progression and therapeutic response[29]. A study investigating tau pathology in HD explored its neuropathological, genetic, and clinical aspects. The researchers identified extensive aggregates of hyperphosphorylated tau in HD brains, some of which co-localized with mHTT, including in cases of young-onset HD. Neurotoxic tau oligomers were also detected, further implicating tau pathology in disease progression. Moreover, genetic variations in the MAPT gene (encoding tau) were linked to cognitive decline in HD patients, highlighting a potential role for tau in modulating HD severity[30]. These findings suggest a significant role for tau in HD pathology and clinical progression, highlighting potential new therapeutic targets for the disease. Another study has demonstrated that HD pathology is associated with increased presence and aggregation of Tau, α-synuclein (α-Syn), and TAR DNA-binding protein 43 (TDP-43) in the brain, alongside abnormal phosphorylation of Tau and altered splicing patterns of Tau isoforms[31]. Further research is warranted to elucidate the clinical implications of these proteinopathies in HD progression.
3.2.2. 24 (S) hydroxycholesterol (24OHC)
24 (S) hydroxycholesterol (24OHC), a significant cholesterol metabolite in the brain, is notably reduced in the plasma of individuals with HD. This decrease correlates with a reduction in caudate volume[32], suggesting that 24OHC may serve as an indicator of progressive neuronal loss in HD[33]. Researchers have analyzed 24OHC levels in plasma from gene-expanded individuals and found notable differences across patient groups. Specifically, 24OHC concentrations decreased with advancing disease stages, with this decline appearing more substantial than changes in cognitive and motor function or neuroimaging alterations. Additionally, reduced levels of cholesterol precursors, such as lanosterol and lathosterol, as well as the bile acid precursor 27-hydroxycholesterol, were observed in HD patients[34]. To validate this metabolite as a reliable biomarker, long-term monitoring of patients is suggested to track changes in metabolic markers throughout HD progression[33].
3.2.3. Neurophysiological Biomarkers (EEG and fMRI)
Electroencephalography (EEG) is a noninvasive method that measures brain oscillatory activity changes, reflecting synaptic dysfunction and progressive neurodegeneration in Huntington's disease (HD). There has documented changes in EEG power among patients with manifest HD, notably an increase in delta power and a decrease in alpha power[35,36,37]. In individuals with premanifest HD (PreHD) and early manifest HD (EMHD), there has been a reported reduction in power within the low alpha band and at the theta-alpha border[35,38,39]. Moreover, research has shown that the number of CAG repeats in the HTT gene correlates with EEG changes and cognitive decline in both preclinical and early manifest HD patients[38,40,41]. A recent cross-sectional observational study aimed to identify neurophysiological alterations in preclinical and early manifest HD by analyzing EEG and fMRI resting-state functional connectivity (rsFC) and examining their interrelationships. This study demonstrated that EEG and fMRI can effectively reveal neurophysiological changes associated with HD. Specifically, it found decreased power in certain EEG frequency ranges in both preclinical (preHD) and early manifest (EMHD) stages compared to healthy controls. These EEG changes were linked with disrupted functional connectivity in fMRI analyses, particularly within frontal, putamen-cortical, and cortico-cerebellar networks. The findings suggest that integrating EEG and fMRI provides valuable insights into HD, with decreased alpha and theta-alpha power correlating with increased connectivity in large-scale brain networks, which may contribute to cognitive decline[42]. While these findings are promising, the utility of EEG as a reliable biomarker for HD is still under investigation, highlighting the need for further research to establish its effectiveness and applicability in clinical settings.
3.2.4. Muscle Atrophy related biomarkers
Transcriptional abnormalities linked to HD extend beyond the central nervous system (CNS) and can also be observed in non-neuronal tissues, including muscles[43,44]. The pathophysiology of HD in skeletal muscle involves high expression levels of mHTT, leading to mitochondrial dysfunction, oxidative stress, inflammation, and impaired protein quality control systems. Addressing these mechanisms through enhanced macroautophagy and proteasome-mediated protein degradation represents a promising therapeutic strategy[45]. A study investigated transcriptional changes related to muscle atrophy in HD and identified a set of biomarkers linked to altered purine metabolism and energy homeostasis in skeletal muscles. Findings included significant upregulation of genes associated with muscle disuse and energy imbalance, such as Ampd3, Prkaa1, and Pdk4, as well as changes in purine metabolism-related genes like Pnp and Xdm. These biomarkers may be useful for tracking disease progression and developing targeted therapies, particularly given their potential applicability to both HD and related conditions like muscle cachexia in cancer models[46]. More recently, a study utilized the YAC128 murine model to investigate mitochondrial dysfunction and bioenergetic failure in the context of Huntington's disease (HD) and aging. The study revealed an increased mitochondrial volume fraction in the axons and dendrites of older mice, along with age- and genotype-dependent variations in mitochondrial DNA copy number (mtDNAcn) in the striatum and skeletal muscle, but not in the cortex. Additionally, mtDNA deletions were observed in striatal and skeletal muscle tissues across various ages and genotypes. These findings underscore the role of mitochondrial dynamics in HD pathogenesis and suggest that mtDNAcn in skeletal muscle may serve as a potential biomarker for further exploration in human HD [47]. Further research is necessary to clarify whether muscle dysfunction in HD is primary or secondary to nerve degeneration, and to elucidate the differential organ susceptibility to mutant Huntingtin's effects across various tissues.
Table 2 provides a comprehensive summary of the different types of biomarkers investigated in Huntington's disease research, including their sources, clinical utility, advantages, and limitations. This detailed overview complements the emerging biomarker subsections by offering a broader perspective on the diverse biomarker landscape in HD, supporting their potential roles in diagnosis, progression monitoring, and therapeutic evaluation. Additionally, Figure 7 highlights how these biomarkers function across various stages of HD, emphasizing their significance in understanding and managing the disease.
4. Discussion
Huntington’s disease poses a serious threat to individuals globally, imposing significant economic and social burdens on nations due to its high management costs and complex symptoms. This highlights the critical need for research into HD to improve understanding and treatment of the disease. The development of fast and reliable methods for diagnosis, progression monitoring and therapeutic success evaluation of neurodegenerative disorders is a key research domain. Biomarkers serving as indicators of disease encompass genetic markers, neuroimaging markers, and biofluid markers, such as proteins, immunological markers, and microRNAs found predominantly in blood and cerebrospinal fluid (CSF) [49]. Early detection of NDs in general and of HD in particular is crucial for the evaluation of targeted medications that may delay the disease progression [65]. Diagnosis of HD in patients with family history is usually straight forward, however around 8% of the patients do not have affected family member, which require quick and reliable diagnostic tools [5]. Biomarkers extend beyond diagnostic purposes; they are also designed for prognostic, predictive, and staging applications. Throughout the disease's progression, biomarkers can detect its presence, assess severity, monitor its advancement, and evaluate treatment responses[65].
While CSF has traditionally been considered the most reliable biofluid for biomarker collection, the difficulty in its collection methods has led researchers to develop blood-based biomarkers, which offer a faster and easier evaluation process. Although blood-based biomarkers have shown promise, further research is needed to refine their accuracy and understand the factors affecting their levels before they can be routinely used in clinical laboratories[49]. This study represents the first bibliometric analysis linking biomarkers with Huntington's disease research, offering a comprehensive overview of research trends over the past decade.
The analysis identified 730 relevant articles published between 2014 and 2024, a substantial number that reflects significant research activity in this area. The substantial number of publications highlights a robust and ongoing commitment to advancing biomarker research, which is vital for improving the diagnosis and management of Huntington's disease. Early detection of neurodegenerative diseases like Huntington's is essential for evaluating therapies that might slow disease progression, with biomarkers playing key roles in diagnosis, prognosis, staging, and monitoring treatment responses[65]. The average of seven co-authors per article highlights the collaborative nature of research in this field. Such collaboration is essential, as it brings together diverse perspectives and expertise, which are crucial for addressing the complex challenges associated with biomarker discovery and validation. The increase in publication numbers, particularly the surge observed in recent years, further highlights the growing recognition of the importance of biomarkers in HD research[91]. This trend suggests that the scientific community is increasingly focused on developing biomarkers that can lead to more targeted therapies and better patient outcomes.
However, the analysis also reveals a significant gap in international collaboration, particularly with countries in Africa. The lack of research contributions from this region is concerning, as it limits the global understanding of HD. While it is possible that the lower prevalence of HD in Africa contributes to this disparity[3], it indicates the need for more inclusive research efforts that involve diverse populations. Greater collaboration with researchers from underrepresented regions could provide valuable insights and enhance the generalizability of biomarker findings.
Future directions in HD biomarker research involve several key areas. First, conducting longitudinal studies is crucial to track disease progression and treatment outcomes over extended periods, ensuring the reliability and clinical relevance of identified biomarkers[23]. Second, extensive research efforts should be focused on exploring emerging biomarkers aimed at deepening our understanding of disease mechanisms and enhancing diagnostic accuracy. Third, collaborative efforts among researchers and clinicians are essential to accelerate biomarker discovery and validation processes. By standardizing protocols and ensuring reproducibility across diverse study populations and settings, these initiatives aim to advance personalized approaches to effectively managing Huntington's disease.
Keyword analysis in this study revealed that terms associated with other neurodegenerative diseases, such as Parkinson's disease (PD), Alzheimer's disease (AD), and amyotrophic lateral sclerosis (ALS), were less frequently used in the context of HD research. This is somewhat surprising, given that HD shares several pathological and clinical features with these disorders[92]. The relatively lower emphasis on these related conditions suggests an area of potential growth in HD research. Exploring the relationships between HD and other neurodegenerative diseases could yield important insights, particularly in identifying common biomarkers or therapeutic targets that span multiple disorders.
6. Study Limitations
This bibliometric analysis has some limitations that should be acknowledged. A primary limitation is the restriction to articles sourced solely from the PubMed database, which may narrow the scope of the findings. Expanding the search to include other databases, such as Scopus or Web of Science, could provide a more comprehensive and nuanced view of the research landscape. Another limitation is the exclusion of non-English publications, which may have led to the omission of valuable insights from studies conducted in other languages and cultural contexts. Additionally, the reliance on bibliometric data does not account for the quality or impact of the publications analyzed, which could influence the interpretation of the findings. Lastly, while the analysis provides a quantitative overview, it does not delve deeply into the specific methodologies or findings of individual studies, which may limit its applicability to guiding specific research directions.
7. Conclusion
In conclusion, this study offers a comprehensive review and bibliometric analysis of HD biomarker research, highlighting the substantial number of publications and the evident collaboration among researchers in this field. However, the lack of representation from certain geographical regions and the limited exploration of connections with other neurodegenerative diseases reveal important gaps that warrant future attention. Addressing these disparities can enhance the global understanding of HD and improve outcomes for HD patients.
Despite advancements, identifying reliable biomarkers for HD remains a significant challenge due to HD heterogeneous presentation, encompassing diverse and variable motor, cognitive, and psychiatric symptoms. Longitudinal observational studies have provided valuable insights into disease progression through imaging techniques and clinical tests, identifying key neurological, behavioral, motor, and cognitive alterations [93,94,95,96]. However, these efforts have yet to yield consistent and dependable HD biomarkers.
Huntington protein involvement in multiple biological pathways—including protein aggregation, neuroinflammation, and mitochondrial dysfunction—complicates biomarker identification. Moreover, factors beyond the primary HTT gene mutations, such as genetic modifiers and environmental influences, add more variability to disease onset, progression, and presentation, further complicating the identification of reliable biomarkers. Therefore, HD presents a multifaceted pathology, underscoring the challenge of finding a single biomarker that accurately reflects the disease process and necessitating the use of a panel of HD biomarkers [23].
These complexities are exacerbated by difficulties in recruiting and retaining patients for long-term studies. Such studies are critical for validating potential biomarkers and ensuring their clinical relevance. Moving forward, focused efforts on addressing these challenges through collaborative research, innovative methodologies, and inclusive study designs will be essential for advancing HD biomarker discovery and improving clinical outcomes.
Author Contributions
Conceptualization, S.A. and A.A.S.; investigation, All authors; resources, A.A.S.; writing—original draft preparation, All authors; writing—review and editing, All Authors; Supervision, A.A.S and W. M. Y. M.; project administration, A.A.S.; funding acquisition, A.A.S. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not Applicable.
Informed Consent Statement
Not Applicable.
Data Availability Statement
Data are contained within the article.
Acknowledgments
The authors acknowledge the support and funding they received from the Qatar University Office of the Vice President for Graduate Studies and Research (office of VPRGS) and the Qatar University Biomedical Research Center (BRC).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Stoker, T.B.; Mason, S.L.; Greenland, J.C.; Holden, S.T.; Santini, H.; Barker, R.A. Huntington's disease: diagnosis and management. Pract Neurol 2022, 22, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Daldin, M.; Fodale, V.; Cariulo, C.; Azzollini, L.; Verani, M.; Martufi, P.; Spiezia, M.C.; Deguire, S.M.; Cherubini, M.; Macdonald, D.; et al. Polyglutamine expansion affects huntingtin conformation in multiple Huntington's disease models. Scientific reports 2017, 7, 5070. [Google Scholar] [CrossRef] [PubMed]
- Medina, A.; Mahjoub, Y.; Shaver, L.; Pringsheim, T. Prevalence and Incidence of Huntington's Disease: An Updated Systematic Review and Meta-Analysis. Movement disorders : official journal of the Movement Disorder Society 2022, 37, 2327–2335. [Google Scholar] [CrossRef] [PubMed]
- Papanna, B.; Lazzari, C.; Rabottini, M. Huntington's disease prevalence in Asia: a systematic review and meta-analysis. Riv Psichiatr 2024, 59, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Walker, F.O. Huntington's disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Rawlins, M.D.; Wexler, N.S.; Wexler, A.R.; Tabrizi, S.J.; Douglas, I.; Evans, S.J.; Smeeth, L. The Prevalence of Huntington's Disease. Neuroepidemiology 2016, 46, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Gillis, C.; Montenigro, P.; Nejati, M.; Maserejian, N. Estimating prevalence of early Alzheimer's disease in the United States, accounting for racial and ethnic diversity. Alzheimer's & Dementia 2023, 19, 1841–1848. [Google Scholar] [CrossRef]
- Rodríguez-Santana, I.; Mestre, T.; Squitieri, F.; Willock, R.; Arnesen, A.; Clarke, A.; D'Alessio, B.; Fisher, A.; Fuller, R.; Hamilton, J.L.; et al. Economic burden of Huntington disease in Europe and the USA: Results from the Huntington's Disease Burden of Illness study. European Journal of Neurology 2023, 30, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Wild, E.; Tabrizi, S. Premanifest and early Huntington's disease; Oxford University Press, 2014. [Google Scholar]
- Mehanna, R.; Jankovic, J. Systemic Symptoms in Huntington's Disease: A Comprehensive Review. Mov Disord Clin Pract 2024, 11, 453–464. [Google Scholar] [CrossRef]
- Ajitkumar; Jesus, D. Ajitkumar; Jesus, D. Huntington Disease. Availabe online: https://www.ncbi.nlm.nih.gov/books/NBK559166/.
- Roth, J. Clinical Symptomatology of Huntington’s Disease. In Pathology, Prevention and Therapeutics of Neurodegenerative Disease; Singh, S., Joshi, N., Eds.; Springer Singapore: Singapore, 2019; pp. 117–131. [Google Scholar] [CrossRef]
- Shafie, A.; Ashour, A.A.; Anwar, S.; Anjum, F.; Hassan, M.I. Exploring molecular mechanisms, therapeutic strategies, and clinical manifestations of Huntington’s disease. Archives of Pharmacal Research 2024. [CrossRef]
- Krause, A.; Anderson, D.G.; Ferreira-Correia, A.; Dawson, J.; Baine-Savanhu, F.; Li, P.P.; Margolis, R.L. Huntington disease-like 2: insight into neurodegeneration from an African disease. Nature Reviews Neurology 2024, 20, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Assaye, M.A.; Gizaw, S.T. Chaperone-Mediated Autophagy and Its Implications for Neurodegeneration and Cancer. International Journal of General Medicine 2022, 15, 5635–5649. [Google Scholar] [CrossRef] [PubMed]
- Jodeiri Farshbaf, M.; Ghaedi, K. Huntington’s Disease and Mitochondria. Neurotoxicity Research 2017, 32, 518–529. [Google Scholar] [CrossRef]
- Rook, M.E.; Southwell, A.L. Antisense Oligonucleotide Therapy: From Design to the Huntington Disease Clinic. BioDrugs 2022, 36, 105–119. [Google Scholar] [CrossRef] [PubMed]
- PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/ (accessed on 23 June 2024).
- Sampaio, C. Huntington disease – Update on ongoing therapeutic developments and a look toward the future. Parkinsonism & Related Disorders 2024, 122, 106049. [Google Scholar] [CrossRef]
- Tippett, L.J.; Waldvogel, H.J.; Snell, R.G.; Vonsattel, J.P.; Young, A.B.; Faull, R.L.M. The Complexity of Clinical Huntington's Disease: Developments in Molecular Genetics, Neuropathology and Neuroimaging Biomarkers. Adv Neurobiol 2017, 15, 129–161. [Google Scholar] [CrossRef]
- Identification of Genetic Factors that Modify Clinical Onset of Huntington's Disease. Cell 2015, 162, 516–526. [CrossRef] [PubMed]
- Martí-Martínez, S.; Valor, L.M. A Glimpse of Molecular Biomarkers in Huntington's Disease. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Cheng, Y.; Shang, H. The updated development of blood-based biomarkers for Huntington's disease. Journal of neurology 2023, 270, 2483–2503. [Google Scholar] [CrossRef]
- Shoulson, I.; Young, A.B. Milestones in huntington disease. Movement disorders : official journal of the Movement Disorder Society 2011, 26, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Jiang, Z. A bibliometrix-based visualization analysis of international studies on conversations of people with aphasia: Present and prospects. Heliyon 2023, 9, e16839. [Google Scholar] [CrossRef]
- Campra, M.; Riva, P.; Oricchio, G.; Brescia, V. Bibliometrix analysis of medical tourism. Health services management research 2022, 35, 172–188. [Google Scholar] [CrossRef]
- Lepinay, E.; Cicchetti, F. Tau: a biomarker of Huntington's disease. Molecular psychiatry 2023, 28, 4070–4083. [Google Scholar] [CrossRef]
- Masnata, M.; Salem, S.; de Rus Jacquet, A.; Anwer, M.; Cicchetti, F. Targeting Tau to Treat Clinical Features of Huntington's Disease. Front Neurol 2020, 11, 580732. [Google Scholar] [CrossRef]
- Rodrigues, F.B.; Byrne, L.; McColgan, P.; Robertson, N.; Tabrizi, S.J.; Leavitt, B.R.; Zetterberg, H.; Wild, E.J. Cerebrospinal fluid total tau concentration predicts clinical phenotype in Huntington's disease. Journal of neurochemistry 2016, 139, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Vuono, R.; Winder-Rhodes, S.; de Silva, R.; Cisbani, G.; Drouin-Ouellet, J.; Spillantini, M.G.; Cicchetti, F.; Barker, R.A. The role of tau in the pathological process and clinical expression of Huntington's disease. Brain : a journal of neurology 2015, 138, 1907–1918. [Google Scholar] [CrossRef]
- St-Amour, I.; Turgeon, A.; Goupil, C.; Planel, E.; Hébert, S.S. Co-occurrence of mixed proteinopathies in late-stage Huntington's disease. Acta neuropathologica 2018, 135, 249–265. [Google Scholar] [CrossRef] [PubMed]
- Leoni, V.; Mariotti, C.; Nanetti, L.; Salvatore, E.; Squitieri, F.; Bentivoglio, A.R.; Bandettini di Poggio, M.; Piacentini, S.; Monza, D.; Valenza, M.; et al. Whole body cholesterol metabolism is impaired in Huntington's disease. Neuroscience letters 2011, 494, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Silajdžić, E.; Björkqvist, M. A Critical Evaluation of Wet Biomarkers for Huntington's Disease: Current Status and Ways Forward. Journal of Huntington's disease 2018, 7, 109–135. [Google Scholar] [CrossRef] [PubMed]
- Leoni, V.; Long, J.D.; Mills, J.A.; Di Donato, S.; Paulsen, J.S. Plasma 24S-hydroxycholesterol correlation with markers of Huntington disease progression. Neurobiol Dis 2013, 55, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Leuchter, M.K.; Donzis, E.J.; Cepeda, C.; Hunter, A.M.; Estrada-Sánchez, A.M.; Cook, I.A.; Levine, M.S.; Leuchter, A.F. Quantitative Electroencephalographic Biomarkers in Preclinical and Human Studies of Huntington's Disease: Are They Fit-for-Purpose for Treatment Development? Front Neurol 2017, 8, 91. [Google Scholar] [CrossRef] [PubMed]
- Odish, O.F.F.; Johnsen, K.; van Someren, P.; Roos, R.A.C.; van Dijk, J.G. EEG may serve as a biomarker in Huntington's disease using machine learning automatic classification. Scientific reports 2018, 8, 16090. [Google Scholar] [CrossRef] [PubMed]
- Delussi, M.; Nazzaro, V.; Ricci, K.; de Tommaso, M. EEG Functional Connectivity and Cognitive Variables in Premanifest and Manifest Huntington's Disease: EEG Low-Resolution Brain Electromagnetic Tomography (LORETA) Study. Frontiers in physiology 2020, 11, 612325. [Google Scholar] [CrossRef] [PubMed]
- Ponomareva, N.; Klyushnikov, S.; Abramycheva, N.; Malina, D.; Scheglova, N.; Fokin, V.; Ivanova-Smolenskaia, I.; Illarioshkin, S. Alpha-theta border EEG abnormalities in preclinical Huntington's disease. Journal of the neurological sciences 2014, 344, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Lazar, A.S.; Panin, F.; Goodman, A.O.; Lazic, S.E.; Lazar, Z.I.; Mason, S.L.; Rogers, L.; Murgatroyd, P.R.; Watson, L.P.; Singh, P.; et al. Sleep deficits but no metabolic deficits in premanifest Huntington's disease. Annals of neurology 2015, 78, 630–648. [Google Scholar] [CrossRef]
- Piano, C.; Mazzucchi, E.; Bentivoglio, A.R.; Losurdo, A.; Calandra Buonaura, G.; Imperatori, C.; Cortelli, P.; Della Marca, G. Wake and Sleep EEG in Patients With Huntington Disease: An eLORETA Study and Review of the Literature. Clinical EEG and neuroscience 2017, 48, 60–71. [Google Scholar] [CrossRef]
- Painold, A.; Anderer, P.; Holl, A.K.; Letmaier, M.; Saletu-Zyhlarz, G.M.; Saletu, B.; Bonelli, R.M. EEG low-resolution brain electromagnetic tomography (LORETA) in Huntington's disease. Journal of neurology 2011, 258, 840–854. [Google Scholar] [CrossRef] [PubMed]
- Ponomareva, N.V.; Klyushnikov, S.A.; Abramycheva, N.; Konovalov, R.N.; Krotenkova, M.; Kolesnikova, E.; Malina, D.; Urazgildeeva, G.; Kanavets, E.; Mitrofanov, A.; et al. Neurophysiological hallmarks of Huntington's disease progression: an EEG and fMRI connectivity study. Frontiers in aging neuroscience 2023, 15, 1270226. [Google Scholar] [CrossRef]
- Strand, A.D.; Aragaki, A.K.; Shaw, D.; Bird, T.; Holton, J.; Turner, C.; Tapscott, S.J.; Tabrizi, S.J.; Schapira, A.H.; Kooperberg, C.; et al. Gene expression in Huntington's disease skeletal muscle: a potential biomarker. Human molecular genetics 2005, 14, 1863–1876. [Google Scholar] [CrossRef] [PubMed]
- Luthi-Carter, R.; Hanson, S.A.; Strand, A.D.; Bergstrom, D.A.; Chun, W.; Peters, N.L.; Woods, A.M.; Chan, E.Y.; Kooperberg, C.; Krainc, D.; et al. Dysregulation of gene expression in the R6/2 model of polyglutamine disease: parallel changes in muscle and brain. Human molecular genetics 2002, 11, 1911–1926. [Google Scholar] [CrossRef] [PubMed]
- Shacham, T.; Sharma, N.; Lederkremer, G.Z. Protein Misfolding and ER Stress in Huntington's Disease. Frontiers in molecular biosciences 2019, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Bozzi, M.; Sciandra, F. Molecular Mechanisms Underlying Muscle Wasting in Huntington's Disease. International journal of molecular sciences 2020, 21. [Google Scholar] [CrossRef]
- Bečanović, K.; Asghar, M.; Gadawska, I.; Sachdeva, S.; Walker, D.; Lazarowski, E.R.; Franciosi, S.; Park, K.H.J.; Côté, H.C.F.; Leavitt, B.R. Age-related mitochondrial alterations in brain and skeletal muscle of the YAC128 model of Huntington disease. NPJ aging and mechanisms of disease 2021, 7, 26. [Google Scholar] [CrossRef]
- Eshraghi, M.; Karunadharma, P.P.; Blin, J.; Shahani, N.; Ricci, E.P.; Michel, A.; Urban, N.T.; Galli, N.; Sharma, M.; Ramírez-Jarquín, U.N.; et al. Mutant Huntingtin stalls ribosomes and represses protein synthesis in a cellular model of Huntington disease. Nature communications 2021, 12, 1461. [Google Scholar] [CrossRef]
- Agnello, L.; Gambino, C.M.; Ciaccio, A.M.; Masucci, A.; Vassallo, R.; Tamburello, M.; Scazzone, C.; Lo Sasso, B.; Ciaccio, M. Molecular Biomarkers of Neurodegenerative Disorders: A Practical Guide to Their Appropriate Use and Interpretation in Clinical Practice. Int J Mol Sci 2024, 25. [Google Scholar] [CrossRef] [PubMed]
- Craufurd, D.; MacLeod, R.; Frontali, M.; Quarrell, O.; Bijlsma, E.K.; Davis, M.; Hjermind, L.E.; Lahiri, N.; Mandich, P.; Martinez, A.; et al. Diagnostic genetic testing for Huntington's disease. Practical neurology 2015, 15, 80–84. [Google Scholar] [CrossRef]
- Byrne, L.M.; Rodrigues, F.B.; Johnson, E.B.; Wijeratne, P.A.; De Vita, E.; Alexander, D.C.; Palermo, G.; Czech, C.; Schobel, S.; Scahill, R.I.; et al. Evaluation of mutant huntingtin and neurofilament proteins as potential markers in Huntington's disease. Science translational medicine 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Wild, E.J.; Boggio, R.; Langbehn, D.; Robertson, N.; Haider, S.; Miller, J.R.; Zetterberg, H.; Leavitt, B.R.; Kuhn, R.; Tabrizi, S.J.; et al. Quantification of mutant huntingtin protein in cerebrospinal fluid from Huntington's disease patients. The Journal of clinical investigation 2015, 125, 1979–1986. [Google Scholar] [CrossRef] [PubMed]
- Southwell, A.L.; Smith, S.E.; Davis, T.R.; Caron, N.S.; Villanueva, E.B.; Xie, Y.; Collins, J.A.; Ye, M.L.; Sturrock, A.; Leavitt, B.R.; et al. Ultrasensitive measurement of huntingtin protein in cerebrospinal fluid demonstrates increase with Huntington disease stage and decrease following brain huntingtin suppression. Scientific reports 2015, 5, 12166. [Google Scholar] [CrossRef] [PubMed]
- Hensman Moss, D.J.; Robertson, N.; Farmer, R.; Scahill, R.I.; Haider, S.; Tessari, M.A.; Flynn, G.; Fischer, D.F.; Wild, E.J.; Macdonald, D.; et al. Quantification of huntingtin protein species in Huntington's disease patient leukocytes using optimised electrochemiluminescence immunoassays. PloS one 2017, 12, e0189891. [Google Scholar] [CrossRef] [PubMed]
- Kuhle, J.; Barro, C.; Andreasson, U.; Derfuss, T.; Lindberg, R.; Sandelius, Å.; Liman, V.; Norgren, N.; Blennow, K.; Zetterberg, H. Comparison of three analytical platforms for quantification of the neurofilament light chain in blood samples: ELISA, electrochemiluminescence immunoassay and Simoa. Clinical chemistry and laboratory medicine 2016, 54, 1655–1661. [Google Scholar] [CrossRef]
- Gaetani, L.; Blennow, K.; Calabresi, P.; Di Filippo, M.; Parnetti, L.; Zetterberg, H. Neurofilament light chain as a biomarker in neurological disorders. Journal of neurology, neurosurgery, and psychiatry 2019, 90, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, F.B.; Byrne, L.M.; Wild, E.J. Biofluid Biomarkers in Huntington's Disease. Methods in molecular biology (Clifton, N.J.) 2018, 1780, 329–396. [Google Scholar] [CrossRef] [PubMed]
- Schumacher-Schuh, A.; Bieger, A.; Borelli, W.V.; Portley, M.K.; Awad, P.S.; Bandres-Ciga, S. Advances in Proteomic and Metabolomic Profiling of Neurodegenerative Diseases. Front Neurol 2021, 12, 792227. [Google Scholar] [CrossRef]
- Byrne, L.M.; Rodrigues, F.B.; Blennow, K.; Durr, A.; Leavitt, B.R.; Roos, R.A.C.; Scahill, R.I.; Tabrizi, S.J.; Zetterberg, H.; Langbehn, D.; et al. Neurofilament light protein in blood as a potential biomarker of neurodegeneration in Huntington's disease: a retrospective cohort analysis. The Lancet. Neurology 2017, 16, 601–609. [Google Scholar] [CrossRef]
- Rodrigues, F.B.; Byrne, L.M.; Tortelli, R.; Johnson, E.B.; Wijeratne, P.A.; Arridge, M.; De Vita, E.; Ghazaleh, N.; Houghton, R.; Furby, H.; et al. Mutant huntingtin and neurofilament light have distinct longitudinal dynamics in Huntington's disease. Science translational medicine 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Zeun, P.; Scahill, R.I.; Tabrizi, S.J.; Wild, E.J. Fluid and imaging biomarkers for Huntington's disease. Molecular and cellular neurosciences 2019, 97, 67–80. [Google Scholar] [CrossRef]
- Morena, E.; Romano, C.; Marconi, M.; Diamant, S.; Buscarinu, M.C.; Bellucci, G.; Romano, S.; Scarabino, D.; Salvetti, M.; Ristori, G. Peripheral Biomarkers in Manifest and Premanifest Huntington's Disease. International journal of molecular sciences 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Walter, N.G. Are non-protein coding RNAs junk or treasure? BioEssays 2024, 46, 2300201. [Google Scholar] [CrossRef]
- Juźwik, C.A.; S, S.D.; Zhang, Y.; Paradis-Isler, N.; Sylvester, A.; Amar-Zifkin, A.; Douglas, C.; Morquette, B.; Moore, C.S.; Fournier, A.E. microRNA dysregulation in neurodegenerative diseases: A systematic review. Progress in neurobiology 2019, 182, 101664. [Google Scholar] [CrossRef]
- Azam, H.M.H.; Rößling, R.I.; Geithe, C.; Khan, M.M.; Dinter, F.; Hanack, K.; Prüß, H.; Husse, B.; Roggenbuck, D.; Schierack, P.; et al. MicroRNA biomarkers as next-generation diagnostic tools for neurodegenerative diseases: a comprehensive review. Frontiers in molecular neuroscience 2024, 17, 1386735. [Google Scholar] [CrossRef]
- Reed, E.R.; Latourelle, J.C.; Bockholt, J.H.; Bregu, J.; Smock, J.; Paulsen, J.S.; Myers, R.H. MicroRNAs in CSF as prodromal biomarkers for Huntington disease in the PREDICT-HD study. Neurology 2018, 90, e264–e272. [Google Scholar] [CrossRef]
- Langfelder, P.; Gao, F.; Wang, N.; Howland, D.; Kwak, S.; Vogt, T.F.; Aaronson, J.S.; Rosinski, J.; Coppola, G.; Horvath, S.; et al. MicroRNA signatures of endogenous Huntingtin CAG repeat expansion in mice. PloS one 2018, 13, e0190550. [Google Scholar] [CrossRef] [PubMed]
- Palaiogeorgou, A.M.; Papakonstantinou, E.; Golfinopoulou, R.; Sigala, M.; Mitsis, T.; Papageorgiou, L.; Diakou, I.; Pierouli, K.; Dragoumani, K.; Spandidos, D.A.; et al. Recent approaches on Huntington's disease (Review). Biomedical reports 2023, 18, 5. [Google Scholar] [CrossRef] [PubMed]
- Murmann, A.E.; Patel, M.; Jeong, S.Y.; Bartom, E.T.; Jennifer Morton, A.; Peter, M.E. The length of uninterrupted CAG repeats in stem regions of repeat disease associated hairpins determines the amount of short CAG oligonucleotides that are toxic to cells through RNA interference. Cell death & disease 2022, 13, 1078. [Google Scholar] [CrossRef]
- CAG Repeat Not Polyglutamine Length Determines Timing of Huntington's Disease Onset. Cell 2019, 178, 887–900e814. [CrossRef] [PubMed]
- Wright, G.E.B.; Collins, J.A.; Kay, C.; McDonald, C.; Dolzhenko, E.; Xia, Q.; Bečanović, K.; Drögemöller, B.I.; Semaka, A.; Nguyen, C.M.; et al. Length of Uninterrupted CAG, Independent of Polyglutamine Size, Results in Increased Somatic Instability, Hastening Onset of Huntington Disease. American journal of human genetics 2019, 104, 1116–1126. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Richman, J.; Langfelder, P.; Wang, N.; Zhang, S.; Bañez-Coronel, M.; Wang, H.B.; Yang, L.; Ramanathan, L.; Deng, L.; et al. Uninterrupted CAG repeat drives striatum-selective transcriptionopathy and nuclear pathogenesis in human Huntingtin BAC mice. Neuron 2022, 110, 1173–1192e1177. [Google Scholar] [CrossRef]
- Rué, L.; Bañez-Coronel, M.; Creus-Muncunill, J.; Giralt, A.; Alcalá-Vida, R.; Mentxaka, G.; Kagerbauer, B.; Zomeño-Abellán, M.T.; Aranda, Z.; Venturi, V.; et al. Targeting CAG repeat RNAs reduces Huntington's disease phenotype independently of huntingtin levels. The Journal of clinical investigation 2016, 126, 4319–4330. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, M.W.; Kennedy, C.J.; Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. Current and Possible Future Therapeutic Options for Huntington's Disease. Journal of central nervous system disease 2022, 14, 11795735221092517. [Google Scholar] [CrossRef] [PubMed]
- Dalrymple, A.; Wild, E.J.; Joubert, R.; Sathasivam, K.; Björkqvist, M.; Petersén, A.; Jackson, G.S.; Isaacs, J.D.; Kristiansen, M.; Bates, G.P.; et al. Proteomic profiling of plasma in Huntington's disease reveals neuroinflammatory activation and biomarker candidates. Journal of proteome research 2007, 6, 2833–2840. [Google Scholar] [CrossRef]
- Björkqvist, M.; Wild, E.J.; Thiele, J.; Silvestroni, A.; Andre, R.; Lahiri, N.; Raibon, E.; Lee, R.V.; Benn, C.L.; Soulet, D.; et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington's disease. The Journal of experimental medicine 2008, 205, 1869–1877. [Google Scholar] [CrossRef]
- Eide, S.; Misztal, M.; Feng, Z.P. Interleukin-6 as a marker of Huntington's disease progression: Systematic review and meta-analysis. Brain, behavior, & immunity - health 2023, 30, 100635. [Google Scholar] [CrossRef]
- Wild, E.; Magnusson, A.; Lahiri, N.; Krus, U.; Orth, M.; Tabrizi, S.J.; Björkqvist, M. Abnormal peripheral chemokine profile in Huntington's disease. PLoS currents 2011, 3, Rrn1231. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.; Li, X.; Luo, J.J.; Wang, H.; Yang, X.F. A Double-edged Sword: Uric Acid and Neurological Disorders. Brain disorders & therapy 2013, 2, 109. [Google Scholar] [CrossRef]
- Wen, M.; Zhou, B.; Chen, Y.H.; Ma, Z.L.; Gou, Y.; Zhang, C.L.; Yu, W.F.; Jiao, L. Serum uric acid levels in patients with Parkinson's disease: A meta-analysis. PloS one 2017, 12, e0173731. [Google Scholar] [CrossRef]
- Auinger, P.; Kieburtz, K.; McDermott, M.P. The relationship between uric acid levels and Huntington's disease progression. Movement disorders : official journal of the Movement Disorder Society 2010, 25, 224–228. [Google Scholar] [CrossRef]
- Corey-Bloom, J.; Haque, A.; Aboufadel, S.; Snell, C.; Fischer, R.S.; Granger, S.W.; Granger, D.A.; Thomas, E.A. Uric Acid as a Potential Peripheral Biomarker for Disease Features in Huntington's Patients. Frontiers in neuroscience 2020, 14, 73. [Google Scholar] [CrossRef]
- Coppen, E.M.; van der Grond, J.; Hafkemeijer, A.; Rombouts, S.A.; Roos, R.A. Early grey matter changes in structural covariance networks in Huntington's disease. NeuroImage. Clinical 2016, 12, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Király, A.; Kincses, Z.T.; Szabó, N.; Tóth, E.; Csete, G.; Faragó, P.; Vécsei, L. Gray matter atrophy in presymptomatic Huntington's patients. Ideggyogyaszati szemle 2016, 69, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Wilson, H.; Dervenoulas, G.; Politis, M.J.I.r.o.n. Structural magnetic resonance imaging in Huntington's disease. International review of neurobiology 2018, 142, 335–380. [Google Scholar]
- Reiner, A.; Dragatsis, I.; Dietrich, P. Genetics and neuropathology of Huntington's disease. International review of neurobiology 2011, 98, 325–372. [Google Scholar] [CrossRef]
- Young, A.B.; Penney, J.B.; Starosta-Rubinstein, S.; Markel, D.S.; Berent, S.; Giordani, B.; Ehrenkaufer, R.; Jewett, D.; Hichwa, R. PET scan investigations of Huntington's disease: cerebral metabolic correlates of neurological features and functional decline. Annals of neurology 1986, 20, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Herben-Dekker, M.; van Oostrom, J.C.; Roos, R.A.; Jurgens, C.K.; Witjes-Ané, M.N.; Kremer, H.P.; Leenders, K.L.; Spikman, J.M. Striatal metabolism and psychomotor speed as predictors of motor onset in Huntington's disease. Journal of neurology 2014, 261, 1387–1397. [Google Scholar] [CrossRef] [PubMed]
- Kuwert, T.; Lange, H.W.; Boecker, H.; Titz, H.; Herzog, H.; Aulich, A.; Wang, B.C.; Nayak, U.; Feinendegen, L.E. Striatal glucose consumption in chorea-free subjects at risk of Huntington's disease. Journal of neurology 1993, 241, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Antonini, A.; Leenders, K.L.; Spiegel, R.; Meier, D.; Vontobel, P.; Weigell-Weber, M.; Sanchez-Pernaute, R.; de Yébenez, J.G.; Boesiger, P.; Weindl, A.; et al. Striatal glucose metabolism and dopamine D2 receptor binding in asymptomatic gene carriers and patients with Huntington's disease. Brain : a journal of neurology 1996, 119 Pt 6, 2085–2095. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, C. Huntington disease - Update on ongoing therapeutic developments and a look toward the future. Parkinsonism Relat Disord 2024, 122, 106049. [Google Scholar] [CrossRef] [PubMed]
- Berth, S.H.; Lloyd, T.E. Disruption of axonal transport in neurodegeneration. The Journal of clinical investigation 2023, 133. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Scahill, R.I.; Durr, A.; Roos, R.A.; Leavitt, B.R.; Jones, R.; Landwehrmeyer, G.B.; Fox, N.C.; Johnson, H.; Hicks, S.L.; et al. Biological and clinical changes in premanifest and early stage Huntington's disease in the TRACK-HD study: the 12-month longitudinal analysis. The Lancet. Neurology 2011, 10, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, J.S.; Langbehn, D.R.; Stout, J.C.; Aylward, E.; Ross, C.A.; Nance, M.; Guttman, M.; Johnson, S.; MacDonald, M.; Beglinger, L.J.; et al. Detection of Huntington's disease decades before diagnosis: the Predict-HD study. Journal of neurology, neurosurgery, and psychiatry 2008, 79, 874–880. [Google Scholar] [CrossRef]
- Dorsey, E.R.; one, H.S.G.C.I.J.P. Characterization of a large group of individuals with Huntington disease and their relatives enrolled in the COHORT study. PloS one 2012, 7, e29522. [Google Scholar] [CrossRef]
- Orth, M.; European Huntington's Disease Network %J Journal of Neurology, N.; Psychiatry. Observing Huntington's disease: the European Huntington's disease network's REGISTRY. Journal of Neurology, Neurosurgery & Psychiatry 2011, 82, 1409–1412. [Google Scholar]
Figure 1.
Huntington’s disease is a hyperkinetic disease with a complex pathophysiology.
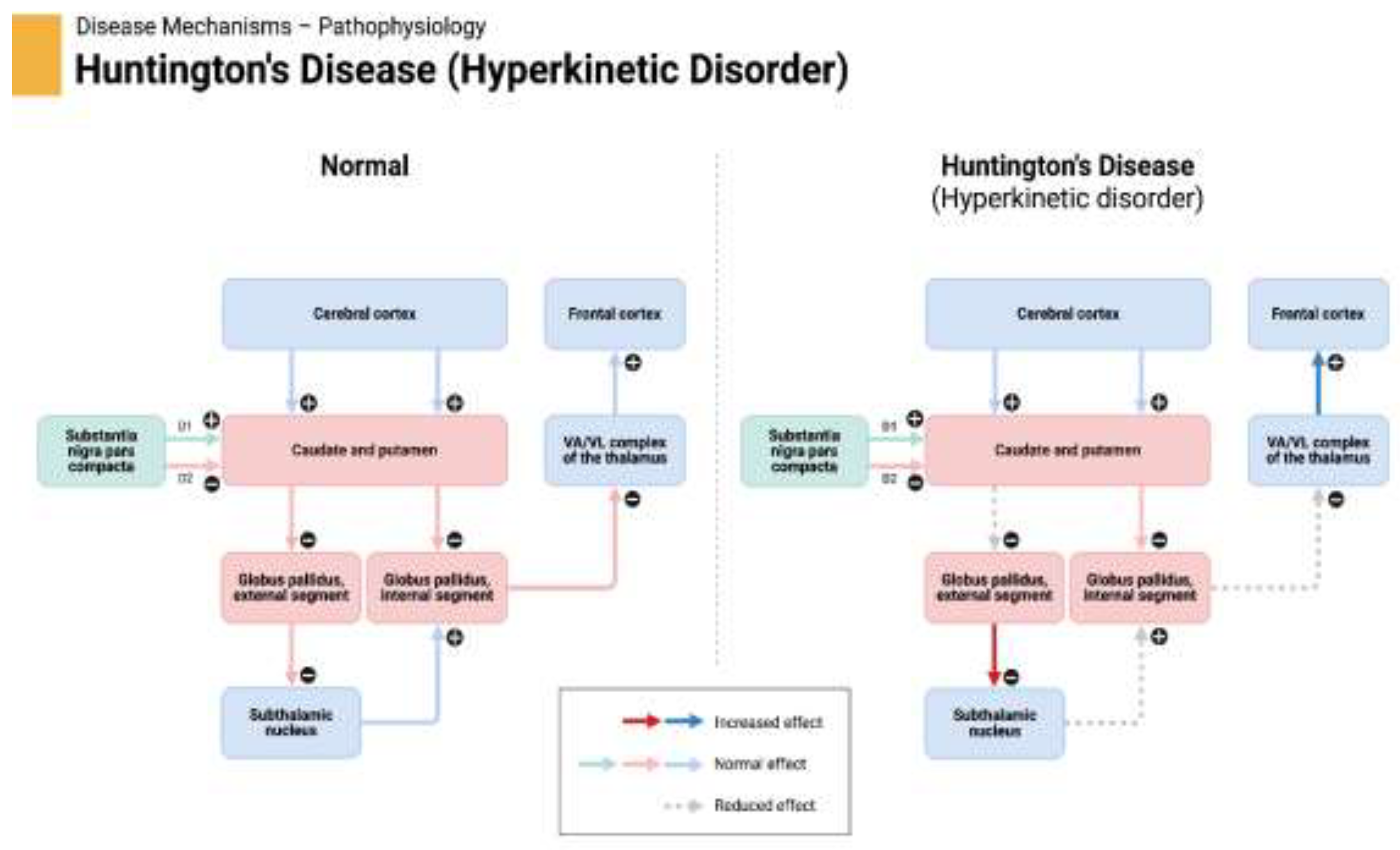
Figure 2.
The intricate molecular pathways and mechanisms underlying pathology of Huntington’s disease.
Figure 2.
The intricate molecular pathways and mechanisms underlying pathology of Huntington’s disease.
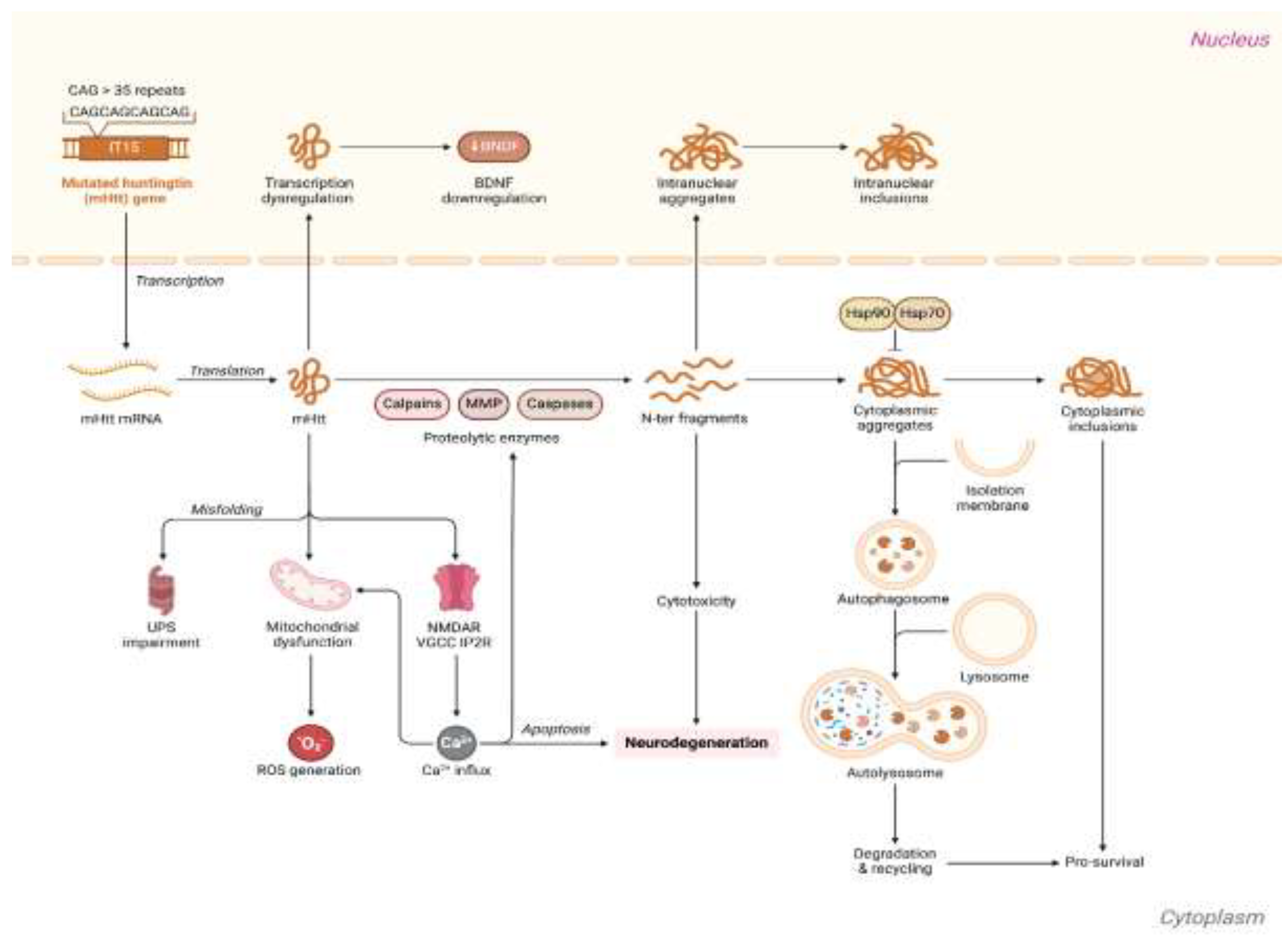
Figure 4.
International collaboration network.

Figure 5.
Most frequently used keywords.
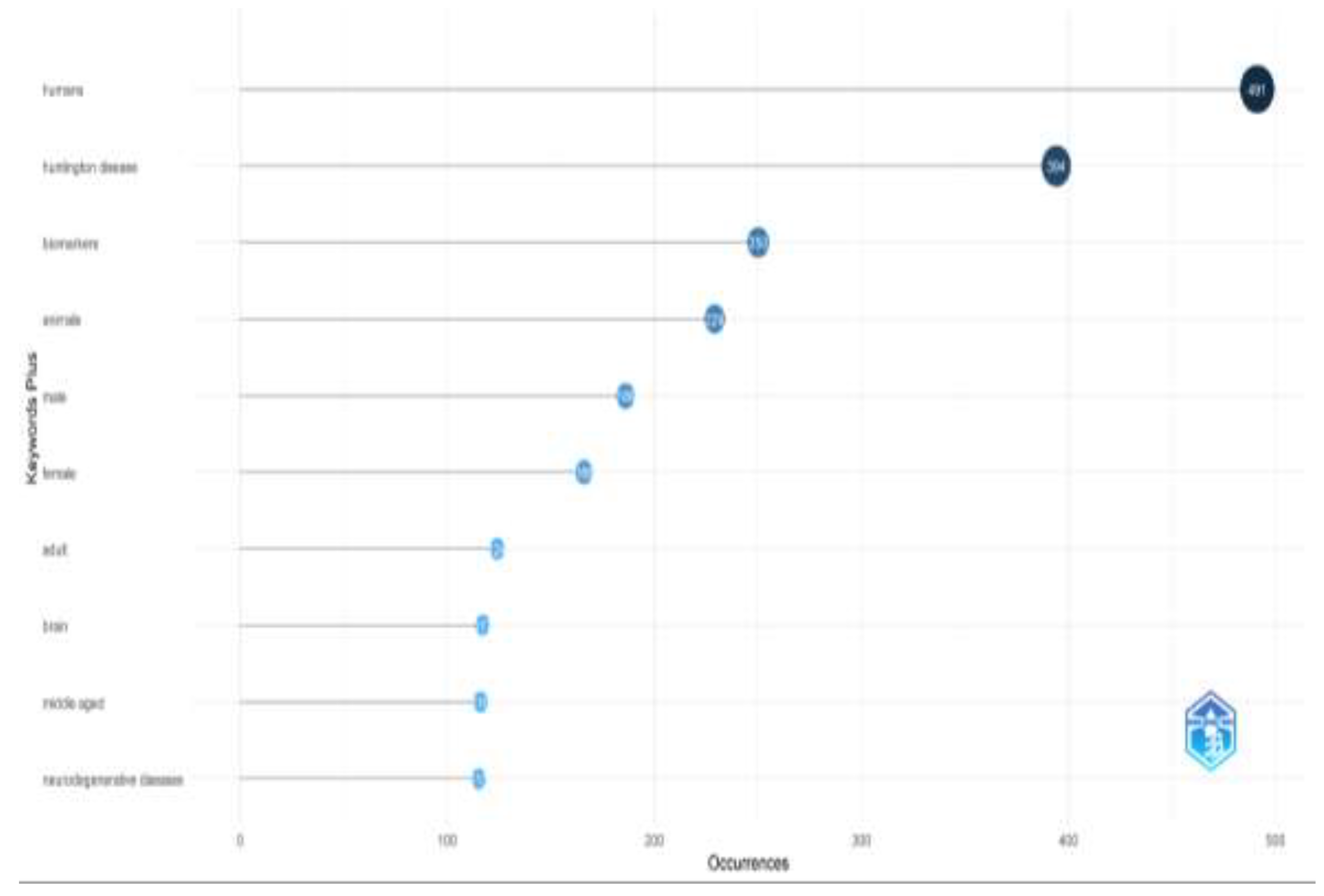
Figure 6.
Network of the 50 most frequent keywords.
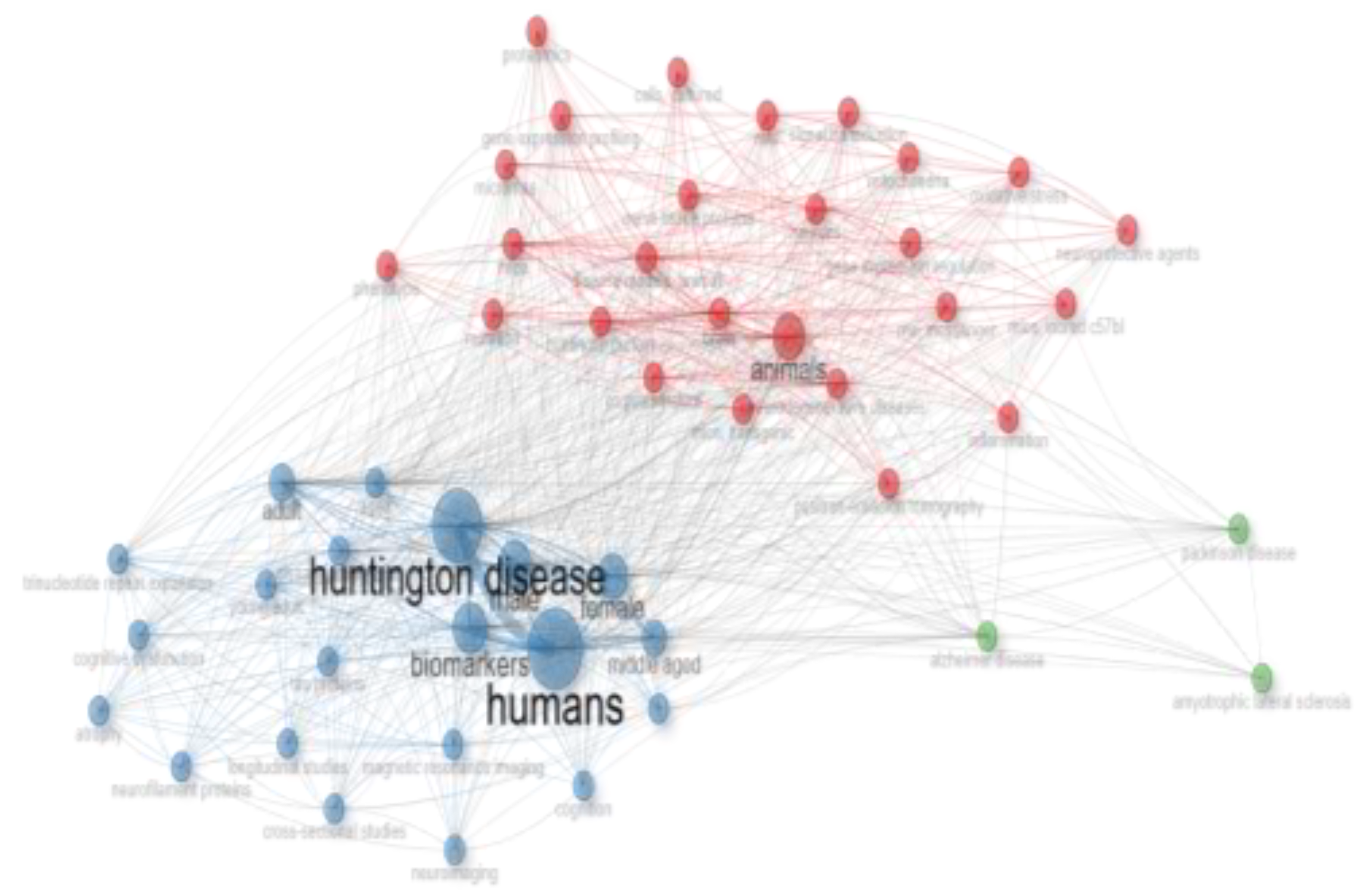
Figure 7.
The role of biomarkers in Huntington's Disease.


Table 1.
Search analysis summary.
| Description | Results |
|---|---|
| Timespan | 2013:2024 |
| Sources (Journals, Books, etc) | 319 |
| Documents | 730 |
| Annual Growth Rate % | 9.13% |
Table 2.
Summary of biomarker types in Huntington's Disease research and their characteristics.
| Biomarker Type | Examples | Source | Clinical Utility | Advantages | Limitations | Reference |
|---|---|---|---|---|---|---|
| Protein Biomarkers | Mutant huntingtin (mHTT) | Blood, CSF | Disease progression monitoring, therapeutic targeting |
Directly linked to HD pathology | Limited sensitivity, challenging quantification | [48,49,50], [51], [52,53], [54] |
| Neurofilament light chain (NfL) | Blood, CSF | Neurodegeneration marker, disease progression |
Reflects neuronal damage | May reflect other neurodegenerative diseases | [55,56,57], [58,59], [60,61], [62] | |
| RNA Biomarkers | microRNAs (miRNAs) | Blood, CSF, brain tissue | Gene expression regulation, disease state indicators |
Non-invasive, early detection potential | Requires complex analysis, variability in expression | [63,64,65], [66,67,68] |
| Mutant HTT mRNA | Blood, CSF | Direct measurement of disease-causing transcript |
Specific to HD | Limited by RNA stability and quantification challenges | [69], [70,71,72], [73,74] | |
| Inflammatory Biomarkers | Cytokines (e.g., IL-6, TNF-α) | Blood, CSF | Inflammation monitoring, disease progression | Indicates neuroinflammation, potential for early intervention |
Non-specific, can be elevated in other conditions | [75,76,77], [76,78] |
| Metabolic Biomarkers | Uric Acid | Blood, Saliva | Potential predictor of disease progression in HD | Associated with slower progression of functional decline in HD | Gender differences in UA levels may complicate interpretation | [79,80], [81,82]. |
| Imaging Biomarkers | MRI (brain volume, atrophy) |
Brain | Structural changes, disease progression | Non-invasive, visualizes brain changes | Expensive, requires specialized equipment | [83,84], [85,86] |
| PET (glucose metabolism, receptor binding) |
Brain | Functional brain imaging, neurotransmitter activity | Provides functional data, specific for HD | Expensive, limited availability, requires radioactive tracers | [87,88,89], [90] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



