
Submitted:
05 December 2024
Posted:
06 December 2024
You are already at the latest version
Abstract
Potassium permanganate (KMnO₄) is a commercially available antiseptic used in bovine intrauterine lavage to manage postpartum infections. Lipopolysaccharides (LPS) are well-studied for their ability to induce inflammation and oxidative stress. While KMnO₄ is known to cause significant irritation, oxidative stress, and toxicity in uterine tissues, its transcriptional impact and potential for inducing similar molecular damage as LPS have not been fully explored.
In this study, we induced oxidative stress in the uterine tissues of Sprague Dawley (SD) rats using KMnO₄ and compared the transcriptional profiles with those treated with LPS. We focused on the differential expression of long noncoding RNAs (lncRNAs) and messenger RNAs (mRNAs) related to oxidative stress, toxicity, and inflammation. RNA sequencing revealed 1125 differentially expressed mRNAs in the KMnO₄-treated group and 989 in the LPS-treated group. Additionally, 1649 lncRNAs were differentially expressed in the KMnO₄ group compared to 1383 in the LPS group.
Gene ontology (GO) and KEGG enrichment analyses showed that 78 pathways were significantly enriched in the KMnO₄ group, while 80 pathways were enriched in the LPS group, with 50 pathways shared between the two. This study offers critical insights into the transcriptional profiles associated with KMnO₄ exposure and its similarities to LPS-induced damage.
Keywords:
Potassium Permanganate
; Lipopolysaccharides
; Oxidative stress
; Inflammation
; Toxicity
; LncRNAs
; mRNAs
1. Introduction
Postpartum infections persist as a significant challenge in obstetrics, even though advancements in hygiene standards and enhanced diagnosis and treatment tools have led to a decrease in maternal mortality from infections in developed countries[1]. During parturition, the uterus can get contaminated with bacteria, leading to postpartum uterine infections such as pruritis, metritis, and endometritis[2]. The incidence of uterine infections exhibits significant variation among different research. Uterine infection is characterized by the attachment of infectious microorganisms to the lining of the uterus, their growth and spread within the tissue, and the production of bacterial toxins, all of which contribute to the development of uterine disease[3].
Previous research has demonstrated the recovery of bacteria from the uterine tissues of infected cows, mainly identified as Escherichia coli (E. coli), Staphylococcus aureus (S. aureus), and Trueperella pyogenes (T. pyogenes) [2,4].E. coli is the most prevalent gram-negative bacterium among all mentioned microorganisms, leading to uterus infection through LPS. It is also used to design many inflammatory models in animals and cells[5,6]
Potassium permanganate (KMnO4) is a potent oxidizing agent with strong antiseptic properties. It has been traditionally used for uterine lavage in cows, especially in cases of postpartum infections like metritis and endometritis [7]. While it is effective as a disinfectant, KMnO4 is also known to cause tissue irritation and oxidative stress, leading to ulcerative injury and apoptosis [8]. Despite these adverse effects, it remains in use in various regions due to its accessibility and antimicrobial properties [9,10]. However, the exact molecular damage caused by KMnO4 in uterine tissues has not been fully explored, particularly in comparison to the well-established inflammatory responses induced by LPS.
RNA molecules longer than 200 nucleotides in length that do not code for proteins but play critical roles in gene expression at both transcriptional and post-transcriptional levels are called long noncoding RNAs (lncRNAs) [11,12]. Research has demonstrated that lncRNAs regulate numerous biological processes, including inflammation, oxidative stress, cell differentiation, apoptosis, and reproductive functions [13] [14]. In the female reproductive system, specific lncRNAs have been shown to modulate critical processes, such as folliculogenesis, spermatogenesis, and embryo implantation [15,16]. For example, NEAT1 has been linked to corpus luteum formation and fertility [17], while lncRNA-H19 has been implicated in regulating endometriosis [18,19]. These findings highlight the crucial regulatory roles that lncRNAs play in uterine biology and disease.
Given that LPS and KMnO4 are both associated with oxidative stress and inflammatory responses in uterine tissues, understanding their transcriptional effects is essential. This study aims to compare the transcriptional profiles of rat uterine tissues exposed to KMnO4 and LPS, focusing on the expression of lncRNAs and mRNAs involved in oxidative stress, toxicity, and inflammation. For the first time, we hypothesize that KMnO4 induces molecular damage similar to that caused by LPS, with both agents activating overlapping pathways related to oxidative stress and inflammation. Additionally, the study seeks to identify potential interactions between lncRNAs and their target mRNAs, paving the way for future research into the regulatory roles of lncRNAs in uterine health.
2. Material and Methods
2.1. Animal Model
The Animal Experiment Centre of Huazhong Agricultural University (Wuhan, P.R. China) provided eighteen adult female Sprague Dawley (SD) rats weighing between 190 and 200g. The rats were housed under standard conditions with a 12-hour light/dark cycle, fed a standard diet, and provided with water ad libitum for an acclimatization period of 7 days. After acclimatization, the rats were randomly assigned to three groups (n=6 per group): a control group, a potassium permanganate (KMnO4) group, and a lipopolysaccharide (LPS) group. The KMnO4 used in the experiment was purchased locally, and a 0.05% solution was prepared by dissolving the powder in distilled water. For dosing, each rat's body weight was used to calculate the appropriate dose of KMnO4 and LPS. A dose of 10 mg/kg body weight was administered to the KMnO4 group. Given that the rats weighed approximately 200g, the dose per rat was calculated as follows:
Dose per rat = Body weight (kg) × Dose (mg/kg) = 0.2kg × 10mg/kg = 2mg
This 2mg of KMnO4 was diluted in distilled water and delivered as an intrauterine injection with a total volume of 20 µL. The LPS group received an intrauterine injection of LPS (50 µL of a 1 mg/mL solution), equivalent to 50 µg of LPS per rat. The control group received an intrauterine instillation of 50 µL of normal saline. Specimens from all groups were collected 48 hours after injection for histopathological analysis to confirm the induction of tissue damage and inflammation, validating the success of the experimental model.
2.2. Histopathology and Wet and Dry Weight (W/D)
Uterine tissue in the fixative solution was removed and rinsed with water for 30 min, dehydrated in a vacuum tissue dehydrator: 30%, 50% and 75% ethanol for 2h, 85%, 90% and 95% for 1.5h, Anhydrous ethanol was dehydrated twice for 1h each time, xylene was transparent twice for 20 min each, and molten paraffin was soaked twice for 1h each. Embedding, remove the uterine tissue from dehydration and put it into the embedding frame, pour in the molten paraffin, remove and trim the wax block after the wax solidifies, and slice it with a thickness of 5 μm. The slices are transparent, rehydrated, and washed with tap water. Hematoxylin & Eosin staining were observed under electron microscopy. In order to determine the wet and dry weight of rat uterine tissues, the tissues were surgically removed and then rinsed with PBS three times. Subsequently, they were placed in an incubator at a temperature of 80℃ for 24h. Following a 24h period, we determined the uterus tissues' wet-to-dry ratio (W/D).
2.3. Myeloperoxidase (MPO), Malondialdehyde (MDA), and Superoxide dismutase (SOD) Assays
Cut an appropriate amount of uterine tissue, weigh it, grind it under liquid nitrogen until crushed, and add isotonic sodium chloride solution according to mass: volume = 1:9 to obtain 10% tissue homogenate, centrifuge at 4°C, 13,000× g for 10 min, collect the supernatant, and measure the MPO, MDA content and activities of SOD (Nanjing Jiancheng Bioengineering Institute, China). The testing steps are strictly following the kit instructions.
2.4. RNA Extraction and cdna Library Construction
After retrieving the cryopreservation tube containing the uterine tissues, place it on ice for 5 min. Next, carefully excise approximately 30 mg of uterine tissue using surgical scissors. Subsequently, the excised tissue is placed into a centrifuge tube with a volume capacity of 2 mL. Add 1 mL of Trizol reagent to the tube containing the uterine tissue. The extraction of total RNA from the control, LPS and KMnO4-treated groups, with two samples per group, was performed using RNAiso Plus (Takara, Dalian, China) following the guidelines provided by the manufacturer. The utilization of total RNA serves as the initial material for the construction of a complementary DNA (cDNA) library. RNA purification, reverse transcription-based cDNA synthesis, adaptor ligation and end repair, PCR amplification, and library construction are the main processes in this procedure. The library is sequenced using the Illumina HiSeq 4000 platform, made in San Diego, California, by Illumina Inc., after quality inspection and preparation.
2.5. Quality Control Data of Sequencing
After the process of base recognition, the initial image file (BCL) received by sequencing was transformed into raw data in the FASTQ format. The initial dataset was evaluated comprehensively to determine its suitability for bioinformatics analysis. The primary components of the quality analysis encompassed the evaluation of sequencing quality and the investigation of base composition—the acquisition of clean data involved eliminating linker and low-quality sequences from the raw data. The evaluation of data cleanliness was subsequently conducted using FASTQC v0.10.1.
2.6. The Alignment of a Reference Genome and the Mapping of Read Distribution Across the Entire Genome
Clean readings were obtained using Trimmomatic (v3.0) after the data was filtered and ribosomal RNA was eliminated using Bowtie2 (v2.2.5). HISAT2 (v2.1.0) was used to align the paired-end clean reads to the reference genome, and Bowtie2 (v2.2.5) was used to create the reference genome index. Using a reference-based methodology, the Scripture (beta2) and Cufflinks (v2.1.1) software produced the assembled transcripts for every sample [20]. Furthermore, each sample's aligned reads were created using a reference-based method with StringTie (v1.3.1) [21].
2.7. Analyzing the Differential Expression
HTSeq (v0.11.2) was used for quantification of genes and transcripts. To find differential expressions in transcriptomic or genomic data, DESeq2 (v1.18.1) was utilized. The thresholds for identifying significant differential expression in abiotic replication are an absolute value of the logarithm base 2 of the fold change <1, and it has been determined that transcripts or genes with a P< 0.05 indicate differential expression in biological replication.
2.8. LncRNA-Gene Interaction Predictions
The mRNA and lncRNA transcripts were screened, followed by quantitative analysis using the StringTie-eB program. This analysis determined transcript abundance, namely the FPKM values, for each sample transcript. In the previous study, the authors employed Cuffdiff (version 2.1.1) to compute the Fragments Per Kilobase of transcript per Million mapped reads (FPKM) for both long non-coding RNAs (lncRNAs) and coding genes across all samples [22]. Transcripts with a p-adjust value of less than 0.05 were classified as differentially expressed.
2.9. KEGG and GO-Based Functional Annotation and Enrichment Analysis
The GOseq R package conducted GO enrichment scores on differentially expressed genes and lncRNA target genes. Significantly enriched GO keywords were those with adjusted P values below 0.05 [23]. The KEGG database is valuable for comprehending biological systems' underlying functions and applications. It offers insights into several aspects, including cells, animals, and ecosystems, by leveraging molecular-level data, particularly about large-scale molecules. The genome sequencing and other high-throughput experimental methods used to collect the dataset used in this investigation are described in the following source: http://www.genome.jp/kegg/. Using the KOBAS software, the enrichment analysis of lncRNA target genes or differentially expressed genes was carried out. [24].
2.10. RT-qPCR Validation
The RNA samples extracted from uterine tissues, including KMnO4-treated, LPS-treated and untreated tissues, were analyzed using reverse transcription-quantitative polymerase chain reaction (RT-qPCR) for lncRNA and mRNA sequencing. Subsequently, complementary DNA (cDNA) was synthesized using a reverse transcriptase reagent following the guidelines provided in the instruction manual. The SYBR Green Plus Reagent Kit with the Light Cycler 96 equipment from Roche, Basel, Switzerland, was used to conduct a quantitative polymerase chain reaction (qPCR). The experiment adhered to the manufacturer's recommendations, using GAPDH as the internal reference gene. The primer sequences employed in the investigation are shown in Table 1.
2.11. Statistical Analysis
This study will use statistical tools to analyze and interpret the acquired data. The statistical studies were conducted utilizing the GraphPad Prism 9 software. A one-way analysis of variance (ANOVA) was used to assess if there were any significant variations in the levels of long non-coding RNAs (lncRNAs) and messenger RNAs (mRNAs) among the groups that were treated with LPS and KMnO4 as well as the control groups. Statistically significant differences were seen when the p-values were below the thresholds of 0.05 and 0.01, respectively.
3. Results
3.1. Histopathological Results of Uterine Tissues
The uterus of SD rats was retrieved and treated with H&E staining. The H&E staining results proved that the control group showed no obvious alterations. However, the uterus treated with KMnO4 and LPS groups displayed evident indications of oedema, endometrial damage, cellular infiltrations, chromatin condensation and necrosis (Figure 1A-C). The wet and dry ratio of normal and KMnO4 and LPS groups were assessed by weighing tissues. The treatment groups exhibited significantly higher ratios than the normal group (p < 0.01; Figure 1D). MPO serves as both a function and activation marker for neutrophils and macrophages. It can be used to forecast the onset of early inflammatory illnesses by assessing the activity level of MPO (Li et al., 2015). The MPO assay was used to assess the infiltration of inflammatory cells, and the results revealed a significant increase in the treatment groups (p < 0.01; Figure 1E). Additional SOD and MDA key markers were employed to examine the oxidative stress in rats. Interestingly, KMnO4 and LPS substantially reduced SOD activity (Figure 1F) and increased MDA content (Figure 1G). These results demonstrate the successful establishment of our model.
3.2. Sequencing Data Statistics of Uterine Tissues
The present investigation involved the construction of a total of six complementary DNA (cDNA) libraries. This was achieved by extracting total RNA from uterine tissues obtained from KMnO4-treated, LPS-treated and control samples. The cDNA library of various samples consisted of the following raw reads: control_1 with a count of 65,468,884, control_2 with a count of 60,513,980, and KMnO4_1 with a count of 64,455,606, and KMnO4_2 with a count of 59,971,680 and LPS _1 732,478,86, LPS _2 87,839,856. The GC content percentages were determined to be as follows: cont1, 50.55%; cont2, 51.83%, and KMnO4-1, 49.93%; KMnO4-2, 51.77% and LPS_1 49.87% and LPS-2 49.64%. The raw data was subjected to a filtering process to obtain clean data. Table 2 presents the counts of clean reads based on various parameters such as GC% and mapped reads (%) and their effective rate (%). Furthermore, the clean reads were matched to the Rat reference genome using the HISAT2 program, which utilizes an enhanced Burrows-Wheeler Transform (BWT) technique known as the FM index. This analysis revealed that the total number of mapped reads or fragments across all samples exceeded 80% and was successfully aligned to the reference genome.
3.3. Chromosomal Distribution of Differentially Expressed Transcripts
In some cases, genes may have a discernible pattern of consistent arrangement along chromosomes, wherein genes located close to one other on a chromosome often demonstrate similarities in their biological functions or participation in the same metabolic pathway. The likelihood of gene regulation is higher for genes close to each other than for genes situated at greater distances. Therefore, the distribution of distinct transcripts on chromosomes and the simultaneous differential expression of adjacent transcripts may have critical biological ramifications. Furthermore, this phenomenon facilitates the precise identification of genes pertinent to the study's objectives. Figure 2 illustrates the specific distribution of differential transcripts across chromosomes. The homogeneous distribution of differentially expressed messenger RNAs (mRNAs) was seen across all chromosomes. The mRNAs that showed up-regulation and downregulation in the Control group,KMnO4-treated and LPS group were found on chromosomes 1, 2, 3, 4, 5, and X. The long non-coding RNAs (lncRNAs) that demonstrated an increase and decrease in expression mainly were situated on chromosomes 2, 3, 6, and 9.
3.4. Differentially Expressed Gene Analysis in KMnO4 treated, LPS treated, and Normal Uterine Tissues
The DESeq2 package was utilized to perform differential expression analysis on genes. The differentially expressed genes between samples were determined based on two criteria: a minimum fold change of ∣log2 Fold Change∣>1 and a significance level of p-value < 0.05 [25,26]. The expression level of the differential gene was quantified using the RPKM/COUNT metric. The present work demonstrates the utilization of hierarchical clustering analysis to examine the patterns of differentially expressed genes, as depicted in Figure 3A. KMnO4 and LPS groups showed a distinct expression pattern compared to the control group. There was no distinct difference between KMnO4 and LPS groups.
The upregulated and downregulated genes were subjected to volcano plots. The data on the number of genes that exhibited differential expressions in the KMnO4-treated, LPS-treated, and control groups were presented in Figure 3B, C). A comprehensive analysis revealed 988 genes exhibiting considerable upregulation and 661 genes displaying significant downregulation in the control vs KMnO4 group. In the control vs LPS group, 655 were upregulated genes, and 728 were downregulated.
3.5. GO Functional Analysis of Differentially Expressed mRNA Enrichment
We conducted GO functional enrichment analysis to enhance comprehension of the regulatory function of target genes in oxidative stress, Inflammation, and the molecular mechanism underlying apoptosis. These enriched GO terms encompassed 20 biological processes, 20 cellular components, and 20 molecular functions. Based on the findings from the GO annotation analysis, we identified several GO terms associated with Inflammation, oxidative stress, apoptosis, negative regulation of glucocorticoid receptor signaling pathway, negative regulation of steroid biosynthesis, metabolic process cell, cytokine receptor activity, and protein transcription factor activity among others (Figure 4A&B).
3.6. KEGG Signaling Pathway Enrichment Analysis of Differentially Expressed mRNA
A KEGG signaling pathway enrichment analysis was conducted on the target genes that exhibited significant differential expressions. The analysis revealed that these targeted genes were enriched in 158 pathways, out of which 78 were enriched in KMnO4 groups and 80 in LPS group. Out of these 50 were the same in both groups. 20 of KMnO4 group and 20 of LPS group pathways, demonstrating considerable enrichment (Figure 5A&B). Several pathways associated with Inflammation and oxidative stress were identified by examining KEGG signalling pathways. These pathways primarily encompass MAPK signaling pathways (RAS, RAF, ERK1/2), cGMP-PKG signaling pathways (ErAK, VASP, PKA, CREB), and necroptotic pathways (RIPK1, RIPK3, MLKL) (Table 3)
3.7. Identification of Differentially Expressed LncRNAs of Control vs KMnO4 and Control vs LPS Groups
In total, 14,546 LncRNAs were acquired, and DESeq2 was used for normalization. A heatmap displayed the expression patterns of these transcripts in various groups. The expression pattern of the KMnO4 and LPS groups, as depicted in Figure 6A, differed from that of the control group. The distinctions between the KMnO4 and LPS groups were indistinct. This suggests differences in the expressions between the groups treated with KMnO4 and LPS. A volcano plot was used to display the expression patterns of these transcripts in the groups treated with KMnO4 and LPS. Figure 6B&C labelled the genes based on the -log10 (p-value).
3.8. Prediction and Functional Analysis of lncRNA Target Genes and mRNAs
The mechanism by which lncRNA regulates target genes can be classified into two distinct categories: cis-regulation (occurring within the same genomic region as the target gene) and trans-regulation (occurring in a separate genomic region from the target gene). Predictions were formulated on the regulatory role of differentially expressed lncRNAs on target genes. To facilitate the comprehensive investigation of the activities of lncRNAs in the samples, a network diagram depicting the interaction between differentially expressed lncRNAs and their target genes was constructed. This map serves as a valuable reference and aid in understanding the overall functionality of lncRNAs. Figure 7A shows that Cacnalc targeted TCONS_00063069, TCONS_00063072,TCONS_00063042,TCONS_00063045,TCONS_00063055,TCONS_00063054, TCONS_00063055, TCONS_00063057, TCONS_00063056, and TCONS_00063066.
On the other hand, IKzf2 targeted TCONS_00089245, TCONS_00089246, TCONS_00089243, TCONS_00089248, and TCONS_00089247 in KMnO4 group. In the LPS group, Pax2 targeted the transcripts TCONS_00010999, TCONS_00010996, TCONS_00010995, TCONS_00011001, and the gene Slc7a7, are known to interact with the transcripts TCONS_00030709, TCONS_00030719, TCONS_00030721, and TCONS_00030713 (Figure 7B).
3.9. Analysis of lncRNA Target Genes via GO Enrichment and KEGG Enrichment
A GO analysis was performed on the target gene, using a significance threshold of P< 0.05 to determine enrichment in GO annotations. The GO concepts for biological processes, cellular components, and molecular functions were presented in Figure 8A and B, each displaying 10 GO terms. The target gene underwent KEGG analysis, with a significance threshold of P< 0.05 used to determine the enrichment of the KEGG pathway and obtain the analysis result. The results of the KEGG pathway analysis are presented in the bubble diagram of the KEGG pathway, as shown in Figure 8C & D. The KEGG enrichment analysis identified target genes linked to oxidative stress and Inflammation that are involved in pathways such as the Wnt signaling system and the cGMP-PKG signaling pathway. Furthermore, the cAMP signaling pathway and the GnRH signaling system involved in hormonal regulation were also recognized.
3.10. Validation of Genes by RT-qPCR
To validate the precision of the sequencing process, this study opted to perform quantitative PCR verification on a subset of differentially expressed mRNAs and lncRNAs. We picked shared mRNA and lncRNAs from both groups (Control vs KMnO4; Control vs LPS). The findings are depicted in Figure 9A-D. qPCR findings exhibit a high degree of concordance with the sequencing outcomes, corroborating the sequencing data's accuracy and reliability.
4. Discussion
Uterine bacterial infections are frequently observed in multiparous cows, causing notable uterine destruction, decreased productivity, and lowered fertility One of the key pathogens in these infections is the endotoxin Escherichia coli (E. coli), which is also linked to inflammatory disorders such as endometritis and acute lung injury [6,27]. Although inflammation is a protective response aimed at fighting infections, excessive or prolonged inflammation can lead to tissue damage, organ dysfunction, and even life-threatening conditions [28,29]. Clinically, antibiotics are the primary treatment for uterine infections, but their overuse comes with significant drawbacks, including negative effects on reproductive health and suppression of immunity in dairy cows [30]. Additionally, the global rise of antibiotic resistance has become a critical concern [31,32].
An alternative treatment, KMnO4, has long been used due to its strong oxidizing properties that make it effective in treating bacterial and fungal infections [33]. However, while KMnO4 is useful in wound disinfection, its oxidizing potential can also cause significant damage to healthy tissues. In cattle, excessive KMnO4 exposure has been associated with ulcers and other tissue injuries. This dual-edged nature of KMnO4 underscores the need to understand its biological effects more thoroughly, especially in sensitive tissues like the uterus.
In this study, we utilized a rat model to compare the effects of KMnO4 and LPS on uterine tissues, focusing on inflammation and oxidative stress at the molecular level. LPS is a well-established inducer of inflammation and oxidative damage, commonly used as a model for reproductive disorders [34]. We sought to investigate the transcriptional changes, specifically in lncRNAs, which play a crucial role in gene regulation and could potentially serve as therapeutic targets.
Histopathology was employed in the present study to ascertain the presence of oxidative stress inside the uterine tissues of rats. Necrosis, edema, and leukocyte infiltration were observed through direct observation. Furthermore, the results of wet and dry weight of uterus tissues also show obvious change. Moreover, biochemical assays measuring MDA, SOD, and MPO further confirm the successful establishment of the animal model.
Following the extraction of total uterine RNA, high-throughput sequencing was conducted on the Illumina platform to elucidate the profiles of long non-coding RNA (lncRNA) and gene expression in KMnO4 treated, comparing LPS group in rat uterine tissues.
LncRNAs are a class of RNA molecules with therapeutic capabilities due to their ability to induce alterations in DNA transcription through methylation and acetylation processes [35]. This work examined the mRNA and lncRNA expression in tissues subjected to oxidative stress. A total of 1649 differentially expressed messenger RNAs (mRNAs) in KMnO4 treated,1383 in LPS treated group were identified by screening and 1125 in KMnO4 treated, 989 in LPS treated groups expressed lncRNAs. The function enrichment analysis revealed that a considerable proportion of the genes that exhibited differential expression were implicated in several biological processes, including reactive oxygen species, signal transduction involved in gene expression regulation, Wnt signaling pathway, regulation of macrophage activation, and various biochemical metabolic processes. The enrichment analysis of the KEGG pathway revealed that the differentially expressed genes primarily engaged in signal pathways associated with the MAPK signaling pathway, cGMP-PKG pathway, Wnt pathway, Necroptosis, and cAMP signaling pathways. Besides genes exhibiting variable expression, most lncRNAs had an ambiguous functional role. The hypothesis in this study posited that lncRNA has a regulatory role in gene expression, as mentioned earlier. Consequently, the function of lncRNA was predicted by considering its associated coding genes, as indicated by previous research [36].
This study estimated target genes of differentially expressed lncRNA using cis and trans methods. Subsequently, GO and KEGG studies were conducted on the identified target genes to explore the potential functional role of the lncRNA[37,38]. Through the utilization of enrichment analysis of GO and KEGG pathways, as well as coding-noncoding co-expression network analysis, we have determined that the differentially expressed target genes primarily exhibit associations with Inflammation and apoptotic signaling pathways.
The lncRNAs TCONS_00030719, TCOS_00078464 and TCONS_00082446 exhibited differential expressions in the Control vs KMnO4 group. They were enriched in various signaling pathways. The signaling pathways strongly correlate with oxidative damage and Inflammation. The identified gene Slc7a7 targets TCONS_00030719, TCONS_00030709, TCONS_00030721, TCONS_00030713. The gene Slc7a7 was found to be implicated in the regulation of lifespan, as well as in the cAMP signaling route and calcium signaling pathway, according to the KEGG pathway enrichment analysis. Prior research has established a strong association between the Slc7a7 gene and human longevity and hypertension. This gene plays a crucial role in regulating the immune response by activating transcription factors, which influence gene expression in immune cells [39,40].
Furthermore, in Control vs LPS, the target lncRNAs predicted for LGR6 include TCONS_00024057,TCONS_00024055,TCONS_00024054,TCONS_00024052,TCONS_00024053,TCONS_00024056. GPRs that are broadly expressed in a variety of cells, including neutrophils, monocytes, cochlear hair cells, and astrocytes, also contain different types such as leucine-rich repeat-containing G protein-coupled receptor 6 (LGR6)[41,42,43]. According to the literature, it has been documented that LGR6 can modulate many inflammatory pathways and contribute to generating reactive oxygen species in the context of oxidative stress [44,45]. According to recent research, in a rat model of subarachnoid hemorrhage, Maresin 1 activates LGR6 to reduce neuroinflammation via the CREB/JMJD3/IRF4 pathway[46]. Maresin 1 reduces diabetic kidney disease by activating the cAMP-SOD2-ROS pathway mediated by LGR6 in another study[45].
In summary, this study provides new insights into the molecular mechanisms underlying KMnO4 and LPS-induced uterine damage, particularly the role of lncRNAs in regulating inflammation and oxidative stress. Our findings reveal that lncRNAs, through their interactions with key genes like Slc7a7 and LGR6, may act as important regulators of the immune response in reproductive tissues. Understanding these regulatory networks could pave the way for the development of targeted molecular therapies to treat uterine.
Author Contributions
TU and DG conceived the idea. TU, HF and FW took part in data collection and manuscript preparation. ZH,CN and ZJ conducted the manuscript preparation and reviewing process. LW,WX, SU, ZU,MA, MN, and QC helped in writing, reviewing and editing the manuscript.
Funding
This study was funded and supported by the National Natural Science Foundation of China (No. 31972744).
Ethics Statement
The animal study was conducted by Laboratory Animal Research Center Hubei province protocols and authorized by the Huazhong Agricultural University Animal Care and Use Committee (HZAURA-2024-0036).
Data Availability
Data will be made available on request.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Ghanem ME, Tezuka E, Devkota B, Izaike Y, Osawa T. Persistence of uterine bacterial infection, and its associations with endometritis and ovarian function in postpartum dairy cows. J Reprod Dev 2015; 61:54-60. [CrossRef]
- Williams EJ, Fischer DP, Pfeiffer DU, England GC, Noakes DE, Dobson H, et al. Clinical evaluation of postpartum vaginal mucus reflects uterine bacterial infection and the immune response in cattle. Theriogenology 2005; 63:102-17. [CrossRef]
- Földi J, Kulcsár M, Pécsi A, Huyghe B, de Sa C, Lohuis JA, et al. Bacterial complications of postpartum uterine involution in cattle. Anim Reprod Sci 2006; 96:265-81. [CrossRef]
- Mandhwani R, Bhardwaz A, Kumar S, Shivhare M, Aich R. Insights into bovine endometritis with special reference to phytotherapy. Vet World 2017; 10:1529-1532. [CrossRef]
- Yin B, Umar T, Ma X, Chen Y, Umar Z, Umer S, et al. Andrograpanin mitigates lipopolysaccharides induced endometritis via TLR4/NF-κB pathway. Reproductive Biology 2022; 22:100606. [CrossRef]
- Umar T, Ma X, Yin B, Umer S, Zahoor A, Akhtar M, et al. miR-424-5p overexpression inhibits LPS-stimulated inflammatory response in bovine endometrial epithelial cells by targeting IRAK2. Journal of Reproductive Immunology 2022; 150:103471. [CrossRef]
- Umar T, Wenjing L, Feng H, Feng W, Bin M, Umar Z, et al. A review on the applications of potassium permanganate in veterinary medicine: toxicity,efficacy, and future considerations. Pakistan Veterinary Journal 2024; 44:214-221. [CrossRef]
- Willhite CC, Bhat VS, Ball GL, McLellan CJ. Emergency Do Not Consume/do Not Use concentrations for potassium permanganate in drinking water. Hum Exp Toxicol 2013; 32:275-98. [CrossRef]
- Manpoong NK, Ahmed K, Bhattacharya DK, Sarma D, Ahmed N, Das A. Efficacy of potassium permanganate and turmeric as antimicrobial agents on the bacterial load and quality of boar semen during preservation at 15 °C. Veterinarski Arhiv 2020; 90:443-451. [CrossRef]
- HOUSE JE. Inorganic Chemistry. 223 Wyman Street, Waltham, MAO2451, USA.: Academic Press, 2013.
- Li X, Wang H, Zhang Y, Zhang J, Qi S, Zhang Y, et al. Overexpression of lncRNA H19 changes basic characteristics and affects immune response of bovine mammary epithelial cells. PeerJ 2019; 7:e6715. [CrossRef]
- Lodde V, Murgia G, Simula ER, Steri M, Floris M, Idda ML. Long Noncoding RNAs and Circular RNAs in Autoimmune Diseases. Biomolecules 2020; 10. [CrossRef]
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol 2010; 11:R106. [CrossRef]
- Qian W, Cao Y. An overview of the effects and mechanisms of m6 A methylation on innate immune cells in sepsis. Front Immunol 2022; 13:1041990. [CrossRef]
- Ganesan G, Rao SM. A novel noncoding RNA processed by Drosha is restricted to nucleus in mouse. Rna 2008; 14:1399-410. [CrossRef]
- Li L, Zhu J, Ye F, Duan Z, Zhou J, Huang Z, et al. Upregulation of the lncRNA SRLR in polycystic ovary syndrome regulates cell apoptosis and IL-6 expression. Cell Biochem Funct 2020; 38:880-885. [CrossRef]
- Nakagawa S, Shimada M, Yanaka K, Mito M, Arai T, Takahashi E, et al. The lncRNA Neat1 is required for corpus luteum formation and the establishment of pregnancy in a subpopulation of mice. Development 2014; 141:4618-27. [CrossRef]
- Liu S, Qiu J, Tang X, Cui H, Zhang Q, Yang Q. LncRNA-H19 regulates cell proliferation and invasion of ectopic endometrium by targeting ITGB3 via modulating miR-124-3p. Experimental Cell Research 2019; 381:215-222. [CrossRef]
- Liu S, Qiu J, Tang X, Li Q, Shao W. Estrogen Regulates the Expression and Function of lncRNA-H19 in Ectopic Endometrium. Int J Womens Health 2022; 14:821-830. [CrossRef]
- Özdemir S, Altun S. Genome-wide analysis of mRNAs and lncRNAs in Mycoplasma bovis infected and non-infected bovine mammary gland tissues. Mol Cell Probes 2020; 50:101512. [CrossRef]
- Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc 2016; 11:1650-67. [CrossRef]
- Chen Y, Jing H, Chen M, Liang W, Yang J, Deng G, et al. Transcriptional Profiling of Exosomes Derived from Staphylococcus aureus-Infected Bovine Mammary Epithelial Cell Line MAC-T by RNA-Seq Analysis. Oxid Med Cell Longev 2021; 2021:8460355. [CrossRef]
- Dennis G, Jr., Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 2003; 4:P3. [CrossRef]
- Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics 2012; 16:284-7. [CrossRef]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014; 15:550. [CrossRef]
- Gao D, Kim J, Kim H, Phang TL, Selby H, Tan AC, et al. A survey of statistical software for analysing RNA-seq data. Hum Genomics 2010; 5:56-60. [CrossRef]
- Jiang K, Guo S, Yang C, Yang J, Chen Y, Shaukat A, et al. Barbaloin protects against lipopolysaccharide (LPS)-induced acute lung injury by inhibiting the ROS-mediated PI3K/AKT/NF-κB pathway. International Immunopharmacology 2018; 64:140-150. [CrossRef]
- Meyer A, Laverny G, Bernardi L, Charles AL, Alsaleh G, Pottecher J, et al. Mitochondria: An Organelle of Bacterial Origin Controlling Inflammation. Frontiers in Immunology 2018; 9. [CrossRef]
- Rojas M, Woods CR, Mora AL, Xu J, Brigham KL. Endotoxin-induced lung injury in mice: structural, functional, and biochemical responses. Am J Physiol Lung Cell Mol Physiol 2005; 288:L333-41. [CrossRef]
- Wang F, Yang L, Sun J, Zheng J, Shi L, Zhang G, et al. Tumor suppressors microRNA-302d and microRNA-16 inhibit human glioblastoma multiforme by targeting NF-κB and FGF2. Mol Biosyst 2017; 13:1345-1354. [CrossRef]
- Bravo A, Ruiz-Cruz S, Alkorta I, Espinosa M. When Humans Met Superbugs: Strategies to Tackle Bacterial Resistances to Antibiotics. Biomol Concepts 2018; 9:216-226. [CrossRef]
- Adegboyega PA, Pei Y, McLarty J. Relationship between eosinophils and chronic endometritis. Human Pathology 2010; 41:33-37. [CrossRef]
- Palaniappan V, Karthikeyan K. Potassium permanganate: a 'desert island drug' in dermatology. Clinical and experimental dermatology 2022; 47:1650-1657. [CrossRef]
- Jiang K, Yang J, Yang C, Zhang T, Shaukat A, Yang X, et al. miR-148a suppresses inflammation in lipopolysaccharide-induced endometritis. J Cell Mol Med 2020; 24:405-417. [CrossRef]
- Winkle M, El-Daly SM, Fabbri M, Calin GA. Noncoding RNA therapeutics - challenges and potential solutions. Nat Rev Drug Discov 2021; 20:629-651. [CrossRef]
- Chen C, Liu C, Niu Z, Li M, Zhang Y, Gao R, et al. RNA-seq analysis of the key long noncoding RNAs and mRNAs related to cognitive impairment after cardiac arrest and cardiopulmonary resuscitation. Aging (Albany NY) 2020; 12:14490-14505. [CrossRef]
- Gene Ontology Consortium: going forward. Nucleic Acids Res 2015; 43:D1049-56.
- Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 2016; 44:D457-62. [CrossRef]
- Tina E, Prosén S, Lennholm S, Gasparyan G, Lindberg M, Göthlin Eremo A. Expression profile of the amino acid transporters SLC7A5, SLC7A7, SLC7A8 and the enzyme TDO2 in basal cell carcinoma. Br J Dermatol 2019; 180:130-140. [CrossRef]
- Zuo T, Chen P, Jing S, Zhang T, Chang L, Xu F, et al. Quantitative Proteomics Reveals the Development of HBV-Associated Glomerulonephritis Triggered by the Downregulation of SLC7A7. J Proteome Res 2020; 19:1556-1564. [CrossRef]
- Chiang N, Libreros S, Norris PC, de la Rosa X, Serhan CN. Maresin 1 activates LGR6 receptor promoting phagocyte immunoresolvent functions. J Clin Invest 2023; 133.
- Jansson L, Ebeid M, Shen JW, Mokhtari TE, Quiruz LA, Ornitz DM, et al. β-Catenin is required for radial cell patterning and identity in the developing mouse cochlea. Proc Natl Acad Sci U S A 2019; 116:21054-21060. [CrossRef]
- Miller SJ, Philips T, Kim N, Dastgheyb R, Chen Z, Hsieh YC, et al. Molecularly defined cortical astroglia subpopulation modulates neurons via secretion of Norrin. Nat Neurosci 2019; 22:741-752. [CrossRef]
- Krishnamoorthy N, Walker KH, Brüggemann TR, Tavares LP, Smith EW, Nijmeh J, et al. The Maresin 1-LGR6 axis decreases respiratory syncytial virus-induced lung inflammation. Proc Natl Acad Sci U S A 2023; 120:e2206480120. [CrossRef]
- Li X, Xu B, Wu J, Pu Y, Wan S, Zeng Y, et al. Maresin 1 Alleviates Diabetic Kidney Disease via LGR6-Mediated cAMP-SOD2-ROS Pathway. Oxid Med Cell Longev 2022; 2022:7177889. [CrossRef]
- Li Z, Yuan W, Yang X, Jiang J, Zhang Q-L, Yan X-X, et al. Maresin 1 Activates LGR6 to Alleviate Neuroinflammation via the CREB/JMJD3/IRF4 Pathway in a Rat Model of Subarachnoid Hemorrhage. Neuroscience 2024; 542:21-32. [CrossRef]
Figure 1.
Histopathological analysis of uterine tissues. (A-C) Histopathological changes demonstrated by H&E staining: (A) Control group, (B) KMnO4 group, and (C) LPS group. (D) Wet-to-dry ratio of uterine tissues in different groups. (E) Myeloperoxidase (MPO) assay results. (F) Malondialdehyde (MDA) activity. (G) Superoxide dismutase (SOD) activities.
Figure 1.
Histopathological analysis of uterine tissues. (A-C) Histopathological changes demonstrated by H&E staining: (A) Control group, (B) KMnO4 group, and (C) LPS group. (D) Wet-to-dry ratio of uterine tissues in different groups. (E) Myeloperoxidase (MPO) assay results. (F) Malondialdehyde (MDA) activity. (G) Superoxide dismutase (SOD) activities.
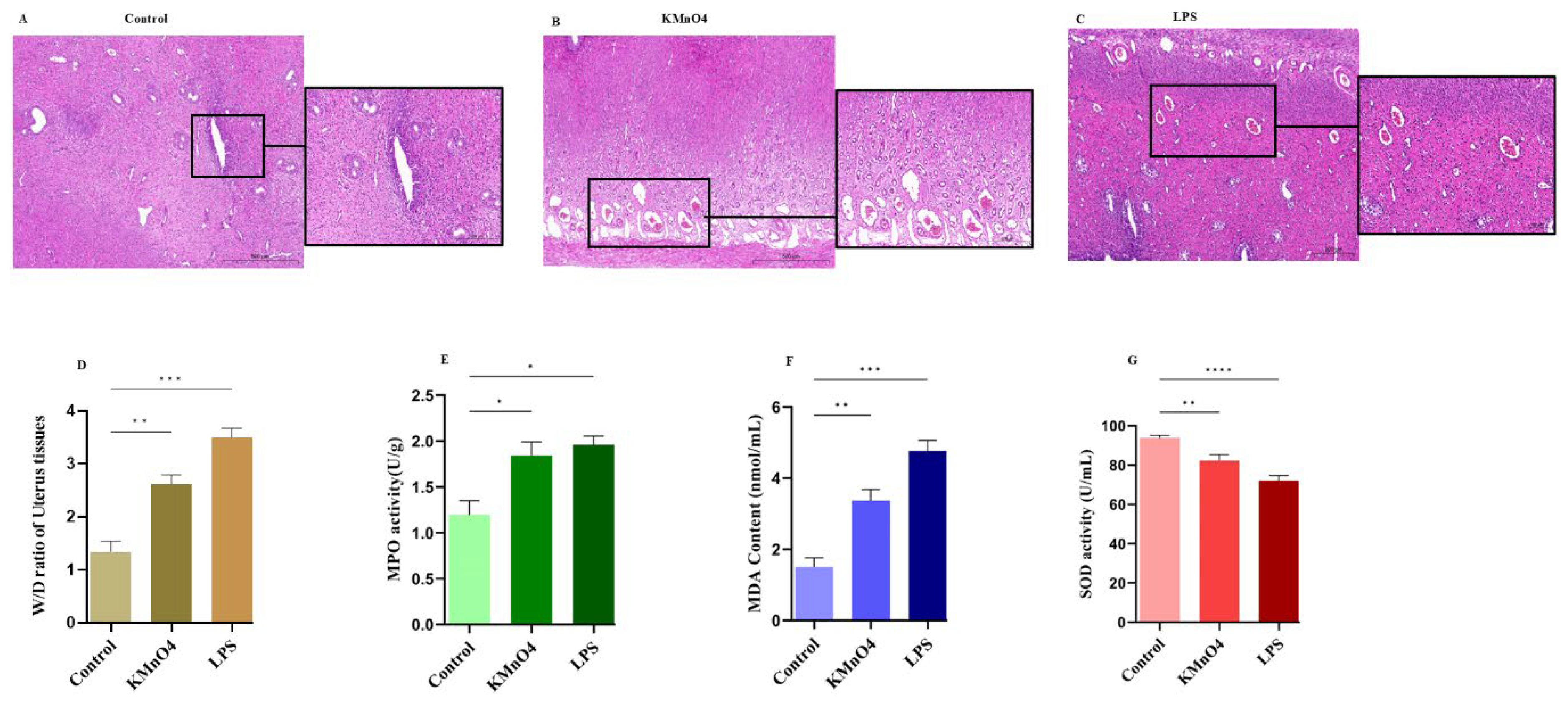
Figure 2.
Chromosomal Distribution of Differentially Expressed Transcripts. This figure represents the chromosomal distribution of differentially expressed transcripts identified across all experimental groups.
Figure 2.
Chromosomal Distribution of Differentially Expressed Transcripts. This figure represents the chromosomal distribution of differentially expressed transcripts identified across all experimental groups.

Figure 3.
Analysis of Differentially Expressed Genes in KMnO4-Treated, LPS-Treated, and Normal Uterine Tissues. (A) Heatmap displaying hierarchical clustering of differentially expressed genes across the experimental groups based on FPKM values. Log10(FPKM+1) transformation is applied for clustering. Genes with higher expression levels are represented in brown, while genes with lower expression are shown in yellow, indicating distinct expression profiles among the groups. (B) Volcano Plot of Differentially Expressed Genes in Control vs. KMnO4 Group illustrating the distribution of differentially expressed genes between the control and KMnO4-treated groups. Genes with significantly higher expression in the KMnO4 group are shown in brown, and genes with lower expression are shown in yellow. (C) Volcano Plot of Differentially Expressed Genes in LPS-Treated Group representing the differentially expressed genes in the LPS-treated group compared to controls. Red dots indicate genes with significantly higher expression, while blue dots represent genes with lower expression, emphasizing the transcriptional shifts induced by LPS treatment.
Figure 3.
Analysis of Differentially Expressed Genes in KMnO4-Treated, LPS-Treated, and Normal Uterine Tissues. (A) Heatmap displaying hierarchical clustering of differentially expressed genes across the experimental groups based on FPKM values. Log10(FPKM+1) transformation is applied for clustering. Genes with higher expression levels are represented in brown, while genes with lower expression are shown in yellow, indicating distinct expression profiles among the groups. (B) Volcano Plot of Differentially Expressed Genes in Control vs. KMnO4 Group illustrating the distribution of differentially expressed genes between the control and KMnO4-treated groups. Genes with significantly higher expression in the KMnO4 group are shown in brown, and genes with lower expression are shown in yellow. (C) Volcano Plot of Differentially Expressed Genes in LPS-Treated Group representing the differentially expressed genes in the LPS-treated group compared to controls. Red dots indicate genes with significantly higher expression, while blue dots represent genes with lower expression, emphasizing the transcriptional shifts induced by LPS treatment.
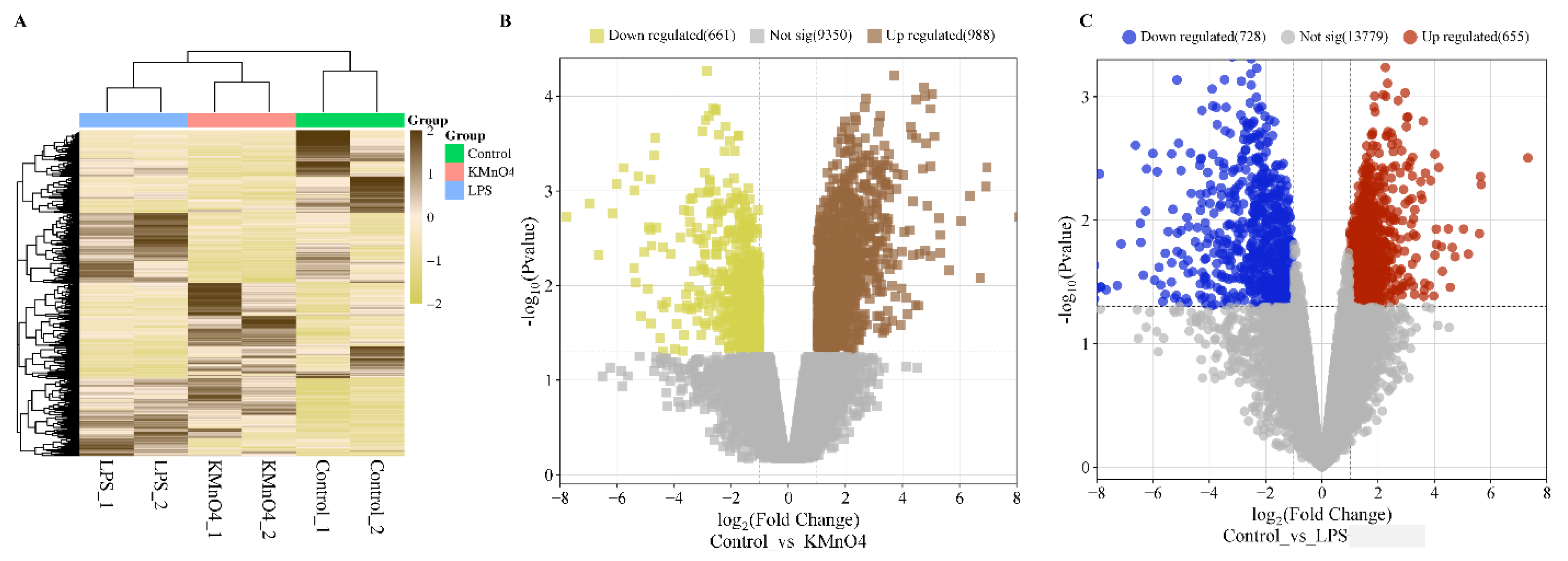
Figure 4.
GO Functional Analysis of Differentially Expressed mRNA Enrichment. (A) GO Enrichment Analysis of Control vs KMnO4 Group for differentially expressed. The analysis highlights significantly enriched biological processes, cellular components, and molecular functions, indicating the primary functional categories impacted by KMnO4 exposure. (B) GO enrichment analysis for differentially expressed mRNAs between the control and LPS-treated groups. The enriched GO terms reflect the key biological processes, cellular components, and molecular functions modulated by LPS treatment.
Figure 4.
GO Functional Analysis of Differentially Expressed mRNA Enrichment. (A) GO Enrichment Analysis of Control vs KMnO4 Group for differentially expressed. The analysis highlights significantly enriched biological processes, cellular components, and molecular functions, indicating the primary functional categories impacted by KMnO4 exposure. (B) GO enrichment analysis for differentially expressed mRNAs between the control and LPS-treated groups. The enriched GO terms reflect the key biological processes, cellular components, and molecular functions modulated by LPS treatment.
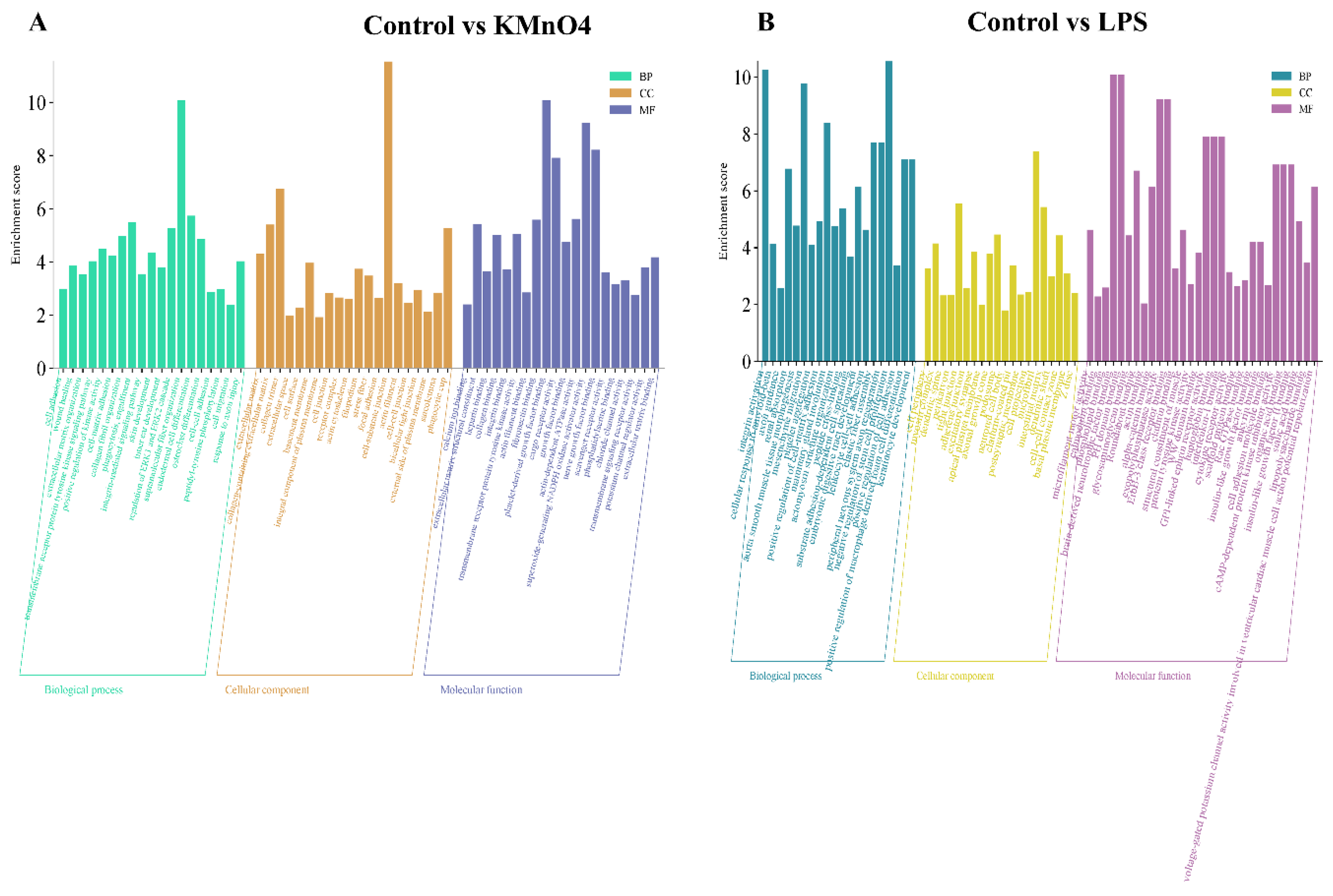
Figure 5.
KEGG Signaling Pathway Enrichment Analysis of Differentially Expressed mRNA. (A) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of differentially expressed mRNAs between the control and KMnO4-treated groups. The pathways enriched in the KMnO4 group reveal the signaling cascades and biological systems affected by KMnO4-induced oxidative stress and toxicity. (B) KEGG pathway enrichment analysis of differentially expressed mRNAs between the control and LPS-treated groups. This analysis uncovers the signaling pathways activated by LPS treatment, providing insight into the inflammatory and immune responses triggered in the uterine tissue.
Figure 5.
KEGG Signaling Pathway Enrichment Analysis of Differentially Expressed mRNA. (A) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of differentially expressed mRNAs between the control and KMnO4-treated groups. The pathways enriched in the KMnO4 group reveal the signaling cascades and biological systems affected by KMnO4-induced oxidative stress and toxicity. (B) KEGG pathway enrichment analysis of differentially expressed mRNAs between the control and LPS-treated groups. This analysis uncovers the signaling pathways activated by LPS treatment, providing insight into the inflammatory and immune responses triggered in the uterine tissue.
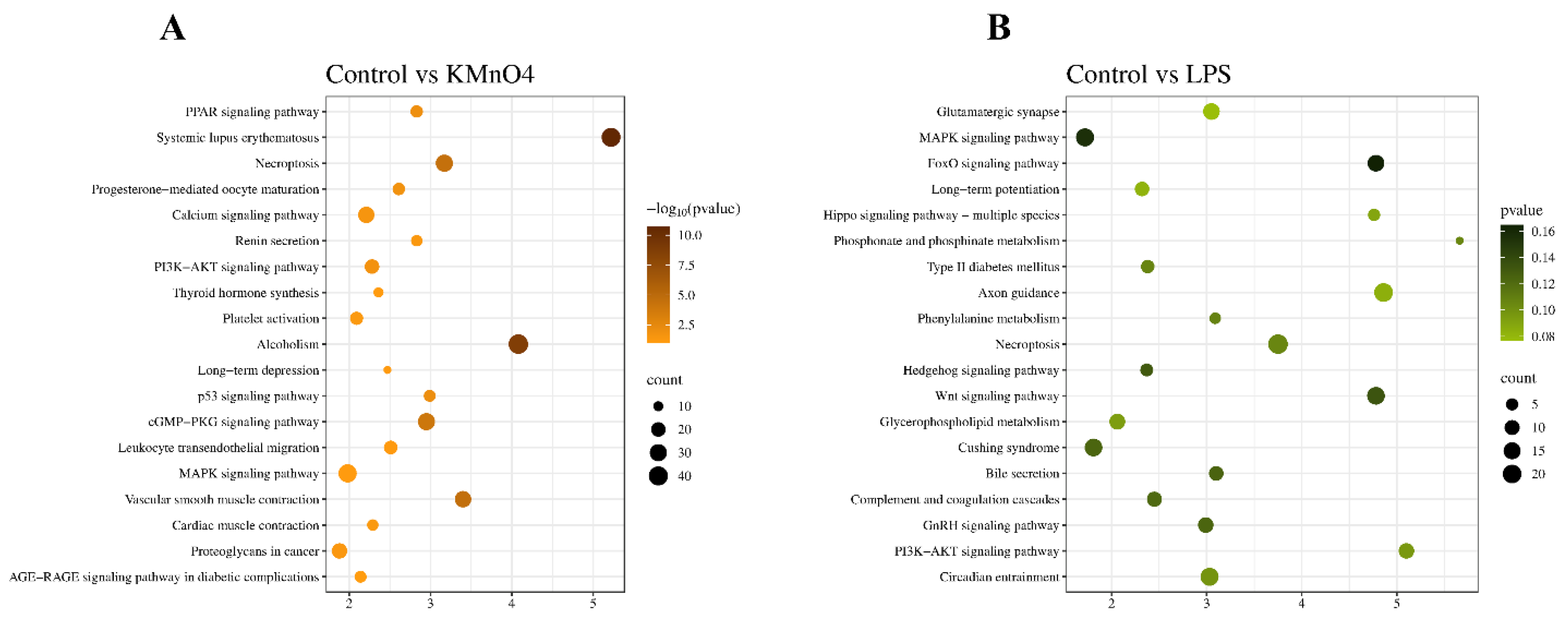
Figure 6.
mRNAs and lncRNAs with differential expressions. (A) LncRNAs with differential expressions grouped together. Based on FPKMs, hierarchical clustering clusters using log10(FPKM+1). Genes with higher expressions are indicated by dark green, and those with lower expressions are indicated light green. (B) A volcano plot of transcripts with differential expressions of mRNA. Genes with higher expressions indicated Red, with lower expressions indicated green. (C) A volcano plot of transcripts with differential expressions of lncRNA. Genes with higher expressions are indicated by yellow, and those with lower expressions are indicated by brown.
Figure 6.
mRNAs and lncRNAs with differential expressions. (A) LncRNAs with differential expressions grouped together. Based on FPKMs, hierarchical clustering clusters using log10(FPKM+1). Genes with higher expressions are indicated by dark green, and those with lower expressions are indicated light green. (B) A volcano plot of transcripts with differential expressions of mRNA. Genes with higher expressions indicated Red, with lower expressions indicated green. (C) A volcano plot of transcripts with differential expressions of lncRNA. Genes with higher expressions are indicated by yellow, and those with lower expressions are indicated by brown.
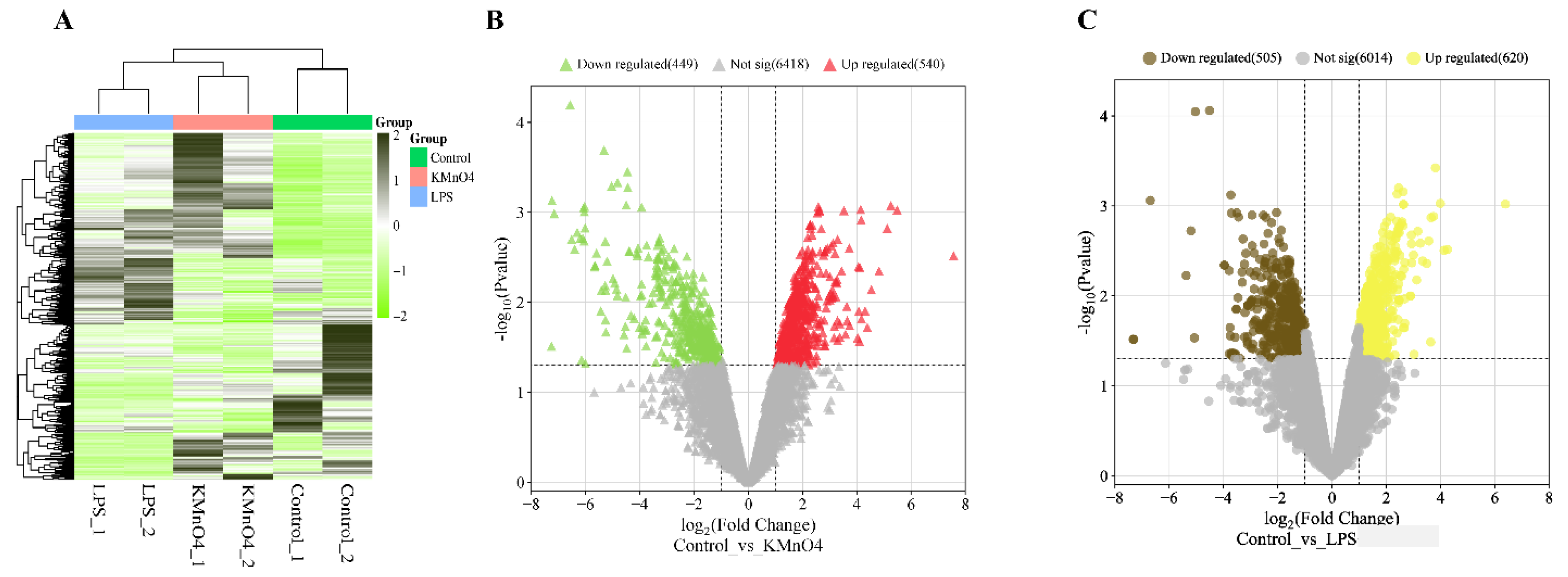
Figure 7.
Prediction and Functional Analysis of lncRNA Target Genes and mRNAs. (A & B) The network diagrams illustrate the interactions between predicted lncRNAs and their target mRNAs in the control group compared to the LPS-treated group (A) and the KMnO4-treated group (B). The visualization highlights key regulatory relationships between lncRNAs and mRNAs, emphasizing the differential gene regulation under treatment conditions.
Figure 7.
Prediction and Functional Analysis of lncRNA Target Genes and mRNAs. (A & B) The network diagrams illustrate the interactions between predicted lncRNAs and their target mRNAs in the control group compared to the LPS-treated group (A) and the KMnO4-treated group (B). The visualization highlights key regulatory relationships between lncRNAs and mRNAs, emphasizing the differential gene regulation under treatment conditions.
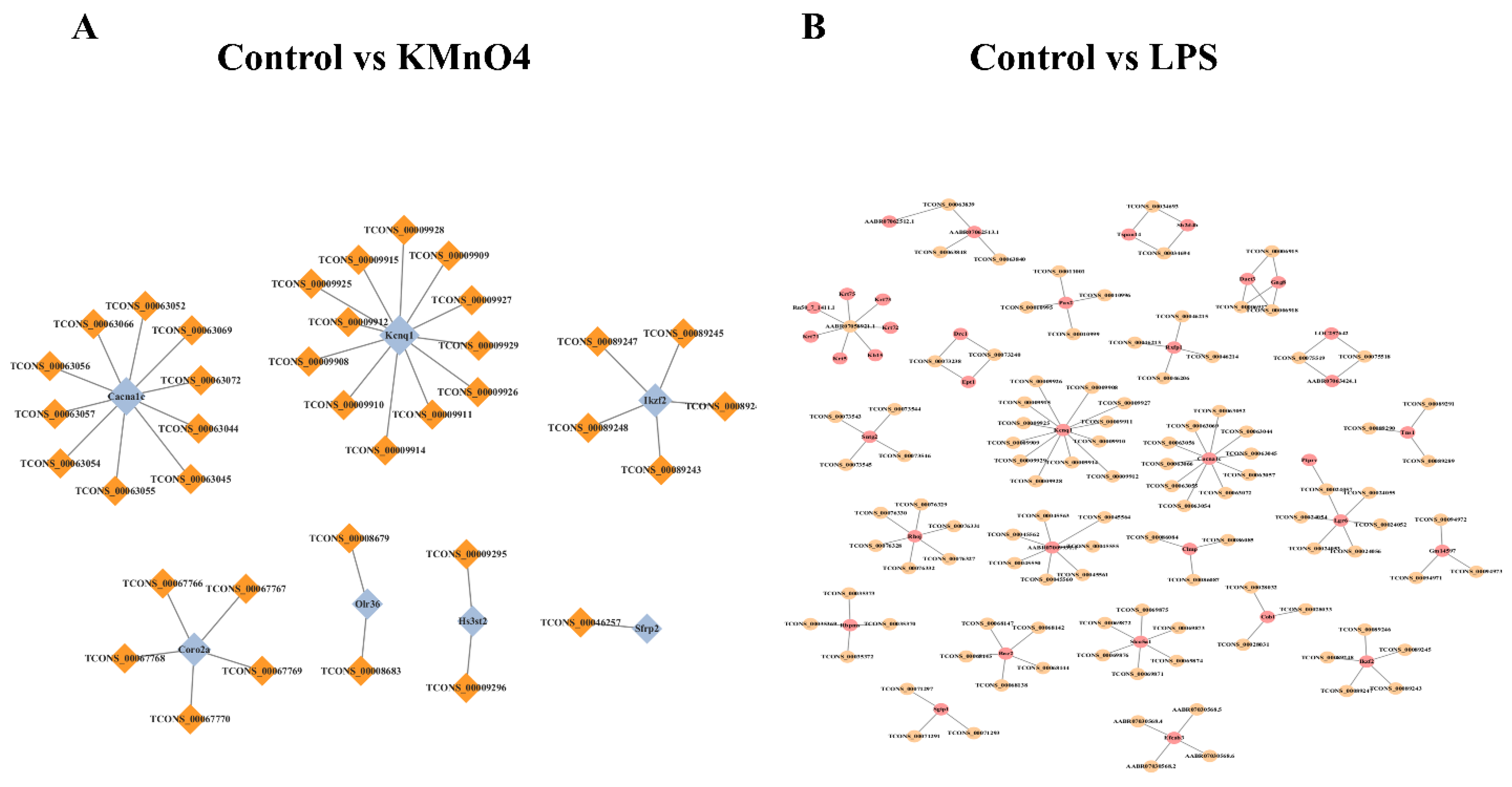
Figure 8.
Analysis of lncRNA Target Genes via GO and KEGG Enrichment. (A & B) These panels present the GO functional enrichment analyses of lncRNA target genes. The comparisons of control vs. KMnO4 (A) and control vs. LPS (B) reveal biological processes, cellular components, and molecular functions.(C & D) The bubble bar plots depict the most enriched KEGG pathways in the comparison of control vs. KMnO4 (C) and control vs. LPS (D). Bubble size represents the gene count, while color intensity indicates the significance of enrichment.
Figure 8.
Analysis of lncRNA Target Genes via GO and KEGG Enrichment. (A & B) These panels present the GO functional enrichment analyses of lncRNA target genes. The comparisons of control vs. KMnO4 (A) and control vs. LPS (B) reveal biological processes, cellular components, and molecular functions.(C & D) The bubble bar plots depict the most enriched KEGG pathways in the comparison of control vs. KMnO4 (C) and control vs. LPS (D). Bubble size represents the gene count, while color intensity indicates the significance of enrichment.
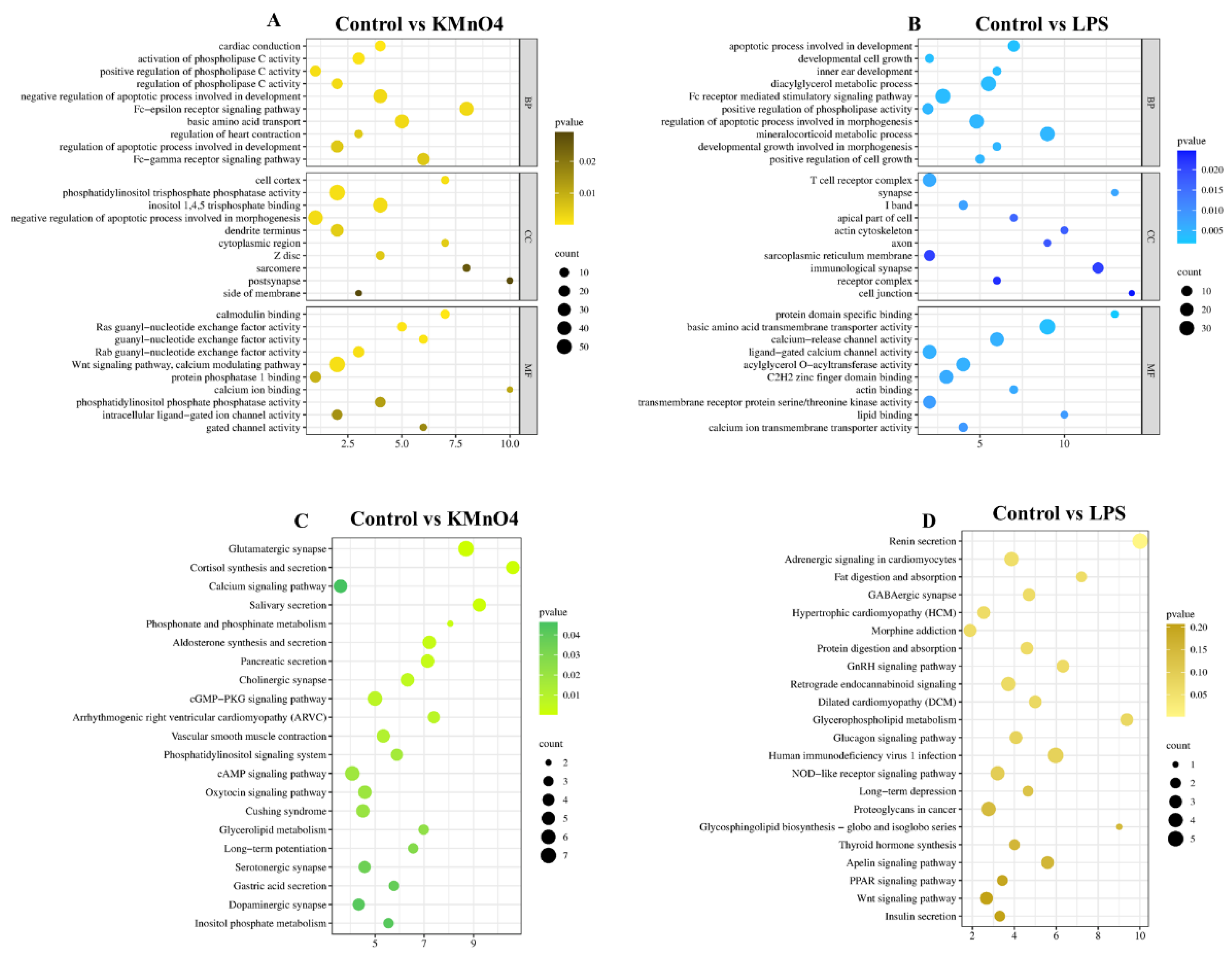
Figure 9.
RT-qPCR Validation of RNA-Seq Results. (A & B) Bar plots comparing the expression levels of up-regulated (A) and down-regulated (B) mRNAs, as determined by RNA-Seq and validated through RT-qPCR. The consistent trends between qPCR and RNA-Seq results confirm the reliability of the transcriptomic data. (C & D) Bar plots showing the differential expression of selected lncRNAs, comparing RNA-Seq data with RT-qPCR validation. Up-regulated (C) and down-regulated (D) lncRNAs exhibit consistent expression patterns across both methods.
Figure 9.
RT-qPCR Validation of RNA-Seq Results. (A & B) Bar plots comparing the expression levels of up-regulated (A) and down-regulated (B) mRNAs, as determined by RNA-Seq and validated through RT-qPCR. The consistent trends between qPCR and RNA-Seq results confirm the reliability of the transcriptomic data. (C & D) Bar plots showing the differential expression of selected lncRNAs, comparing RNA-Seq data with RT-qPCR validation. Up-regulated (C) and down-regulated (D) lncRNAs exhibit consistent expression patterns across both methods.
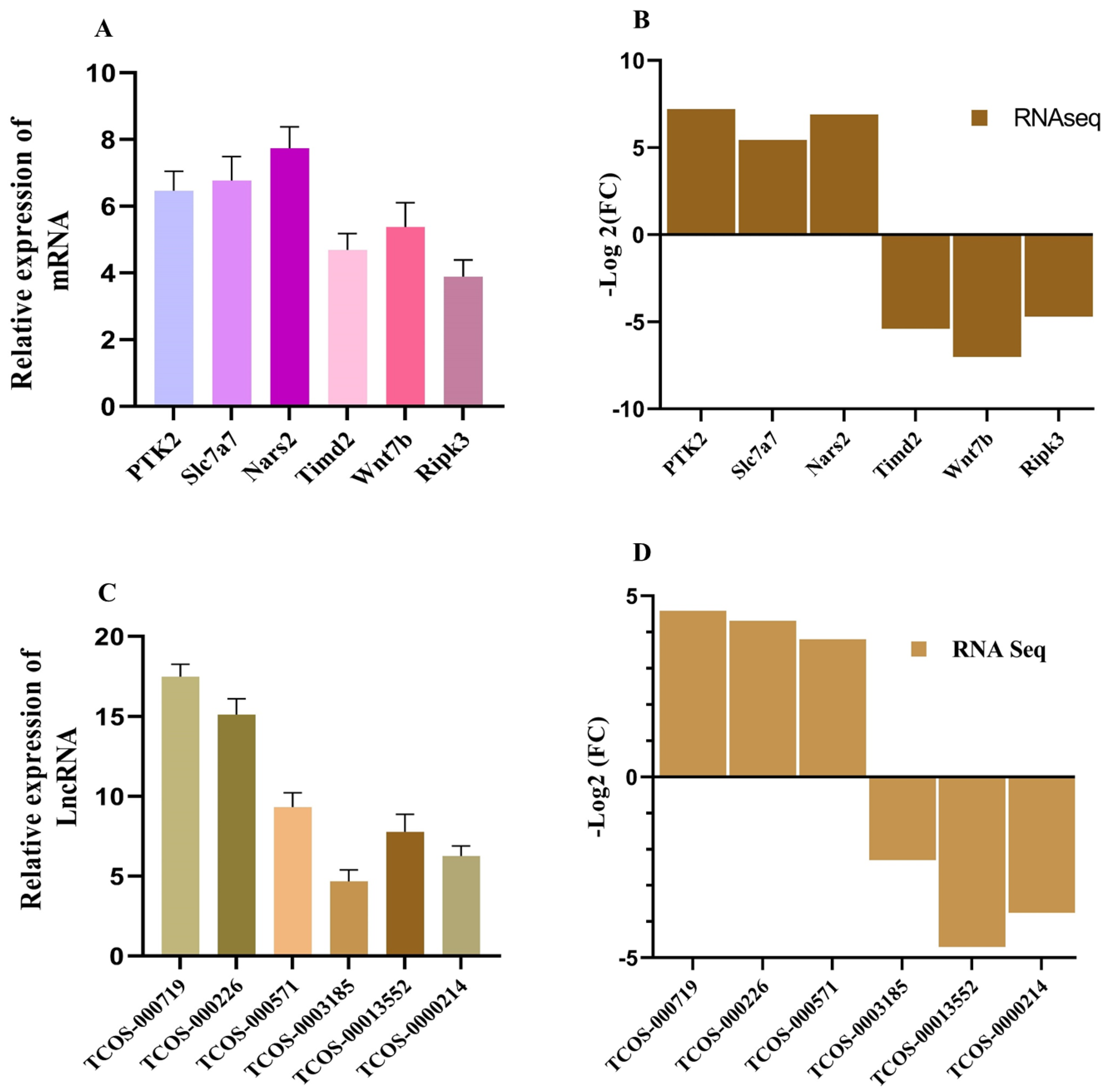

Table 1.
qPCR Primers for LncRNAs and mRNAs.
| Name | Product Size (bp) | Primer Sequence(5’-3’) |
|---|---|---|
| PTK2 | 246 |
Forward: CGTGTGGATGTTTGGTGTGT Reverse: TGCACCTTCTCTTCCTCCAG |
| Slc7a7 | 190 |
Forward: CCTGTTCTTCCCCATCGTCT Reverse: TGTGGTAGACGCTACGATCC |
| Nars2 | 235 |
Forward: GTGGATCCGGTCAGTCAGAT Reverse: AAATGCCTTGGCTTCACAGG |
| Timd2 | 223 |
Forward: ACTGAAGCAATCCCTCCACA Reverse: CTTCAATTCTGGCCTGCTCC |
| Wnt7b | 161 |
Forward: TTCACAACAATGAGGCAGGC Reverse: AGCTGCGTTGTACTTCTCCT |
| RIPK3 | 182 |
Forward: TCCACATTTCAGGGAGGGTC Reverse: ACCACCTCAGCTTCTCTTCC |
| TCOS_000719 | 216 |
Forward: AACAAGGAACAAGGCCACAC Reverse: GGCTTTCCCAGGTCTTAGGT |
| TCOS_000226 | 234 |
Forward: CTGTCTGAAGGGCACATGTG Reverse: TCCAGGATCTCAGCCACAAA |
| TCOS_000571 | 197 |
Forward: GCTGGATGTTGAGAGCACAG Reverse: TTCTAGCTCCCCACTCCTCT |
| TCOS_0003185 | 250 |
Forward: ACTGTGTGTGCTGGGTATGA Reverse: TGGCAAGTTTCGACTGTGTG |
| TCOS_13552 | 207 |
Forward: ACAACACTACCAGGGGACAG Reverse: GCAGAGGCCCATAGTCTAGG |
| TCOS_000214 | 201 |
Forward: GTTCCCAAGGCTTGACCCAA Reverse: AAGAATTGCCTGGTGTGTCCT |
Table 2.
Summary of Reads.
| Sample Name | Clean Reads | Clean GC (%) | Mapped Reads(%) | Effective rate(%) |
|---|---|---|---|---|
| Control_1 | 57300644 | 50.09 | 93.54 | 87.53 |
| Control_2 | 51742860 | 50.69 | 91.38 | 85.51 |
| KMnO4_1 | 57128986 | 49.36 | 94.43 | 88.36 |
| KMnO4_2 | 51422446 | 50.56 | 93.09 | 88.63 |
| LPS | 65824496 | 49.65 | 92.68 | 85.74 |
| LPS | 78704690 | 49.41 | 92.91 | 87.48 |
Table 3.
KEGG pathways and their Genes.
| KEGG Pathways | Enrichment | Genes |
|---|---|---|
| MAPK signaling pathway | 1.98 | Rac3;Cacnla;Cacnalc;Mef2c;Dusp8 |
| cGMP-PKG signaling pathway | 2.95 | Wos3;Kcnj8;Pde3a;Mylk;Kcnmal;Mef2c;Pde5a; Kcnmb1 |
| Necroptosis | 3.17 | RIPK1;RIPK3;MLKL;H2afx |
| FOXO signalling pathway | 4.78 | Cdkn2b;Tgfb2;Pck1;Fbxo32;PlKI;Ccdn2;Ccnb1;Irs2;Slc2a4 |
| GnRH pathway | 2.99 | Plcb1;Cacnalc;Plcb4;Adcy3;Calml3;Camk2g;Adcy9 |
| Wnt pathway | 4 | SfrD2;Plcbl;Rspo3;Camk2g;Nfatc4;Rac3;Sfrp4;Ccdn2;Plcb4 |
| PI3K-AKT signaling pathway | 5.1 | PI3K;PTEN;AKT;FOXO;CytokineR |
| P53 signaling pathway | 2.99 | CHK2;P53;Fas;Caspase8 |
| TNF signaling pathway | 2.21 | TNFR1;TRADD;TRAF;TAK1;TAB1/2;MKK;IL18R |
| JAK-STAT signaling pathway | 2.38 | JAK;STAT;IL13ra;IL6r;IL11r;IL4r; |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



