
Submitted:
28 December 2024
Posted:
30 December 2024
You are already at the latest version
Abstract
The inherited genetic disorder β-thalassemia affects the hematopoietic system and is caused by low production or absence of adult hemoglobin (HbA). Ineffective erythropoiesis is the hallmark of β-thalassemia pathophysiology and is characterized by an erythropoietin-driven substantial increase in erythroblast proliferation, coupled with an increase in late-stage precursor apoptosis, which results in low levels of circulating mature red blood cells (RBCs) and chronic anemia. Mitochondrial dysfunction commonly occurs in these cells because of the increased demand for energy production and the need to manage abnormal hemoglobin chain synthesis. Moreover, several studies have highlighted the importance of gradual mitochondrial clearance for mature erythroid cell production. In this review, we presented the available information on the role of mitochondria in vital cellular processes, which makes them promising pharmacological targets for maintaining the health and function of RBCs.
Keywords:
β-thalassemia
; mitochondria
; ineffective erythropoiesis
1. Introduction
Thalassemia is a very common genetic autosomal recessive inherited disorder worldwide and is characterized by the abnormal production of hemoglobin, leading to anemia [1]. Specifically, β-thalassemia is caused by the absence of or a reduction in the synthesis of β-globin chains. The imbalance of α/β globin chains and the relative excess of unpaired α-globin chains results in the formation of cytotoxic intracellular precipitates that compromise erythroid cell production and maturation in the bone marrow, leading to ineffective erythropoiesis, the hallmark of β-thalassemia pathophysiology. Moreover, oxidative stress and membrane damage in circulating erythroid cells lead to chronic hemolysis, which leads to the release of free hemoglobin (Hb) and free heme in the bloodstream [2]. Both ineffective erythropoiesis and increased hemolysis lead to deregulated iron homeostasis in patients, with the release of erythroid factors that suppress hepcidin production and cause iron overload, which is also worsened by regular transfusions [3].
The clinical manifestations of the disease are extremely heterogeneous, varying from very mild to more severe forms, called non-transfusion-dependent thalassemia (NTDT) and transfusion-dependent thalassemia (TDT), respectively [4]. The considerable variability and severity in clinical expression depend on the specific genotypes due to different β+ or β° alleles, known as primary modifiers, and on the co-presence of other independent genetic factors that may worsen or alleviate the phenotype [5]. However, individuals with identical β-thalassemia genotypes can have variable clinical severities [6], because of the high complexity of the genetic background associated with the disease [7] or acquired and ambient factors [8].
Two secondary key modifiers, i.e., the coinheritance of α-thalassemia and the innate ability to produce fetal hemoglobin (HbF), can ameliorate the imbalance of α/β globin chains, affecting the pathophysiology of β-thalassemia at the primary level [9]. Common DNA polymorphisms and rare mutations in cis-regulatory elements modulating expression of the HBG2 [10,11], BCL11A [12], HBS1L [13], MYB [14,15] and KLF1 [16] genes may be associated with a delay in the fetal-to-adult hemoglobin switch and/or an increase in the synthesis of HbF in adults [17]. Other studies focusing on different human populations reported that these single nucleotide polymorphisms (SNPs) are deeply geographically structured and that other loci, including KCNK10, GPR65, RNASE2, RNASE3, and C/EBPE, are also involved in the regulation of HbF expression [18,19].
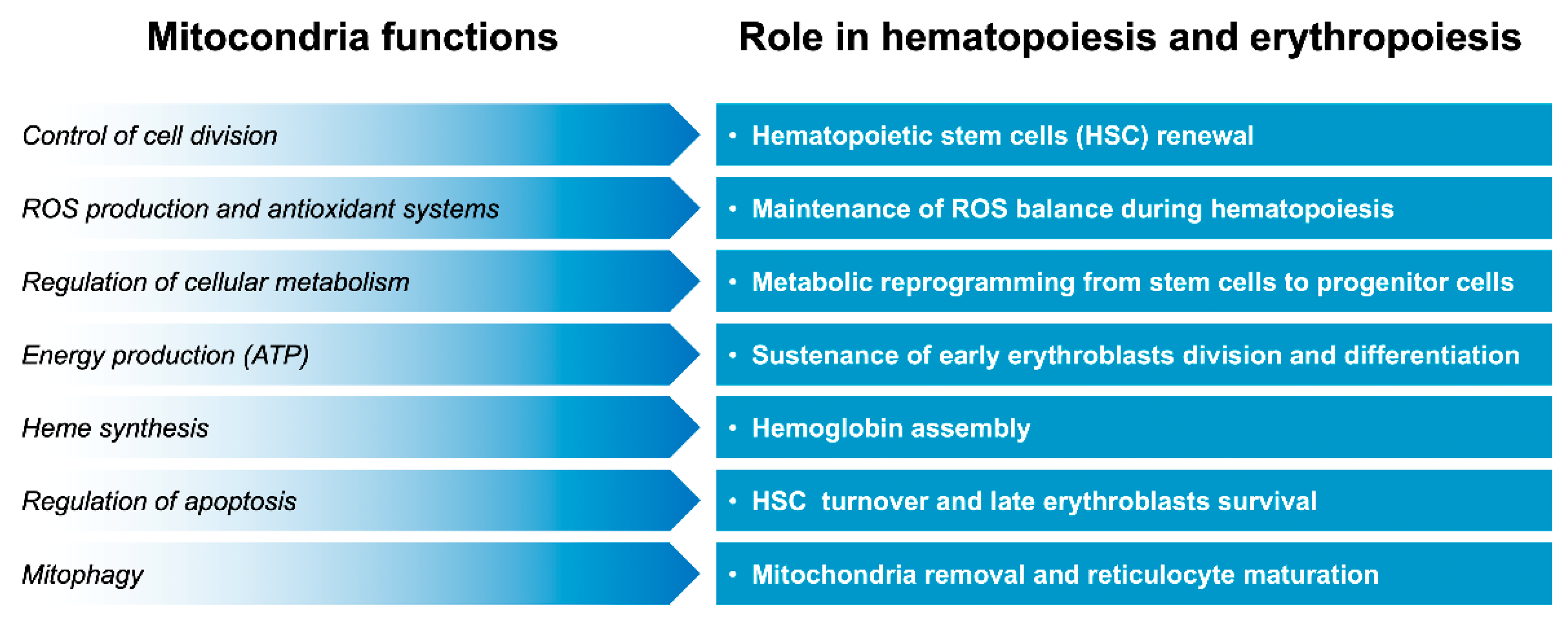
The upregulation of peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) can induce fetal hemoglobin synthesis in human primary erythroblasts [20]. PGC-1α belongs to a family of coactivator proteins that play a key role in regulating various signaling pathways [21]. Along with its effects on the liver, neurons, and muscle [22], studies have suggested that PGC-1α also plays an important role in the maturation and survival of erythroid cells [23]. PGC-1α also regulates mitochondria biogenesis and functions [24]. An increase in mitochondrial activity facilitated by increased PGC-1α levels can create a cellular environment conducive to erythropoiesis and the synthesis of hemoglobin, including HbF. In another study, PGC1β-depleted erythroid progenitor cells showed an increase in mitochondrial mass, suggesting impaired mitochondrial clearance [25]. These findings suggest that mitochondria play a crucial role in β-thalassemia pathophysiology and also contribute to β-thalassemia-related complications. In this review, we summarized evidence concerning the role of mitochondria, especially in maintaining red blood cell production and function (Figure 1).
2. Mitochondria in Physiology
Mitochondria are essential organelles in eukaryotic cells because they play an important role in producing adenosine triphosphate (ATP) through oxidative phosphorylation (OXPHOS) and providing energy for the activities in the cell [26]. They also play critical roles in several other cellular functions, including the regulation of different metabolic pathways (such as glycolysis, beta-oxidation, and amino acid metabolism) to produce ATP, which is crucial for nearly all cellular activities [27,28]. Moreover, mitochondria control intracellular calcium levels, which are crucial for cellular signaling, muscle contraction, neurotransmitter release, and cell division [29]. They also modulate cell survival by regulating programmed cell death. When a cell is damaged or stressed, mitochondria can release proteins such as cytochrome c into the cytoplasm, which activates caspases—enzymes that drive apoptosis. This ensures that damaged or unnecessary cells are efficiently eliminated, maintaining tissue homeostasis [30].
In certain cells, such as those in brown adipose tissue, mitochondria can produce heat instead of ATP through thermogenesis [31]. This process involves the uncoupling of oxidative phosphorylation, where energy created by the electron transport chain is released as heat instead of being used to produce ATP. This process helps regulate body temperature, especially in newborns and during cold exposure [32]. Mitochondria also support biosynthetic pathways, such as those associated with the synthesis of steroid hormones, including glucocorticoids, sex hormones, and mineralocorticoids, primarily in endocrine cells. The enzymes required for these processes are located in the mitochondrial membrane and convert cholesterol into steroid precursors [33].
Mitochondria can replicate and increase in number in response to the energy demands of the cell through a process called mitochondrial biogenesis [34,35]. This involves the activation of transcription factors such as PGC-1α, which stimulate the expression of genes related to mitochondrial function [21]. This process is important in cells with high-energy demands, such as muscle cells and neurons, and helps the metabolism of the cell to adapt to different environmental or energy states [24]. Mitochondria are highly dynamic organelles that can undergo fusion (combination of two mitochondria) and fission (splitting of one mitochondrion into two). These processes help maintain mitochondrial health by allowing the exchange of mitochondrial contents and the removal of damaged parts [36]. Fusion helps in the mixing of mitochondrial contents, such as mitochondrial DNA and proteins, which is beneficial for repairing damaged mitochondria. Fission facilitates mitochondrial division and replication, ensuring that cells can produce enough mitochondria to meet their energy needs [37,38].
Moreover, mitochondria can be selectively degraded through mitophagy, a form of autophagy in which damaged or dysfunctional mitochondria are engulfed and broken down by lysosomes in cells. This helps control mitochondrial quality and protect the cell from stress caused by defective mitochondria [39]. Mitochondria are the primary site of reactive oxygen species (ROS) production as byproducts of the electron transport chain. Although ROS can damage cellular components such as proteins, lipids, and DNA, they also play roles in cell signaling and defense against pathogens [40,41]. Mitochondria have antioxidant systems (such as superoxide dismutase and glutathione peroxidase) to neutralize excess ROS and protect the cell from oxidative damage [42].
Mitochondria participate in the synthesis of iron-sulfur clusters, which are essential for many enzymes involved in cellular metabolism [43]. They also play a central role in heme synthesis, which is crucial for the production of hemoglobin in red blood cells [44]. Mitochondria help incorporate iron into porphyrin rings to produce heme, a key component of hemoglobin.
3. Mitochondria in Pathology
As mitochondria are necessary for cellular activities, their dysfunction leads to various human pathological conditions [45]. Mitochondria have a 16-kilobase mitochondrial genome (mtDNA), which is distinct from the nuclear DNA (nDNA) found in the nucleus of cells. This DNA is circular and encodes only a few proteins that are required for mitochondrial function, most of which are involved in oxidative phosphorylation, whose expression and function are strictly coordinated by the nuclear DNA [46].
Mutations in mtDNA or nDNA that affect mitochondrial function can lead to various inherited diseases, such as Leber’s hereditary optic neuropathy (LHON), Kearns-Sayre syndrome (KSS), mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS), mitochondrial myopathy, and Leigh syndrome [47,48]. These diseases are often characterized by defects in energy production, leading to tissue dysfunction, particularly in high-energy-demanding organs such as muscles and the brain. Besides lactic acidosis, impaired ATP production results in neurological and muscle problems. The symptoms include muscle weakness, exercise intolerance, fatigue, and progressive loss of vision due to degeneration of retinal cells, reduced cardiac contractility, and heart failure [49,50,51].
Mitochondrial dysfunction is also associated with several neurodegenerative diseases, particularly those that involve the central nervous system (CNS), due to the high-energy demands of neurons. These diseases include Parkinson’s disease [52], Alzheimer’s disease [53], Huntington’s disease [54] and amyotrophic lateral sclerosis (ALS) [55]. These conditions often involve mitochondrial damage due to the loss of mitochondrial maintenance systems or the accumulation of proteins that interfere with mitochondrial function, leading to energy deficits and an increase in oxidative stress.
However, mitochondrial functions decline with age, leading to a decrease in ATP production and an increase in ROS production [56]. Moreover, mtDNA is highly vulnerable to oxidative damage due to its proximity to ROS produced in mitochondria [57]. The common 4977-bp mitochondrial deletion (ΔmtDNA4977) increases in frequency with oxidative stress [58] and accumulates in aging tissues, serving as a biomarker of mtDNA damage [59]. This contributes to cellular senescence, tissue degeneration, and age-related diseases, including osteoporosis, macular degeneration, and sarcopenia (muscle loss). The expression of antioxidants can ameliorate age-dependent anemia and decrease ineffective erythropoiesis. This effect is mediated by the activation of the nuclear factor erythroid-2-related factor (Nrf2) function by peroxiredoxin-2, which ensures erythroid maturation and growth during aging [60].
Mitochondria also regulate glucose and lipid metabolism, and their dysfunction can exacerbate metabolic disorders [61]. Additionally, mitochondrial dysfunction is often observed in cancer cells [62]. Excessive ROS production contributes to DNA damage and genomic instability, which is a hallmark of cancer development. Cancer cells may also undergo metabolic reprogramming (known as the Warburg effect) to favor glycolysis over oxidative phosphorylation, even in the presence of oxygen, a process known as aerobic glycolysis. This helps cancer cells produce energy and biosynthetic precursors more rapidly, supporting rapid cell growth [63]. Dysfunctional mitochondria can release damage-associated molecular patterns (DAMPs), which trigger the immune system and contribute to autoimmune diseases such as rheumatoid arthritis and lupus. Finally, mitochondrial stress plays a role in activating inflammatory pathways associated with chronic inflammatory diseases [64].
4. Mitochondria in Hematopoiesis and Erythropoiesis
Hematopoiesis involves the differentiation of hematopoietic stem cells (HSCs) into various blood cell lineages, including red blood cells (RBCs), white blood cells (WBCs), and platelets. Mitochondria play crucial roles in this process, considering that they are involved in energy production, regulation of cellular metabolism, and maintenance of cell survival [65]. During hematopoiesis, the shift from stem cells to differentiated progenitor cells and specialized blood cells is accompanied by metabolic reprogramming [66], which is orchestrated by mitochondria to meet the energy and biosynthetic demands of each type of cells[67] [68]. Early-stage hematopoietic progenitor cells rely more on glycolysis (anaerobic metabolism) for energy, but as they mature into specialized blood cells, they increasingly rely on oxidative phosphorylation, which is a more energy-efficient process [69]. The early commitment of the erythroid lineage can be affected via the block of mitochondrial respiration [70,71].
In hematopoietic cells, mitochondrial dynamics are also important for cell division and differentiation. Fusion facilitates the mixing of mitochondrial contents, helping cells cope with metabolic stress, whereas fission enables the production of new mitochondria to support rapidly dividing cells [72]. Moreover, the regulation of apoptosis, a type of programmed cell death, is especially important in hematopoiesis, where proper cell turnover is necessary. This process ensures that only healthy cells survive, maintaining the proper balance of blood cell populations. Mitochondrial dysfunction can result in defects in the self-renewal and differentiation of HSCs, potentially leading to bone marrow failure or hematological diseases such as leukemia [73]. Additionally, the ROS generated in mitochondria regulate the self-renewal and differentiation of HSCs. Low ROS levels can stimulate the function and differentiation of HSCs, whereas excessive ROS can cause DNA damage and lead to hematopoietic cell death. Thus, mitochondria help maintain a balance of ROS that support normal hematopoiesis [74].
Erythropoiesis is the process by which RBCs are produced. This process occurs mainly in the bone marrow and involves several stages of proliferation, differentiation, and terminal maturation from erythroid progenitor cells to highly specialized oxygen-transporting cells filled with hemoglobin (erythrocytes). Mitochondria play a crucial role, particularly in the early phases of erythropoiesis. Early erythroblasts (precursors to red blood cells) rely on mitochondria for energy, as they undergo rapid division and differentiation [67]. At this stage, mitochondrial biogenesis, respiration, and metabolism are enhanced at least partly through mTORC1 (mammalian target of rapamycin complex 1)-mediated protein translation [75]. A mitochondrial deficiency induced by loss of a major mitochondrial transcriptional regulator, mitochondrial transcription factor A (TFAM), in erythroid cells leads to Inhibition of class I and II histone deacetylases (HDACs) causing increased histone acetylation and persistent expression of HSPC-associated genes to impair erythroid differentiation. These findings are consistent with the role of mitochondria in the modulation of key metabolites associated with epigenetic regulation [71]. Similarly, defective mitochondrial function impairs erythroid cell differentiation and leads to anemia due to insufficient production of mature red blood cells [76]. Moreover, conditions such as mitochondrial myopathy or Leber’s hereditary optic neuropathy are associated with defects in mitochondrial DNA that affect blood cell production [77].
As erythroblasts mature, their mitochondria become less active, and the cells start extruding their nuclei (reticulocytes) and finalizing their maturation into erythrocytes, which lack mitochondria. The molecular pathways involved in mitochondrial degradation in reticulocytes need to be fully elucidated, although researchers have made several important findings regarding this process, indicating that autophagy is important for mitochondrial clearance during terminal erythroid maturation. [78]. NIX and ULK1, two critical regulators of autophagy, play major roles in organelle clearance during reticulocyte maturation [79,80]. Nutrient and energy deprivation can result in the activation of ULK1 and autophagy induction by two sensor molecules, mTOR and AMP-activated protein kinase (AMPK) [81]. An increase in autophagy leads to a decrease in apoptosis during β-thalassemic mouse and patient erythropoiesis, suggesting that a lack of autophagy might be associated with high apoptosis of erythroblasts, a hallmark of ineffective erythropoiesis [82]. The loss of autophagy in erythroid cells leads to the defective removal of mitochondria and severe anemia in vivo. This is important since abnormal RBC retention in damaged mitochondria is responsible for the high frequency of death of erythrocytes [83]. Moreover, analyses of transcriptomic, proteomic, and metabolomic changes between hematopoietic stem/progenitor cells and differentiating erythroid cells in mice and humans have shown that mitochondria are critical regulators of erythropoiesis and participate in erythroid cell maturation, erythroblast enucleation, and RBC production [84].
Finally, the key function of mitochondria during erythropoiesis involves the synthesis of heme, the iron-containing molecule that binds oxygen in RBCs. Mitochondria are closely involved in the production of porphyrins, which are then used to produce heme. However, mitochondria also participate in iron metabolism, which must be regulated to prevent excess iron accumulation, as it can be toxic to cells. Mitochondrial iron is required for heme synthesis, and proper handling of iron is essential for the production of hemoglobin and the development of functional red blood cells [76].
5. Mitochondria in β-thalassemia
In β-thalassemia, excess globin chains that accumulate in red blood cell precursors can directly interact with mitochondria, impairing their function. Moreover, the inefficient production of hemoglobin causes an erythropoietin-driven expansion of early-stage erythroid precursors, leading to an increase in metabolic load [67]. This can cause mitochondrial dysfunction, as these cells support the increased energy demands related to the ineffective production of red blood cells [85].
When mitochondria become dysfunctional, they produce excessive ROS, leading to oxidative stress. ROS can further damage mitochondrial membranes, proteins, and DNA, impairing general cell function. A substantially different redox state, resulting from the differing levels of EPO, was reported in newly isolated CD34+ cells from β-thalassemia/Hb E patients compared to those from normal controls [86]. The effects on mitochondria were observed by the seventh day of differentiation, and significant deficits in activity were observed on day 10, coincident with significant levels of globin chain synthesis [87]. The greater number of mitochondria present in cells from thalassemia patients on day 10 suggested that the effect was magnified, with the damage to mitochondria at this point being coincident with the onset of apoptosis and ineffective erythropoiesis [88].
Moreover, a marked increase in mtDNA to nuclear DNA copy number (Mt/N) and ΔmtDNA4977 was observed in blood cells from adult individuals with transfusion-dependent thalassemia [89]. These findings supported an adaptive mechanism to secondary oxidant stress from uncontrolled labile plasma iron [90]. However, sickle cell disease (SCD) erythroid cells abnormally retain their mitochondria, a potential source of cell-free circulating DNA in this disease [91]. The retention of functional mitochondria in mature erythroid cells becomes a source of ROS, which further causes mtDNA damage and degradation [92]. Similarly, stress erythropoiesis or defects in mitophagy may be the source of mitochondrial retention in β-thalassemia [93].
6. Mitochondria-Targeting therapy
The recently approved drug luspatercept (ACE-536), which acts as a ligand trap for TGF β-like molecules, increases the differentiation of late erythroblasts, reduces hemichromes, and ameliorates anemia in a dose-dependent manner [94;95]. However, the metabolic pathways relevant to disease pathophysiology and the underlying mechanisms are poorly understood. In contrast, Mitapivat, a pyruvate kinase activator, has positive metabolic effects on animal models and patients [96]. Oral administration of drugs was found to ameliorate ineffective erythropoiesis in β-thalassemia model mice, resulting in increased ATP levels, reduced ROS levels, and improved mitochondrial clearance [97,98]. Moreover, early studies using a mouse model of SCD showed that the inhibition of lysine-specific demethylase-1 (LSD1) and mammalian target of rapamycin (mTOR) via RN-1 and sirolimus, respectively, reduced mitochondrial retention with a concomitant reduction in ROS in RBCs from SCD mice [99]. The inhibition of mTORC also led to the induction of autophagy due to an increase in Ulk1 expression, a decrease in the α-globin chain, and an increase in HbF in β-thalassemia patients [100]. An adequate understanding of the processes regulating normal and disordered erythropoiesis has important implications for therapeutic interventions. In this context, the evaluation of metabolic pathways involving mitochondria as potential targets for treating β-thalassemia or other hematologic disorders associated with ineffective erythropoiesis is of interest.
7. Conclusions
To summarize, while thalassemia is primarily a disorder of hemoglobin production, mitochondrial dysfunction due to oxidative stress, iron overload, and energy demands plays a significant role in the pathology of the disease, contributing to its complications. The end stages of the erythroid cell maturation process include nuclear expulsion followed by the clearance of mitochondria and other organelles to generate mature RBCs. If these processes are improperly regulated, apoptosis may occur, resulting in ineffective erythropoiesis. Studies have suggested that mitochondria actively participate in erythroid cell maturation, erythroblast enucleation, and RBC production. Thus, mitochondria may serve as a potential target for developing new drugs to treat β-thalassemia.
Author Contributions
Conceptualization, EDP and GG; writing—original draft preparation, EDP; writing—review GG; editing, VDS and MMDA. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable
Data Availability Statement
Suggested Data Availability Statements are available in section “MDPI Research Data Policies” at https://www.mdpi.com/ethics.
Acknowledgments
We acknowledge Luigi Ghilardini for graphical support in preparing the image.
Conflicts of Interest
The authors declare no conflicts of interest
References
- Weatherall DJ: Thalassemia as a global health problem: recent progress toward its control in the developing countries. Ann N Y Acad Sci 2010, 1202, 17–23. [CrossRef] [PubMed]
- Higgs DR, Engel JD, Stamatoyannopoulos G: Thalassaemia. Lancet 2012, 379, 373–383. [CrossRef] [PubMed]
- Taher A: Iron overload in thalassemia and sickle cell disease. Semin Hematol 2005, 42, S5–S9. [CrossRef] [PubMed]
- Musallam KM, Cappellini MD, Viprakasit V, Kattamis A, Rivella S, Taher AT: Revisiting the non-transfusion-dependent (NTDT) vs. transfusion-dependent (TDT) thalassemia classification 10 years later. Am J Hematol 2021, 96, E54–E56.
- Dadheech S, Jain S, Madhulatha D, Sharma V, Joseph J, Jyothy A, Munshi A: Association of Xmn1 -158 gammaG variant with severity and HbF levels in beta-thalassemia major and sickle cell anaemia. Mol Biol Rep 2014, 41, 3331–3337. [CrossRef]
- Thein SL: Genetic association studies in beta-hemoglobinopathies. Hematology Am Soc Hematol Educ Program 2013, 2013, 354–361. [CrossRef]
- Giardine B, Borg J, Viennas E, Pavlidis C, Moradkhani K, Joly P, Bartsakoulia M, Riemer C, Miller W, Tzimas G, Wajcman H, Hardison RC, Patrinos GP: Updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Res 2014, 42, D1063–D1069. [CrossRef]
- Sankaran VG, Weiss MJ: Anemia: progress in molecular mechanisms and therapies. Nat Med 2015, 21, 221–230. [CrossRef]
- Weatherall DJ: Phenotype-genotype relationships in monogenic disease: lessons from the thalassaemias. Nat Rev Genet 2001, 2, 245–255. [CrossRef]
- Gilman JG, Huisman TH: DNA sequence variation associated with elevated fetal G gamma globin production. Blood 1985, 66, 783–787. [CrossRef]
- Bianchi N, Cosenza LC, Lampronti I, Finotti A, Breveglieri G, Zuccato C, Fabbri E, Marzaro G, Chilin A, De AG, Borgatti M, Gallucci C, Alfieri C, Ribersani M, Isgro A, Marziali M, Gaziev J, Morrone A, Sodani P, Lucarelli G, Gambari R, Paciaroni K: Structural and Functional Insights on an Uncharacterized Agamma-Globin-Gene Polymorphism Present in Four beta0-Thalassemia Families with High Fetal Hemoglobin Levels. Mol Diagn Ther 2016, 20, 161–173.
- Sebastiani P, Farrell JJ, Alsultan A, Wang S, Edward HL, Shappell H, Bae H, Milton JN, Baldwin CT, Al-Rubaish AM, Naserullah Z, Al-Muhanna F, Alsuliman A, Patra PK, Farrer LA, Ngo D, Vathipadiekal V, Chui DH, Al-Ali AK, Steinberg MH: BCL11A enhancer haplotypes and fetal hemoglobin in sickle cell anemia. Blood Cells Mol Dis 2015, 54, 224–230.
- Lai Y, Zhou L, Yi S, Chen Y, Tang Y, Yi S, Yang Z, Wei H, Zheng C, He S: The association between four SNPs (rs7482144, rs4671393, rs28384513 and rs4895441) and fetal hemoglobin levels in Chinese Zhuang beta-thalassemia intermedia patients. Blood Cells Mol Dis 2017, 63, 52–57. [CrossRef] [PubMed]
- Stadhouders R, Aktuna S, Thongjuea S, Aghajanirefah A, Pourfarzad F, van IW, Lenhard B, Rooks H, Best S, Menzel S, Grosveld F, Thein SL, Soler E: HBS1L-MYB intergenic variants modulate fetal hemoglobin via long-range MYB enhancers. J Clin Invest 2014, 124, 1699–1710. [CrossRef]
- Thein SL, Menzel S, Peng X, Best S, Jiang J, Close J, Silver N, Gerovasilli A, Ping C, Yamaguchi M, Wahlberg K, Ulug P, Spector TD, Garner C, Matsuda F, Farrall M, Lathrop M: Intergenic variants of HBS1L-MYB are responsible for a major quantitative trait locus on chromosome 6q23 influencing fetal hemoglobin levels in adults. Proc Natl Acad Sci U S A 2007, 104, 11346–11351.
- Liu D, Zhang X, Yu L, Cai R, Ma X, Zheng C, Zhou Y, Liu Q, Wei X, Lin L, Yan T, Huang J, Mohandas N, An X, Xu X: KLF1 mutations are relatively more common in a thalassemia endemic region and ameliorate the severity of beta-thalassemia. Blood 2014, 124, 803–811. [CrossRef]
- Uda M, Galanello R, Sanna S, Lettre G, Sankaran VG, Chen W, Usala G, Busonero F, Maschio A, Albai G, Piras MG, Sestu N, Lai S, Dei M, Mulas A, Crisponi L, Naitza S, Asunis I, Deiana M, Nagaraja R, Perseu L, Satta S, Cipollina MD, Sollaino C, Moi P, Hirschhorn JN, Orkin SH, Abecasis GR, Schlessinger D, Cao A: Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc Natl Acad Sci U S A 2008, 105, 1620–1625.
- Mejri A, Mansri M, Hadj FS, Ouali F, Bibi A, Hafsia R, Messaoud T, Siala H: First description of the rs45496295 polymorphism of the C/EBPE gene in beta-thalassemia intermedia patients. Hemoglobin 2016, 40, 411–416. [CrossRef]
- Sherva R, Sripichai O, Abel K, Ma Q, Whitacre J, Angkachatchai V, Makarasara W, Winichagoon P, Svasti S, Fucharoen S, Braun A, Farrer LA: Genetic modifiers of Hb E/beta0 thalassemia identified by a two-stage genome-wide association study. BMC Med Genet 2010, 11, 51.
- Sun Y, Habara A, Le CQ, Nguyen N, Chen R, Murphy GJ, Chui DHK, Steinberg MH, Cui S: Pharmacologic induction of PGC-1alpha stimulates fetal haemoglobin gene expression. Br J Haematol 2022, 197, 97–109. [CrossRef]
- Lin J, Handschin C, Spiegelman BM: Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab 2005, 1, 361–370. [CrossRef] [PubMed]
- Liang H, Balas B, Tantiwong P, Dube J, Goodpaster BH, O’Doherty RM, DeFronzo RA, Richardson A, Musi N, Ward WF: Whole body overexpression of PGC-1alpha has opposite effects on hepatic and muscle insulin sensitivity. Am J Physiol Endocrinol Metab 2009, 296, E945–E954. [CrossRef] [PubMed]
- Cui S, Tanabe O, Lim KC, Xu HE, Zhou XE, Lin JD, Shi L, Schmidt L, Campbell A, Shimizu R, Yamamoto M, Engel JD: PGC-1 coactivator activity is required for murine erythropoiesis. Mol Cell Biol 2014, 34, 1956–1965. [CrossRef] [PubMed]
- Scarpulla RC: Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim Biophys Acta 2011, 1813, 1269–1278. [CrossRef] [PubMed]
- Sen T, Chen J, Singbrant S: Decreased PGC1beta expression results in disrupted human erythroid differentiation, impaired hemoglobinization and cell cycle exit. Sci Rep 2021, 11, 17129. [CrossRef]
- Siekevitz P: Powerhouse of the Cell. Sci Am 1957, 197, 131–144. [CrossRef]
- Spinelli JB, Haigis MC: The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol 2018, 20, 745–754. [CrossRef]
- Lee C, Zeng J, Drew BG, Sallam T, Martin-Montalvo A, Wan J, Kim SJ, Mehta H, Hevener AL, de CR, Cohen P: The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab 2015, 21, 443–454. [CrossRef]
- Rizzuto R, De SD, Raffaello A, Mammucari C: Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol 2012, 13, 566–578. [CrossRef]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X: Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 1996, 86, 147–157. [CrossRef]
- de ML, Arruda AP, da Costa RM, Benchimol M: Identification of a Ca2+-ATPase in brown adipose tissue mitochondria: regulation of thermogenesis by ATP and Ca2+. J Biol Chem 2006, 281, 16384–16390. [CrossRef] [PubMed]
- Yau WW, Yen PM: Thermogenesis in Adipose Tissue Activated by Thyroid Hormone. Int J Mol Sci 2020, 21.
- Melchinger P, Garcia BM: Mitochondria are midfield players in steroid synthesis. Int J Biochem Cell Biol 2023, 160, 106431.
- Popov LD: Mitochondrial biogenesis: An update. J Cell Mol Med 2020, 24, 4892–4899. [CrossRef]
- Jornayvaz FR, Shulman GI: Regulation of mitochondrial biogenesis. Essays Biochem 2010, 47, 69–84. [CrossRef]
- Seo BJ, Yoon SH, Do JT: Mitochondrial Dynamics in Stem Cells and Differentiation. Int J Mol Sci 2018, 19.
- Roy M, Reddy PH, Iijima M, Sesaki H: Mitochondrial division and fusion in metabolism. Curr Opin Cell Biol 2015, 33, 111–118. [CrossRef]
- Mishra P, Chan DC: Metabolic regulation of mitochondrial dynamics. J Cell Biol 2016, 212, 379–387. [CrossRef]
- Ploumi C, Daskalaki I, Tavernarakis N: Mitochondrial biogenesis and clearance: a balancing act. FEBS J 2017, 284, 183–195. [CrossRef]
- Chen Y, Zhou Z, Min W: Mitochondria, Oxidative Stress and Innate Immunity. Front Physiol 2018, 9, 1487. [CrossRef]
- Chen S, Liao Z, Xu P: Mitochondrial control of innate immune responses. Front Immunol 2023, 14, 1166214. [CrossRef] [PubMed]
- Cox AG, Pearson AG, Pullar JM, Jonsson TJ, Lowther WT, Winterbourn CC, Hampton MB: Mitochondrial peroxiredoxin 3 is more resilient to hyperoxidation than cytoplasmic peroxiredoxins. Biochem J 2009, 421, 51–58. [CrossRef] [PubMed]
- Cardenas-Rodriguez M, Chatzi A, Tokatlidis K: Iron-sulfur clusters: from metals through mitochondria biogenesis to disease. J Biol Inorg Chem 2018, 23, 509–520. [CrossRef] [PubMed]
- Belot A, Puy H, Hamza I, Bonkovsky HL: Update on heme biosynthesis, tissue-specific regulation, heme transport, relation to iron metabolism and cellular energy. Liver Int 2024, 44, 2235–2250. [CrossRef] [PubMed]
- La Morgia C, Maresca A, Caporali L, Valentino ML, Carelli V: Mitochondrial diseases in adults. J Intern Med 2020, 287, 592–608. [CrossRef] [PubMed]
- Kozhukhar N, Alexeyev MF: 35 Years of TFAM Research: Old Protein, New Puzzles. Biology (Basel) 2023, 12.
- Tuppen HA, Blakely EL, Turnbull DM, Taylor RW: Mitochondrial DNA mutations and human disease. Biochim Biophys Acta 2010, 1797, 113–128. [CrossRef]
- Taylor RW, Turnbull DM: Mitochondrial DNA mutations in human disease. Nat Rev Genet 2005, 6, 389–402. [CrossRef]
- Yuan J, Zhao J, Ye C, Pang L, Zhang X, Luk A, Du Y, Fan KY, Zhang X, Li B, Chen C: Leber’s Hereditary Optic Neuropathy with Mitochondrial DNA Mutation G11778A: A Systematic Literature Review and Meta-Analysis. Biomed Res Int 2023, 2023, 1107866.
- Gao R, Gu L, Zuo W, Wang P: Long-term prognostic factors and outcomes in mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes: a clinical and biochemical marker analysis. Front Neurol 2024, 15, 1491283. [CrossRef]
- Zhao Y, Hou Y, Zhao X, Liufu T, Yu M, Zhang W, Xie Z, Zhang VW, Yuan Y, Wang Z: The clinical, myopathological, and genetic analysis of 155 Chinese mitochondrial ophthalmoplegia patients with mitochondrial DNA single large deletions. Mol Genet Genomic Med 2024, 12, e2328. [CrossRef] [PubMed]
- Pyle A, Anugrha H, Kurzawa-Akanbi M, Yarnall A, Burn D, Hudson G: Reduced mitochondrial DNA copy number is a biomarker of Parkinson’s disease. Neurobiol Aging 2016, 38, 216.
- Podlesniy P, Figueiro-Silva J, Llado A, Antonell A, Sanchez-Valle R, Alcolea D, Lleo A, Molinuevo JL, Serra N, Trullas R: Low cerebrospinal fluid concentration of mitochondrial DNA in preclinical Alzheimer disease. Ann Neurol 2013, 74, 655–668. [CrossRef] [PubMed]
- Alqahtani T, Deore SL, Kide AA, Shende BA, Sharma R, Dadarao CR, Nemade LS, Kishor KN, Borah S, Shrikant DS, Behera A, Dhawal BD, Gaikwad N, Kalam AA, Ghosh A: Mitochondrial dysfunction and oxidative stress in Alzheimer’s disease, and Parkinson’s disease, Huntington’s disease and Amyotrophic Lateral Sclerosis -An updated review. Mitochondrion 2023, 71, 83–92.
- Xu M, Li T, Liu X, Islam B, Xiang Y, Zou X, Wang J: Mechanism and Clinical Application Prospects of Mitochondrial DNA Single Nucleotide Polymorphism in Neurodegenerative Diseases. Neurochem Res 2024, 50, 61.
- He YH, Lu X, Wu H, Cai WW, Yang LQ, Xu LY, Sun HP, Kong QP: Mitochondrial DNA content contributes to healthy aging in Chinese: a study from nonagenarians and centenarians. Neurobiol Aging 2014, 35, 1779–4.
- Gao X, Campian JL, Qian M, Sun XF, Eaton JW: Mitochondrial DNA damage in iron overload. J Biol Chem 2009, 284, 4767–4775. [CrossRef]
- Nie H, Chen G, He J, Zhang F, Li M, Wang Q, Zhou H, Lyu J, Bai Y: Mitochondrial common deletion is elevated in blood of breast cancer patients mediated by oxidative stress. Mitochondrion 2016, 26, 104–112. [CrossRef]
- Mohamed SA, Hanke T, Erasmi AW, Bechtel MJ, Scharfschwerdt M, Meissner C, Sievers HH, Gosslau A: Mitochondrial DNA deletions and the aging heart. Exp Gerontol 2006, 41, 508–517. [CrossRef]
- Mbiandjeu SCT, Siciliano A, Matte A, Federti E, Perduca M, Melisi D, Andolfo I, Amoresano A, Iolascon A, Valenti MT, Turrini F, Bovi M, Pisani A, Recchiuti A, Mattoscio D, Riccardi V, Dalle CL, Brugnara C, Mohandas N, De FL: Nrf2 Plays a Key Role in Erythropoiesis during Aging. Antioxidants (Basel) 2024, 13.
- Chien MC, Huang WT, Wang PW, Liou CW, Lin TK, Hsieh CJ, Weng SW: Role of mitochondrial DNA variants and copy number in diabetic atherogenesis. Genet Mol Res 2012, 11, 3339–3348. [CrossRef] [PubMed]
- Zhang G, Qu Y, Dang S, Yang Q, Shi B, Hou P: Variable copy number of mitochondrial DNA (mtDNA) predicts worse prognosis in advanced gastric cancer patients. Diagn Pathol 2013, 8, 173. [CrossRef] [PubMed]
- Hu Y, Liu W, Fang W, Dong Y, Zhang H, Luo Q: Tumor energy metabolism: implications for therapeutic targets. Mol Biomed 2024, 5, 63.
- Lyu Y, Wang T, Huang S, Zhang Z: Mitochondrial Damage-Associated Molecular Patterns and Metabolism in the Regulation of Innate Immunity. J Innate Immun 2023, 15, 665–679. [CrossRef]
- Filippi MD, Ghaffari S: Mitochondria in the maintenance of hematopoietic stem cells: new perspectives and opportunities. Blood 2019, 133, 1943–1952. [CrossRef]
- Papoin J, Yan H, Leduc M, Le GM, Narla A, Palis J, Steiner LA, Gallagher PG, Hillyer CD, Gautier EF, Mohandas N, Blanc L: Phenotypic and proteomic characterization of the human erythroid progenitor continuum reveal dynamic changes in cell cycle and in metabolic pathways. Am J Hematol 2024, 99, 99–112.
- Lyu J, Ni M, Weiss MJ, Xu J: Metabolic regulation of erythrocyte development and disorders. Exp Hematol 2024, 131, 104153. [CrossRef]
- Fontenay M, Cathelin S, Amiot M, Gyan E, Solary E: Mitochondria in hematopoiesis and hematological diseases. Oncogene 2006, 25, 4757–4767. [CrossRef]
- Simsek T, Kocabas F, Zheng J, DeBerardinis RJ, Mahmoud AI, Olson EN, Schneider JW, Zhang CC, Sadek HA: The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 2010, 7, 380–390. [CrossRef]
- Rossmann MP, Hoi K, Chan V, Abraham BJ, Yang S, Mullahoo J, Papanastasiou M, Wang Y, Elia I, Perlin JR, Hagedorn EJ, Hetzel S, Weigert R, Vyas S, Nag PP, Sullivan LB, Warren CR, Dorjsuren B, Greig EC, Adatto I, Cowan CA, Schreiber SL, Young RA, Meissner A, Haigis MC, Hekimi S, Carr SA, Zon LI: Cell-specific transcriptional control of mitochondrial metabolism by TIF1gamma drives erythropoiesis. Science 2021, 372, 716–721.
- Anso E, Weinberg SE, Diebold LP, Thompson BJ, Malinge S, Schumacker PT, Liu X, Zhang Y, Shao Z, Steadman M, Marsh KM, Xu J, Crispino JD, Chandel NS: The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nat Cell Biol 2017, 19, 614–625. [CrossRef] [PubMed]
- Luis TC, Lawson H, Kranc KR: Divide and Rule: Mitochondrial Fission Regulates Quiescence in Hematopoietic Stem Cells. Cell Stem Cell 2020, 26, 299–301. [CrossRef] [PubMed]
- Zhang A, Liu W, Qiu S: Mitochondrial genetic variations in leukemia: a comprehensive overview. Blood Sci 2024, 6, e00205. [CrossRef] [PubMed]
- Samimi A, Khodayar MJ, Alidadi H, Khodadi E: The Dual Role of ROS in Hematological Malignancies: Stem Cell Protection and Cancer Cell Metastasis. Stem Cell Rev Rep 2020, 16, 262–275. [CrossRef] [PubMed]
- Liu X, Zhang Y, Ni M, Cao H, Signer RAJ, Li D, Li M, Gu Z, Hu Z, Dickerson KE, Weinberg SE, Chandel NS, DeBerardinis RJ, Zhou F, Shao Z, Xu J: Regulation of mitochondrial biogenesis in erythropoiesis by mTORC1-mediated protein translation. Nat Cell Biol 2017, 19, 626–638.
- Gonzalez-Ibanez AM, Ruiz LM, Jensen E, Echeverria CA, Romero V, Stiles L, Shirihai OS, Elorza AA: Erythroid Differentiation and Heme Biosynthesis Are Dependent on a Shift in the Balance of Mitochondrial Fusion and Fission Dynamics. Front Cell Dev Biol 2020, 8, 592035.
- Finsterer J: Hematological manifestations of primary mitochondrial disorders. Acta Haematol 2007, 118, 88–98. [CrossRef]
- Chaichompoo P, Svasti S, Smith DR: The Roles of Mitophagy and Autophagy in Ineffective Erythropoiesis in beta-Thalassemia. Int J Mol Sci 2022, 23.
- Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J: Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008, 454, 232–235. [CrossRef]
- Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, Selak MA, Ney PA, Thompson CB: Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 2008, 112, 1493–1502. [CrossRef]
- Liu GY, Sabatini DM: mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol 2020, 21, 183–203. [CrossRef] [PubMed]
- Chaichompoo P, Nithipongvanitch R, Kheansaard W, Tubsuwan A, Srinoun K, Vadolas J, Fucharoen S, Smith DR, Winichagoon P, Svasti S: Increased autophagy leads to decreased apoptosis during beta-thalassaemic mouse and patient erythropoiesis. Sci Rep 2022, 12, 18628. [CrossRef] [PubMed]
- Mortensen M, Ferguson DJ, Edelmann M, Kessler B, Morten KJ, Komatsu M, Simon AK: Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc Natl Acad Sci U S A 2010, 107, 832–837. [CrossRef]
- Menon V, Slavinsky M, Hermine O, Ghaffari S: Mitochondrial regulation of erythropoiesis in homeostasis and disease. Br J Haematol 2024, 205, 429–439. [CrossRef]
- Lee HC, Yin PH, Lu CY, Chi CW, Wei YH: Increase of mitochondria and mitochondrial DNA in response to oxidative stress in human cells. Biochem J 2000, 348 Pt 2, 425–432.
- Khungwanmaythawee K, Sornjai W, Paemanee A, Jaratsittisin J, Fucharoen S, Svasti S, Lithanatudom P, Roytrakul S, Smith DR: Mitochondrial Changes in beta0-Thalassemia/Hb E Disease. PLoS One 2016, 11, e0153831.
- Leecharoenkiat A, Wannatung T, Lithanatudom P, Svasti S, Fucharoen S, Chokchaichamnankit D, Srisomsap C, Smith DR: Increased oxidative metabolism is associated with erythroid precursor expansion in beta0-thalassaemia/Hb E disease. Blood Cells Mol Dis 2011, 47, 143–157. [CrossRef]
- Mathias LA, Fisher TC, Zeng L, Meiselman HJ, Weinberg KI, Hiti AL, Malik P: Ineffective erythropoiesis in beta-thalassemia major is due to apoptosis at the polychromatophilic normoblast stage. Exp Hematol 2000, 28, 1343–1353. [CrossRef]
- Liu CS, Tsai CS, Kuo CL, Chen HW, Lii CK, Ma YS, Wei YH: Oxidative stress-related alteration of the copy number of mitochondrial DNA in human leukocytes. Free Radic Res 2003, 37, 1307–1317. [CrossRef]
- Lal A, Gomez E, Calloway C: Increased mitochondrial DNA deletions and copy number in transfusion-dependent thalassemia. JCI Insight 2016, 1.
- Moriconi C, Dzieciatkowska M, Roy M, D’Alessandro A, Roingeard P, Lee JY, Gibb DR, Tredicine M, McGill MA, Qiu A, La CF, Francis RO, Hod EA, Thomas T, Picard M, Akpan IJ, Luckey CJ, Zimring JC, Spitalnik SL, Hudson KE: Retention of functional mitochondria in mature red blood cells from patients with sickle cell disease. Br J Haematol 2022, 198, 574–586.
- Esperti S, Nader E, Stier A, Boisson C, Carin R, Marano M, Robert M, Martin M, Horand F, Cibiel A, Renoux C, Van BR, Blans C, Dargaud Y, Joly P, Gauthier A, Poutrel S, Romana M, Roussel D, Connes P: Increased retention of functional mitochondria in mature sickle red blood cells is associated with increased sickling tendency, hemolysis and oxidative stress. Haematologica 2023, 108, 3086–3094.
- Martino S, Arlet JB, Odievre MH, Jullien V, Moras M, Hattab C, Lefebvre T, Gouya L, Ostuni MA, Lefevre SD, Le Van KC: Deficient mitophagy pathways in sickle cell disease. Br J Haematol 2021, 193, 988–993. [CrossRef]
- Suragani RN, Cadena SM, Cawley SM, Sako D, Mitchell D, Li R, Davies MV, Alexander MJ, Devine M, Loveday KS, Underwood KW, Grinberg AV, Quisel JD, Chopra R, Pearsall RS, Seehra J, Kumar R: Transforming growth factor-beta superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat Med 2014, 20, 408–414.
- Dussiot M, Maciel TT, Fricot A, Chartier C, Negre O, Veiga J, Grapton D, Paubelle E, Payen E, Beuzard Y, Leboulch P, Ribeil JA, Arlet JB, Cote F, Courtois G, Ginzburg YZ, Daniel TO, Chopra R, Sung V, Hermine O, Moura IC: An activin receptor IIA ligand trap corrects ineffective erythropoiesis in beta-thalassemia. Nat Med 2014, 20, 398–407.
- Matte A, Wilson AB, Gevi F, Federti E, Recchiuti A, Ferri G, Brunati AM, Pagano MA, Russo R, Leboeuf C, Janin A, Timperio AM, Iolascon A, Gremese E, Dang L, Mohandas N, Brugnara C, De FL: Mitapivat reprograms the RBC metabolome and improves anemia in a mouse model of hereditary spherocytosis. JCI Insight 2023, 8.
- Matte A, Federti E, Kung C, Kosinski PA, Narayanaswamy R, Russo R, Federico G, Carlomagno F, Desbats MA, Salviati L, Leboeuf C, Valenti MT, Turrini F, Janin A, Yu S, Beneduce E, Ronseaux S, Iatcenko I, Dang L, Ganz T, Jung CL, Iolascon A, Brugnara C, De FL: The pyruvate kinase activator mitapivat reduces hemolysis and improves anemia in a beta-thalassemia mouse model. J Clin Invest 2021, 131.
- Quezado ZMN, Kamimura S, Smith M, Wang X, Heaven MR, Jana S, Vogel S, Zerfas P, Combs CA, Almeida LEF, Li Q, Quezado M, Horkayne-Szakaly I, Kosinski PA, Yu S, Kapadnis U, Kung C, Dang L, Wakim P, Eaton WA, Alayash AI, Thein SL: Mitapivat increases ATP and decreases oxidative stress and erythrocyte mitochondria retention in a SCD mouse model. Blood Cells Mol Dis 2022, 95, 102660.
- Jagadeeswaran R, Vazquez BA, Thiruppathi M, Ganesh BB, Ibanez V, Cui S, Engel JD, Diamond AM, Molokie RE, DeSimone J, Lavelle D, Rivers A: Pharmacological inhibition of LSD1 and mTOR reduces mitochondrial retention and associated ROS levels in the red blood cells of sickle cell disease. Exp Hematol 2017, 50, 46–52.
- Lechauve C, Keith J, Khandros E, Fowler S, Mayberry K, Freiwan A, Thom CS, Delbini P, Romero EB, Zhang J, Motta I, Tillman H, Cappellini MD, Kundu M, Weiss MJ: The autophagy-activating kinase ULK1 mediates clearance of free alpha-globin in beta-thalassemia. Sci Transl Med 2019, 11.
Figure 1.
Overview of mitocondria functions in maintaining red blood cell production and function.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



