
Submitted:
05 January 2025
Posted:
06 January 2025
You are already at the latest version
Abstract
Atherosclerosis (AS) is a chronic inflammatory vascular disease closely tied to cellular metabolism. Recent genome-wide association study data have suggested that the significant roles of endothelial cells, smooth muscle cells, and macrophages in the regression and exacerbation of AS. However, the impact of cellular crosstalk and cellular metabolic derangements on disease progression in AS is vaguely understood. In this review, we analyze the roles of the three cell types in AS. We also summarize the crosstalk between the two of them, and the associated molecules and consequences involved. In addition, we emphasize potential therapeutic targets and highlight the importance of the three-cell co-culture model and extracellular vesicles in AS-related research, providing ideas for future studies.
Keywords:
atherosclerosis
; endothelial cells
; smooth muscle cells
; macrophages
1. Introduction
With the improvement of people's living standard and concern for life and health, cardiovascular diseases have been increasingly emphasized. Atherosclerosis (AS), as an important cardiovascular disease, is a chronic inflammatory vascular disease that usually occurs in large and medium-sized arteries, and even leads to cardiovascular diseases such as coronary heart disease, cerebral infarction and peripheral vascular disease in severe cases. Various reasons such as heredity, poor dietary habits, chronic diseases, etc. can cause AS, which produces stimulation to human cells. The exchange of information between cells causes changes in cellular physiological activity, which in turn alters the body's integrated response to the external environment and the process of growth and development. Therefore, it is of great significance to study the pathogenesis of AS and to explore the mechanism of intercellular interaction for the treatment of cardiovascular diseases.
The traditional theory of lipid infiltration believes that molecules such as excess low-density lipoprotein (LDL) can penetrate arterial tissue and undergo a series of reactions. Oxidized low-density lipoproteins (ox-LDL) and others formed in this process activate endothelial cells (ECs) to recruit monocytes and help convert monocytes that enter the intima into macrophages. While macrophages phagocytose ox-LDL, lipid deposition due to excess ox-LDL causes macrophages to form foam cells. ox-LDL also stimulates phenotypic conversion of smooth muscle cells (SMCs), which results in foam cells. Releasing lysosomal enzymes and lipids, necrotic rupture of foam cells consequently encourages the development of atheromatous plaque.
Russell Ross proposed the doctrine of endothelial damage and inflammation in 1999. He clarified that the development of AS is accompanied by the involvement of various inflammatory cells and inflammatory factors, among which the damage of arterial endothelium is the main reason for the development of AS. ECs release adhesion factors that adhere to monocytes into the subendothelial space, so that they phagocytose oxidized lipids and form foam cells. The accumulation of lipid plaques leads to inflammation, causing the body to develop a series of immune-inflammatory responses in which ECs, SMCs, and macrophages play an important role. According to a research using public genome-wide association study data[1],genes for cardiovascular disease susceptibility were determined to be enriched in diseased macrophages, ECs, and SMCs. This undoubtedly shows the importance of these three cells in cardiovascular disease.
In this review, we discuss the roles of ECs, SMCs, and macrophages in AS based on the latest research advances, and summarize the signaling communication process between the three cell types two by two for further research and exploration.
2. Roles of Endothelial Cells, smooth Muscle Cells and Macrophages in Atherosclerosis
2.1. Endothelial Cells
It is commonly known that ECs are mainly involved in constituting the inner wall of arterial vessels and participate in the process of selective fluid osmotic exchange between blood and tissue fluids. Meanwhile, their intercellular junctions and transmembrane transport of vesicles contribute to the transport of substances and exchange of information in blood and tissue cells. Normally, ECs in the human vasculature are in a relatively stable state, but in pathological states like injury or hypoxia, the ECs can be activated rapidly and undergo neovascularization that provides oxygen and nutrients to the tissues in order to slow down further damage[2]. However, the continuous activation of the EC can lead to a decompensated phase, resulting in endothelial dysfunction and AS.
2.2. Smooth Muscle Cells
One of the key features of AS is the proliferation and migration of SMCs, which are found in the tunica media. SMCs are highly plastic and can undergo phenotypic transformation through signaling pathways such as Wnt, Notch, and transforming growth factor (TGF)-β. In normal individuals, the SMCs in the arterial vessel wall mostly exhibit a contractile phenotype, which have limited proliferative and migratory potential, and serve to maintain vascular elasticity and constrict blood vessels. However, SMCs in pathological conditions transform into a synthetic phenotype with enhanced capabilities for cell migration and proliferation, which can be specifically classified into proliferative-migratory, inflammatory, and ossification/osteogenic phenotypes, etc. This phenotype is able to synthesize a large amount of extracellular matrix (ECM), collagen, and bone-bridging proteins, among other things. Currently, some studies suggest that in the early stage of AS, abnormally proliferating SMCs can promote plaque formation, whereas in the late stage, SMCs have the ability to prevent the rupture of the fibrous cap to stabilize the plaque.[3]Thus, it is clear that SMCs play different roles at different periods of AS.
2.3. Endothelial Cells
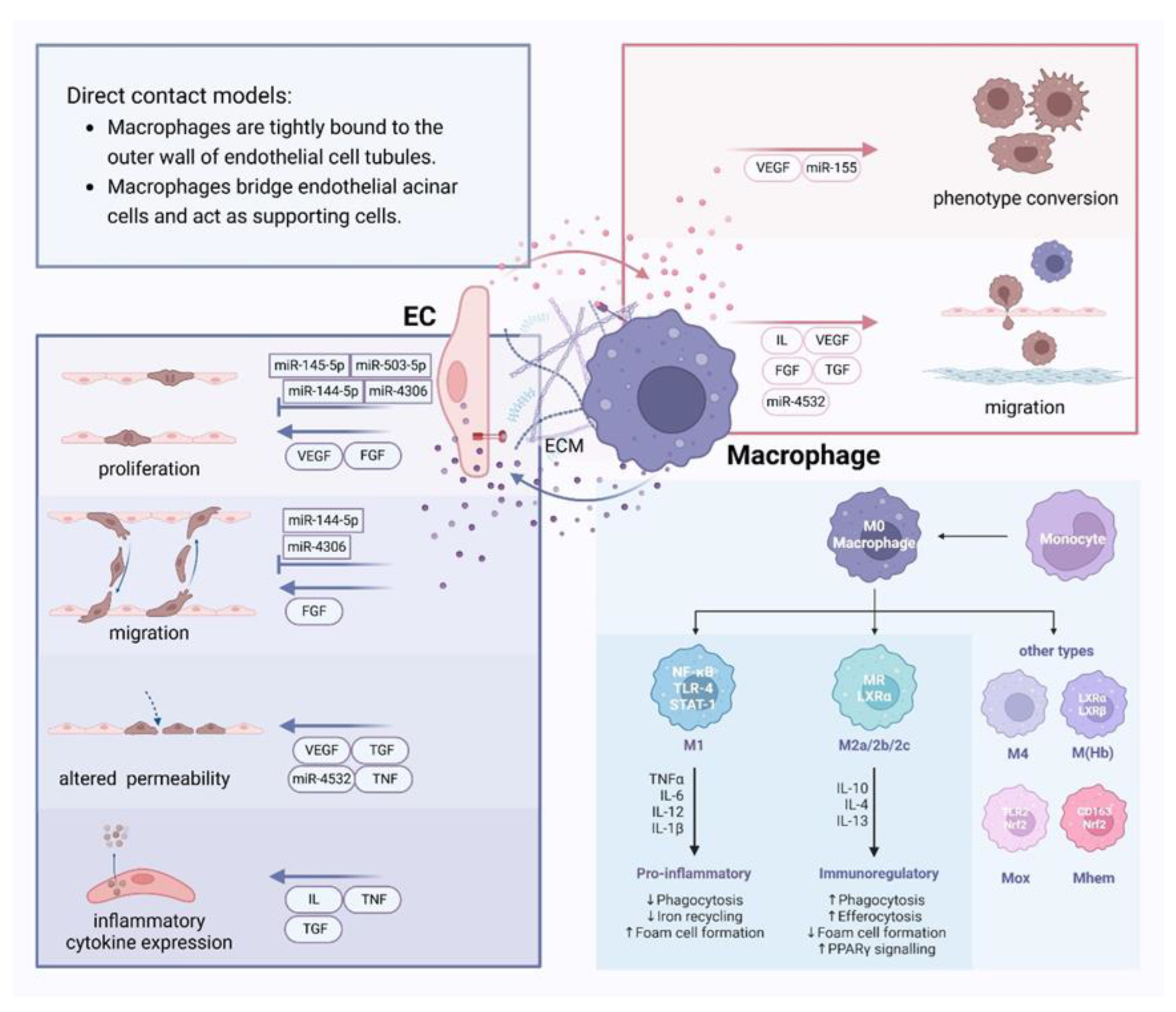
Macrophages are mainly differentiated from different kinds of cells, such as monocytes, ECs and SMCs, and can also be proliferated from macrophages in local tissues. They can generally be divided into three groups: recruited macrophages, tissue-resident macrophages, and perivascular macrophages[4]. These cells become activated when they are stimulated, which results in increased cell size, more active cell metabolism, stronger phagocytosis, etc. There are two main phenotypes of macrophages: M1 and M2. It is now believed that the M1 phenotype stimulates the production of pro-inflammatory factors like tumor necrosis factor (TNF) -α, interleukin (IL)-1β, and nitric oxide (NO), which play a critical part in the initial process of AS. On the other hand, the M2 phenotype secretes pro-angiogenic anti-inflammatory cytokines like TGF-β and IL -10, which support tissue repair and angiogenesis[5]. In addition to the two phenotypes, M1 and M2, cellular phenotypes such as M(Hb), Mhem, HA-mac, Mox, and M4[6, 7] have different characteristics and are all important in AS.
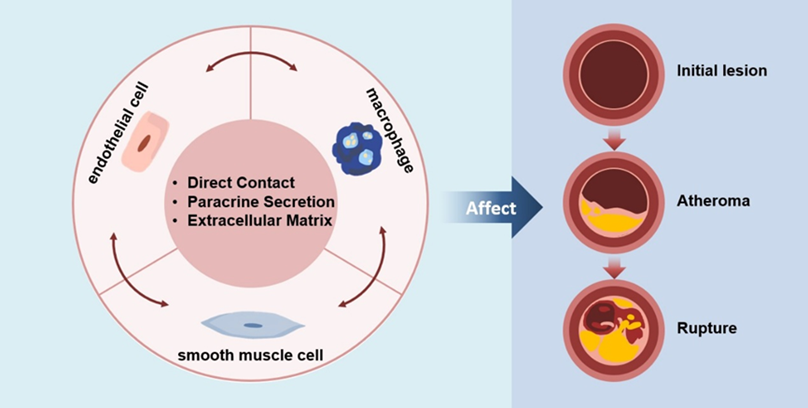
Figure 1.
Intercellular Communication Affects the Progression of Atherosclerosis In atherosclerosis, initial changes at the cellular and molecular levels lead to endothelial injury, which may not immediately cause luminal narrowing or impairment of tissues and organs. The foam cells then gather below the endothelium cells and appear as yellow fat splotches. This is followed by the accumulation of lipids within and outside the cells, generating lipid pools that disrupt the intimal structure and deform the arterial wall. Smooth muscle cells migrate to participate in the formation of a fibrous cap, leading to the emergence of white plaques that protrude into the arterial lumen, causing narrowing and the formation of atherosclerotic and fibrous plaques. On the basis of these plaques, secondary lesions such as hemorrhage, necrosis, ulceration, calcification, and mural thrombosis can occur, potentially triggering acute cardiovascular events. In this process, endothelial cells, smooth muscle cells and macrophages all play important roles.
Figure 1.
Intercellular Communication Affects the Progression of Atherosclerosis In atherosclerosis, initial changes at the cellular and molecular levels lead to endothelial injury, which may not immediately cause luminal narrowing or impairment of tissues and organs. The foam cells then gather below the endothelium cells and appear as yellow fat splotches. This is followed by the accumulation of lipids within and outside the cells, generating lipid pools that disrupt the intimal structure and deform the arterial wall. Smooth muscle cells migrate to participate in the formation of a fibrous cap, leading to the emergence of white plaques that protrude into the arterial lumen, causing narrowing and the formation of atherosclerotic and fibrous plaques. On the basis of these plaques, secondary lesions such as hemorrhage, necrosis, ulceration, calcification, and mural thrombosis can occur, potentially triggering acute cardiovascular events. In this process, endothelial cells, smooth muscle cells and macrophages all play important roles.
3. Crosstalk
Crosstalk between the three cell types can be broadly classified into three categories: direct cell-to-cell communication, paracrine secretion of vasoactive substances and extracellular vesicles (EVs), and cell-matrix interactions.
3.1. Crosstalk Between Endothelial Cells and Smooth Muscle Cells
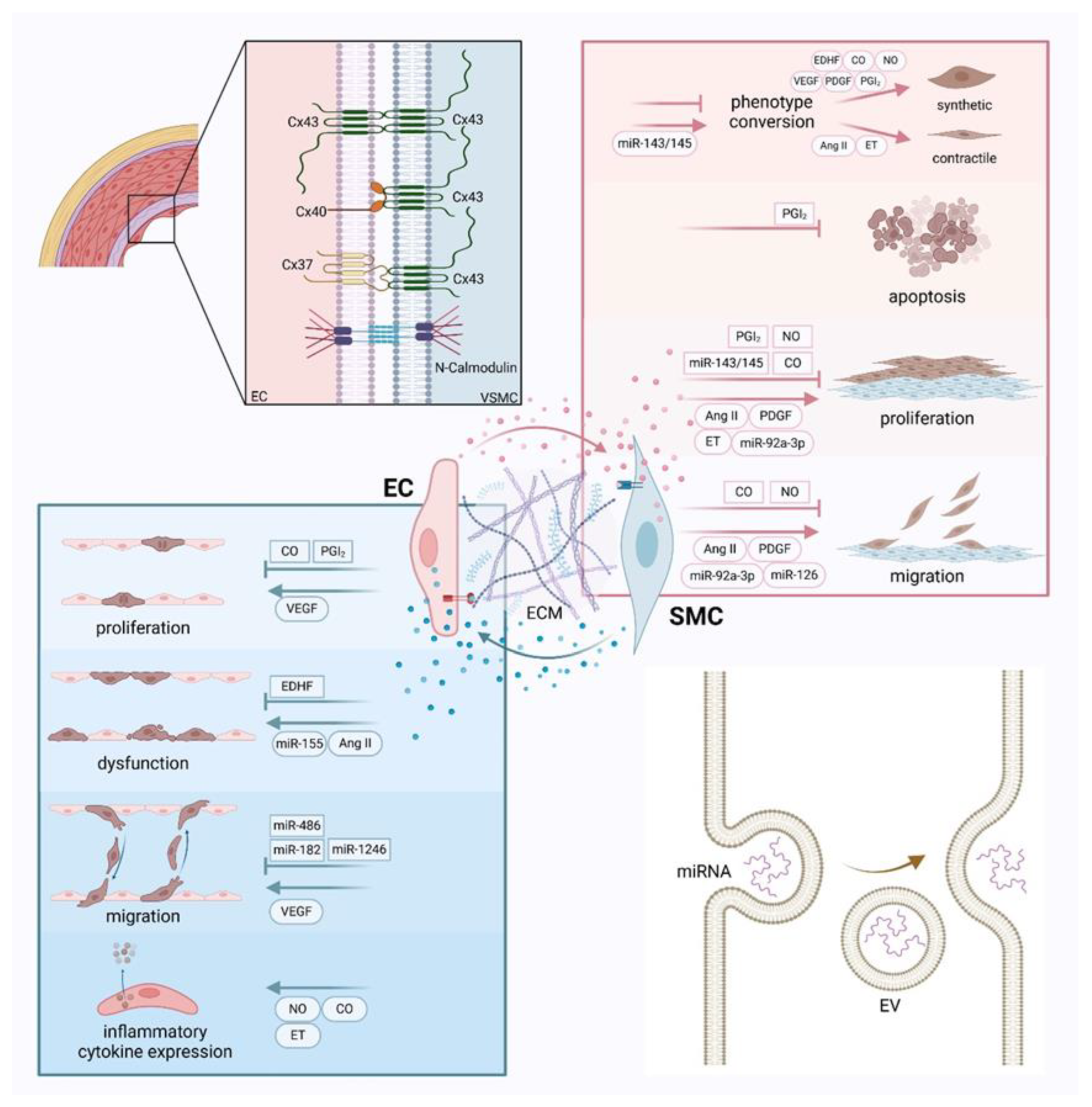
Endothelial cells (ECs) and smooth muscle cells (SMCs) communicate directly through gap junctions and adhesion junctions, involving proteins such as Cx37, Cx40, Cx43, and N-cadherin. The two cells can also influence each other through various vasoactive substances and extracellular vesicles. Extracellular vesicles are formed from the plasma membrane and transport molecules such as miRNAs and peptides. These chemicals regulate EC proliferation, dysfunction, migration, and expression of inflammatory factors. They can also alter SMC phenotypic transition, apoptosis, proliferation, migration, and expression of inflammatory factors. Furthermore, the extracellular matrix can mediate communication between ECs and SMCs through its protein or carbohydrate components and mechanical transmission.
3.1.1. Direct Contact
The direct communication process between ECs and SMCs that are in contact with each other can occur through gap junctions and adhesion junctions.
In gap junctions, six identical connexin proteins form a connexon, and the connexons on the surfaces of adjacent cells connect to form channels through which signaling molecules can pass. This allows metabolic coupling between cells, playing a crucial role in cell growth and proliferation. The gap junctions that have been investigated between EC and SMC are primarily myoendothelial gap junctions (MEGJs), which are located at the interface of adjacent protrusions formed by ECs and SMCs, and involved in the regulation of vascular tension. The major connexins in the vascular system include Cx37, Cx40, Cx43, and Cx45. All connexins except Cx45 are present in the MEGJs between ECs and SMCs[8]. Since ECs predominantly express Cx37, Cx40, and Cx43, and SMCs mainly express Cx43 and Cx45[8], the MEGJ between them is primarily composed of Cx43/Cx43, Cx40/Cx43, and Cx37/Cx43, with the specific type also related to individual species and vascular types. In the ECs covering late-stage atherosclerotic plaques, the expression of Cx40 and Cx37 is undetectable. However, these connexins are highly expressed in the endothelium around the plaque and during the development of AS[9]. Blocking the expression of Cx43 induced by ox-LDL can activate SMCs autophagy by inhibiting the PI3K/AKT/mTOR signaling pathway, thereby inhibiting SMC hyperproliferation and migration[10], as well as foam cell formation[11]. Additionally, Cx37 in injured arteries can promote gap junction communication by phosphorylating Cx43 via the Akt pathway, which in turn limits SMC proliferation and intimal hyperplasia[12, 13]. Gap junctions containing Cx43 can also mediate signal transmission from SMCs to ECs to regulate vascular tone. When the intracellular Ca2+ levels in SMCs increase, gap junction inhibitors can suppress the passage of IP3, thus preventing the elevation of Ca2+ levels in ECs[14]. Furthermore, the function of connexins is regulated by cytokines; for instance, in vitro, bacterial lipopolysaccharide, tumor necrosis factor-α, and IL-1β can selectively inhibit intercellular communication between human vascular gap junction cells[15]. The impact of changes in the expression of different connectivity proteins during AS development on AS is yet to be fully elucidated.
Figure 2.
Crosstalk between endothelial cells and smooth muscle cells and extracellular vesicle transfer process.
Figure 2.
Crosstalk between endothelial cells and smooth muscle cells and extracellular vesicle transfer process.
Besides gap junctions, ECs and SMCs are also connected by adhesion junctions. N-cadherin, an adhesion molecule that can mediate the connection between cells[16], is present in the elastic membrane between the EC and SMC layers, suggesting that N-cadherin may mediate EC-SMC interactions[17]. New studies have shown that inhibiting N-cadherin function can inhibit SMC migration and promote EC survival, thereby delaying endothelial thickening[18, 19]. Additionally, VE-cadherin participates in the adhesion junctions between ECs, but EC proliferation induced by mechanical stretch necessitates contact with SMCs, whereas SMC proliferation occurs without the need for interaction with other cells[20]. This highlights the significance of direct cell-to-cell communication between the two cell types for AS development.
3.1.2. Paracrine Secretion
Vasoactive Substance
Endothelium-derived diastolic factors such as NO, carbon monoxide (CO), prostacyclin (PGI2), and endothelium-derived hyperpolarizing factor (EDHF) lead to SMC and vasodilatory relaxation. Conversely, angiotensin II (AngII), and endothelin (ET), are endothelium-derived contracting factors that promote SMC and vasoconstriction. These factors can also be secreted by SMCs and consequently influence the growth and metabolic processes of ECs. Moreover, growth factors such as platelet-derived growth factor (PDGF) and vascular endothelial growth factor (VEGF) also regulate cell activity.
Endothelial nitric oxide synthase (eNOS) can oxidize L-arginine to promote the production of NO after external or physiological stimulation[21]. The newly formed NO diffuses rapidly to the cell membrane of ECs, thereby changing the concentration of cyclic guanosine monophosphate (cGMP) [22], which causes vasodilation in SMCs[23, 24], and reversibly inhibits SMCs migration[25] and proliferation[26]. This process also promotes the transformation of SMCs from a contractile to a synthetic phenotype through the soluble guanylyl cyclase (sGC)-PRKG1 signaling pathway[27]. Moreover, recent studies have shown that eNOS can also be expressed in SMCs[28], and it has been demonstrated that the interaction of eNOS with cell adhesion molecules (CAMs) in SMCs is promoted by ECs. This suggests that in the process of new vessel formation, SMCs may compensate for the production of NO by ECs to regulate vascular tone.
CO can be produced by heme oxygenase catalysis[29]. CO shares many similarities with NO, as it can also bind to and activate sGC, leading to an increase in cGMP levels in cells and subsequently causing vasodilation of SMCs. In addition to these, CO can inhibit SMC proliferation and migration. For example, CO inhibits SMC proliferation and neointimal formation by activating p38 mitogen-activated protein kinase (MAPK) and p21 (Waf1 / Cip1) [30, 31] or through the TGF-β1 pathway[32]. It also inhibits SMC migration stimulated by PDGF by reducing the enzyme activity of Nox1[33]. On the other hand, CO released by SMCs can increase endothelial cGMP levels and reduce the expression of mitogens, ET-1, and PDGF-B under hypoxia, which in turn inhibits SMC proliferation[34]. CO can also restore TNF-α-induced downregulation of eNOS and endothelial dysfunction by inhibiting NF-κB[35]. However, the specific role of CO in AS needs further research.
PGI2 is a class of fatty acids produced by the metabolism of arachidonic acid via the cyclooxygenase (COX) pathway. In response to proinflammatory mediators and mechanical stress, PGI2 produced by ECs can bind to SMC surface receptors, activate adenylate cyclase, and generate cAMP. This subsequently activates protein kinase A, inducing relaxation of both blood vessels and SMCs, and inhibiting platelet aggregation[36, 37]. PGI2 also can reduce SMC apoptosis and maintain the contractile phenotype through PPARα and 14-3-3 signaling pathways[38, 39]. Studies have also shown that PGI2 receptor deficiency or inhibition of PGI2 production by inhibiting COX-2 can accelerate AS and thrombosis in mice[40]. Therefore, severe endothelial injury leads to insufficient PGI2 production, which results in SMC apoptosis, proliferation, and intimal hyperplasia. Moreover, PGI2 promotes the release of NO from ECs[41] while NO can also enhance the effects of PGI2 on SMCs.
EDHF is a class of active substances that dilate blood vessels, distinct from NO and PGI2. It does not refer to a specific substance but includes cytochrome P450 metabolites, hydrogen peroxide, cyclic adenosine monophosphate, and C-type natriuretic peptide, among others[42]. EDHF may serve as a compensatory pathway for NO-dependent endothelial vascular dilation, promoting vascular relaxation before the development of AS in the aorta[43, 44]. Similarly, in elderly obese mice on a long-term high-fat diet, a reduction in EDHF-mediated vascular relaxation has been found. This reduction may potentially contribute to endothelial dysfunction in small resistance arteries[45, 46]. In addition, the absence of the Cx40 gene slows down the EDHF-mediated spread of hyperpolarization from ECs to SMCs[47]. Therefore, EDHF also plays a role in vascular relaxation through gap junctions.
Ang II is a peptide with potent vasoconstrictive properties that is typically converted from angiotensinogen by renin and angiotensin-converting enzyme (ACE), or produced via a non-ACE-dependent system[48]. Ang II can be recognized by two different G protein-coupled receptors, the AngII type 1 receptor (AT1R) and the AngII type 2 receptor (AT2R). AT1R usually mediates vasoconstriction and inflammatory responses, while AT2R is generally associated with vasodilation and apoptosis[49]. Ang II can advance the progression of AS by affecting ECs through AT1R or by mediating oxidative stress[49]. For instance, Ang II, when bound to AT1R, activates the JNK pathway, which promotes the expression of ACE in ECs. Ang II-mediated oxidative stress can lead to dysregulation of NO production in ECs, resulting in endothelial dysfunction[50]. Additionally, Ang II can promote the transition of SMCs from a contractile to a synthetic phenotype, proliferation, migration, and alterations in ECM components through AT1R[50]. It can also facilitate the transformation of SMCs into foam cells by increasing the expression of the lipoprotein receptor LRP1[51]. Moreover, AT2R inhibits the lectin-like oxidized low-density lipoprotein receptor LOX-1, and the expression of inflammatory mediators reduced in plaques from AS-simulated mice with overexpression of AT2R[52], indicating a certain anti-inflammatory effect of AT2R. Although both ECs and SMCs can produce Ang II, most studies focus on the individual effects of Ang II on ECs or SMCs, with fewer investigations into their communication through Ang II. In vitro experiments suggest that endothelial progenitor cells may inhibit Ang II-induced proliferation of vascular SMCs by blocking MAPKs and NF-κB signaling pathways and the expression of related factors, with early endothelial progenitor cells showing a more significant inhibitory effect compared to human umbilical vein ECs [53]. In summary, the crosstalk between ECs and SMCs mediated by Ang II and the role of AT2R in AS deserve further research.
ET is produced from big endothelin-1 through the action of peptidases and endothelin-converting enzymes. There are three isoforms of ET: ET-1, ET-2, and ET-3[54]. Among them, ET-1 plays a significant role in regulating cardiovascular function. Both ECs and SMCs can secrete ET-1, which binds to two types of ET receptors, ETA and ETB, present in the human body to exert its effects. ETA receptors are primarily found on SMCs and mediate cell proliferation and vasoconstriction[55]. When ET-1 binds to ETA, it activates phospholipase C, promotes the opening of Ca2+ channels, leading to SMC contraction. Additionally, ET-1 activates the transcription of related genes through a cascade amplification signaling system, promoting cell proliferation and differentiation[56]. ETB receptors are mainly present on ECs and mediate vasodilation by promoting NO release[57]. ETB can utilize cytoskeletal rearrangement and Rho kinase to increase the expression of CHSY-1 in aortic ECs through the transduction of TGF-β type I receptors[58].
PDGF is a bioactive peptide growth factor that can be produced by ECs and SMCs. It can be divided into PDGF-A, PDGF-B, PDGF-C, and PDGF-D subtypes, which can form homodimers or heterodimers PDGF-AA, PDGF-BB, PDGF-AB, PDGF-CC, and PDGF-DD[59] through disulfide bond linkage, and they function by binding to the ligand and platelet-derived growth factor receptor (PDGFR). PDGF-B was predominantly present in ECs and PDGF-A in SMCs. Influenced by some pro-inflammatory factors[60, 61] and low shear stress[62], the expression of PDGF-B in ECs increases and induces the proliferation, migration, and phenotypic switching of SMCs. Similarly, decreased PDGF-B expression in ECs prevents SMC migration and proliferation, which lessens the damage-induced neointimal hyperplasia[63]. Although PDGF-A is often used as an indicator for AS development, research on its role in cell communication is relatively limited.
VEGF is a dimeric cationic glycoprotein whose family members include VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E, VEGF-F, placental growth factor, and endocrine gland-derived vascular endothelial growth factor (EG-VEGF) [64, 65]. These proteins can bind to vascular endothelial growth factor receptors (VEGFRs) to exert their effects. Inducing angiogenesis and increasing vascular permeability, VEGF-A plays a critical function in the proliferation and migration of tissue vascular ECs[66]. It can also upregulate endothelial eNOS through VEGFR-2/KDR, facilitating NO release and vascular relaxation[67]. Furthermore, research has demonstrated that PDGF can stimulate EC proliferation in vitro by upregulating VEGF expression in SMCs via the ERK-1/2 and AP-1 signaling pathways[68]. It can be seen that different signaling pathways can influence each other, suggesting that blocking the ERK-1/2 and AP-1 signaling pathways may provide a potential approach for treating AS.
Extracellular Vesicle
EVs are closed membrane vesicles formed by a phospholipid bilayer. They serves as carriers of proteins, lipids, DNA, and mRNA, representing a novel pathway for intercellular communication. EVs can be classified into three types: exosomes, microvesicles, and apoptotic bodies. Exosomes typically have a diameter of 30-100nm and are produced through the endosomal pathway, collecting intraluminal vesicles in multivesicular bodies, which then fuse with the plasma membrane to release exosomes[69, 70]. Microvesicles have a diameter of 100-1000nm and are formed by budding from the plasma membrane. Apoptotic bodies, with a diameter of approximately 50-5000nm, are formed by the formation of bubbles in the plasma membrane of apoptotic cells[71]. The production and release of EVs depend on external stimuli and changes in the body's internal environment, such as environmental stress and calcium concentration, which can alter the physiological activities of target cells. ECs, SMCs, and macrophages can all produce EVs.
As one of the key molecules transported by EVs, miRNAs are short single-stranded RNA molecules composed of 18-22 nucleotides[72]. Most miRNAs in circulating plasma are found in plasma exosomes and microvesicles[73]. The miRNAs in exosomes are involved in a range of physiological and pathological processes, including AS. They play a crucial role in the communication between ECs and SMCs and in cell growth and metabolism in AS[74], such as miRNA-126, miRNA-214, miRNA-143/145, miR-92a-3p, miRNA-155, and miRNA-1246, miRNA-182, and miRNA-486, among others[75]. Under the effect of laminar shear stress in AS lesions, EC-released miR-126 can stimulate SMC migration[76]. ECs can also secrete exosomes rich in miR-214, which mediates an anti-angiogenic effect by downregulating the protein Quaking and inhibiting the expression of pro-angiogenic growth factors[77]. However, it has also been shown that miR-214 stimulates AS by inhibiting the expression of mutated ataxia telangiectasia mutated in neighboring target cells[78]. The opposite effects may be due to differences in experimental materials or mediating pathways. miR-143/145 can promote SMC differentiation and inhibit their proliferation[79]. According to studies, the shear response transcription factor Krüppel-like factor 2 stimulates ECs to secrete EVs containing miRNA-143/145 to SMCs, which inhibits the expression of miR-143/145 target genes and dedifferentiation-related genes to reduce the formation of AS lesions[74]. Exosomes from ECs can transfer miR-92a-3p to SMCs, inducing cell proliferation and migration and exacerbating inflammatory responses[80]. On the other hand, Krüppel-like factor 5 mediates SMC-derived EVs to deliver miRNA-155 to ECs, leading to increased endothelial permeability and accelerating AS progression[81]. Decreased expression of miR-1246, miR-182, and miR-486 in SMC exosomes under the influence of PDGF promotes EC migration[82].
In addition to miRNAs, which are the main focus of this paper in EV, there are other substances (e.g., circRNAs and heat shock proteins, etc.) that are also involved in EC-SMC communication and play distinct roles in AS formation.
3.1.3. Extracellular Matrix
The ECM is a complex network composed of collagen, non-collagenous proteins, elastin, proteoglycans, and glycosaminoglycans, etc. Both ECs and SMCs are capable of synthesizing and secreting ECM[83, 84]. The vascular endothelium can form an endothelial glycocalyx, which blocks the adhesion of ECs to leukocytes. Under inflammatory stimulation, ECs and leukocytes can secrete related cytokines to remodel the glycocalyx barrier, thus promoting the interaction between ECs and leukocytes. During the development of AS, SMCs migrate and produce ECM, forming a fibrous cap that covers the plaque surface, which can maintain the stability of the plaque.
ECM not only supports and connects cells but also plays an extremely important role in cell growth and proliferation as well as metabolism. Firstly, the ECM can regulate cell activity through growth factors such as insulin-like growth factors and fibroblast growth factors (FGF)[85]. In most cases, the functions of these cytokines depend entirely on the adhesion of cells to the ECM. For instance, when ECs are prevented from adhering to the ECM, they can induce cell apoptosis and growth arrest[86]. Secondly, many physiological processes regulated by the ECM are achieved through the continuous remodeling of actin and the cytoskeleton[87]. Cadherins and integrins play important roles in it, primarily responsible for the mechanical integrity of tissues[88]. Cadherins mediate the connection between cells, which have been mentioned above. Whereas integrins mediate adhesion between cells and ECM[16], their signalling with other cell surface receptors such as growth factors, cytokines and G-protein coupled receptors is also closely linked. Moreover, changes in the ECM can also alter the activity of different integrins[89], thereby regulating cell activities.
The components of the ECM, such as collagen, proteoglycans, and hyaluronic acid (HA), mediate or influence the communication between ECs and SMCs. Research has found that collagen V secreted by SMCs into the ECM may inhibit the recovery of adjacent endothelial damage caused by inflammation[90]. The ECM component Mindin/spondin 2 can prevent intimal thickening and vascular proliferation by inhibiting abnormal SMC proliferation, migration, and phenotypic transformation through an AKT-dependent manner[91]. Nevertheless, the expression of mindin is significantly downregulated in SMCs and vascular tissues stimulated by PDGF-BB and guidewire injury respectively. Furthermore, HA is highly present in the vascular media and new intima, facilitating SMC dedifferentiation, migration, and proliferation[92], thereby strongly contributing to vascular wall thickening. New research has further shown that treatment of SMC with PDGF-BB results in a decrease in the expression of proteoglycan link protein 1 (HAPLN1) [93]. Meanwhile, as a molecule that can assist in maintaining the stability of HA and proteoglycans in the ECM, exogenous HAPLN1 can significantly inhibit the proliferation, migration, and dedifferentiation of human aortic SMCs stimulated by PDGF-BB[94]. The balance and specific mechanisms of the effects of PDGF-BB and HAPLN1, as well as HA and HAPLN1 on SMCs, still need to be further elucidated.
The ECM can also mediate mechanical communication between ECs and SMCs, allowing the two cell types to communicate by responding to changes in the mechanical properties of the ECM induced by adjacent cells[95]. Abnormal mechanical communication can lead to the development of diseases such as AS[96]. The matrix protein thrombospondin-1 (Thbs1) acts as an extracellular mediator of matrix mechanical transmission and may protect cells under mechanical stress by increasing cell stiffness through the Thbs1 / integrins / YAP signaling pathway. Simultaneously, inhibiting this pathway can reduce the proliferation of neointimal cells during carotid ligation[97, 98].
3.2. Crosstalk Between Endothelial Cells and Macrophages
3.2.1. Direct Contact
There are two modes of direct contact between EC and macrophages[99]: macrophages are tightly bound to the outer wall of endothelial tubules; and macrophages bridge endothelial tip cells and act as supportive cells.
The conditioned medium of M1-type macrophages promotes angiogenesis, but when M1-type macrophages are directly cocultured with ECs, angiogenesis is inhibited[100], indicating that direct contact between ECs and macrophages suppresses vascular formation. Conversely, the Notch ligand Dll1 expressed by vascular ECs can mediate macrophage maturation and inhibit the proliferation of M1-type macrophages[101]. Still, more research is necessary to completely understand the precise chemicals behind these processes and how they affect vascular formation. ECs have the ability to successfully stimulate macrophage differentiation and M2-like polarization in vitro[102], and the close association between M2-type macrophages and ECs is conducive to promoting angiogenesis.
Macrophages can secrete VEGF-C to activate the Notch signaling pathway[103], thereby bridging endothelial tip cells at the post-vascular connections and providing the possibility for communication between ECs of different vascular segments[104]. Therefore, some scholars also believe that macrophages act mainly through molecular pathways rather than direct mechanical means.
Figure 3.
Crosstalk between endothelial cells and macrophages and different phenotypes of macrophages Different modes of direct contact between endothelial cells (ECs) and macrophages provide avenues for communication between these two cell types. Vasoactive substances as well as miRNAs contained within extracellular vesicles can lead to proliferation, dysfunction, changes in permeability and expression of inflammatory factors in ECs. Moreover, those can also induce phenotypic transformation and migration in macrophages. Additionally, components of the extracellular matrix can influence their communication. Macrophages are classified into two main phenotypes: M1, which secretes pro-inflammatory factors, and M2, which secretes anti-inflammatory cytokines. Beyond these, other phenotypes such as M(Hb), Mhem, HA-mac, Mox, and M4 also exist.
Figure 3.
Crosstalk between endothelial cells and macrophages and different phenotypes of macrophages Different modes of direct contact between endothelial cells (ECs) and macrophages provide avenues for communication between these two cell types. Vasoactive substances as well as miRNAs contained within extracellular vesicles can lead to proliferation, dysfunction, changes in permeability and expression of inflammatory factors in ECs. Moreover, those can also induce phenotypic transformation and migration in macrophages. Additionally, components of the extracellular matrix can influence their communication. Macrophages are classified into two main phenotypes: M1, which secretes pro-inflammatory factors, and M2, which secretes anti-inflammatory cytokines. Beyond these, other phenotypes such as M(Hb), Mhem, HA-mac, Mox, and M4 also exist.
3.2.2. Paracrine Secretion
Vasoactive Substance
Substances that mediate intercellular communication between the two types of cells, EC and macrophages, include VEGF, IL, TGF, TNF and FGF.
VEGF is now believed to stimulate EC proliferation, increase vascular permeability, and induce macrophage migration. Within the hemorrhagic regions of plaques, M(Hb) macrophages can enhance vascular endothelial permeability and promote plaque inflammation by promoting the expression of vascular cell adhesion molecule (VCAM) on plaque endothelium through VEGF-A[105]. Additionally, both in vitro[106] and in vivo[107] experiments have proven that macrophages expressing stable overexpression of VEGF are capable of transdifferentiating into endothelial-like cells (ELCs). These cells can impede the advancement of AS by mending early-damaged artery ECs and encouraging repair through the release of VEGF and localized NO production. This finding provides insights for future macrophage-targeted therapies against AS. Furthermore, a study using ECs specifically deficient in the glycolysis regulator pfkfb3 revealed that ECs can secrete lactic acid to signal macrophages towards an M2-like phenotype polarization through a mechanism dependent on monocarboxylate transporter 1 s(MCT1) [108], thereby promoting VEGF secretion and angiogenesis. Subsequently, VEGF derived from macrophages reinforces this circuit, leading to the activation of angiogenic potential in microvascular endothelium (mEC).
IL was originally produced by leukocytes and functioned as a cytokine between leukocytes, all of which are small molecule peptides or glycoproteins. More than 40 members of IL have been identified now[109]. Among them, IL-1 is a significant factor in the IL family that mediates inflammation and can be produced by both ECs and macrophages. IL-1 can promote the production of IL-8 by ECs[110], which may recruit monocytes and other cells to the site of inflammation, and thus promoting the formation of new blood vessels[111]. In the early stages of AS, the subtype IL-1β can induce the expression of adhesion molecules in ECs, activate and enhance macrophage activity, and induce the onset of inflammatory responses[112]. On the other hand, IL-1β secreted by macrophages in the early stages of AS can induce inflammation in ECs, such as increasing the expression of adhesion factors and chemokines[113].
TGF is a peptide that regulates cell growth and differentiation, primarily produced by macrophages. It is divided into TGF-αand TGF-β. M1-type macrophage-derived foam cells can induce endothelial-to-mesenchymal transition by upregulating chemokine ligand 4 (CCL-4, a member of the TGF family), increasing endothelial permeability, and promoting monocyte adhesion[114]. Inhibiting the TGF-β signaling pathway in ECs can reduce the expression of VCAM-1 and intercellular adhesion molecule-1 (ICAM-1) on ECs and the recruitment of leukocytes, thereby inhibiting the progression of AS[115]. Some studies have also suggested that TGF-β can carry out pro-angiogenic and anti-angiogenic effects through different pathways, which may have different effects on EC proliferation and migration, etc.
TNF is a small molecule protein that drives inflammatory responses and is typically produced in individuals with inflammation or disease. Specifically, TNF-α, predominantly secreted by macrophages, exerts significant influence on the function of ECs. TNF-α can induce endothelial dysfunction associated with increased oxidative stress and decreased eNOS protein expression[116, 117]. It also promotes the expression of CAMs on the surface of vascular ECs, such as E-selectin, VCAM-1, and ICAM-1[118]. Furthermore, TNF-α disrupts the stability of EC cytoskeleton, triggering cytoskeletal rearrangements that lead to functional impairment of the ECs.
FGF is a polypeptide of about 150-200 amino acids, with at least 20 members in the family. The main member involved in atherosclerotic lesions is the basic fibroblast growth factor (bFGF). bFGF is secreted by SMCs and macrophages, which exerts its activity by binding to different types of cell surface FGF receptors (FGFRs) on cells, including ECs and SMCs. This process produces signals that promote cell motility, proliferation, and survival, leading to angiogenesis. Activated endothelial FGF-R2 signaling promotes the synthesis of adhesion molecules in ECs, facilitating the migration of inflammatory cells to the subendothelial space to promote the development of AS[119]. Additionally, in vivo studies[120] have demonstrated that by inhibiting FGFR1 phosphorylation and blocking bFGF signaling, the migration, proliferation, and angiogenesis of ECs can be reduced. This ultimately decreases intralesional angiogenesis and plaque hemorrhage.
Extracellular Vesicle
It has been proposed that ox-LDL-stimulated macrophages can inhibit EC growth by secreting exosomes[121]. New researches further reveal that macrophage-derived EVs carrying miR-144-5p, miR-145-5p, miR-223, miR-503-5p, miR-4532, miR-4306, and other miRNAs can affect EC.
A study using miR-144-5p mimics for vitro experiments surmised that macrophage-derived miR-144-5p can inhibit EC proliferation and migration, and induce apoptosis[122]. Under this, miR-145-5p may suppress LPS-induced EC proliferation by regulating macrophage polarization to the M2 phenotype[123]. Microvesicles released by activated human macrophages were able to transport miR-223 to ECs to exert its effects[124]. Macrophages can also secrete exosomes containing miR-503-5p to inhibit human coronary artery EC proliferation and angiogenic function and promote AS formation[125]. In response to ox-LDL stimulation, macrophage-derived exosomes carrying miR-4532 can cause EC damage by targeting SP1 and activating downstream NF-κB P65, while positive feedback increased the attraction to macrophages, exacerbating foam cell formation and the transfer of exosomal miR-4532 to ECs[126] and promoting AS formation. ox-LDL can also stimulate macrophages to release EVs containing miR-4306 to inhibit proliferation and migration and angiogenesis of EC from coronary vessels in vitro[127]. Further studies on how macrophages balance the release of these two miRNAs to regulate the course of AS are warranted.
Correspondingly, EC-derived EVs carrying miR-10a, miR-155, and other miRNAs can also affect macrophage function. Resting healthy ECs can secrete EVs carrying miR-10a to transfer to monocytes, and target factors such as IRAK4, β-TRC, and MAP3K7 in the NF-κB pathway to inhibit inflammatory signaling[128]. Ox-LDL stimulation can induce ECs to produce EVs rich in miR-155 to promote monocyte activation and shift monocytes/macrophages from anti-inflammatory M2 to pro-inflammatory M1 phenotypes. EVs secreted by human coronary artery ECs expressing Krüppel-like factor 2 can inhibit inflammation and monocyte activation, and AS formation[129]. So different exosomes may have different pro-inflammatory and anti-inflammatory effects.
3.2.3. Extracellular Matrix
Macrophages can secrete substances that degrade ECM proteins, such as matrix metalloproteinases (MMPs). MMPs are a family of zinc-dependent endopeptidases[130], and their production is closely related to the rupture of the fibrous cap in atherosclerotic plaque. Proteinase-activated receptors (PARs) are important targets for MMPs. Activation of PAR-2 stimulates endothelial NO production and promotes vasodilation via the Ser1177-eNOS phosphorylation pathway[131]. EC-produced MMP-10 can regulate macrophage migration and invasion[132]. Most studies have focused on the changes in MMP expression in plaques and their role as biomarkers, with few studies reporting on MMP-mediated cell communication and its specific mechanisms. On the other hand, macrophages can also synthesize molecules that contribute to the formation and stability of the ECM, such as the proteoglycan APLP2 and serglycin[133]. These molecules play a role in the ECM changes that occur during AS development. However, how they contribute to the development of AS, and whether they aid in pro- or anti-inflammatory processes, remains to be determined.
Moreover, chemokines on ECs and their secreted ECM components primarily promote the adhesion and transfer of monocytes. For example, HA in the ECM produced by ECs can mediate macrophage translocation through the HA receptor Lyve-1, indicating their position through the EC gap.
3.3. Crosstalk Between Smooth Muscle Cells and Macrophages
3.3.1. Direct Contact
Evidence indicates that two cell types may engage in interactions mediated by membrane surface proteins including ICAM-1, VCAM-1, and CX3CL1[134]. ICAM-1 and VCAM-1 are largely expressed on ECs and macrophages, with a detectable presence on SMCs, where they significantly contribute to the adhesion and recruitment of macrophages. R M Hashem et al. used mesenchymal stem cell therapies to replace SMCs in the neointima, reducing the expression levels of adhesion molecules and markers of oxidative and inflammatory stress, such as COX-2, iNOS, and TNF-α[135]. This reduction subsequently inhibited the migration of monocytes and the formation of foam cells by macrophages, underscoring the intricate relationship between macrophages and SMCs in the AS process. Furthermore, the engagement between SMCs and monocyte/macrophages can also augment the expression of TNF-α, IL-1β, IL-6, and MMP through the CX3CL1/CX3CR1 pathway[136], suggesting a potential mechanism by which these cells contribute to the promotion of AS.
Figure 4.
Crosstalk between smooth muscle cells and macrophages and atherosclerotic scenarios in the vasculature Membrane surface proteins like ICAM-1, VCAM-1, and CX3CL1 can mediate the interaction between smooth muscle cells (SMCs) and macrophages, or they can interact directly through signaling pathways like Dll4/Notch. Vasoactive substances such as IL, TGF, TNF, IFN, and PDGF play crucial roles in the crosstalk between SMCs and macrophages. Factors secreted by both types of cells can influence SMC phenotypic switching, apoptosis, proliferation, migration, and changes in inflammatory factor expression, as well as affect macrophage phenotypic conversion and the expression of inflammatory factors in atherosclerosis. miRNAs such as miR-503-5p and miR-145-5p in extracellular vesicles also play a significant role in this process. The extracellular matrix, which constitutes the cellular environment, can be altered by the communication between macrophages and SMCs.
Figure 4.
Crosstalk between smooth muscle cells and macrophages and atherosclerotic scenarios in the vasculature Membrane surface proteins like ICAM-1, VCAM-1, and CX3CL1 can mediate the interaction between smooth muscle cells (SMCs) and macrophages, or they can interact directly through signaling pathways like Dll4/Notch. Vasoactive substances such as IL, TGF, TNF, IFN, and PDGF play crucial roles in the crosstalk between SMCs and macrophages. Factors secreted by both types of cells can influence SMC phenotypic switching, apoptosis, proliferation, migration, and changes in inflammatory factor expression, as well as affect macrophage phenotypic conversion and the expression of inflammatory factors in atherosclerosis. miRNAs such as miR-503-5p and miR-145-5p in extracellular vesicles also play a significant role in this process. The extracellular matrix, which constitutes the cellular environment, can be altered by the communication between macrophages and SMCs.
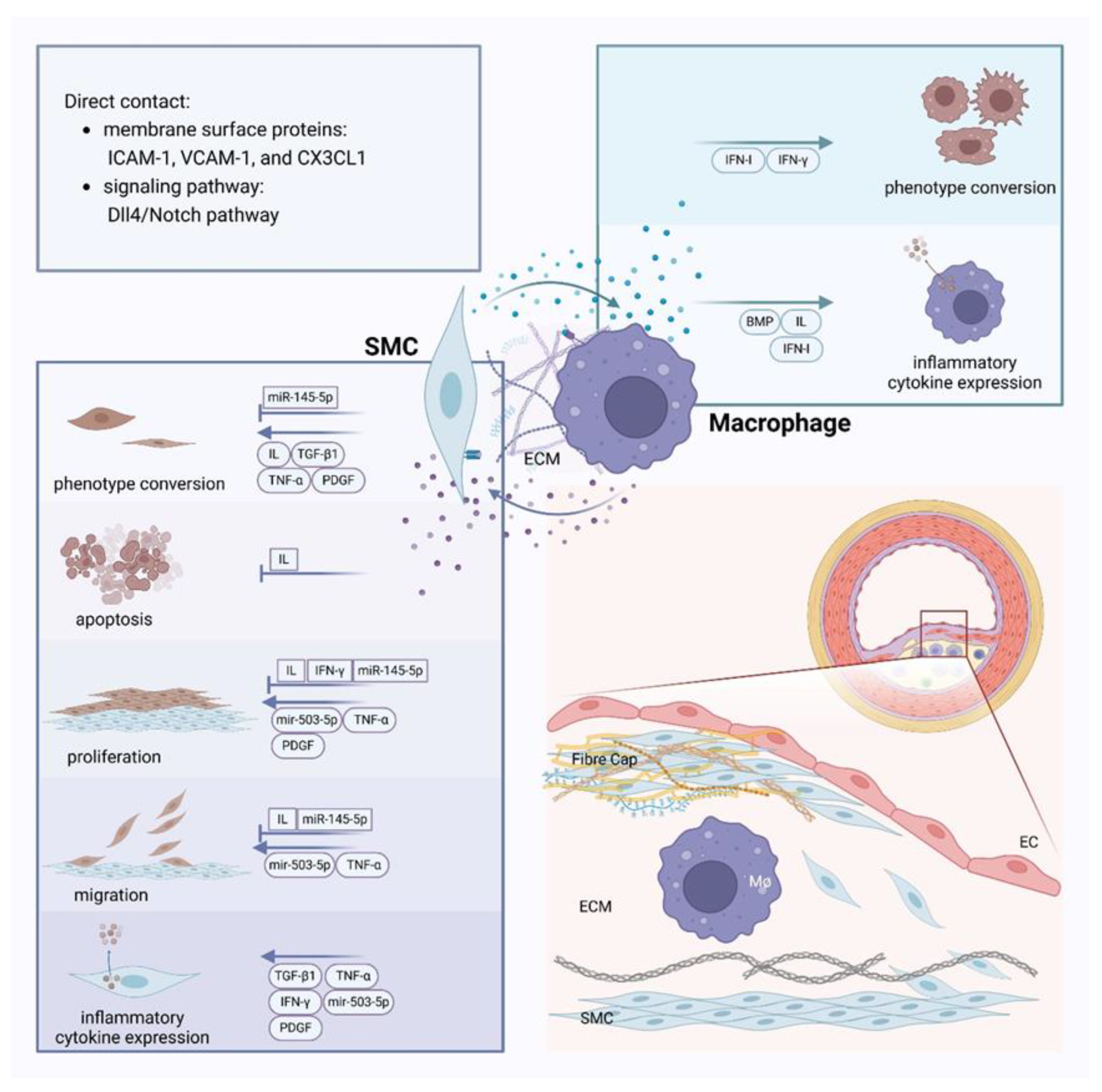
Furthermore, a study has shown that in a direct coculture system of SMCs and macrophages, M1-type macrophages can facilitate the transition of SMCs from a contractile to a synthetic phenotype via the Dll4/Notch pathway[137]. This transition is known to enhance the migration of SMCs and promote the progression of AS. Moreover, macrophages can induce apoptosis in SMCs within plaques through direct contact, a process influenced by Fas/Fas-L interaction[138] and NO[139].
Consequently, researches on the direct contact between SMCs and macrophages mainly occur in vivo or in direct coculture systems, regulating the inflammatory process of AS through various pathways. However, scholars including Pinhao Xiang[140] have found that in human AS lesions, SMCs and macrophages are relatively segregated and maintain distinct positions. In contrast, in mouse models of the disease, SMCs migrate to the intima only after the induction of AS and rapidly mix with macrophages. This suggests that findings from different disease models may vary, indicating a need for further exploration in this field.
3.3.2. Paracrine Secretion
Vasoactive Substance
ILs, TGF, TNF, interferon (IFN), and platelet-derived growth factor (PDGF) are all important factors that mediate communication between macrophages and SMCs.
Macrophages are capable of secreting ILs that induce metabolic and morphological changes in SMCs. In co-culture systems of macrophages and SMCs, IL-5 overexpressing macrophages have been shown to inhibit Ang II-induced apoptosis in SMCs[141]. IL-10 secreted by macrophages can bind to IL-10 receptors on rat vascular SMCs, directly inhibiting SMC proliferation and migration[142]. Studies have linked coronary artery calcification to increased levels of IL-18. SMCs can be activated by IL-18 through the TRPM7 channel, differentiating into an osteoblastic phenotype[143], thereby promoting vascular calcification. SMCs also secrete ILs to communicate with macrophages; for instance, in diseases such as allergic asthma, IL-11 secreted by SMCs can inhibit macrophage activity[144]. Additionally, treatment with an anti-IL11 antibody in SMCs has been associated with decreased levels of common macrophage markers such as LGALS3 and LAMP2[145]. However, research on the role of these interactions in AS is currently limited.
TGF-β1 produced by macrophages can promote SMC secretion of IL11, which in turn induces an extracellular regulated protein kinase (ERK)-dependent shift of SMC to the synthetic phenotype[145]. It also stimulates ECM expression and exerts an anti-inflammatory effect[146]. TGF-β1 can inhibit SMC activation and the inflammatory response in SMCs by mediating the Smad3 pathway[147]. Moreover, bone morphogenetic protein (BMP), a member of the TGF-β family, is produced by SMCs and has been shown to be closely related to macrophages recruitment. In early AS formation in ApoE-/- mice, the expression of BMP-2 and BMP-4 genes in SMCs is upregulated, promoting monocyte recruitment and inflammation[148]. This highlights the dual nature of TGF-β, and how it balances pro-inflammatory and anti-inflammatory effects during AS development still remains to be investigated.
In AS, TNF-α secreted by macrophages can induce proliferation, migration and synthetic behaviour in SMCs[149]. It induces the differentiation of SMCs into osteoblast-like cells, leading to calcium deposition and arterial sclerosis[150]. TNF-α also upregulates the activity of NOX enzyme complex in human aortic SMCs, contributing to the generation of oxidative stress and vascular dysfunction[151]. Consequently, TNF-α plays an important role in promoting the progression of AS.
IFNs were initially identified as protein substances that interfere with viral replication and involved in physiological activities such as cell proliferation and immune response. They are primarily classified into three types: Type I, Type II, and Type III, with Type II IFN consisting only of the IFN-γ subtype. IFN-γ inhibits the proliferation of rat SMC by inducing the production of NO by NO synthase[152]. Meanwhile, IFN-γ stimulates the differentiation of monocytes to macrophages and promotes macrophage activation and foam cell formation[153]. Long-term in vivo studies have suggested that IFN-I can promote the recruitment of monocytes, and these monocytes/macrophages may mature into SMC-like macrophages or macrophage-like SMCs[154], thereby contributing to the development of AS. However, it has also been shown that IFN-β, an IFN-I isoform, attenuates Ang II-induced AS in ApoE-/- mice and exerts anti-inflammatory effects[155]. This may be associated with different downstream signaling pathways of IFN-I, such as the NF-κB pathway, the PI3K/AKT pathway, and the MAPK pathway.
Increased PDGF receptor β signaling in SMCs promotes the production of chemotactic factors[156], which induce SMC dedifferentiation and proliferation, ECM secretion and monocyte recruitment. This not only facilitates the formation of advanced plaque morphology but also promotes the formation of new plaques in the thoracic aorta. Correspondingly, in SMCs in Pdgfrb knockout mice with a long-term Western diet, there is a decrease in collagen content in the fibrous cap and an increase in intraplaque hemorrhage[157]. This means that long-term absence of PDGF receptor β in SMCs leads to reduced stability of the fibrous cap and plaques.
Extracellular Vesicle
Macrophages deliver miR-503-5p to human coronary SMC via EV, which downregulates Smad7, smurf1, and smurf2 and elevates TGF-β1 to promote SMC proliferation and migration and exacerbate AS[125]. In addition, overexpression of miR-145-5p inhibits PDGF-induced proliferation, migration, and phenotypic transformation of VSMCs[158], closely associated with the Smad4 and TGF-β signaling cascade. In contrast, EVs produced by macrophages treated with lipopolysaccharide can induce inflammation and oxidative stress in SMCs, exacerbating the process of vascular calcification[159]. Most studies have focused on in vitro experiments[160], and more research is needed to combine in vitro and in vivo experiments to clarify whether the role of miRNAs in vivo differs from that observed in vitro, thus holding promise for the diagnosis and treatment of diseases.
3.3.3. Extracellular Matrix
Communication between macrophages and SMCs may alter the composition of ECM and the expression of factors related to neoangiogenesis. For instance, in macrophage and SMC co-cultures, the expression of ECM proteins, such as collagen I, collagen III, and elastin, is decreased in SMC, while the expression and activity of MMP-9 and MMP-1 are increased[161].
SMCs not only secrete components of the ECM but also release various MMPs that can digest ECM proteins. MMP-induced degradation of ECM proteins may lead to plaque instability[162]; however, the ability of MMPs to promote SMC migration and proliferation may contribute to the growth and stability of atherosclerotic plaque caps[163]. Thus, MMPs exhibit a dual role in this process, yet most scholars still believe that MMP-induced destruction of the ECM weakens the protective fibrous cap.
Peripheral blood monocytes and macrophages constitutively secrete MMP-9, which is crucial for SMC proliferation and migration to the endothelium, as well as its involvement in the reorganization of the collagen matrix[164]. It significantly impacts the progression of AS and vascular restenosis. In addition to MMP-induced degradation of ECM proteins and thinning of the fibrous cap, macrophage secretions (e.g., TGF-β) also lead to a reduction in collagen synthesis by SMCs[165], thereby thinning the fibrous cap and destabilizing the plaque.
It is clear that the ECM is the site of cellular survival and and establishes the conditions for cellular activity, and in turn, cells actively remodel their surrounding ECM to maintain microenvironment stability.
4. Discussion and Perspective
The communication between ECs, SMCs, macrophages, and related molecules—which either stimulate or inhibit AS through various pathways—under the effect of ox-LDL, shear stress, free radicals, and mechanical stress is summarized in this review. Currently, more research has been done on the interactions between ECs and SMCs, but less has been explored on the crosstalk between the two cells and macrophages[166], particularly when it comes to direct contact and the ECM. Not only may EC, SMC, and macrophages communicate with each other to affect each other's growth and metabolism, but they can also use autocrine regulation to alter the behavior of the same cells. But no research has yet clearly defined the conditions in which these cells must communicate, or under what circumstances both autocrine and communication are required to fulfil normal physiological activities. Investigations into these issues are still pending.
Although the involvement of different molecules in cellular communication and their impact on the development of AS have been understood, the specific mechanisms by which these molecules function in AS are still not fully elucidated. Meanwhile, a variety of molecules have distinct functions during different phases of the illness, which could be connected to the disease's fluctuating cellular makeup. The exact reasons for this phenomenon remain to be explored. We should not limit our attention to a single cell type or substance because the progression of AS always involves numerous cell types and is accompanied by complicated molecular signaling. Rather, we ought to look into the interactions between cells and the influence of different factors on each other. In AS, the substances secreted by cells are always under dynamic change, and the imbalance between pro-inflammatory and anti-inflammatory substances may lead to changes in the direction of AS development. This could provide a new perspective for studying the causes of AS as well as its treatment.
What’s more, Subtle differences in the selection of experimental models may contribute to the significant disparities between in vitro experimental results and clinical trial outcomes. Factors such as racial differences (between humans and mice) mentioned above, as well as gender differences and variations in blood vessel types, can influence the results. According to recent research, the Y chromosome in humans has an impact on the immune system and inflammatory responses. This leads to the fact that more pronounced pro-inflammatory gene expression in male M1 macrophages during LDL internalization, while female M1 macrophages secrete more substances associated with cell damage[167]. Moreover, a study compared the commonly used model in experiments with the actual human atherosclerotic process for the first time. They found that the histological features of the different stages of AS in human coronary arteries and aorta were similar, but there was no intraplaque hemorrhage in aortic lesions[168]. All these differences need to be carefully considered and addressed in future experiments to provide references for clinical treatment.
Several studies have constructed in vitro three-cell co-culture models of EC, macrophages and vascular SMCs, with different models focusing on different aspects[169]. Aspects including lipid accumulation[170], macrophage migration and foam cell formation[171], and immune-vascular interactions[172] are the subject of many models. A recent study introduced a new human AS three-cell direct co-culture model, which mimics the early to mid-stage of the natural history of AS[173]. This model has low shear stress and LDL conditions that are comparable to those in human AS. Meanwhile, the model's construction took only 24 hours, greatly improving experimental efficiency and offering a valuable method for researching the pathogenesis of AS. We may keep investigating the communication between three cells based on this remarkable three-cell co-culture method.
These three cells are not the only immune cells that impact the AS process; T cells, B cells, neutrophils, and other cells also release various cytokines that impact the course of AS. These cells may also exhibit different forms of communication and crosstalk with each other. It would be worthwhile to explore whether analyzing the communication of other cell types can lead to a more profound understanding of AS progression.
Researching the communication mechanism can yield more insights into potential future therapies for AS disorders. For instance, blocking intercellular communication or modifying the targeting of intercellular communicating substances could be therapeutic approaches. As previously noted, miRNAs contained in EVs play an important role in intercellular communication. A recent study has discovered that a unique sequence on miRNAs in cells, called motif, controls whether or not those miRNAs are loaded into vesicles and passed on to other cells[174]. Recoding the motif of cells through RNA editing techniques may represent a viable option for AS treatment.
In summary, communication between ECs, SMCs and macrophages plays a crucial role in the progression of AS. Human intervention in their communication process can offer insights into potential therapeutic approaches for the disease in the future.
Author Contributions
Sihe Gong: Investigation, Writing - Original Draft, Writing - Review &Editing, Visualization; Yanni Li: Investigation, Resources; Kaijie Yan: Investigation, Resources; Zhonghong Shi: Investigation, Resources; Jing Leng: Supervision, Project administration; Yimin Bao: Supervision, Project administration; Ke Ning: Resources, Supervision, Project administration, Writing – review & editing, Funding acquisition.
Funding
This work was supported by the National Natural Science Foundation of China (No. 82274133, 82104430 to Ke Ning), Shanghai Sailing Program (No. 21YF1447600 to Ke Ning), Future Plan for Traditional Chinese Medicine development of Science and Technology of Shanghai Municipal Hospital of Traditional Chinese Medicine (WL-HBQN-2022002K to Jing Leng), the Shanghai University of Traditional Chinese Medicine Students’ Innovation Program, and the “Pei-Ran Plan” of Shanghai University of Traditional Chinese Medicine.
Institutional Review Board Statement
The manuscript does not contain clinical studies or patient data.
Informed Consent Statement
Not applicable. All the authors had complete access to the manuscript and agreed to submit it for publication.
Data Availability Statement
Not applicable.
Acknowledgments
The Figures were created by Biorender (https://www.biorender.com/).
Conflicts of Interest
All authors declare that they have no conflict of interest.
References
- Depuydt, M.A.; Prange, K.H.; Slenders, L.; Örd, T.; Elbersen, D.; Boltjes, A.; de Jager, S.C.; Asselbergs, F.W.; de Borst, G.J.; Aavik, E.; et al. Microanatomy of the Human Atherosclerotic Plaque by Single-Cell Transcriptomics. Circ. Res. 2020, 127, 1437–1455. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Ilyas, I.; Little, P.J.; Li, H.; Kamato, D.; Zheng, X.; Luo, S.; Li, Z.; Liu, P.; Han, J.; et al. Endothelial Dysfunction in Atherosclerotic Cardiovascular Diseases and Beyond: From Mechanism to Pharmacotherapies. Pharmacol. Rev. 2021, 73, 924–967. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Lyu, Q.R.; Ilyas, I.; Tian, X.-Y.; Weng, J. Vascular homeostasis in atherosclerosis: A holistic overview. Front. Immunol. 2022, 13, 976722. [Google Scholar] [CrossRef] [PubMed]
- Corliss, B.A.; Azimi, M.S.; Munson, J.M.; Peirce, S.M.; Murfee, W.L. Macrophages: An Inflammatory Link Between Angiogenesis and Lymphangiogenesis. Microcirculation 2015, 23, 95–121. [Google Scholar] [CrossRef]
- Wolf, M.P.; Hunziker, P. Atherosclerosis: Insights into Vascular Pathobiology and Outlook to Novel Treatments. J. Cardiovasc. Transl. Res. 2020, 13, 744–757. [Google Scholar] [CrossRef]
- Jinnouchi, H.; Guo, L.; Sakamoto, A.; Torii, S.; Sato, Y.; Cornelissen, A.; Kuntz, S.; Paek, K.H.; Fernandez, R.; Fuller, D.; et al. Diversity of macrophage phenotypes and responses in atherosclerosis. Cell. Mol. Life Sci. 2019, 77, 1919–1932. [Google Scholar] [CrossRef]
- Mushenkova, N.V.; Nikiforov, N.G.; Melnichenko, A.A.; Kalmykov, V.; Shakhpazyan, N.K.; Orekhova, V.A.; Orekhov, A.N. Functional Phenotypes of Intraplaque Macrophages and Their Distinct Roles in Atherosclerosis Development and Atheroinflammation. Biomedicines 2022, 10, 452. [Google Scholar] [CrossRef]
- Pogoda, K.; Kameritsch, P. Molecular regulation of myoendothelial gap junctions. Curr Opin Pharmacol 2019, 45, 16–22. [Google Scholar] [CrossRef]
- Kwak, B.R.; Mulhaupt, F.; Veillard, N.; Gros, D.B.; Mach, F. Altered Pattern of Vascular Connexin Expression in Atherosclerotic Plaques. Arter. Thromb. Vasc. Biol. 2002, 22, 225–230. [Google Scholar] [CrossRef]
- Wang, M.; Wu, Y.; Yu, Y.; Fu, Y.; Yan, H.; Wang, X.; Li, T.; Peng, W.; Luo, D. Rutaecarpine prevented ox-LDL-induced VSMCs dysfunction through inhibiting overexpression of connexin 43. Eur. J. Pharmacol. 2019, 853, 84–92. [Google Scholar] [CrossRef]
- Qin, X.; He, W.; Yang, R.; Liu, L.; Zhang, Y.; Li, L.; Si, J.; Li, X.; Ma, K. Inhibition of Connexin 43 reverses ox-LDL-mediated inhibition of autophagy in VSMC by inhibiting the PI3K/Akt/mTOR signaling pathway. PeerJ 2022, 10, e12969. [Google Scholar] [CrossRef] [PubMed]
- Allagnat, F.; Dubuis, C.; Lambelet, M.; Le Gal, L.; Alonso, F.; Corpataux, J.-M.; Déglise, S.; Haefliger, J.-A. Connexin37 reduces smooth muscle cell proliferation and intimal hyperplasia in a mouse model of carotid artery ligation. Cardiovasc. Res. 2017, 113, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Dunn, C.A.; Lampe, P.D. Injury-triggered Akt phosphorylation of Cx43: a ZO-1-driven molecular switch that regulates gap junction size. J. Cell Sci. 2013, 127, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Sedovy, M.W.; Leng, X.; Leaf, M.R.; Iqbal, F.; Payne, L.B.; Chappell, J.C.; Johnstone, S.R. Connexin 43 across the Vasculature: Gap Junctions and Beyond. J. Vasc. Res. 2022, 60, 101–113. [Google Scholar] [CrossRef]
- Hu, J.; A Cotgreave, I. Differential regulation of gap junctions by proinflammatory mediators in vitro. J. Clin. Investig. 1997, 99, 2312–2316. [Google Scholar] [CrossRef]
- Mui, K. L.; Chen, C. S.; Assoian, R. K. , The mechanical regulation of integrin-cadherin crosstalk organizes cells, signaling and forces. J Cell Sci 2016, 129, (6), 1093–100. [Google Scholar] [CrossRef]
- Gilbertson-Beadling, S.K.; Fisher, C. A potential role for N-cadherin in mediating endothelial cell-smooth muscle cell interactions in the rat vasculature. .Lab Invest 1993, 69, 203–9. [Google Scholar]
- Lyon, C.A.; Koutsouki, E.; Aguilera, C.M.; Blaschuk, O.W.; George, S.J. Inhibition of N-cadherin retards smooth muscle cell migration and intimal thickening via induction of apoptosis. J. Vasc. Surg. 2010, 52, 1301–1309. [Google Scholar] [CrossRef]
- Lyon, C.A.; Wadey, K.S.; George, S.J. Soluble N-cadherin: A novel inhibitor of VSMC proliferation and intimal thickening. Vasc. Pharmacol. 2016, 78, 53–62. [Google Scholar] [CrossRef]
- Liu, W.F.; Nelson, C.M.; Tan, J.L.; Chen, C.S. Cadherins, RhoA, and Rac1 Are Differentially Required for Stretch-Mediated Proliferation in Endothelial Versus Smooth Muscle Cells. Circ. Res. 2007, 101, e44–e52. [Google Scholar] [CrossRef]
- Zhang, Y.; Janssens, S.P.; Wingler, K.; Schmidt, H.H.H.W.; Moens, A.L. Modulating endothelial nitric oxide synthase: a new cardiovascular therapeutic strategy. Am. J. Physiol. Circ. Physiol. 2011, 301, H634–H646. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, P.M.; Shimokawa, H.; Feletou, M.; Tang, E.H.C. Endothelial dysfunction and vascular disease - a 30th anniversary update. Acta Physiol. 2017, 219, 22–96. [Google Scholar] [CrossRef]
- Hong, F.-F.; Liang, X.-Y.; Liu, W.; Lv, S.; He, S.-J.; Kuang, H.-B.; Yang, S.-L. Roles of eNOS in atherosclerosis treatment. Inflamm. Res. 2019, 68, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.M.J.; Ferrige, A.G.; Moncada, S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 1987, 327, 524–526. [Google Scholar] [CrossRef]
- Sarkar, R.; Meinberg, E.G.; Stanley, J.C.; Gordon, D.; Webb, R.C. Nitric Oxide Reversibly Inhibits the Migration of Cultured Vascular Smooth Muscle Cells. Circ. Res. 1996, 78, 225–230. [Google Scholar] [CrossRef]
- Gao, Y.; Zhu, P.; Xu, S.-F.; Li, Y.-Q.; Deng, J.; Yang, D.-L. Ginsenoside Re inhibits PDGF-BB-induced VSMC proliferation via the eNOS/NO/cGMP pathway. Biomed. Pharmacother. 2019, 115, 108934. [Google Scholar] [CrossRef]
- Jiang, H.; Jiang, Y.; Qu, Y.; Lv, J.; Zeng, H. sGC agonist BAY1021189 promotes thoracic aortic dissection formation by accelerating vascular smooth muscle cell phenotype switch. Eur. J. Pharmacol. 2023, 952, 175789. [Google Scholar] [CrossRef]
- Stencel, M. G.; VerMeer, M.; Giles, J.; Tran, Q. K. , Endothelial regulation of calmodulin expression and eNOS-calmodulin interaction in vascular smooth muscle. Mol Cell Biochem 2022, 477, (5), 1489–1498. [Google Scholar] [CrossRef]
- Christodoulides, N.; Durante, W.; Kroll, M.H.; Schafer, A.I. Vascular Smooth Muscle Cell Heme Oxygenases Generate Guanylyl Cyclase–Stimulatory Carbon Monoxide. Circulation 1995, 91, 2306–2309. [Google Scholar] [CrossRef]
- Ou, H.S.; Yan, L.M.; Fu, M.G.; Wang, X.H.; Pang, Y.Z.; Su, J.Y.; Tang, C.S. [The role of endogenous CO in the regulation of endothelin-induced VSMC proliferation and MAPK activity]. .Sheng Li Xue Bao 1999, 51, 315–20. [Google Scholar]
- Kim, H. P.; Wang, X.; Nakao, A.; Kim, S. I.; Murase, N.; Choi, M. E.; Ryter, S. W.; Choi, A. M. , Caveolin-1 expression by means of p38beta mitogen-activated protein kinase mediates the antiproliferative effect of carbon monoxide. Proc Natl Acad Sci U S A 2005, 102, 11319–24. [Google Scholar] [CrossRef]
- A Tulis, D.; Keswani, A.N.; Peyton, K.J.; Wang, H.; I Schafer, A.; Durante, W. Local administration of carbon monoxide inhibits neointima formation in balloon injured rat carotid arteries. .Cell Mol Biol (Noisy-le-grand) 2005, 51, 441–6. [Google Scholar] [PubMed]
- Rodriguez, A.I.; Gangopadhyay, A.; Kelley, E.E.; Pagano, P.J.; Zuckerbraun, B.S.; Bauer, P.M. HO-1 and CO Decrease Platelet-Derived Growth Factor-Induced Vascular Smooth Muscle Cell Migration Via Inhibition of Nox1. Arter. Thromb. Vasc. Biol. 2010, 30, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Morita, T.; Kourembanas, S. Endothelial cell expression of vasoconstrictors and growth factors is regulated by smooth muscle cell-derived carbon monoxide. J. Clin. Investig. 1995, 96, 2676–2682. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Kim, J.; Kim, J. H.; Lee, D. K.; Park, W.; Park, M.; Kim, S.; Hwang, J. Y.; Won, M. H.; Choi, Y. K.; Ryoo, S.; Ha, K. S.; Kwon, Y. G.; Kim, Y. M. , Carbon monoxide prevents TNF-α-induced eNOS downregulation by inhibiting NF-κB-responsive miR-155-5p biogenesis. Exp Mol Med 2017, 49, (11), e403. [Google Scholar] [CrossRef]
- Chu, L.-Y.; Liou, J.-Y.; Wu, K.K. Prostacyclin protects vascular integrity via PPAR/14-3-3 pathway. Prostaglandins Other Lipid Mediat. 2015, 118-119, 19–27. [Google Scholar] [CrossRef]
- Narumiya, S.; Sugimoto, Y.; Ushikubi, F. Prostanoid Receptors: Structures, Properties, and Functions. Physiol. Rev. 1999, 79, 1193–1226. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Chu, L.-Y.; Yang, S.-F.; Chen, H.-L.; Yet, S.-F.; Wu, K.K. Prostacyclin and PPARα Agonists Control Vascular Smooth Muscle Cell Apoptosis and Phenotypic Switch through Distinct 14-3-3 Isoforms. PLOS ONE 2013, 8, e69702. [Google Scholar] [CrossRef]
- Tsai, M. C.; Chen, L.; Zhou, J.; Tang, Z.; Hsu, T. F.; Wang, Y.; Shih, Y. T.; Peng, H. H.; Wang, N.; Guan, Y.; Chien, S.; Chiu, J. J. , Shear stress induces synthetic-to-contractile phenotypic modulation in smooth muscle cells via peroxisome proliferator-activated receptor alpha/delta activations by prostacyclin released by sheared endothelial cells. Circ Res 2009, 105, 471–80. [Google Scholar] [CrossRef]
- Cheng, Y.; Wang, M.; Yu, Y.; Lawson, J.; Funk, C. D.; Fitzgerald, G. A. , Cyclooxygenases, microsomal prostaglandin E synthase-1, and cardiovascular function. J Clin Invest 2006, 116, 1391–9. [Google Scholar] [CrossRef]
- Shimokawa, H.; Flavahan, N.A.; Lorenz, R.R.; Vanhoutte, P.M. Prostacyclin releases endothelium-derived relaxing factor and potentiates its action in coronary arteries of the pig. Br. J. Pharmacol. 1988, 95, 1197–1203. [Google Scholar] [CrossRef] [PubMed]
- Flammer, A.J.; Lüscher, T.F. Human endothelial dysfunction: EDRFs. Pfl?gers Arch. Eur. J. Physiol. 2010, 459, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Taddei, S.; Ghiadoni, L.; Virdis, A.; Buralli, S.; Salvetti, A. Vasodilation to Bradykinin Is Mediated by an Ouabain-Sensitive Pathway as a Compensatory Mechanism for Impaired Nitric Oxide Availability in Essential Hypertensive Patients. Circulation 1999, 100, 1400–1405. [Google Scholar] [CrossRef] [PubMed]
- Csányi, G.; Gajda, M.; Franczyk-Zarow, M.; Kostogrys, R.; Gwoźdź, P.; Mateuszuk, L.; Sternak, M.; Wojcik, L.; Zalewska, T.; Walski, M.; Chlopicki, S. , Functional alterations in endothelial NO, PGI₂ and EDHF pathways in aorta in ApoE/LDLR-/- mice. Prostaglandins Other Lipid Mediat 2012, 98, 107–15. [Google Scholar] [CrossRef]
- Dunn, S.M.; Hilgers, R.H.P.; Das, K.C. Decreased EDHF-mediated relaxation is a major mechanism in endothelial dysfunction in resistance arteries in aged mice on prolonged high-fat sucrose diet. Physiol. Rep. 2017, 5, e13502. [Google Scholar] [CrossRef]
- Ozkor, M.A.; Quyyumi, A.A. Endothelium-Derived Hyperpolarizing Factor and Vascular Function. Cardiol. Res. Pr. 2011, 2011, 1–12. [Google Scholar] [CrossRef]
- Mather, S.; Dora, K.A.; Sandow, S.L.; Winter, P.; Garland, C.J. Rapid Endothelial Cell–Selective Loading of Connexin 40 Antibody Blocks Endothelium-Derived Hyperpolarizing Factor Dilation in Rat Small Mesenteric Arteries. Circ. Res. 2005, 97, 399–407. [Google Scholar] [CrossRef]
- Montezano, A.C.; Cat, A.N.D.; Rios, F.J.; Touyz, R.M. Angiotensin II and Vascular Injury. Curr. Hypertens. Rep. 2014, 16, 1–11. [Google Scholar] [CrossRef]
- St Paul, A.; Corbett, C. B.; Okune, R.; Autieri, M. V. , Angiotensin II, Hypercholesterolemia, and Vascular Smooth Muscle Cells: A Perfect Trio for Vascular Pathology. Int J Mol Sci 2020, 21, (12). [Google Scholar] [CrossRef]
- Nakashima, H.; Suzuki, H.; Ohtsu, H.; Chao, J.Y.; Utsunomiya, H.; Frank, G.D.; Eguchi, S. Angiotensin II Regulates Vascular and Endothelial Dysfunction: Recent Topics of Angiotensin II Type-1 Receptor Signaling in the Vasculature. Curr. Vasc. Pharmacol. 2006, 4, 67–78. [Google Scholar] [CrossRef]
- Sendra, J.; Llorente-Cortés, V.; Costales, P.; Huesca-Gómez, C.; Badimon, L. Angiotensin II upregulates LDL receptor-related protein (LRP1) expression in the vascular wall: a new pro-atherogenic mechanism of hypertension. Cardiovasc. Res. 2008, 78, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Dandapat, A.; Chen, J.; Liu, Y.; Hermonat, P.L.; Carey, R.M.; Mehta, J.L. Over-expression of angiotensin II type 2 receptor (agtr2) reduces atherogenesis and modulates LOX-1, endothelial nitric oxide synthase and heme-oxygenase-1 expression. Atherosclerosis 2008, 199, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Chen, M.-F.; Xiao, Z.-L.; Yu, G.-L.; Chen, X.-B.; Xie, X.-M. The Effect of Endothelial Progenitor Cells on Angiotensin II–induced Proliferation of Cultured Rat Vascular Smooth Muscle Cells. J. Cardiovasc. Pharmacol. 2011, 58, 617–625. [Google Scholar] [CrossRef]
- Inoue, A.; Yanagisawa, M.; Kimura, S.; Kasuya, Y.; Miyauchi, T.; Goto, K.; Masaki, T. The human endothelin family: three structurally and pharmacologically distinct isopeptides predicted by three separate genes. Proc. Natl. Acad. Sci. 1989, 86, 2863–2867. [Google Scholar] [CrossRef] [PubMed]
- Neylon, C.B. VASCULAR BIOLOGY OF ENDOTHELIN SIGNAL TRANSDUCTION. Clin. Exp. Pharmacol. Physiol. 1999, 26, 149–153. [Google Scholar] [CrossRef]
- Zamora, M.A.; Dempsey, E.C.; Walchak, S.J.; Stelzner, T.J. BQ123, an ETA Receptor Antagonist, Inhibits Endothelin-1-mediated Proliferation of Human Pulmonary Artery Smooth Muscle Cells. Am. J. Respir. Cell Mol. Biol. 1993, 9, 429–433. [Google Scholar] [CrossRef]
- Khimji, A. K.; Rockey, D. C. , Endothelin--biology and disease. Cell Signal 2010, 22, 1615–25. [Google Scholar] [CrossRef]
- Seif, F.; Little, P.J.; Niayesh-Mehr, R.; Zamanpour, M.; Babaahmadi-Rezaei, H. Endothelin-1 increases CHSY-1 expression in aortic endothelial cells via transactivation of transforming growth factor β type I receptor induced by type B receptor endothelin-1. J. Pharm. Pharmacol. 2019, 71, 988–995. [Google Scholar] [CrossRef]
- Fredriksson, L.; Li, H.; Eriksson, U. The PDGF family: four gene products form five dimeric isoforms. Cytokine Growth Factor Rev. 2004, 15, 197–204. [Google Scholar] [CrossRef]
- Nair, D.G.; Miller, K.G.; Lourenssen, S.R.; Blennerhassett, M.G. Inflammatory cytokines promote growth of intestinal smooth muscle cells by induced expression of PDGF-Rβ. J. Cell. Mol. Med. 2014, 18, 444–454. [Google Scholar] [CrossRef]
- Zhang, D.; Chen, Y.; Xie, X.; Liu, J.; Wang, Q.; Kong, W.; Zhu, Y. Homocysteine activates vascular smooth muscle cells by DNA demethylation of platelet-derived growth factor in endothelial cells. J. Mol. Cell. Cardiol. 2012, 53, 487–496. [Google Scholar] [CrossRef]
- Mondy, J.S.; Lindner, V.; Miyashiro, J.K.; Berk, B.C.; Dean, R.H.; Geary, R.L.; T, Z.; J, L.; X, C.; J, P.; et al. Platelet-Derived Growth Factor Ligand and Receptor Expression in Response to Altered Blood Flow In Vivo. Circ. Res. 1997, 81, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, T.; Liu, J.; Chen, X.; Pi, J.; Kuang, Y.; Wang, Y.; Tomlinson, B.; Chan, P.; Zhang, Q.; Li, Y.; et al. Cell-Specific Effects of GATA (GATA Zinc Finger Transcription Factor Family)-6 in Vascular Smooth Muscle and Endothelial Cells on Vascular Injury Neointimal Formation. Arter. Thromb. Vasc. Biol. 2019, 39, 888–901. [Google Scholar] [CrossRef] [PubMed]
- LeCouter, J.; Ferrara, N. EG-VEGF and the concept of tissue-specific angiogenic growth factors. Semin. Cell Dev. Biol. 2002, 13, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Tammela, T.; Enholm, B.; Alitalo, K.; Paavonen, K. The biology of vascular endothelial growth factors. Cardiovasc Res 2005, 65, 550–563. [Google Scholar] [CrossRef]
- Ahmad, A.; Nawaz, M.I. Molecular mechanism of VEGF and its role in pathological angiogenesis. J. Cell. Biochem. 2022, 123, 1938–1965. [Google Scholar] [CrossRef]
- Kroll, J.; Waltenberger, J. VEGF-A Induces Expression of eNOS and iNOS in Endothelial Cells via VEGF Receptor-2 (KDR). Biochem. Biophys. Res. Commun. 1998, 252, 743–746. [Google Scholar] [CrossRef]
- Chang, H.J.; Park, J.S.; Kim, M.H.; Hong, M.H.; Kim, K.M.; Kim, S.M.; Shin, B.A.; Ahn, B.W.; Jung, Y.D. Extracellular signal-regulated kinases and AP-1 mediate the up-regulation of vascular endothelial growth factor by PDGF in human vascular smooth muscle cells. Int. J. Oncol. 2006, 28, 135–141. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Extracellular vesicles and atherosclerotic disease. Cell. Mol. Life Sci. 2015, 72, 2697–2708. [Google Scholar] [CrossRef]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Poon, I.K.H.; Chiu, Y.-H.; Armstrong, A.J.; Kinchen, J.M.; Juncadella, I.J.; Bayliss, D.A.; Ravichandran, K.S. Unexpected link between an antibiotic, pannexin channels and apoptosis. Nature 2014, 507, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Tabaei, S.; Tabaee, S.S. Implications for MicroRNA involvement in the prognosis and treatment of atherosclerosis. Mol. Cell. Biochem. 2021, 476, 1327–1336. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, D.; Chen, X.; Li, J.; Li, L.; Bian, Z.; Sun, F.; Lu, J.; Yin, Y.; Cai, X.; et al. Secreted Monocytic miR-150 Enhances Targeted Endothelial Cell Migration. Mol. Cell 2010, 39, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Hergenreider, E.; Heydt, S.; Tréguer, K.; Boettger, T.; Horrevoets, A.J.G.; Zeiher, A.M.; Scheffer, M.P.; Frangakis, A.S.; Yin, X.; Mayr, M.; et al. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat. Cell Biol. 2012, 14, 249–256. [Google Scholar] [CrossRef]
- Lu, M.; Yuan, S.; Li, S.; Li, L.; Liu, M.; Wan, S. The Exosome-Derived Biomarker in Atherosclerosis and Its Clinical Application. J. Cardiovasc. Transl. Res. 2018, 12, 68–74. [Google Scholar] [CrossRef]
- Zhou, J.; Li, Y. S.; Nguyen, P.; Wang, K. C.; Weiss, A.; Kuo, Y. C.; Chiu, J. J.; Shyy, J. Y.; Chien, S. , Regulation of vascular smooth muscle cell turnover by endothelial cell-secreted microRNA-126: role of shear stress. Circ Res 2013, 113, 40–51. [Google Scholar] [CrossRef]
- van Mil, A.; Grundmann, S.; Goumans, M.-J.; Lei, Z.; Oerlemans, M.I.; Jaksani, S.; Doevendans, P.A.; Sluijter, J.P. MicroRNA-214 inhibits angiogenesis by targeting Quaking and reducing angiogenic growth factor release. Cardiovasc. Res. 2012, 93, 655–665. [Google Scholar] [CrossRef]
- van Balkom, B.W.M.; de Jong, O.G.; Smits, M.; Brummelman, J.; Ouden, K.D.; de Bree, P.M.; van Eijndhoven, M.A.J.; Pegtel, D.M.; Stoorvogel, W.; Würdinger, T.; et al. Endothelial cells require miR-214 to secrete exosomes that suppress senescence and induce angiogenesis in human and mouse endothelial cells. Blood 2013, 121, 3997–4006. [Google Scholar] [CrossRef]
- Cordes, K.R.; Sheehy, N.T.; White, M.P.; Berry, E.C.; Morton, S.U.; Muth, A.N.; Lee, T.-H.; Miano, J.M.; Ivey, K.N.; Srivastava, D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 2009, 460, 705–710. [Google Scholar] [CrossRef]
- Wang, H.; Xie, Y.; Salvador, A.M.; Zhang, Z.; Chen, K.; Li, G.; Xiao, J. Exosomes: Multifaceted Messengers in Atherosclerosis. Curr. Atheroscler. Rep. 2020, 22, 1–9. [Google Scholar] [CrossRef]
- Zheng, B.; Yin, W. N.; Suzuki, T.; Zhang, X. H.; Zhang, Y.; Song, L. L.; Jin, L. S.; Zhan, H.; Zhang, H.; Li, J. S.; Wen, J. K. , Exosome-Mediated miR-155 Transfer from Smooth Muscle Cells to Endothelial Cells Induces Endothelial Injury and Promotes Atherosclerosis. Mol Ther 2017, 25, (6), 1279–1294. [Google Scholar] [CrossRef]
- Heo, J.; Yang, H.C.; Rhee, W.J.; Kang, H. Vascular Smooth Muscle Cell-Derived Exosomal MicroRNAs Regulate Endothelial Cell Migration Under PDGF Stimulation. Cells 2020, 9, 639. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Barbero, N.; Gutiérrez-Muñoz, C.; Blanco-Colio, L.M. Cellular Crosstalk between Endothelial and Smooth Muscle Cells in Vascular Wall Remodeling. Int. J. Mol. Sci. 2021, 22, 7284. [Google Scholar] [CrossRef] [PubMed]
- Mongiat, M.; Andreuzzi, E.; Tarticchio, G.; Paulitti, A. Extracellular Matrix, a Hard Player in Angiogenesis. Int. J. Mol. Sci. 2016, 17, 1822. [Google Scholar] [CrossRef] [PubMed]
- Taipale, J.; Keski-Oja, J. , Growth factors in the extracellular matrix. Faseb j 1997, 11, 51–9. [Google Scholar] [CrossRef]
- Davis, G. E.; Senger, D. R. , Endothelial extracellular matrix: biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ Res 2005, 97, 1093–107. [Google Scholar] [CrossRef]
- Romani, P.; Valcarcel-Jimenez, L.; Frezza, C.; Dupont, S. Crosstalk between mechanotransduction and metabolism. Nat. Rev. Mol. Cell Biol. 2020, 22, 22–38. [Google Scholar] [CrossRef]
- Hadjisavva, R.; Anastasiou, O.; Ioannou, P.S.; Zheltkova, M.; Skourides, P.A. Adherens junctions stimulate and spatially guide integrin activation and extracellular matrix deposition. Cell Rep. 2022, 40, 111091. [Google Scholar] [CrossRef]
- Murphy, J.M.; Jeong, K.; Lim, S.-T.S. FAK Family Kinases in Vascular Diseases. Int. J. Mol. Sci. 2020, 21, 3630. [Google Scholar] [CrossRef]
- Underwood, P.; Bean, P.; Whitelock, J. Inhibition of endothelial cell adhesion and proliferation by extracellular matrix from vascular smooth muscle cells: role of type V collagen. Atherosclerosis 1998, 141, 141–152. [Google Scholar] [CrossRef]
- Zhu, L.-H.; Huang, L.; Zhang, X.; Zhang, P.; Zhang, S.-M.; Guan, H.; Zhang, Y.; Zhu, X.-Y.; Tian, S.; Deng, K.; et al. Mindin regulates vascular smooth muscle cell phenotype and prevents neointima formation. Clin. Sci. 2015, 129, 129–145. [Google Scholar] [CrossRef] [PubMed]
- Parnigoni, A.; Viola, M.; Karousou, E.; Rovera, S.; Giaroni, C.; Passi, A.; Vigetti, D. Hyaluronan in pathophysiology of vascular diseases: specific roles in smooth muscle cells, endothelial cells, and macrophages. Am. J. Physiol. Physiol. 2022, 323, C505–C519. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Urabe, G.; Marcho, L.; Stratton, M.; Guo, L.-W.; Kent, C.K. ALDH1A3 Regulations of Matricellular Proteins Promote Vascular Smooth Muscle Cell Proliferation. iScience 2019, 19, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Ha, H. C.; Yang, G.; Jang, J. M.; Park, B. K.; Fu, Z.; Shin, I. C.; Kim, D. K. , Hyaluronic acid and proteoglycan link protein 1 suppresses platelet derived growth factor-BB-induced proliferation, migration, and phenotypic switching of vascular smooth muscle cells. BMB Rep 2023, 56, 445–450. [Google Scholar] [CrossRef]
- Sapir, L.; Tzlil, S. , Talking over the extracellular matrix: How do cells communicate mechanically? Semin Cell Dev Biol 2017, 71, 99–105. [Google Scholar] [CrossRef]
- Yamashiro, Y.; Yanagisawa, H. The molecular mechanism of mechanotransduction in vascular homeostasis and disease. Clin. Sci. 2020, 134, 2399–2418. [Google Scholar] [CrossRef]
- Yamashiro, Y.; Thang, B.Q.; Ramirez, K.; Shin, S.J.; Kohata, T.; Ohata, S.; Nguyen, T.A.V.; Ohtsuki, S.; Nagayama, K.; Yanagisawa, H. Matrix mechanotransduction mediated by thrombospondin-1/integrin/YAP in the vascular remodeling. Proc. Natl. Acad. Sci. 2020, 117, 9896–9905. [Google Scholar] [CrossRef]
- Moura, R.; Tjwa, M.; Vandervoort, P.; Van Kerckhoven, S.; Holvoet, P.; Hoylaerts, M. F. , Thrombospondin-1 deficiency accelerates atherosclerotic plaque maturation in ApoE-/- mice. Circ Res 2008, 103, 1181–9. [Google Scholar] [CrossRef]
- Wang, Z.; Fan, P.; Zhou, J.; Wang, J.; Feng, B. , Role of macrophages in angiogenesis. Chinese Journal of Immunology 2021, 37, 2710–2714. [Google Scholar]
- Jetten, N.; Verbruggen, S.; Gijbels, M.J.; Post, M.J.; De Winther, M.P.J.; Donners, M.M.P.C. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis 2014, 17, 109–118. [Google Scholar] [CrossRef]
- Krishnasamy, K.; Limbourg, A.; Kapanadze, T.; Gamrekelashvili, J.; Beger, C.; Häger, C.; Lozanovski, V.J.; Falk, C.S.; Napp, L.C.; Bauersachs, J.; et al. Blood vessel control of macrophage maturation promotes arteriogenesis in ischemia. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Xu, J.; Warren, C.M.; Duan, D.; Li, X.; Wu, L.; Iruela-Arispe, M.L. Endothelial cells provide an instructive niche for the differentiation and functional polarization of M2-like macrophages. Blood 2012, 120, 3152–3162. [Google Scholar] [CrossRef] [PubMed]
- Tammela, T.; Zarkada, G.; Nurmi, H.; Jakobsson, L.; Heinolainen, K.; Tvorogov, D.; Zheng, W.; Franco, C.A.; Murtomäki, A.; Aranda, E.; et al. VEGFR-3 controls tip to stalk conversion at vessel fusion sites by reinforcing Notch signalling. Nat. Cell Biol. 2011, 13, 1202–1213. [Google Scholar] [CrossRef] [PubMed]
- Fantin, A.; Vieira, J.M.; Gestri, G.; Denti, L.; Schwarz, Q.; Prykhozhij, S.; Peri, F.; Wilson, S.W.; Ruhrberg, C. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood 2010, 116, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Akahori, H.; Harari, E.; Smith, S.L.; Polavarapu, R.; Karmali, V.; Otsuka, F.; Gannon, R.L.; Braumann, R.E.; Dickinson, M.H.; et al. CD163+ macrophages promote angiogenesis and vascular permeability accompanied by inflammation in atherosclerosis. J. Clin. Investig. 2018, 128, 1106–1124. [Google Scholar] [CrossRef]
- Yan, D.; He, Y.; Dai, J.; Yang, L.; Wang, X.; Ruan, Q. Vascular endothelial growth factor modified macrophages transdifferentiate into endothelial-like cells and decrease foam cell formation. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef]
- Yan, D.; Zhang, D.; Lu, L.; Qiu, H.; Wang, J. Vascular endothelial growth factor-modified macrophages accelerate reendothelialization and attenuate neointima formation after arterial injury in atherosclerosis-prone mice. J. Cell. Biochem. 2019, 120, 10652–10661. [Google Scholar] [CrossRef]
- Zhang, J.; Muri, J.; Fitzgerald, G.; Gorski, T.; Gianni-Barrera, R.; Masschelein, E.; D’hulst, G.; Gilardoni, P.; Turiel, G.; Fan, Z.; et al. Endothelial Lactate Controls Muscle Regeneration from Ischemia by Inducing M2-like Macrophage Polarization. Cell Metab. 2020, 31, 1136–1153.e7. [Google Scholar] [CrossRef]
- Catalan-Dibene, J.; McIntyre, L. L.; Zlotnik, A. , Interleukin 30 to Interleukin 40. J Interferon Cytokine Res 2018, 38, 423–439. [Google Scholar] [CrossRef]
- Sica, A.; Matsushima, K.; Vandamme, J.; Wang, J.; Polentarutti, N.; Dejana, E.; Colotta, F.; Mantovani, A. IL-1 TRANSCRIPTIONALLY ACTIVATES THE NEUTROPHIL CHEMOTACTIC FACTOR IL-8 GENE IN ENDOTHELIAL-CELLS. 1990, 69, 548–553.
- Zhao, X.; Zhang, W.; Xing, D.; Li, P.; Fu, J.; Gong, K.; Hage, F.G.; Oparil, S.; Chen, Y.-F.; Giordano, S.; et al. Endothelial cells overexpressing IL-8 receptor reduce cardiac remodeling and dysfunction following myocardial infarction. Am. J. Physiol. Circ. Physiol. 2013, 305, H590–H598. [Google Scholar] [CrossRef]
- Mai, W.; Liao, Y. Targeting IL-1β in the Treatment of Atherosclerosis. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Bevilacqua, M.; Pober, J.; Wheeler, M.; Cotran, R.; Gimbrone, M. INTERLEUKIN-1 ACTIVATION OF VASCULAR ENDOTHELIUM - EFFECTS ON PROCOAGULANT ACTIVITY AND LEUKOCYTE ADHESION. 1985, 121, 394–403.
- Yang, Y.; Luo, N.-S.; Ying, R.; Xie, Y.; Chen, J.-Y.; Wang, X.-Q.; Gu, Z.-J.; Mai, J.-T.; Liu, W.-H.; Wu, M.-X.; et al. Macrophage-derived foam cells impair endothelial barrier function by inducing endothelial-mesenchymal transition via CCL-4. Int. J. Mol. Med. 2017, 40, 558–568. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-Y.; Qin, L.; Li, G.; Wang, Z.; Dahlman, J.E.; Malagon-Lopez, J.; Gujja, S.; Cilfone, N.A.; Kauffman, K.J.; Sun, L.; et al. Endothelial TGF-β signalling drives vascular inflammation and atherosclerosis. Nat. Metab. 2019, 1, 912–926. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; You, B.; Chen, S.-P.; Liao, J.K.; Sun, J. Tumor Necrosis Factor-α Downregulates Endothelial Nitric Oxide Synthase mRNA Stability via Translation Elongation Factor 1-α 1. Circ. Res. 2008, 103, 591–597. [Google Scholar] [CrossRef]
- Steyers, C. M.; Miller, F. J. , Endothelial dysfunction in chronic inflammatory diseases. Int J Mol Sci 2014, 15, 11324–49. [Google Scholar] [CrossRef]
- Kleinbongard, P.; Heusch, G.; Schulz, R. TNFα in atherosclerosis, myocardial ischemia/reperfusion and heart failure. Pharmacol. Ther. 2010, 127, 295–314. [Google Scholar] [CrossRef]
- Che, J.; Okigaki, M.; Takahashi, T.; Katsume, A.; Adachi, Y.; Yamaguchi, S.; Matsunaga, S.; Takeda, M.; Matsui, A.; Kishita, E.; et al. Endothelial FGF receptor signaling accelerates atherosclerosis. Am. J. Physiol. Circ. Physiol. 2011, 300, H154–H161. [Google Scholar] [CrossRef]
- Parma, L.; Peters, H.A.B.; Sluiter, T.J.; Simons, K.H.; Lazzari, P.; de Vries, M.R.; Quax, P.H.A. bFGF blockade reduces intraplaque angiogenesis and macrophage infiltration in atherosclerotic vein graft lesions in ApoE3*Leiden mice. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Huang, C.; Huang, Y.; Zhou, Y.; Nie, W.; Pu, X.; Xu, X.; Zhu, J. Exosomes derived from oxidized LDL-stimulated macrophages attenuate the growth and tube formation of endothelial cells. Mol. Med. Rep. 2018, 17, 4605–4610. [Google Scholar] [CrossRef]
- Fu, W.; Liu, Z.; Zhang, J.; Shi, Y.; Zhao, R.; Zhao, H. Effect of microRNA-144-5p on the proliferation, invasion and migration of human umbilical vein endothelial cells by targeting SMAD1. Exp. Ther. Med. 2019, 19, 165–171. [Google Scholar] [CrossRef]
- Su, D. Up-regulation of MiR-145-5p promotes the growth and migration in LPS-treated HUVECs through inducing macrophage polarization to M2. J. Recept. Signal Transduct. 2020, 41, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Ismail, N.; Wang, Y.; Dakhlallah, D.; Moldovan, L.; Agarwal, K.; Batte, K.; Shah, P.; Wisler, J.; Eubank, T.D.; Tridandapani, S.; et al. Macrophage microvesicles induce macrophage differentiation and miR-223 transfer. Blood 2013, 121, 984–995. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, Z.; Wang, X.; Zheng, J.; Peng, L.; Zhou, Y.; Song, Y.; Lu, Z. Extracellular-vesicle containing miRNA-503-5p released by macrophages contributes to atherosclerosis. Aging 2021, 13, 12239–12257. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wang, S.; Wang, G.; Zhao, M.; Du, F.; Li, K.; Wang, L.; Wu, H.; Chen, J.; Yang, Y.; Su, G. , Macrophage-derived exosomal miR-4532 promotes endothelial cells injury by targeting SP1 and NF-κB P65 signalling activation. J Cell Mol Med 2022, 26, (20), 5165–5180. [Google Scholar] [CrossRef]
- Yang, Y.; Luo, H.; Zhou, C.; Zhang, R.; Liu, S.; Zhu, X.; Ke, S.; Liu, H.; Lu, Z.; Chen, M. Regulation of capillary tubules and lipid formation in vascular endothelial cells and macrophages via extracellular vesicle-mediated microRNA-4306 transfer. J. Int. Med Res. 2018, 47, 453–469. [Google Scholar] [CrossRef] [PubMed]
- Njock, M.-S.; Cheng, H.S.; Dang, L.T.; Nazari-Jahantigh, M.; Lau, A.C.; Boudreau, E.; Roufaiel, M.; Cybulsky, M.I.; Schober, A.; Fish, J.E. Endothelial cells suppress monocyte activation through secretion of extracellular vesicles containing antiinflammatory microRNAs. Blood 2015, 125, 3202–3212. [Google Scholar] [CrossRef]
- He, S.; Wu, C.; Xiao, J.; Li, D.; Sun, Z.; Li, M. Endothelial extracellular vesicles modulate the macrophage phenotype: Potential implications in atherosclerosis. Scand. J. Immunol. 2018, 87, e12648. [Google Scholar] [CrossRef]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef]
- Maruyama, K.; Kagota, S.; McGuire, J.J.; Wakuda, H.; Yoshikawa, N.; Nakamura, K.; Shinozuka, K. Enhanced Nitric Oxide Synthase Activation via Protease-Activated Receptor 2 Is Involved in the Preserved Vasodilation in Aortas from Metabolic Syndrome Rats. J. Vasc. Res. 2015, 52, 232–243. [Google Scholar] [CrossRef]
- Murray, M.Y.; Birkland, T.P.; Howe, J.D.; Rowan, A.D.; Fidock, M.; Parks, W.C.; Gavrilovic, J. Macrophage Migration and Invasion Is Regulated by MMP10 Expression. PLOS ONE 2013, 8, e63555. [Google Scholar] [CrossRef]
- Chang, M. Y.; Chan, C. K.; Braun, K. R.; Green, P. S.; O'Brien, K. D.; Chait, A.; Day, A. J.; Wight, T. N. , Monocyte-to-macrophage differentiation: synthesis and secretion of a complex extracellular matrix. J Biol Chem 2012, 287, 14122–35. [Google Scholar] [CrossRef] [PubMed]
- Beck-Joseph, J.; Lehoux, S. Molecular Interactions Between Vascular Smooth Muscle Cells and Macrophages in Atherosclerosis. Front. Cardiovasc. Med. 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Hashem, R.; Rashed, L.; Abdelkader, R.; Hashem, K. Stem cell therapy targets the neointimal smooth muscle cells in experimentally induced atherosclerosis: involvement of intracellular adhesion molecule (ICAM) and vascular cell adhesion molecule (VCAM). Braz. J. Med Biol. Res. 2021, 54, e10807. [Google Scholar] [CrossRef] [PubMed]
- Butoi, E. D.; Gan, A. M.; Manduteanu, I.; Stan, D.; Calin, M.; Pirvulescu, M.; Koenen, R. R.; Weber, C.; Simionescu, M. , Cross talk between smooth muscle cells and monocytes/activated monocytes via CX3CL1/CX3CR1 axis augments expression of pro-atherogenic molecules. Biochim Biophys Acta 2011, 1813, 2026–35. [Google Scholar] [CrossRef]
- Xing, Y.; Pan, S.; Zhu, L.; Cui, Q.; Tang, Z.; Liu, Z.; Liu, F. Advanced Glycation End Products Induce Atherosclerosis via RAGE/TLR4 Signaling Mediated-M1 Macrophage Polarization-Dependent Vascular Smooth Muscle Cell Phenotypic Conversion. Oxidative Med. Cell. Longev. 2022, 2022, 1–11. [Google Scholar] [CrossRef]
- Boyle, J. J.; Bowyer, D. E.; Weissberg, P. L.; Bennett, M. R. , Human blood-derived macrophages induce apoptosis in human plaque-derived vascular smooth muscle cells by Fas-ligand/Fas interactions. Arterioscler Thromb Vasc Biol 2001, 21, 1402–7. [Google Scholar] [CrossRef]
- Boyle, J.J.; Weissberg, P.L.; Bennett, M.R. Human Macrophage-Induced Vascular Smooth Muscle Cell Apoptosis Requires NO Enhancement of Fas/Fas-L Interactions. Arter. Thromb. Vasc. Biol. 2002, 22, 1624–1630. [Google Scholar] [CrossRef]
- Xiang, P.; Blanchard, V.; Francis, G. A. , Smooth Muscle Cell-Macrophage Interactions Leading to Foam Cell Formation in Atherosclerosis: Location, Location, Location. Front Physiol 2022, 13, 921597. [Google Scholar] [CrossRef]
- Ren, W.; Wang, Z.; Wang, J.; Wu, Z.; Ren, Q.; Yu, A.; Ruan, Y. IL-5 overexpression attenuates aortic dissection by reducing inflammation and smooth muscle cell apoptosis. Life Sci. 2020, 241, 117144. [Google Scholar] [CrossRef]
- Mazighi, M.; Pellé, A.; Gonzalez, W.; Mtairag el, M.; Philippe, M.; Hénin, D.; Michel, J. B.; Feldman, L. J. , IL-10 inhibits vascular smooth muscle cell activation in vitro and in vivo. Am J Physiol Heart Circ Physiol 2004, 287, H866–71. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, Y.; Feng, W.; Chen, R.; Chen, J.; Touyz, R.M.; Wang, J.; Huang, H. Interleukin-18 Enhances Vascular Calcification and Osteogenic Differentiation of Vascular Smooth Muscle Cells Through TRPM7 Activation. Arter. Thromb. Vasc. Biol. 2017, 37, 1933–1943. [Google Scholar] [CrossRef] [PubMed]
- Akdis, M.; Burgler, S.; Crameri, R.; Eiwegger, T.; Fujita, H.; Gomez, E.; Klunker, S.; Meyer, N.; O'Mahony, L.; Palomares, O.; Rhyner, C.; Ouaked, N.; Schaffartzik, A.; Van De Veen, W.; Zeller, S.; Zimmermann, M.; Akdis, C. A. , Interleukins, from 1 to 37, and interferon-γ: receptors, functions, and roles in diseases. J Allergy Clin Immunol 2011, 127, 701–21e1. [Google Scholar] [PubMed]
- Lim, W.-W.; Corden, B.; Ng, B.; Vanezis, K.; D’agostino, G.; Widjaja, A.A.; Song, W.-H.; Xie, C.; Su, L.; Kwek, X.-Y.; et al. Interleukin-11 is important for vascular smooth muscle phenotypic switching and aortic inflammation, fibrosis and remodeling in mouse models. Sci. Rep. 2020, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Yurdagul, A. Crosstalk Between Macrophages and Vascular Smooth Muscle Cells in Atherosclerotic Plaque Stability. Arter. Thromb. Vasc. Biol. 2022, 42, 372–380. [Google Scholar] [CrossRef]
- Feinberg, M. W.; Watanabe, M.; Lebedeva, M. A.; Depina, A. S.; Hanai, J.; Mammoto, T.; Frederick, J. P.; Wang, X. F.; Sukhatme, V. P.; Jain, M. K. , Transforming growth factor-beta1 inhibition of vascular smooth muscle cell activation is mediated via Smad3. J Biol Chem 2004, 279, 16388–93. [Google Scholar] [CrossRef]
- Sato, A.Y.S.; Bub, G.L.; Campos, A.H. BMP-2 and -4 produced by vascular smooth muscle cells from atherosclerotic lesions induce monocyte chemotaxis through direct BMPRII activation. Atherosclerosis 2014, 235, 45–55. [Google Scholar] [CrossRef]
- Chen, S.; Ding, Y.; Tao, W.; Zhang, W.; Liang, T.; Liu, C. Naringenin inhibits TNF-α induced VSMC proliferation and migration via induction of HO-1. Food Chem. Toxicol. 2012, 50, 3025–3031. [Google Scholar] [CrossRef]
- Moe, S.M.; Chen, N.X. Pathophysiology of Vascular Calcification in Chronic Kidney Disease. Circ. Res. 2004, 95, 560–567. [Google Scholar] [CrossRef]
- Lamb, F.S.; Choi, H.; Miller, M.R.; Stark, R.J. TNFα and Reactive Oxygen Signaling in Vascular Smooth Muscle Cells in Hypertension and Atherosclerosis. Am. J. Hypertens. 2020, 33, 902–913. [Google Scholar] [CrossRef]
- Nunokawa, Y.; Ishida, N.; Tanaka, S. Cloning of Inducible Nitric Oxide Synthase in Rat Vascular Smooth Muscle Cells. Biochem. Biophys. Res. Commun. 1993, 191, 89–94. [Google Scholar] [CrossRef]
- Schroder, K.; Hertzog, P. J.; Ravasi, T.; Hume, D. A. , Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol 2004, 75, 163–89. [Google Scholar] [CrossRef] [PubMed]
- Diao, Y.; Mohandas, R.; Lee, P.; Liu, Z.; Sautina, L.; Mu, W.; Li, S.; Wen, X.; Croker, B.; Segal, M.S.; et al. Effects of Long-Term Type I Interferon on the Arterial Wall and Smooth Muscle Progenitor Cells Differentiation. Arter. Thromb. Vasc. Biol. 2016, 36, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-N.; Velichko, S.; Vincelette, J.; Fitch, R.M.; Vergona, R.; Sullivan, M.E.; Croze, E.; Wang, Y.-X. Interferon-beta attenuates angiotensin II-accelerated atherosclerosis and vascular remodeling in apolipoprotein E deficient mice. Atherosclerosis 2008, 197, 204–211. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Medley, S.C.; Hu, T.; Hinsdale, M.E.; Lupu, F.; Virmani, R.; Olson, L.E. PDGFRβ signalling regulates local inflammation and synergizes with hypercholesterolaemia to promote atherosclerosis. Nat. Commun. 2015, 6, 1–14. [Google Scholar] [CrossRef]
- Newman, A.A.C.; Serbulea, V.; Baylis, R.A.; Shankman, L.S.; Bradley, X.; Alencar, G.F.; Owsiany, K.; Deaton, R.A.; Karnewar, S.; Shamsuzzaman, S.; et al. Multiple cell types contribute to the atherosclerotic lesion fibrous cap by PDGFRβ and bioenergetic mechanisms. Nat. Metab. 2021, 3, 166–181. [Google Scholar] [CrossRef]
- Li, L.; Mao, D.; Li, C.; Li, M. miR-145-5p Inhibits Vascular Smooth Muscle Cells (VSMCs) Proliferation and Migration by Dysregulating the Transforming Growth Factor-b Signaling Cascade. Med Sci. Monit. 2018, 24, 4894–4904. [Google Scholar] [CrossRef]
- Yaker, L.; Tebani, A.; Lesueur, C.; Dias, C.; Jung, V.; Bekri, S.; Guerrera, I.C.; Kamel, S.; Ausseil, J.; Boullier, A. Extracellular Vesicles From LPS-Treated Macrophages Aggravate Smooth Muscle Cell Calcification by Propagating Inflammation and Oxidative Stress. Front. Cell Dev. Biol. 2022, 10, 823450. [Google Scholar] [CrossRef]
- Li, X.; Wang, J.; Wu, C.; Lu, X.; Huang, J. MicroRNAs involved in the TGF-β signaling pathway in atherosclerosis. Biomed. Pharmacother. 2021, 146, 112499. [Google Scholar] [CrossRef]
- Butoi, E.; Gan, A.; Tucureanu, M.; Stan, D.; Macarie, R.; Constantinescu, C.; Calin, M.; Simionescu, M.; Manduteanu, I. Cross-talk between macrophages and smooth muscle cells impairs collagen and metalloprotease synthesis and promotes angiogenesis. Biochim. et Biophys. Acta (BBA) - Mol. Cell Res. 2016, 1863, 1568–1578. [Google Scholar] [CrossRef]
- Galis, Z.S.; Sukhova, G.K.; Lark, M.W.; Libby, P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J. Clin. Investig. 1994, 94, 2493–2503. [Google Scholar] [CrossRef]
- Wang, X.; Khalil, R. A. , Matrix Metalloproteinases, Vascular Remodeling, and Vascular Disease. Adv Pharmacol 2018, 81, 241–330. [Google Scholar] [PubMed]
- Thomas, A.C.; Newby, A.C. Effect of Matrix Metalloproteinase-9 Knockout on Vein Graft Remodelling in Mice. J. Vasc. Res. 2009, 47, 299–308. [Google Scholar] [CrossRef]
- Xu, H.; Jiang, J.; Chen, W.; Li, W.; Chen, Z. Vascular Macrophages in Atherosclerosis. J. Immunol. Res. 2019, 2019, 4354786. [Google Scholar] [CrossRef]
- Li, M.; Qian, M.; Kyler, K.; Xu, J. Endothelial–Vascular Smooth Muscle Cells Interactions in Atherosclerosis. Front. Cardiovasc. Med. 2018, 5, 151. [Google Scholar] [CrossRef] [PubMed]
- Nambo-Venegas, R.; Palacios-González, B.; Mas-Oliva, J.; Aurioles-Amozurrutia, A.K.; Cruz-Rangel, A.; Moreno, A.; Hidalgo-Miranda, A.; Rodríguez-Dorantes, M.; Vadillo-Ortega, F.; Xicohtencatl-Cortes, J.; et al. Conversion of M1 Macrophages to Foam Cells: Transcriptome Differences Determined by Sex. Biomedicines 2023, 11, 490. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, R.A.; Kleemann, R.; Schaapherder, A.F.; Bogaerdt, A.v.D.; Hedin, U.; Matic, L.; Lindeman, J.H. Validating human and mouse tissues commonly used in atherosclerosis research with coronary and aortic reference tissue: similarities but profound differences in disease initiation and plaque stability. JVS: Vasc. Sci. 2023, 4, 100118. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, X.; Millican, R.; Lynd, T.; Gangasani, M.; Malhotra, S.; Sherwood, J.; Hwang, P.T.; Cho, Y.; Brott, B.C.; et al. Recent Progress in in vitro Models for Atherosclerosis Studies. Front. Cardiovasc. Med. 2022, 8, 790529. [Google Scholar] [CrossRef]
- Navab, M.; Hough, G.P.; Stevenson, L.W.; Drinkwater, D.C.; Laks, H.; Fogelman, A.M. Monocyte migration into the subendothelial space of a coculture of adult human aortic endothelial and smooth muscle cells. J. Clin. Investig. 1988, 82, 1853–1863. [Google Scholar] [CrossRef]
- Takaku, M.; Wada, Y.; Jinnouchi, K.; Takeya, M.; Takahashi, K.; Usuda, H.; Naito, M.; Kurihara, H.; Yazaki, Y.; Kumazawa, Y.; et al. An In Vitro Coculture Model of Transmigrant Monocytes and Foam Cell Formation. Arter. Thromb. Vasc. Biol. 1999, 19, 2330–2339. [Google Scholar] [CrossRef]
- Noonan, J.; Grassia, G.; MacRitchie, N.; Garside, P.; Guzik, T.J.; Bradshaw, A.C.; Maffia, P. A Novel Triple-Cell Two-Dimensional Model to Study Immune-Vascular Interplay in Atherosclerosis. Front. Immunol. 2019, 10, 849. [Google Scholar] [CrossRef]
- Liu, M.; Samant, S.; Vasa, C.H.; Pedrigi, R.M.; Oguz, U.M.; Ryu, S.; Wei, T.; Anderson, D.R.; Agrawal, D.K.; Chatzizisis, Y.S. Co-culture models of endothelial cells, macrophages, and vascular smooth muscle cells for the study of the natural history of atherosclerosis. PLOS ONE 2023, 18, e0280385. [Google Scholar] [CrossRef]
- Garcia-Martin, R.; Wang, G.; Brandão, B.B.; Zanotto, T.M.; Shah, S.; Patel, S.K.; Schilling, B.; Kahn, C.R. MicroRNA sequence codes for small extracellular vesicle release and cellular retention. Nature 2021, 601, 446–451. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



