
Submitted:
06 February 2025
Posted:
06 February 2025
You are already at the latest version
Abstract
Background: Exocrine pancreatic insufficiency in Cystic Fibrosis (CF) increases fecal choline losses, but the postnatal course of plasma choline and its metabolites in these patients is un-known. While choline homeostasis is crucial for cellular, bile, and lipoprotein metabolism, via phosphatidylcholine (PC) and via betaine as a methyl donor, choline deficiency is associated with impaired lung and liver function, including hepatic steatosis. Objective: To assess plasma levels of choline, betaine, trimethylamine oxide (TMAO), PC, and PC subclasses in CF patients from infancy to adulthood, comparing those with exocrine pancreatic insufficiency (EPI) and sufficiency (EPS). Methods: Retrospective analysis of target parameters in plasma samples (7/2015-11/2023) of CF patients (0.64-24.6 years) with tandem mass spectrometry. Results: A total of 477 samples from 162 CF patients were analyzed. In CF patients with EPI (N=148), plasma choline and betaine concentrations were lower, and decreased with age compared to EPS pa-tients showing normal values. TMAO concentrations, indicating intestinal choline degradation by bacterial colonization, were frequently elevated in EPI from infancy onwards, and inversely related to plasma choline and betaine levels. PC-containing linoleic acid levels were lower in EPI, but arachidonic and docosahexaenoic acid content was similar in both patient groups. Con-clusion: CF patients with EPI are at risk of choline and betaine deficiency compared to exocrine pancreas-sufficient CF patients. Elevated TMAO concentrations in EPI patients indicate in-creased bacterial colonization leading to choline degradation before absorption. These findings indicate that laboratory testing of choline, betaine and TMAO, and clinical trials on choline supplementation are warranted in CF patients.
Keywords:
betaine
; choline deficiency
; cystic fibrosis
; exocrine pancreas insufficiency
; hepatosteatosis
; PEMT
; phosphatidylcholine
; SIBO
; TMAO
1. Introduction
Cystic Fibrosis (CF) is a recessive autosomal disease with an incidence of 1:3300 to 1:4800 in the Caucasian population. It is caused by mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene, mostly characterized by the homozygous or compound heterozygous F508del genotype, comprising about 80% of patients in the Caucasian population. There are approximately 200 new CF cases per year in Germany, with a prevalence of 6.000-7.000 patients. Prevalence is 30.000-35.000 in all Europe as well as in North America, but lower in non-Caucasians [1,4,23]. Organ manifestations, such as impaired structure and function of the exocrine pancreas, intestine and lungs begin to develop in utero [5,6]. Postnatally, development is impaired despite state-of-the-art therapies, including digestive enzyme substitution in exocrine pancreatic insufficient patients. Lung disease is the most frequent, and CF-associated liver disease (CFALD) the second-most cause of early death [7,8,9].
85-87% of CF patients develop exocrine pancreas insufficiency (EPI), mostly in utero or in early infancy, and there is a strong link between exocrine pancreas and lung function [10]. Notably, CF patients with EPI show increased fecal loss of the essential nutrient choline, and frequently low plasma concentrations of choline and PC [11,12]. Hence, choline deficiency potentially impacts on ~5,000-6,000 CF patients in Germany and ~25,000-30,000 in Europe as well as in the USA. Pancreatic enzyme substitution, adequate nutrition and antibiotic treatment have increased the median life expectancy of these patients to 50 years [13,14]. Although pulmonary failure is the most frequent cause of death [15], CFALD, a synopsis of alterations from hepatic steatosis to biliary cirrhosis with potentially fatal consequences, is the second-most frequent cause of death and associated with pancreatic insufficiency [16,17,18,19,20]. As hepatic steatosis and/or cholestasis are known consequences of choline deficiency, particularly in choline-free parenteral nutrition (PNALD=parenteral nutrition associated liver disease) [21,22,23,24], chronic choline deficiency may contribute to CFALD as well. However, only limited data on the choline status of CF patients in relation to age, and no systematic investigation comparing patients with and without exocrine pancreas insufficiency, are available [11].
CFTR functions as a transporter of chloride (Cl−), hydrogen carbonate (HCO3−), sphingosine-1-phosphate (S1P) and γ-glutamyl-cysteinyl-glycine (reduced glutathione, GSH). These are all altered in CF and related to choline metabolism: via HCO3−, requiring CFTR-mediated Cl− transport for the Cl−/HCO3− antiporter, CFTR is crucial for physiological pH regulation in the small intestine, mucin solubilization, and the optimal function of pancreatic phospholipase A2 IB (sPLA2IB). This enzyme is essential for the enterohepatic cycle of bile PC and, therefore, EPI impacts on choline homeostasis [11,25,26,27,28]. Moreover, via S1P and other components, CFTR contributes to sphingolipid metabolism and, therefore, to choline-containing sphingomyelin (SPH) and apoptosis. Finally, homeostasis of the antioxidant GSH, which is also decreased in CF, is linked to choline via betaine and its downstream metabolites as methyl donors for the methylation of homocysteine to methionine. Its activation to S-adenosylmethionine (SAM) is required for stimulating the hepatic transsulfuration pathway resulting in cysteine and GSH formation [29,30,31,32].
Quantitatively, choline as an essential nutrient is predominantly required for parenchymal homeostasis in the form of membrane phospholipids, such as PC and SPH, and for the formation of betaine as a methyl donor. SPH is high in cell membranes and high-density lipoproteins, whereas PC dominates in bile and very low-density lipoproteins (VLDL). Hepatic PC turnover amounts to ~50% of its PC pool per day via bile secretion, along with the PC moiety of VLDL for hepatic triglyceride export [33,34]. The irreversible synthesis and turnover of betaine as a methyl donor account for ~40% of ingested choline which significantly contributes to choline requirement [27,35,36].
Key to choline deficiency in CF patients is the loss of bile PC via feces due to EPI [11,37]. Plasma concentrations of choline and betaine are low in these CF patients compared to fasting controls (6.4±0.3 vs. 7.8±0.3 µmol/L and 20.9±2.1 vs. 30.3±4.2µmol/L, respectively) [12], and may be extremely low (~2µmol/L) in some individuals with severe hepatic steatosis [60]. A pilot interventional study conducted on adult CF patients confirmed the relationship between choline homeostasis, liver steatosis, and pulmonary function; the observed clinical improvements were most significant in patients whose organs were more severely affected [38].
From a pathophysiological perspective, decreased small intestinal pH and impaired function of the highly pH-sensitive pancreatic sPLA2IB result in a lack of PC cleavage to lyso-PC, thereby blunting choline re-uptake via the enterohepatic cycle in the terminal ileum and resulting in fecal choline loss. About 50% of the hepatic PC pool is secreted daily into the duodenum, accounting for ~11-12 of 23g PC in a 1500g adult liver [39], and equaling ~1.6g choline, i. e. the 3-4fold of the adequate intake [AI] of adults [400-550mg/d] [40,41]. Hence, dysfunction of the enterohepatic cycle due to exocrine pancreas insufficiency may result in choline deficiency. Finally, as the AI of choline is mostly not achieved by the population, its enterohepatic turnover may exceed the value of 3-4fold of its daily intake [42,43].
In addition, PC is essential for hepatic triglyceride secretion and the transport of long-chain poly-unsaturated fatty acids (LC-PUFA) via VLDL, comprising 20% PC [44,45]. Hence, via the PC moiety of VLDL, choline and LC-PUFA metabolism of the liver are linked to that in other organs including the lungs [46,47,48,49].
Endogenous hepatic PC synthesis via the phosphatidylethanolamine N-methyltransferase (PEMT) pathway (requiring SAM) is not sufficient to meet choline requirements [40,41,50]. Moreover, decreased PEMT activity is characteristic of CF patients [12]. Additionally, frequent single nucleotide polymorphisms (SNPs) in the PEMT gene further increase exogenous choline requirements and are associated with (non-alcoholic) fatty liver disease [51,52,53]. Among the “modifier genes” influencing the clinical course of CF [54] may be other frequent SNPs in choline and one-carbon metabolism as well, linking this nutrient with liver disease in CF as it does in the overall population [55].
Finally, non-genetic factors such as small intestinal bacterial overgrowth (SIBO) may contribute to choline deficiency via the intraluminal degradation of choline prior to its absorption. This is suggested by increased plasma concentrations of trimethylamine oxide (TMAO) in relation to the intestinal microbiota [56].
However, no systematic overview of choline homeostasis, including that of its metabolites, exists to date for CF patients of different ages. Although an association between exocrine pancreatic function and choline status has been suggested [11,37], it has never been evaluated on a routine basis in patients of different ages nor in comparison with pancreas-sufficient CF patients. However, this is important as pancreatic insufficiency - and poor sPLA2IB function - develops very early, and correcting for choline deficiency may be important for the development of these patients [27,57]. Therefore, we retrospectively investigated the plasma concentrations of choline and its metabolites in relation to exocrine pancreatic function in CF patients from infancy to adulthood.
2. Materials and Methods
2.1. Study Population
Retrospective analysis of patient data was approved by the Institutional Review Board (121/2020BO2). As a pseudonymized, retrospective study, written consent of patients was not required. Routine assessment of choline and fatty acid status in patients with CF was introduced in our outpatient clinic in 2015. Exclusion criteria for participation in this study were absence of genetic verification of CF, acute liver damage with hepatocyte decay (prior to liver transplant), verified intake of choline as a prescribed choline supplement or as part of medication, age 25y or beyond and missing choline data (Table 1, Figure 1).
2.2. Exclusion of Additional Choline Intake
Additional choline intake via medication (3000mg choline chloride in adults) or via dietary supplements (such as Fortimel® [Danone Deutschland GmbH, Frankfurt, Germany], Combiotic® [HiPP, Pfaffenhofen, Germany], or Energea P Kid [metaX, Friedberg, Germany], only the latter being free of choline) was retrieved from the patient files. Plasma choline concentrations showed a weak correlation with the additional intake of choline supplements or medication (0-220mg/d, r=0.1470, p=0.0007) (supplementary Figure S1). Therefore, although at the given choline amounts via formula or supplements plasma effects were low compared to interventions with 2200mg/d [38,58], samples during any kind of choline supplementation were excluded. For further patient and inclusion characteristics, see the flow chart (Figure 1) and Table 1.

Table 1.
Inclusion and exclusion parameters of study patients.
| Inclusion parameter | Genetically verified Cystic Fibrosis with or without exocrine pancreas insufficiency; Age 0 to <25 y |
| Exclusion Parameters | Acute liver failure Pre-transplantation status choline supplementation via prescribed choline or dietary supplements 25 y or older No data on choline parameters |
2.3. Sample Parameters and Numbers
477 of 611 EDTA plasma samples (7/2015 to 11/2023) from 162 CF patients aged 0 to < 25 years, without (N=14) or with (N=148) exocrine pancreatic insufficiency, were included in the study (see flow sheet Figure 1). Patients were routinely screened for plasma concentrations of choline, betaine, TMAO, PC, lyso-PC, SPH and ceramides. PC was further differentiated into sub-classes as defined by their fatty acid content, that is, comprising either two saturated fatty acids (disat.-PC) or an oleic acid (OA) (C18:1-PC), linoleic acid (LA) (C18:2-PC), arachidonic acid (ARA) (C20:4-PC), eicosapentaenoic acid (EPA) (C20:5-PC), or docosahexaenoic acid (DHA) residue (C22:6-PC), together with that of a saturated fatty acid (mainly palmitic or stearic acid) [38].
2.4. Plasma Collection
EDTA blood (1-2.7mL) was harvested from venous puncture after >4h fasting, kept on crash ice and centrifuged within 1h at 1000 * g for 10min at room temperature. Plasma supernatant was immediately aspirated and aliquoted in 100µL samples, frozen at -20°C and transferred to a -80°C freezer within 5 days until analysis.
2.5. Mass Spectrometry
Plasma samples were processed using established standard procedures as previously described [38,58,59,60]. In brief, plasma was spiked with internal standard (D4-choline chloride), and extracted with chloroform: methanol according to Bligh & Dyer [61]. The upper water-methanol phase, containing the water-soluble metabolites (choline, betaine, TMAO, and others), was separated from the lipid-containing chloroform phase. Diarachidoyl-PC (PC20:0/20:0) as a mass spectrometry standard was added after phase separation to an aliquot of the chloroform extract as Bligh & Dyer extraction is quantitative [61], to obtain sample aliquots without additional arachidic acid for potential gas chromatography.
Equipment for analysis comprised a TSQ Quantum Discovery Ultra tandem mass spectrometer, Finnigan Surveyor Autosampler Plus, and Finnigan Surveyor MS Pump Plus (Thermo Fisher Scientific, Dreieich, Germany). Choline, D4-choline, betaine and TMAO were separated on a ZORBAX HILIC Plus Narrow Bore RR column (2.1x100mm inner diameter; 3.5µm particle size) (Agilent Tecnologies, Waldbronn, Germany) at 35°C. Elution was performed at 0.4mL/min with solvent A (acetonitril:water:50mM ammonium formate; 74:21:5; v/v) and B (water: 50mM ammonium formate, 95:5; v/v): Gradient conditions were 100% A (0-0.1min) → 32% B (0.1-5.25min) → 100% A (5.25-5.4min) → 100% A (5.4-12min), and components were analyzed at positive ionization [59]. PC, lyso-PC and SPH were separated isocratically on a Polaris Si-A (2x150mm i.d.; 2µm; Agilent Technologies) with chloroform:methanol:300mM ammonium acetate (60:38:2%, v/v) as the mobile phase. Phosphorylcholine (mass/charge [m/z]=+184) served as the diagnostic fragment [60].
2.6. Clinical Parameters
Clinical data were extracted from the hospital’s software SAP (Systemanalyse Programmentwicklung; Version 2020 & 2021, SAP SE & Co. KG, Walldorf, Germany) and the documentation system of the Mucoviszidosis outpatient clinic ARDIS2 (Arthritis und Rheumatologie Dokumentations- und Informationssystem 2; Version 1.52.2, axaris-software & systeme GmbH, Dornstadt, Deutschland, 2009-2022). Lung function parameters measured were forced volume vital capacity (FVC), percentage of predicted forced expiratory volume in 1 s relative to FVC (ppFEV1), and forced expiratory flow at 25% and at 25–75% of the pulmonary volume (FEF25, FEF 25–75).
2.7. Statistics
Data management was performed using Microsoft Office Excel version 2021. To eliminate unbalanced documentation of individual patients with multiple determinations (2-9) and to reduce errors due to single extremely low or high values, the medians of an individual’s data were formed, if more than one sample was available, resulting in 162 data points of which 14 were from pancreas-sufficient and 148 from pancreas-insufficient patients. All analytical and clinical data are expressed as medians and interquartile ranges. Significance values were determined by non-parametric testing for group comparison (Mann-Whitney U-statistic) and non-parametric correlation (Spearman's rank correlation coefficient), using GraphPad InStat, version 3.10 (STATCON GmbH, Witzenhausen, Germany). P-values <0.05 were considered significant.
3. Results
Biometric, routine laboratory, and lung function data are shown in Table 2. The study groups showed similar median ages and age ranges, regardless of exocrine pancreatic (in-)sufficiency (EPI or EPS), with no significant statistical difference (p>0.05). A significant difference was observed in the genotype distribution, with EPI patients predominantly exhibiting F508del homozygous or compound heterozygous, whereas EPS patients were largely characterized by other CFTR genetic variants (Table 2). Most clinical parameters showed no differences between CF patients with and without exocrine pancreatic-insufficiency, except the earlier diagnosis (p=0.0009), higher alanine aminotransferase (ALA) (p=0.0021) and decreased FEF25-75 (p=0.045) in EPI patients. The lower median FEF25 did not reach significance and ppFEV1 as well as PEF were identical in EPI compared to EPS patients.
3.1. Parameters of Choline Homeostasis in the Whole Study Group
Plasma concentrations of both choline and betaine were significantly lower in the EPI group (Table 3A). By contrast, concentrations of trimethylamine oxide (TMAO), showing a large variability, were significantly higher in these patients, whereas in EPS CF patients TMAO was <3µmol/L throughout. Phosphatidylcholine (PC) concentrations, other components containing a choline headgroup (lyso-PC, sphingomyelin [SPH]) and ceramides did not differ between the patient groups (Table 3B). However, the pattern of PC subclasses was different (Table 3C): in EPI patients, PC species containing two saturated fatty acid residues (disaturated PC), an oleic acid (OA/C18:1) or an eicosapentaenoic acid (EPA/C20:5) residue were slightly increased, which was at the expense of PCs comprising a linoleic acid (LA/C18:2) residue or other (minor) PC compounds (Table 3B). Notably, there were no differences in PC subclasses comprising an arachidonic (ARA/C20:4) or docosahexaenoic acid (DHA/C22:6) residue.
3.2. Age-Related Changes of Water-Soluble Choline Compounds in Plasma
Figure 2 A-C shows the median individual concentrations of plasma choline, betaine and trimethylamine oxide (TMAO) in relation to age (0.64-24.6 years) in CF patients with and without exocrine pancreas insufficiency. There was no age-dependent change in plasma choline or betaine levels in pancreas-sufficient patients, whereas in CF patients with exocrine pancreatic insufficiency, both choline and betaine (Figure 2A+B) as well as their sum (supplementary Figure s2) decreased with age. TMAO remained low in exocrine pancreas sufficiency throughout (<3µmol/L), but in insufficient patients the median was higher (Table 3A), but with a large range of values, from near zero to more than 10µmol/L. There was no correlation with age (p=0.1993), and values were significantly increased (7.8 and 12.7µmol/L) even in 2 infants (0.64 and 0.96 years, respectively). There was a direct correlation between plasma concentrations of betaine and choline but an inverse correlation between plasma TMAO and choline concentrations (Figure 2D). This correlation similarly applied to betaine alone and the sum of choline+betaine vs. TMAO (supplementary Figure s3)
3.3. Age-Related Changes of PC and Its Sub-Groups
Figure 3A shows that PC concentrations were identical in CF individuals with or without exocrine pancreatic insufficiency and did not change with age. However, there were distinct differences in the molecular composition of PC, that is, its fatty acid pattern: whereas in EPI, the fraction of disaturated PC increased with age (Figure 3B), that of PC containing an oleic acid (OA) residue (C18:1-PC) remained constant (Figure 3C). By contrast, PC containing a linoleic acid (LA) residue (C18:2-PC) decreased with age in CF patients with EPI only (Figure 3D).
Among the PC species containing a long-chain poly-unsaturated (LC-PUFA) fatty acid residue, those comprising an arachidonic (ARA), eicosapentaenoic (EPA) or docosahexaenoic acid (DHA) residue (C20:4n-6-PC, C20:5n-3-PC and C22:6n-3-PC, respectively) are shown in Figure 4A-C. There was no difference between CF patient groups, or change with age, in C20:4n-6-PC and C22:6n-3-PC. However, with age, EPA-containing C20:5n-3-PC increased, which was statistically significant only in patients with pancreatic insufficiency (Figure 4B). There was no difference in the ratio between PC comprising the omega-6 fatty acid ARA versus PC containing omega-3 fatty acids (EPA, DHA) throughout (Figure 4D).
4. Discussion
This study addresses the plasma concentrations of choline and its derivatives, as indicators of choline deficiency, in CF patients with (EPI) and without (EPS) exocrine pancreatic insufficiency. Imbalances of other metabolites related to choline and betaine homeostasis, such as increased homocysteine and decreased methionine, SAM and glutathione, are well described in patients with EPI, which constitutes the majority of CF patients [37,62,63,64]. Choline deficiency was attributed to chronic fecal choline losses, resulting from these patients’ inefficient enterohepatic circulation of PC released into the duodenum via biliary secretion [11]. A clinical impact of choline deficiency and altered PC/lipoprotein metabolism in CF has been suggested so far in only two pilot studies and one case report [12,37]. A dose of 2200mg rather than 550mg per day for healthy male adults (Institute of Medicine/National Academy of Medicine) was effective in improving lung function and resolving severe hepatic steatosis [58]. Importantly, choline was most effective on lung function improvement if ppFEV1 values were low [38], whereas actually lung function of CF patients is mostly very good, particularly owing to recent advances in modulator treatment (see Table 2 and [65]). Nevertheless, during choline deficiency, choline drains from the lungs to the liver via high-density lipoproteins which in the long run may affect lung parenchyma homeostasis and repair via impaired sphingolipid homeostasis [27,66,67]. Interestingly, within our cohort of CF patients, those with pancreatic insufficiency demonstrated significantly lower FEF25 measurements (see Table 2).
4.1. Plasma Choline and Betaine Levels in Relation to Exocrine Pancreas Function
In our study, we showed that only CF patients with EPI had decreased plasma levels of choline, mostly F508del compound heterozygous (53%) or homozygous (36%) (Table 2). Notably, all patients with EPI in our group received pancreatic enzyme replacement therapy (PERT). This observation highlights that PERT alone may not be effective in preventing choline deficiency. 92% of our studied patients suffered from EPI, so that only 14 pancreas sufficient CF patients could be investigated. While this impairs the statistical significance for some parameters, such as the increase in EPA with age, which was significant only for the 148 EPI patients, the data clearly show that low plasma levels of choline and betaine are characteristic for the EPI patient group. Moreover, the correlation between choline and betaine levels demonstrates that low choline is an indicator of low availability of betaine as an essential methyl donor for homocysteine degradation and for the generation of SAM as the most important methyl group donor involved in glutathione (GSH) and creatine synthesis [37]. In this context, it was shown that choline supplementation increased creatine concentrations in muscle tissue [38].
4.2. The Impact of Small Intestinal Bacterial Colonization and PEMT Genetics
Moreover, variable but frequently high plasma levels of TMAO in EPI CF patients from infancy onwards, and its inverse correlation with choline and betaine levels, suggest that small intestinal bacterial colonization or overgrowth (SIBO) affects choline bioavailability at all ages. In addition to fecal and bacterial choline depletion, impaired endogenous (indirect) choline synthesis by methylation of phosphatidylethanolamine (PE), which requires SAM and PE-N-methyltransferase (PEMT), may contribute to choline deficiency in EPI of CF patients: while the PEMT pathway is generally decreased in these patients [12], frequent single nucleotide polymorphisms (SNPs) of the PEMT gene (such as rs12325817) [21,51,52] may further impact the severity of the clinical consequences of choline deficiency. This was shown in a CF patient with very low plasma choline (2µmol/L) and progressive liver steatosis from preschool age onwards, which was effectively treated with choline chloride. This patient not only suffered from SIBO but was also homozygous for the frequent PEMT SNP rs12325817 (25-44% of the population) [58].
The impact of choline deficiency on liver disease has been supported by other studies, demonstrating that low plasma choline concentrations are associated with liver disease, particularly hepatic steatosis, in parenterally fed patients, and is resolved by parenteral or enteral choline supplementation [32,68]. Despite no routine liver function test in our study cohort, CF-associated liver disease cannot be excluded. There are varying recommendations for screening for liver involvement in CF including laboratory-based scores, histopathology and imaging [69]. However, in our retrospective study we could not systematically assess and evaluate for the presence of liver disease such as hepatic steatosis. Therefore, we were not able to compare EPI and EPS patients with regard to clinical signs of CFALD. Future studies are needed to assess for both the presence of choline deficiency in CFALD with and without pancreatic insufficiency, and for the effect of choline supplementation.
4.3. Effects of Age on Plasma Choline and TMAO Levels
Reference values in relation to age show that plasma choline levels are high after birth, but postnatally rapidly decrease from 48±15µmol/L at birth to 12.8±2.0µmol/L at 3 years and 8.4±3.1µmol/L in adults [59,70]. These values were achieved by EPS, but not by EPI CF patients (Table 3). Betaine levels were similarly decreased in patients with EPI, whereas sufficient patients were similar to healthy controls [12,75]. The increased levels of TMAO demonstrate that the homeostasis of choline and betaine in EPI patients is linked to an overall altered microbial intestinal colonization: TMAO is an indicator of non-quantitative choline absorption, due to the presence of increased numbers of choline-cleaving bacteria in the small intestine, i.e. small intestinal bacterial overgrowth (SIBO). Here, bacteria cleave choline (and related compounds with trifold methylated ammonium groups, such as betaine and carnitine) prior to absorption. They release trimethylamine, which is subsequently absorbed and oxidized to TMAO in the liver, indicating increased intestinal choline degradation [71,72].
Expression of the responsible flavin-containing trimethylamine monooxygenase (FMO3; EC 1.14.13.148) starts postnatally [73], so that TMAO formation is nearly absent in preterm infants [74,75]. However, it is unclear whether the increase in postnatal FMO3 expression in CF patients is as rapid as in healthy infants and identical in all patients. Therefore, it is unclear whether low TMAO levels represent the absence of SIBO in CF patients, and to which degree differences in the individual microbiota contribute to TMAO levels. Nevertheless, the inverse correlations between TMAO and choline and betaine concentrations suggest that impaired choline bioavailability due to intestinal degradation starts at infant age and persists until adulthood (see results and [38,58]). Moreover, it is unclear whether early SIBO and TMAO formation is associated with the development of CF-associated liver disease (CFALD) development.
4.4. Plasma Phospholipids and Long-Chain Poly-Unsaturated Fatty Acids (LC-PUFA) in CF
While choline and betaine values were decreased, and those of TMAO were increased in CF patients with EPI, plasma concentrations of phospholipids (PL), that is PC, lyso-PC and SPH, were similar in both CF groups (1.2 to 1.5mmol/L). Hence, the characteristically lower PL values of CF patients compared to non-CF infants and healthy adults [12,60] applies to all CF patients. Previous research on patients receiving parenteral nutrition demonstrated that choline deficiency as indicated by low free plasma choline concentrations can occur independently of normal or elevated plasma phospholipid (PL) levels. Notably, symptoms of choline deficiency, such as hepatic steatosis, were not alleviated by high PC concentrations in plasma, but only by free choline [76,77]. In line with this, animal experiments have shown that the delivery of free plasma choline from PC stores is low [35].
Nevertheless, the fatty acid pattern of PC was different in CF patients with EPI compared to those with EPS. Notably, there was no difference in PC containing ARA or DHA residues. In contrast, LA-PC was significantly decreased in EPI patients (see Figure 2 and 3). The fraction of LA-PC decreased with age in pancreas-insufficient CF patients, whereas EPA increased. ARA and DHA remained constant and were not related to exocrine pancreas function. As LA is partly and EPA, ARA and DHA are primarily transported via PC in plasma [45], these data show that the EPA status increases but that of LA decreases with age, whereas DHA, ARA and the omega-6 to omega-3 ratio does not change with age in EPI patients. These clinical data are in contradictory to CF animal experiments, describing an increase in the ARA/DHA and a decrease of the EPA/LA ratio [78]. In contrast, our data confirm that from infancy to adulthood, irrespective of exocrine pancreas function, there are no general alterations in ARA and DHA compared to non-CF patients [12].
5. Conclusions
Low plasma concentrations of choline and betaine, together with high TMAO, are characteristic for CF patients with exocrine pancreatic insufficiency despite PERT. In EPI patients, plasma phospholipid concentrations were lower than in healthy controls, but not different from pancreas-sufficient CF patients. There are age-dependent changes in the fatty acid profile of plasma PC, primarily concerning LA and EPA, but not ARA and DHA. LA continuously decreased, whereas EPA increased with age, with no age-dependent alteration in the ARA/EPA+DHA ratio. Low plasma concentrations of choline and betaine, together with high TMAO levels, in CF patients with EPI, and clinical data on the successful treatment of CF-related and -unrelated steatosis by choline supplementation suggest the implementation of choline/betaine/TMAO determination into clinical routine and randomized clinical trials on choline supplementation in patients with CF-related liver disease.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Figure S1: Plasma concentrations of choline in relation to the intake of choline via food supplements; Figure S2: Plasma concentrations of the sum of choline + betaine in relation to the age of CF patients.
Author Contributions
Conceptualization and supervision, W.B., Data collection and analysis, A.S., J.B., writing—original draft preparation, W.B.; writing—review and editing, W.B., U.G-M., J.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
No. 121/2020BO2.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank the staff of the Mucoviscidosis Outpatient Clinic of Children’s Hospital, Medical Faculty, Eberhard-Karls-University, Tübingen.
Conflicts of Interest
W.B receives investigator-initiated trial and consultancy funding by HiPP Inc., Pfaffenhofen, Germany. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Naehrig, S.; Chao, C.-M.; Naehrlich, L. Mukoviszidose - Diagnose und Therapie (Cystic fibrosis—diagnosis and treatment). Dtsch Arztebl Int 2017, 114, 564–574. [Google Scholar] [CrossRef]
- Kosorok, M.R.; Wei, W.H.; Farrell, P.M. The incidence of cystic fibrosis. Stat Med 1996, 15, 449–462. [Google Scholar] [CrossRef]
- Raskin S, Pereira-Ferrari L, Reis FC, Abreu F, Marostica P, Rozov T, Cardieri J, Ludwig N, Valentin L, Rosario-Filho NA, Camargo Neto E, Lewis E, Giugliani R, Diniz EM, Culpi L, Phillip JA 3rd, Chakraborty R. Incidence of cystic fibrosis in five different states of Brazil as determined by screening of p.F508del, mutation at the CFTR gene in newborns and patients. J Cyst Fibros 2008, 7, 15–22.
- Scotet, V.; Duguépéroux, I.; Saliou, P.; Rault, G.; Roussey, M.; Audrézet, M.P.; Férec, C. Evidence for decline in the incidence of cystic fibrosis: a 35-year observational study in Brittany, France. Orphanet J Rare Dis 2012, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Hoo AF, Thia LP, Nguyen TT, Bush A, Chudleigh J, Lum S, Ahmed D, Balfour Lynn I, Carr SB, Chavasse RJ, Costeloe KL, Price J, Shankar A, Wallis C, Wyatt HA, Wade A, Stocks J; London Cystic Fibrosis Collaboration. Lung function is abnormal in 3-month-old infants with cystic fibrosis diagnosed by newborn screening. Thorax 2012, 67, 874–881.
- Korten I, Kieninger E, Yammine S, Cangiano G, Nyilas S, Anagnostopoulou P, Singer F, Kuehni CE, Regamey N, Frey U, Casaulta C, Spycher BD, Latzin P; SCILD; BILD study group. Respiratory rate in infants with cystic fibrosis throughout the first year of life and association with lung clearance index measured shortly after birth. J Cyst Fibros 2019, 18, 118–126. [CrossRef]
- Colombo, C.; Battezzati, P.M.; Crosignani, A.; Morabito, A.; Costantini, D.; Padoan, R.; Giunta, A. Liver disease in cystic fibrosis: a prospective study on incidence, risk factors, and outcome. Hepatology 2002, 36, 1374–1382. [Google Scholar] [CrossRef]
- Williams, S.M.; Goodman, R.; Thomson, R.; Mchugh, K.; Lindsell, D.R.M. Ultrasound evaluation of liver disease in cystic fibrosis as part of an annual assessment clinic: a 9-year review. Clin. Radiol. 2002, 57, 365–370. [Google Scholar] [CrossRef]
- Debray, D.; Kelly, D.; Houwen, R.; Strandvik, B.; Colombo, C. Best practice guidance for the diagnosis and management of cystic fibrosis- associated liver disease. J Cyst Fibros 2011, 10 (Suppl 2), 29–36. [Google Scholar] [CrossRef]
- Gaskin, K.; Gurwitz, D.; Durie, P.; Corey, M.; Levison, H.; Forstner, G. Improved respiratory prognosis in patients with cystic fibrosis with normal fat absorption. J Pediatr 1982, 100, 857–862. [Google Scholar] [CrossRef]
- Chen, A.H.; Innis, S.M.; Davidson, A.G.; James, S.J. Phosphatidylcholine and lysophosphatidylcholine excretion is increased in children with cystic fibrosis and is associated with plasma homocysteine, S-adenosylhomocysteine, and S-adenosylmethionine. Am J Clin Nutr 2005, 81, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Grothe, J.; Riethmüller, J.; Tschürtz, S.M.; Raith, M.; Pynn, C.J.; Stoll, D.; Bernhard, W. Plasma phosphatidylcholine alterations in cystic fibrosis patients: impaired metabolism and correlation with lung function and inflammation. Cell Physiol Biochem 2015, 35, 1437–1453. [Google Scholar] [CrossRef] [PubMed]
- Cystic Fibrosis Foundation Patient Registry. Annual Data Report. Bethesda, Maryland, 2010: http://www.cff.org/UploadedFiles/LivingWithCF/CareCenterNetwork/PatientRegistry/2010-Patient-Registry-Report.pdf.
- Canadian Cystic Fibrosis Foundation Registry. Annual Report. Toronto, Ontario,CAN, 2011: http://www.cysticfibrosis.ca/wp-content/uploads/2013/10/Registry2011FINALOnlineEN.pdf.
- Lindblad, A.; Glaumann, H.; Strandvik, B. Natural history of liver disease in cystic fibrosis. Hepatology 1999, 30, 1151–1158. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, N.A. Intracellular CFTR: Localization and function. Physiol Rev 2011, 79, S175–S191. [Google Scholar] [CrossRef]
- Colombo, C.; Battezzati, P.M.; Crosignani, A.; Morabito, A.; Costantini, D.; Padoan, R.; Giunta, A. Liver disease in cystic fibrosis: a prospective study on incidence, risk factors, and outcome. Hepatology 2002, 36, 1374–1382. [Google Scholar] [CrossRef]
- Williams, S.M.; Goodman, R.; Thomson, R.; Mchugh, K.; Lindsell, D.R.M. Ultrasound evaluation of liver disease in cystic fibrosis as part of an annual assessment clinic: a 9-year review. Clin Radiol 2002, 57, 365–370. [Google Scholar] [CrossRef]
- Herrmann, U.; Dockter, G.; Lammert, F. Cystic fibrosis-associated liver disease. Best Pract Res Clin Gastroenterol 2010, 24, 585–592. [Google Scholar] [CrossRef]
- Freudenberg, F.; Broderick, A.L.; Yu, B.B.; Leonard, M.R.; Glickman, J.N.; Carey, M.C. Pathophysiological basis of liver disease in cystic fibrosis employing a DeltaF508 mouse model. Am J Physiol Gastrointest Liver Physiol 2008, 294, G1411–G1420. [Google Scholar] [CrossRef]
- Fischer, L.M.; da Costa, K.A.; Kwock, L.; Galanko, J.; Zeisel, S.H. Dietary choline requirements of women: effects of estrogen and genetic variation. Am J Clin Nutr 2010, 92, 1113–1119. [Google Scholar] [CrossRef]
- Buchman, A.L.; Dubin, M.; Jenden, D.; Moukarzel, A.; Roch, M.H.; Rice, K.; Gornbein, J.; Ament, M.E.; Eckhert, C.D. Lecithin increases plasma free choline and decreases hepatic steatosis in long-term total parenteral nutrition patients. Gastroenterology 1992, 102, 1363–1370. [Google Scholar] [CrossRef]
- Demetriou, A.A. Lecithin increases plasma free choline and decreases hepatic steatosis in long-term total parenteral nutrition patients. JPEN J Parenter Enteral Nutr 1992, 16, 487–488. [Google Scholar] [CrossRef] [PubMed]
- Buchman, A.L.; Dubin, M.D.; Moukarzel, A.A.; Jenden, D.J.; Roch, M.; Rice, K.M.; Gornbein, J.; Ament, M.E. Choline deficiency: a cause of hepatic steatosis during parenteral nutrition that can be reversed with intravenous choline supplementation. Hepatology 1995, 22, 1399–1403. [Google Scholar] [PubMed]
- Boujaoude, L.C.; Bradshaw-Wilder, C.; Mao, C.; Cohn, J.; Ogretmen, B.; Hannun, Y.A.; Obeid, L.M. Cystic fibrosis transmembrane regulator regulates uptake of sphingoid base phosphates and lysophosphatidic acid: modulation of cellular activity of sphingosine 1-phosphate. J Biol Chem 2001, 276, 35258–35264. [Google Scholar] [CrossRef]
- Scheele GA, Fukuoka SI, Kern HF, Freedman SD (1996) Pancreatic dysfunction in cystic fibrosis occurs as a result of impairments in luminal pH, apical trafficking of zymogen granule membranes, and solubilization of secretory enzymes. Pancreas 1996, 12, 1–9. [CrossRef] [PubMed]
- Bernhard WCholine in cystic fibrosis: relations to pancreas insufficiency, enterohepatic cycle, PEMT and intestinal microbiota. Eur J Nutr 2021, 60, 1737–1759. [CrossRef]
- Linsdell, P.; Hanrahan, J.W. Glutathione permeability of CFTR. Am J Physiol 1998, 275, C323–C326. [Google Scholar] [CrossRef]
- Borowitz, D. CFTR, bicarbonate, and the pathophysiology of cystic fibrosis. Pediatr Pulmonol 2015, 50 (Suppl 40), S24–S30. [Google Scholar] [CrossRef]
- Kogan I, Ramjeesingh M, Li C, Kidd JF, Wang Y, Leslie EM, Cole SP, Bear CE. CFTR directly mediates nucleotide-regulated glutathione flux. EMBO J 2003, 22, 1981–1989.
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Regulators of the transsulfuration pathway. Br J Pharmacol 2019, 176, 583–593. [Google Scholar] [CrossRef]
- Bernhard, W.; Böckmann, K.A.; Minarski, M.; Wiechers, C.; Busch, A.; Bach, D.; Poets, C.F.; Franz, A.R. Evidence and Perspectives for Choline Supplementation during Parenteral Nutrition-A Narrative Review. Nutrients 2024, 16, 1873. [Google Scholar] [CrossRef]
- Northfield, T.C.; Hofmann, A.F. Biliary lipid output during three meals and an overnight fast. I. Relationship to bile acid pool size and cholesterol saturation of bile in gallstone and control subjects. Gut 1975, 16, 1–11. [Google Scholar] [PubMed]
- Nilsson Å, Duan RD. Pancreatic and mucosal enzymes in choline phospholipid digestion. Am J Physiol Gastrointest Liver Physiol 2019, 316, G425–G445. [CrossRef]
- Bernhard, W.; Raith, M.; Shunova, A.; Lorenz, S.; Böckmann, K.; Minarski, M.; Poets, C.F.; Franz, A.R. Choline Kinetics in Neonatal Liver, Brain and Lung-Lessons from a Rodent Model for Neonatal Care. Nutrients 2022, 14, 720. [Google Scholar] [CrossRef] [PubMed]
- Day, C.R.; Kempson, S.A. Betaine chemistry, roles, and potential use in liver disease. Biochim Biophys Acta 2016, 1860, 1098–1106. [Google Scholar] [CrossRef]
- Innis, S.M.; Davidson, A.G.; Melynk, S.; James, S.J. Choline-related supplements improve abnormal plasma methionine-homocysteine metabolites and glutathione status in children with cystic fibrosis. Am J Clin Nutr 2007, 85, 702–708. [Google Scholar] [CrossRef]
- Bernhard, W.; Lange, R.; Graepler-Mainka, U.; Engel, C.; Machann, J.; Hund, V.; Shunova, A.; Hector, A.; Riethmüller, J. Choline Supplementation in Cystic Fibrosis-The Metabolic and Clinical Impact. Nutrients 2019, 11, 656. [Google Scholar] [CrossRef]
- Northfield, T.C.; Hofmann, A.F. Biliary lipid output during three meals and an overnight fast. I. Relationship to bile acid pool size and cholesterol saturation of bile in gallstone and control subjects. Gut 1975, 16, 1–11. [Google Scholar] [CrossRef]
- Natl Acad Sci, U.S.A. Dietary Standing Committee on the Scientific Evaluation of Dietary Reference Intakes and its Panel on Folate, Other B Vitamins, and Choline (1998) Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. National Academies Press (US), Washington (DC). https://www.ncbi.nlm.nih.gov/books/NBK114310/pdf/ Bookshelf_NBK114310.pdf.
- EFSA Panel on Dietetic Products, Nutrition and Allergies (NDA). Dietary Reference Values for choline. EFSA Journal 2016, 14, 4484. [CrossRef]
- Bailey RL, Pac SG, Fulgoni VL 3rd, Reidy KC, Catalano PM. Estimation of Total Usual Dietary Intakes of Pregnant Women in the United States. JAMA Netw Open 2019, 2, e195967. [CrossRef]
- Probst, Y.; Guan, V.; Neale, E. Development of a Choline Database to Estimate Australian Population Intakes. Nutrients 2019, 11, 913. [Google Scholar] [CrossRef]
- The LipidWeb (2020) Plasma lipoproteins. https://lipidmaps.org/resources/lipidweb/lipidweb_html/lipids/simple/lipoprot/index.htm (acc. 2024.11.08).
- Bernhard, W.; Maas, C.; Shunova, A.; Mathes, M.; Böckmann, K.; Bleeker, C.; Vek, J.; Poets, C.F.; Schleicher, E.; Franz, A.R. Transport of long-chain polyunsaturated fatty acids in preterm infant plasma is dominated by phosphatidylcholine. Eur J Nutr 2018, 57, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.R.; Tao, J.Q.; Yu, K.J.; Borok, Z.; Crandall, E.D.; Collins, H.L.; Rothblat, G.H. Expression and biological activity of ABCA1 in alveolar epithelial cells. Am J Respir Cell Mol Biol 2008, 38, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Agassandian, M.; Miakotina, O.L.; Andrews, M.; Mathur, S.N.; Mallampalli, R, K. Pseudomonas aeruginosa and sPLA2 IB stimulate ABCA1-mediated phospholipid efflux via ERK-activation of PPARalpha-RXR. Biochem J, 2007, 403, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Agellon, L.B.; Vance, D.E. Choline redistribution during adaptation to choline deprivation. J Biol Chem 2007, 282, 10283–10289. [Google Scholar] [CrossRef]
- Li, Z.; Agellon, L.B.; Vance, D.E. Phosphatidylcholine homeostasis and liver failure. J Biol Chem 2005, 280, 37798–37802. [Google Scholar] [CrossRef]
- EFSA Panel on Nutrition, Novel Foods and Food allergens (NDA), Turck D, Bohn T, Castenmiller J, De Henauw S, Hirsch-Ernst KI, Knutsen HK, Maciuk A, Mangelsdorf I, McArdle HJ, Naska A, Pentieva K, Thies F, Tsabouri S, Vinceti M, Bresson J-L, Fiolet T, Siani A. Choline and contribution to normal liver function of the foetus and exclusively breastfed infants: evaluation of a health claim pursuant to Article 14 of Regulation (EC) No 1924/2006. EFSA Journal 2023, 21. [CrossRef]
- Resseguie, M.E.; da Costa, K.A.; Galanko, J.A.; Patel, M.; Davis, I.J.; Zeisel, S.H. Aberrant estrogen regulation of PEMT results in choline deficiency-associated liver dysfunction. J Biol Chem 2011, 286, 1649–1658. [Google Scholar] [CrossRef]
- da Costa KA, Corbin KD, Niculescu MD, Galanko JA, Zeisel SH. Identification of new genetic polymorphisms that alter the dietary requirement for choline and vary in their distribution across ethnic and racial groups. FASEB J 2014, 28, 2970–2978. [CrossRef]
- Piras, I.S.; Raju, A.; Don, J.; Schork, N.J.; Gerhard, G.S.; DiStefano, J.K. Hepatic PEMT Expression Decreases with Increasing NAFLD Severity. Int J Mol Sci 2022, 23, 9296. [Google Scholar] [CrossRef]
- Corvol, H.; Blackman, S.M.; Boëlle, P.Y.; Gallins, P.J.; Pace, R.G.; Stonebraker, J.R.; Accurso, F.J.; Clement, A.; Collaco, J.M.; Dang, H.; Dang, A.T.; Franca, A.; Gong, J.; Guillot, L.; Keenan, K.; Li, W.; Lin, F.; Patrone, M.V.; Raraigh, K.S.; Sun, L.; Zhou, Y.H.; O'Neal, W.K.; Sontag, M.K.; Levy, H.; Durie, P.R.; Rommens, J.M.; Drumm, M.L.; Wright, F.A.; Strug, L.J.; Cutting, G.R.; Knowles, M.R. Genome-wide association meta-analysis identifies five modifier loci of lung disease severity in cystic fibrosis. Nat Commun 2015, 6, 8382. [Google Scholar] [CrossRef]
- da Costa KA, Kozyreva OG, Song J, Galanko JA, Fischer LM, Zeisel SH. Common genetic polymorphisms affect the human requirement for the nutrient choline. FASEB J 2006, 20, 1336–1344.
- Meyer KA, Benton TZ, Bennett BJ, Jacobs DR Jr, Lloyd-Jones DM, Gross MD, Carr JJ, Gordon-Larsen P, Zeisel SH. Microbiota-Dependent Metabolite Trimethylamine N-Oxide and Coronary Artery Calcium in the Coronary Artery Risk Development in Young Adults Study (CARDIA). J Am Heart Assoc 2016, 5, e003970. [CrossRef]
- Bernhard, W.; Poets, C.F.; Franz, A.R. Choline and choline-related nutrients in regular and preterm infant growth. Eur J Nutr 2019, 58, 931–945. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, W.; Shunova, A.; Machann, J.; Grimmel, M.; Haack, T.B.; Utz, P.; Graepler-Mainka, U. Resolution of severe hepatosteatosis in a cystic fibrosis patient with multifactorial choline deficiency: A case report. Nutrition 2021, 89, 111348. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, W.; Raith, M.; Kunze, R.; Koch, V.; Heni, M.; Maas, C.; Abele, H.; Poets, C.F.; Franz, A.R. Choline concentrations are lower in postnatal plasma of preterm infants than in cord plasma. Eur J Nutr 2015, 54, 733–741. [Google Scholar] [CrossRef]
- Bernhard, W.; Raith, M.; Koch, V.; Maas, C.; Abele, H.; Poets, C.F.; Franz, A.R. Developmental changes in polyunsaturated fetal plasma phospholipids and feto-maternal plasma phospholipid ratios and their association with bronchopulmonary dysplasia. Eur J Nutr 2016, 55, 2265–2274. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Roum, J.H.; Buhl, R.; McElvaney, N.G.; Borok, Z.; Crystal, R.G. Systemic deficiency of glutathione in cystic fibrosis. J. Appl. Physiol. 1993, 75, 2419–2424. [Google Scholar] [CrossRef]
- Linsdell, P.; Hanrahan, J.W. Glutathione permeability of CFTR. Am J Physiol 1998, 275, C323–C326. [Google Scholar] [CrossRef]
- Kogan I, Ramjeesingh M, Li C, Kidd JF, Wang Y, Leslie EM, Cole SP, Bear CE. CFTR directly mediates nucleotide-regulated glutathione flux. EMBO J. 2003, 22, 1981–1989.
- Ratjen, F. The future of cystic fibrosis: A global perspective. Pediatr Pulmonol. 2024 Oct 17. [CrossRef] [PubMed]
- Li, Z.; Agellon, L.B.; Vance, D.E. Choline redistribution during adaptation to choline deprivation. J Biol Chem 2007, 282, 10283–10289. [Google Scholar] [CrossRef] [PubMed]
- Kleuser, B.; Schumacher, F.; Gulbins, E. New Therapeutic Options in Pulmonal Diseases: Sphingolipids and Modulation of Sphingolipid Metabolism. Handb Exp Pharmacol 2024, 284, 289–312. [Google Scholar] [CrossRef] [PubMed]
- Buchman, A.L.; Ament, M.E.; Sohel, M.; Dubin, M.; Jenden, D.J.; Roch, M.; Pownall, H.; Farley, W.; Awal, M.; Ahn, C. Choline deficiency causes reversible hepatic abnormalities in patients receiving parenteral nutrition: proof of a human choline requirement: a placebo-controlled trial. JPEN J Parenter Enteral Nutr 2001, 25, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Narkewicz, M.R. Integrating Clinical Ultrasound Into Screening for Cystic Fibrosis Liver Disease. J Pediatr Gastroenterol Nutr 2019, 69, 394–395. [Google Scholar] [CrossRef] [PubMed]
- Buchman, A.L.; Sohel, M.; Moukarzel, A.; Bryant, D.; Schanler, R.; Awal, M.; Burns, P.; Dorman, K.; Belfort, M.; Jenden, D.J.; Killip, D.; Roch, M. Plasma choline in normal newborns, infants, toddlers, and in very-low-birth-weight neonates requiring total parenteral nutrition. Nutrition 2001, 17, 18–21. [Google Scholar] [CrossRef]
- al-Waiz M, Mikov M, Mitchell SC, Smith RL. The exogenous origin of trimethylamine in the mouse. Metabolism 1992, 41, 135–136.
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; Wu, Y.; Schauer, P.; Smith, J.D.; Allayee, H.; Tang, W.H.; DiDonato, J.A.; Lusis, A.J.; Hazen, S.L. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef]
- Lang, D.H.; Yeung, C.K.; Peter, R.M.; Ibarra, C.; Gasser, R.; Itagaki, K.; Philpot, R.M.; Rettie, A.E. Isoform specificity of trimethylamine N-oxygenation by human flavin-containing monooxygenase (FMO) and P450 enzymes: selective catalysis by FMO3. Biochem Pharmacol 1998, 56, 1005–1012. [Google Scholar] [CrossRef]
- Bernhard, W.; Böckmann, K.; Maas, C.; Mathes, M.; Hövelmann, J.; Shunova, A.; Hund, V.; Schleicher, E.; Poets, C.F.; Franz, A.R. Combined choline and DHA supplementation: a randomized controlled trial. Eur J Nutr 2020, 59, 729–739. [Google Scholar] [CrossRef]
- Böckmann, K.A.; Franz, A.R.; Minarski, M.; Shunova, A.; Maiwald, C.A.; Schwarz, J.; Gross, M.; Poets, C.F.; Bernhard, W. Differential metabolism of choline supplements in adult volunteers. Eur J Nutr 2022, 61, 219–230. [Google Scholar] [CrossRef]
- Buchman, A. The addition of choline to parenteral nutrition. Gastroenterology 2009, 137, S119–S128. [Google Scholar] [CrossRef] [PubMed]
- Kumpf, V.J. Parenteral nutrition-associated liver disease in adult and pediatric patients. Nutr Clin Pract 21 2006, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, N.; Rout-Pitt, N.; McCarron, A.; Jackson, C.A.; Bulmer, A.C.; McAinch, A.J.; Donnelley, M.; Parsons, D.W.; Hryciw, D.H. Changes in Essential Fatty Acids and Ileal Genes Associated with Metabolizing Enzymes and Fatty Acid Transporters in Rodent Models of Cystic Fibrosis. Int J Mol Sci 2023, 24, 7194. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Flow chart of sample and patient inclusion.
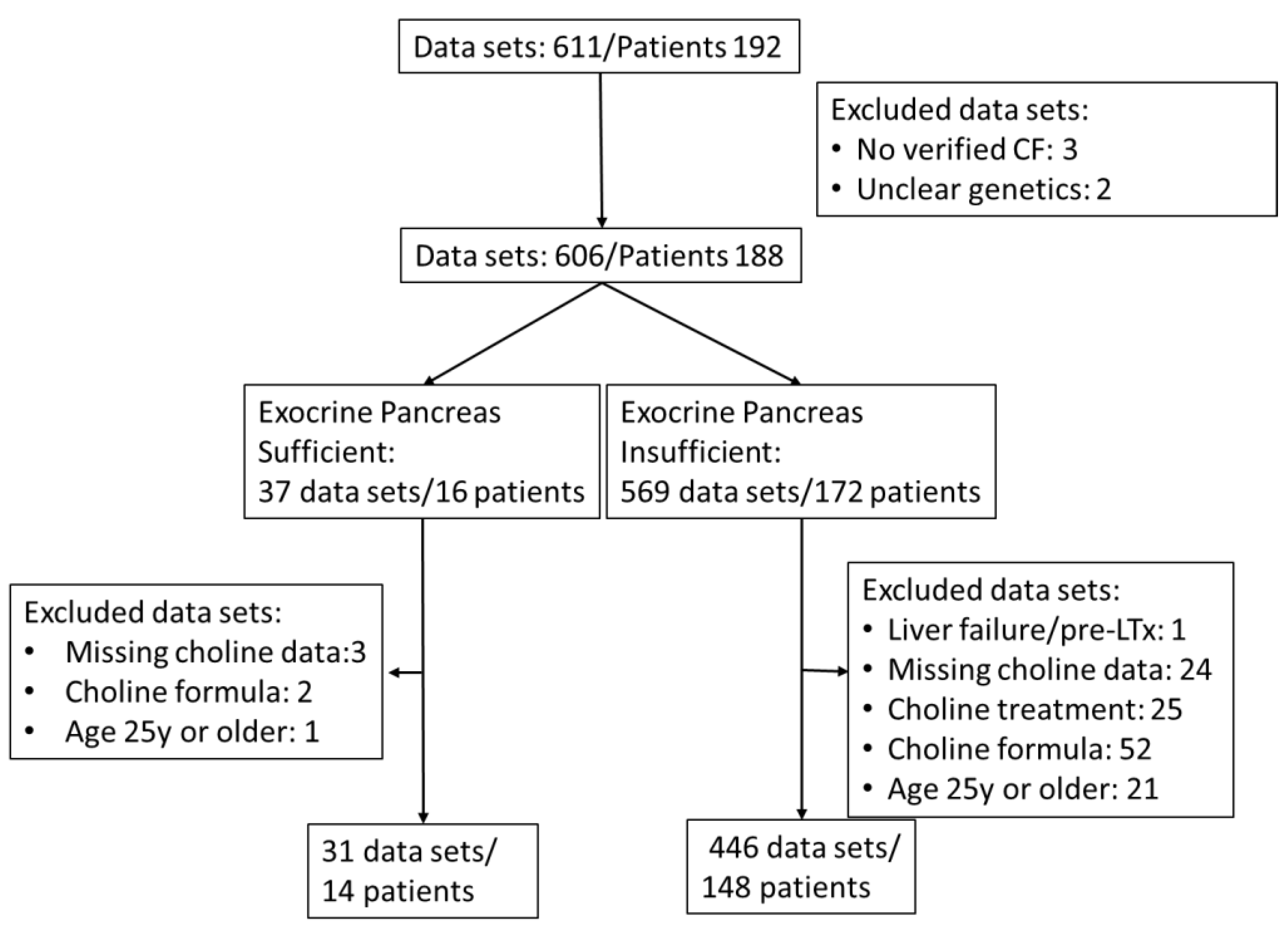
Figure 2.
Plasma concentrations of choline (A), betaine (B) and trimethylamine oxide (TMAO) (C), and the relations of betaine and TMAO versus plasma choline (D) in CF patients. Data are median values of individual CF patients with (N=148) and without (N=14) exocrine pancreas insufficiency. Abbreviations: , Spearman’s correlation coefficient; p, significance level.
Figure 2.
Plasma concentrations of choline (A), betaine (B) and trimethylamine oxide (TMAO) (C), and the relations of betaine and TMAO versus plasma choline (D) in CF patients. Data are median values of individual CF patients with (N=148) and without (N=14) exocrine pancreas insufficiency. Abbreviations: , Spearman’s correlation coefficient; p, significance level.
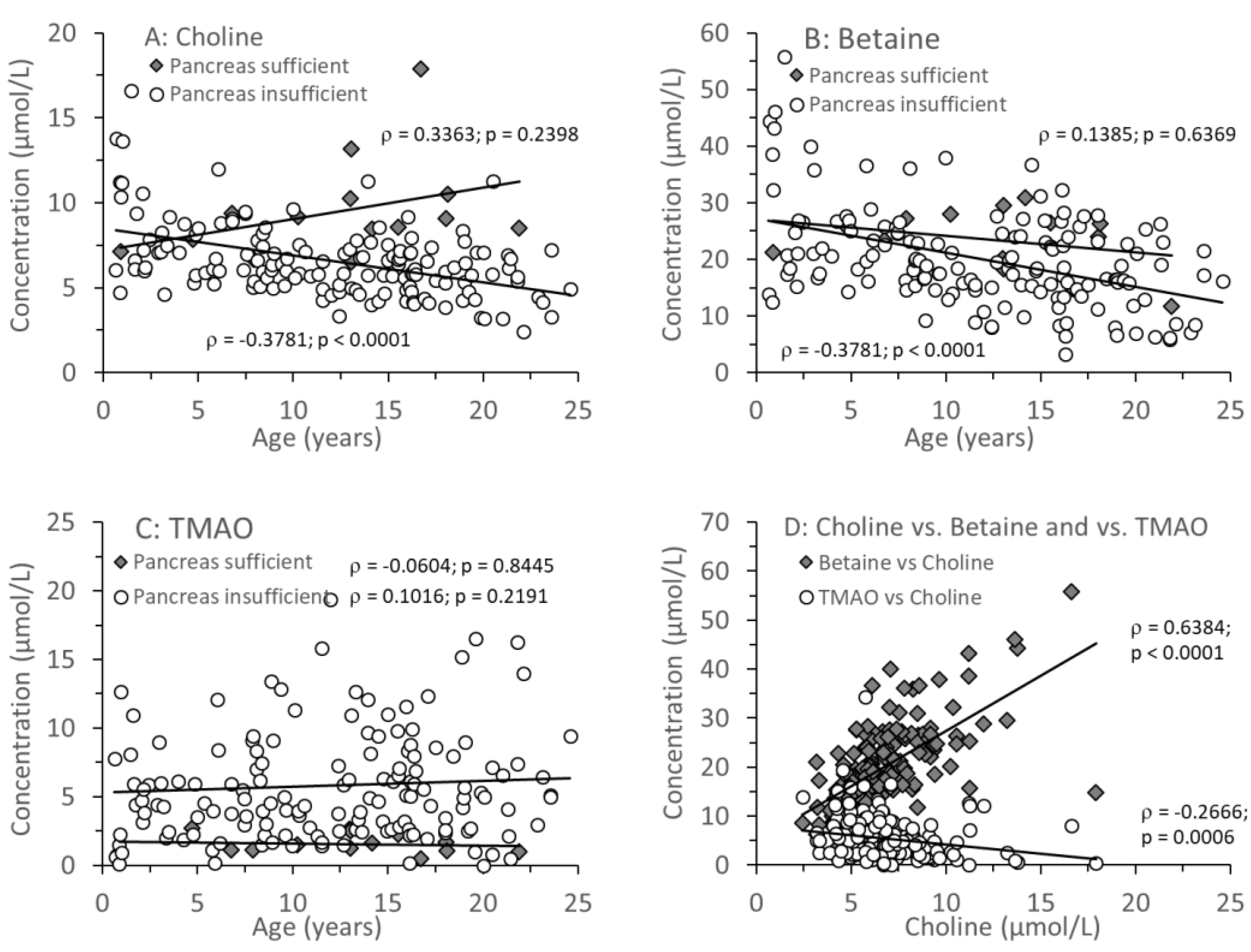
Figure 3.
Plasma concentrations of phosphatidylcholine (PC; A) and fractions of PC containing two saturated (B) or a mono- (C) or di-unsaturated (D) fatty acid residue. Data are median values of individual CF patients with (N=148) and without (N=14) exocrine pancreas insufficiency. Abbreviations: , Spearman correlation coefficient; p, significance level.
Figure 3.
Plasma concentrations of phosphatidylcholine (PC; A) and fractions of PC containing two saturated (B) or a mono- (C) or di-unsaturated (D) fatty acid residue. Data are median values of individual CF patients with (N=148) and without (N=14) exocrine pancreas insufficiency. Abbreviations: , Spearman correlation coefficient; p, significance level.
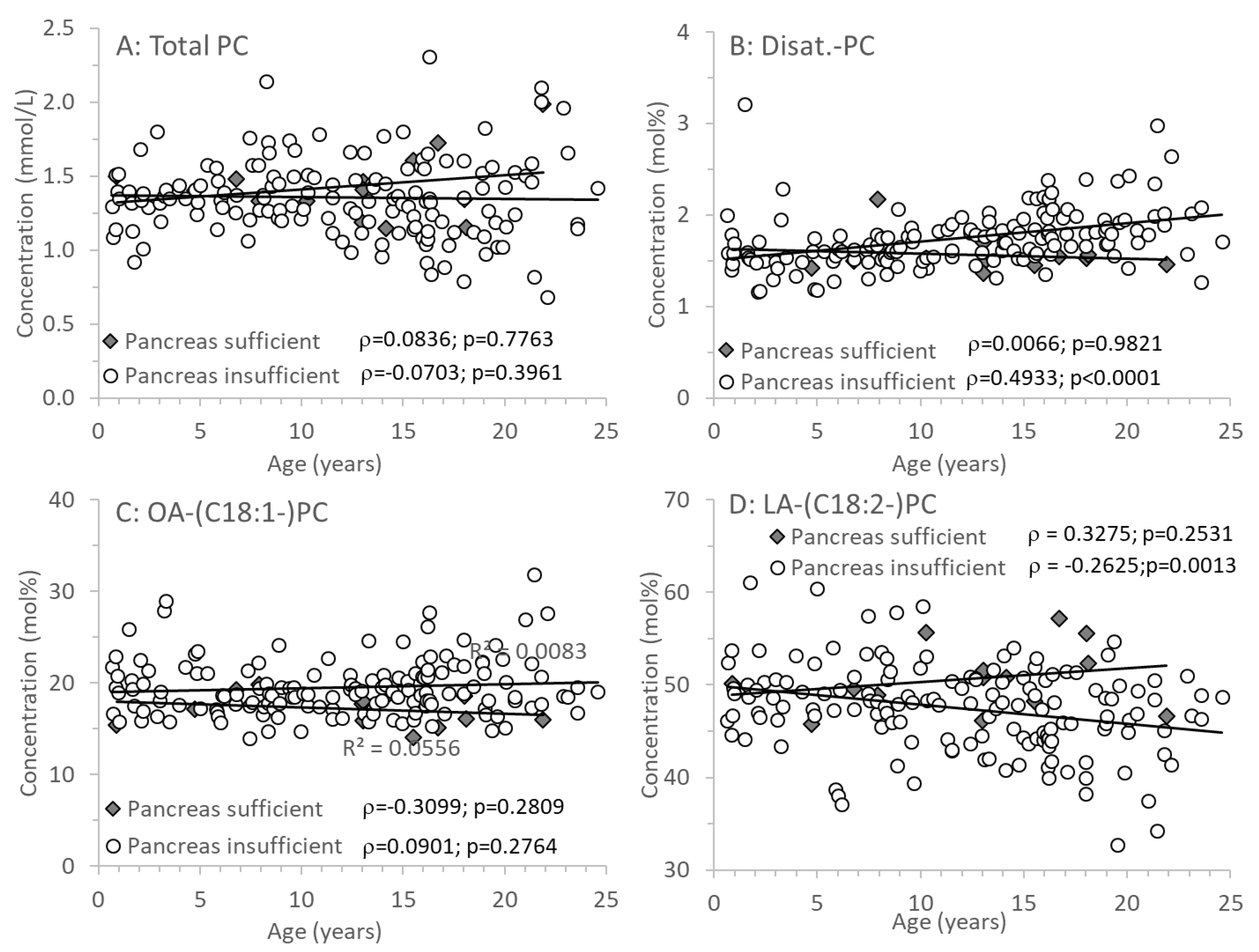
Figure 4.
PC containing long-chain polyunsaturated fatty acid (LC-PUFA) residues in plasma of CF patients. Fractions of PC species comprising an arachidonic acid (C20:4-PC; A), eicosapentaenoic acid (C20:5-PC; B) and docosahexaenoic acid (C22:6-PC, C) are shown. D shows the ratio of n-6 LC-PUFA (ARA-PC) to n-3 LC-PUFA-PC (EPA-PC+DHA-PC). Data are median values of individual CF patients with (N=148) and without (N=14) exocrine pancreas insufficiency. Abbreviations: , Spearman correlation coefficient; p, significance level.
Figure 4.
PC containing long-chain polyunsaturated fatty acid (LC-PUFA) residues in plasma of CF patients. Fractions of PC species comprising an arachidonic acid (C20:4-PC; A), eicosapentaenoic acid (C20:5-PC; B) and docosahexaenoic acid (C22:6-PC, C) are shown. D shows the ratio of n-6 LC-PUFA (ARA-PC) to n-3 LC-PUFA-PC (EPA-PC+DHA-PC). Data are median values of individual CF patients with (N=148) and without (N=14) exocrine pancreas insufficiency. Abbreviations: , Spearman correlation coefficient; p, significance level.
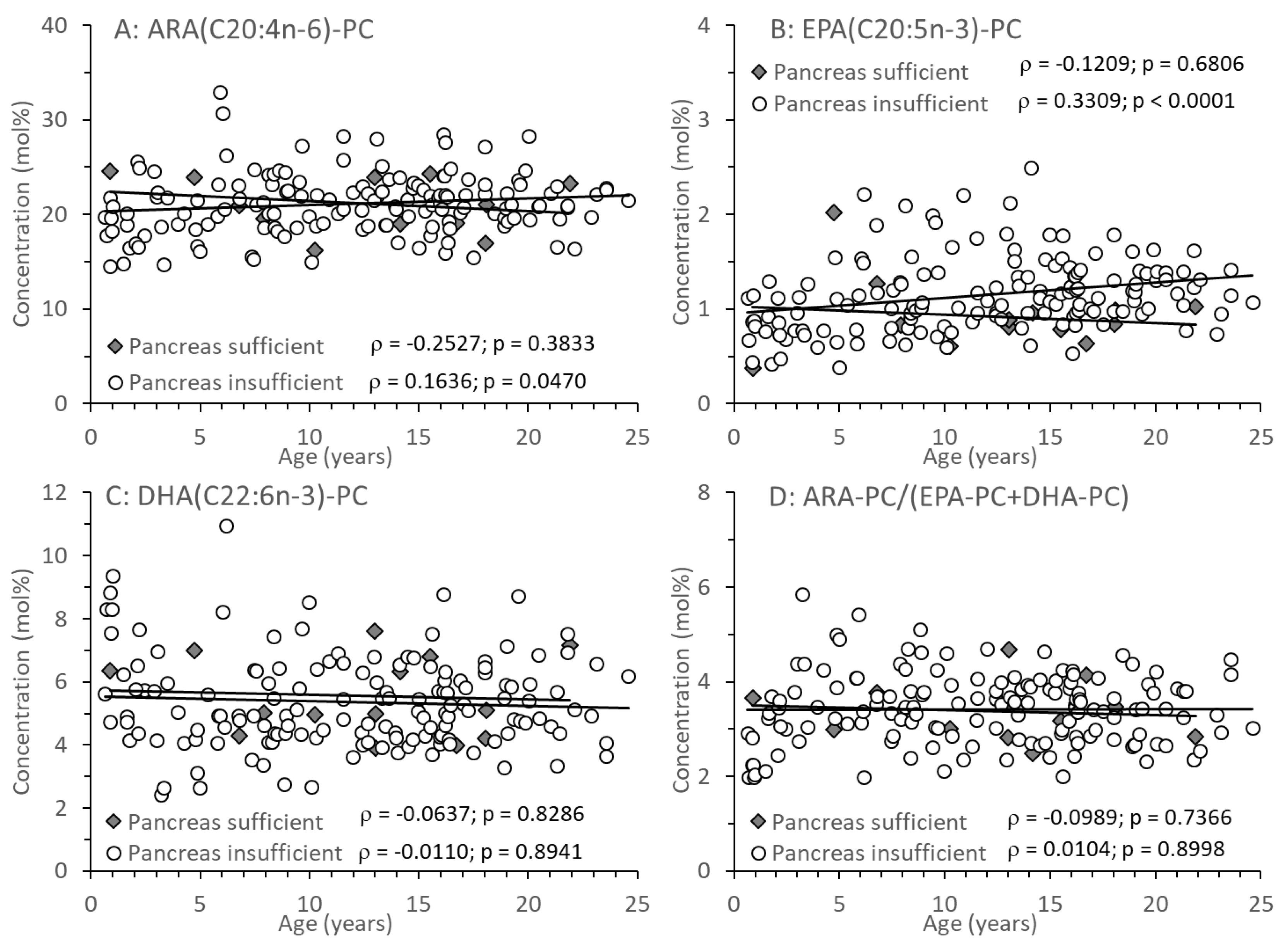
Table 2.
Biometric and clinical data of pancreas sufficient and insufficient CF patients.
| Parameter | Exocrine pancreatic sufficiency | Exocrine pancreatic insufficiency |
| Number of Patients (and Data Sets) | 14 (31) | 148 (446) |
| F508del compound heterocygous F508del homocygous Other |
4 0 10 |
78 53 17 |
| Median Age at CF Diagnosis (y) | 1.04 (0.92-5.34) | 0.27 (0.10-1.67) p=0.0009 |
| Age at Measurements (y) | 13.03 (8.50-16.42) [0.89-21.90] |
12.66 (7.22-16.36) [0.64-24.58] p=0.(8208 |
| Sex (male/female) | 8/6 | 68/80; p=0.4258 |
| Body weight (kg) | 44.9 (28.8-56.8) | 38.1 (22.2-54.6) p=0.3634 |
| Body length (cm) | 156.0 (131.2-161.5) (71-181) | 150,9 (117.0-165.0) (61-188) p=0.7324 |
| (BMI (kg/m2) | 18.2 (17.2-21.4) | 17.2 (15.8-20.1) p=0.2199 |
| BMI Percentile (%) | 57,5 (26.8-73.3) | 40.0 (18.6-64.8) p=0.3051 |
| Coagulation (Quick) [70–120] | 95 (88-103) | 89 (79-100) p=0.0808 |
| Thrombocytes (103/µL) | 281 (256-298) | 304 (260-355) p=0.2566 |
| Cholesterol (mg/dL) [130–190] | 137(126-152) | 128 (105-143) p=0.3272 |
| Triglycerides (mg/dL) [<200] | 68 (55-74) | 83 (62-117) p=0.1218 |
| Albumin (g/dL) [3.0-5.0] | 4.3 (4.1-4.4) | 4.0 (3.8-4.2) p=0.0016 |
| AST [<39 U/L] | 19 (16-32) | 25 (19-36) p=0.0879 |
| ALT [<39U/L] | 17 (13-20) | 25 (20-34) p=0.0021 |
| AP [130-400] | 225 (134-267) | 225(150-279) p=0.7037 |
| gGT [<30U/L] | 12 (10-13) | 12 (10-20) p=0.2901 |
| Lipid-soluble vitamins A (µmol/L) [1.1-2.7] |
1.50 (1.15-1.70) |
1.40 (1.20-1.60) p=0.6669 |
| E (µmol/L) [10-40] | 24.3 (20.7-24.6) | 20.4 (15.7-24.8) p=0.0518 |
| D (nmol/L) [50-175] | [130–40054.0 (46.6-59.0) | 51.0 (35.0-63.8) p=0.5444 |
| Lung function parameters ppFEV1 (%)* FVC (%)* FEF 25 (%)* FEF25-75 (%)* |
97 (88-101) 103 (87-107) 104 (75-125) 93 (74-117) |
96 (83-104) p=0.9754 100 (93-107) p=0.8155 80 (57-106) p=0.0450 84 (64-100) p=0.0843 |
Data are medians and interquartile ranges of 14 (pancreas sufficient) and 148 (pancreas insufficient) patients. Abbreviations: AST, serum aspartate aminotransferase; ALT, serum alanine aminotransferase; AP, alkaline phosphatase; gGT, gamma glutamyl transferase; ppFEV1, forced expiratory volume percent predicted; FVC, forced vital capacity; FEF25, forced expiratory flow at 25% of predicted FVC; FEF2575, forced expiratory flow at 25-75% of predicted FVC; *, median values of up to 6 independent measurements are indicated.
Table 3.
Choline compounds in plasma of study groups.
| Parameter | EPS | EPI | P-level |
| A: Choline and water-soluble derivatives | |||
| Choline (µmol/L) | 8.8 (8.0-10.0) | 6.1 (5.2-7.4) | < 0.0001 |
| Betaine (µmol/L) | 24.9 (21.4-27.1) | 18.6 (14.8-24.6) | 0.0287 |
| Choline + Betaine | 33.3 (30.9-35.6) | 25.3 (20.5-31.9) | 0.0020 |
| TMAO (µmol/L) | 1.4 (1.1-2.1) | 4.9 (2.6-8.0) | < 0.0001 |
| Betaine/Choline | 2.64 (2.36-3.31) | 3.00 (2.473.66) | 0.0772 |
| TMAO/Choline | 0.18 (0.13-0.20) | 0.71 (0.44-1.35) | < 0.0001 |
| B: Phospholipids | |||
| Phosphatidylcholine (PC) (mmol/L) | 1.41 (1.33-1.50) | 1.35 (1.19-1.51) | 0.2927 |
| Lyso-PC (% of PC) | 2.57 (1.90-3.41) | 2.56 (2.01-3.09) | 0.7318 |
| SPH (% of PC) | 25.2 (21.1-29.3) | 23.2 (20.6-26.2) | 0.2344 |
| Ceramides (% of PC) | 0.27 (0.17-0.31) | 0.24 (0.19-0.30) | 0.6938 |
| C: PC sub-groups | |||
| Disaturated PC | 1.53 (1.47-1.57) | 1.70 (1.54-1.88) | 0.0056 |
| Oleyl (C18:1)-PC | 17.4 (15.9-18.6) | 18.9 (17.4-21.1) | 0.0031 |
| Linoleoyl (C18:2)-PC | 50.5 (48.4-52.1) | 47.9 (44.5-50.5) | 0.0121 |
| Arachidonoyl (C20:4)-PC | 21.3 (19.2-23.8) | 20.9 (18.9-23.0) | 0.7543 |
| Eicosapentaenoyl (C20:5)-PC | 0.86 (0.79-0.98) | 1.11 (0.88-1.37) | 0.0119 |
| Docosahexaenoyl: C22:6-PC | 5.06 (4.46-6.69) | 5.02 (4.34-6.29) | 0.4949 |
| Other PC* | 2.55 (2.37-2.96) | 2.43 (2.88-4.22) | 0.0005 |
Data show medians and interquartile ranges of parameters in CF patients with exocrine pancreas sufficiency (EPS) compared to exocrine pancreas insufficient patients (EPI). Abbreviatins: PC, phosphatidylcholine; SPH, sphingomyelin; TMAO trimethylamine oxide.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



